 Hong Nan
Hong Nan Richard A. Ludlow2
Richard A. Ludlow2 Min Lu
Min Lu- 1College of Agriculture, Guizhou University, Guiyang, China
- 2School of Biosciences, Cardiff University, Cardiff, United Kingdom
Dof (DNA binding with one finger) proteins play important roles in plant development and defense regulatory networks. In the present study, we report a genome-wide analysis of rose Dof genes (RchDof), including phylogenetic inferences, gene structures, chromosomal locations, gene duplications, and expression diversity. A total of 24 full-length RchDof genes were identified in Rosa chinensis, which were assigned to nine distinct subgroups. These RchDof genes were unevenly distributed on rose chromosomes. The genome-scale analysis of synteny indicated that segmental duplication events may have played a major role in the evolution of the RchDof gene family. Analysis of cis-acting elements revealed putative functions of Dofs in rose during development as well as under numerous biotic and abiotic stress conditions. Moreover, the expression profiles derived from qRT-PCR experiments demonstrated distinct expression patterns in various tissues, and gene expression divergence existed among the duplicated RchDof genes, suggesting a fundamentally functional divergence of the duplicated Dof paralogs in rose. The gene expression analysis of RchDofs under drought and salt stress conditions was also performed. The present study offered novel insights into the evolution of RchDofs and can aid in the further functional characterization of its candidate genes.
Introduction
Plants have developed diverse molecular mechanisms to survive against various types of biotic and abiotic stress conditions. Numerous transcription factors have been identified in plants, which confer tolerance to a broad range of stress conditions. They are important regulators for adjusting gene expression by binding to specific DNA sequences at their promoter region (Wray et al., 2003). The Dof (DNA binding with one finger) gene family is one of the plant-specific transcription factors that is widespread in higher plants. Since the first Dof gene (Zmdof1) was isolated from maize (Yanagisawa and Izui, 1993), numerous Dof genes have been studied in other plants (Jin et al., 2014; Yang et al., 2018; Zhang et al., 2018; Hong et al., 2019).
The Dof gene family contains a highly conserved Dof domain at the N-terminus of approximately 52 residues in length. The Dof domain has a C2–C2 finger structure (CX2CX21CX2C) that can specifically bind to a core sequence (AT/AAAAG) in plant gene promoters and regulate downstream genes (Yanagisawa, 2002; Wang et al., 2019). However, some Dof proteins, such as AOBP in pumpkin, can bind to AGTA motif but have lost the capability to interact with the AT/AAAAG motif (Kisu et al., 1998). In addition to the Dof domain, Dof proteins harbor a bipartite nuclear localization signal that partly overlaps with the conserved Dof domain (Krebs et al., 2010) and a variable C-terminal transcriptional regulation domain (Yanagisawa, 2001). Moreover, the Dof domain and some specific amino acids in the C-terminal region also bind to specific DNA sequences to regulate various physiological activities (Yanagisawa and Schmidt, 1999).
DNA binding with one finger members have been reported to participate in the regulation of gene expression in diverse physiological processes. For example, the Dof transcription factor MdDof24 identified in apple was reported to be associated with flower development and the regulation of metabolic pathways (Yang et al., 2018). PbDof9.2 in pear (Pyrus bretschneideri) was reported to regulate flowering time. Overexpression of PbDof9.2 in Arabidopsis could delay flowering time via interactions with the promoters of PbTFL1a and PbTFL1b (Liu et al., 2020). The peach Dof transcription factor FaDof2 positively regulates eugenol biosynthesis by interacting with FaEOBII (Molina-Hidalgo et al., 2017). In addition, Dof transcription factors are also involved in abiotic and biotic stress responses, including heat, salt, drought, and pathogen attack. A total of 60 Dof genes were recently identified in the apple genome, and the expression levels of most MdDof members were upregulated by heat and salt stress conditions, revealing the important function of Dof genes in abiotic stress tolerance (Zhang et al., 2018). Overexpression of tomato SlCDF1 (SlDof25) and SlCDF3 (SlDof26) genes in Arabidopsis can increase tolerance to salt and drought stress (Corrales et al., 2014). The transient expression of BBF1-related Dof genes in tobacco enhanced the expression profiles of the mosaic viral resistance gene N and defense-related genes (Sasaki et al., 2015).
The Chinese rose (Rosa chinensis Jacq.) is an economically important flower crop of the Rosaceae family, which is widely cultivated in China (Feng et al., 2015). Although the cultivation of R. chinensis is increasing, it suffers from various biotic and abiotic conditions of stress. Drought and salt stress, which limit the growth and productivity of R. chinensis, had become the most harmful factors in the irrigated areas of China (Tian et al., 2018, 2019). Therefore, a better understanding of the molecular basis of drought and salt tolerance is required to breed new varieties with desirable traits. Despite the important role of Dof genes in plant drought and salt stress resistance, their exact functions have not yet been well studied in rose. The recently sequenced rose genome provides a framework for the identification and functional characterization of gene families (Raymond et al., 2018). Here we comprehensively characterized the number, structure, chromosomal locations, and phylogenetic associations of the Dof gene family throughout the rose genome. We also examined the expression differences of Dof genes in different tissues and in response to drought and salt stress conditions. The present study will form the foundation for further functional analysis of the Dof genes in rose.
Materials and Methods
Identification of Putative Dof Genes
The Dof genes of A. thaliana were obtained from tair1. The rose (R. chinensis “Old Blush”) genome sequences were downloaded from a rose website2. To comprehensively identify the Dof genes, the HMM file (PF02701) of the Dof domain was obtained from the Pfam database3 and used to perform the HMMER search (version 3.34) with an E-value < 1e–5. The resulting Dof sequences were then adopted for TBLASTN as described before with default parameters (Altschul et al., 1997; Zou et al., 2019). Finally, following the removal of incorrect and redundant predicted sequences, the sequences of all candidate Dofs were further confirmed using ScanProsite5 and InterProScan6. The molecular weight (MW) and isoelectric point (pI) of the Dof proteins were evaluated using the ExPASy-ProtParam online software7. The subcellular localization of the Dof proteins was predicted using Plant-mPLoc8 (Chou and Shen, 2010). As a control, the apple and pear genome sequences were downloaded from GDR9 and Pear Genome Project10, respectively. The Dof transcription factors of these two Rosaceae species were also identified using the same method as described above.
Phylogenetic Analysis of RchDofs
To investigate the phylogenetic associations among Dofs, a multiple sequence alignment including RchDof protein sequences and those from Arabidopsis, apple, and pear was performed using MUSCLE with default parameters in MEGA7 (Kumar et al., 2016). Subsequently, a maximum likelihood (ML) tree based on the above-mentioned alignment was constructed. The reliability of the obtained phylogenetic tree was tested using a bootstrap value of 1,000 iterations. To further determine the best-fit substitution model for the phylogeny tree, the ProtTest program (version 3.4) was used. Based on the multiple alignments of the Dof proteins and the classification of Lijavetzky, the RchDof genes were assigned to nine subgroups (Lijavetzky et al., 2003).
Gene Structure Analysis and Identification of Conserved Motifs
To understand the structures of the RchDof genes, the GSDS (version 2.011) online software was used to characterize the exon–intron structures. The motifs of each deduced RchDof protein were analyzed by MEME (version 4.12.012) (Bailey et al., 2009), with the maximum number set to 30. The 1,500-bp upstream sequences of the RchDof genes were extracted with an in-house Perl script to predict cis-elements using the PlantCARE13 (Lescot et al., 2002).
Determination of Chromosomal Distribution, Gene Duplication, and Synteny
The chromosomal location information of RchDofs was obtained from the GFF3 file. The synteny, segmental duplication, and tandem duplication were analyzed using a previously reported method (Nan and Gao, 2019) and visualized (including gene positions) by Circos (version 0.69) (Krzywinski et al., 2009). To estimate the duplication events of RchDof genes, Ka and Ks values were measured using the maximum likelihood method implemented in codeml program (Yang, 2007). The Ks values were subsequently used to approximately date the duplication event according to T = Ks/2λ, assuming clock-like rates (λ) of synonymous substitution of 1.5 × 10–8 (Zhang et al., 2018).
Ortholog Dof Gene Identification
The orthologs of the candidate RchDofs in Arabidopsis were identified using Ensembl Plants (release 46,14). In addition, the orthologs in the tomato plant for each RchDof gene were analyzed as described previously (Nan and Gao, 2019), and the well-categorized tomato Dof sequences were obtained from Corrales (Corrales et al., 2014).
Plant Material and Treatments
In this study, 3-year-old R. chinensis “Old Blush” plants were used as experimental materials, which were grown in the greenhouse at 22/18°C day/night temperature and 16/8 h day/night photoperiod. Two tissues, including matured leaves and fully blooming flowers, were collected for tissue-specific gene expression analysis. The drought stress treatment was performed with 20% PEG600, and matured leaves were collected at 0, 2, 4, 8, and 24 h following treatments (Li et al., 2016, 2017). Rose plants grown without drought stress were used as an unstressed control. A salinity stress treatment was carried out by irrigating the plants with 200 mM NaCl, followed by sampling matured leaves at 0, 2, 4, 8, and 24 h following treatments (Li et al., 2016, 2017). The plants irrigated with sterile water were used as a control. All samples were immediately frozen in liquid nitrogen and stored at −80°C until use. Three biological replicates were performed for each treatment.
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction
Total RNA was extracted with Trizol reagent (Invitrogen) and treated with RNase-free DNase I (Transgen). Subsequently, 1 μg of total RNA was reverse-transcribed to cDNA using an All-in-One First-Strand cDNA Synthesis Kit (Transgen) according to the manufacturer’s instructions. Gene-specific primers of the Dof genes were designed using the Primer Premier 5.0 software and are presented in Supplementary Table 1. The GAPDH gene was selected as a reference gene according to a previous study (Tian et al., 2018), and three technical replicates were conducted for each sample. Real-time PCR was performed with a CFX96 real-time PCR detection system (Bio-Rad). Each reaction was carried out in a final volume of 10 μl. The reaction mixture contained the following reagents: 1.0 μl cDNA, 0.4 μl of each primer pair, 5 μl SYBR, and 3.6 μl ddH2O. The RT-PCR cycle was set as follows: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s, and a final extension at 60°C for 30 s. A melting curve analysis for assessing specific amplification was performed by heating the products from 65°C to 95°C with 0.5°C increments. The 2–Δ Δ CT method (Livak and Schmittgen, 2001) was used to analyze real-time PCR data, and the time point 0 h was used as an untreated control (expression = 1.0) to estimate the fold change in the expression levels of the relevant genes.
