 Mengqi Wang
Mengqi Wang Eveline M. Ibeagha-Awemu
Eveline M. Ibeagha-Awemu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 23 February 2021
Sec. Livestock Genomics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.613636
This article is part of the Research TopicEpigenetic Variation Influences on Livestock Production and Disease TraitsView all 24 articles
The dynamic changes in the epigenome resulting from the intricate interactions of genetic and environmental factors play crucial roles in individual growth and development. Numerous studies in plants, rodents, and humans have provided evidence of the regulatory roles of epigenetic processes in health and disease. There is increasing pressure to increase livestock production in light of increasing food needs of an expanding human population and environment challenges, but there is limited related epigenetic data on livestock to complement genomic information and support advances in improvement breeding and health management. This review examines the recent discoveries on epigenetic processes due to DNA methylation, histone modification, and chromatin remodeling and their impacts on health and production traits in farm animals, including bovine, swine, sheep, goat, and poultry species. Most of the reports focused on epigenome profiling at the genome-wide or specific genic regions in response to developmental processes, environmental stressors, nutrition, and disease pathogens. The bulk of available data mainly characterized the epigenetic markers in tissues/organs or in relation to traits and detection of epigenetic regulatory mechanisms underlying livestock phenotype diversity. However, available data is inadequate to support gainful exploitation of epigenetic processes for improved animal health and productivity management. Increased research effort, which is vital to elucidate how epigenetic mechanisms affect the health and productivity of livestock, is currently limited due to several factors including lack of adequate analytical tools. In this review, we (1) summarize available evidence of the impacts of epigenetic processes on livestock production and health traits, (2) discuss the application of epigenetics data in livestock production, and (3) present gaps in livestock epigenetics research. Knowledge of the epigenetic factors influencing livestock health and productivity is vital for the management and improvement of livestock productivity.
Increasing animal food demand by an ever-expanding human population as well as the challenges of global climate change is a clarion call for the sustainable development of the food animal industry, with the expectation of increased supply of high-quality animal proteins with minimal environmental impacts. In response, recent research efforts are geared towards developing different approaches to improve livestock production efficiency, decrease production cost, and develop environmentally friendly livestock production systems (Capper and Bauman, 2013; Scott, 2018). In recent years, the application of modern technologies including advanced sequencing technologies, genotype analysis, and genome profiling has promoted important changes in livestock genetic breeding programs and gains in important livestock traits like milk yield/quality in dairy cattle and goat, meat quality in beef cattle and swine, egg yield/quality in chickens, etc. Continued technological advances have further promoted the implementation of genomic selection in livestock production (Georges et al., 2019; Gurgul et al., 2019). Sequence analysis of livestock genomes uncovered the general molecular and regulatory mechanisms of the coding and non-coding genome underlying production and health traits, which have supported advances in trait improvement (Kamath et al., 2016; Do et al., 2017a; Wara et al., 2019). These factors still fall short of accounting for the optimal level of variation that is required to achieve continued improvements in livestock health and productivity. The epigenome, which responds to internal and external environmental cues, is less explored but contains additional levels of variation that could be exploited for livestock trait improvement.
Epigenetics is defined as the study of heritable molecular modifications responsible for the regulation of genome activities and gene expression, resulting in phenotypic differences without alterations to the basic DNA sequence (Nicoglou and Merlin, 2017; Greally, 2018). Epigenetic processes, which include DNA methylation, histone modification, chromatin remodeling, and non-coding RNA (ncRNA) regulation, regulate gene expression and, thus, play significant roles in genome function and stability (Bird, 2002; Kouzarides, 2007; Morris and Mattick, 2014; Do and Ibeagha-Awemu, 2017). The epigenome, which encompasses these epigenetic processes, is dynamic during the whole lifetime and is significantly associated with the interaction between genetic activities and environmental stimulation (Bernstein et al., 2007; Monk et al., 2019). Sufficient evidence from epigenetics-related studies in humans and animals have demonstrated the distinctive roles of epigenetic mechanisms in various biological processes, such as growth, development, metabolism, and health (Ibeagha-Awemu and Zhao, 2015; Paiva et al., 2019). The occurrence of epigenetic mechanisms with important roles at specific key times of development or pathological conditions may be key to further exploration of the intricacies of diseases. In addition, the awareness and use of epigenetic mechanisms could be advantageous to the understanding of quantitative traits and in achieving advancements in the improvement of livestock productivity and disease resistance (Ibeagha-Awemu and Zhao, 2015; Ibeagha-Awemu and Khatib, 2017; Banta and Richards, 2018; Panzeri and Pospisilik, 2018). The important contribution of epigenetic processes to phenotypic outcome in livestock is beginning to attract attention implying that the impacts of these processes may soon find application in advancing livestock productivity and health (Doherty et al., 2014; Goddard and Whitelaw, 2014; Meirelles et al., 2014; Ibeagha-Awemu and Zhao, 2015; Triantaphyllopoulos et al., 2016; Ibeagha-Awemu and Khatib, 2017; Yakovlev, 2018; Paiva et al., 2019). This review has been categorized into sections that concentrate on discussing the epigenetics processes and impacts on gene regulation; evidence of the impacts of epigenetic regulatory mechanisms underlying productivity and health in different species of livestock animals, such as bovine, swine, sheep, goat, poultry, and other species; application of epigenetics data in livestock production; and research gaps and future perspectives. Evidence of epigenetic impacts on livestock reproduction and epigenetic alterations due to assisted reproduction technologies have been addressed in several recent reviews (Weksberg et al., 2007; Das et al., 2017; Franco, 2017; Khezri et al., 2020; Rivera, 2020; Wang et al., 2020e) and will not be discussed here.
DNA methylation is thus far the most stable and characterized epigenetic modification in most mammalian genomes. DNA methylation principally occurs in the fifth carbon of cytosine residues (addition of a methyl or hydroxymethyl group, denoted as 5mC or 5hmC, respectively) in DNA sequence and mostly at cytosine-phosphate-guanosine (CpG) dinucleotides and to a lesser extent at cytosine-phosphate-adenosine (CpA), cytosine-phosphate-thymine (CpT), and cytosine-phosphate-cytosine (CpC) dinucleotides. The formation of DNA methylation patterns is catalyzed by the activities of a class of enzymes known as DNA methyltransferases (DNMTs). While DNMT3A and DNMT3B are responsible for the establishment of DNA methylation patterns during embryonic development or in response to environmental challenges, DNMT1 maintains DNA methylation during cell division (Edwards et al., 2017; Luo et al., 2018; Schmitz et al., 2019). DNMT3L, which acts as a co-factor of DNMT3A and DNMT3B during de novo DNA methylation, is required for mammalian genome imprinting (Hanna and Kelsey, 2014; Basu, 2016; Veland et al., 2019). There are other DNMTs which act in different biological processes. For example, DNMT3C is responsible for the silencing of young retrotransposons (Barau et al., 2016), while DNMT2 plays active roles in RNA methylation and the expression of small ncRNAs (Raddatz et al., 2013; Jeltsch et al., 2017; Zhang et al., 2018b). However, when established DNA methylation is not maintained, the process of passive or active demethylation sets in. Passive demethylation is through the activities of TET (ten–eleven translocation methylcytosine dioxygenases) proteins (e.g., TET1, TET2, and TET3) which mediate the oxidation of 5mC to produce 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC; He et al., 2011; Wu and Zhang, 2017). Active demethylation is when replication-dependent dilution of 5hmC, 5fC, and 5caC or thymine DNA glycosylase (TDG)-mediated excision of 5fC and 5caC is coupled with base excision repair (Wu and Zhang, 2017). Other TET–TDG-independent mechanisms are also proposed to mediate active DNA demethylation (Wu and Zhang, 2010, 2014; Bochtler et al., 2017).
The impact of DNA methylation on gene expression is associated with its genome distribution. CpG-rich regions (also known as CpG islands) are frequently distributed in the promoter regions (usually extends to 5′UTR) and usually non-methylated, whereby its abnormal methylation may cause the repression of corresponding transcription and gene silencing (Deaton and Bird, 2011; Smith et al., 2020). About 72% of promoters are within CpG islands and nearly unmethylated, and their activities might be regulated by DNA methylation (Saxonov et al., 2006; Deaton and Bird, 2011). Promoter CpG islands have differential susceptibility to methylation during normal development or during disease progression (e.g., carcinogenesis), which might be influenced by intrinsic sequence properties (Feltus et al., 2006). Most promoter CpG islands (usually unmethylated) escape from de novo methylation during all developmental stages, and the activity of promoters with intermediate to high CpG content was negatively correlated with their DNA methylation status (Weber et al., 2007). However, there are still a small number of methylated CpG islands in gene promoters, such as at germline imprinting control regions, or on the inactive X chromosome in female somatic cells (Proudhon et al., 2012). DNA methylation could perturb gene expression activities through direct inhibition of transcription factor (TF) binding or indirect mediation by methyl-binding domain (MBD) proteins that recruit chromatin-modifying activities to methylated DNA (Razin and Kantor, 2005; Zhu et al., 2016; Yin et al., 2017; Greenberg and Bourc’his, 2019). It has been noted that TFs are likely to induce local epigenetic remodeling (Wapinski et al., 2013). DNA methylation in recognition sequences of some TFs was revealed to alter their binding specificity (Zhu et al., 2016). It was reported that DNA methylation of target sequences diminished the binding activity of numerous TFs in the human genome, whereas some TFs of the extended homeodomain family preferred CpG methylated sequences (Yin et al., 2017). The identification of MBDs, such as methyl-CpG binding protein 1 (MeCP1) and methyl-CpG binding protein 2 (MeCP2), revealed that DNA methylation is connected with chromatin structure and gene expression. Except for binding to CpG-rich heterochromatin, some MBDs contain a transcriptional repressor complex that may induce histone deacetylation and chromatin remodeling, contributing to gene silencing (Razin and Kantor, 2005; Greenberg and Bourc’his, 2019).
DNA methylation is also found in the gene body, especially in introns, and is prone to high levels of methylation (Maunakea et al., 2010). DNA methylation in gene body is highly conserved across eukaryotes and has been positively correlated with transcription (Lister et al., 2009; Varley et al., 2013), indicating potential functions other than gene silencing. Two hypotheses underlying the function of DNA methylation in gene bodies have been suggested (Greenberg and Bourc’his, 2019). On the one hand, DNA methylation enriched at exons influences splicing and gene expression (Gelfman et al., 2013; Yang et al., 2014; Shayevitch et al., 2018). DNA methylation was found to facilitate exon exclusion by preventing CCCTC-binding factor (CTCF) binding or contribute to exon inclusion by recruitment of MeCP2 or splicing factors (Shukla et al., 2011; Maunakea et al., 2013; Yearim et al., 2015). However, these mechanisms could only explain a small portion of alternative splicing events. On the other hand, gene body DNA methylation suppresses intragenic promoters consistent with the possible role of DNA methylation as a transcriptional repressor. It was reported that binding of ADD domain to H3K36me3 released the inhibition of DNMT3 enzymes thereby promoting the establishment of de novo DNA methylation and, consequently, inhibition of cryptic promoters (Carrozza et al., 2005; Greenberg and Bourc’his, 2019). Indeed, methylation of CpG island in gene body suppressed promoter activity, and altered methylation contributed to the regulation of transcription initiation in a tissue- and cell-type-specific mechanism in mammals (Maunakea et al., 2010). Compared with its well-established repressive function at regulatory elements (such as the promoter region), less is known about DNA methylation regulation and function(s) at intergenic regions. Intergenic regions are mainly populated by regulatory ncRNA genes and other regulatory elements yet to be described, and the DNA methylation at these regions potentially regulates these factors. Downstream regions of genes contain miRNA binding sites, which may interact with DNA methylation and regulate gene expression. For example, the interaction between piwi RNA and DNA methylation is dedicated to silencing transposable elements in the germline (Manakov et al., 2015; Barau et al., 2016).
Histone modification is another important epigenetic mechanism with significant impacts on chromatin regulation and regulation of transcription processes (Adamczyk, 2019). Histones are a family of proteins (H1/H5, H2A, H2B, H3, and H4) that park and order the DNA molecule into structural units called nucleosomes. The N-terminal tails of histones are subjected to various posttranscriptional or posttranslational modifications with more than 100 forms (e.g., lysine acetylation, lysine methylation, ubiquitination, serine/threonine phosphorylation, crotonylation, sumoylation, etc.) with varying effects on transcriptional activities described (Kouzarides, 2007; Zhao and Shilatifard, 2019; Roma-Rodrigues et al., 2020). Histone acetylation and methylation frequently occurs in the lysine of the N-terminal tails resulting from the interaction of associated enzymes or factors (Srivastava et al., 2016). Histone acetyltransferases are responsible for histone acetylation, which play important roles in releasing chromatin structure (histone–DNA interaction) and promoting transcriptional activities, while histone deacetylases cause deacetylation to repress gene expression (Dose et al., 2011; Schmauss, 2017). Similarly, the dynamic changes of histone methylation, which are generally classified into tri-, di-, and monomethylation of lysine residues and the monomethylation of arginine residues, are regulated by histone methyltransferases and demethylases (Ye et al., 2017). The impact of histone methylation on transcriptional activity is complex and depends on both modified residues and the state of methylation (Jambhekar et al., 2019). Additionally, histone phosphorylation, ubiquitylation, and ADP ribosylation are involved in the regulation of DNA damage and transcriptional activities (Liu C. et al., 2017; Alhamwe et al., 2018; Shanmugam et al., 2018).
Chromatin structure dynamics always correspond with the instructive gene expression pattern for cellular differentiation and lineage specification during development (Kishi and Gotoh, 2018; Quan et al., 2020). In addition to the covalent modification of DNA and histone, the remodeling of nucleosome is another important determinant of chromatin structure. Nucleosome formation, which is crucial for the compaction of genomic DNA into chromatin, has intrinsic dynamic properties regulated by chromatin remodeling complexes to ensure genomic DNA functions in chromatin (Zhou C. Y. et al., 2016; Kobayashi and Kurumizaka, 2019). Increasingly, reports of copious chromatin remodeling complexes and their essential regulatory potentials related to transcription activities and gene expression during development and disease processes have emerged (Bhattacharjee et al., 2016; Kim and Kaang, 2017; Stachowiak et al., 2020). Adenosine triphosphate (ATP)-dependent chromatin remodeling complexes are particularly known to utilize the energy derived from ATP hydrolysis to change nucleosome structure and consequently regulate DNA accessibility to TFs (Hota and Bruneau, 2016). As one representative of ATP-dependent complexes, BRM/BRG1-associated factor (BAF) complexes, also known as SWI/SNF (switch/sucrose non-fermentable) complexes, have various roles in gene activation and repression during mammalian development and in the development of disease (Clapier et al., 2017; Alfert et al., 2019; Hota et al., 2019). BAF complexes consist of over 15 different subunits with varied roles at different stages of mammalian development, including embryogenesis, neural development, cardiovascular development, skeletal muscle development, immune cell development, etc. (Hota and Bruneau, 2016; Nguyen et al., 2016; Sokpor et al., 2017; Sun et al., 2018). Homologous to the BAF complex, the RSC (remodel structure of chromatin) remodeling complex is an abundant and fundamental nuclear protein complex with important roles in transcriptional and other cellular processes, including initiation and elongation of transcription as well as replication, segregation, and chromosome repair (Klein-Brill et al., 2019; Ye et al., 2019). The RSC complex can partially disrupt histone–DNA interaction by stable binding to nucleosomes or enhancer elements and can also disassemble or slide nucleosome through a DNA-sequence-dependent system, that is required for nucleosome-free region formation by removing nucleosome from upstream of transcription start sites (TSSs; Spain et al., 2014; Lorch and Kornberg, 2017; Kubik et al., 2018). Besides, nucleosome remodeling and deacetylation (NuRD) complex (highly conserved in mammals), initially defined as a transcriptional repressor, has been reported to link histone modifications to nucleosome remodeling and interaction with numerous TFs (Feng and Zhang, 2003; Liang et al., 2017). Genome-wide data revealed the presence of the NuRD complex at all active enhancers and promoters in diverse cells, and also that NuRD could deposit histone modifications at enhancers and promoters of active genes and thereby trigger their repression (Miller et al., 2016; Yang et al., 2016a; Bornelöv et al., 2018).
In addition to these classic epigenetic processes (DNA methylation, histone modification, and chromatin remodeling), ncRNAs also play important regulatory roles in gene expression and chromatin modification impacting livestock production and health (Do et al., 2017a; Benmoussa et al., 2020). ncRNAs are a class of RNA species that mediate their functions as RNA (are not translated into proteins) and generally regulate gene expression at the transcriptional and posttranscriptional levels. ncRNAs with epigenetic-related functions interfere with transcription, mRNA stability, or translation and include small interfering RNA (siRNA), piwi-interacting RNA (piRNA), microRNA (miRNA), and long non-coding RNA (lncRNA; Kaikkonen et al., 2011; Wei et al., 2017). For example, some miRNAs regulated by DNMT1 are involved in the regulation of mammary gland development and lactation in dairy cattle (Do et al., 2017b; Melnik and Schmitz, 2017a). The binding of miRNA to a specific target, resulting in degradation or blockage of mRNA transcription, may induce a feedback modification related to DNA methylation (Lamouille et al., 2014). Besides, ncRNAs have been found to be involved in the regulation of epigenetic alterations in both DNA and histones (Sabin et al., 2013). Furthermore, it is speculated that some transcripts initiating from gene body CpG islands are regulated by ncRNAs whose presence or absence affects the expression of the associated protein-coding genes or nearby genes (Mercer et al., 2009). For example, AIR is a ncRNA that initiates at a CpG island within intron 2 of IGF2R and is essential for silencing of the paternal allele (Sleutels et al., 2002). Similarly, analysis of a CpG island in intron 10 of the imprinted KCNQ1 gene identified it as the origin of a non-coding transcript (KCNQ1OT1) that is required for imprinting of several genes within this domain (Mancini-DiNardo et al., 2006). ncRNA regulation impacts on livestock productivity will not be discussed in this review as it has been adequately covered in the literature and in recent reviews (Do and Ibeagha-Awemu, 2017; Wara et al., 2019; Kosinska-Selbi et al., 2020).
Dynamic epigenetic modifications are essential for normal growth and development through involvement in numerous biological processes, especially in response to environmental stimulus (Del Corvo et al., 2020; Thompson et al., 2020). The identification of epigenomic patterns in different tissues helps in the further understanding of epigenetic regulatory roles in livestock development and health. The impacts of epigenetic regulatory processes on livestock production in response to different impact factors or the exposome and the phenotypic outcomes are summarized in Figure 1. The important regulatory roles and impacts of epigenetic processes on placental and embryo development of livestock have been discussed in detail in many excellent reviews (Das et al., 2017; Franco, 2017; Hwang et al., 2017).
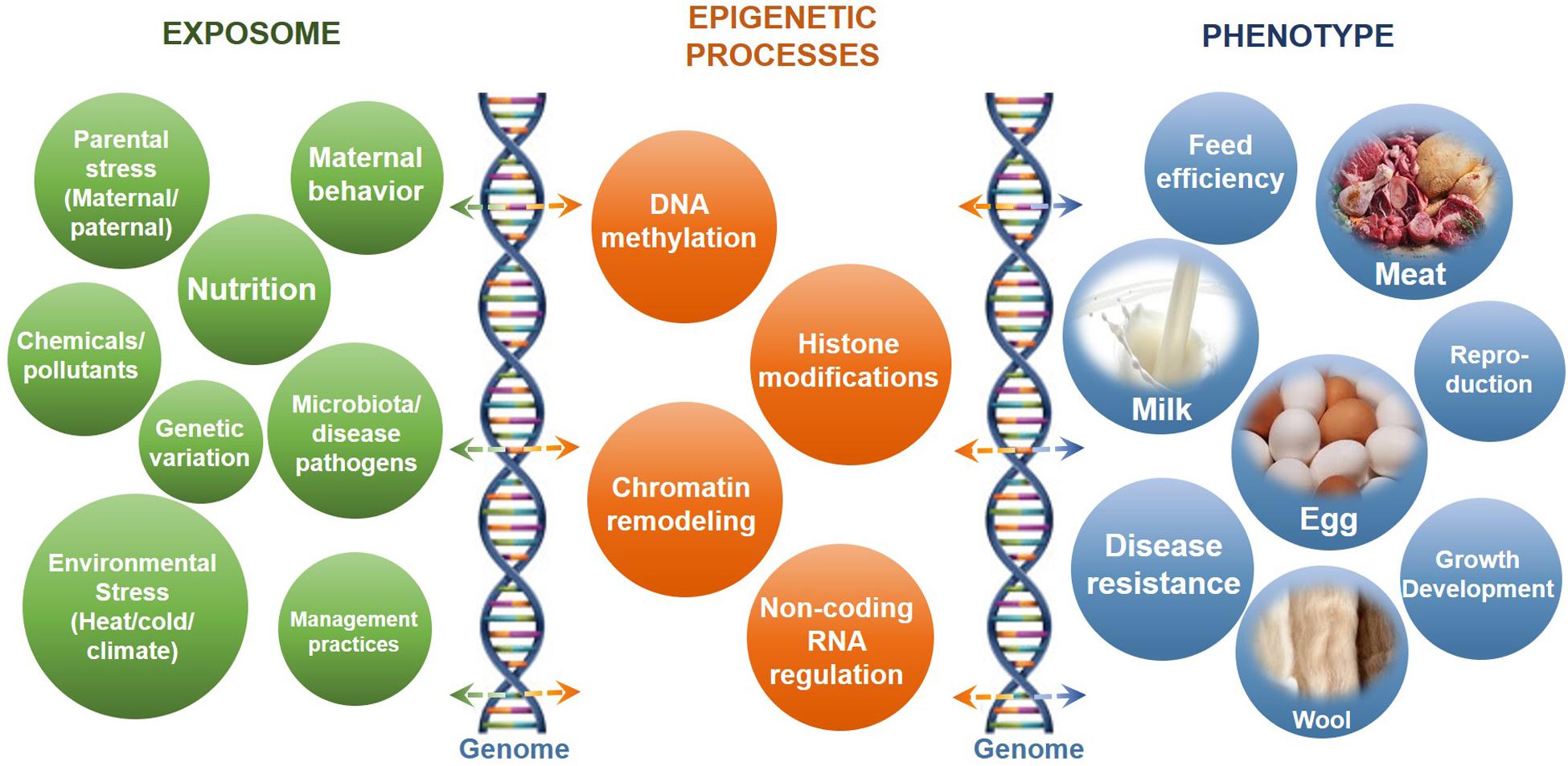
Figure 1. The impacts of epigenetic regulatory processes on livestock production in response to different impact factors or the exposome. Shown are the common impact factors or exposome (left) that interact with epigenetic processes (center) and the genome (vertical DNA helix structures) to influence phenotypic outcome (right).
