 Junxing Yang1†
Junxing Yang1† Lin Zhou
Lin Zhou Qingjiong Zhang
Qingjiong Zhang- 1State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China
- 2Department of Ophthalmology, West China Hospital, Sichuan University, Chengdu, China
Purpose: RPGR is the most common cause of X-linked retinitis pigmentosa (RP), of which female carriers are also frequently affected. The aim of the current study was to explore the RPGR variation spectrum and associated phenotype based on the data from our lab and previous studies.
Methods: Variants in RPGR were selected from exome sequencing data of 7,092 probands with different eye conditions. The probands and their available family members underwent comprehensive ocular examinations. Similar data were collected from previous reports through searches in PubMed, Web of Science, and Google Scholar. Systematic analyses of genotypes, phenotypes and their correlations were performed.
Results: A total of 46 likely pathogenic variants, including nine missense and one in-frame variants in RCC1-like domain and 36 truncation variants, in RPGR were detected in 62 unrelated families in our in-house cohort. In addition, a total of 585 variants, including 491 (83.9%) truncation variants, were identified from the literature. Systematic analysis of variants from our in-house dataset, literature, and gnomAD suggested that most of the pathogenic variants of RPGR were truncation variants while pathogenic missense and in-frame variants were enriched in the RCC1-like domain. Phenotypic variations were present between males and female carriers, including more severe refractive error but better best corrected visual acuity (BCVA) in female carriers than those in males. The male patients showed a significant reduction of BCVA with increase of age and males with exon1-14 variants presented a better BCVA than those with ORF15 variants. For female carriers, the BCVA also showed significant reduction with increase of age, but BCVA in females with exon1-14 variants was not significant difference compared with those with ORF15 variants.
Conclusion: Most pathogenic variants of RPGR are truncations. Missense and in-frame variants located outside of the RCC1-like domain might be benign and the pathogenicity criteria for these variants should be considered with greater caution. The BCVA and refractive error are different between males and female carriers. Increase of age and location of variants in ORF15 contribute to the reduction of BCVA in males. These results are valuable for understanding genotypes and phenotypes of RPGR.
Introduction
Retinitis pigmentosa (RP) is a common type of inherited retinal degenerations (IRD) characterized by impaired dark adaptation and night blindness, progressive visual field defects and pigmentary retinopathy, affecting approximately one in 3,500–4,000 people worldwide (Berger et al., 2010; Traboulsi, 2010; Sundaram et al., 2012; Zhang, 2016). RP can be inherited as an autosomal dominant, autosomal recessive, or X-linked trait, with these categories accounting for approximately 30–40%, 50–60%, and 5–15% of RP patients, respectively (Bunker et al., 1984; Grondahl, 1987; Hartong et al., 2006).
X-linked RP is one of the most severe forms of human retinal degeneration (Bird, 1975). Affected males usually suffer nyctalopia and severe and rapid progressive loss of peripheral vision with an early onset, followed by progressive central visual loss during the second to fourth decades of life, while female carriers may present a wide range of phenotypes, ranging from asymptomatic to severe phenotype (Bird, 1975; Fishman et al., 1986; Banin et al., 2007). Additionally, the phenotype of X-linked RP generally shows great phenotypic heterogeneity, including interfamily heterogeneity, in terms of the age of onset, clinical severity, rate of progression, and prevailing damage to rods and cones (Fahim et al., 2011). Variants in retinitis pigmentosa GTPase regulator (RPGR, OMIM 312610) account for 70–80% (Sharon et al., 2003; Pelletier et al., 2007; Shu et al., 2007) of X-linked RP cases. This protein localizes to the connecting cilium in photoreceptors and is thought to play a role in protein transport (Roepman et al., 2000; Hong et al., 2003).
In 2007, a study provided an overview of RPGR genotypes and the associated phenotypic variation (Shu et al., 2007). However, the widespread application of next-generation sequencing (NGS) in recent years has increased the number of variants identified in RPGR and expanded the known phenotypic spectrum of patients. Further comprehensive analysis of RPGR genotype–phenotype relationships would be expected. In addition, most of the patients previously reported to show variants in RPGR were recruited from America or Europe.
In this study, we performed a summary of the genotypes and corresponding phenotypes in RPGR from our database and the literature. The pathogenicity of the variants in RPGR and genotype–phenotype correlations were further assessed and summarized.
Materials and Methods
Samples
In an ongoing study of genetic eye diseases, we recruited 7092 probands with different eye conditions from the pediatric and Genetic Eye Clinic of the Zhongshan Ophthalmic Center, and we collected the available clinical data of the probands and their available family members with RPGR variations. This study was performed in accordance with the Declaration of Helsinki, and written informed consent was obtained from participating individuals or their guardians. Our study was approved by the Institutional Review Board of Zhong Shan Ophthalmic Center. All patients included in the study underwent exome sequencing [whole-exome sequencing (WES) and targeted exome sequencing (TES)]. The rare variants were defined as variants with a minor allelic frequency of less than 0.01 in general population from gnomAD database and patients with likely pathogenic variants were subsequently discriminated from the rare variants of RPGR in this study. Genotype–phenotype correlation was investigated by statistical analyses on different groups of patients with likely pathogenic variants of RPGR according to the variants in certain regions. In addition, patients with rare variants in RPGR were summarized based on our data (Tables 1, 2).
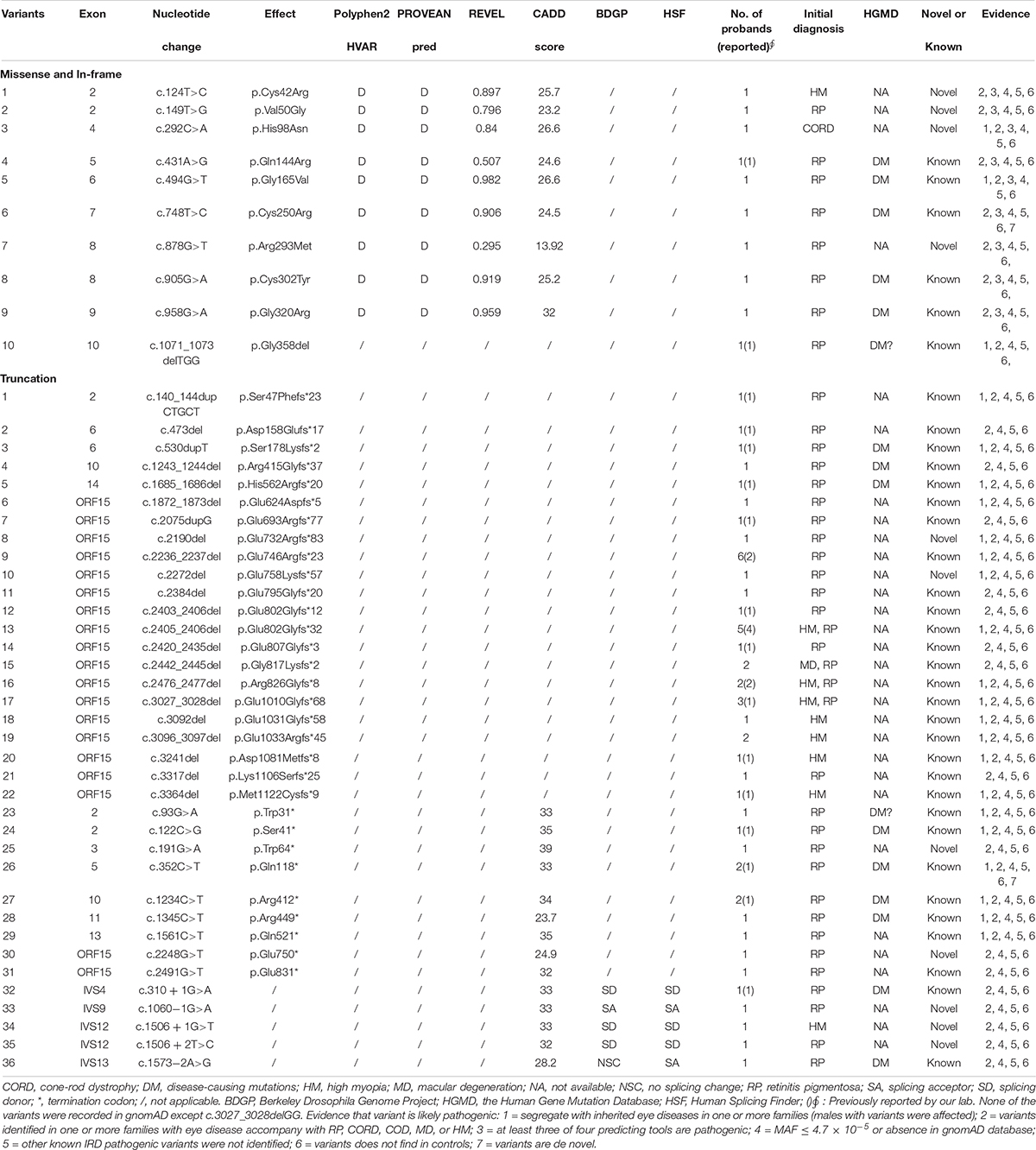
Table 1. 46 likely pathogenic variants in RPGR from 62 unrelated families (based on NM_001034853).
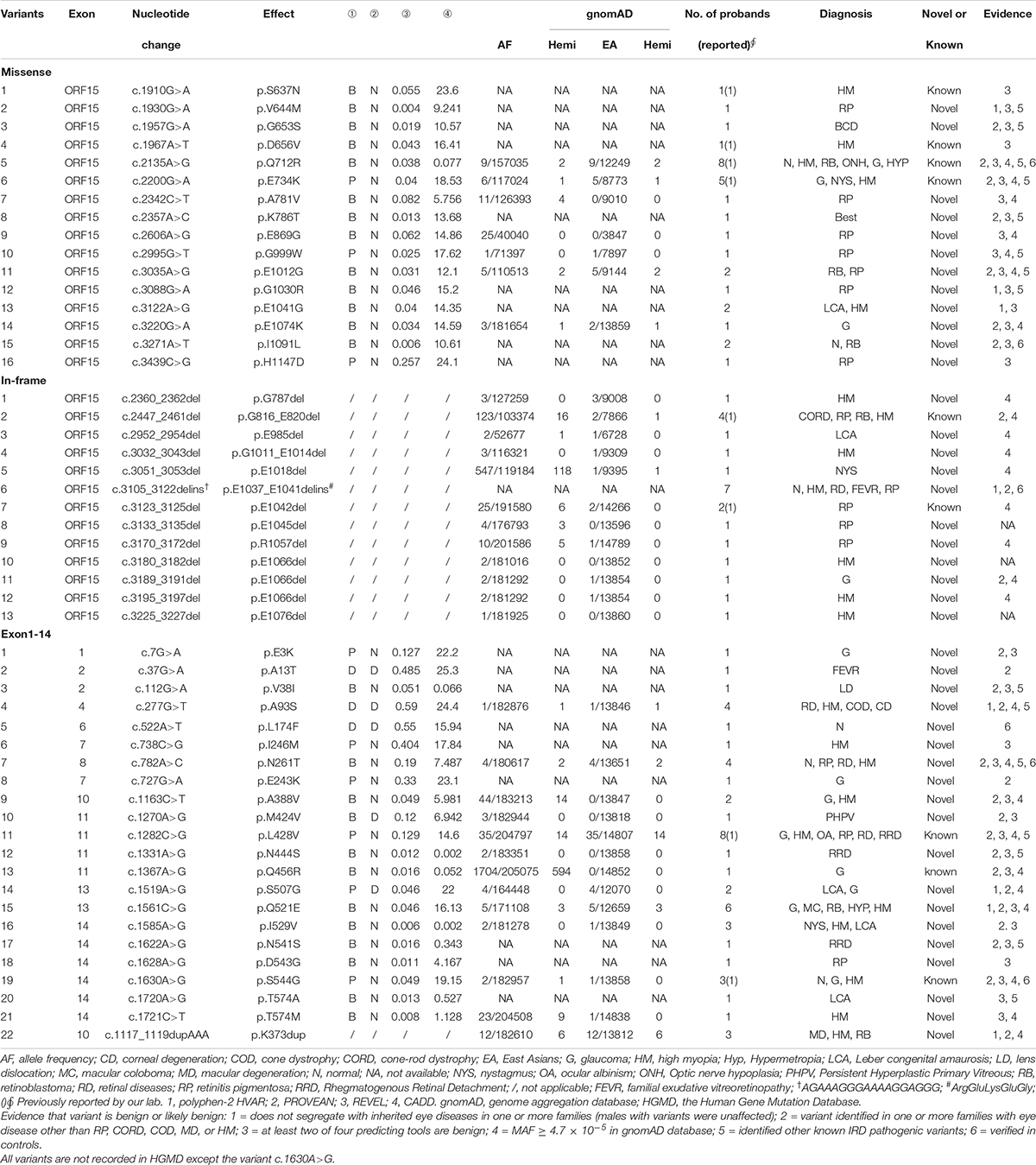
Table 2. 51 benign or likely benign variants in RPGR from 101 unrelated families (based on NM_001034853).