Statistical Analyses
The experiment was performed in three biological replicates. All data were expressed as mean ± standard deviations (SD) following normalization. Statistical analysis was performed using SPSS software (version 18.0). Data were analyzed using Fisher’s least significant difference analysis, and significant differences were reported at P < 0.05 level.
Results
The Dof Gene Family in the Rose Genome
Using the consensus sequences of the Dof domains, we screened the rose genome assembly. We identified 24 non-redundant RchDof genes and named them as RchDof1 to RchDof24 based on the order of the gene IDs (Supplementary Table 2). The open reading frame lengths of the RchDof genes ranged from 173 to 531 amino acids, with the pI of the resultant proteins predicted to range from 4.82 to 9.48 and the MW from 19.52 to 56.83 kDa (Supplementary Table 2). The predicted grand average of hydropathicity values of the RchDof proteins varied from −0.888 (RchDof14) to −0.397 (RchDof4), suggesting that they were hydrophilic. Furthermore, the predicted subcellular localizations indicated that all RchDof proteins were located in the nucleus. In addition, multiple sequence alignment of RchDofs revealed a highly conserved Cys2/Cys2 Zn2+ DNA binding domain (Figure 1), which was designated as the Dof domain. The distribution of amino acid residues at the corresponding positions of the rose Dof domain revealed that it was very similar to that of Arabidopsis (Lijavetzky et al., 2003), apple (Hong et al., 2019), tomato (Cai et al., 2013), pear (Liu et al., 2020), cassava (Zou et al., 2019), and pepper (Wu et al., 2016), indicating that the Dof domain was highly conserved among different higher plants. The detailed information of these RchDof genes regarding the type of genes and the Dof domains is shown in Supplementary Table 2 and Figure 1.
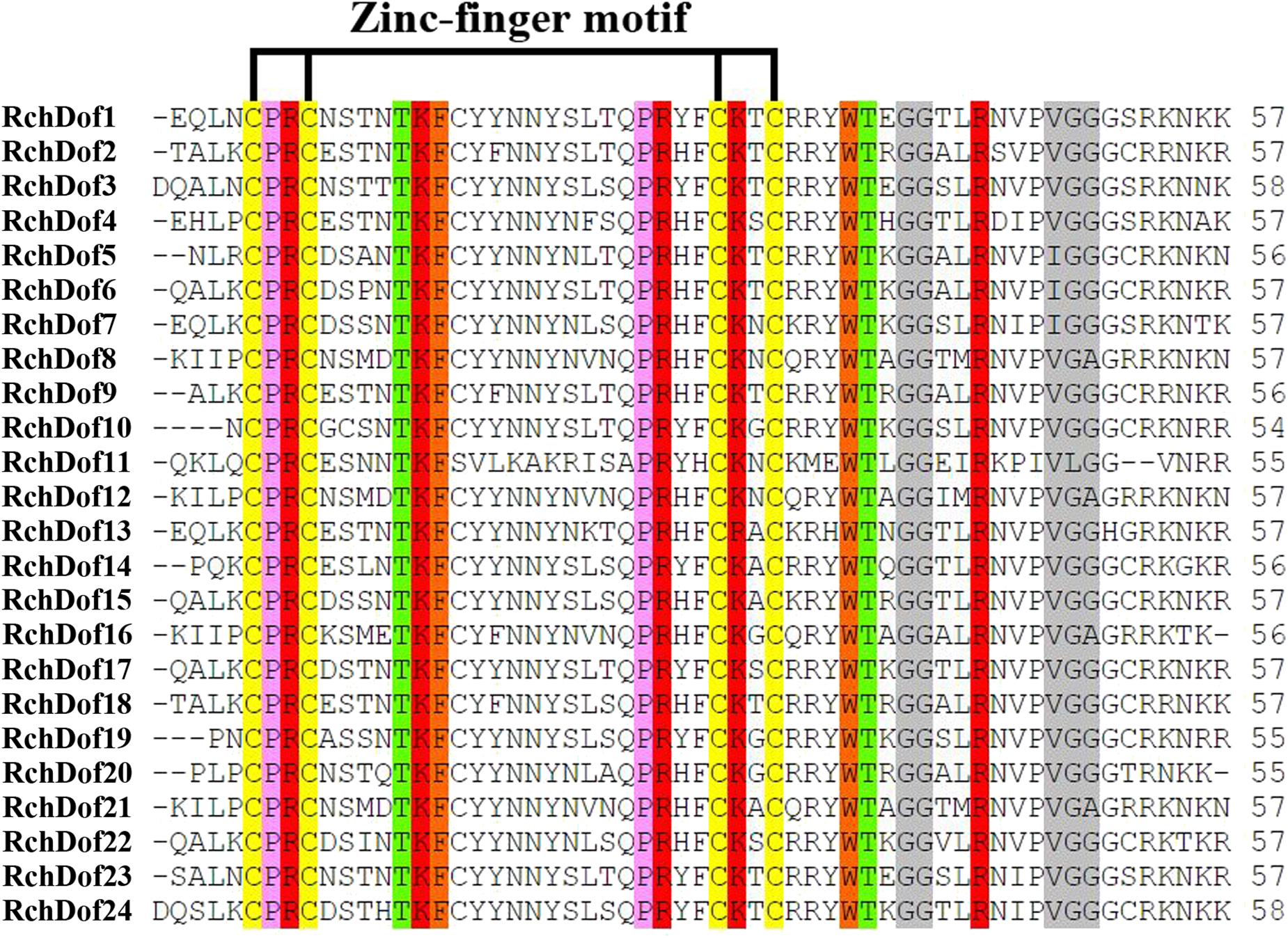
Figure 1. Dof domain sequence alignment of rose Dof proteins. Only the conserved Dof domain residue is currently shown. The four cysteine residues putatively responsible of the zinc-finger structure are indicated. Identical and similar amino acids are highlighted.
Phylogenetic Analysis of RchDof Genes
To further examine the evolutionary relationships in the Dof genes, an un-rooted ML phylogenetic tree was constructed using R. chinensis and other species (Arabidopsis, apple, and pear). Using the ProtTest program, we found that JTT + G was the best substitution model. As shown in Figure 2, the Dof proteins in the four species were classified into nine groups, namely, A, B1, B2, C1, C2.1, C2.2, C3, D1, and D2, respectively. The 24 RchDof genes were unevenly distributed in the nine subgroups. Class B1 was the largest subfamily, which contained five RchDof factors. Class C3 was the smallest class, containing only one member. Furthermore, classes A, B2, C2.2, and D2 were present at the same proportion of 8.33% (Figure 2). The phylogenetic tree showed that all the nine classes were monophyletic, except for B2. Group B2 could be divided into two clades, one of which was the largest and clustered with class B1, and the other was clustered with class A. These results were consistent with the past results of physic nut and castor bean (Zou and Zhang, 2019). In addition, the phylogenetic analysis also indicated that most Dof genes of rose were clustered with the Dof members of apple and pear, suggesting a close relationship among the three Rosaceae species. When the number of Dof genes was compared among these three species, we observed that the number in apple and pear was greatly increased by almost 2.5 and 1.9 times than that of rose. The results indicated that a large-scale expansion of Dof members in apple and pear seemingly occurred after the divergence of the three Rosaceae species, evidenced by the Dof members of apple and pear exclusively clustered together in most groups, respectively.
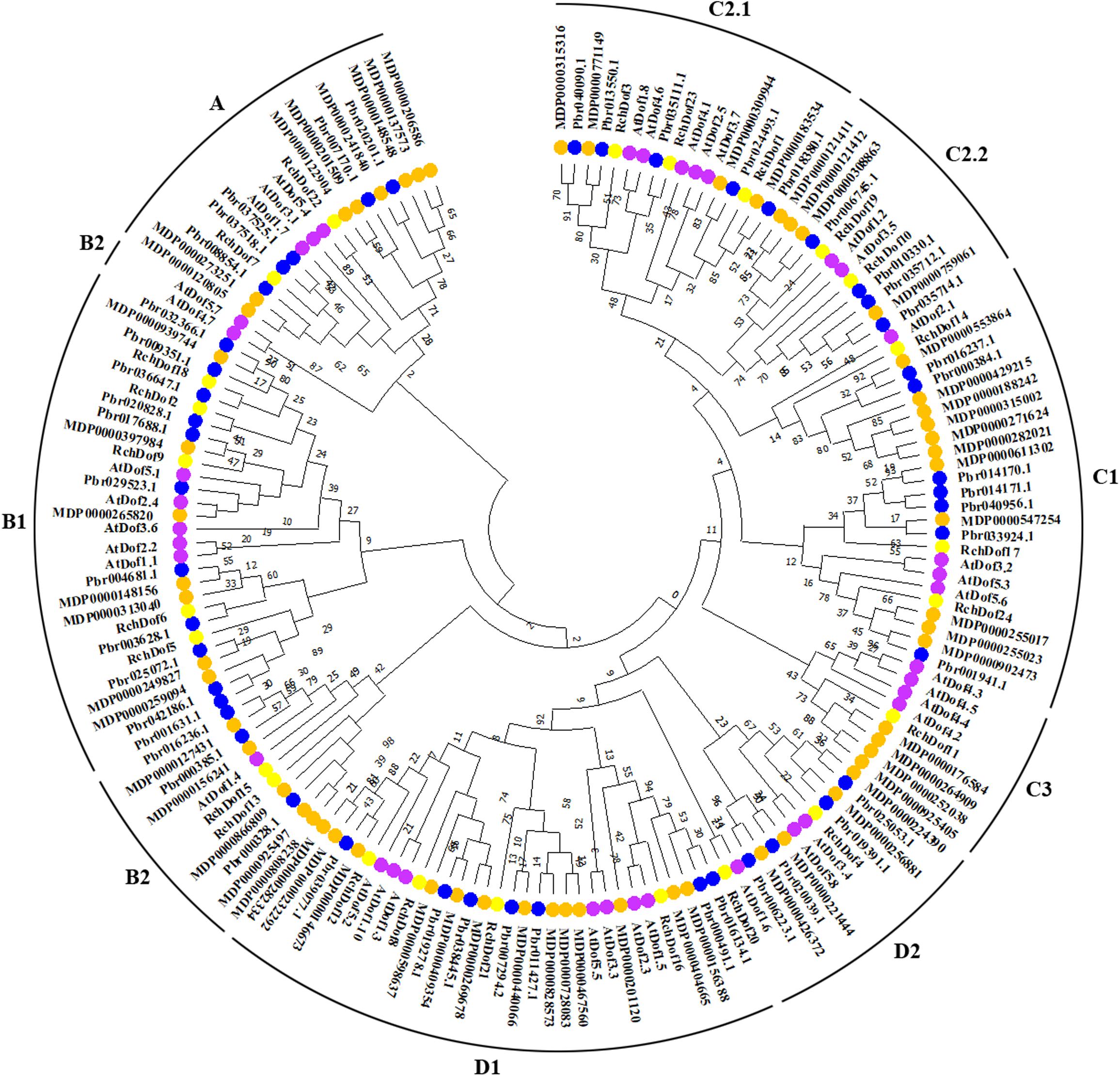
Figure 2. Phylogenetic tree based on Dof proteins from rose, Arabidopsis, apple, and pear. The phylogenetic tree was constructed using the maximum likelihood method. The reliability of the predicted tree was tested using bootstrapping with 1,000 replicates.