The genomic DNA methylation profiles of several tissues in multiple livestock species including cattle, chicken, swine, goat, sheep, etc. have been characterized (Korkmaz and Kerr, 2017; Lee K. H. et al., 2017; Zhang Y. et al., 2017; McKay et al., 2018; Sevane et al., 2019; Liu et al., 2020; Wang et al., 2020b). DNA methylation pattern screening in embryo at different embryonic stages of development, especially at the early stage when two major epigenetic reprogramming occur, indicated important regulatory roles of DNA methylation in embryo viability and fetus development relating to various metabolic and differentiation processes (Ispada et al., 2018; Salilew-Wondim et al., 2018; Duan et al., 2019; Cao P. et al., 2020; Ivanova et al., 2020). DNA methylation changes were found to partly explain the poor performance of offspring caused by maternal stressors, such as heat stress, metabolic disorder, and negative energy balance (Desmet et al., 2016; Akbarinejad et al., 2017). Differentially methylated cytosines (DMCs) in the liver from calves under maternal heat stress or cooling treatment (fans and water soakers) were found in genes involved in immune function, cell cycle, development, and enzyme activity, while DMCs in mammary gland tissues were enriched in biological functions, including protein binding, phosphorylation, enzyme and cell activation, and cell signaling (Skibiel et al., 2018). It was observed recently that sperm DNA methylome was characterized by generally low methylation levels compared with somatic tissue DNA methylome (Perrier et al., 2018; Zhou et al., 2018). Moreover, dysregulation of DNA methylation in sperm (Kropp et al., 2017; Perrier et al., 2018; Fang et al., 2019b) and histone modifications, including histone acetylation and methylation (Kutchy et al., 2017, 2018), were found to impact male fertility and related traits. For instance, sperm whole-genome bisulfite sequence (WGBS) data from high- and low-fertile bulls indicated high methylation differences (1,765 DMCs) and 10 candidate genes for the prediction of bull fertility (Gross et al., 2020). Another sperm WGBS dataset from 28 bulls further classified the sperm methylome into conversed, variable, and highly variable methylated regions (Liu S. et al., 2019). The highly variable methylated regions associated significantly with reproduction traits and were enriched for glycosyltransferase genes that are crucial for spermatogenesis and fertilization, while the variable methylated regions were co-localized with genes with functions in sperm motility (Liu S. et al., 2019). In addition, methylation analysis of high and low motile bull sperm populations found methylation variations in genes involved in chromatin remodeling and repetitive element activities in pericentric regions, which is indicative of crucial epigenetic regulatory functions in sperm functionality and fertility (Capra et al., 2019). Methylation alteration was also predicted as one potential epigenetic regulatory mechanism underlying sperm fertility differences caused by age and other stressors such as heat or oxidative stress (Lambert et al., 2018; Rahman et al., 2018; Wyck et al., 2018; Takeda et al., 2019). Moreover, epigenomic profiling of somatic tissues such as the liver, brain, and mammary gland tissues revealed that epigenetic modifications impact bovine development, health, and productivity (Zhou Y. et al., 2016; Kweh et al., 2019; Wang et al., 2020c). For example, DNA methylation is involved in the regulation of SIRT6 promoter activity during bovine adipocyte differentiation (Hong et al., 2018).
Analysis of epigenetic processes in different porcine tissues, including tooth, brain, small intestine, and longissimus dorsi muscle, indicated their significant regulatory roles during the growth and development of pigs (Su et al., 2016; Larsen et al., 2018). The DNA methylation patterns of porcine tooth germ from different developmental stages (embryonic day 50 and day 60) revealed 2,469 differentially methylated genes (DMGs), including 104 DMGs with potential key regulatory roles in porcine tooth development (Su et al., 2016). Using the whole-genome DNA methylation approach to profile DNA methylation of porcine longissimus dorsi muscles from heat-stressed and non-stressed pigs, Hao and colleagues identified 57,147 differentially methylated regions (DMRs) and corresponding DMGs (n = 1,422) with functions in energy and lipid metabolism, cellular defense, and stress responses, indicating the roles of DNA methylation in heat stress processes (Hao et al., 2016). The expressions of DNMT1, DNMT3A, and DNMT3B were found to decrease in brain tissues during the middle stage of gestation, indicating the potential of DNA methylation to regulate brain development of piglets (Larsen et al., 2018). Daily oral boluses of broad-spectrum antibiotics after preterm birth decreased bacterial density, diversity, and fermentation and altered the DNA methylation profile in the small intestine, which is indicative of the influence of epigenetic process on bacterial colonization of preterm neonate’s intestine (Pan et al., 2018). Furthermore, transcriptional N6-methyladenosine (m6A) profiling in porcine liver at different ages (0 day, 21 days, and 2 years) demonstrated that m6A modified about 33% of transcribed genes with roles in the regulation of growth and development and metabolic and protein catabolic processes implying that m6A methylation may be vital for the regulation of nutrient metabolism in porcine liver (He et al., 2017). Moreover, abundant m6A modifications were identified in granulosa cells, which are crucial for follicle development with potential associations with steroidogenesis and folliculogenesis in pigs (Cao Z. et al., 2020).
DNA methylation and histone modification patterns have been characterized in various chicken strains in recent years, indicating the potential roles of epigenetic processes in the development and evolution of chicken (Li et al., 2015; Sarah-Anne et al., 2017). Epigenetic analysis of various tissues including the brain, retina, cornea, liver, and muscle strongly revealed the involvement of DNA methylation in the growth and development of chicken (Liu et al., 2016, Liu Z. et al., 2019; Lee I. et al., 2017). For example, DNA methylation profiles of broilers and layers at different embryonic stages revealed lower methylation levels in broilers, and enriched gene ontology terms related to muscle development by corresponding genes suggest a potential contribution of DNA methylation to embryonic muscle development (Liu Z. et al., 2019). ACC and MTTP showed abundant expression and were negatively correlated with lower DNA methylation at their promoter regions in the liver of chickens with fatty liver syndrome, linking DNA methylation to fat metabolism (Liu et al., 2016). The global DNA methylation profiling of strongly and weakly inbred chickens identified various DMRs and DMGs enriched in reproduction pathways, indicating the regulatory roles of DNA methylation in the repressed development of the reproduction system of inbred chickens (Han et al., 2020). Besides, chicken erythrocyte epigenome analysis identified more than 100 highly transcribed genes located in dynamical highly acetylated, salt-soluble chromatin domains, which were associated with H3K4me3 and H3K27ac, and also produced distinct antisense transcripts (Jahan et al., 2016). Results of comparing histone H1 subtypes between five avian species (chicken, gray partridge, quail, duck, and pheasant) indicated the potential involvement of histone modification in chromatin structure and function in the development of poultry (Kowalski and Pałyga, 2017).
In addition to the main livestock species discussed above, some epigenetics-related data have been reported in other livestock species. For instance, DNA methylation was predicted to be an age-dependent process in domestic horses (Andraszek et al., 2016). Different from the generally methylated mammalian genomes, honeybees have unique methylation patterns that concentrate in gene bodies and are associated with gene expression (Wedd and Maleszka, 2016; Harris et al., 2019). DNA methylation is involved in the learning and memory processes of honeybees, crucial traits for honey production (Li et al., 2017). Besides, epigenetic modifications, including DNA methylation, histone methylation, and phosphorylation, represent new possible mechanisms of sex and caste determination process in honeybees (Cardoso-Júnior et al., 2017; Yagound et al., 2019). Moreover, WGBS data showed differential methylation levels between honeybee queen larvae and worker larvae as well as 38 DMGs with functions in specific organ differentiation, morphology, reproduction, and vision differentiation during caste determination (Wang et al., 2020a). Furthermore, the identification of allele-specific DNA methylation patterns in honeybees provided a relatively reliable theory of genomic imprinting underlying parent-of-organ effects caused by reciprocal crosses (Remnant et al., 2016).
Nutrition represents the principal environmental determinant of an individual’s growth and development. In order to enhance livestock health and welfare, reduce production cost, and adapt to global warming, efforts have been concentrated on adjusting nutritional supplements to livestock animals and their related impacts (McGuffey, 2017; Bobeck, 2020). Growing evidence supports the notion that permanent alterations in the epigenome of germline cells or embryos could be transferred to offspring, referred to as intergeneration or transgenerational epigenetic inheritance (Heard and Martienssen, 2014; Miska and Ferguson-Smith, 2016). It is well accepted that nutrition-induced epigenetic alterations can be heritable; however, the underlying mechanism is still controversial. On one side, epigenetic changes caused by consistent nutritional stimulus were identified in somatic tissues, indicating an undefined indirect mechanism of inheritance (Xue et al., 2016). On the other side, continuous stimulus as a result of the effects of nutritional factors on health and diseases of livestock could be transmitted between generations through altered epigenetic state of germline cells (Ideraabdullah and Zeisel, 2018). For example, the excessively high or excessive lack of nutrition (hyper-/hyponutrition, respectively) or nutrition component deficiencies could lead to epigenetic alterations (DNA methylation, histone modifications, and ncRNAs) in germ cells and transmission to subsequent generations (Guo T. et al., 2020). A number of studies, both in vivo and in vitro, showed that nutritional stimulus, including methionine, choline, and energy restriction, could induce epigenetic modifications causing the alteration of gene expression (Murdoch et al., 2016; Chavatte-Palmer et al., 2018; Elolimy et al., 2019). Data on the epigenetic modifications in response to nutritional stimulus in livestock are summarized in Table 1.
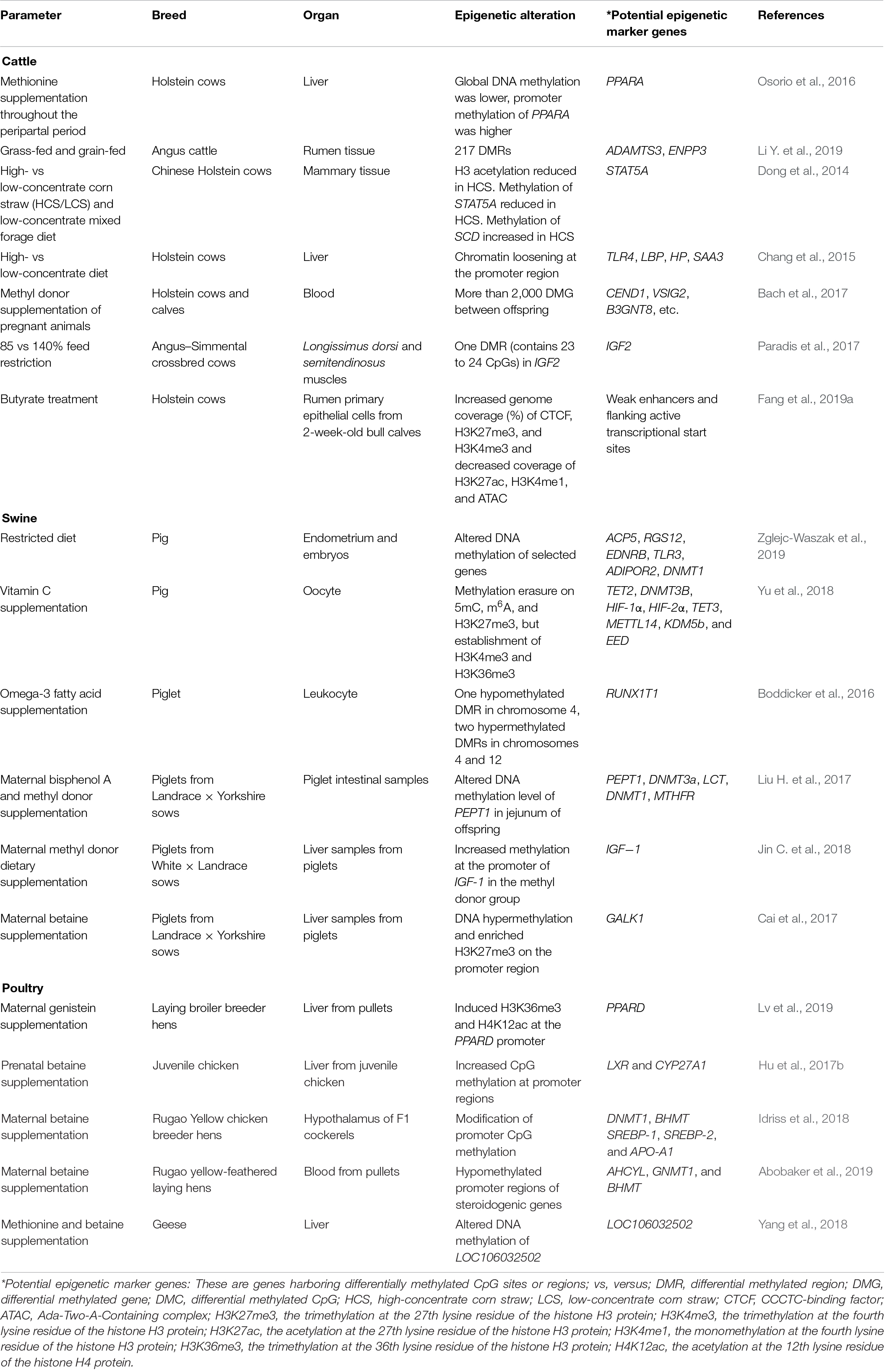
Table 1. Epigenetic alterations in response to nutritional stimulus in livestock.
DNA methylation modifications in response to nutritional stimulus or environmental changes may cause alteration in production performance or disease susceptibility (Jang and Serra, 2014; Block and El-Osta, 2017; Maugeri and Barchitta, 2020). The interaction between changes in feed composition and epigenetic regulatory mechanisms has been reported. Liver tissues of methionine (Met)-supplemented Holsteins were found to have lower general DNA methylation levels compared with that of Holsteins without Met supplementation (Osorio et al., 2016). In the same study, the overall gene expression levels of PPARα and its target genes were upregulated in Met-supplemented Holsteins, which was related to improved metabolism and immune functions (Osorio et al., 2016). Additionally, differential expression of ADAMTS3 and ENPP3 genes (have roles related to the biosynthesis and regulation of glycosyltransferase activity, respectively) between grass-fed and grain-fed Angus cattle were associated with the methylation abundance of corresponding DMRs (Li Y. et al., 2019). Meanwhile, DNA methylation was involved in the regulation of altered gene expression in response to high-concentrate diets resulting in the downregulation of immune-related genes (TLR-4, LBP, HP, and SAA3) in mammary and liver tissues of cows (Dong et al., 2014; Chang et al., 2015; Xu et al., 2018). In addition to DNA methylation, other epigenetic modifications have been reported to respond to nutritional stimulus. Histone H3 acetylation was significantly reduced in mammary gland tissue and also correlated negatively with lipopolysaccharide (LPS) concentrations in the mammary arterial blood of Chinese Holstein cows fed a high-concentrate corn straw diet (Dong et al., 2014). Linseed oil supplementation of Holstein cows in mid lactation, which resulted in 30% reduction of milk fat yield, significantly repressed the expression of histone acetylases (HDAC2, HDAC3, SIRT2, and KAT2A) and histone methyltransferase (EHMT2), suggesting potential epigenetic regulation of milk fatty acid synthesis (Li and Ibeagha-Awemu, 2017). Additionally, chromatin loosening was found to contribute to the upregulation of some immune-related genes in the liver of dairy cows in response to a high-concentrate diet (Chang et al., 2015). Furthermore, butyrate treatment of rumen epithelial cells revealed increased genome coverage (percentage) of CTCF, H3K27me3, and H3K4me3, but decreased coverage of H3K27ac, H3K4me1, and ATAC (Fang et al., 2019a). In the same study, 15 distinct chromatin states were defined according to the combination of identified epigenomic markers in genomic regions, which revealed that weak enhancers flanking active transcriptional start sites could be possible mechanisms underlying gene expression regulation by epigenomic markers (Fang et al., 2019a).
Nutritional supplementation during pregnancy caused epigenetic alterations in the fetus with long-term influences on the development and productivity of the offspring. Embryos (at 6.5 days) of dairy cows showed lower DNA methylation level in response to Met supplementation during the preimplantation period, which probably enhanced its survival capacity (Acosta et al., 2016). The supplementation of methyl donors to Holstein dams during pregnancy significantly altered the methylome of their offspring, and the DMCs affected the expression of genes involved in various biological processes, such as immune function, regulation of cell growth, and kinase activity (Bach et al., 2017). Maternal methyl donor supplementation was also found to alter the hepatic metabolism program of calves by maintaining Met homeostasis, DNA methylation, energy metabolism, etc., which potentially contributed to better nutrient utilization efficiency of calves and promoted their growth and development performance (Alharthi et al., 2019). Moreover, energy restriction significantly impacted the DNA methylation level of a DMR in IGF2 in fetal longissimus dorsi of beef cattle, where IGF2 expression was negatively associated with fetus weight in Angus–Simmental crossbred cows (Paradis et al., 2017). Furthermore, offsprings’ weight was affected by their mothers’ high-fat diet (offspring were obese), and individual differences of obesity were potentially regulated by epigenetic modifications (Keleher et al., 2018; Glendining and Jasoni, 2019). Also, 5hmC and 5mC were found to be negatively and positively correlated with body weight in offspring, respectively, and altered CpG methylation in the proopiomelanocortin (POMC) promoter region induced histone modification through binding of MBD1 to 5mC, which reduced POMC expression (Marco et al., 2016). Furthermore, it was reported that changes in diet during pregnancy can also influence the reproduction ability of female offspring, which may be regulated by epigenetic modifications (Noya et al., 2019; Shah and Chauhan, 2019).
In pigs, dietary changes, such as feed restriction and vitamin C supplemental feeding, were reported to induce modifications of DNA methylation during development of porcine germline cell and embryo (Yu et al., 2018; Zglejc-Waszak et al., 2019). Prenatal and postnatal dietary omega-3 fatty acid supplementation resulted in altered global DNA methylation patterns and probable implication in the growth and inflammatory processes of piglets (Boddicker et al., 2016). In addition, supplementation of methyl donors during gestation could improve intestinal digestion and absorption and the growth rate of offspring piglets, and these attributes were associated with DNA methylation modifications in specific genes and their corresponding regulated expression (Liu H. et al., 2017; Jin C. et al., 2018). Furthermore, repressed expression of GALK1 gene by DNA hypermethylation and histone trimethylation in the liver was associated with low serum concentration of galactose in neonatal pigs in response to betaine-supplemental feeding of sows (Cai et al., 2017).
Epigenetic modifications have been reported to regulate the impacts of parental feed additive supplementation on offspring pullets. For example, histones H3K36me3 and H4K12ac in the promoter region of PPARδ gene were involved in the regulation of altered lipid metabolism and growth performance following maternal genistein supplementation (Lv et al., 2019). Betaine is a frequently used supplement in the chicken industry, and its impact on intercellular metabolism is probably influenced by DNA methylation (Hu et al., 2017a). DNA methylation alteration was found to regulate gene expression related to cholesterol and corticosteroid synthesis of offspring pullets in response to maternal betaine supplementation (Hu et al., 2017b; Idriss et al., 2018; Abobaker et al., 2019). Additionally, DNA methylation was associated with the regulation of transcriptional regulatory network in response to dietary methionine and betaine supplementation in goose (Yang et al., 2018).
Milk production is the most important economic trait of the bovine dairy industry, which is affected by multitudinous factors including genetics, nutrition, health, farm management, and environmental conditions (Pragna et al., 2017; Waterman et al., 2017; Sørensen et al., 2018). As summarized in Table 2, epigenetic modifications have been identified as important regulatory mechanisms of milk production in dairy cows and other livestock species (Singh et al., 2010, 2011; Ibeagha-Awemu and Zhao, 2015). Significant differences of global DNA methylation levels in blood were reported between lactating dairy cows with high and low milk yield, indicating the association between milk yield and DNA methylation (Dechow and Liu, 2018; Wang H. et al., 2019). Particularly, abnormal DNA methylation around the STAT5-binding enhancer in the αS1-casein promoter negatively regulated αS1-casein synthesis in milk during lactation, which could be affected by foreign stimulus, such as mastitis and daily milking times (Platenburg et al., 1996; Vanselow et al., 2006; Nguyen et al., 2014). Additionally, the DNA methylation of EEF1D, a gene strongly related to milk production, regulates its spatial expression (Liu X. et al., 2017). Meanwhile, differential DNA methylation levels of milk-related genes (e.g., PPARα, RXRα, and NPY) in the mammary glands of dairy goats at dry and lactation periods indicated important regulatory roles of DNA methylation in goat lactation (Zhang X. et al., 2017). Moreover, inhibition of miR-145 expression impaired fatty acid synthesis in goat milk by increased methylation levels of some lipid-related genes (FASN, SCD1, PPARG, and SREBF1) (Wang et al., 2017b). In addition, higher promoter DNA methylation of ACACA and SCD downregulated their expression and were associated with decreased milk fat of dairy goats in response to a high grain diet (Tian et al., 2017).