Exome Sequencing
Exome sequencing, including WES and TES, was conducted in the patients included in our study. Whole-exome sequencing was performed on 5,307 probands using a commercial service as described in our previous study (Li et al., 2015). Genomic DNA from the probands was sheared and fragments of an approximate 150 bp were selected. Exome was captured by an Agilent SureSelect Human All Exon Enrichment Kit (Agilent, Santa Clara, CA, United States). Library quality was assessed using an Agilent 2100 Bioanalyzer and were then sequenced on the Illumina HiSeq platform (Illumina, San Diego, CA, United States) with an average depth of at least 125-fold. After filtering out low quality reads, and remaining clean data was verified by aligning the sequencing with the UCSC hg191 reference using the Burrows-Wheeler Aligner (BWA2). Variants were detected by SAMTOOLS3 and were annotated and predicted by SnpEff4, ANNOVAR5, and dbNSFP6, respectively.
Targeted-exome sequencing was conducted on 1,785 probands by our lab as described in our previous study (Wang et al., 2019). Approximately 200 bp fragments were obtained from genomic DNA using a Bioruptor Plus (Diagenode, Liege, Belgium). A paired-end library was prepared using a KAPA HTP Library Preparation kit (Roche, Basel, Switzerland). Targeted exome was captured using a designed NimbleGen SeqCap EZ Prime Choice kit (Roche, Basel, Switzerland). Library quality was assessed using an Agilent 2100 Bioanalyzer and were then sequenced on an Illumina Nextseq550 Analyzer using the Illumina NextSeq550 Mild output v2 kit (150 PE) (Illumina, San Diego, CA, United States) with an average depth of 250-fold. Variant calling and annotation were analyzed using the StrandNGS software (Karnataka, India) according to the manufacturer’s instructions. The UCSC Genome Browser on Human hg19 Assembly was used as an alignment reference. The dbNSFP was used for predictions of missense variants. The list of 126 target genes, including RPGR, in TES has been described in our previous study (Wang et al., 2019). Variants in RPGR identified through WES and TES were selected and filtered via multistep bioinformatics analyses as previously reported (Xu et al., 2014; Li et al., 2015; Sun et al., 2015; Zhou L. et al., 2018). Additionally, we used CADD7 and REVEL8 to further predict the severity of the missense variants in RPGR. Data from the Genome Aggregation Database (gnomAD9) and Human Genome Mutation Database (HGMD10) were included as references for evaluating the pathogenicity of the variants in RPGR. Selected remaining variants were verified by Sanger sequencing. The pedigrees and sequence diagrams of potential likely pathogenic variants are shown in Supplementary Figures 1, 2.
Phenotype Analysis in Our Lab
Probands and available family members with variants in RPGR were recruited for further comprehensive ocular examinations. All of the examinations were performed by the same experienced team of ophthalmologists. A detailed family and ophthalmic history were obtained. The comprehensive ocular examinations included best corrected visual acuity (BCVA), refractive error (RE), and spectral domain-optical coherence tomography (SD-OCT).
Refractive error was measured using an autorefractometer (Topcon KR-8000, Paramus, NJ, United States) after mydriasis with tropicamide (Mydrin-P, Santen Pharmaceutical, Japan). An optical biometer (IOL master V5.0, Carl Zeiss Meditec AG, Germany) was used to detect the ocular biometric axial length. Full-field electroretinogram (ERG) responses were recorded in patients in accordance with the standards of the International Society for Clinical Electrophysiology of Vision for evaluating retinal disorders, using an Espion ERG system (Diagnosys LLC, United States). Optical coherence tomography of the macular and optic disks was performed via SD-OCT (Optovue, Inc., United States).
Literature Review of RPGR Variants and Ophthalmologic Outcomes
The variants and clinical data of patients with RPGR were obtained by searching the literature for the keyword RPGR in three databases: PubMed11, Web of Science12, and Google Scholar13 (Meindl et al., 1996; Roepman et al., 1996; Andreasson et al., 1997, 2003; Buraczynska et al., 1997; Fujita et al., 1997; Jacobson et al., 1997; Weleber et al., 1997; Bauer et al., 1998; Fishman et al., 1998a,b; Miano et al., 1998, 1999; Dry et al., 1999; Flaxel et al., 1999; Rosenberg et al., 1999; Zito et al., 1999, 2000, 2003; Liu et al., 2000, 2002; Vervoort et al., 2000; Guevara-Fujita et al., 2001; Yokoyama et al., 2001; Zhao et al., 2001, 2020; Aguirre et al., 2002; Ayyagari et al., 2002; Breuer et al., 2002; Demirci et al., 2002, 2004, 2005, 2006; Pusch et al., 2002; Rozet et al., 2002; Yang et al., 2002, 2014; Bader et al., 2003; Barnes et al., 2003; Iannaccone et al., 2003, 2008; Koenekoop et al., 2003; Lorenz et al., 2003; Rebello et al., 2003; Sharon et al., 2003; Wegscheider et al., 2004; Adamian et al., 2005; Ebenezer et al., 2005; Jin et al., 2005, 2006, 2007a,b, 2008; Wang et al., 2005, 2015; Chakarova et al., 2006; Garcia-Hoyos et al., 2006; Moore et al., 2006; Sullivan et al., 2006, 2013; Aleman et al., 2007; Banin et al., 2007; Chang et al., 2007; Duncan et al., 2007; Neidhardt et al., 2007, 2008; Pelletier et al., 2007; Prokisch et al., 2007; Sandberg et al., 2007; Shu et al., 2007; Walia et al., 2008; Al-Maskari et al., 2009; Ruddle et al., 2009; Ji et al., 2010; Sheng et al., 2010; Wu et al., 2010; Bowne et al., 2011; Fahim et al., 2011, 2020; Glaus et al., 2011; Li N. et al., 2011; Li Z.L. et al., 2011; Liskova et al., 2011; Thiadens et al., 2011; Branham et al., 2012, 2018; O’Sullivan et al., 2012; Acton et al., 2013; Bukowy-Bieryllo et al., 2013; Churchill et al., 2013; Eisenberger et al., 2013; Huang et al., 2013, 2014, 2015a,b, 2019; Kousal et al., 2013, 2014; Liu and Zack, 2013; Pyo Park et al., 2013; Zahid et al., 2013; Glockle et al., 2014; Gonzalez-del Pozo et al., 2014; Hu et al., 2014; Oishi et al., 2014; Pierrottet et al., 2014; Wang F. et al., 2014; Wang J. et al., 2014; Xu et al., 2014, 2019; Almoguera et al., 2015; Chassine et al., 2015; Consugar et al., 2015; Fernandez-San Jose et al., 2015; Ge et al., 2015; Kastner et al., 2015; Ogino et al., 2015; Sharon and Banin, 2015; Sun et al., 2015; Haddad et al., 2016; Li et al., 2016; Parmeggiani et al., 2016; Tiwari et al., 2016; Bellingrath et al., 2017; Hendriks et al., 2017; Kalitzeos et al., 2017; Stone et al., 2017; Tee et al., 2017; Birtel et al., 2018a,b; Chiang et al., 2018; Nanda et al., 2018; Talib et al., 2018, 2019; Wawrocka et al., 2018; Zhou L. et al., 2018; Zhou Q. et al., 2018; Gill et al., 2019; Koyanagi et al., 2019; Mawatari et al., 2019, 2020; Sanchez Tocino et al., 2019; Tang et al., 2019; Zhang Z. et al., 2019; Dan et al., 2020; Foote et al., 2020; Menghini et al., 2020; Nguyen et al., 2020; Rodriguez-Munoz et al., 2020; Salvetti et al., 2020; Zampaglione et al., 2020) on July 01, 2020. The papers were limited to English-language reports of definitive variants in RPGR. Variant descriptions based on the work of Meindl et al. (1996) were converted to descriptions based on NM_001034853. Variants in RPGR previously reported to be likely pathogenic were summarized in Supplementary Table 1 based on the literature.
Clinical data were collected to perform further comparisons between genders, ages, locations and variation types. Spherical equivalent refraction (SER) was calculated by adding spherical refraction to half the cylindrical refraction.
Statistical Analysis
Analyses were performed using R software and SPSS version 23. Logistic regression was used to screen out the factors influencing BCVA in males and females. Median (IQR, interquartile range) were used for continuous parameters. Mann–Whitney U test was used to compare the BCVA and refractive error among groups, namely (1) patients with variants in exon1-14; (2) patients with variants in ORF15; (3) patients with variants in RCC1-like domain; (4) patients with missense and in-frame variants; (5) patients with truncation variants. The corrected significant P-value for this study should be less than 0.017 (α = 0.05/3) according to the Bonferroni method.
Results
Identification of RPGR Variants in 7,092 Probands With Different Eye Conditions in Our Lab
A total of 121 variants, including 15 polymorphisms, eight 3′UTR variants, one synonymous variant and 97 rare variants, were detected in 7,092 probands. Of the 97 rare variants, 46 likely pathogenic variants (11 novels) and 51 likely benign variants were identified. Among the 46 likely pathogenic variants, nine missense variants, one in-frame variant and 17 truncation variants were located in exon1-14, and the remaining 19 truncation variants were located in ORF15 (Table 1). The 46 likely pathogenic variants were identified in 62 families, of which truncation variants were identified in 52 (83.9%, 52/62), while missense and in-frame variants were identified in nine (14.5%, 9/62) and one (1.6%, 1/62) family, respectively. Of the other 51 likely benign variants, 21 missense variants and one in-frame variant, were identified in exon1-14, while 16 missense and 13 in-frame variants were detected in ORF15 (Table 2).
Review of RPGR Genotypes From Our Lab and Previous Literature
A total of 585 variants have been reported in previous literature, including 491 truncations, 84 missenses, and 10 in-frame variants. Of the 94 missense and in-frame variants, 81 were located in the RCC1-like domain, while the remaining 13 were located outside the domain (Supplementary Table 1). A total of 585 previously reported variants, combining 46 likely pathogenic variants with our laboratory data, a total of 606 variants were analyzed (25 variants were repetitive).
Pathogenicity Evaluation of Missense and In-Frame Variants Located Outside of the RCC1
A total of 57 missense and in-frame variants were located outside of the RCC1 region, including 45 variants from our in-house cohort and 13 from literature were identified (one variant was repetitive) (Table 2 and Supplementary Table 1). The following lines of evidence suggested that these variants in RPGR might not be disease causing. (1) Missense and in-frame variants were significantly enriched outside of the RCC1 region according to the gnomAD database, and the frequency was obviously high (Figure 1). (2) Most of these variants were identified in one or more probands with different eye conditions other than RP or closely relative early onset high myopia (HM), cone-rod dystrophy (CORD), cone-dystrophy (COD), or macular degeneration (MD) (Table 2). (3) All but two missense variants (c.37G > A and c.1519A > G) located outside of the RCC1 were predicted to be benign by at least two of four prediction tools (90% cutoff score: 0.29 in REVEL and 21.5 in CADD) (Table 2). (4) A few patients showed variants in other known IRD genes, and some variants were verified in unaffected controls. (5) Segregation analysis contributed further evidence that missense and in-frame variants in non-RCC1 regions are not disease causing, and the corresponding pedigrees are shown in Supplementary Figure 3. (6) A previous study reported frequent in-frame deletions of 3–36 bp in healthy controls, suggesting that in-frame variants are benign (Karra et al., 2006). In addition, Zhang Q. et al. (2019) developed an in vitro assay illustrating that some variations located outside of the RCC1 region might be non-disease-causing polymorphisms.