Protein Structure of the RchDof Gene Family
To reveal the structural variation of the RchDof genes in rose, we predicted putative motifs using the program MEME (Bailey et al., 2009) and identified a total of 30 distinct motifs. The schematic distribution of these motifs among different gene groups is described (Figure 3), representing their relative locations within the proteins. The multi-level consensus sequences were produced among these motifs (Table 1). Among the 30 identified motifs, motif 1 was considered the Dof domain and was uniformly observed across all the RchDof proteins. In contrast to motif 1, majority of subgroups of RchDofs exhibited several special motifs at their C-terminal regions, and little was known about these structures. Motifs 16 and 21 were widely present in most members of group B1, motif 12 was limited to group C2.1, motif 19 was present in all members of group C2.2, motif 9 was limited to group D1, and motif 20 was present in all members of group D2 (Figure 3). Moreover, as expected, the majority of closely associated members in the phylogenetic tree possessed common motif compositions, suggesting a functional similarity among the Dof proteins within the same subfamily (Figure 3). Almost all members in group C2.1 possessed motifs 12, 13, and 28, group B1 usually contained motifs 3, 5, 13, 16, and 21, and group D1 harbored motifs 8, 7, 17, 6, 4, 10, 15, 2, and 9 in order. The motif distribution differences among the different groups or subgroups indicated the functional divergence of the Dof gene family. The protein structure of the Dof genes corroborated with the ML phylogenetic tree.
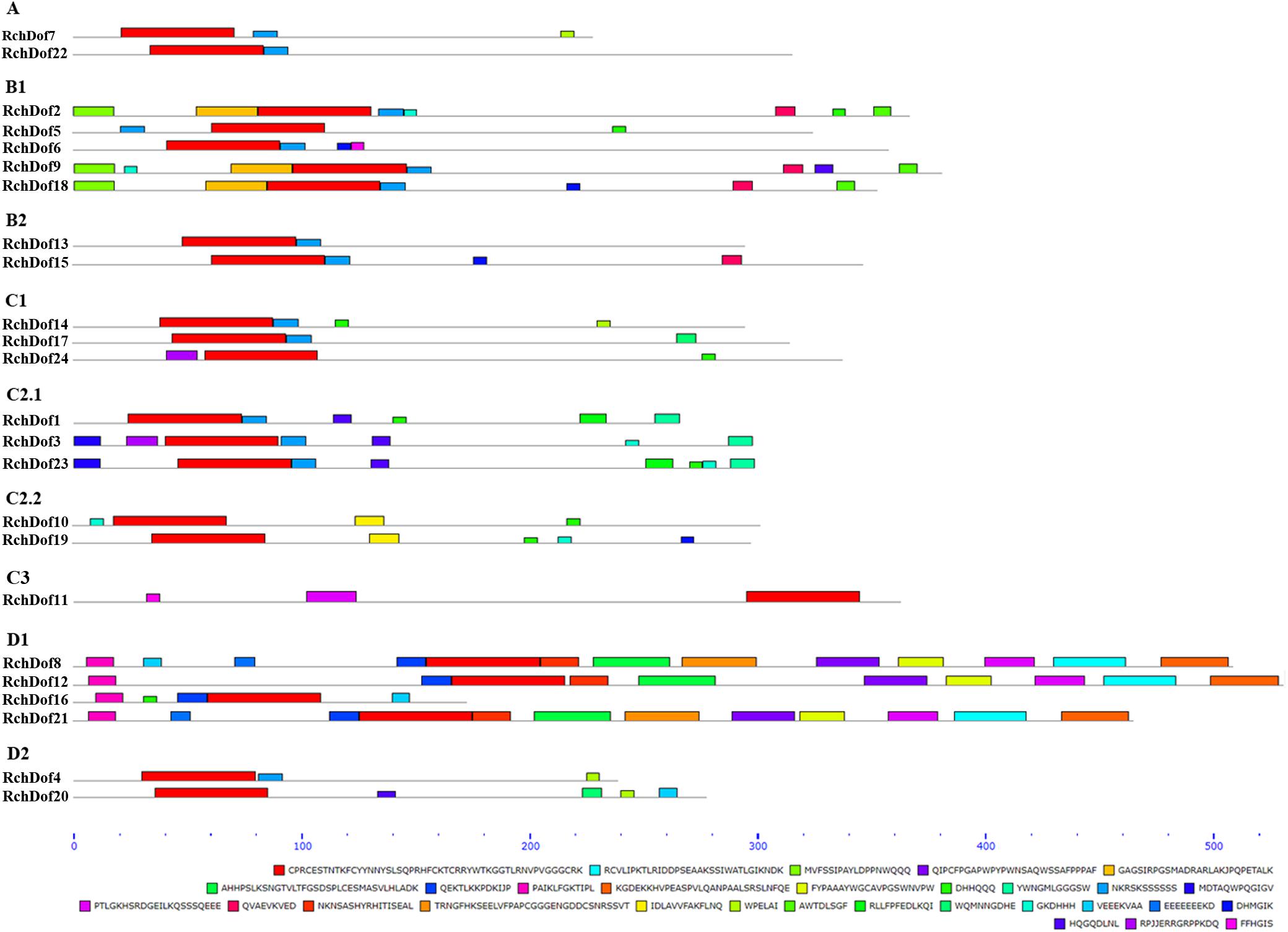
Figure 3. Conserved motif compositions of the RchDof gene family. The conserved motifs were detected using MEME software and represented by colored boxes. The length of RchDof proteins can be estimated using the scale at the bottom, and the conserved motifs are shown in Table 1.
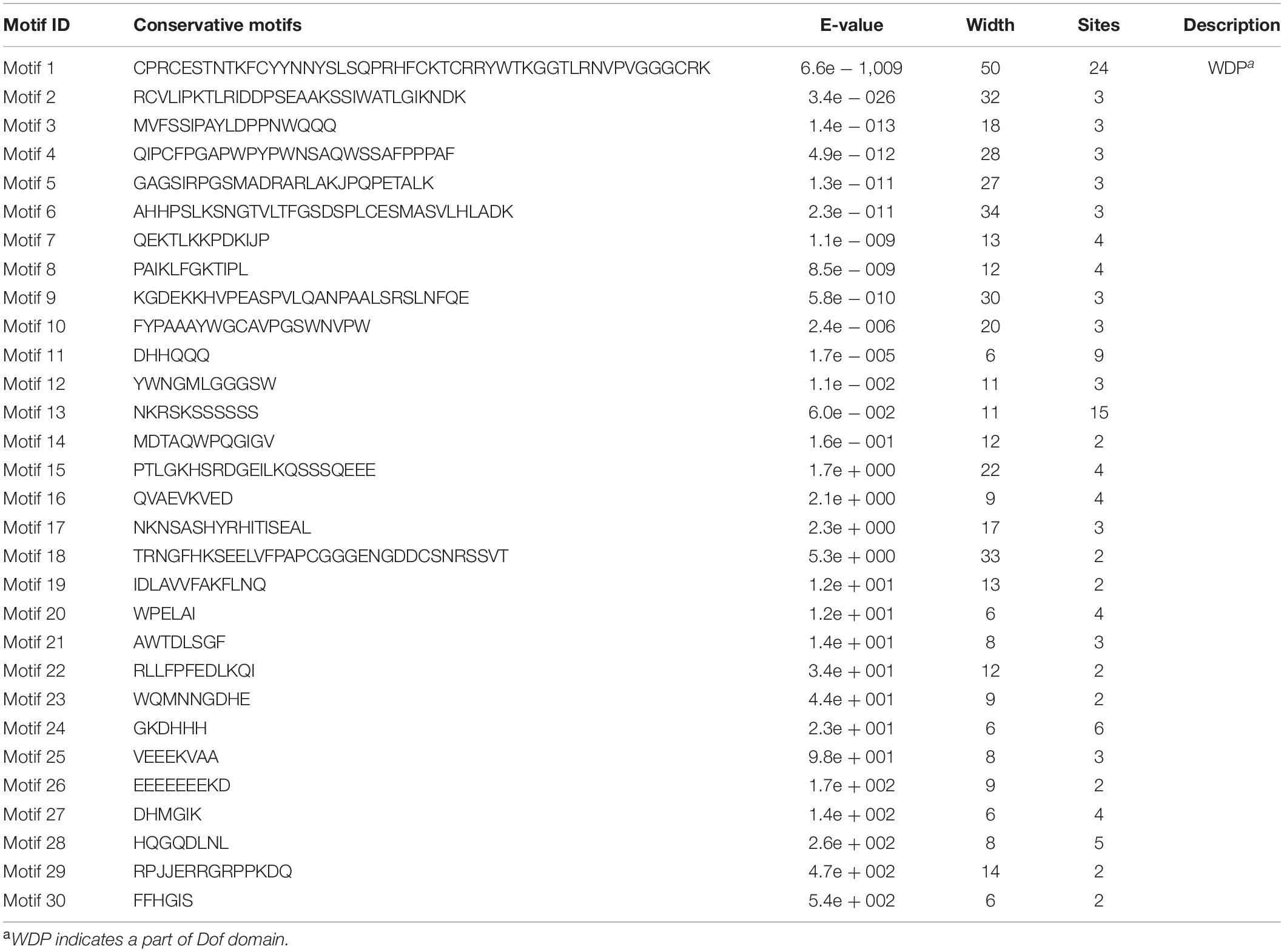
Table 1. Conserved motifs of RchDof proteins in the Rosa genome.