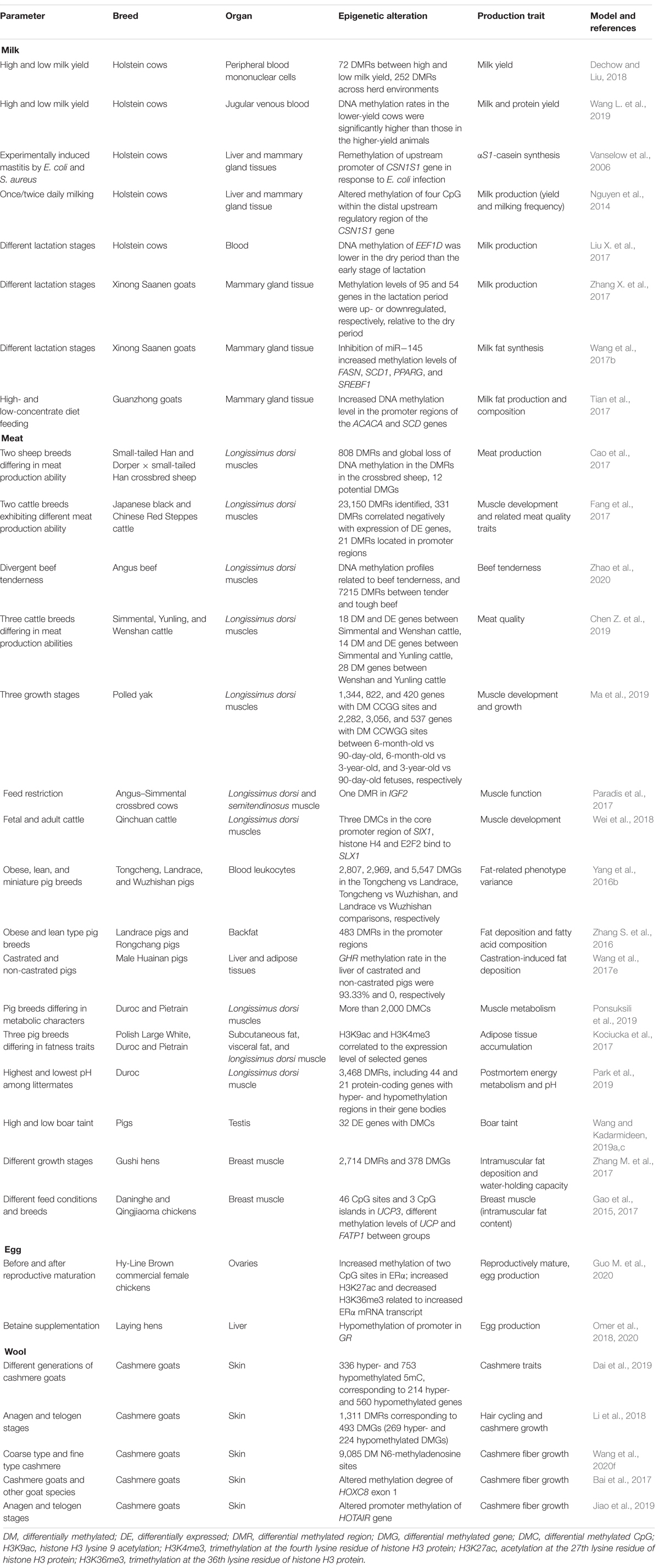
Table 2. Alteration of epigenetic markers in relation to livestock production traits.
Interesting reports of how DNA methylation interacts with miRNA expression and function to regulate milk production have emerged. MiR-152 and miR-29s and their respective target genes DNMT1, and DNMT3A, and DNMT3B are inversely expressed during lactation (Bian et al., 2015; Melnik et al., 2016). For example, miR-148a and miR-152 as well as miRNA-29s impact bovine mammary gland epithelial cell activities and milk synthesis by reducing the mRNA expression levels of DNMT1 as well as DNMT3A and DNMT3B, respectively (Wang et al., 2014; Melnik and Schmitz, 2017a; Liang et al., 2018). Specifically, over- or forced expression of miR-152 resulted in marked reduction of DNMT1 expression (both mRNA and protein), decrease in global DNA methylation levels, increase in the expression of two lactation-related genes (AKT and PPARγ), and enhanced viability and multiplication capacity of mammary epithelial cells (Wang et al., 2014). These effects were reversed by inhibition of miR-152 expression (Wang et al., 2014). Similarly, inhibition of miR-29s triggered global DNA hypermethylation and increased methylation levels of the promoters of some important lactation-related genes, such as CSN1S1, ElF5, SREBP1, PPARγ, and GLUT1, and consequently decreased the secretion of triglycerides, lactose, and lactoprotein by cow mammary gland epithelial cells (Bian et al., 2015). MiRNAs targeting DNMTs were also found to decrease the methylation of core CpG islands at the promoter regions of genes (such as FTO, INS, IGF1, CAV1, etc.) involved in the activation or regulation of various genes with roles in metabolism and milk synthesis (Melnik and Schmitz, 2017b). Conversely, induced methylation at the 5′ terminal of miR-183 inhibited its expression, consequently affecting milk lipid metabolism of dairy cows (Jiao et al., 2020). Furthermore, milk exosomes, regarded as epigenetic regulators that transfer specific regulatory molecules to consumers, regulate the expression of DNMTs and affect human health, especially milk allergy (Melnik et al., 2016; Paparo et al., 2016; Melnik and Schmitz, 2017b).
DNA methylation is one of the most studied epigenetic mechanisms involved in the regulation of gene expression related to muscle development (Table 2; Baik et al., 2014; Gotoh, 2015; Chen Z. et al., 2019). The genome-wide DNA methylation profiles of longissimus dorsi muscles from different breeds of sheep provided insight on the epigenetic regulatory mechanisms modulating the expression of genes involved in the regulation of muscle development, such as DLK1, NR4A1, TGFB3, ACSL1, RYR1, ACOX2, PPARG2, NTN1, and MAPRE1 (Couldrey et al., 2014; Cao et al., 2017; Fan et al., 2020). Meanwhile, a number of DMRs on genes associated with important biological processes such as lipid translocation and lipid transport were identified in latissimus dorsi muscle from different breeds of beef cattle with diverse meat quality traits (Fang et al., 2017). For instance, DNA methylation profiling in relation to beef tenderness in Angus cattle revealed 7,215 DMRs between tender and tough beef (Zhao et al., 2020). The DMRs were significantly enriched in ATP binding cassette subfamily and myosin-related genes, including ABCA1, ABCA7, and ABCG1, with roles in beef tenderness and fatty acid metabolism (Zhao et al., 2020). Besides, demethylation of a DMR in the SIRT4 promoter promoted its transcriptional activity through CMYB mediation or inhibited its transcriptional activity through NRF1 mediation, thereby shedding light on the role of an epigenetic process in the transcriptional regulation of the expression of SIRT4 during bovine adipocyte differentiation (Hong et al., 2019). In addition, important genes with DMRs, including TMEM8C, IGF2, FASN, CACNA1S, FADS6, and MUSTN1, were significantly associated with differences in muscle development and meat quality in several cattle (beef) breeds (Chen Z. et al., 2019; Ma et al., 2019). The methylation level of IGF2, which negatively correlated with its expression, was found to change more in longissimus dorsi muscle than in semitendinosus muscle in response to feed restriction (Paradis et al., 2017). Besides, differential expression of some important DNA methylation genes (DNMT3A, DNMT3B, and DNMT1) were significantly associated with meat and carcass quality traits such as carcass weight, flank thickness, and chuck short rib score in Wagyu × Limousin × Fuzhou yellow crossbred beef cattle (Guo et al., 2012; Liu et al., 2015). DNA methylation in the core promoter region of SIX1 gene in muscle tissues was potentially regulated by histone H4 and E2F2 and shown to impact muscle development in Qinchuan cattle (Wei et al., 2018). Expression and splicing quantitative trait loci mapping analyses for meat quality traits in longissimus dorsi muscle found that the expression of PHF14, an important epigenetic regulator of organ development, was influenced by multigenic effects (Leal-Gutiérrez et al., 2020). The PHF14 protein has many plant homeodomain fingers that are able to recognize specific epigenetic markers on histone tails and thus regulate gene expression, indicating its important roles in skeletal muscle growth and development.
Meat quality is also a trait of high interest in the pig industry. Genome-wide DNA methylation analysis identified numerous DMRs and DMGs between obese and lean pigs, revealing vital roles of DNA methylation in lipogenesis in pigs (Yang et al., 2016b). Specifically, the back fat of Landrace pigs (leaner) had higher global DNA methylation level than the fatty Rongchang pigs, indicating that some identified DMRs may affect lipid metabolism (Zhang S. et al., 2016). Altered methylation in lipid metabolism-related genes was also identified in diverse tissues from different pig breeds (Wang et al., 2017e; Ponsuksili et al., 2019). Additionally, histone modifications were found to affect adipose tissue accumulation by regulating corresponding gene expression (Kociucka et al., 2017). Moreover, DNA methylation pattern scanning revealed its potential involvement in other meat quality traits, such as pH, meat color, and carcasses’ traits (Te Pas et al., 2017; Park et al., 2019). Boar taint, an unpleasant odor that affects pork acceptability, was found to be regulated by epigenetic processes. Genome-wide methylation analysis of the testis of pigs with high, medium, and low boar taint associated DMCs and candidate genes with pig reproduction (e.g., DICER1, PCK1, SS18, and TGFB3) and boar taint (e.g., CAPN10, FTO, HSD17B2, IGF2, SALL4, FASN, PEMT, CRYL1, DNMT3A, and EGFR) thereby revealing important regulatory roles of DNA methylation in boar taint formation (Wang and Kadarmideen, 2019a,b).
In chickens, DNA methylation was reported as one of the regulatory mechanisms modulating crucial meat traits such as intramuscular fat deposition and skeletal muscle development (Zhang M. et al., 2017, Zhang et al., 2020). The whole-genome DNA methylation profiles of later laying-period hen and juvenile hens with differential intramuscular fat deposition and water-holding capacities identified 378 DMRs related to muscle development (Zhang M. et al., 2017). Further research indicated that DNA methylation affected the intramuscular fat deposition by regulating the expression of some key genes, such as ABCA1, COL6A1, and GSTT1L (Zhang M. et al., 2017, Zhang et al., 2020). Moreover, different breeds or feed condition significantly affected the methylation levels of UCP3 and FATP1 genes in chicken breast muscle, which enhanced the reliability of these genes as important candidate genes of intramuscular fat deposition in chicken meat (Gao et al., 2015, 2017). The alterations of epigenetic markers in relation to livestock products (milk, meat, egg, and wool) are summarized in Table 2.
Egg laying in poultry relies on the reproductive maturation of the ovaries, where epigenetic mechanisms play important regulatory roles (He et al., 2018). Epigenetic modifications in ERα were identified during ovarian development and maturation, whereby higher DNA methylation rates in specific CpG sites, higher histone H3K27ac, and lower H3K36me3 associated the abundance of ERα expression with important roles in egg laying (Guo M. et al., 2020). In addition, changes in DNA methylation were identified in response to betaine supplementation and were associated with improved egg laying performance in hens (Xing and Jiang, 2012). Supplemental betaine potentially caused hypomethylation of the promoter of GR, followed by enhanced expression of GR and GR/ERα interaction (contributed to increase VTGII expression in the liver), which partly supported improved egg production in betaine-supplemented laying hens (Omer et al., 2018). Furthermore, promoter region methylation could be the possible regulatory mechanism underlying altered expression of liver lipid synthesis and transport-related genes in response to betaine supplementation, which supported the synthesis and release of yolk precursor substances in the liver and consequently promoted egg laying performance (Omer et al., 2020).
Wool is an economic product of high regard with increasing value in the goat industry, but the limited yield of cashmere wool (or cashmere) was recently speculated to be potentially regulated by epigenetic modifications (Wang et al., 2017f, 2020d). DNA methylation and histone acetylation were found to actively contribute to the regulation of goat fetal fibroblast cells, which is critical for cashmere production (Wang et al., 2017f; Palazzese et al., 2018). Recently, DNA methylation was associated to the genetic stability of cashmere traits between generations of cashmere goats (Dai et al., 2019). Besides, genome-wide scanning revealed potential regulatory roles of DNA methylation and RNA m6A methylation in the growth and development of cashmere fibers in cashmere goats (Li et al., 2018; Wang et al., 2020f). Moreover, epigenetic modifications of some specific genes were reported to affect cashmere traits. For instance, methylation of HOXC8 is involved in regulating the growth of cashmere fiber in cashmere goat (Bai et al., 2017). Promoter methylation of HOTAIR gene and related suppressed expression were found to regulate the reconstruction of secondary hair follicles in cashmere goat (Jiao et al., 2019). Furthermore, crucial regulatory roles for DNA methylation in wool fiber development and transformation of fur with special characters and production purpose, such as curly wool with beautiful white color or high-quality brush hair, have been observed (Qiang et al., 2018; Xiao et al., 2019).
Environmental stressors including heat stress, pathogens, dietary changes, etc. are the greatest determinants of individual health and productivity. As summarized in Table 3, epigenetic alterations in response to environmental stressors have been reported in livestock animals. Heat stress has negative impacts on animal production and health, which may continue to be of great concern due to increasing global temperatures. The important roles of DNA and histone methylation on heat-shock proteins under heat stress and heat acclimation and their involvement in host response to heat stress were summarized recently (Wu et al., 2020a). Heat stress, potentially regulated by epigenetic modifications, such as DNA methylation, DNA hydroxymethylation, and histone modifications, was reported to significantly affect bovine embryonic development and fertility (Mendes et al., 2017; de Barros and Paula-Lopes, 2018; Diaz et al., 2019; Sun et al., 2019). Epigenetic regulation was associated with the effects of betaine on heat stress reduction in poultry (Nayak et al., 2016; Saeed et al., 2017). In addition, DNA methylation and histone H3K27me3 and H3K4me3 changes were identified to partly regulate the adaption of chickens to embryonic thermal manipulation, which is crucial for improving their thermal adaptability to heat stress in postnatal life (Kisliouk et al., 2017; Vinoth et al., 2018; David et al., 2019). DMRs and associated genes with roles in energy and lipid metabolism, cellular defense, and stress responses were identified in longissimus dorsi muscles of heat-stressed pigs indicative of roles of epigenetic regulation of pig muscle development, meat quality, and heat stress processes in pigs (Hao et al., 2016). Increased m6A RNA methylation level and increased expression levels of m6A-related enzymes and heat stress proteins were observed in the liver of sheep after heat stress, indicating involvement of m6A in the regulation of host response to heat stress (Lu et al., 2019).

Table 3. Epigenetic changes impacting livestock health.
Hypoxic stress is an important environmental stressor affecting porcine growth, especially in high-altitude regions. Genome-wide DNA methylation profiles of porcine tissues from pigs raised in regions of different altitudes revealed important regulatory roles of DNA methylation in porcine hypoxia adaptation (Jin L. et al., 2018; Zhang et al., 2019). For example, some DMGs identified in heart tissues of Tibetan pigs from high- and low-altitude regions were significantly enriched in hypoxia-inducible factor (HIF) 1 signaling pathway suggesting impact on hypoxia-related processes (Zhang et al., 2019). Particularly, DNA methylation mediated the expression of SIN3A and CACNG6 in longissimus dorsi muscle of Tibetan pigs during low-altitude acclimation (Jin L. et al., 2018). Moreover, methylation changes in hypoxia genes, such as higher methylation levels in HIF-1α, HIF-3α, and EPO and lower methylation level in HIF-1, were identified in the heart, liver, lungs, kidney, muscle, and brain tissues of plateau goat and sheep, suggesting the involvement of epigenetic regulatory mechanisms of hypoxia resistance of plateau animals (Wang et al., 2017g).
In cattle, maternal stress due to transportation was identified as a potential factor that induced methylome changes in Brahman bull calves, whereby thousands of hyper- and hypomethylated CpG sites were identified compared with non-transported control calves (Littlejohn et al., 2018). The methylome changes were through increased DNA methylation sites at promoter regions of genes enriched in pathways related to behavior, stress response, metabolism, and immune response, which induced the repression of their transcriptional activities (Littlejohn et al., 2018). In goat, lowered global DNA methylation level was thought to be involved in upregulated activity of caspase-3 and caspase-8 enzymes, increased expression of inflammatory cytokines (IL-10, IL-1β, and iNOS2) and activation of TLR-4 and NF-κB pathways in response to chronic stress induced by long-term application of low doses of dexamethasone in colonic epithelium of goats (Cai et al., 2019).
Epigenetic modifications are known to significantly affect the dynamic regulation of immune responses to infection and other stressors (Emam et al., 2019; Safi-Stibler and Gabory, 2019). Studies on DNA methylation and the immune response have described the methylation of immune-related genes and the global DNA methylation patterns in response to varied disease pathogens (Table 3). For example, DNA methylation was found to directly affect gene expression in CD4+ T cells during an infection of Mycobacterium bovis in cattle (Doherty et al., 2016). The promoter region of miR-29b showed significant decreased methylation level in Madin–Darby bovine kidney (MDBK) cell line infected with bovine viral diarrhea virus (BVDV; Fu et al., 2017). Moreover, silencing of DNMT1 expression in MDBK significantly decreased miR-29b promoter methylation and upregulated its expression, as well as repressed BVDV replication, supporting the interaction between DNA methylation and miRNA in the regulation of livestock health (Fu et al., 2017). Bacterial LPS stimulation of endometrial cells resulted in increased expression of immune-related genes (IL-6 and IL-8), which was enhanced by the inhibition of DNA methylation (Wang et al., 2018). In bovine mammary epithelial cells, altered methylome (mainly hypermethylation) in response to lower doses of LPS (1–10 EU/ml) impacted the expression of genes (e.g., ACACA, ACSS2, and S6K1) related to milk production (lipid and amino acid metabolism), while high LPS doses (>10 EU/ml) induced hypomethylation of genes in immune response pathways (Chen J. et al., 2019). DNA methylation was reported to regulate the expression of IL-6R rather than genetic mutations in response to mastitic pathogen (Zhang et al., 2018a). Moreover, the co-stimulation of bovine mammary epithelial cells with LPS, peptidoglycan (PGN), and lipoteichoic acid (LTA) significantly increased DNA hypomethylation compared with LPS stimulation, indicating that the additive effects of co-stimulation decreased methylation levels resulting in increased transcriptome changes and inflammatory responses (Wu et al., 2020c). The hypermethylation of the CD4 promoter was reported to repress its gene expression in Holstein cows with clinical mastitis (Wang et al., 2013; Usman et al., 2016). Recently, NCKAP5 and transposon MTD were found to be differentially methylated in a mouse model of mastitis, indicating their potential effects on the development of Staphylococcus aureus mastitis and their potential as epigenetic markers of S. aureus mastitis (Di Wang et al., 2020). Furthermore, a plethora of DMRs were identified in bovine mammary gland tissues in response to mastitis caused by different pathogens, including Escherichia coil and S. aureus, revealing crucial regulatory roles of DNA methylation in mammary immunity during mastitis (Sajjanar et al., 2019; Wu et al., 2020b). Moreover, genome-wide DNA methylation alteration in the format of CmCGG was significantly related to the immune response to S. aureus-induced mastitis, and several genes including IL-6R, TNF, BTK, IL-1R2, and TNFSF8 were identified as potential epigenetic markers of S. aureus mastitis (Wang et al., 2020c). DMRs were also identified in peripheral blood from mastitis-infected cattle, further demonstrating the importance of DNA methylation in host immune response (Song et al., 2016; Ju et al., 2020).
In addition to DNA methylation, histone modifications also contribute to mammary gland immunity (Silva et al., 2018; Wu et al., 2020b). For example, inhibition of histone deacetylase increased the expression of β-defensin and possibly improved host resistance to intramammary infections (Kweh et al., 2019). Besides, H3K27me3 in the upstream region of key genes like IL-10, PTX3, etc. regulated their expression in bovine lymphocytes in response to S. aureus mastitis (He et al., 2016). Recently, integration of chromatin immunoprecipitation sequencing (ChIP-seq), RNA sequencing, and miRNA sequencing data from M. bovis-infected macrophage revealed that bovine alveolar macrophage transcriptional reprogramming arises through discrepant distribution of H3K4me3 and RNA polymerase II at key immune genes (Hall et al., 2019). However, no differences were found between the methylomes of healthy and M. bovis-infected bovine alveolar macrophage 24 h post infection suggesting that DNA methylation may be less involved in the early host response to M. bovis (O’Doherty et al., 2019).