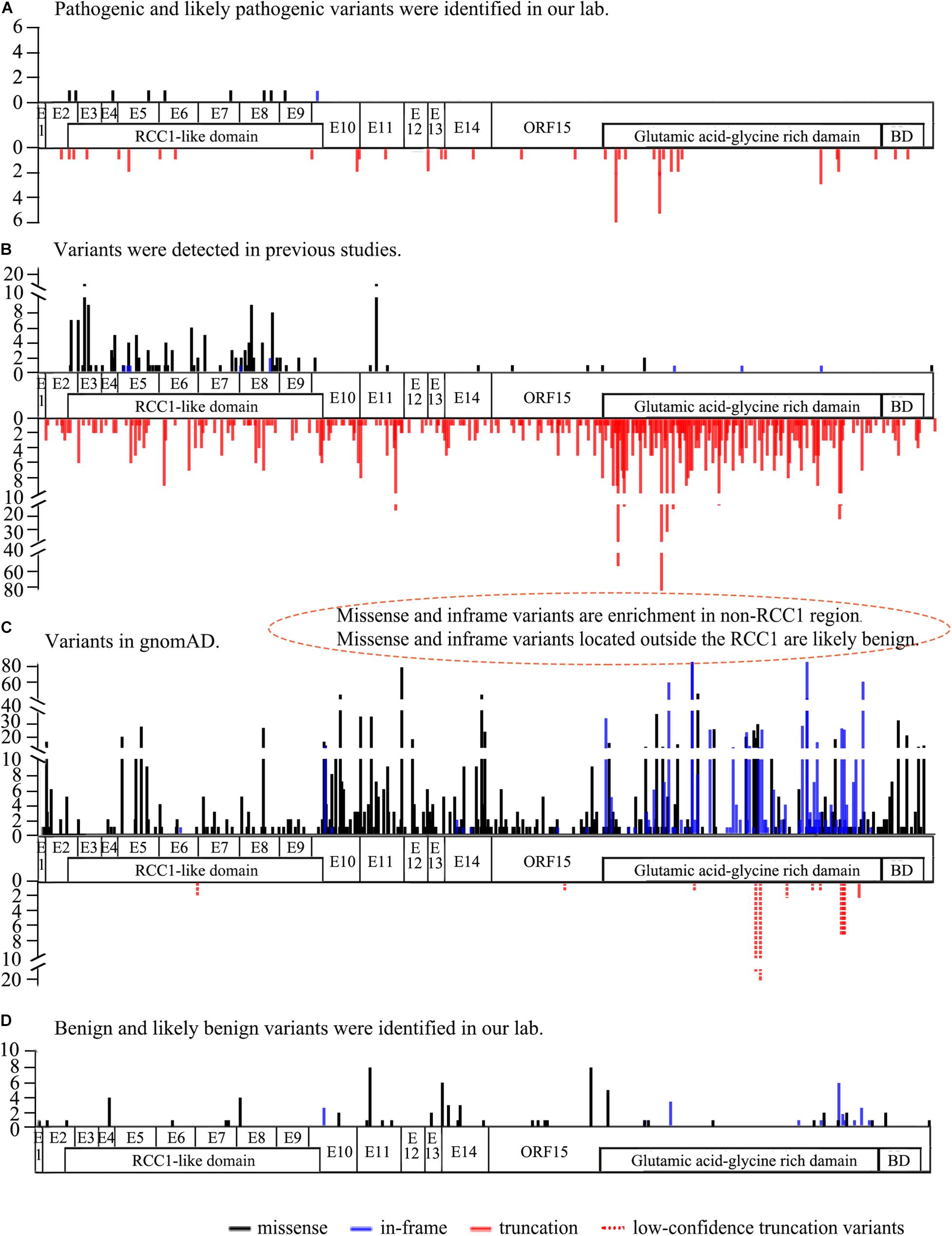
Figure 1. The frequency and location of the variants from our lab, previous studies, and the gnomAD database (Ref. NM_001034853). (A) The frequency and location of pathogenic and likely pathogenic RPGR variants detected in our lab. Missense and in-frame variants are distributed above the structure, and truncation variants are shown below the structure. (B) The frequency and location of RPGR variants identified in previous studies. Missense and in-frame variants enriched in the RCC1-like domain are shown above the structure, and truncation variants are indicated below the structure. Gross deletion variants are not shown here. (C) The frequency and location of RPGR variants from the gnomAD database. Missense and in-frame variants are significantly enriched in the non-RCC1-like domain above the structure. Truncation variants in all coding regions below the structure. Of the 11 truncation variants, 10 were low confidence truncations (dotted line). (D) The frequency and location of benign and likely benign RPGR variants identified in our lab. The white regions represent the coding regions. RCC1-like domain: p.38∼367, BD: basic domain p.1086-1139, Glutamic acid-glycine-rich domain: p.728∼1084.
RPGR-Associated Phenotype Analysis of Based on Our Data and the Literature
BCVA in Patients With RPGR Variations
The clinical data of the probands and available families with pathogenic variants from our database and previous studies are summarized in Supplementary Tables 2, 3. The statistical results table were shown in Supplementary Table 4. BCVA showed a significant reduction with increase of age in both males and females (r = 0.479 and r = 0.216, respectively) (Figure 2C). Better BCVA in female carriers (0.10 [0.00, 0.30] logMAR) than in male patients (0.40 [0.17, 0.60] logMAR) (P = 7.41E-25) (Figure 2A). Logistic regression was used to screen out the factors influencing BCVA in males and females, and the receiver operating characteristic (ROC) curves suggested that our model showed high sensitivity and specificity in distinguishing the different degrees of BCVA (Figure 3D). For males, the variation type was not associated with BCVA (P = 0.183) (Figure 3C). The BCVA of male patients with variants in exon1-14 (0.36 [0.17, 0.48] logMAR) was significantly better than that of male patients with variants in ORF15 (0.40 [0.20, 0.70] logMAR) (P = 0.005) after age adjustment, however, the comparison between RCC1 and ORF15 was no significant difference (P = 0.048) (Figures 3A,B). BCVA was not associated with location or variation type in female carriers (all P > 0.05, respectively) (Supplementary Figures 5A–C).
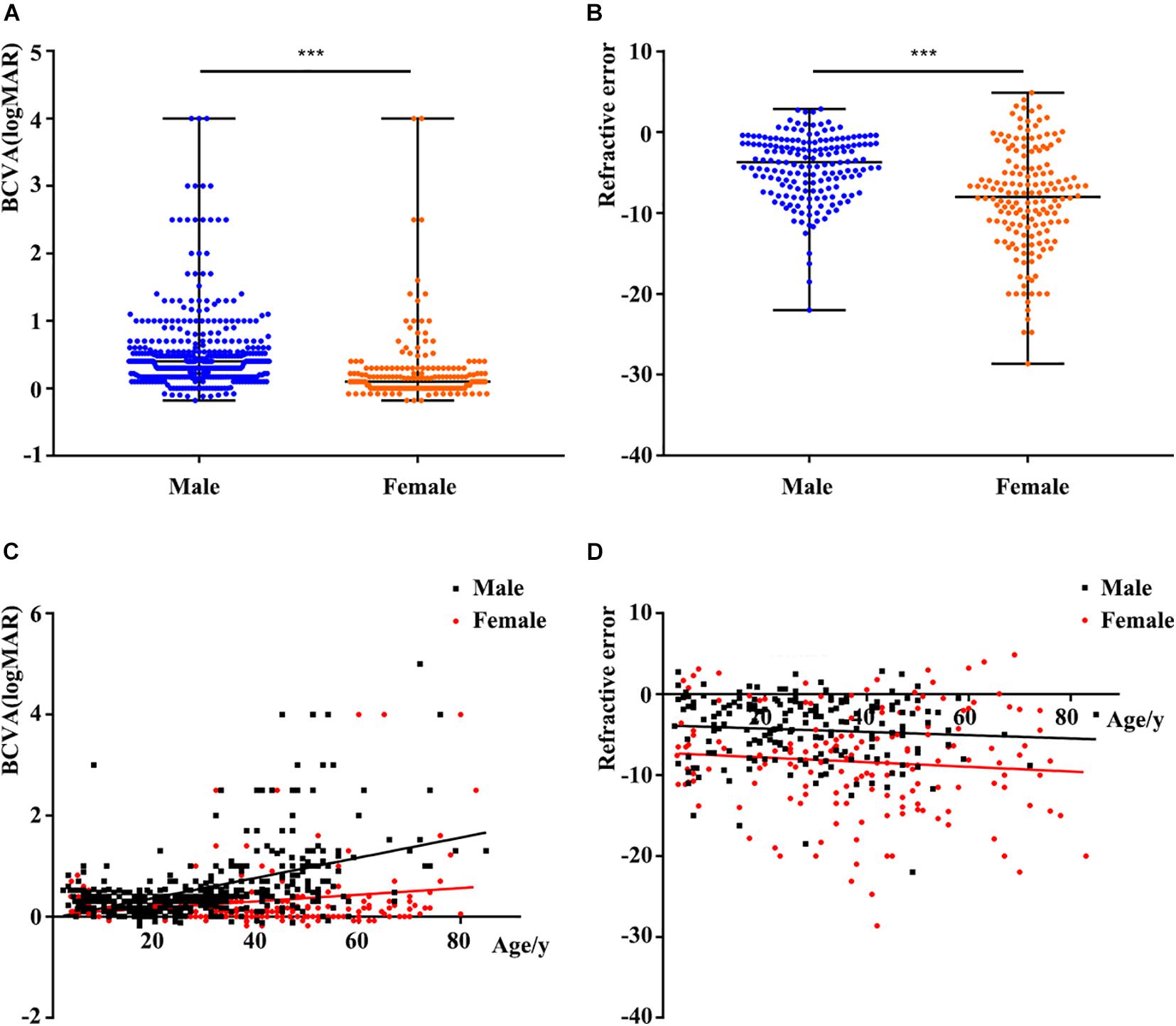
Figure 2. Comparison of phenotypes according to different factors. (A) Comparison of logMAR BCVA between males and females. The BCVA of female carriers was better than that of male patients. (B) Comparison of refractive error (RE) between males and females. Spherical equivalent refraction represents the severity of RE. The RE of female carriers was more serious than that of males. (C) Scatterplots of logMAR BCVA and age, the two fitted lines correspond to male (black) and female (red) patients. A significant reduction of BCVA with increase of age in both males and females. (D) Scatterplots of RE and age, the two fitted lines correspond to male (black) and female (red) patients. The trends of the two lines are basically smooth. BCVA, best corrected visual acuity. ∗∗∗, P value less than 0.001.