Exon–Intron Organization of the RchDof Genes
To further examine the evolution of the RchDof genes, we investigated their exon–intron structures. As illustrated in Figure 4, the exon numbers of RchDof genes ranged from one to eight. Nearly all the RchDof genes exhibited no intron or a single intron, except for RchDof11. Similar exon–intron structures were also observed in the Dof genes of Arabidopsis, rice, castor bean, and cassava (Lijavetzky et al., 2003; Jin et al., 2014; Zou et al., 2019). Moreover, the data showed that the RchDof genes in the same group exhibited similar exon–intron compositions. For example, the majority of C2.1 and D1 contained one intron, whereas groups C2.2 and D2 had no intron. Interestingly, we found that there were no significant differences among RchDof genes in the intron phase, except for RchDof11, which contained phase 0, 2, 0, 1, 0, 0, 2, and 2 introns.
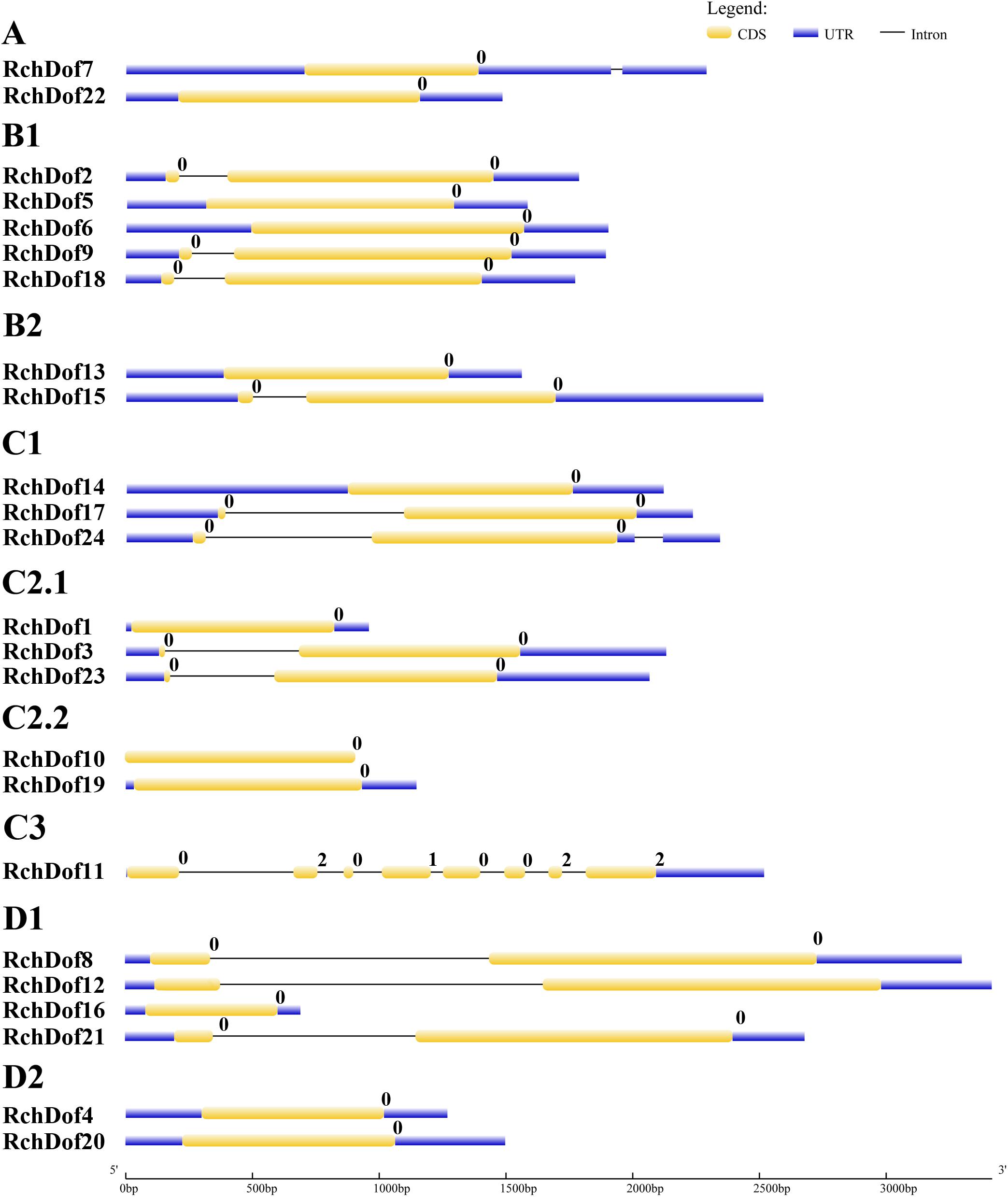
Figure 4. Exon–intron structures of RchDof genes. The exons and introns are indicated by yellow rectangles and black lines, respectively. The length of RchDof proteins can be estimated using the scale at the bottom. The intron phases are indicated as numbers 0, 1, and 2.
Stress-Related Cis-Elements in Promoters of RchDof Genes
In order to investigate the evolution and functional divergence of the RchDof genes, the upstream 1.5-kb promoter regions of all the RchDof members were extracted and analyzed using the PlantCARE online software. Various cis-acting regulatory elements were analyzed, including 10 elements related to plant development and 11 motifs associated with stress response (Supplementary Table 3). As shown in Supplementary Table 3, all members contained more than one cis-element, and the majority of the RchDof genes possessed box4, G-box, CGTCA motif, and ABRE. It is interesting to note that two cis-regulation elements, including the ACE and GARE motif, were only contained in RchDof8 and RchDof4, respectively. A total of eight RchDofs possessed the W-box (TTGACC), which regulates gene expression by binding to the WRKY transcription factors, suggesting that these genes may be cross-regulated by other proteins. MBS is a MYB-binding site involved in drought response and was identified in eight genes, indicating that these RchDofs participated in drought stress response.
Chromosomal Location, Gene Duplication, and Genomic Synteny of RchDof Genes
To examine the genomic distribution of the RchDof genes, their chromosomal locations were searched against the rose genome database. The results indicated that 24 RchDofs were unevenly distributed on the seven pseudo-chromosomes. As shown in Figure 5, chromosome 5 exhibited the largest number of the RchDof genes, followed by chromosome 2, which had five, and chromosome 4 that harbored only one.
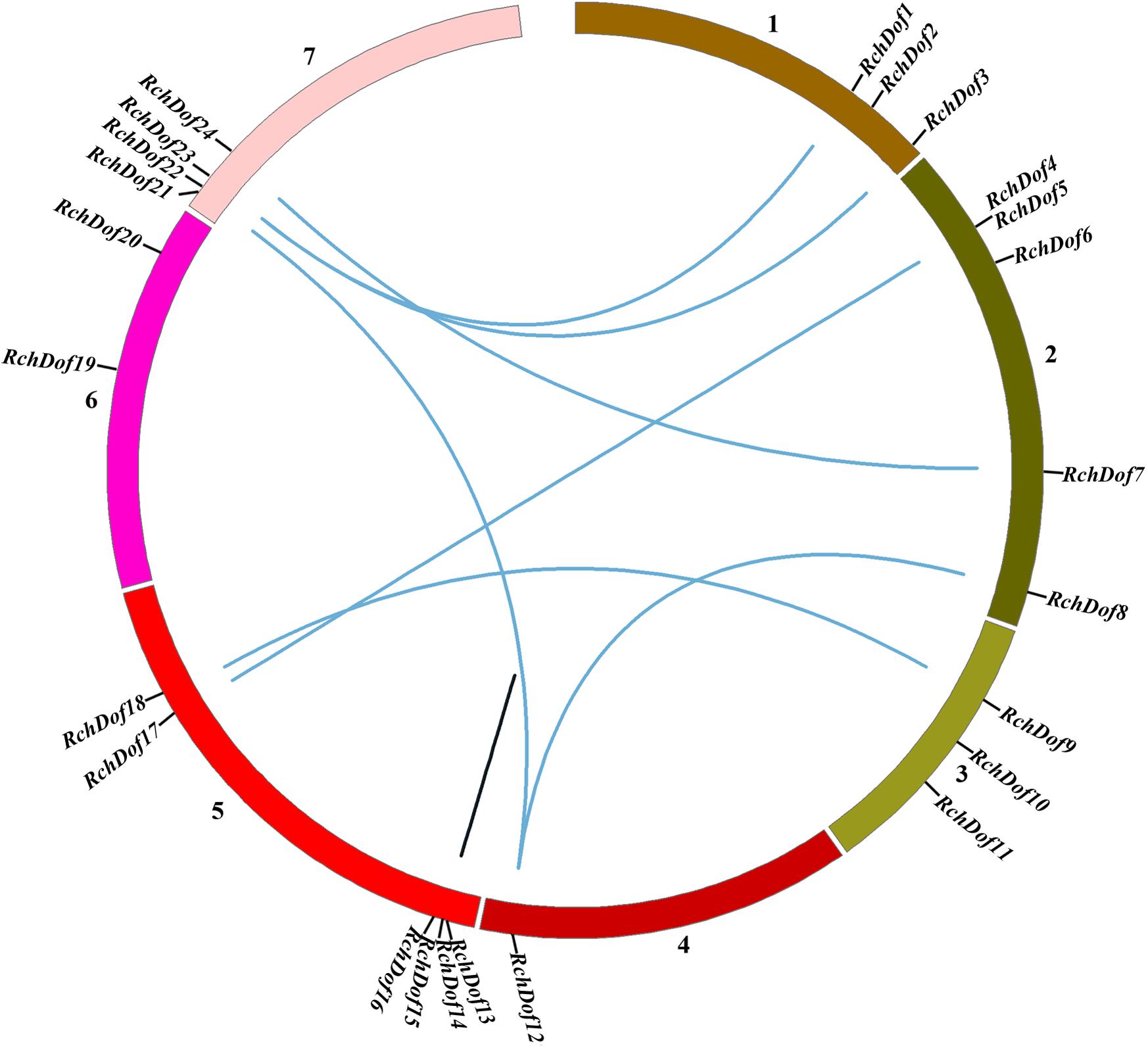
Figure 5. Chromosome distribution and synteny analysis of rose Dof genes. Chromosomes 1–7 are shown with different colors. The approximate location of each RchDof gene is marked with a short black line. Blue lines indicate segmentally duplicated RchDof genes, and black lines in the circle indicate tandemly duplicated genes.