In dairy goats, reduced promoter methylation contributed to the regulated expression of key genes related to inflammation and apoptosis in the liver during subacute ruminal acidosis induced by high-concentrate diets (Chang et al., 2018). Additionally, abnormal DNA methylation levels of genes with roles in signaling and transportation and their involvement in the pathogenesis of scrapie were identified in sheep with scrapie compared with healthy controls (Hernaiz et al., 2019). Dynamic DNA methylation changes have also been reported to impact porcine immune responses by regulating the expression of immune-related genes. A high number of DMRs showing inverse association with gene expression were identified in peripheral blood mononuclear cells in response to poly I:C stimulation, as well as 70 differently methylated and expressed genes with related functions in the regulation of the immune system and leukocyte activation (Wang et al., 2017c). Differential gene expression in response to poly I:C and LPS stimulation was also reported to be significantly associated with H3K27ac alteration at active regulatory regions enriched for TF binding motifs of TFs with roles in the inflammation response (Herrera-Uribe et al., 2020). In addition, involvement of DNA methylation in the regulation of the expression of intestinal immune metabolism-related genes during bacteria colonization immediately after birth and the subsequent influence on newborn intestinal immune development has been reported (Pan et al., 2018, 2020). Promoter methylation level of BPI gene in Yorkshire, Sutai, and Meishan pigs was negatively associated with its gene expression and contributed to intestinal immunity and disease susceptibility (Wang et al., 2017d). Promoter methylation in PACSNI1 repressed its expression and indirectly promoted the production of IL−6, IL−8, and TNFα, indicating its potential to mediate porcine response to disease pathogens (Feng et al., 2019). In addition to regulatory roles in porcine immune responses, epigenetic mechanisms, including DNA methylation and histone modifications, were frequently observed to play roles in porcine diseases. E. coli-induced DNA methylation alteration in the form of DMCs in porcine mammary epithelial cells was mapped to the regulatory regions of immune-related genes, such as SDF4, SRXN1, CSF1, and CXCL14 (Sajjanar et al., 2019). A total of 1,885 H3K4me3 associated with 1,723 genes were identified in the jejunum of piglets with porcine epidemic diarrhea virus, revealing a positive correlation between higher H3K4me3 deposition and increased expression levels of some antiviral genes, including AS1, OAS2, EFNB2, and CKS1B (Wang H. et al., 2019). The overexpression of HDAC6 enhanced host resistance to porcine reproductive and respiratory syndrome virus (PRRSV) infection, resulting in repressed PRRSV production in vitro and lower viral load in the lung and less clinical symptoms in vivo (Lu et al., 2017).
Epigenetic modifications also play important regulatory roles in the immune response of chickens. The whole genome-wide DNA methylation patterns of lungs from two chicken lines differing in genetic resistance to multiple pathogens revealed many immune-related gene ontology terms enriched by genes within DMRs, suggesting DNA methylation as a possible regulatory mechanism underlying the immune response differences (Li et al., 2015). A dynamic unstable chromatin structure with nucleosome-free regions, that intermingled with H3K4me3- and H3K27ac-modified nucleosomes, was identified in the body of some genes participating in the innate immune response of chickens (Jahan et al., 2019). Also, 5hmC was associated with B-cell death during the immune response to infectious bursal disease virus infection in chickens (Ciccone et al., 2017). In addition, DNA methylation, histone modifications, and other epigenetic signatures were reported during the immune response to diverse infectious diseases in chickens. The global DNA methylation level of immune organs, including thymus and bursa, was significantly upregulated in chickens with avian influenza virus infection (Zhang Y. et al., 2016). The blood methylome showed slightly higher methylation levels around the transcription start and termination sites in Salmonella enterica-infected chickens than healthy controls, and the differentially methylated peaks in the promoter regions were vastly correlated with immune-related genes (Wang et al., 2017a). Marek’s disease virus induced various temporal chromatin signatures to bursa of Fabricius chickens at different stages of Marek’s disease development, and the differential H3K27me3 was significantly enriched in pathways related to the immune response (Mitra et al., 2015; Song, 2016). The response of two genetically distinct highly inbred layer chicken lines (Leghorns and Fayoumis) to Newcastle disease virus (DNV) infection while under heat stress revealed greater differences in histone modification (H3K27ac and H3K4me1) levels in Leghorns than Fayoumis, and the associated genes were enriched in biological processes gene ontology terms related to cell cycle and receptor signaling of lymphocytes, thereby revealing the possible cellular mechanisms underlying the development of genetic variation in NDV resistance (Chanthavixay et al., 2020a). Furthermore, epigenetic reprogramming in the form of histone trimethylation and acetylation is possibly involved in the regulation of gene expression related to improved innate immune system conditioning following vaccination of laying hens (Kang et al., 2019a,b).
A biomarker is a factor or distinctive property or character that can be measured and evaluated as an indicator or gauge of normal biological and pathological processes. The Food and Agricultural Organization defines a biomarker as any substance, structure, or process which impacts or predicts the incidence of disease or its consequences, and could be quantified (World Health Organization [WHO], 2001). Biomarkers are classified into many specific types, including diagnostic, prognostic, predictive, therapy monitoring, and risk biomarkers (Bock, 2009). For clinical application, biomarkers are expected to be specific, sensitive, and stable and could be validated in abundant samples by different labs (Mishra and Verma, 2010).
According to the properties of biomarkers, an epigenetic biomarker is defined as any epigenetic mark or changed epigenetic mechanism that is measurable in different tissues or body fluids and can delineate a disease condition (detection), predict the outcome of disease (prognostic biomarker) or response to therapy or treatment (predictive biomarker) or a monitor of treatment response (therapy monitoring biomarker), or forecast the risk of future disease development (risk biomarker) (García-Giménez et al., 2016). Since epigenetic markers respond to different types of internal (e.g., maternal environment, etc.) and external environmental cues (e.g., nutrition, management practices, disease pathogens, etc.) as directed by the underlying genetic composition during a lifetime, epigenetic biomarkers may represent the evolution of individual phenotype variations and can contribute to improved disease and production management. In addition, the dynamic changes due to extra- or intraenvironmental cellular conditions and disease progression or evolution in response to environmental factors are one advantage of epigenetic biomarkers when compared with stable (not changing) genetic biomarkers based on gene sequence (García-Giménez et al., 2016). Association of genetic biomarkers to phenotypes is often inconsistent across studies, while epigenetic markers are promising substitutes for the timely diagnosis and monitoring of diseases (Rahat et al., 2020). Furthermore, epigenetic markers being tissue specific reflect the pattern of disease progression (Zeng et al., 2019). Moreover, epigenetic markers, especially methylated DNA and miRNA, have high stability in a variety of samples (e.g., tissues, blood, urine, plasma, milk, etc.) and are stable over a range of conditions. Also, a higher spontaneous epimutation rate (three orders of magnitude) than genetic mutation rate in Arabidopsis thaliana has been reported (Schmitz et al., 2011), implying a higher spontaneous mutation rate and availability of more raw materials for genetic improvement due to epimutations than genetic or nucleotide mutations. An epimutation, which is different from DNA mutation, is generally defined as a heritable change in gene activity that is linked to gain or loss of DNA methylation or modifications of chromatin (Oey and Whitelaw, 2014). Epimutations have been further separated into primary (occurs in the absence of DNA sequence change) and secondary (occurs secondary to a DNA mutation in a cis- or trans-acting factor) categories (Horsthemke, 2006). Moreover, epimutations have been described as constitutional, meaning that they are derived from the germline and consequently should be present in all of the tissues of an individual or somatic (arise in cells in somatic tissues) (Hitchins and Ward, 2009). Evidence of how epimutations induced by endocrine disrupting chemicals impact gene expression, potentially leading to the development of heritable disease conditions in humans have been summarized recently (Lehle and Mccarrey, 2020).
To enable application, biomarkers must be characterized and validated. In farm animals, however, epigenetic research is still at the exploratory level, compared with extensive work in humans and model organisms that has enabled the detection of epigenetic biomarkers and application in various conditions. In humans, epigenome-wide association studies (EWAS) have facilitated the identification of epigenetic biomarkers and their association with phenotype of complex traits, such as human longevity, disease predisposition, diseases, etc. (Abbring et al., 2019; Szymczak et al., 2020). Besides, growing EWAS evidence supports the application of epigenetic biomarkers in human disease diagnosis and treatment (Birney et al., 2016; Carnero-Montoro and Alarcón-Riquelme, 2018; Edris et al., 2019). Various epigenetic biomarkers have been identified for different diseases, such as tumors, colorectal cancer, cardiovascular diseases, etc., revealing their potential use in prognostic, prediction, and even treatment (Kamińska et al., 2019; Soler-Botija et al., 2019; Jung et al., 2020). A DNA methylation assay based on SEPT9 was the first Food and Drug Administration (FDA)-approved cancer test based on DNA methylation and showed high sensitivity (71.1–95.6%) and specificity (81.5–99%) to colorectal cancer, the leading cause of cancer deaths (Tanić and Beck, 2017). In addition, a GSTP1 methylation assay based on a hypermethylated CpG island in the promoter of GSTP1 and frequently reported in tumor tissues from prostate cancer patients is under clinical test to improve the detection sensitivity, specificity, and accuracy of early prostate cancer diagnosis (Martignano et al., 2016; Markou et al., 2017). Recently, the discovery of epigenetic drugs promoted the further development of sensitive epigenetic biomarkers for predicting or dealing with disease evaluation (Sistare and DeGeorge, 2007). For instance, DNMT inhibitors, including 5-azacytidine and 5-aza-2’-deoxycytidine, were approved by the FDA and demonstrated to be highly efficient in the treatment of hematological malignancies (Kantarjian et al., 2012; Adès et al., 2013). DNMT inhibitors (azacitidine and decitabine) were reported to significantly improve the survival of patients with myelodysplastic syndromes; however, only about 50% of patients showed good clinical responses that were measurable or visible after 4–6 months of treatment (Lee et al., 2013). To deal with the silent 4–6-month stage, predictive epigenetic biomarkers would have great clinical value to reduce the possible effects of ineffective treatments that may cause side effects, unnecessary cost, and time wastage (Treppendahl et al., 2016).
As discussed in the sections above, diverse alterations of epigenetic markers have been revealed to be significantly associated with livestock health, suggesting their potential as epigenetic biomarkers that could be used for diagnostic, prognostic, predictive, or therapy monitoring. Moreover, environmental factors, such as living or farm environment, feed quality/quantity, pathogens, parental stress, environmental stress, chemicals, etc., directly affect livestock productivity, and these effects captured through epigenetic markers can be included in animal health management. For example, dynamic alterations of epigenetic mechanisms in response to parental nourishment and environmental factors or perturbations, especially at the stage of embryo development during pregnancy, have been demonstrated (Dean et al., 2005; Luo et al., 2018), and their identification and consideration during critical stages of offspring development could lead to healthier pregnancies by including them in farm management strategies. In addition, the identification of possible epigenetic biomarkers underlying these effects could contribute to the evaluation of health and productivity of offspring early in life paving the way for early intervention. Epigenetics biomarkers could be particularly suitable for the detection and management of chronic, silent (no obvious clinical symptoms) livestock diseases, such as metabolic disorders, porcine muscular degenerative disease, chronic mastitis, subacute ruminal acidosis, and paratuberculosis.
The contribution of epigenetic modifications to livestock phenotype variation, supported by growing evidence, is gaining importance and supports the potential application of epigenetic biomarkers, especially DNA methylation in livestock breeding programs (González-Recio et al., 2015; Ibeagha-Awemu and Zhao, 2015; Triantaphyllopoulos et al., 2016; Ibeagha-Awemu and Khatib, 2017; Paiva et al., 2019). The potential usefulness of epigenetic biomarkers in livestock breeding is further emphasized by the fact that phenotypic expression is not only a reflection of an individual’s DNA composition or sequence but also a reflection of how the genome is copied and regulated by the epigenome taking into account both past and present environmental influences or information (Ibeagha-Awemu and Khatib, 2017). Furthermore, epigenetic inheritance (also known as non-genetic inheritance or transgenerational epigenetic effects) refers to any modification in offspring phenotype that is due to the transmission of factors other than DNA sequence information from parents or ancestors (Bonduriansky and Day, 2009). Epigenetic inheritance has been reported to play crucial roles in phenotypic variation during one’s own and offspring development (Triantaphyllopoulos et al., 2016; Nilsson et al., 2018). Therefore, epigenetic inheritance, including intragenerational and transgenerational inheritance, underscores the notion that individual phenotype modifications could at least partly result from the environmental effects on founder generations during key developmental stages of germline cells (Skinner, 2011; Nilsson et al., 2018; Skinner et al., 2018). Therefore, the transmission of epigenetic biomarkers, such as DNA methylation, histone modifications, and ncRNAs, between generations plays a part in epigenetic inheritance in livestock animals (Feeney et al., 2014; Triantaphyllopoulos et al., 2016; Thompson et al., 2020). The current genetic data used for livestock breeding could only explain a portion of phenotypic variance or trait heritability, and supplementing genetic data with epigenetic biomarkers could improve the prediction accuracy of breeding values (González-Recio et al., 2015; Ibeagha-Awemu and Khatib, 2017; Yakovlev, 2018).
As discussed in the sections above, epigenetic variations ranging from single sites to epigenome-wide maps and their regulatory mechanisms of gene expression have been reported in different tissues of several mammalian species (Ibeagha-Awemu and Zhao, 2015; Chavatte-Palmer et al., 2018; Ahmad et al., 2019; Morales-Nebreda et al., 2019; Choi et al., 2020). Furthermore, association between single nucleotide polymorphisms (SNPs) and differential DNA methylation has been reported, indicating that methylation alteration leads to variable expression of related genes and thereby phenotype determination (Banovich et al., 2014; Imgenberg-Kreuz et al., 2018). The alteration of CpG sites caused by SNP suggested one possible mechanism that SNP impacts gene expression by the altered epigenetic patterns, thereby suggesting the possible application of epigenetic biomarkers in livestock improvement breeding (Zhi et al., 2013; Maldonado et al., 2019). However, data to support the exploration and application of epigenetic biomarkers in livestock breeding is currently limited. For instance, data on the contribution of epigenetic alterations to the heritability of livestock health and production traits are not available. Moreover, statistical methods are urgently needed to support quantification of the exact contribution of epigenetic biomarkers to phenotype variation. It was proposed recently that the effects of genetic and non-genetic inheritance should be dissected and considered in the estimation of trait heritability (Danchin et al., 2011). Therefore, the animal or mixed-effects models or statistical approaches that support the simultaneous evaluation of several variance components can be expanded to include the non-genetic or epigenetic components of variation for the purpose of livestock breeding (Tal et al., 2010; Ibeagha-Awemu and Khatib, 2017). Thence, the relationship between DNA methylation biomarkers with production traits, identified through EWAS, could be included in the development of new breeding methods to enable quantification of the epigenetic contribution to the prediction of breeding values.
Next-generation sequencing methods, such as WGBS, reduced representation bisulfite sequencing (RRBS), ChIP-Seq, etc., have supported the profiling of epigenetic markers at a genome-wide scale in livestock species. However, only a limited number of samples can be profiled at a time due to the cost associated with using these technologies. Furthermore, data generated on a limited number of samples is not adequate for use in improvement management/breeding. Therefore, less expensive tools that support application in a large number of samples are needed to support the application of epigenetic information in livestock production. In humans for example, array-based DNA methylation arrays have been developed to support EWAS and for further application in disease diagnosis and treatment (Birney et al., 2016; Carnero-Montoro and Alarcón-Riquelme, 2018). The Infinium® HumanMethylation450 BeadChip methylation array (450K) is popularly used to detect methylation changes at 450,000 CpG sites in the human epigenome, and was recently updated to Infinium MethylationEPIC BeadChip array (850K) with doubled coverage (over 850,000 CpG sites) of methylation sites (Sandoval et al., 2011; Moran et al., 2016). The high accuracy and reliability of DNA methylation measurement and association with biological traits, in hundreds to thousands of samples based on arrays, promoted the wide application of DNA methylation in EWAS in humans (Li M. et al., 2019). The lack of commercially available epigenome analysis assays severely restricts the application of EWAS for uncovering the epigenetic biomarkers associated with livestock health and production traits. The development of epigenome-wide arrays for epigenetic pattern identification in large samples becomes a prerequisite for the application of epigenetic biomarkers in livestock breeding and production management. Therefore, there is an urgent need for the development of livestock-specific assays based on epigenetic mechanisms (especially DNA methylation) with high reliability and commercial availability. Besides, livestock epigenetics research is developing with the potential to improve the reliability and accuracy of breeding values estimation with possible application in livestock management, breeding, and selection.
Genome editing technologies which have been successfully used to modify livestock phenotype through the introduction of useful alleles for heat tolerance, disease resistance (e.g., tuberculosis, mastitis, bovine respiratory disease), production (e.g., production of male-only offspring, myostatin gene knockout), elimination of allergens (e.g., beta-lactoglobulin gene knockout), and welfare (e.g., introduction of polled or hornlessness) into livestock populations [reviewed by Mueller et al. (2019); Van Eenennaam (2019), and Bishop and Van Eenennaam (2020)] hold great promise for furthering the application of epigenetic modifications in livestock improvement. Moreover, application of epigenetic editing at specific loci of interest epitomizes an innovative procedure that might selectively and heritably alter gene expression (Vojta et al., 2016).
As demonstrated in the sections above, epigenetic markers contribute to livestock phenotype variation. Moreover, the epigenome responds to the exposome (nutrition, pathogens, chemicals, maternal behavior, parental environment, climatic conditions, environment, management practices, etc.) in a tissue- and cell-specific manner. With current advances in sequencing and data management technologies, the possibility for genome-wide analysis of epigenetic modifications in specific livestock tissues in response to the exposome is enormous. However, compared with human and model organisms, epigenetic studies in livestock is less developed. Many possible reasons have been advanced to explain this, including limited available funding, research tools and epigenetic research activities on livestock, insufficient recognition of the contribution of epigenetic variation to livestock phenotype diversity, and limited involvement of a considerable number of research professionals in livestock epigenetic research (Ibeagha-Awemu and Zhao, 2015). Efforts of the international consortium on functional annotation of animal genomes (FAANG Project, www.faang.org) and various genome-wide DNA methylation or histone modifications profiling (mentioned in the sections above) have reported epigenetic variation in diverse but limited tissues of livestock species (Foissac et al., 2019; Halstead et al., 2020). Therefore, more efforts are needed to explore the epigenetic variations and biomarkers in different livestock organs/tissues under varying conditions and their contribution to phenotypic expression. Furthermore, monitoring of the dynamic changes of epigenetic markers in response to environmental factors, such as nutritional changes and diseases, could be used to develop new health and disease detection and prediction tools.
Even with mounting evidence supporting the contribution of epigenetic modifications to livestock phenotype variation, there is limited recognition and exploitation of the contribution of epigenetic biomarkers to phenotype variation in livestock management and breeding. Development of advanced statistical methods is required to enhance the understanding of how epigenetic markers interact with genetic factors to influence phenotype diversity of production and health traits in livestock. Furthermore, potential epigenetic regulation has been explored only in a handful of traits and conditions. Moreover, most recent investigations paid attention to the epigenetic modifications in response to single factors, but majority of livestock traits are modulated by the interaction of multiple factors, therefore deserving holistic approaches. Therefore, identification of the epigenetic contributions to livestock traits under the influence of multiple factors is needed and will bring added value for the utilization of epigenetic biomarkers in livestock management. Moreover, limited studies have focused on the linkage between epigenetic modifications and developmental outcomes. Thus, in-depth exploration linking epigenetics and related physiologic responses may further our understanding of the mechanisms underlying livestock productivity and health.
Among the epigenetic studies carried out in livestock, a limited number focused on epigenetic inherence and epigenetic transgenerational biomarkers detection (Triantaphyllopoulos et al., 2016). A major challenge in the examination of epigenetic inherence and its potential application in livestock breeding is the ability to trace epigenetic variations between generations. Accumulating evidence indicates that environmentally induced epigenetic biomarkers could be acquired and used to form transgenerational memory that is partly responsible for environmentally induced heritable traits (Heijmans et al., 2008; Daxinger and Whitelaw, 2010). As summarized in Tables 1–3, many factors, such as pathogens, nutrition, etc., are associated with epigenetic alterations during individual development, especially in germline cells. It has been reported that environmentally induced epigenetic transgenerational inherence of DNA methylation changes in sperm promoted genome instability such as changes of copy number variations in next generations (Skinner et al., 2015). However, the effects of environmental variables on offspring still cannot be fully and directly estimated. The stochastic changes of epigenetic biomarkers act as potential intermedium between environmental variables and related phenotypic variation. In addition, stochastic epigenetic changes can generate tissue- or cell-specific epigenetic variability over time without changes in DNA sequence, contributing to explain the phenotypic variation that cannot be explained by genetic mechanisms (Petronis, 2010). Therefore, recognition of the significance of transgenerational epigenetic inheritance for animal breeding purposes will also facilitate further exploration of currently identified epigenetic effects and their applications in livestock production (Feeney et al., 2014).
With the development of livestock epigenetic research, reliable epigenetic biomarkers related to productivity and health could be identified and used in livestock management (Franco, 2017; Lin et al., 2019). For example, epigenetic biomarkers in embryo biopsies, placenta, or newborn blood could be discovered and used to develop predictive biomarkers for future phenotypes of interest later in life. Moreover, epigenetic biomarkers in sperm could be used for the selection of sires and sperm quality. Possible monitoring of dynamic epigenetic biomarkers during individual development has potential for use to predict responses to environmental exposure and stressors before observable phenotypic changes. Epigenetic biomarkers could be used to improve the efficiency of different diets, disease diagnosis, and treatments and determine cost-saving avenues (time and money) for precision livestock management.
This review summarized recent epigenetic reports in livestock and discussed the potential application of epigenetic processes in livestock productivity and health management. The wealth of epigenetic modification data constantly being discovered in livestock has the potential to contribute to enhanced livestock productivity and health. A better understanding of epigenetic modifications, such as DNA methylation, is expected to compliment information on genome processes, including molecular, cellular, biological, and immune responses, and provide deeper insights on how they interact to define phenotypic outcome. Given the high dependence of humans on foods of animal origin and the need to protect the environment, information on epigenetic regulatory processes has the potential to support the development of strategies for increased productivity of livestock animals with minimal environmental impacts. With regard to livestock reproduction, development, growth, productivity, product quality, health, and the immune response, the role of epigenetics and the underlying mechanisms remains to be fully clarified. Knowledge of epigenetic impacts on livestock health can potentially support the development of strategies to lower disease incidence and increase disease resistance in livestock. It also can increase the suitability and efficiency of diagnostic measures, control approaches, such as vaccination, and treatments. To promote the successful application of epigenetics information in livestock management and improvement, more studies and tools are needed to examine the epigenetic effects and to develop strategies for implementation. However, the current comprehension and exploration of epigenetic mechanisms and their potentials in livestock health and production management is far from complete. More studies are therefore needed to get a better understanding of the epigenetic mechanisms underlying phenotypic variation in livestock production and health.