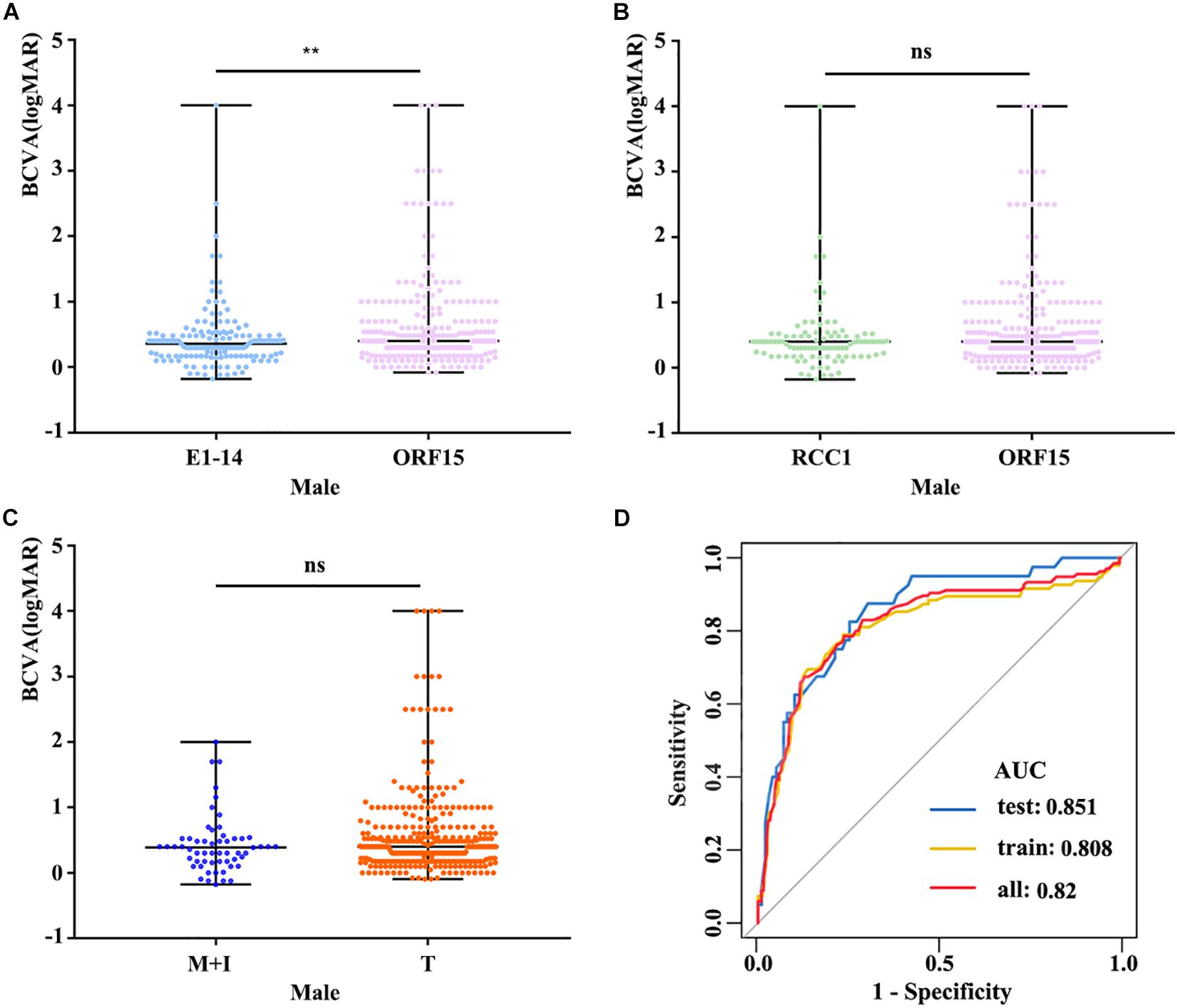
Figure 3. (A) The logMAR BCVA of male patients with variants in exon1-14 and ORF15 showed that patients with variants in exon1-14 have a better visual acuity. (B) Patients with variants in RCC1-like domain were no significant difference compared to those in ORF15. (C) Comparison of logMAR BCVA between M + I and T, there was no significant difference in variation type. (D) ROC curves suggested that our model shows high sensitivity and specificity in distinguishing different degrees of BCVA. The datasets used for AUC analysis were from available males’ data and were randomly divided into two independent datasets (training and test datasets) by the R-software. BCVA, best corrected visual acuity; E1-14, exon1-exon14; RCC1, RCC1-like domain; M + I, missense and in-frame; T, truncation. ns, no statistical significance; ∗∗, P value less than 0.01.
Refractive Error in Patients With RPGR Variations
Spherical equivalent refraction was used to assess the severity of the RE. The percentage of female carriers with high myopia was significantly greater than that of males (109/165 and 51/179, respectively). Females with variants in RPGR showed a more serious of SER than males (−8.00 [−12.00, −4.19] in female carriers and −3.72 [−6.99, −1.28] in male patients, P = 5.46E-10) (Figure 2B). Logistic regression showed that RE was unrelated to age, location or variation type in both male patients and female carriers (all P > 0.05) (Figure 2D and Supplementary Figures 4, 5D–F).
In addition, the fundus changes vary widely among patients with RPGR variants, including gray-white fundal spots, tessellated fundus, retinal degeneration to macular degeneration in males and female carriers.
Discussion
In this study, 97 rare RPGR variations were detected in our in-house exome sequence data. A total of 585 variants were identified from previous studies. All in-house data and previous literature data were combined for further genotype–phenotype analysis.
Enrichment and the frequency analyses showed that the benign variants were enriched in non-RCC1 regions. Multistep bioinformatics analyses provided evidence that the corresponding prediction scores were lower than those of variants in the RCC1 region. In addition, segregation and phenotypic consistency analyses further confirmed the benign nature of the variants. A few families also showed variants in other known IRD genes, and some variants were verified in unaffected controls. In previous studies, three families with compound heterozygous variants in RPGR, one allele was an in-frame variant in ORF15, and the other allele was a truncation variant (Pelletier et al., 2007; Neidhardt et al., 2008). Moreover, in-frame variants in ORF15 (spanning 3–36 bp) in healthy individuals were reported in a previous study, suggesting that at least some in-frame variants in ORF15 of RPGR might not be causative (Karra et al., 2006). An in vitro assay developed in a previous study illustrated that some variations located outside of the RCC1 regions might be non-disease-causing polymorphisms (Zhang Q. et al., 2019). Taken together, these findings suggest that at least some missense changes and in-frame variants in the non-RCC1 region might not be pathogenic. Interestingly, several truncation variants at C-terminal region of RPGR had a high frequency in the gnomAD database, but all of them were low-confidence. If the high frequency of these truncations were validated, the pathogenicity of truncations around and downstream of these variants should be considered with greater caution.
More than 85% of the patients with pathogenic RPGR variants had RP. The remainder were diagnosed with a variety of X-linked retinal diseases, including IRD, CORD, COD, high myopia, and MD, among others. The BCVA of the probands with RPGR was age depended, and the BCVA of female carriers was better than that of male patients. In addition to age, the location of the variants in RPGR might play important roles in male patients with BCVA but not in female patients. Male patients with variants in exon1-14 retained better BCVA.
Based on our analysis, there were no significant differences in the SER with regard to the variation type, location or age in either males or females. These results suggest that progression of myopia is relatively slow in patients with variants in RPGR. Because some probands exhibited high myopia in the early stage, the specific screening of RPGR was initially not carried out in many of these patients. This emphasizes the importance of performing a comprehensive examination of patients with early-onset high myopia and of considering the possibility that RPGR variants may exist in these patients. RE was only associated with gender and was more serious in females than in males.
In summary, the results of this study suggested that missense and in-frame variants located outside the RCC1-like domain are likely benign. The pathogenicity criteria for RPGR should be considered with greater caution. Increase of age and location of variants in ORF15 contribute to the reduction of BCVA in males. These results are valuable for understanding genotypes and phenotypes of RPGR.
Data Availability Statement
The data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Zhong Shan Ophthalmic Center. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
XX, SL, and QZ recruited patients. JY, LZ, WS, XX, and SL collected the clinical data. XX and QZ performed whole exome analysis. QZ, JY, and LZ performed the bioinformatic analysis and designed the study. JY, LZ, JO, WS, and QZ discussed the results and wrote the manuscript. All authors reviewed and approved the manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81371058 and 81970837) and the Fundamental Research Funds of the State Key Laboratory of Ophthalmology.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank all patients and family members for their participation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.600210/full#supplementary-material
Supplementary Figure 1 | Pedigrees of 51 families with likely pathogenic variants in RPGR. Previously reported of RPGR variants identified in families by Sanger sequencing are not shown. The family ID is provided above each pedigree. The probands and available family members were analyzed by Sanger sequencing. Arrows, probands of each family; filled symbols, patients with different eye diseases; M, mutation; +, wild type; square, male; circle, female.
Supplementary Figure 2 | Sanger sequencing of 51 unrelated families with likely pathogenic variants. Pedigrees are shown in the left column. Diagrams of the mutant sequence and the corresponding normal control sequence diagram are shown in the columns on the right. Sites of sequence changes are shown above the sequence and indicated by a black arrow.
Supplementary Figure 3 | Pedigrees with likely benign variants. The family ID is provided above each pedigree. The probands and available family members were identified by Sanger sequencing. Arrows, probands of each family; filled symbols, patients with different eye diseases; M, mutation; +, wild type; square, male; circle, female.
Supplementary Figure 4 | Comparison of phenotypes according to different factors in male patients. (A–C) Refractive error were not associated with location and variation type, there was no statistical significance. M + I, missense and in-frame; T, truncation; E1-14, exon1-exon14; ns, no statistical significance; RCC1, RCC1-like domain.
Supplementary Figure 5 | Comparison of phenotypes according to different factors in female carriers. (A–F) The severity of BCVA and refractive error show no correlation with different location, variation type, there was no statistical significance. M + I, missense and in-frame; T, truncation; E1-14, exon1-exon14; BCVA, best corrected visual acuity; ns, no statistical significance; RCC1, RCC1-like domain.
Abbreviations
AF, allele frequency; All, all population; BCVA, best corrected visual acuity; CD, corneal degeneration; COD, cone dystrophy; CORD, cone-rod dystrophy; DM, disease-causing mutations; EA, East Asians; G, glaucoma; HM, high myopia; Hyp, hypermetropia; IRD, inherited retinal degenerations; LCA, Leber congenital amaurosis; LD, lens dislocation; MC, macular coloboma; MD, macular degeneration; N, normal; NA, not available; NYS, nystagmus; OA, ocular albinism; ONH, Optic nerve hypoplasia; PHPV, Persistent Hyperplastic Primary Vitreous; RB, retinoblastoma; RD, retinal diseases; RE, refractive error; RP, retinitis pigmentosa; RRD, Rhegmatogenous Retinal Detachment; SA, splicing acceptor; SD, splicing donor; SER, spherical equivalent refraction. BDGP Berkeley Drosophila Genome Project; gnomAD, genome aggregation database; HGMD, the Human Gene Mutation Database; HSF, Human Splicing Finder.
Footnotes
- ^ http://genome.ucsc.edu/
- ^ http://bio-bwa.sourceforge.net/
- ^ http://samtools.sourceforge.net/
- ^ http://snpeff.sourceforge.net/
- ^ http://annovar.openbioinformatics.org/en/latest/
- ^ http://varianttools.sourceforge.net/Annotation/DbNSFP
- ^ https://cadd.gs.washington.edu/info
- ^ https://sites.google.com/site/revelgenomics/
- ^ http://gnomad.broadinstitute.org/
- ^ http://www.hgmd.cf.ac.uk/ac/index.php
- ^ https://www.ncbi.nlm.nih.gov/pubmed/
- ^ http://apps.webofknowledge.com/
- ^ https://scholar.google.com/
References
Acton, J. H., Greenberg, J. P., Greenstein, V. C., Marsiglia, M., Tabacaru, M., Smith, R. T., et al. (2013). Evaluation of multimodal imaging in carriers of X-linked retinitis pigmentosa. Exp. Eye Res. 113, 41–48. doi: 10.1016/j.exer.2013.05.003
Adamian, M., Pawlyk, B., Hong, D. H., and Berson, E. L. (2005). Rod and cone opsin mislocalization in an autopsy eye from a female carrier of X-linked RP with a Gly436Asp mutation in the RPGR gene. Investig. Ophthalmol. Vis. Sci. 46:3400.