It has been reported that gene duplication events are a primary source of genetic novelty and the main effect in gene family expansion (Moore and Purugganan, 2005). Duplications are classified into three types: segmental, tandem, and dispersed (Li et al., 2012). In order to trace the origins of the RchDof genes in rose, we performed syntenic analysis with MCScanX. The collinear relationships of the duplicated pairs in the RchDof genes are shown in Table 2. Out of the 24 RchDof genes, seven pairs (12 RchDof genes) might have resulted from segmental duplication events, while the remaining one cluster (RchDof14 and RchDof15) probably originated from tandem duplication events. The results indicate that segmental duplication has played a predominant role in the evolution of the RchDof genes. In other plants, the majority of Dof genes were also found to derive from segmental duplication events, such as in apple (Zhang et al., 2018), cassava (Zou et al., 2019), tomato (Cai et al., 2013), and cotton (Li et al., 2018). In addition, the majority of the segmentally duplicated genes belonged to the same RchDof subgroup, with the exception of two clusters (RchDof5/RchDof17 and RchDof7/RchDof24) (Table 2). This phenomenon may result from the gain or loss of certain motif structures. As shown in Figure 5, chromosomes 2 and 7 contained the most segmentally duplicated RchDof genes, while chromosome 6 harbored none. The tandem-duplicated gene pair was located on chromosome 5. In addition, the two tandem-duplicated genes in one pair belonged to different classes (Table 2). These results indicate that the tandem-duplicated pairs may have undergone functional divergence.
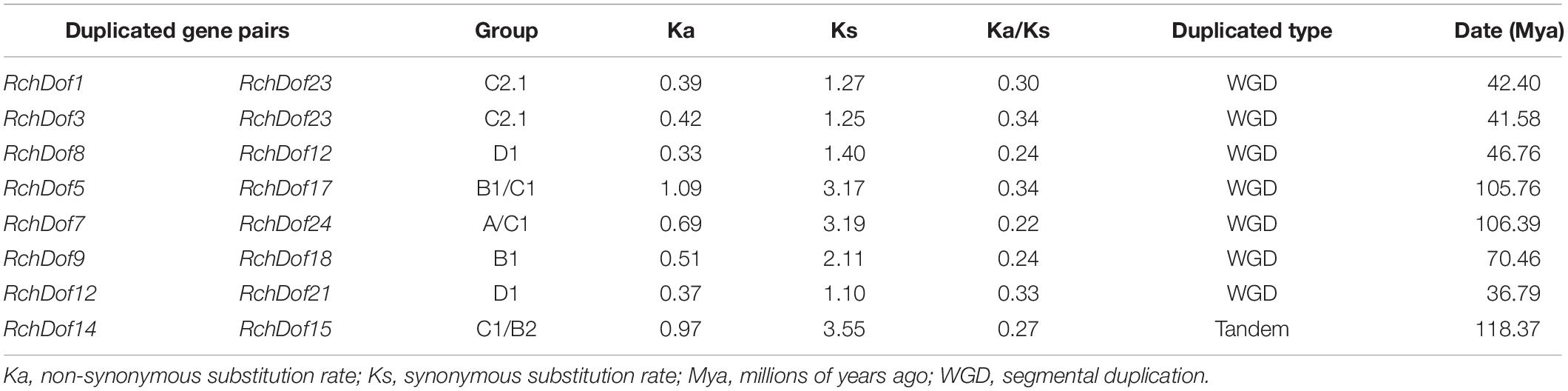
Table 2. Ka/Ks calculation and divergence times of the duplicated RchDof gene pairs in syntenic blocks.
As the ratio Ka/Ks is a good indicator of the selective pressure occurring at the protein level, we used the PAML software to estimate the Ks (synonymous) and Ka (non-synonymous) values as well as the Ka/Ks ratio. These duplication events were approximately dated using the formula: T = Ks/2r (Table 2). The duplication-derived RchDof genes spanned from 36.79 to 118.37 Mya (millions of years ago). It is often assumed that the values Ka/Ks < 1, Ka/Ks = 1, and Ka/Ks > 1 indicate negative selection, neutral evolution, and positive selection, respectively (Li, 1993; Yang and Bielawski, 2000). All duplicated RchDofs from the seven segmentally duplicated gene pairs had Ka/Ks < 1, ranging from 0.22 to 0.34, while the Ka/Ks of the tandem-duplicated gene pair was 0.27. The results suggest that all of the duplicated gene pairs are under strong purifying selection, which corroborates with observations in other plants, such as apple and tomato (Cai et al., 2013; Zhang et al., 2018).
Profiling of RchDof Gene Expression
The expression patterns of the 24 RchDof genes in leaves and flowers were examined using real-time reverse transcription-PCR (qRT-PCR) experiments. The primer pairs for all genes are listed in Supplementary Table 1. The results indicated that the RchDof genes were differentially expressed in these two tissues (Figure 6). Some RchDof genes, such as RchDof5 (group B1), RchDof7 (group A), RchDof8 (group D1), RchDof12 (group D1), and RchDof15 (group B2), were highly expressed at maximum levels in leaves. The remaining RchDof genes, such as RchDof9 (group B1), RchDof13 (group B2), and RchDof17 (group C1), exhibited a low expression in leaves, whereas they were highly expressed in flowers. Interestingly, the results suggested that RchDof genes that were classified as of the same subgroup could exhibit distinctive expression patterns among tissues, such as RchDof13 (group B2) and RchDof15 (group B2). In addition, gene expression divergence was also investigated by comparing the levels of the duplicated RchDof genes. The results indicated that the expression divergence was also present among duplicated RchDof paralogs. For example, RchDof1 and RchDof23 were segmentally duplicated gene pairs. RchDof1 expression was upregulated in the leaves and downregulated in the flowers, while the expression of its paralog RchDof23 was downregulated in the leaves and upregulated in the flowers. Previous studies reported that tissue-specific expression divergence is one of the most important indicators of functional differentiation between genes (Makova and Li, 2003; Li et al., 2005). Therefore, the same subgroup and the expanded Dof genes may result in novel biological function during plant evolution, which is beneficial to regulate various physiological processes by removing their redundancy.
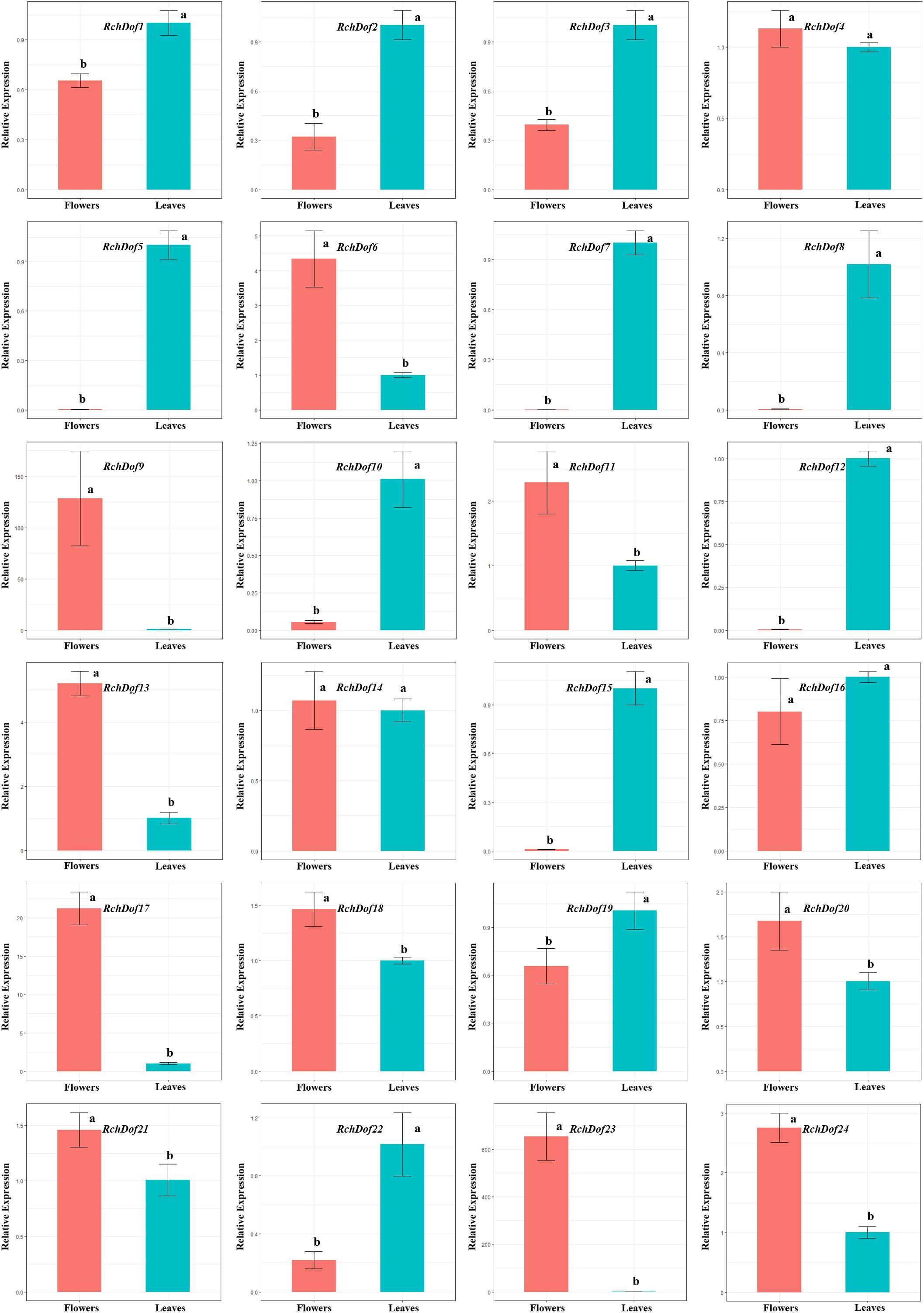
Figure 6. Expression patterns of RchDof genes in tissues of rose. The x-axis shows different tissues, while the y-axis represents the relative expression levels of RchDof genes compared with GAPDH gene using 2− ΔΔCT method. Error bars indicate the standard deviations (mean ± SD) of three independent replicates. The histogram bars labeled with different letters (a and b) above them are significantly different (least significant difference test, P < 0.05).