EI-A conceptualized the study and obtained funding. MW and EI-A made substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding for this study was provided by Agriculture and Agri-Food Canada.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
5hmC, 5-hydroxymethylcytosine; 5mC, 5-methylcytosine; ABCA1, subfamily A, member 1; ABCA7, subfamily A, member 7; ABCG1, ATP binding cassette subfamily G member 1; ACACA, acetyl-coenzyme A carboxylase alpha; ACC, acetyl-CoA carboxylase; ACOX2, acyl-CoA oxidase 2; ACSL1, acyl-CoA synthetase long chain family member 1; ACSS2, acyl-CoA synthetase short-chain family member 2; ADAMTS3, ADAM metallopeptidase with thrombospondin type 1 motif 3; Akt, serine/threonine protein kinase Akt; ATAC, Ada-Two-A-Containing complex; BAF, BRM/BRG1-associated factor complexes; BTK, Bruton tyrosine kinase; BPI, bactericidal/permeability-increasing protein; BVDV, bovine viral diarrhea virus; CACNA1S, calcium voltage-gated channel subunit alpha1 S; CACNG6, calcium voltage-gated channel auxiliary subunit gamma 6; CAPN10, calpain 10; LOC101896713, caspase-3; CASP8, caspase-8; CAV1, caveolin-1; CD4, CD4 molecule; CKS1B, CDC28 protein kinase regulatory subunit 1B; COL6A1, collagen type VI alpha 1 chain; CpG, cytosine-phosphate-guanosine; CRYL1, crystallin lambda 1; CSF1, colony stimulating factor 1; CSN1S1, alpha s1 casein; CTCF, CCCTC-binding factor; CXCL14, C-X -C motif chemokine ligand 14; DICER1, dicer 1, ribonuclease III; DLK1, delta like non-canonical Notch ligand 1; DMC, differentially methylated cytosines; DMG, differentially methylated gene; DMR, differentially methylated region; DNMTs, DNA methyltransferases; DNMT1, DNA methyltransferase 1; DNMT2, DNA methyltransferase 2; DNMT3A, DNA methyltransferase 3 A; DNMT3B, DNA methyltransferase 3 B; DNMT3C, DNA methyltransferase 3 C; DNMT3L, DNA methyltransferase 3 L; E. coli, Escherichia coli; E2F2, E2F transcription factor 2; EEF1D, eukaryotic translation elongation factor 1 delta; EFNB2, ephrin B2; EGFR, epidermal growth factor receptor; EHMT2, euchromatic histone lysine methyltransferase 2; ElF5, E74-like factor 5; ENPP3, ectonucleotide pyrophosphatase/phosphodiesterase 3; EPO, erythropoietin; ER α , estrogen receptor 1 (alpha); EWAS, epigenome-wide association studies; FADS6, fatty acid desaturase 6; FASN, fatty acid synthase; FAT-1, FAT atypical cadherin 1; FATP1, fatty acid transport protein 1; FDFT1, farnesyl-diphosphate farnesyltransferase 1; FTO, fat mass- and obesity-associated protein; GALK1, galactokinase 1; GLUT1, glucose transporter 1; GR, glucocorticoid receptor; GSTP1, glutathione S-transferase Pi 1; GSTT1L, glutathione S-transferase theta 1-like; GWAS, genomic-wide association studies; H3K27ac, the acetylation at the 27th lysine residue of the histone H3 protein; H3K26me3, the trimethylation at the 26th lysine residue of the histone H3 protein; H3K27me3, the trimethylation at the 27th lysine residue of the histone H3 protein; H3K36me3, the trimethylation at the 36th lysine residue of the histone H3 protein; H3K4me1, the monomethylation at the fourth lysine residue of the histone H3 protein; H3K4me3, the trimethylation at the fourth lysine residue of the histone H3 protein; H4K12ac, the acetylation at the 12th lysine residue of the histone H4 protein; HDAC2, histone deacetylase 2; HDAC3, histone deacetylase 3; HDAC6, histone deacetylase 6; HIF-1 α , hypoxia-inducible factor 1 subunit alpha; HIF-3 α , hypoxia-inducible factor 3 subunit alpha; HOTAIR, HOX transcript antisense RNA; HOXC8, homeobox C8; HP, haptoglobin; HSD17B2, hydroxysteroid 17-beta dehydrogenase 2; IDH2, isocitrate dehydrogenase (NADP(+)) 2; IGF1, insulin-like growth factor 1; IGF2, insulin-like growth factor 2; IGF2R, insulin-like growth factor 2 receptor; IGFBP4, insulin-like growth factor binding protein 4; IL-10, interleukin-10; IL-1 β , interleukin-1 beta; IL − 6, interleukin − 6; IL-6R, interleukin-6 receptor; IL − 8, interleukin − 8; IL-1R2, interleukin 1 receptor type 2; iNOS2, inducible nitric oxide synthase 2; INS, insulin; KAT2A, lysine acetyltransferase 2A; Kcnq1, potassium voltage-gated channel subfamily Q member 1; KCNQ3, potassium voltage-gated channel subfamily Q member 3; LBP, lipopolysaccharide binding protein; LPS, bacterial lipopolysaccharide; MAPRE1, microtubule associated protein RP/EB family member 1; MBD, methyl-binding domain; MBD1, methyl binding domain 1; MDBK, Madin–Darby bovine kidney; MeCP1, methyl-CpG binding protein 1; MeCP2, methyl-CpG binding protein 2; Met, methionine; MTTP, microsomal triglyceride transfer protein; MUSTN1, musculoskeletal, embryonic nuclear protein 1; MYB, myb proto-oncogene protein; NCKAP5, NCK-associated protein 5; ncRNA, non-coding RNA; NDV, Newcastle disease virus; NPY, neuropeptide Y; NR4A1, nuclear receptor subfamily 4 group A member 1; NRF1, nuclear respiratory factor 1; NTN1, netrin 1; NuRD, nucleosome remodeling and deacetylation complex; OAS1, 2 ′ –5 ′ -oligoadenylate synthetase 1; OAS2, 2 ′ –5 ′ -oligoadenylate synthetase 2; PACSNI1, protein kinase C and casein kinase substrate in neurons 1; PCK1, phosphoenolpyruvate carboxykinase 1; PEMT, phosphatidylethanolamine N-methyltransferase; PHF14, PHD Finger Protein 14; PITX1, paired-like homeodomain transcription factor 1; POMC, proopiomelanocortin; PPARG2, peroxisome proliferator-activated receptor gamma; PPAR α , peroxisome proliferator-activated receptor alpha; PPAR δ , peroxisome proliferator-activated receptor delta; PPAR γ , peroxisome proliferator-activated receptor gamma; PRRSV, porcine reproductive and respiratory syndrome virus; PTX3, pentraxin 3RRBS: reduced representation bisulfite sequencing; RSC, remodel the structure of chromatin; RXR α , retinoid X receptor alpha; RYR1, ryanodine receptor 1; S6K1, ribosomal protein S6 kinase 1; S. aureus, Staphylococcus aureus; SAA3, serum amyloid A 3; SALL4, spalt-like transcription factor 4; SCD1, stearoyl-CoA desaturase 1; SDF4, stromal cell derived factor 4; SEPT9, Septin 9; SIN3A, SIN3 transcription regulator family member A; SIRT2, sirtuin 2; SIRT4, sirtuin 4; SIX1, SIX homeobox 1; SNP, single nucleotide polymorphisms; SNX25, sorting nexin 25; SREBF1, sterol regulatory element binding transcription factor 1; SREBP1, sterol regulatory element binding protein 1; SRXN1, sulfiredoxin 1; SS18, SS18 subunit of BAF chromatin remodeling complex; SWI/SNF, switch/sucrose non-fermentable complexes; TCF7L2, type 2 diabetes gene; TET, ten-eleven translocation methylcytosine dioxygenases; TGFB3, transforming growth factor beta 3; TLR-4, toll-like receptor 4; TMEM8C, transmembrane protein 8c; TNF, tumor necrosis factor α; TNF, tumor necrosis factor; TNFSF8, TNF superfamily member 8; TSS, transcriptional start sites; UCP3, uncoupling protein 3; VTGII, vitellogenin 2; WGBS, whole-genome bisulfite sequencing; WHO, United Nations World Health Organization.
Abbring, S., Wolf, J., Ayechu-Muruzabal, V., Diks, M. A., Alashkar Alhamwe, B., Alhamdan, F., et al. (2019). Raw cow’s milk reduces allergic symptoms in a murine model for food allergy—a potential role for epigenetic modifications. Nutrients 11:1721. doi: 10.3390/nu11081721
Abobaker, H., Hu, Y., Omer, N. A., Hou, Z., Idriss, A. A., and Zhao, R. (2019). Maternal betaine suppresses adrenal expression of cholesterol trafficking genes and decreases plasma corticosterone concentration in offspring pullets. J. Anim. Sci. Biotechnol. 10:87.
Acosta, D., Denicol, A., Tribulo, P., Rivelli, M., Skenandore, C., Zhou, Z., et al. (2016). Effects of rumen-protected methionine and choline supplementation on the preimplantation embryo in Holstein cows. Theriogenology 85, 1669–1679. doi: 10.1016/j.theriogenology.2016.01.024
Adamczyk, S. (2019). Role of Chromatin Structure in Regulating Transcription. Doctoral dissertation, Oakland: Oakland University.
Adès, L., Sekeres, M. A., Wolfromm, A., Teichman, M. L., Tiu, R. V., Itzykson, R., et al. (2013). Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk. Res. 37, 609–613. doi: 10.1016/j.leukres.2013.01.004
Ahmad, S. F., Shanaz, S., Kumar, A., Dar, A. H., Mohmad, A., and Bhushan, B. (2019). Crosstalk of epigenetics with biological rhythmicity in animal kingdom. Biol. Rhythm Res. 1–9. doi: 10.1080/09291016.2019.1607218
Akbarinejad, V., Gharagozlou, F., and Vojgani, M. (2017). Temporal effect of maternal heat stress during gestation on the fertility and anti-Müllerian hormone concentration of offspring in bovine. Theriogenology 99, 69–78. doi: 10.1016/j.theriogenology.2017.05.018
Alfert, A., Moreno, N., and Kerl, K. (2019). The BAF complex in development and disease. Epigenetics Chromatin 12:19.
Alhamwe, B. A., Khalaila, R., Wolf, J., von Bülow, V., Harb, H., Alhamdan, F., et al. (2018). Histone modifications and their role in epigenetics of atopy and allergic diseases. Allergy Asthma Clin. Immunol. 14:39.
Alharthi, A., Coleman, D., Liang, Y., Batistel, F., Elolimy, A., Yambao, R., et al. (2019). Hepatic 1-carbon metabolism enzyme activity, intermediate metabolites, and growth in neonatal Holstein dairy calves are altered by maternal supply of methionine during late pregnancy. J. Dairy Sci. 102, 10291–10303. doi: 10.3168/jds.2019-16562
Andraszek, K., Gryzińska, M., Danielewicz, A., Batkowska, J., and Smalec, E. (2016). Age-dependent stability of nucleoli and global DNA methylation level in spermatocytes of the domestic horse (Equus caballus). Can. J. Anim. Sci. 96, 215–220. doi: 10.1139/cjas-2015-0076
Bach, A., Aris, A., and Guasch, I. (2017). Consequences of supplying methyl donors during pregnancy on the methylome of the offspring from lactating and non-lactating dairy cattle. PLoS One 12:e0189581. doi: 10.1371/journal.pone.0189581
Bai, W. L., Wang, J. J., Yin, R. H., Dang, Y. L., Wang, Z. Y., Zhu, Y. B., et al. (2017). Molecular characterization of HOXC8 gene and methylation status analysis of its exon 1 associated with the length of cashmere fiber in liaoning cashmere goat. Genetica 145, 115–126. doi: 10.1007/s10709-017-9950-5
Baik, M., Vu, T., Piao, M., and Kang, H. (2014). Association of DNA methylation levels with tissue-specific expression of adipogenic and lipogenic genes in longissimus dorsi muscle of Korean cattle. Asian-Australas. J. Anim. Sci. 27, 1493–1498. doi: 10.5713/ajas.2014.14283
Banovich, N. E., Lan, X., McVicker, G., Van de Geijn, B., Degner, J. F., Blischak, J. D., et al. (2014). Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 10:e1004663. doi: 10.1371/journal.pgen.1004663
Banta, J. A., and Richards, C. L. (2018). Quantitative epigenetics and evolution. Heredity 121, 210–224. doi: 10.1038/s41437-018-0114-x
Barau, J., Teissandier, A., Zamudio, N., Roy, S., Nalesso, V., Hérault, Y., et al. (2016). The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science 354, 909–912. doi: 10.1126/science.aah5143
Benmoussa, A., Laugier, J., Beauparlant, C. J., Lambert, M., Droit, A., and Provost, P. (2020). Complexity of the microRNA transcriptome of cow milk and milk-derived extracellular vesicles isolated via differential ultracentrifugation. J. Dairy Sci. 103, 16–29. doi: 10.3168/jds.2019-16880
Bernstein, B. E., Meissner, A., and Lander, E. S. (2007). The mammalian epigenome. Cell 128, 669–681. doi: 10.1016/j.cell.2007.01.033
Bhattacharjee, D., Shenoy, S., and Bairy, K. L. (2016). DNA methylation and chromatin remodeling: the blueprint of cancer epigenetics. Scientifica 2016:e6072357.
Bian, Y., Lei, Y., Wang, C., Wang, J., Wang, L., Liu, L., et al. (2015). Epigenetic regulation of miR-29s affects the lactation activity of dairy cow mammary epithelial cells. J. Cell. Physiol. 230, 2152–2163. doi: 10.1002/jcp.24944
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. doi: 10.1101/gad.947102
Birney, E., Smith, G. D., and Greally, J. M. (2016). Epigenome-wide association studies and the interpretation of disease-omics. PLoS Genet. 12:e1006105. doi: 10.1371/journal.pgen.1006105
Bishop, T. F., and Van Eenennaam, A. L. (2020). Genome editing approaches to augment livestock breeding programs. J. Exp. Biol. 223(Suppl. 1), jeb207159. doi: 10.1242/jeb.207159
Block, T., and El-Osta, A. (2017). Epigenetic programming, early life nutrition and the risk of metabolic disease. Atherosclerosis 266, 31–40. doi: 10.1016/j.atherosclerosis.2017.09.003
Bobeck, E. A. (2020). Functional nutrition in livestock and companion animals to modulate the immune response. J. Anim. Sci. 98:skaa035.
Bochtler, M., Kolano, A., and Xu, G. L. (2017). DNA demethylation pathways: additional players and regulators. Bioessays 39, 1–13.
Boddicker, R., Koltes, J., Fritz-Waters, E., Koesterke, L., Weeks, N., Yin, T., et al. (2016). Genome-wide methylation profile following prenatal and postnatal dietary omega-3 fatty acid supplementation in pigs. Anim. Genet. 47, 658–671. doi: 10.1111/age.12468
Bonduriansky, R., and Day, T. (2009). Nongenetic inheritance and its evolutionary implications. Annu. Rev. Ecol. Evol. Syst. 40, 103–125. doi: 10.1146/annurev.ecolsys.39.110707.173441
Bornelöv, S., Reynolds, N., Xenophontos, M., Gharbi, S., Johnstone, E., Floyd, R., et al. (2018). The nucleosome remodeling and deacetylation complex modulates chromatin structure at sites of active transcription to fine-tune gene expression. Mol. Cell. 71, 56–72. doi: 10.1016/j.molcel.2018.06.003
Cai, D., Yuan, M., Liu, H., Han, Z., Pan, S., Yang, Y., et al. (2017). Epigenetic and SP1-mediated regulation is involved in the repression of galactokinase 1 gene in the liver of neonatal piglets born to betaine-supplemented sows. Eur. J. Nutr. 56, 1899–1909. doi: 10.1007/s00394-016-1232-y
Cai, L., Hua, C., Geng, Y., Chen, Q., Niu, L., Tao, S., et al. (2019). Chronic dexamethasone exposure activates the TLR4-Mediated inflammation pathway and induces epithelial apoptosis in the goat colon. Biochem. Biophys. Res. Commun. 518, 7–13. doi: 10.1016/j.bbrc.2019.07.071
Cao, P., Li, H., Zuo, Y., and Nashun, B. (2020). Characterization of DNA methylation patterns and mining of epigenetic markers during genomic reprogramming in SCNT embryos. Front. Cell Dev. Biol. 8:570107. doi: 10.3389/fcell.2020.570107
Cao, Z., Zhang, D., Wang, Y., Tong, X., Avalos, L. F. C., Khan, I. M., et al. (2020). Identification and functional annotation of m6A methylation modification in granulosa cells during antral follicle development in pigs. Anim. Reprod. Sci. 29:106510. doi: 10.1016/j.anireprosci.2020.106510
Cao, Y., Jin, H.-G., Ma, H.-H., and Zhao, Z.-H. (2017). Comparative analysis on genome-wide DNA methylation in longissimus dorsi muscle between small tailed han and dorper× small tailed han crossbred sheep. Asian-Australas. J. Anim. Sci. 30, 1529–1539. doi: 10.5713/ajas.17.0154
Capper, J. L., and Bauman, D. E. (2013). The role of productivity in improving the environmental sustainability of ruminant production systems. Annu. Rev. Anim. Biosci. 1, 469–489. doi: 10.1146/annurev-animal-031412-103727
Capra, E., Lazzari, B., Turri, F., Cremonesi, P., Portela, A. M. R., Ajmone-Marsan, P., et al. (2019). Epigenetic analysis of high and low motile sperm populations reveals methylation variation in satellite regions within the pericentromeric position and in genes functionally related to sperm DNA organization and maintenance in Bos taurus. BMC Genomics 20:940. doi: 10.1186/s12864-019-6317-6
Cardoso-Júnior, C. A., Fujimura, P. T., Santos-Júnior, C. D., Borges, N. A., Ueira-Vieira, C., Hartfelder, K., et al. (2017). Epigenetic modifications and their relation to caste and sex determination and adult division of labor in the stingless bee Melipona scutellaris. Genet. Mol. Biol. 40, 61–68. doi: 10.1590/1678-4685-gmb-2016-0242
Carnero-Montoro, E., and Alarcón-Riquelme, M. E. (2018). Epigenome-wide association studies for systemic autoimmune diseases: the road behind and the road ahead. Clin. Immunol. 196, 21–33. doi: 10.1016/j.clim.2018.03.014
Carrozza, M. J., Li, B., Florens, L., Suganuma, T., Swanson, S. K., Lee, K. K., et al. (2005). Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592. doi: 10.1016/j.cell.2005.10.023
Chang, G., Liu, X., Ma, N., Yan, J., Dai, H., Roy, A. C., et al. (2018). Dietary addition of sodium butyrate contributes to attenuated feeding-induced hepatocyte apoptosis in dairy goats. J. Agric. Food Chem. 66, 9995–10002. doi: 10.1021/acs.jafc.8b03526
Chang, G., Zhang, K., Xu, T., Jin, D., Guo, J., Zhuang, S., et al. (2015). Epigenetic mechanisms contribute to the expression of immune related genes in the livers of dairy cows fed a high concentrate diet. PLoS One 10:e0123942. doi: 10.1371/journal.pone.0123942
Chanthavixay, G., Kern, C., Wang, Y., Saelao, P., Lamont, S. J., Gallardo, R. A., et al. (2020a). Integrated transcriptome and histone modification analysis reveals NDV infection under heat stress affects bursa development and proliferation in susceptible chicken line. Front. Genet. 11:567812. doi: 10.3389/fgene.2020.567812
Chanthavixay, G., Kern, C., Wang, Y., Saelao, P., Lamont, S. J., Gallardo, R. A., et al. (2020b). Integrated transcriptome and histone modification analysis reveals NDV infection under heat stress affects bursa development and proliferation in susceptible chicken line. Front. Genet. 11:1176.
Chavatte-Palmer, P., Velazquez, M., Jammes, H., and Duranthon, V. (2018). Epigenetics, developmental programming and nutrition in herbivores. Animal 12, s363–s371.