Aguirre, G. D., Yashar, B. M., John, S. K., Smith, J. E., Breuer, D. K., Hiriyanna, S., et al. (2002). Retinal histopathology of an XLRP carrier with a mutation in the RPGR exon ORF15. Exp. Eye Res. 75, 431–443.
Aleman, T. S., Cideciyan, A. V., Sumaroka, A., Schwartz, S. B., Roman, A. J., Windsor, E. A. M., et al. (2007). Inner retinal abnormalities in X-linked retinitis pigmentosa with RPGR mutations. Investig. Ophthalmol. Vis. Sci. 48, 4759–4765. doi: 10.1167/iovs.07-0453
Al-Maskari, A., O’Grady, A., Pal, B., and McKibbin, M. (2009). Phenotypic progression in X-linked retinitis pigmentosa secondary to a novel mutation in the RPGR gene. Eye 23, 519–521. doi: 10.1038/eye.2008.427
Almoguera, B., Li, J., Fernandez-San Jose, P., Liu, Y., March, M., Pellegrino, R., et al. (2015). Application of whole exome sequencing in six families with an initial diagnosis of autosomal dominant retinitis pigmentosa: lessons learned. PLoS One 10:e0133624. doi: 10.1371/journal.pone.0133624
Andreasson, S., Breuer, D. K., Eksandh, L., Ponjavic, V., Frennesson, C., Hiriyanna, S., et al. (2003). Clinical studies of X-linked retinitis pigmentosa in three Swedish families with newly identified mutations in the RP2 and RPGR-ORF15 genes. Ophthalmic Genet. 24, 215–223.
Andreasson, S., Ponjavic, V., Abrahamson, M., Ehinger, B., Wu, W., Fujita, R., et al. (1997). Phenotypes in three Swedish families with X-linked retinitis pigmentosa caused by different mutations in the RPGR gene. Am. J. Ophthalmol. 124, 95–102.
Ayyagari, R., Demirci, F. Y., Liu, J., Bingham, E. L., Stringham, H., Kakuk, L. E., et al. (2002). X-linked recessive atrophic macular degeneration from RPGR mutation. Genomics 80, 166–171.
Bader, I., Brandau, O., Achatz, H., Apfelstedt-Sylla, E., Hergersberg, M., Lorenz, B., et al. (2003). X-linked retinitis pigmentosa: RPGR mutations in most families with definite X linkage and clustering of mutations in a short sequence stretch of exon ORF15. Invest. Ophthalmol. Vis. Sci. 44, 1458–1463.
Banin, E., Mizrahi-Meissonnier, L., Neis, R., Silverstein, S., Magyar, I., Abeliovich, D., et al. (2007). A non-ancestral RPGR missense mutation in families with either recessive or semi-dominant X-linked retinitis pigmentosa. Am. J. Med. Genet. A 143A, 1150–1158. doi: 10.1002/ajmg.a.31642
Barnes, R. M., Holder, G. E., Smith, J. M. A., Drizen, P. R., Winchester, E., Mantel, I., et al. (2003). Phenotype of heterozygotes with mutations in the ORF 15 (open reading frame 15) of RPGR. Investig. Ophthalmol. Vis. Sci. 44, U679–U679.
Bauer, S., Fujita, R., Buraczynska, M., Abrahamson, M., Ehinger, B., Wu, W., et al. (1998). Phenotype of an X-linked retinitis pigmentosa family with a novel splice defect in the RPGR gene. Invest. Ophthalmol. Vis. Sci. 39, 2470–2474.
Bellingrath, J. S., Ochakovski, G. A. I, Seitz, P., Kohl, S., Zrenner, E., Hanig, N., et al. (2017). High symmetry of visual acuity and visual fields in RPGR-linked retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 58, 4457–4466. doi: 10.1167/iovs.17-22077
Berger, W., Kloeckener-Gruissem, B., and Neidhardt, J. (2010). The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 29, 335–375. doi: 10.1016/j.preteyeres.2010.03.004
Birtel, J., Eisenberger, T., Gliem, M., Muller, P. L., Herrmann, P., Betz, C., et al. (2018a). Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 8:4824. doi: 10.1038/s41598-018-22096-0
Birtel, J., Gliem, M., Mangold, E., Muller, P. L., Holz, F. G., Neuhaus, C., et al. (2018b). Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS One 13:e0207958. doi: 10.1371/journal.pone.0207958
Bowne, S. J., Sullivan, L. S., Koboldt, D. C., Ding, L., Fulton, R., Abbott, R. M., et al. (2011). Identification of disease-causing mutations in autosomal dominant retinitis pigmentosa (adRP) using next-generation DNA sequencing. Invest. Ophthalmol. Vis. Sci. 52, 494–503. doi: 10.1167/iovs.10-6180
Branham, K., Guru, A. A., Kozak, I., Biswas, P., Othman, M., Kishaba, K., et al. (2018). Identification of novel deletions as the underlying cause of retinal degeneration in two pedigrees. Adv. Exp. Med. Biol. 1074, 229–236. doi: 10.1007/978-3-319-75402-4_28
Branham, K., Othman, M., Brumm, M., Karoukis, A. J., Atmaca-Sonmez, P., Yashar, B. M., et al. (2012). Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Invest. Ophthalmol. Vis. Sci. 53, 8232–8237. doi: 10.1167/iovs.12-11025
Breuer, D. K., Yashar, B. M., Filippova, E., Hiriyanna, S., Lyons, R. H., Mears, A. J., et al. (2002). A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 70, 1545–1554. doi: 10.1086/340848
Bukowy-Bieryllo, Z., Zietkiewicz, E., Loges, N. T., Wittmer, M., Geremek, M., Olbrich, H., et al. (2013). RPGR mutations might cause reduced orientation of respiratory cilia. Pediatr. Pulmonol. 48, 352–363. doi: 10.1002/ppul.22632
Bunker, C. H., Berson, E. L., Bromley, W. C., Hayes, R. P., and Roderick, T. H. (1984). Prevalence of retinitis pigmentosa in Maine. Am. J. Ophthalmol. 97, 357–365.
Buraczynska, M., Wu, W., Fujita, R., Buraczynska, K., Phelps, E., Andreasson, S., et al. (1997). Spectrum of mutations in the RPGR gene that are identified in 20% of families with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 61, 1287–1292. doi: 10.1086/301646
Chakarova, C. F., Cherninkova, S., Tournev, I., Waseem, N., Kaneva, R., Jordanova, A., et al. (2006). Molecular genetics of retinitis pigmentosa in two Romani (Gypsy) families. Mol. Vis. 12, 909–914.
Chang, W., Ding, Q., Tang, Z., Liu, P., Jiang, F., Ke, T., et al. (2007). A novel de novo frameshift mutation of RPGR ORF15 is associated with X-linked retinitis pigmentosa in a Chinese family. Mol. Vis. 13, 1548–1554.
Chassine, T., Bocquet, B., Daien, V., vila-Fernandez, A. A., Ayuso, C., Collin, R. W., et al. (2015). Autosomal recessive retinitis pigmentosa with RP1 mutations is associated with myopia. Br. J. Ophthalmol. 99, 1360–1365. doi: 10.1136/bjophthalmol-2014-306224
Chiang, J. P. W., Lamey, T. M., Wang, N. K., Duan, J., Zhou, W., McLaren, T. L., et al. (2018). Development of high-throughput clinical testing of RPGR ORF15 using a large inherited retinal dystrophy cohort. Invest. Ophthalmol. Vis. Sci. 59, 4434–4440. doi: 10.1167/iovs.18-24555
Churchill, J. D., Bowne, S. J., Sullivan, L. S., Lewis, R. A., Wheaton, D. K., Birch, D. G., et al. (2013). Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 54, 1411–1416. doi: 10.1167/iovs.12-11541
Consugar, M. B., Navarro-Gomez, D., Place, E. M., Bujakowska, K. M., Sousa, M. E., Fonseca-Kelly, Z. D., et al. (2015). Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet. Med. 17, 253–261. doi: 10.1038/gim.2014.172
Dan, H., Huang, X., Xing, Y., and Shen, Y. (2020). Application of targeted panel sequencing and whole exome sequencing for 76 Chinese families with retinitis pigmentosa. Mol. Genet. Genomic Med. 8:e1131. doi: 10.1002/mgg3.1131
Demirci, F. Y., Gupta, N., Radak, A. L., Rigatti, B. W., Mah, T. S., Milam, A. H., et al. (2005). Histopathologic study of X-linked cone-rod dystrophy (CORDX1) caused by a mutation in the RPGR exon ORF15. Am. J. Ophthalmol. 139, 386–388. doi: 10.1016/j.ajo.2004.08.041
Demirci, F. Y., Radak, A. L., Rigatti, B. W., Mah, T. S., and Gorin, M. B. (2004). A presumed missense mutation of RPGR causes abnormal RNA splicing with exon skipping. Am. J. Ophthalmol. 138, 504–505. doi: 10.1016/j.ajo.2004.04.019
Demirci, F. Y., Rigatti, B. W., Mah, T. S., and Gorin, M. B. (2006). A novel RPGR exon ORF15 mutation in a family with X-linked retinitis pigmentosa and Coats’-like exudative vasculopathy. Am. J. Ophthalmol. 141, 208–210. doi: 10.1016/j.ajo.2005.07.077
Demirci, F. Y., Rigatti, B. W., Wen, G., Radak, A. L., Mah, T. S., Baic, C. L., et al. (2002). X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am. J. Hum. Genet. 70, 1049–1053. doi: 10.1086/339620
Dry, K. L., Manson, F. D., Lennon, A., Bergen, A. A., Van Dorp, D. B., and Wright, A. F. (1999). Identification of a 5’ splice site mutation in the RPGR gene in a family with X-linked retinitis pigmentosa (RP3). Hum. Mutat. 13, 141–145. doi: 10.1002/(SICI)1098-1004199913:2<141::AID-HUMU6<3.0.CO;2-Q
Duncan, J. L., Zhang, Y., Gandhi, J., Nakanishi, C., Othman, M., Branham, K. E., et al. (2007). High-resolution imaging with adaptive optics in patients with inherited retinal degeneration. Invest. Ophthalmol. Vis. Sci. 48, 3283–3291. doi: 10.1167/iovs.06-1422
Ebenezer, N. D., Michaelides, M., Jenkins, S. A., Audo, I., Webster, A. R., Cheetham, M. E., et al. (2005). Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest. Ophthalmol. Vis. Sci. 46, 1891–1898. doi: 10.1167/iovs.04-1482
Eisenberger, T., Neuhaus, C., Khan, A. O., Decker, C., Preising, M. N., Friedburg, C., et al. (2013). Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS One 8:e78496. doi: 10.1371/journal.pone.0078496
Fahim, A. T., Bowne, S. J., Sullivan, L. S., Webb, K. D., Williams, J. T., Wheaton, D. K., et al. (2011). Allelic heterogeneity and genetic modifier loci contribute to clinical variation in males with X-linked retinitis pigmentosa due to RPGR mutations. PLoS One 6:e23021. doi: 10.1371/journal.pone.0023021
Fahim, A. T., Sullivan, L. S., Bowne, S. J., Jones, K. D., Wheaton, D. K. H., Khan, N. W., et al. (2020). X-chromosome inactivation is a biomarker of clinical severity in female carriers of RPGR-associated X-linked retinitis pigmentosa. Ophthalmol. Retina 4, 510–520. doi: 10.1016/j.oret.2019.11.010
Fernandez-San Jose, P., Corton, M., Blanco-Kelly, F., Avila-Fernandez, A., Lopez-Martinez, M. A., Sanchez-Navarro, I., et al. (2015). Targeted next-generation sequencing improves the diagnosis of autosomal dominant retinitis pigmentosa in spanish patients. Invest. Ophthalmol. Vis. Sci. 56, 2173–2182. doi: 10.1167/iovs.14-16178
Fishman, G. A., Grover, S., Buraczynska, M., Wu, W., and Swaroop, A. (1998a). A new 2-base pair deletion in the RPGR gene in a black family with X-linked retinitis pigmentosa. Arch. Ophthalmol. 116, 213–218.