Expression Profiling of RchDof Gene Under Drought and Salt Stress
The functions of the rose Dof genes in response to abiotic stress are largely unknown. In order to clarify the potential functions of the RchDof genes in response to abiotic stress conditions, we analyzed their expression patterns under specific stress conditions, including drought and salinity treatment. The analysis was performed using qRT-PCR in leaves.
As shown in Figure 7, RchDof genes were sensitive to drought stress, the majority of which exhibited different expression patterns. Some genes were evidently up-regulated after 8 h and followed by a decrease, such as RchDof5, RchDof11, RchDof12, and RchDof21. Some were gradually induced, peaked at 2 h, and then decreased in 4, 8, and 24 h (RchDof6 and RchDof22). The greatest increase in expression (nearly 19-fold) occurred for the RchDof7 gene at 4 h. The results indicated that the expression levels of RchDofs were responsive to drought stress. The expression divergence was also examined by comparing the expression levels of the duplicated RchDof genes. For example, the pair of duplicated genes RchDof9 and RchDof18 was a case of segmental duplication. RchDof9 indicated the higher expression following 4 h, whereas RchDof18 reached the higher level in the leaves following 8 h.
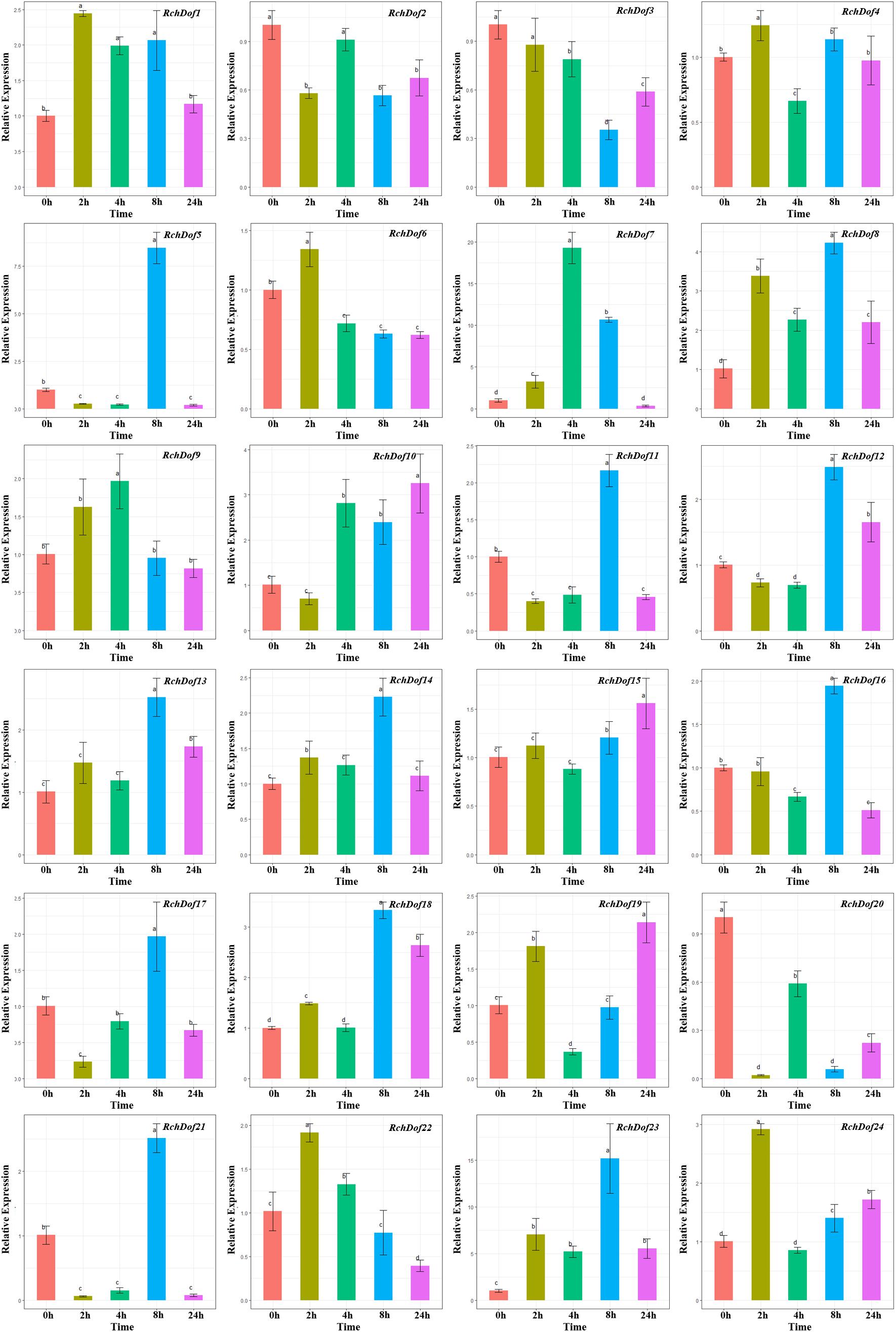
Figure 7. Expression patterns of RchDof genes under drought stress. Samples were collected at 0, 2, 4, 8, and 24 h after treatment. Error bars indicate the standard deviations (mean ± SD) of three independent replicates. The histogram bars labeled with different letters (a–d) above them are significantly different (least significant difference test, P < 0.05).
To investigate the expression pattern of the RchDof genes under salt stress conditions, the plants were irrigated with 200 mM NaCl solution. Salt treatment resulted in a wide variety of RchDof gene expression profiles. As described in Figure 8, the gene expression levels of RchDof9, RchDof10, RchDof17, and RchDof20 were rapidly increased by 4.8, 3, 6.6, and 6.7 times following 2 h, respectively. After that time period, they decreased. The expression levels of RchDof23 sharply decreased at 2 h and reached the lowest levels after 4 h. In addition, the greatest increase in expression levels was noted for RchDof7 (nearly 163-fold).
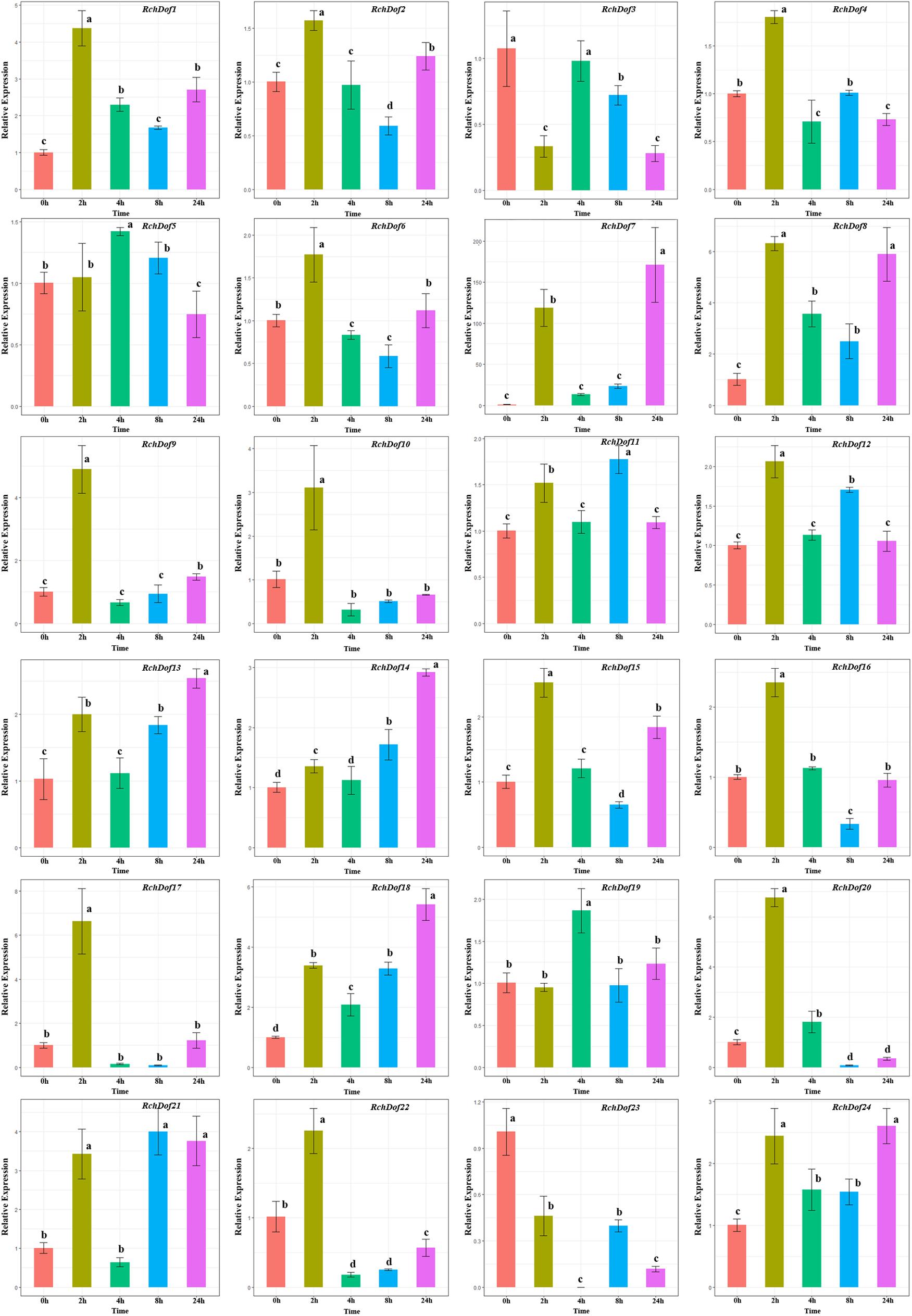
Figure 8. Expression patterns of RchDof genes under salt stress. Samples were collected at 0, 2, 4, 8, and 24 h after treatment. Error bars indicate the standard deviations (mean ± SD) of three independent replicates. The histogram bars labeled with different letters (a–d) above them are significantly different (least significant difference test, P < 0.05).