Chen, J., Wu, Y., Sun, Y., Dong, X., Wang, Z., Zhang, Z., et al. (2019). Bacterial lipopolysaccharide induced alterations of genome-wide DNA methylation and promoter methylation of lactation-related genes in bovine mammary epithelial cells. Toxins 11:298. doi: 10.3390/toxins11050298
Chen, Z., Chu, S., Xu, X., Jiang, J., Wang, W., Shen, H., et al. (2019). Analysis of longissimus muscle quality characteristics and associations with DNA methylation status in cattle. Genes Genomics 41, 1147–1163. doi: 10.1007/s13258-019-00844-4
Choi, J., Lyons, D. B., Kim, M. Y., Moore, J. D., and Zilberman, D. (2020). DNA Methylation and Histone H1 Jointly Repress Transposable Elements and Aberrant Intragenic Transcripts. Mol. Cell. 77, 310–323. doi: 10.1016/j.molcel.2019.10.011
Ciccone, N. A., Smith, L. P., Mwangi, W., Boyd, A., Broadbent, A. J., Smith, A. L., et al. (2017). Early pathogenesis during infectious bursal disease in susceptible chickens is associated with changes in B cell genomic methylation and loss of genome integrity. Dev. Comp. Immunol. 73, 169–174. doi: 10.1016/j.dci.2017.03.014
Clapier, C. R., Iwasa, J., Cairns, B. R., and Peterson, C. L. (2017). Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 18, 407–422. doi: 10.1038/nrm.2017.26
Couldrey, C., Brauning, R., Bracegirdle, J., Maclean, P., Henderson, H. V., and McEwan, J. C. (2014). Genome-wide DNA methylation patterns and transcription analysis in sheep muscle. PLoS One 9:e101853. doi: 10.1371/journal.pone.0101853
Dai, B., Zhang, M., Yuan, J.-L., Ren, L.-Q., Han, X.-Y., and Liu, D.-J. (2019). Integrative analysis of methylation and transcriptional profiles to reveal the genetic stability of cashmere traits in the Tβ4 overexpression of cashmere goats. Animals 9:1002. doi: 10.3390/ani9121002
Danchin, É, Charmantier, A., Champagne, F. A., Mesoudi, A., Pujol, B., and Blanchet, S. (2011). Beyond DNA: integrating inclusive inheritance into an extended theory of evolution. Nat. Rev. Genet. 12, 475–486. doi: 10.1038/nrg3028
Das, L., Parbin, S., Pradhan, N., Kausar, C., and Patra, S. K. (2017). Epigenetics of reproductive infertility. Front. Biosci. 9:509–535. doi: 10.2741/s497
David, S.-A., Vitorino Carvalho, A., Gimonnet, C., Brionne, A., Hennequet-Antier, C., Piégu, B., et al. (2019). Thermal manipulation during embryogenesis impacts H3K4me3 and H3K27me3 histone marks in chicken hypothalamus. Front. Genet. 10:1207. doi: 10.3389/fgene.2019.01207
Daxinger, L., and Whitelaw, E. (2010). Transgenerational epigenetic inheritance: more questions than answers. Genome Res. 20, 1623–1628. doi: 10.1101/gr.106138.110
de Barros, F. R. O., and Paula-Lopes, F. F. (2018). Cellular and epigenetic changes induced by heat stress in bovine preimplantation embryos. Mol. Reprod. Dev. 85, 810–820. doi: 10.1002/mrd.23040
Dean, W., Lucifero, D., and Santos, F. (2005). DNA methylation in mammalian development and disease. Birth Defects Res. 75, 98–111.
Deaton, A. M., and Bird, A. (2011). CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022. doi: 10.1101/gad.2037511
Dechow, C. D., and Liu, W.-S. (2018). DNA methylation patterns in peripheral blood mononuclear cells from holstein cattle with variable milk yield. BMC Genomics 19:744. doi: 10.1186/s12864-018-5124-9
Del Corvo, M., Bongiorni, S., Stefanon, B., Sgorlon, S., Valentini, A., Ajmone Marsan, P., et al. (2020). Genome-wide DNA methylation and gene expression profiles in cows subjected to different stress level as assessed by cortisol in milk. Genes 11:850. doi: 10.3390/genes11080850
Desmet, K. J., Van Hoeck, V., Gagné, D., Fournier, E., Thakur, A., O’doherty, A., et al. (2016). Exposure of bovine oocytes and embryos to elevated non-esterified fatty acid concentrations: integration of epigenetic and transcriptomic signatures in resultant blastocysts. BMC Genomics 17:1004. doi: 10.1186/s12864-016-3366-y
Di Wang, Y. W., Shi, L., Khan, M. Z., Fan, L., Wang, Y., and Yu, Y. (2020). Genome-wide DNA methylation pattern in a mouse model reveals two novel genes associated with Staphylococcus aureus mastitis. Asian-Australas. J. Anim. Sci. 33, 203–211. doi: 10.5713/ajas.18.0858
Diaz, F., Gutierrez, E., Foster, B., Hardin, P., and Bondioli, K. (2019). 114 Effect of in vivo heat stress on DNA methylation and DNA hydroxymethylation of bovine oocytes. Reprod. Fertil. Dev. 31:183. doi: 10.1071/rdv31n1ab114
Do, D. N., Dudemaine, P.-L., Fomenky, B., and Ibeagha-Awemu, E. M. (2017a). Transcriptome Analysis of Non-Coding RNAs in Livestock Species: Elucidating the Ambiguity. Applications of RNA-Seq and Omics Strategies: From Microorganisms to Human Health. London: IntechOpen, 103.
Do, D. N., Li, R., Dudemaine, P.-L., and Ibeagha-Awemu, E. M. (2017b). MicroRNA roles in signalling during lactation: an insight from differential expression, time course and pathway analyses of deep sequence data. Sci. Rep. 7:44605.
Do, D. N., and Ibeagha-Awemu, E. M. (2017). Non-Coding RNA Roles in Ruminant Mammary Gland Development and Lactation. London: IntechOpen, 55–81.
Doherty, R., Farrelly, C. O., and Meade, K. G. (2014). Comparative epigenetics: relevance to the regulation of production and health traits in cattle. Anim. Genet. 45, 3–14. doi: 10.1111/age.12140
Doherty, R., Whiston, R., Cormican, P., Finlay, E. K., Couldrey, C., Brady, C., et al. (2016). The CD4+ T cell methylome contributes to a distinct CD4+ T cell transcriptional signature in Mycobacterium bovis-infected cattle. Sci. Rep. 6:31014.
Dong, G., Qiu, M., Ao, C., Zhou, J., Wang, X., Zhang, Z., et al. (2014). Feeding a high-concentrate corn straw diet induced epigenetic alterations in the mammary tissue of dairy cows. PLoS One 9:e107659. doi: 10.1371/journal.pone.0107659
Dose, A., Liokatis, S., Theillet, F.-X., Selenko, P., and Schwarzer, D. (2011). NMR profiling of histone deacetylase and acetyl-transferase activities in real time. ACS Chem. Biol. 6, 419–424. doi: 10.1021/cb1003866
Duan, J., Jiang, Z., Alqahtani, F., Mandoiu, I., Hong, D., Zheng, X., et al. (2019). Methylome dynamics of bovine gametes and in vivo early embryos. Front. Genet. 10:512. doi: 10.3389/fgene.2019.00512
Edris, A., den Dekker, H. T., Melén, E., and Lahousse, L. (2019). Epigenome-wide association studies in asthma: A systematic review. Clin. Exp. Allergy 49, 953–968. doi: 10.1111/cea.13403
Edwards, J. R., Yarychkivska, O., Boulard, M., and Bestor, T. H. (2017). DNA methylation and DNA methyltransferases. Epigenetics Chromatin 10:23.
Elolimy, A., Vailati-Riboni, M., Liang, Y., and Loor, J. J. (2019). Cellular mechanisms and epigenetic changes: role of nutrition in livestock. Vet. Clin. Food Anim. Pract. 35, 249–263.
Emam, M., Livernois, A., Paibomesai, M., Atalla, H., and Mallard, B. (2019). Genetic and epigenetic regulation of immune response and resistance to infectious diseases in domestic ruminants. Vet. Clin. Food Anim. Pract. 35, 405–429. doi: 10.1016/j.cvfa.2019.07.002
Fan, Y., Liang, Y., Deng, K., Zhang, Z., Zhang, G., Zhang, Y., et al. (2020). Analysis of DNA methylation profiles during sheep skeletal muscle development using whole-genome bisulfite sequencing. BMC Genomics 21:327. doi: 10.1186/s12864-020-6751-5
Fang, L., Liu, S., Liu, M., Kang, X., Lin, S., Li, B., et al. (2019a). Functional annotation of the cattle genome through systematic discovery and characterization of chromatin states and butyrate-induced variations. BMC Biol. 17:68. doi: 10.1186/s12915-019-0687-8
Fang, L., Zhou, Y., Liu, S., Jiang, J., Bickhart, D. M., Null, D. J., et al. (2019b). Integrating signals from sperm methylome analysis and genome-wide association study for a better understanding of male fertility in cattle. Epigenomes 3:10. doi: 10.3390/epigenomes3020010
Fang, X., Zhao, Z., Yu, H., Li, G., Jiang, P., Yang, Y., et al. (2017). Comparative genome-wide methylation analysis of longissimus dorsi muscles between Japanese black (Wagyu) and Chinese Red Steppes cattle. PLoS One 12:e0182492. doi: 10.1371/journal.pone.0182492
Feeney, A., Nilsson, E., and Skinner, M. K. (2014). Epigenetics and transgenerational inheritance in domesticated farm animals. J. Anim. Sci. Biotechnol. 5:48. doi: 10.1186/2049-1891-5-48
Feltus, F. A., Lee, E. K., Costello, J. F., Plass, C., and Vertino, P. M. (2006). DNA motifs associated with aberrant CpG island methylation. Genomics 87, 572–579. doi: 10.1016/j.ygeno.2005.12.016
Feng, Q., and Zhang, Y. (2003). “The NuRD complex: linking histone modification to nucleosome remodeling,” in Protein Complexes that Modify Chromatin. Current Topics in Microbiology and Immunology, Vol. 274, ed. J. L. Workman (Berlin: Springer), 269–290. doi: 10.1007/978-3-642-55747-7_10
Feng, W., Zhou, L., Wang, H., Hu, Z., Wang, X., Fu, J., et al. (2019). Functional analysis of DNA methylation of the PACSIN1 promoter in pig peripheral blood mononuclear cells. J. Cell. Biochem. 120, 10118–10127. doi: 10.1002/jcb.28295
Foissac, S., Djebali, S., Munyard, K., Vialaneix, N., Rau, A., Muret, K., et al. (2019). Multi-species annotation of transcriptome and chromatin structure in domesticated animals. BMC Biol. 17:108. doi: 10.1186/s12915-019-0726-5
Franco, M. M. (2017). Epigenetics in genetic improvement and animal reproduction. Arch. Latinoam. Producción Anim. 25, 75–80.
Fu, Q., Shi, H., and Chen, C. (2017). Roles of bta-miR-29b promoter regions DNA methylation in regulating miR-29b expression and bovine viral diarrhea virus NADL replication in MDBK cells. Arch. Virol. 162, 401–408. doi: 10.1007/s00705-016-3107-1
Gao, G., Wang, H., Zhao, X., Li, Q., Li, J., Li, Q., et al. (2015). Feeding conditions and breed affect the level of DNA methylation of the mitochondrial uncoupling protein 3 gene in chicken breast muscle. J. Anim. Sci. 93, 1522–1534. doi: 10.2527/jas.2014-8431
Gao, G., Wang, H., Zhao, X., Li, Q., Wang, C., Li, J., et al. (2017). Effect of feeding conditions on the methylation status of Fatp1 gene in chicken breast muscle. Braz. J. Poult. Sci. 19, 55–58. doi: 10.1590/1806-9061-2016-0367
García-Giménez, J. L., Ushijima, T., and Tollefsbol, T. O. (2016). “Epigenetic biomarkers: new findings, perspectives, and future directions in diagnostics,” in Epigenetic Biomarkers and Diagnostics, ed. J. L. García-Giménez (Amsterdam: Elsevier), 1–18. doi: 10.1016/b978-0-12-801899-6.00001-2
Gelfman, S., Cohen, N., Yearim, A., and Ast, G. (2013). DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon–intron structure. Genome Res. 23, 789–799. doi: 10.1101/gr.143503.112
Georges, M., Charlier, C., and Hayes, B. (2019). Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 20, 135–156. doi: 10.1038/s41576-018-0082-2
Glendining, K. A., and Jasoni, C. L. (2019). Maternal high fat diet-induced obesity modifies histone binding and expression of oxtr in offspring hippocampus in a sex-specific manner. Int. J. Mol. Sci. 20:329. doi: 10.3390/ijms20020329
Goddard, M. E., and Whitelaw, E. (2014). The use of epigenetic phenomena for the improvement of sheep and cattle. Front. Genet. 5:247. doi: 10.3389/fgene.2014.00247
González-Recio, O., Toro, M. A., and Bach, A. (2015). Past, present and future of epigenetics applied to livestock breeding. Front. Genet. 6:305. doi: 10.3389/fgene.2015.00305
Gotoh, T. (2015). Potential of the application of epigenetics in animal production. Anim. Prod. Sci. 55, 145–158. doi: 10.1071/an14467
Greally, J. M. (2018). A user’s guide to the ambiguous word’epigenetics’. Nat. Rev. Mol. Cell Biol. 19, 207–208. doi: 10.1038/nrm.2017.135
Greenberg, M. V., and Bourc’his, D. (2019). The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20, 590–607. doi: 10.1038/s41580-019-0159-6
Gross, N., Peñagaricano, F., and Khatib, H. (2020). Integration of whole-genome DNA methylation data with RNA sequencing data to identify markers for bull fertility. Anim. Genet. 51, 502–510. doi: 10.1111/age.12941
Guo, M., Chen, Y., Chen, Q., Guo, X., Yuan, Z., Kang, L., et al. (2020). Epigenetic changes associated with increased estrogen receptor alpha mRNA transcript abundance during reproductive maturation in chicken ovaries. Anim. Reprod. Sci. 214:106287. doi: 10.1016/j.anireprosci.2020.106287
Guo, T., Luo, F., and Lin, Q. (2020). You are affected by what your parents eat: Diet, epigenetics, transgeneration and intergeneration. Trends Food Sci. Technol. 100, 248–261. doi: 10.1016/j.tifs.2020.04.021
Guo, X., Liu, X., Xu, X., Wu, M., Zhang, X., Li, Q., et al. (2012). The expression levels of DNMT3a/3b and their relationship with meat quality in beef cattle. Mol. Biol. Rep. 39, 5473–5479. doi: 10.1007/s11033-011-1349-2
Gurgul, A., Miksza-Cybulska, A., Szmatoła, T., Jasielczuk, I., Piestrzyńska-Kajtoch, A., Fornal, A., et al. (2019). Genotyping-by-sequencing performance in selected livestock species. Genomics 111, 186–195. doi: 10.1016/j.ygeno.2018.02.002
Hall, T. J., Vernimmen, D., Browne, J. A., Mullen, M. P., Gordon, S. V., MacHugh, D. E., et al. (2019). Alveolar macrophage chromatin is modified to orchestrate host response to Mycobacterium bovis infection. Front. Genet. 10:e01386. doi: 10.3389/fgene.2019.01386
Halstead, M. M., Kern, C., Saelao, P., Wang, Y., Chanthavixay, G., Medrano, J. F., et al. (2020). A comparative analysis of chromatin accessibility in cattle, pig, and mouse tissues. BMC Genomics 21:698. doi: 10.1186/s12864-020-07078-9
Han, W., Xue, Q., Li, G., Yin, J., Zhang, H., Zhu, Y., et al. (2020). Genome-wide analysis of the role of DNA methylation in inbreeding depression of reproduction in Langshan chicken. Genomics 112, 2677–2687. doi: 10.1016/j.ygeno.2020.02.007
Hanna, C., and Kelsey, G. (2014). The specification of imprints in mammals. Heredity 113, 176–183. doi: 10.1038/hdy.2014.54
Hao, Y., Cui, Y., and Gu, X. (2016). Genome-wide DNA methylation profiles changes associated with constant heat stress in pigs as measured by bisulfite sequencing. Sci. Rep. 6:27507.
Harris, K. D., Lloyd, J. P., Domb, K., Zilberman, D., and Zemach, A. (2019). DNA methylation is maintained with high fidelity in the honey bee germline and exhibits global non-functional fluctuations during somatic development. Epigenetics Chromatin 12:62.
He, S., Wang, H., Liu, R., He, M., Che, T., Jin, L., et al. (2017). mRNA N6-methyladenosine methylation of postnatal liver development in pig. PLoS One 12:e0173421. doi: 10.1371/journal.pone.0173421
He, Y., Song, M., Zhang, Y., Li, X., Song, J., Zhang, Y., et al. (2016). Whole-genome regulation analysis of histone H3 lysin 27 trimethylation in subclinical mastitis cows infected by Staphylococcus aureus. BMC Genomics 17:565. doi: 10.1186/s12864-016-2947-0
He, Y., Zuo, Q., Edwards, J., Zhao, K., Lei, J., Cai, W., et al. (2018). DNA methylation and regulatory elements during chicken germline stem cell differentiation. Stem Cell Rep. 10, 1793–1806. doi: 10.1016/j.stemcr.2018.03.018
He, Y.-F., Li, B.-Z., Li, Z., Liu, P., Wang, Y., Tang, Q., et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. doi: 10.1126/science.1210944
Heard, E., and Martienssen, R. A. (2014). Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157, 95–109. doi: 10.1016/j.cell.2014.02.045
Heijmans, B. T., Tobi, E. W., Stein, A. D., Putter, H., Blauw, G. J., Susser, E. S., et al. (2008). Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. U.S.A. 105, 17046–17049. doi: 10.1073/pnas.0806560105
Hernaiz, A., Sentre, S., Bolea, R., López-Pérez, O., Sanz, A., Zaragoza, P., et al. (2019). “Epigenetic changes in the central nervous system of sheep naturally infected with scrapie,” in Proceedings of the 18th Jornadas Sobre Producción Animal, Zaragoza, España, 7 y 8 de mayo de 2019, Zaragoza, 507–509.
Herrera-Uribe, J., Liu, H., Byrne, K. A., Bond, Z. F., Loving, C. L., and Tuggle, C. K. (2020). Changes in H3K27ac at gene regulatory regions in porcine alveolar macrophages following LPS or PolyIC exposure. Front. Genet. 11:817. doi: 10.3389/fgene.2020.00817
Hitchins, M. P., and Ward, R. L. (2009). Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J. Med. Genet. 46, 793–802. doi: 10.1136/jmg.2009.068122
Hong, J., Wang, X., Mei, C., Wang, H., and Zan, L. (2019). DNA methylation and transcription factors competitively regulate SIRT4 promoter activity in bovine adipocytes: roles of NRF1 and CMYB. DNA Cell Biol. 38, 63–75. doi: 10.1089/dna.2018.4454
Hong, J.-Y., Mei, C.-G., Li, S.-J., Wang, H.-B., Zhao, C.-P., and Zan, L.-S. (2018). Coordinate regulation by transcription factors and DNA methylation in the core promoter region of SIRT6 in bovine adipocytes. Arch. Biochem. Biophys. 659, 1–12. doi: 10.1016/j.abb.2018.09.018
Horsthemke, B. (2006). Epimutations in human disease. Curr. Top. Microbiol. Immunol. 310, 45–59. doi: 10.1007/3-540-31181-5_4
Hota, S. K., and Bruneau, B. G. (2016). ATP-dependent chromatin remodeling during mammalian development. Development 143, 2882–2897. doi: 10.1242/dev.128892
Hota, S. K., Johnson, J. R., Verschueren, E., Thomas, R., Blotnick, A. M., Zhu, Y., et al. (2019). Dynamic BAF chromatin remodeling complex subunit inclusion promotes temporally distinct gene expression programs in cardiogenesis. Development 146:dev174086. doi: 10.1242/dev.174086
Hu, Y., Sun, Q., Liu, J., Jia, Y., Cai, D., Idriss, A. A., et al. (2017a). In ovo injection of betaine alleviates corticosterone-induced fatty liver in chickens through epigenetic modifications. Sci. Rep. 7:40251.
Hu, Y., Sun, Q., Zong, Y., Liu, J., Idriss, A. A., Omer, N. A., et al. (2017b). Prenatal betaine exposure alleviates corticosterone-induced inhibition of CYP27A1 expression in the liver of juvenile chickens associated with its promoter DNA methylation. Gen. Comp. Endocrinol. 246, 241–248. doi: 10.1016/j.ygcen.2016.12.014
Hwang, J. H., An, S. M., Kwon, S., Park, D. H., Kim, T. W., Kang, D. G., et al. (2017). DNA methylation patterns and gene expression associated with litter size in Berkshire pig placenta. PLoS One 12:e0184539. doi: 10.1371/journal.pone.0184539
Ibeagha-Awemu, E. M., Bhattarai, S., Dudemaine, P.-L., Wang, M., Mckay, S. D., Zhao, X., et al. (2020a). 63 DNA methylome wide profile associates differentially methylated loci and regions with cow’s ileal lymph node response to Mycobacterium avium subsp. paratuberculosis. J. Anim. Sci. 98, 39–40. doi: 10.1093/jas/skaa278.071
Ibeagha-Awemu, E. M., Bhattarai, S., Dudemaine, P.-L., Wang, M., Mckay, S. D., Zhao, X., et al. (2020b). PSVIII-15 Genome wide DNA methylation analysis reveals role of DNA methylation in cow’s ileal response to Mycobacterium avium subsp. paratuberculosis. J. Anim. Sci. 98, 260–261. doi: 10.1093/jas/skaa278.471
Ibeagha-Awemu, E. M., and Khatib, H. (2017). “Epigenetics of livestock breeding,” in Handbook of Epigenetics, 2nd Edn, Chap. 29. ed. T. O. Tollefsbol (Cambridge: Academic Press), 441–463.
Ibeagha-Awemu, E. M., and Zhao, X. (2015). Epigenetic marks: regulators of livestock phenotypes and conceivable sources of missing variation in livestock improvement programs. Front. Genet. 6:302. doi: 10.3389/fgene.2015.00302
Ideraabdullah, F. Y., and Zeisel, S. H. (2018). Dietary modulation of the epigenome. Physiol. Rev. 98, 667–695. doi: 10.1152/physrev.00010.2017
Idriss, A. A., Hu, Y., Hou, Z., Hu, Y., Sun, Q., Omer, N. A., et al. (2018). Dietary betaine supplementation in hens modulates hypothalamic expression of cholesterol metabolic genes in F1 cockerels through modification of DNA methylation. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 217, 14–20. doi: 10.1016/j.cbpb.2017.12.001
Imgenberg-Kreuz, J., Almlöf, J. C., Leonard, D., Alexsson, A., Nordmark, G., Eloranta, M.-L., et al. (2018). DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 77, 736–743. doi: 10.1136/annrheumdis-2017-212379
Ispada, J., De Lima, C. B., Sirard, M.-A., Fontes, P. K., Nogueira, M. F. G., Annes, K., et al. (2018). Genome-wide screening of DNA methylation in bovine blastocysts with different kinetics of development. Epigenetics Chromatin 11:1.