Fishman, G. A., Grover, S., Jacobson, S. G., Alexander, K. R., Derlacki, D. J., Wu, W., et al. (1998b). X-linked retinitis pigmentosa in two families with a missense mutation in the RPGR gene and putative change of glycine to valine at codon 60. Ophthalmology 105, 2286–2296. doi: 10.1016/S0161-6420(98)91231-3
Fishman, G. A., Weinberg, A. B., and McMahon, T. T. (1986). X-linked recessive retinitis pigmentosa. Clinical characteristics of carriers. Arch. Ophthalmol. 104, 1329–1335.
Flaxel, C. J., Jay, M., Thiselton, D. L., Nayudu, M., Hardcastle, A. J., Wright, A., et al. (1999). Difference between RP2 and RP3 phenotypes in X linked retinitis pigmentosa. Br. J. Ophthalmol. 83, 1144–1148.
Foote, K. G., Wong, J. J., Boehm, A. E., Bensinger, E., Porco, T. C., Roorda, A., et al. (2020). Comparing cone structure and function in RHO- and RPGR-associated retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 61:42. doi: 10.1167/iovs.61.4.42
Fujita, R., Buraczynska, M., Gieser, L., Wu, W., Forsythe, P., Abrahamson, M., et al. (1997). Analysis of the RPGR gene in 11 pedigrees with the retinitis pigmentosa type 3 genotype: paucity of mutations in the coding region but splice defects in two families. Am. J. Hum. Genet. 61, 571–580. doi: 10.1086/515523
Garcia-Hoyos, M., Garcia-Sandoval, B., Cantalapiedra, D., Riveiro, R., Lorda-Sanchez, I., Trujillo-Tiebas, M. J., et al. (2006). Mutational screening of the RP2 and RPGR genes in Spanish families with X-linked retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 47, 3777–3782. doi: 10.1167/iovs.06-0323
Ge, Z., Bowles, K., Goetz, K., Scholl, H. P., Wang, F., Wang, X., et al. (2015). NGS-based Molecular diagnosis of 105 eyeGENE((R)) probands with Retinitis Pigmentosa. Sci. Rep. 5:18287. doi: 10.1038/srep18287
Gill, J. S., Georgiou, M., Kalitzeos, A., Moore, A. T., and Michaelides, M. (2019). Progressive cone and cone-rod dystrophies: clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 103, 711–720. doi: 10.1136/bjophthalmol-2018-313278
Glaus, E., Schmid, F., Da Costa, R., Berger, W., and Neidhardt, J. (2011). Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol. Ther. 19, 936–941. doi: 10.1038/mt.2011.7
Glockle, N., Kohl, S., Mohr, J., Scheurenbrand, T., Sprecher, A., Weisschuh, N., et al. (2014). Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 22, 99–104. doi: 10.1038/ejhg.2013.72
Gonzalez-del Pozo, M., Mendez-Vidal, C., Bravo-Gil, N., Vela-Boza, A., Dopazo, J., Borrego, S., et al. (2014). Exome sequencing reveals novel and recurrent mutations with clinical significance in inherited retinal dystrophies. PLoS One 9:e0116176. doi: 10.1371/journal.pone.0116176
Grondahl, J. (1987). Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway. Clin. Genet. 31, 255–264.
Guevara-Fujita, M., Fahrner, S., Buraczynska, K., Cook, J., Wheaton, D., Cortes, F., et al. (2001). Five novel RPGR mutations in families with X-linked retinitis pigmentosa. Hum. Mutat. 17:151. doi: 10.1002/1098-1004(200102)17:2<151::AID-HUMU7<3.0.CO;2-W
Haddad, M. F., Khabour, O. F., Abuzaideh, K. A., and Shihadeh, W. (2016). Screening for mutations in RPGR and RP2 genes in Jordanian families with X-linked retinitis pigmentosa. Genet. Mol. Res. 15:gmr7842. doi: 10.4238/gmr.15027842
Hartong, D. T., Berson, E. L., and Dryja, T. P. (2006). Retinitis pigmentosa. Lancet 368, 1795–1809. doi: 10.1016/S0140-6736(06)69740-7
Hendriks, M., Verhoeven, V. J. M., Buitendijk, G. H. S., Polling, J. R., Meester-Smoor, M. A., Hofman, A., et al. (2017). Development of refractive errors-what can we learn from inherited retinal dystrophies? Am. J. Ophthalmol. 182, 81–89. doi: 10.1016/j.ajo.2017.07.008
Hong, D. H., Pawlyk, B., Sokolov, M., Strissel, K. J., Yang, J., Tulloch, B., et al. (2003). RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest. Ophthalmol. Vis. Sci. 44, 2413–2421.
Hu, F., Zeng, X. Y., Liu, L. L., Lao, Y. L., Jiang, Y. P., Wang, H., et al. (2014). Genetic analysis of Chinese families reveals a novel truncation allele of the retinitis pigmentosa GTPase regulator gene. Int. J. Ophthalmol. 7, 753–758. doi: 10.3980/j.issn.2222-3959.2014.05.02
Huang, J. T., Heckenlively, J. R., Jayasundera, K. T., and Branham, K. E. (2014). The ophthalmic experience: unanticipated primary findings in the era of next generation sequencing. J. Genet. Counsel. 23, 588–593. doi: 10.1007/s10897-013-9679-y
Huang, L., Zhang, Q., Li, S., Guan, L., Xiao, X., Zhang, J., et al. (2013). Exome sequencing of 47 chinese families with cone-rod dystrophy: mutations in 25 known causative genes. PLoS One 8:e65546. doi: 10.1371/journal.pone.0065546
Huang, X., Liu, Y., Yu, X., Huang, Q., Lin, C., Zeng, J., et al. (2019). The clinical application of preimplantation genetic diagnosis for X-linked retinitis pigmentosa. J. Assist. Reprod. Genet. 36, 989–994. doi: 10.1007/s10815-019-01434-9
Huang, X. F., Huang, F., Wu, K. C., Wu, J., Chen, J., Pang, C. P., et al. (2015a). Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 17, 271–278. doi: 10.1038/gim.2014.138
Huang, X. F., Wu, J., Lv, J. N., Zhang, X., and Jin, Z. B. (2015b). Identification of false-negative mutations missed by next-generation sequencing in retinitis pigmentosa patients: a complementary approach to clinical genetic diagnostic testing. Genet. Med. 17, 307–311. doi: 10.1038/gim.2014.193
Iannaccone, A., Breuer, D. K., Wang, X. F., Kuo, S. F., Normando, E. M., Filippova, E., et al. (2003). Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J. Med. Genet. 40:e118.
Iannaccone, A., Othman, M. I., Cantrell, A. D., Jennings, B. J., Branham, K., and Swaroop, A. (2008). Retinal phenotype of an X-linked pseudo-Usher syndrome in association with the G173R mutation in the RPGR gene. Adv. Exp. Med. Biol. 613, 221–227.
Jacobson, S. G., Buraczynska, M., Milam, A. H., Chen, C., Jarvalainen, M., Fujita, R., et al. (1997). Disease expression in X-linked retinitis pigmentosa caused by a putative null mutation in the RPGR gene. Invest. Ophthalmol. Vis. Sci. 38, 1983–1997.
Ji, Y., Wang, J., Xiao, X., Li, S., Guo, X., and Zhang, Q. (2010). Mutations in RPGR and RP2 of Chinese patients with X-linked retinitis pigmentosa. Curr. Eye Res. 35, 73–79. doi: 10.3109/02713680903395299
Jin, Z. B., Gu, F., Ma, X., and Nao-i, N. (2007a). Identification of a novel RPGR exon ORF15 mutation in a family with X-linked retinitis pigmentosa. Arch. Ophthalmol. 125, 1407–1412. doi: 10.1001/archopht.125.10.1407
Jin, Z. B., Gu, F., Matsuda, H., Yukawa, N., Ma, X., and Nao-i, N. (2007b). Somatic and gonadal mosaicism in X-linked retinitis pigmentosa. Am. J. Med. Genet. A 143A, 2544–2548. doi: 10.1002/ajmg.a.31984
Jin, Z. B., Liu, X. Q., Hayakawa, M., Murakami, A., and Nao-i, N. (2006). Mutational analysis of RPGR and RP2 genes in Japanese patients with retinitis pigmentosa: identification of four mutations. Mol. Vis. 12, 1167–1174.
Jin, Z. B., Liu, X. Q., Uchida, A., Vervoort, R., Morishita, K., Hayakawa, M., et al. (2005). Novel deletion spanning RCC1-like domain of RPGR in Japanese X-linked retinitis pigmentosa family. Mol. Vis. 11, 535–541.
Jin, Z. B., Mandai, M., Yokota, T., Higuchi, K., Ohmori, K., Ohtsuki, F., et al. (2008). Identifying pathogenic genetic background of simplex or multiplex retinitis pigmentosa patients: a large scale mutation screening study. J. Med. Genet. 45, 465–472. doi: 10.1136/jmg.2007.056416
Kalitzeos, A., Samra, R., Kasilian, M., Tee, J. J. L., Strampe, M., Langlo, C., et al. (2017). Cellular imaging of the tapetal-like reflex in carriers of Rpgr-associated retinopathy. Retina 39, 570–580. doi: 10.1097/IAE.0000000000001965
Karra, D., Jacobi, F. K., Broghammer, M., Blin, N., and Pusch, C. M. (2006). Population haplotypes of exon ORF15 of the retinitis pigmentosa GTPase regulator gene in Germany - Implications for screening for inherited retinal disorders. Mol. Diagn. Ther. 10, 115–123. doi: 10.1007/Bf03256451
Kastner, S. I, Thiemann, J., Dekomien, G., Petrasch-Parwez, E., Schreiber, S., Akkad, D. A., et al. (2015). Exome sequencing reveals AGBL5 as novel candidate gene and additional variants for retinitis pigmentosa in five turkish families. Invest. Ophthalmol. Vis. Sci. 56, 8045–8053. doi: 10.1167/iovs.15-17473
Koenekoop, R. K., Loyer, M., Hand, C. K., Al Mahdi, H., Dembinska, O., Beneish, R., et al. (2003). Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families. Am. J. Ophthalmol. 136, 678–687.
Kousal, B., Skalicka, P., Diblik, P., Kuthan, P., Langrova, H., and Liskova, P. (2013). [Clinical findings in members of a Czech family with retinitis pigmentosa caused by the c.2426_2427delAG mutation in RPGR]. Cesk. Slov. Oftalmol. 69, 8–15.
Kousal, B., Skalicka, P., Valesova, L., Colclough, T., Hart-Holden, N., O’Grady, A., et al. (2014). Severe retinal degeneration in females with c.2543del mutation in the RPGR gene. Acta Ophthalmol. 92:253. doi: 10.1111/j.1755-3768.2014.T070.x
Koyanagi, Y., Akiyama, M., Nishiguchi, K. M., Momozawa, Y., Kamatani, Y., Takata, S., et al. (2019). Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. J. Med. Genet. 56, 662–670. doi: 10.1136/jmedgenet-2018-105691
Li, J., Jiang, D., Xiao, X., Li, S., Jia, X., Sun, W., et al. (2015). Evaluation of 12 myopia-associated genes in Chinese patients with high myopia. Invest. Ophthalmol. Vis. Sci. 56, 722–729. doi: 10.1167/iovs.14-14880
Li, J., Tang, J., Feng, Y., Xu, M., Chen, R., Zou, X., et al. (2016). Improved diagnosis of inherited retinal dystrophies by high-fidelity PCR of ORF15 followed by next-generation sequencing. J. Mol. Diagn. 18, 817–824. doi: 10.1016/j.jmoldx.2016.06.007
Li, N., Dai, S., Zhang, L., Mei, H., and Wang, L. (2011). A novel mutation of RPGR gene in an X-linked Chinese family with retinitis pigmentosa. Mol. Genet. Metab. 102, 488–493. doi: 10.1016/j.ymgme.2010.12.006
Li, Z. L., Zhuang, W. J., Zhao, W., Zhang, X. F., Wang, J., Meng, R. H., et al. (2011). [Novel RPGR gene mutation in a Chinese family with X-linked recessive retinitis pigmentosa]. Zhonghua Yan Ke Za Zhi 47, 516–520.