Discussion
Dof genes play essential roles in various plant physiological processes as well as diverse abiotic and biotic stress responses (Cai et al., 2013; Corrales et al., 2014; Wu et al., 2016). Despite roses (R. chinensis Jacq.) being the most commercially important plant in the Rosaceae family, Dof genes in rose have not been comprehensively characterized, and their exact functions remain unknown. In the current study, a search for Dof genes in the rose genome resulted in the identification of 24 members, which were named from RchDof1 to RchDof24 based on their gene IDs. In addition, an analysis of their structure, duplication events, and expression diversity was conducted with regard to drought and salt stress.
Compared with rose (24 RchDofs, genome size 560 Mb) (Yokoya, 2000), a comparable number of Dofs was identified in castor bean (24 Dofs, genome size 320 Mb) (Chan et al., 2010; Zou and Zhang, 2019) and grape (25 Dofs, genome size 487 Mb) (Jaillon et al., 2007; da Silva et al., 2016), although the number of Dofs was greater in Arabidopsis (36 Dofs, genome size 125 Mb) (Yanagisawa, 2002; Ma et al., 2015) and Chinese cabbage (76 Dofs, genome size 485 Mb) (Ma et al., 2015). This suggests that the number of Dof genes is not associated with the genome size. The gene structure, protein composition, exon–intron organization, and phylogenetic relationships support the conclusion that these 24 RchDof proteins can be divided into four major groups (A, B, C, and D) as previously described in other plant species (Liu and Ekramoddoullah, 2009; Xiao et al., 2017). Furthermore, in the present study, class B2 could be obviously divided into two clusters (Figure 2). Thereby, we updated the classification as follows: class 1 (A), 2 (B1), 3 (B2), 4 (B2), 5 (C1), 6 (C2.1), 7 (C2.2), 8 (C3), 9 (D1), 10 (D2), and 11 (D2).
Gene duplication has long been regarded as a key contributor to plant gene evolution (Li et al., 2005). Our results indicate that seven pairs of RchDof duplicated genes were derived from segmental duplication events, and one pair of Dof genes was generated by tandem duplications. A number of studies suggested that genes arising through segmental duplication events may more often be retained due to sub-functionalization without increasing the likelihood of gene rearrangement (Zhang, 2003). Segmental duplication may have played a more important role than tandem duplication in driving Dof gene family evolution, as suggested by previous findings in A. thaliana, rice, and apple (Cannon et al., 2004; Wang et al., 2014; Huang et al., 2015). The spatiotemporal expression changes are the main indicators of functional divergence in duplicated genes (Makova and Li, 2003; Hellsten et al., 2007). The 24 candidate RchDof genes in leaves and flowers displayed markedly different expression profiles, of which even genes in the same subgroup were divergently expressed. It is interesting to note that six pairs of duplicated genes (RchDof1/RchDof23, RchDof3/RchDof23, RchDof5/RchDof17, RchDof7/RchDof24, RchDof9/RchDof18, and RchDof12/RchDof21) arising from segmental duplication events exhibited exceptionally different expression patterns, indicating that the functional divergence may have provided genetic sources with novel biological functions during the evolution of the RchDof gene family. These results indicate that the expanded RchDof genes might result in novel biological complexity in order to remove function redundancy.
Drought and high salinity cause abiotic stress that influences plant growth and development. However, as described previously, roses are often grown under non-stressed conditions, and natural selection has unintentionally narrowed the genetic variability of abiotic stress tolerance. Therefore, the understanding of the mechanism by which roses respond and develop tolerance to drought and salt stress is the first step toward improving the adaptation of commercial rose cultivars to stressful environments. It was reported that Dof genes were involved in a wide variety of biological processes, including drought and salt stress responses (Corrales et al., 2014; Cai et al., 2016; Renau-Morata et al., 2017). Here we demonstrated that the expression levels of all the RchDof genes were upregulated/downregulated following drought and salinity treatments, suggesting that they may be involved in drought and salt stress responses. These results are supported by previous findings, as CDF3 (AT3G47500), the orthologs gene of RchDof21 (Supplementary Table 2), is well studied for its role in salinity stress (Renau-Morata et al., 2017). In addition, the orthologs gene of RchDof12 and RchDof21 in tomato (SlCDF1) (Supplementary Table 2) demonstrated increased drought and salt tolerance (Corrales et al., 2014). Therefore, our findings suggest that rose Dof genes have a role in response to drought and salt stress. The functional characterization of RchDof12 and RchDof21 will aid in the exploration of the mechanism of drought and salt tolerance in future studies. Moreover, in contrast to RchDof12 and RchDof21, RchDof7 showed the highest increase in expression following drought and salinity treatment. Cis-element analysis indicated that the RchDof7 exhibited a MYB binding site, which is involved in the drought response. These results support our hypothesis that the RchDof7 gene may play a key role in drought and salt responses. The results also provide a number of RchDof candidate genes for improving drought and salt tolerance in rose.
Conclusion
In this study, a total of 24 RchDof genes were identified in the rose genome assembly. Phylogenetic analysis suggested that the RchDof transcription factors were conserved as demonstrated by the identification of highly conserved motifs and gene structures. Genomic synteny analysis suggested that segmental duplications may have played a major role in RchDof gene family evolution. The cis-element analysis suggested that the majority of the RchDof genes were involved in various processes as well as stress responses, which provides a basis for the functional characterization of RchDof genes. The data showed the tissue-specific expression of the RchDofs and differential expression in response to drought and salt stress conditions, suggesting the existence of a complicated molecular regulatory network response to drought and salt stress conditions in rose. This provides novel insights into the evolutionary and functional divergence of the Dof gene family, which can aid in functional genomic studies of candidate Dof genes in order to genetically improve commercially important rose cultivars.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
HN and ML performed the data analysis and experiments. HN drafted the manuscript. ML and HA served as the principal investigator, facilitated the project, and revised the manuscript. RL revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (32060657), the Joint Fund of the National Natural Science Foundation of China and the Karst Science Research Center of Guizhou Province (U1812401), and the Talent Project of Guizhou Province (20164016).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate the reviewers for their comments on this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.538733/full#supplementary-material
Footnotes
- ^ https://www.arabidopsis.org/
- ^ https://lipm-browsers.toulouse.inra.fr/pub/RchiOBHm-V2/
- ^ http://pfam.xfam.org/
- ^ http://hmmer.janelia.org/
- ^ http://www.expasy.ch/tools/scanprosite/
- ^ http://www.ebi.ac.uk/Tools/InterProScan/
- ^ http://web.expasy.org/protparam/
- ^ http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/
- ^ https://www.rosaceae.org/
- ^ http://peargenome.njau.edu.cn
- ^ http://gsds.gao-lab.org/
- ^ http://meme-suite.org/tools/meme
- ^ http://bioinformatics.psb.ugent.be/webtools/plantcare/html/
- ^ http://plants.ensembl.org/index.html
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37(Suppl._2), W202–W208. doi: 10.1093/nar/gkp335
Cai, X., Zhang, C., Shu, W., Ye, Z., Li, H., and Zhang, Y. (2016). The transcription factor SlDof22 involved in ascorbate accumulation and salinity stress in tomato. Biochem. Biophs. Res. Commun. 474, 736–741. doi: 10.1016/j.bbrc.2016.04.148
Cai, X., Zhang, Y., Zhang, C., Zhang, T., Hu, T., Ye, J., et al. (2013). Genome-wide analysis of plant-specific Dof transcription factor family in tomato. J. Integr. Plant Biol. 55, 552–566. doi: 10.1111/jipb.12043
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., and May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4:10. doi: 10.1186/1471-2229-4-10
Chan, A. P., Crabtree, J., Zhao, Q., Lorenzi, H., Orvis, J., Puiu, D., et al. (2010). Draft genome sequence of the oilseed species Ricinus communis. Nat. Biotechnol. 28, 951–956. doi: 10.1038/nbt.1674
Chou, K. C., and Shen, H. B. (2010). Plant-mPLoc: a top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS One 5:e11335. doi: 10.1371/journal.pone.0011335
Corrales, A. R., Nebauer, S. G., Carrillo, L., Fernández-Nohales, P., Marqués, J., Renau-Morata, B., et al. (2014). Characterization of tomato cycling Dof factors reveals conserved and new functions in the control of flowering time and abiotic stress responses. J. Exp. Bot. 65, 995–1012. doi: 10.1093/jxb/ert451
da Silva, D. C., da Silveira Falavigna, V., Fasoli, M., Buffon, V., Porto, D. D., and Pappas, G. J. Jr. et al., (2016). Transcriptome analyses of the Dof-like gene family in grapevine reveal its involvement in berry, flower and seed development. Hortic. Res. 3:16042. doi: 10.1038/hortres.2016.42
Feng, L., Ding, H., Wang, J., Wang, M., Xia, W., Zang, S., et al. (2015). Molecular cloning and expression analysis of RrNHX1 and RrVHA-c genes related to salt tolerance in wild Rosa rugosa. Saudi J. Biol. Sci. 22, 417–423. doi: 10.1016/j.sjbs.2015.01.008
Hellsten, U., Khokha, M. K., Grammer, T. C., Harland, R. M., Richardson, P., and Rokhsar, D. S. (2007). Accelerated gene evolution and subfunctionalization in the pseudotetraploid frog Xenopus laevis. BMC Biol. 5:31. doi: 10.1186/1741-7007-5-31
Hong, K., Xian, J., Jia, Z., Hou, X., and Zhang, L. (2019). Genome-wide identification of Dof transcription factors possibly associated with internal browning of postharvest pineapple fruits. Sci. Hortic. 251, 80–87. doi: 10.1016/j.scienta.2019.03.007
Huang, X., Li, K., Xu, X., Yao, Z., Jin, C., and Zhang, S. (2015). Genome-wide analysis of WRKY transcription factors in white pear (Pyrus bretschneideri) reveals evolution and patterns under drought stress. BMC Genom. 16:1104. doi: 10.1186/s12864-015-2233-6
Jaillon, O., Aury, J. M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467. doi: 10.1038/nature06148
Jin, Z., Chandrasekaran, U., and Liu, A. (2014). Genome-wide analysis of the Dof transcription factors in castor bean (Ricinus communis L.). Genes Genom. 36, 527–537. doi: 10.1007/s13258-014-0189-6
Kisu, Y., Ono, T., Shimofurutani, N., Suzuki, M., and Esaka, M. (1998). Characterization and expression of a new class of zinc finger protein that binds to silencer region of ascorbate oxidase gene. Plant Cell Physiol. 39, 1054–1064. doi: 10.1093/oxfordjournals.pcp.a029302
Krebs, J., Mueller-Roeber, B., and Ruzicic, S. (2010). A novel bipartite nuclear localization signal with an atypically long linker in DOF transcription factors. J. Plant Physiol. 167, 583–586. doi: 10.1016/j.jplph.2009.11.016
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327.