Ivanova, E., Canovas, S., Garcia-Martínez, S., Romar, R., Lopes, J. S., Rizos, D., et al. (2020). DNA methylation changes during preimplantation development reveal inter-species differences and reprogramming events at imprinted genes. Clin. Epigenetics 12:64.
Jahan, S., Beacon, T. H., Xu, W., and Davie, J. R. (2019). Atypical chromatin structure of immune-related genes expressed in chicken erythrocytes. Biochem. Cell Biol. 98, 171–177. doi: 10.1139/bcb-2019-0107
Jahan, S., Xu, W., He, S., Gonzalez, C., Delcuve, G. P., and Davie, J. R. (2016). The chicken erythrocyte epigenome. Epigenetics Chromatin 9:19.
Jambhekar, A., Dhall, A., and Shi, Y. (2019). Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 20, 625–641. doi: 10.1038/s41580-019-0151-1
Jang, H., and Serra, C. (2014). Nutrition, epigenetics, and diseases. Clin. Nutr. Res. 3, 1–8. doi: 10.1007/978-3-319-31143-2_66-1
Jeltsch, A., Ehrenhofer-Murray, A., Jurkowski, T. P., Lyko, F., Reuter, G., Ankri, S., et al. (2017). Mechanism and biological role of Dnmt2 in nucleic acid methylation. RNA Biol. 14, 1108–1123. doi: 10.1080/15476286.2016.1191737
Jiao, P., Yuan, Y., Zhang, M., Sun, Y., Wei, C., Xie, X., et al. (2020). PRL/microRNA-183/IRS1 pathway regulates milk fat metabolism in cow mammary epithelial cells. Genes 11:196. doi: 10.3390/genes11020196
Jiao, Q., Yin, R. H., Zhao, S. J., Wang, Z. Y., Zhu, Y. B., Wang, W., et al. (2019). Identification and molecular analysis of a lncRNA-HOTAIR transcript from secondary hair follicle of cashmere goat reveal integrated regulatory network with the expression regulated potentially by its promoter methylation. Gene 688, 182–192. doi: 10.1016/j.gene.2018.11.084
Jin, C., Zhuo, Y., Wang, J., Zhao, Y., Xuan, Y., Mou, D., et al. (2018). Methyl donors dietary supplementation to gestating sows diet improves the growth rate of offspring and is associating with changes in expression and DNA methylation of insulin-like growth factor-1 gene. J. Anim. Physiol. Anim. Nutr. 102, 1340–1350. doi: 10.1111/jpn.12933
Jin, L., Mao, K., Li, J., Huang, W., Che, T., Fu, Y., et al. (2018). Genome-wide profiling of gene expression and DNA methylation provides insight into low-altitude acclimation in Tibetan pigs. Gene 642, 522–532. doi: 10.1016/j.gene.2017.11.074
Ju, Z., Jiang, Q., Wang, J., Wang, X., Yang, C., Sun, Y., et al. (2020). Genome-wide methylation and transcriptome of blood neutrophils reveal the roles of DNA methylation in affecting transcription of protein-coding genes and miRNAs in E. coli-infected mastitis cows. BMC Genomics 21:102. doi: 10.1186/s12864-020-6526-z
Jung, G., Hernández-Illán, E., Moreira, L., Balaguer, F., and Goel, A. (2020). Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 17, 111–130. doi: 10.1038/s41575-019-0230-y
Kaikkonen, M. U., Lam, M. T., and Glass, C. K. (2011). Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 90, 430–440. doi: 10.1093/cvr/cvr097
Kamath, P. L., Foster, J. T., Drees, K. P., Luikart, G., Quance, C., Anderson, N. J., et al. (2016). Genomics reveals historic and contemporary transmission dynamics of a bacterial disease among wildlife and livestock. Nat. Commun. 7:11448.
Kamińska, K., Nalejska, E., Kubiak, M., Wojtysiak, J., Żołna, Ł, Kowalewski, J., et al. (2019). Prognostic and predictive epigenetic biomarkers in oncology. Mol. Diagn. Ther. 23, 83–95. doi: 10.1007/s40291-018-0371-7
Kang, Y., Nii, T., Isobe, N., and Yoshimura, Y. (2019a). Effects of Salmonella enteritidis vaccination on the expression of innate immune molecules and histone modifications in the follicular theca of laying hens. J. Poult. Sci. 56, 298–307. doi: 10.2141/jpsa.0190034
Kang, Y., Nii, T., Isobe, N., and Yoshimura, Y. (2019b). Effects of the routine multiple vaccinations on the expression of innate immune molecules and induction of histone modification in ovarian cells of layer chicks. Poult. Sci. 98, 5127–5136. doi: 10.3382/ps/pez214
Kantarjian, H. M., Thomas, X. G., Dmoszynska, A., Wierzbowska, A., Mazur, G., Mayer, J., et al. (2012). Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J. Clin. Oncol. 30:2670. doi: 10.1200/jco.2011.38.9429
Keleher, M. R., Zaidi, R., Shah, S., Oakley, M. E., Pavlatos, C., El Idrissi, S., et al. (2018). Maternal high-fat diet associated with altered gene expression, DNA methylation, and obesity risk in mouse offspring. PLoS One 13:e0192606. doi: 10.1371/journal.pone.0192606
Khezri, A., Narud, B., Stenseth, E.-B., Zeremichael, T. T., Myromslien, F. D., Wilson, R. C., et al. (2020). Sperm DNA hypomethylation proximal to reproduction pathway genes in maturing elite Norwegian Red bulls. Front. Genet. 11:922. doi: 10.3389/fgene.2020.00922
Kim, S., and Kaang, B.-K. (2017). Epigenetic regulation and chromatin remodeling in learning and memory. Exp. Mol. Med. 49:e281. doi: 10.1038/emm.2016.140
Kishi, Y., and Gotoh, Y. (2018). Regulation of chromatin structure during neural development. Front. Neurosci. 12:874. doi: 10.3389/fnins.2018.00874
Kisliouk, T., Cramer, T., and Meiri, N. (2017). Methyl CpG level at distal part of heat-shock protein promoter HSP 70 exhibits epigenetic memory for heat stress by modulating recruitment of POU 2F1-associated nucleosome-remodeling deacetylase (Nu RD) complex. J. Neurochem. 141, 358–372. doi: 10.1111/jnc.14014
Klein-Brill, A., Joseph-Strauss, D., Appleboim, A., and Friedman, N. (2019). Dynamics of chromatin and transcription during transient depletion of the RSC chromatin remodeling complex. Cell Rep. 26, 279–292. doi: 10.1016/j.celrep.2018.12.020
Kobayashi, W., and Kurumizaka, H. (2019). Structural transition of the nucleosome during chromatin remodeling and transcription. Curr. Opin. Struct. Biol. 59, 107–114. doi: 10.1016/j.sbi.2019.07.011
Kociucka, B., Stachecka, J., Szydlowski, M., and Szczerbal, I. (2017). Rapid communication: the correlation between histone modifications and expression of key genes involved in accumulation of adipose tissue in the pig. J. Anim. Sci. 95, 4514–4519. doi: 10.2527/jas2017.2010
Korkmaz, F. T., and Kerr, D. E. (2017). Genome-wide methylation analysis reveals differentially methylated loci that are associated with an age-dependent increase in bovine fibroblast response to LPS. BMC Genomics 18:405. doi: 10.1186/s12864-017-3796-1
Kosinska-Selbi, B., Mielczarek, M., and Szyda, J. (2020). Long non-coding RNA in livestock. Animal 10, 2003–2013.
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Kowalski, A., and Pałyga, J. (2017). Distribution of non-allelic histone H1 subtypes in five avian species. Ann. Anim. Sci. 17, 385–398. doi: 10.1515/aoas-2016-0063
Kropp, J., Carrillo, J. A., Namous, H., Daniels, A., Salih, S. M., Song, J., et al. (2017). Male fertility status is associated with DNA methylation signatures in sperm and transcriptomic profiles of bovine preimplantation embryos. BMC Genomics 18:280. doi: 10.1186/s12864-017-3673-y
Kubik, S., O’Duibhir, E., de Jonge, W. J., Mattarocci, S., Albert, B., Falcone, J.-L., et al. (2018). Sequence-directed action of RSC remodeler and general regulatory factors modulates+ 1 nucleosome position to facilitate transcription. Mol. Cell. 71, 89–102. doi: 10.1016/j.molcel.2018.05.030
Kutchy, N., Menezes, E., Chiappetta, A., Tan, W., Wills, R., Kaya, A., et al. (2018). Acetylation and methylation of sperm histone 3 lysine 27 (H3K27ac and H3K27me3) are associated with bull fertility. Andrologia 50:e12915. doi: 10.1111/and.12915
Kutchy, N. A., Velho, A., Menezes, E. S., Jacobsen, M., Thibaudeau, G., Wills, R. W., et al. (2017). Testis specific histone 2B is associated with sperm chromatin dynamics and bull fertility-a pilot study. Reprod. Biol. Endocrinol. 15:59.
Kweh, M. F., Merriman, K. E., and Nelson, C. D. (2019). Inhibition of DNA methyltransferase and histone deacetylase increases β-defensin expression but not the effects of lipopolysaccharide or 1, 25-dihydroxyvitamin D3 in bovine mammary epithelial cells. J. Dairy Sci. 102, 5706–5712. doi: 10.3168/jds.2018-16141
Lambert, S., Blondin, P., Vigneault, C., Labrecque, R., Dufort, I., and Sirard, M.-A. (2018). Spermatozoa DNA methylation patterns differ due to peripubertal age in bulls. Theriogenology 106, 21–29. doi: 10.1016/j.theriogenology.2017.10.006
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15:178.
Larsen, K., Kristensen, K. K., and Callesen, H. (2018). DNA methyltransferases and tRNA methyltransferase DNMT2 in developing pig brain-expression and promoter methylation. Gene Rep. 11, 42–51. doi: 10.1016/j.genrep.2018.02.003
Leal-Gutiérrez, J. D., Elzo, M. A., and Mateescu, R. G. (2020). Identification of eQTLs and sQTLs associated with meat quality in beef. BMC Genomics 21:104. doi: 10.1186/s12864-020-6520-5
Lee, I., Rasoul, B. A., Holub, A. S., Lejeune, A., Enke, R. A., and Timp, W. (2017). Whole genome DNA methylation sequencing of the chicken retina, cornea and brain. Sci. Data 4:170148.
Lee, K. H., Song, Y., O’Sullivan, M., Pereira, G., Loh, R., and Zhang, G. B. (2017). The implications of DNA methylation on food allergy. Int. Arch. Allergy Immunol. 173, 183–192. doi: 10.1159/000479513
Lee, Y. G., Kim, I., Yoon, S. S., Park, S., Cheong, J. W., Min, Y. H., et al. (2013). Comparative analysis between azacitidine and decitabine for the treatment of myelodysplastic syndromes. Br. J. Haematol. 161, 339–347.
Lehle, J. D., and Mccarrey, J. R. (2020). Differential susceptibility to endocrine disruptor-induced epimutagenesis. Environ. Epigenet. 6:dvaa016. doi: 10.1093/eep/dvaa016
Li, C., Li, Y., Zhou, G., Gao, Y., Ma, S., Chen, Y., et al. (2018). Whole-genome bisulfite sequencing of goat skins identifies signatures associated with hair cycling. BMC Genomics 19:638. doi: 10.1186/s12864-018-5002-5
Li, J., Li, R., Wang, Y., Hu, X., Zhao, Y., Li, L., et al. (2015). Genome-wide DNA methylome variation in two genetically distinct chicken lines using MethylC-seq. BMC Genomics 16:851. doi: 10.1186/s12864-015-2098-8
Li, M., Zou, D., Li, Z., Gao, R., Sang, J., Zhang, Y., et al. (2019). EWAS atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 47, D983–D988.
Li, Y., Carrillo, J. A., Ding, Y., He, Y., Zhao, C., Liu, J., et al. (2019). DNA methylation, microRNA expression profiles and their relationships with transcriptome in grass-fed and grain-fed angus cattle rumen tissue. PLoS One 14:e0214559. doi: 10.1371/journal.pone.0214559
Li, R., and Ibeagha-Awemu, E. M. (2017). Altered gene expression of epigenetic modifying enzymes in response to dietary supplementation with linseed oil. J. Dairy Res. 84:119. doi: 10.1017/s002202991700022x
Li, Y., Zhang, L.-Z., Yi, Y., Hu, W.-W., Guo, Y.-H., Zeng, Z.-J., et al. (2017). Genome-wide DNA methylation changes associated with olfactory learning and memory in Apis mellifera. Sci. Rep. 7:17017.
Liang, S., Nie, Z.-W., Guo, J., Niu, Y.-J., Shin, K.-T., Ock, S. A., et al. (2018). Overexpression of microRNA-29b decreases expression of DNA methyltransferases and improves quality of the blastocysts derived from somatic cell nuclear transfer in cattle. Microsc. Microanal. 24, 29–37. doi: 10.1017/s1431927618000016
Liang, Z., Brown, K. E., Carroll, T., Taylor, B., Vidal, I. F., Hendrich, B., et al. (2017). A high-resolution map of transcriptional repression. Elife 6:e22767.
Lin, S., Fang, L., Liu, G. E., and Li, C.-J. (2019). Epigenetics and Heritable Phenotypic Variations in Livestock. Amsterdam: Elsevier, 283–313.
Lister, R., Pelizzola, M., Dowen, R. H., Hawkins, R. D., Hon, G., Tonti-Filippini, J., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. doi: 10.1038/nature08514
Littlejohn, B. P., Price, D. M., Neuendorff, D. A., Carroll, J. A., Vann, R. C., Riggs, P. K., et al. (2018). Prenatal transportation stress alters genome-wide DNA methylation in suckling Brahman bull calves. J. Anim. Sci. 96, 5075–5099. doi: 10.1093/jas/sky350
Liu, C., Vyas, A., Kassab, M. A., Singh, A. K., and Yu, X. (2017). The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 45, 8129–8141. doi: 10.1093/nar/gkx565
Liu, H., Wang, J., Mou, D., Che, L., Fang, Z., Feng, B., et al. (2017). Maternal methyl donor supplementation during gestation counteracts the bisphenol a-induced impairment of intestinal morphology, disaccharidase activity, and nutrient transporters gene expression in newborn and weaning pigs. Nutrients 9:423. doi: 10.3390/nu9050423
Liu, X., Yang, J., Zhang, Q., and Jiang, L. (2017). Regulation of DNA methylation on EEF1D and RPL8 expression in cattle. Genetica 145, 387–395. doi: 10.1007/s10709-017-9974-x
Liu, S., Fang, L., Zhou, Y., Santos, D. J., Xiang, R., Daetwyler, H. D., et al. (2019). Analyses of inter-individual variations of sperm DNA methylation and their potential implications in cattle. BMC Genomics 20:888. doi: 10.1186/s12864-019-6228-6
Liu, Z., Han, S., Shen, X., Wang, Y., Cui, C., He, H., et al. (2019). The landscape of DNA methylation associated with the transcriptomic network in layers and broilers generates insight into embryonic muscle development in chicken. Int. J. Biol. Sci. 15, 1404–1418. doi: 10.7150/ijbs.35073
Liu, S., Yu, Y., Zhang, S., Cole, J. B., Tenesa, A., Wang, T., et al. (2020). Epigenomics and genotype-phenotype association analyses reveal conserved genetic architecture of complex traits in cattle and human. BMC Biol. 18:80. doi: 10.1186/s12915-020-00792-6
Liu, X., Usman, T., Wang, Y., Wang, Z., Xu, X., Wu, M., et al. (2015). Polymorphisms in epigenetic and meat quality related genes in fourteen cattle breeds and association with beef quality and carcass traits. Asian-Australas. J. Anim. Sci. 28, 467–475. doi: 10.5713/ajas.13.0837
Liu, Z., Li, Q., Liu, R., Zhao, G., Zhang, Y., Zheng, M., et al. (2016). Expression and methylation of microsomal triglyceride transfer protein and acetyl-CoA carboxylase are associated with fatty liver syndrome in chicken. Poult. Sci. 95, 1387–1395. doi: 10.3382/ps/pew040
Lorch, Y., and Kornberg, R. D. (2017). Chromatin-remodeling for transcription. Q. Rev. Biophys. 50:e5.
Lu, T., Song, Z., Li, Q., Li, Z., Wang, M., Liu, L., et al. (2017). Overexpression of histone deacetylase 6 enhances resistance to porcine reproductive and respiratory syndrome virus in pigs. PLoS One 12:e0169317. doi: 10.1371/journal.pone.0169317
Lu, Z., Ma, Y., Li, Q., Liu, E., Jin, M., Zhang, L., et al. (2019). The role of N 6-methyladenosine RNA methylation in the heat stress response of sheep (Ovis aries). Cell Stress Chaperones 24, 333–342. doi: 10.1007/s12192-018-00965-x
Luo, C., Hajkova, P., and Ecker, J. R. (2018). Dynamic DNA methylation: In the right place at the right time. Science 361, 1336–1340. doi: 10.1126/science.aat6806
Lv, Z., Fan, H., Song, B., Li, G., Liu, D., and Guo, Y. (2019). Supplementing genistein for breeder hens alters the fatty acid metabolism and growth performance of offsprings by epigenetic modification. Oxid. Med. Cell. Longev. 2019:e9214209.
Ma, X., Jia, C., Chu, M., Fu, D., Lei, Q., Ding, X., et al. (2019). Transcriptome and DNA methylation analyses of the molecular mechanisms underlying with longissimus dorsi muscles at different stages of development in the polled Yak. Genes 10:970. doi: 10.3390/genes10120970
Maldonado, M. B., de Rezende Neto, N. B., Nagamatsu, S. T., Carazzolle, M. F., Hoff, J. L., Whitacre, L. K., et al. (2019). Identification of bovine CpG SNPs as potential targets for epigenetic regulation via DNA methylation. PLoS One 14:e0222329. doi: 10.1371/journal.pone.0222329
Manakov, S. A., Pezic, D., Marinov, G. K., Pastor, W. A., Sachidanandam, R., and Aravin, A. A. (2015). MIWI2 and MILI have differential effects on piRNA biogenesis and DNA methylation. Cell Rep. 12, 1234–1243. doi: 10.1016/j.celrep.2015.07.036
Mancini-DiNardo, D., Steele, S. J., Levorse, J. M., Ingram, R. S., and Tilghman, S. M. (2006). Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 20, 1268–1282. doi: 10.1101/gad.1416906
Marco, A., Kisliouk, T., Tabachnik, T., Weller, A., and Meiri, N. (2016). DNA CpG methylation (5mC) and its derivative (5hmC) alter histone post translational modifications at the POMC promoter, affecting the impact of perinatal diet on leanness and obesity of the offspring. Diabetes 65, 2258–2267. doi: 10.2337/db15-1608
Markou, A. N., Paraskevopoulos, P., Lazaridou, M., Chen, S., Kroneis, T., Świerczewska, M., et al. (2017). GSTP1 Promoter Methylation in In-Vivo Isolated CTCs from High-Risk Prostate Cancer Patients. Washington, DC: American Association for Cancer Research.
Martignano, F., Gurioli, G., Salvi, S., Calistri, D., Costantini, M., Gunelli, R., et al. (2016). GSTP1 methylation and protein expression in prostate cancer: diagnostic implications. Dis. Markers 2016:e4358292.
Maugeri, A., and Barchitta, M. (2020). How dietary factors affect DNA methylation: lesson from epidemiological studies. Medicina 56:374. doi: 10.3390/medicina56080374
Maunakea, A. K., Chepelev, I., Cui, K., and Zhao, K. (2013). Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269. doi: 10.1038/cr.2013.110
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., D’Souza, C., Fouse, S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. doi: 10.1038/nature09165
McGuffey, R. (2017). A 100-year review: metabolic modifiers in dairy cattle nutrition. J. Dairy Sci. 100, 10113–10142. doi: 10.3168/jds.2017-12987
McKay, S., Betancourt, F., Bhattarai, S., Buttolph, T., White, S., Lachance, H., et al. (2018). 115 Profiling conservation of DNA methylation in cattle. J. Anim. Sci. 96(Suppl._3), 370–370. doi: 10.1093/jas/sky404.812
Meirelles, F. V., Bressan, F. F., Smith, L. C., Perecin, F., Chiaratti, M. R., and Ferraz, J. B. S. (2014). Cytoplasmatic inheritance, epigenetics and reprogramming DNA as tools in animal breeding. Livest. Sci. 166, 199–205. doi: 10.1016/j.livsci.2014.05.024
Melnik, B. C., John, S. M., and Schmitz, G. (2016). Milk: an epigenetic inducer of FoxP3 expression. J. Allergy Clin. Immunol. 138, 937–938. doi: 10.1016/j.jaci.2016.04.039
Melnik, B. C., and Schmitz, G. (2017a). DNA methyltransferase 1-targeting miRNA-148a of dairy milk: a potential bioactive modifier of the human epigenome. Funct. Foods Health Dis. 7, 671–687. doi: 10.31989/ffhd.v7i9.379
Melnik, B. C., and Schmitz, G. (2017b). Milk’s role as an epigenetic regulator in health and disease. Diseases 5:12. doi: 10.3390/diseases5010012
Mendes, V., Dias, R., Souza, J., Souza, E., Quintao, C., Batista, R., et al. (2017). 184 effect of histone deacetylase inhibitor on development of embryos derived from heat-shocked bovine oocytes. Reprod. Fertil. Dev. 29, 200–201. doi: 10.1071/rdv29n1ab184
Mercer, T. R., Dinger, M. E., and Mattick, J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159.