Liskova, P., Colclough, T., Hart-Holden, N., Chakarova, C. F., O’Grady, A., Kondrova, L., et al. (2011). Molecular genetic cause of X-linked retinitis pigmentosa in a Czech family. Acta Ophthalmol. 89, e213–e215. doi: 10.1111/j.1755-3768.2009.01802.x
Liu, L., Chen, H., Liu, M., Jin, L., Wei, Y., Wu, X., et al. (2002). Two novel mutations of the retinitis pigmentosa GTPase regulator gene in two Chinese families with X-linked retinitis pigmentosa. Chin. Med. J. 115, 833–836.
Liu, L., Jin, L., Liu, M., Wei, Y., Wu, X., Liu, Y., et al. (2000). Identification of two novel mutations (E332X and c1536delC) in the RPGR gene in two Chinese families with X-linked retinitis pigmentosa. Hum. Mutat. 15:584. doi: 10.1002/1098-1004(200006)15:6<584::AID-HUMU26<3.0.CO;2-O
Liu, M. M., and Zack, D. J. (2013). Alternative splicing and retinal degeneration. Clin. Genet. 84, 142–149. doi: 10.1111/cge.12181
Lorenz, B., Andrassi, M., and Kretschmann, U. (2003). Phenotype in two families with RP3 associated with RPGR mutations. Ophthalmic Genet. 24, 89–101.
Mawatari, G., Fujinami, K., Liu, X., Yang, L., Fujinami-Yokokawa, Y., Komori, S., et al. (2020). Correction to: clinical and genetic characteristics of 14 patients from 13 Japanese families with RPGR-associated retinal disorder: report of eight novel variants. Hum. Genome Var. 7:3. doi: 10.1038/s41439-019-0086-2
Mawatari, G., Fujinami, K., Liu, X., Yang, L., Yokokawa, Y. F., Komori, S., et al. (2019). Clinical and genetic characteristics of 14 patients from 13 Japanese families with RPGR-associated retinal disorder: report of eight novel variants. Hum. Genome Var. 6:34. doi: 10.1038/s41439-019-0065-7
Meindl, A., Dry, K., Herrmann, K., Manson, F., Ciccodicola, A., Edgar, A., et al. (1996). A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat. Genet. 13, 35–42. doi: 10.1038/ng0596-35
Menghini, M., Jolly, J. K., Nanda, A., Wood, L., Cehajic-Kapetanovic, J., and MacLaren, R. E. (2020). Early cone photoreceptor outer segment length shortening in RPGR X-linked retinitis pigmentosa. Ophthalmologica [Epub ahead of print]. doi: 10.1159/000507484
Miano, M. G., Testa, F., Strazzullo, M., Trujillo, M., De Bernardo, C., Grammatico, B., et al. (1999). Mutation analysis of the RPGR gene reveals novel mutations in south European patients with X-linked retinitis pigmentosa. Eur. J. Hum. Genet. 7, 687–694. doi: 10.1038/sj.ejhg.5200352
Miano, M. G., Valverde, D., Solans, T., Grammatico, B., Migliaccio, C., Cirigliano, V., et al. (1998). Two novel mutations in the retinitis pigmentosa GTPase regulator (RPGR) gene in X-linked retinitis pigmentosa (RP3). Mutations in brief no. 172. Hum. Mutat. 12, 212–213.
Moore, A., Escudier, E., Roger, G., Tamalet, A., Pelosse, B., Marlin, S., et al. (2006). RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J. Med. Genet. 43, 326–333. doi: 10.1136/jmg.2005.034868
Nanda, A., Salvetti, A. P., Clouston, P., Downes, S. M., and MacLaren, R. E. (2018). Exploring the variable phenotypes of RPGR carrier females in assessing their potential for retinal gene therapy. Genes 9:643. doi: 10.3390/genes9120643
Neidhardt, J., Glaus, E., Barthelmes, D., Zeitz, C., Fleischhauer, J., and Berger, W. (2007). Identification and characterization of a novel RPGR isoform in human retina. Hum. Mutat. 28, 797–807. doi: 10.1002/humu.20521
Neidhardt, J., Glaus, E., Lorenz, B., Netzer, C., Li, Y., Schambeck, M., et al. (2008). Identification of novel mutations in X-linked retinitis pigmentosa families and implications for diagnostic testing. Mol. Vis. 14, 1081–1093.
Nguyen, X. T., Talib, M., van Schooneveld, M. J., Brinks, J., Ten Brink, J., Florijn, R. J., et al. (2020). RPGR-associated dystrophies: clinical, genetic, and histopathological features. Int. J. Mol. Sci. 21:835. doi: 10.3390/ijms21030835
Ogino, K., Oishi, M., Oishi, A., Morooka, S., Sugahara, M., Gotoh, N., et al. (2015). Radial fundus autofluorescence in the periphery in patients with X-linked retinitis pigmentosa. Clin. Ophthalmol. 9, 1467–1474. doi: 10.2147/Opth.S89371
Oishi, M., Oishi, A., Gotoh, N., Ogino, K., Higasa, K., Iida, K., et al. (2014). Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Invest. Ophthalmol. Vis. Sci. 55, 7369–7375. doi: 10.1167/iovs.14-15458
O’Sullivan, J., Mullaney, B. G., Bhaskar, S. S., Dickerson, J. E., Hall, G., O’Grady, A., et al. (2012). A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J. Med. Genet. 49, 322–326. doi: 10.1136/jmedgenet-2012-100847
Parmeggiani, F., Barbaro, V., De Nadai, K., Lavezzo, E., Toppo, S., Chizzolini, M., et al. (2016). Identification of novel X-linked gain-of-function RPGR-ORF15 mutation in Italian family with retinitis pigmentosa and pathologic myopia. Sci. Rep. 6:39179. doi: 10.1038/srep39179
Pelletier, V., Jambou, M., Delphin, N., Zinovieva, E., Stum, M., Gigarel, N., et al. (2007). Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: genotype-phenotype correlations and impact on genetic counseling. Hum. Mutat. 28, 81–91. doi: 10.1002/humu.20417
Pierrottet, C. O., Zuntini, M., Digiuni, M., Bazzanella, I., Ferri, P., Paderni, R., et al. (2014). Syndromic and non-syndromic forms of retinitis pigmentosa: a comprehensive Italian clinical and molecular study reveals new mutations. Genet. Mol. Res. 13, 8815–8833. doi: 10.4238/2014.October.27.23
Prokisch, H., Hartig, M., Hellinger, R., Meitinger, T., and Rosenberg, T. (2007). A population-based epidemiological and genetic study of X-linked retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 48, 4012–4018. doi: 10.1167/iovs.07-0071
Pusch, C. M., Broghammer, M., Jurklies, B., Besch, D., and Jacobi, F. K. (2002). Ten novel ORF15 mutations confirm mutational hot spot in the RPGR gene in European patients with X-linked retinitis pigmentosa. Hum. Mutat. 20:405. doi: 10.1002/humu.9072
Pyo Park, S. I, Hwan, Hong, Tsang, S. H., and Chang, S. (2013). Cellular imaging demonstrates genetic mosaicism in heterozygous carriers of an X-linked ciliopathy gene. Eur. J. Hum. Genet. 21, 1240–1248. doi: 10.1038/ejhg.2013.21
Rebello, G., Vorster, A., Greenberg, J., Coutts, N., Roberts, L., Ehrenreich, L., et al. (2003). Analysis of RPGR in a South African family with X-linked retinitis pigmentosa: research and diagnostic implications. Clin. Genet. 64, 137–141.
Rodriguez-Munoz, A., Aller, E., Jaijo, T., Gonzalez-Garcia, E., Cabrera-Peset, A., Gallego-Pinazo, R., et al. (2020). Expanding the clinical and molecular heterogeneity of nonsyndromic inherited retinal dystrophies. J. Mol. Diagn. 22, 532–543. doi: 10.1016/j.jmoldx.2020.01.003
Roepman, R., Bernoud-Hubac, N., Schick, D. E., Maugeri, A., Berger, W., Ropers, H. H., et al. (2000). The retinitis pigmentosa GTPase regulator (RPGR) interacts with novel transport-like proteins in the outer segments of rod photoreceptors. Hum. Mol. Genet. 9, 2095–2105.
Roepman, R., van Duijnhoven, G., Rosenberg, T., Pinckers, A. J., Bleeker-Wagemakers, L. M., Bergen, A. A., et al. (1996). Positional cloning of the gene for X-linked retinitis pigmentosa 3: homology with the guanine-nucleotide-exchange factor RCC1. Hum. Mol. Genet. 5, 1035–1041.
Rosenberg, T., Schwahn, U., Feil, S., and Berger, W. (1999). Genotype-phenotype correlation in X-linked retinitis pigmentosa 2 (RP2). Ophthalmic Genet. 20, 161–172.
Rozet, J., Perrault, J., Gigarel, N., Souied, E., Ghazi, I., Gerber, S., et al. (2002). Dominant X-linked RP is frequently accounted for by truncating mutations in the exon ORF15 of the RPGR gene. Investig. Ophthalmol. Vis. Sci. 43:U169.
Ruddle, J. B., Ebenezer, N. D., Kearns, L. S., Mulhall, L. E., Mackey, D. A., and Hardcastle, A. J. (2009). RPGR ORF15 genotype and clinical variability of retinal degeneration in an Australian population. Br. J. Ophthalmol. 93, 1151–1154. doi: 10.1136/bjo.2008.153908
Salvetti, A. P., Nanda, A., and MacLaren, R. E. (2020). RPGR-Related X-Linked Retinitis Pigmentosa Carriers with a Severe “Male Pattern”. Ophthalmologica 244, 60–67. doi: 10.1159/000503687
Sanchez Tocino, H., Diez Montero, C., Villanueva Gomez, A., Lobo Valentin, R., and Montero-Moreno, J. A. (2019). Phenotypic high myopia in X-linked retinitis pigmentosa secondary to a novel mutation in the RPGR gene. Ophthalmic Genet. 40, 170–176. doi: 10.1080/13816810.2019.1605385
Sandberg, M. A., Rosner, B., Weigel-DiFranco, C., Dryja, T. P., and Berson, E. L. (2007). Disease course of patients with X-linked retinitis pigmentosa due to RPGR gene mutations. Investig. Ophthalmol. Vis. Sci. 48, 1298–1304. doi: 10.1167/iovs.06-0971
Sharon, D., and Banin, E. (2015). Nonsyndromic retinitis pigmentosa is highly prevalent in the Jerusalem region with a high frequency of founder mutations. Mol. Vis. 21, 783–792.
Sharon, D., Sandberg, M. A., Rabe, V. W., Stillberger, M., Dryja, T. P., and Berson, E. L. (2003). RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 73, 1131–1146. doi: 10.1086/379379
Sheng, X., Li, Z., Zhang, X., Wang, J., Ren, H., Sun, Y., et al. (2010). A novel mutation in retinitis pigmentosa GTPase regulator gene with a distinctive retinitis pigmentosa phenotype in a Chinese family. Mol. Vis. 16, 1620–1628.