Li, H., Dou, L., Li, W., Wang, P., Zhao, Q., Xi, R., et al. (2018). Genome-wide identification and expression analysis of the Dof transcription factor gene family in Gossypium hirsutum L. Agronomy 8:186. doi: 10.3390/agronomy8090186
Li, H., Huang, W., Liu, Z. W., Wang, Y. X., and Zhuang, J. (2016). Transcriptome-based analysis of Dof family transcription factors and their responses to abiotic stress in tea plant (Camellia sinensis). Int. J. Genom. 2016:5614142. doi: 10.1155/2016/5614142
Li, H., Wang, Y., Wu, M., Li, L., Li, C., Han, Z., et al. (2017). Genome-wide identification of AP2/ERF transcription factors in cauliflower and expression profiling of the ERF family under salt and drought stresses. Front. Plant Sci. 8:946. doi: 10.3389/fpls.2017.00946
Li, W. H. (1993). Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J. Mol. Evol. 36, 96–99.
Li, W. H., Yang, J., and Gu, X. (2005). Expression divergence between duplicate genes. Trends Genet. 21, 602–607. doi: 10.1016/j.tig.2005.08.006
Li, Y., Xiao, J., Wu, J., Duan, J., Liu, Y., Ye, X., et al. (2012). A tandem segmental duplication (TSD) in green revolution gene Rht-D1b region underlies plant height variation. New Phytol. 196, 282–291. doi: 10.1111/j.1469-8137.2012.04243.x
Lijavetzky, D., Carbonero, P., and Vicente-Carbajosa, J. (2003). Genome-wide comparative phylogenetic analysis of the rice and Arabidopsis Dof gene families. BMC Evol. Biol. 3:17. doi: 10.1186/1471-2148-3-17
Liu, J. J., and Ekramoddoullah, A. K. (2009). Identification and characterization of the WRKY transcription factor family in Pinus monticola. Genome 52, 77–88. doi: 10.1139/G08-106
Liu, X., Liu, Z., Hao, Z., Chen, G., Qi, K., Zhang, H., et al. (2020). Characterization of Dof family in Pyrus bretschneideri and role of PbDof9.2 in flowering time regulation. Genomics 112, 712–720. doi: 10.1016/j.ygeno.2019.05.005
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–Δ Δ CT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Ma, J., Li, M. Y., Wang, F., Tang, J., and Xiong, A. S. (2015). Genome-wide analysis of Dof family transcription factors and their responses to abiotic stresses in Chinese cabbage. BMC Genom. 16:33. doi: 10.1186/s12864-015-1242-9
Makova, K. D., and Li, W. H. (2003). Divergence in the spatial pattern of gene expression between human duplicate genes. Genome Res. 13, 1638–1645. doi: 10.1101/gr.1133803
Molina-Hidalgo, F. J., Medina-Puche, L., Canete-Gomez, C., Franco-Zorrilla, J. M., Lopez-Vidriero, I., Solano, R., et al. (2017). The fruit-specific transcription factor FaDOF2 regulates the production of eugenol in ripe fruit receptacles. J. Exp. Bot. 68, 4529–4543. doi: 10.1093/jxb/erx257
Moore, R. C., and Purugganan, M. D. (2005). The evolutionary dynamics of plant duplicate genes. Curr. Opin. Plant Biol. 8, 122–128. doi: 10.1016/j.pbi.2004.12.001
Nan, H., and Gao, L. Z. (2019). Genome-wide analysis of WRKY genes and their response to hormone and mechanic stresses in carrot. Front. Genet. 10:363. doi: 10.3389/fgene.2019.00363
Raymond, O., Gouzy, J., Just, J., Badouin, H., Verdenaud, M., Lemainque, A., et al. (2018). The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 50, 772–777. doi: 10.1038/s41588-018-0110-3
Renau-Morata, B., Molina, R. V., Carrillo, L., Cebolla-Cornejo, J., Sánchez-Perales, M., Pollmann, S., et al. (2017). Ectopic expression of CDF3 genes in tomato enhances biomass production and yield under salinity stress conditions. Front. Plant Sci. 8:660. doi: 10.3389/fpls.2017.00660
Sasaki, N., Matsumaru, M., Odaira, S., Nakata, A., Nakata, K., Nakayama, I., et al. (2015). Transient expression of tobacco BBF1-related Dof proteins, BBF2 and BBF3, upregulates genes involved in virus resistance and pathogen defense. Physiol. Mol. Plant Pathol. 89, 70–77. doi: 10.1016/j.pmpp.2014.12.005
Tian, X., Wang, Z., Zhang, Q., Ci, H., Wang, P., Yu, L., et al. (2018). Genome-wide transcriptome analysis of the salt stress tolerance mechanism in Rosa chinensis. PLoS One 13:e0200938. doi: 10.1371/journal.pone.0200938
Tian, X., Wang, Z., Zhang, Q., Ci, H., Wang, P., Yu, L., et al. (2019). Identification of salt stress response genes in Rosa chinensis leaves by comparative RNA-seq analysis of transcriptome dynamics. Russ. J. Plant Physl. 66, 119–127. doi: 10.1134/s1021443719010175
Wang, M., Vannozzi, A., Wang, G., Liang, Y. H., Tornielli, G. B., Zenoni, S., et al. (2014). Genome and transcriptome analysis of the grapevine (Vitis vinifera L.) WRKY gene family. Hortic. Res. 1:14016. doi: 10.1038/hortres.2014.16
Wang, S., Bai, Y., Li, P., Yang, L., and Wang, X. (2019). Genome-wide identification and expression analysis of the dof (DNA binding with one finger) protein family in monocot and dicot species. Physiol. Mol. Plant Pathol. 108:101431. doi: 10.1016/j.pmpp.2019.101431
Wray, G. A., Hahn, M. W., Abouheif, E., Balhoff, J. P., Pizer, M., Rockman, M. V., et al. (2003). The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol. 20, 1377–1419. doi: 10.1093/molbev/msg140
Wu, Z., Cheng, J., Cui, J., Xu, X., Liang, G., Luo, X., et al. (2016). Genome-wide identification and expression profile of Dof transcription factor gene family in pepper (Capsicum annuum L.). Front. Plant Sci. 7:574. doi: 10.3389/fpls.2016.00574
Xiao, Y., Zhou, L., Lei, X., Cao, H., Wang, Y., Dou, Y., et al. (2017). Genome-wide identification of WRKY genes and their expression profiles under different abiotic stresses in Elaeis guineensis. PLoS One 12:e0189224. doi: 10.1371/journal.pone.0189224
Yanagisawa, S. (2001). The transcriptional activation domain of the plant-specific Dof1 factor functions in plant, animal, and yeast cells. Plant Cell Physiol. 42, 813–822. doi: 10.1093/pcp/pce105
Yanagisawa, S. (2002). The Dof family of plant transcription factors. Trends Plant Sci. 7, 555–560. doi: 10.1016/S1360-1385(02)02362-2
Yanagisawa, S., and Izui, K. (1993). Molecular cloning of two DNA-binding proteins of maize that are structurally different but interact with the same sequence motif. J. Biol. Chem. 268, 16028–16036. doi: 10.1016/S1360-1385(02)02362-2
Yanagisawa, S., and Schmidt, R. J. (1999). Diversity and similarity among recognition sequences of Dof transcription factors. Plant J. 17, 209–214. doi: 10.1046/j.1365-313X.1999.00363.x
Yang, Q., Chen, Q., Zhu, Y., and Li, T. (2018). Identification of MdDof genes in apple and analysis of their response to biotic or abiotic stress. Funct. Plant Biol. 45, 528–541. doi: 10.1071/FP17288
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z. H., and Bielawski, J. P. (2000). Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15, 496–503.
Zhang, J. (2003). Evolution by gene duplication: an update. Trends Ecol. Evol. 18, 292–298. doi: 10.1016/s0169-5347(03)00033-8
Zhang, Z., Yuan, L., Liu, X., Chen, X., and Wang, X. (2018). Evolution analysis of Dof transcription factor family and their expression in response to multiple abiotic stresses in Malus domestica. Gene 639, 137–148. doi: 10.1016/j.gene.2017.09.039
Zou, Z., and Zhang, X. (2019). Genome-wide identification and comparative evolutionary analysis of the Dof transcription factor family in physic nut and castor bean. PeerJ 7:e6354. doi: 10.7717/peerj.6354
Keywords: Dof transcription factor, rose (Rosa chinensis), phylogenetic analysis, synteny analysis, expression divergence, salt and drought stress
Citation: Nan H, Ludlow RA, Lu M and An H (2021) Genome-Wide Analysis of Dof Genes and Their Response to Abiotic Stress in Rose (Rosa chinensis). Front. Genet. 12:538733. doi: 10.3389/fgene.2021.538733
Received: 09 March 2020; Accepted: 26 January 2021;
Published: 04 March 2021.
Edited by:
Vijay Kumar Tiwari, University of Maryland, College Park, United StatesReviewed by:
Ze Peng, University of Florida, United StatesAmita Mohan, University of Pennsylvania, United States
Copyright © 2021 Nan, Ludlow, Lu and An. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Lu, NDgxODEyNjZAcXEuY29t; Huaming An, YW5odWFtaW5nQGhvdG1haWwuY29t