Shu, X., Black, G. C., Rice, J. M., Hart-Holden, N., Jones, A., O’Grady, A., et al. (2007). RPGR mutation analysis and disease: an update. Hum. Mutat. 28, 322–328. doi: 10.1002/humu.20461
Stone, E. M., Andorf, J. L., Whitmore, S. S., DeLuca, A. P., Giacalone, J. C., Streb, L. M., et al. (2017). Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology 124, 1314–1331. doi: 10.1016/j.ophtha.2017.04.008
Sullivan, L. S., Bowne, S. J., Birch, D. G., Hughbanks-Wheaton, D., Heckenlively, J. R., Lewis, R. A., et al. (2006). Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest. Ophthalmol. Vis. Sci. 47, 3052–3064. doi: 10.1167/iovs.05-1443
Sullivan, L. S., Bowne, S. J., Reeves, M. J., Blain, D., Goetz, K., Ndifor, V., et al. (2013). Prevalence of mutations in eyeGENE probands with a diagnosis of autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 54, 6255–6261. doi: 10.1167/iovs.13-12605
Sun, W., Huang, L., Xu, Y., Xiao, X., Li, S., Jia, X., et al. (2015). Exome sequencing on 298 probands with early-onset high myopia: approximately one-fourth show potential pathogenic mutations in RetNet genes. Invest. Ophthalmol. Vis. Sci. 56, 8365–8372. doi: 10.1167/iovs.15-17555
Sundaram, V., Moore, A. T., Ali, R. R., and Bainbridge, J. W. (2012). Retinal dystrophies and gene therapy. Eur. J. Pediatr. 171, 757–765. doi: 10.1007/s00431-011-1615-2
Talib, M., van Schooneveld, M. J., Thiadens, A. A., Fiocco, M., Wijnholds, J., Florijn, R. J., et al. (2019). Clinical and genetic characteristics of male patients with rpgr-associated retinal dystrophies: a long-term follow-up study. Retina 39, 1186–1199. doi: 10.1097/IAE.0000000000002125
Talib, M., van Schooneveld, M. J., Van Cauwenbergh, C., Wijnholds, J., Ten Brink, J. B., Florijn, R. J., et al. (2018). The spectrum of structural and functional abnormalities in female carriers of pathogenic variants in the RPGR gene. Invest. Ophthalmol. Vis. Sci. 59, 4123–4133. doi: 10.1167/iovs.17-23453
Tang, P. H., Jauregui, R., Tsang, S. H., Bassuk, A. G., and Mahajan, V. B. (2019). Optical coherence tomography angiography of RPGR-associated retinitis pigmentosa suggests foveal avascular zone is a biomarker for vision loss. Ophthalmic Surg. Lasers Imaging Retina 50, e44–e48. doi: 10.3928/23258160-20190129-18
Tee, J. J. L., Carroll, J., Webster, A. R., and Michaelides, M. (2017). Quantitative analysis of retinal structure using spectral-domain optical coherence tomography in RPGR-associated retinopathy. Am. J. Ophthalmol. 178, 18–26. doi: 10.1016/j.ajo.2017.03.012
Thiadens, A. A., Soerjoesing, G. G., Florijn, R. J., Tjiam, A. G., den Hollander, A. I., van den Born, L. I., et al. (2011). Clinical course of cone dystrophy caused by mutations in the RPGR gene. Graefes Arch. Clin. Exp. Ophthalmol. 249, 1527–1535. doi: 10.1007/s00417-011-1789-3
Tiwari, A., Bahr, A., Bahr, L., Fleischhauer, J., Zinkernagel, M. S., Winkler, N., et al. (2016). Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 6:28755. doi: 10.1038/srep28755
Traboulsi, E. I. (2010). The Marshall M. Parks memorial lecture: making sense of early-onset childhood retinal dystrophies–the clinical phenotype of Leber congenital amaurosis. Br. J. Ophthalmol. 94, 1281–1287. doi: 10.1136/bjo.2009.165654
Vervoort, R., Lennon, A., Bird, A. C., Tulloch, B., Axton, R., Miano, M. G., et al. (2000). Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 25, 462–466. doi: 10.1038/78182
Walia, S., Fishman, G. A., Swaroop, A., Branham, K. E., Lindeman, M., Othman, M., et al. (2008). Discordant phenotypes in fraternal twins having an identical mutation in exon ORF15 of the RPGR gene. Arch. Ophthalmol. 126, 379–384. doi: 10.1001/archophthalmol.2007.72
Wang, D. Y., Chan, W. M., Tam, P. O., Baum, L., Lam, D. S., Chong, K. K., et al. (2005). Gene mutations in retinitis pigmentosa and their clinical implications. Clin. Chim. Acta 351, 5–16. doi: 10.1016/j.cccn.2004.08.004
Wang, F., Wang, H., Tuan, H. F., Nguyen, D. H., Sun, V., Keser, V., et al. (2014). Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum. Genet. 133, 331–345. doi: 10.1007/s00439-013-1381-5
Wang, H., Wang, X., Zou, X., Xu, S., Li, H., Soens, Z. T., et al. (2015). Comprehensive molecular diagnosis of a large chinese leber congenital amaurosis cohort. Invest. Ophthalmol. Vis. Sci. 56, 3642–3655. doi: 10.1167/iovs.14-15972
Wang, J., Zhang, V. W., Feng, Y., Tian, X., Li, F. Y., Truong, C., et al. (2014). Dependable and efficient clinical utility of target capture-based deep sequencing in molecular diagnosis of retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 55, 6213–6223. doi: 10.1167/iovs.14-14936
Wang, P., Li, S., Sun, W., Xiao, X., Jia, X., Liu, M., et al. (2019). An ophthalmic targeted exome sequencing panel as a powerful tool to identify causative mutations in patients suspected of hereditary eye diseases. Transl. Vis. Sci. Technol. 8:21. doi: 10.1167/tvst.8.2.21
Wawrocka, A., Skorczyk-Werner, A., Wicher, K., Niedziela, Z., Ploski, R., Rydzanicz, M., et al. (2018). Novel variants identified with next-generation sequencing in Polish patients with cone-rod dystrophy. Mol. Vis. 24, 326–339.
Wegscheider, E., Preising, M. N., and Lorenz, B. (2004). Fundus autofluorescence in carriers of X-linked recessive retinitis pigmentosa associated with mutations in RPGR, and correlation with electrophysiological and psychophysical data. Graefes Arch. Clin. Exp. Ophthalmol. 242, 501–511. doi: 10.1007/s00417-004-0891-1
Weleber, R. G., Butler, N. S., Murphey, W. H., Sheffield, V. C., and Stone, E. M. (1997). X-linked retinitis pigmentosa associated with a 2-base pair insertion in codon 99 of the RP3 gene RPGR. Arch. Ophthalmol. 115, 1429–1435.
Wu, D. M., Khanna, H., Atmaca-Sonmez, P., Sieving, P. A., Branham, K., Othman, M., et al. (2010). Long-term follow-up of a family with dominant X-linked retinitis pigmentosa. Eye 24, 764–774. doi: 10.1038/eye.2009.270
Xu, K., Xie, Y., Sun, T., Zhang, X., Chen, C., and Li, Y. (2019). Genetic and clinical findings in a Chinese cohort with Leber congenital amaurosis and early onset severe retinal dystrophy. Br. J. Ophthalmol. 104, 932–937. doi: 10.1136/bjophthalmol-2019-314281
Xu, Y., Guan, L., Shen, T., Zhang, J., Xiao, X., Jiang, H., et al. (2014). Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 133, 1255–1271. doi: 10.1007/s00439-014-1460-2
Yang, L., Yin, X., Feng, L., You, D., Wu, L., Chen, N., et al. (2014). Novel mutations of RPGR in Chinese retinitis pigmentosa patients and the genotype-phenotype correlation. PLoS One 9:e85752. doi: 10.1371/journal.pone.0085752
Yang, Z. L., Peachey, N. S., Moshfeghi, D. M., Thirumalaichary, S., Chorich, L., Shugart, Y. Y., et al. (2002). Mutations in the RPGR gene cause X-linked cone dystrophy. Hum. Mol. Genet. 11, 605–611. doi: 10.1093/hmg/11.5.605
Yokoyama, A., Maruiwa, F., Hayakawa, M., Kanai, A., Vervoort, R., Wright, A. F., et al. (2001). Three novel mutations of the RPGR gene exon ORF15 in three Japanese families with X-linked retinitis pigmentosa. Am. J. Med. Genet. 104, 232–238.
Zahid, S., Khan, N., Branham, K., Othman, M., Karoukis, A. J., Sharma, N., et al. (2013). Phenotypic conservation in patients with X-linked retinitis pigmentosa caused by RPGR mutations. JAMA Ophthalmol. 131, 1016–1025. doi: 10.1001/jamaophthalmol.2013.120
Zampaglione, E., Kinde, B., Place, E. M., Navarro-Gomez, D., Maher, M., Jamshidi, F., et al. (2020). Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet. Med. 22, 1079–1087. doi: 10.1038/s41436-020-0759-8
Zhang, Q. (2016). Retinitis pigmentosa: progress and perspective. Asia Pac. J. Ophthalmol. 5, 265–271. doi: 10.1097/APO.0000000000000227
Zhang, Q., Giacalone, J. C., Searby, C., Stone, E. M., Tucker, B. A., and Sheffield, V. C. (2019). Disruption of RPGR protein interaction network is the common feature of RPGR missense variations that cause XLRP. Proc. Natl. Acad. Sci. U.S.A. 116, 1353–1360. doi: 10.1073/pnas.1817639116
Zhang, Z., Dai, H., Wang, L., Tao, T., Xu, J., Sun, X., et al. (2019). Novel mutations of RPGR in Chinese families with X-linked retinitis pigmentosa. BMC Ophthalmol. 19:240. doi: 10.1186/s12886-019-1250-7
Zhao, K., Wang, L., Wang, L., Wang, L., Zhang, Q., and Wang, Q. (2001). Novel deletion of the RPGR gene in a Chinese family with X-linked retinitis pigmentosa. Ophthalmic Genet. 22, 187–194.
Zhao, P. Y., Hovland, P. G., and Fahim, A. T. (2020). Cystoid macular edema precipitated by altitude in a patient with X-linked retinitis pigmentosa. Ophthalmic Genet. 41, 275–278. doi: 10.1080/13816810.2020.1762901
Zhou, L., Xiao, X., Li, S., Jia, X., and Zhang, Q. (2018). Frequent mutations of RetNet genes in eoHM: further confirmation in 325 probands and comparison with late-onset high myopia based on exome sequencing. Exp. Eye Res. 171, 76–91. doi: 10.1016/j.exer.2018.02.007
Zhou, Q., Yao, F., Wang, F., Li, H., Chen, R., and Sui, R. (2018). A heterozygous mutation in RPGR associated with X-linked retinitis pigmentosa in a patient with Turner syndrome mosaicism (45,X/46,XX). Am. J. Med. Genet. A 176, 214–218. doi: 10.1002/ajmg.a.38501
Zito, I., Downes, S. M., Patel, R. J., Cheetham, M. E., Ebenezer, N. D., Jenkins, S. A., et al. (2003). RPGR mutation associated with retinitis pigmentosa, impaired hearing, and sinorespiratory infections. J. Med. Genet. 40, 609–615.
Zito, I., Gorin, M. B., Plant, C., Bird, A. C., Bhattacharya, S. S., and Hardcastle, A. J. (2000). Novel mutations of the RPGR gene in RP3 families. Hum. Mutat. 15:386. doi: 10.1002/(SICI)1098-1004(200004)15:4<386::AID-HUMU23<3.0.CO;2-4
Keywords: RPGR, retinitis pigmentosa, genotype, phenotype, exome sequencing
Citation: Yang J, Zhou L, Ouyang J, Xiao X, Sun W, Li S and Zhang Q (2021) Genotype–Phenotype Analysis of RPGR Variations: Reporting of 62 Chinese Families and a Literature Review. Front. Genet. 12:600210. doi: 10.3389/fgene.2021.600210
Received: 29 August 2020; Accepted: 27 April 2021;
Published: 23 June 2021.
Edited by:
Musharraf Jelani, Islamia College University, PakistanReviewed by:
Nelson L. S. Tang, The Chinese University of Hong Kong, ChinaAsifullah Khan, Abdul Wali Khan University Mardan, Pakistan
Copyright © 2021 Yang, Zhou, Ouyang, Xiao, Sun, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingjiong Zhang, emhhbmdxamlAbWFpbC5zeXN1LmVkdS5jbg==; emhhbmdxaW5namlvbmdAZ3p6b2MuY29t
†These authors have contributed equally to this work