Miller, A., Ralser, M., Kloet, S. L., Loos, R., Nishinakamura, R., Bertone, P., et al. (2016). Sall4 controls differentiation of pluripotent cells independently of the Nucleosome Remodelling and Deacetylation (NuRD) complex. Development 143, 3074–3084. doi: 10.1242/dev.139113
Mishra, A., and Verma, M. (2010). Cancer biomarkers: are we ready for the prime time? Cancers 2, 190–208. doi: 10.3390/cancers2010190
Miska, E. A., and Ferguson-Smith, A. C. (2016). Transgenerational inheritance: Models and mechanisms of non–DNA sequence–based inheritance. Science 354, 59–63. doi: 10.1126/science.aaf4945
Mitra, A., Luo, J., He, Y., Gu, Y., Zhang, H., Zhao, K., et al. (2015). Histone modifications induced by MDV infection at early cytolytic and latency phases. BMC Genomics 16:311. doi: 10.1186/s12864-015-1492-6
Monk, D., Mackay, D. J., Eggermann, T., Maher, E. R., and Riccio, A. (2019). Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 20, 235–248. doi: 10.1038/s41576-018-0092-0
Morales-Nebreda, L., McLafferty, F. S., and Singer, B. D. (2019). DNA methylation as a transcriptional regulator of the immune system. Transl. Res. 204, 1–18. doi: 10.1016/j.trsl.2018.08.001
Moran, S., Arribas, C., and Esteller, M. (2016). Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 8, 389–399. doi: 10.2217/epi.15.114
Morris, K. V., and Mattick, J. S. (2014). The rise of regulatory RNA. Nat. Rev. Genet. 15, 423–437. doi: 10.1038/nrg3722
Mueller, M., Cole, J., Sonstegard, T., and Van Eenennaam, A. (2019). Comparison of gene editing versus conventional breeding to introgress the POLLED allele into the US dairy cattle population. J. Dairy Sci. 102, 4215–4226. doi: 10.3168/jds.2018-15892
Murdoch, B. M., Murdoch, G. K., Greenwood, S., and McKay, S. (2016). Nutritional influence on epigenetic marks and effect on livestock production. Front. Genet. 7:182. doi: 10.3389/fgene.2016.00182
Nayak, N., Bhanja, S., Ahmad, S. F., Mehra, M., Goel, A., Sahu, A. R., et al. (2016). Role of epigenetic modifications in improving the thermo-tolerance, growth and immuno-competence in poultry: current status and future applications. Indian J. Poul. Sci. 51, 1–9. doi: 10.5958/0974-8180.2016.00015.5
Nguyen, H., Sokpor, G., Pham, L., Rosenbusch, J., Stoykova, A., Staiger, J. F., et al. (2016). Epigenetic regulation by BAF (mSWI/SNF) chromatin remodeling complexes is indispensable for embryonic development. Cell Cycle 15, 1317–1324. doi: 10.1080/15384101.2016.1160984
Nguyen, M., Boutinaud, M., Pétridou, B., Gabory, A., Pannetier, M., Chat, S., et al. (2014). DNA methylation and transcription in a distal region upstream from the bovine AlphaS1 casein gene after once or twice daily milking. PLoS One 9:e111556. doi: 10.1371/journal.pone.0111556
Nicoglou, A., and Merlin, F. (2017). Epigenetics: A way to bridge the gap between biological fields. Stud. Hist. Philos. Sci. C 66, 73–82. doi: 10.1016/j.shpsc.2017.10.002
Nilsson, E. E., Sadler-Riggleman, I., and Skinner, M. K. (2018). Environmentally induced epigenetic transgenerational inheritance of disease. Environ. Epigenet. 4:dvy016.
Noya, A., Casasús, I., Ferrer, J., and Sanz, A. (2019). Effects of developmental programming caused by maternal nutrient intake on postnatal performance of beef heifers and their calves. Animals 9:1072. doi: 10.3390/ani9121072
O’Doherty, A. M., Rue-Albrecht, K. C., Magee, D. A., Ahting, S., Irwin, R. E., Hall, T. J., et al. (2019). The bovine alveolar macrophage DNA methylome is resilient to infection with Mycobacterium bovis. Sci. Rep. 9:1510. doi: 10.1038/s41598-018-37618-z
Oey, H., and Whitelaw, E. (2014). On the meaning of the word ‘epimutation’. Trends Genet. 30, 519–520. doi: 10.1016/j.tig.2014.08.005
Omer, N. A., Hu, Y., Hu, Y., Idriss, A. A., Abobaker, H., Hou, Z., et al. (2018). Dietary betaine activates hepatic VTGII expression in laying hens associated with hypomethylation of GR gene promoter and enhanced GR expression. J. Anim. Sci. Biotechnol. 9:2.
Omer, N. A., Hu, Y., Idriss, A. A., Abobaker, H., Hou, Z., Yang, S., et al. (2020). Dietary betaine improves egg-laying rate in hens through hypomethylation and glucocorticoid receptor–mediated activation of hepatic lipogenesis-related genes. Poult. Sci. 99, 3121–3132. doi: 10.1016/j.psj.2020.01.017
Osorio, J., Jacometo, C., Zhou, Z., Luchini, D., Cardoso, F., and Loor, J. J. (2016). Hepatic global DNA and peroxisome proliferator-activated receptor alpha promoter methylation are altered in peripartal dairy cows fed rumen-protected methionine. J. Dairy Sci. 99, 234–244. doi: 10.3168/jds.2015-10157
Paiva, J., Resende, M., Resende, R., Oliveira, H., Silva, H., Caetano, G., et al. (2019). Epigenetics: mechanisms, inheritance and implications in animal breeding. Arch. Zootec. 68, 304–311.
Palazzese, L., Czernik, M., Iuso, D., Toschi, P., and Loi, P. (2018). Nuclear quiescence and histone hyper-acetylation jointly improve protamine-mediated nuclear remodeling in sheep fibroblasts. PLoS One 13:e0193954. doi: 10.1371/journal.pone.0193954
Pan, X., Gong, D., Nguyen, D. N., Zhang, X., Hu, Q., Lu, H., et al. (2018). Early microbial colonization affects DNA methylation of genes related to intestinal immunity and metabolism in preterm pigs. DNA Res. 25, 287–296. doi: 10.1093/dnares/dsy001
Pan, X., Thymann, T., Gao, F., and Sangild, P. T. (2020). Rapid gut adaptation to preterm birth involves feeding-related DNA methylation reprogramming of intestinal genes in pigs. Front. Immunol. 11:565. doi: 10.3389/fimmu.2020.00565
Panzeri, I., and Pospisilik, J. A. (2018). Epigenetic control of variation and stochasticity in metabolic disease. Mol. Metab. 14, 26–38. doi: 10.1016/j.molmet.2018.05.010
Paparo, L., Nocerino, R., Cosenza, L., Aitoro, R., D’Argenio, V., Del Monaco, V., et al. (2016). Epigenetic features of FoxP3 in children with cow’s milk allergy. Clin. Epigenetics 8:86.
Paradis, F., Wood, K. M., Swanson, K. C., Miller, S. P., McBride, B. W., and Fitzsimmons, C. (2017). Maternal nutrient restriction in mid-to-late gestation influences fetal mRNA expression in muscle tissues in beef cattle. BMC Genomics 18:632. doi: 10.1186/s12864-017-4051-5
Park, H., Seo, K.-S., Lee, M., and Seo, S. (2019). Identification of meat quality-related differentially methylated regions in the DNA of the longissimus dorsi muscle in pig. Anim. Biotechnol. 31, 189–194. doi: 10.1080/10495398.2019.1604378
Perrier, J.-P., Sellem, E., Prézelin, A., Gasselin, M., Jouneau, L., Piumi, F., et al. (2018). A multi-scale analysis of bull sperm methylome revealed both species peculiarities and conserved tissue-specific features. BMC Genomics 19:404. doi: 10.1186/s12864-018-4764-0
Petronis, A. (2010). Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 465, 721–727. doi: 10.1038/nature09230
Platenburg, G. J., Vollebregt, E. J., Karatzas, C. N., Kootwijk, E. P., De Boer, H. A., and Strijker, R. (1996). Mammary gland-specific hypomethylation ofHpa II sites flanking the bovine αS1-casein gene. Transgenic Res. 5, 421–431. doi: 10.1007/bf01980207
Ponsuksili, S., Trakooljul, N., Basavaraj, S., Hadlich, F., Murani, E., and Wimmers, K. (2019). Epigenome-wide skeletal muscle DNA methylation profiles at the background of distinct metabolic types and ryanodine receptor variation in pigs. BMC Genomics 20:492. doi: 10.1186/s12864-019-5880-1
Pragna, P., Archana, P., Aleena, J., Sejian, V., Krishnan, G., Bagath, M., et al. (2017). Heat stress and dairy cow: impact on both milk yield and composition. Int. J. Dairy Sci. 12, 1–11. doi: 10.3923/ijds.2017.1.11
Proudhon, C., Duffié, R., Ajjan, S., Cowley, M., Iranzo, J., Carbajosa, G., et al. (2012). Protection against de novo methylation is instrumental in maintaining parent-of-origin methylation inherited from the gametes. Mol. Cell. 47, 909–920. doi: 10.1016/j.molcel.2012.07.010
Qiang, W., Guo, H., Li, Y., Shi, J., Yin, X., and Qu, J. (2018). Methylation analysis of CMTM3 and DUSP1 gene promoters in high-quality brush hair in the yangtze river delta white goat. Gene 668, 166–173. doi: 10.1016/j.gene.2018.05.031
Quan, H., Yang, Y., Liu, S., Tian, H., Xue, Y., and Gao, Y. Q. (2020). Chromatin structure changes during various processes from a DNA sequence view. Curr. Opin. Struct. Biol. 62, 1–8. doi: 10.1016/j.sbi.2019.10.010
Raddatz, G., Guzzardo, P. M., Olova, N., Fantappié, M. R., Rampp, M., Schaefer, M., et al. (2013). Dnmt2-dependent methylomes lack defined DNA methylation patterns. Proc. Natl. Acad. Sci. U.S.A. 110, 8627–8631. doi: 10.1073/pnas.1306723110
Rahat, B., Ali, T., Sapehia, D., Mahajan, A., and Kaur, J. (2020). Circulating cell-free nucleic acids as epigenetic biomarkers in precision medicine. Front. Genet. 11:844. doi: 10.3389/fgene.2020.00844
Rahman, M. B., Schellander, K., Luceño, N. L., and Van Soom, A. (2018). Heat stress responses in spermatozoa: Mechanisms and consequences for cattle fertility. Theriogenology 113, 102–112. doi: 10.1016/j.theriogenology.2018.02.012
Razin, A., and Kantor, B. (2005). DNA methylation in epigenetic control of gene expression. Prog. Mol. Subcell. Biol. 38, 151–167. doi: 10.1007/3-540-27310-7_6
Remnant, E. J., Ashe, A., Young, P. E., Buchmann, G., Beekman, M., Allsopp, M. H., et al. (2016). Parent-of-origin effects on genome-wide DNA methylation in the Cape honey bee (Apis mellifera capensis) may be confounded by allele-specific methylation. BMC Genomics 17:226. doi: 10.1186/s12864-016-2506-8
Rivera, R. M. (2020). Consequences of assisted reproductive techniques on the embryonic epigenome in cattle. Reprod. Fertil. Dev. 32, 65–81. doi: 10.1071/rd19276
Roma-Rodrigues, C., Rivas-García, L., Baptista, P. V., and Fernandes, A. R. (2020). Gene therapy in cancer treatment: why go nano? Pharmaceutics 12:233. doi: 10.3390/pharmaceutics12030233
Sabin, L. R., Delás, M. J., and Hannon, G. J. (2013). Dogma derailed: the many influences of RNA on the genome. Mol. Cell. 49, 783–794. doi: 10.1016/j.molcel.2013.02.010
Saeed, M., Babazadeh, D., Naveed, M., Arain, M. A., Hassan, F. U., and Chao, S. (2017). Reconsidering betaine as a natural anti-heat stress agent in poultry industry: a review. Trop. Anim. Health Prod. 49, 1329–1338. doi: 10.1007/s11250-017-1355-z
Safi-Stibler, S., and Gabory, A. (2019). Epigenetics and the Developmental Origins of Health and Disease: Parental Environment Signalling to the Epigenome, Critical Time Windows and Sculpting the Adult Phenotype. Amsterdam: Elsevier.
Sajjanar, B., Trakooljul, N., Wimmers, K., and Ponsuksili, S. (2019). DNA methylation analysis of porcine mammary epithelial cells reveals differentially methylated loci associated with immune response against Escherichia coli challenge. BMC Genomics 20:623. doi: 10.1186/s12864-019-5976-7
Salilew-Wondim, D., Saeed-Zidane, M., Hoelker, M., Gebremedhn, S., Poirier, M., Pandey, H. O., et al. (2018). Genome-wide DNA methylation patterns of bovine blastocysts derived from in vivo embryos subjected to in vitro culture before, during or after embryonic genome activation. BMC Genomics 19:424. doi: 10.1186/s12864-018-4826-3
Sandoval, J., Heyn, H., Moran, S., Serra-Musach, J., Pujana, M. A., Bibikova, M., et al. (2011). Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 6, 692–702. doi: 10.4161/epi.6.6.16196
Sarah-Anne, D., Mersch, M., Foissac, S., Collin, A., Pitel, F., and Coustham, V. (2017). Genome-wide epigenetic studies in chicken: a review. Epigenomes 1:20. doi: 10.3390/epigenomes1030020
Saxonov, S., Berg, P., and Brutlag, D. L. (2006). A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. U.S.A. 103, 1412–1417. doi: 10.1073/pnas.0510310103
Schmauss, C. (2017). The roles of class I histone deacetylases (HDACs) in memory, learning, and executive cognitive functions: a review. Neurosci. Biobehav. Rev. 83, 63–71. doi: 10.1016/j.neubiorev.2017.10.004
Schmitz, R. J., Lewis, Z. A., and Goll, M. G. (2019). DNA methylation: shared and divergent features across eukaryotes. Trends Genet. 35, 818–827. doi: 10.1016/j.tig.2019.07.007
Schmitz, R. J., Schultz, M. D., Lewsey, M. G., O’Malley, R. C., Urich, M. A., Libiger, O., et al. (2011). Transgenerational epigenetic instability is a source of novel methylation variants. Science 334, 369–373. doi: 10.1126/science.1212959
Scott, C. (2018). Sustainably sourced junk food? Big food and the challenge of sustainable diets. Glob. Environ. Polit. 18, 93–113. doi: 10.1162/glep_a_00458
Sevane, N., Martínez, R., and Bruford, M. W. (2019). Genome-wide differential DNA methylation in tropically adapted Creole cattle and their Iberian ancestors. Anim. Genet. 50, 15–26. doi: 10.1111/age.12731
Shah, S. M., and Chauhan, M. S. (2019). Modern biotechnological tools for enhancing reproductive efficiency in livestock. Indian J. Genet. 79(1 Suppl. 241), 249.
Shanmugam, M. K., Arfuso, F., Arumugam, S., Chinnathambi, A., Jinsong, B., Warrier, S., et al. (2018). Role of novel histone modifications in cancer. Oncotarget 9, 11414–11426.
Shayevitch, R., Askayo, D., Keydar, I., and Ast, G. (2018). The importance of DNA methylation of exons on alternative splicing. RNA 24, 1351–1362. doi: 10.1261/rna.064865.117
Shukla, S., Kavak, E., Gregory, M., Imashimizu, M., Shutinoski, B., Kashlev, M., et al. (2011). CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74–79. doi: 10.1038/nature10442
Silva, L. G., Ferguson, B. S., Avila, A. S., and Faciola, A. P. (2018). Sodium propionate and sodium butyrate effects on histone deacetylase (HDAC) activity, histone acetylation, and inflammatory gene expression in bovine mammary epithelial cells. J. Anim. Sci. 96, 5244–5252.
Singh, K., Erdman, R. A., Swanson, K. M., Molenaar, A. J., Maqbool, N. J., Wheeler, T. T., et al. (2010). Epigenetic regulation of milk production in dairy cows. J. Mammary Gland. Biol. Neoplasia 15, 101–112.
Singh, K., Molenaar, A., Swanson, K., Gudex, B., Arias, J., Erdman, R., et al. (2011). Epigenetics: a possible role in acute and transgenerational regulation of dairy cow milk production. Animal 6, 375–381. doi: 10.1017/s1751731111002564
Sistare, F., and DeGeorge, J. (2007). Preclinical predictors of clinical safety: opportunities for improvement. Clin. Pharmacol. Ther. 82, 210–214. doi: 10.1038/sj.clpt.6100243
Skibiel, A., Peñagaricano, F., Amorín, R., Ahmed, B., Dahl, G., and Laporta, J. (2018). In utero heat stress alters the offspring epigenome. Sci. Rep. 8:14609.
Skinner, M. K. (2011). Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics 6, 838–842. doi: 10.4161/epi.6.7.16537
Skinner, M. K., Guerrero-Bosagna, C., and Haque, M. M. (2015). Environmentally induced epigenetic transgenerational inheritance of sperm epimutations promote genetic mutations. Epigenetics 10, 762–771. doi: 10.1080/15592294.2015.1062207
Skinner, M. K., Maamar, M. B., Sadler-Riggleman, I., Beck, D., Nilsson, E., McBirney, M., et al. (2018). Alterations in sperm DNA methylation, non-coding RNA and histone retention associate with DDT-induced epigenetic transgenerational inheritance of disease. Epigenetics Chromatin 11, 1–24. doi: 10.1080/15592294.2020.1853319
Sleutels, F., Zwart, R., and Barlow, D. P. (2002). The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 415, 810–813. doi: 10.1038/415810a
Smith, J., Sen, S., Weeks, R. J., Eccles, M. R., and Chatterjee, A. (2020). Promoter DNA hypermethylation and paradoxical gene activation. Trends Cancer 6, 392–406. doi: 10.1016/j.trecan.2020.02.007
Sokpor, G., Xie, Y., Rosenbusch, J., and Tuoc, T. (2017). Chromatin remodeling BAF (SWI/SNF) complexes in neural development and disorders. Front. Mol. Neurosci. 10:243. doi: 10.3389/fnmol.2017.00243
Soler-Botija, C., Gálvez-Montón, C., and Bayes Genis, A. (2019). Epigenetic biomarkers in cardiovascular diseases. Front. Genet. 10:950. doi: 10.3389/fgene.2019.00950
Song, J. (2016). Genome-wide assessment of inbred chicken lines indicates epigenetics signatures of resistance to Marek’s disease. J. Anim. Sci. 94:47. doi: 10.2527/jas2016.94supplement447x
Song, M., He, Y., Zhou, H., Zhang, Y., Li, X., and Yu, Y. (2016). Combined analysis of DNA methylome and transcriptome reveal novel candidate genes with susceptibility to bovine Staphylococcus aureus subclinical mastitis. Sci. Rep. 6:29390.
Sørensen, J. T., Winckler, C., Wallenbeck, A., Spengler, A., Knierim, U., Kargo, M., et al. (2018). Improving Animal Health and Welfare in Organic Cattle Milk Production through Breeding and Management. Aarhus: Aarhus University.
Spain, M. M., Ansari, S. A., Pathak, R., Palumbo, M. J., Morse, R. H., and Govind, C. K. (2014). The RSC complex localizes to coding sequences to regulate Pol II and histone occupancy. Mol. Cell. 56, 653–666. doi: 10.1016/j.molcel.2014.10.002
Srivastava, R., Singh, U. M., and Dubey, N. K. (2016). Histone Modifications by different histone modifiers: insights into histone writers and erasers during chromatin modification. J. Biol. Sci. Med. 2, 45–54.
Stachowiak, M., Szymanski, M., Ornoch, A., Jancewicz, I., Rusetska, N., Chrzan, A., et al. (2020). SWI/SNF chromatin remodeling complex and glucose metabolism are deregulated in advanced bladder cancer. IUBMB Life 72, 1175–1188. doi: 10.1002/iub.2254
Su, Y., Fan, Z., Wu, X., Li, Y., Wang, F., Zhang, C., et al. (2016). Genome-wide DNA methylation profile of developing deciduous tooth germ in miniature pigs. BMC Genomics 17:134. doi: 10.1186/s12864-016-2485-9
Sun, X., Hota, S. K., Zhou, Y.-Q., Novak, S., Miguel-Perez, D., Christodoulou, D., et al. (2018). Cardiac-enriched BAF chromatin-remodeling complex subunit Baf60c regulates gene expression programs essential for heart development and function. Biol. Open 7:bio029512. doi: 10.1242/bio.029512
Sun, X., Li, J., Fu, L., Jiang, J., Zhao, J., and Wang, G. (2019). The epigenetic modification in mammals under heat stress. World J. Vet. Sci. 1:1005.
Szymczak, S., Dose, J., Torres, G. G., Heinsen, F.-A., Venkatesh, G., Datlinger, P., et al. (2020). DNA methylation QTL analysis identifies new regulators of human longevity. Hum. Mol. Genet. 29, 1154–1167. doi: 10.1093/hmg/ddaa033
Takeda, K., Kobayashi, E., Nishino, K., Imai, A., Adachi, H., Hoshino, Y., et al. (2019). Age-related changes in DNA methylation levels at CpG sites in bull spermatozoa and in vitro fertilization-derived blastocyst-stage embryos revealed by combined bisulfite restriction analysis. J. Reprod. Dev. 65, 305–312. doi: 10.1262/jrd.2018-146
Tal, O., Kisdi, E., and Jablonka, E. (2010). Epigenetic contribution to covariance between relatives. Genetics 184, 1037–1050. doi: 10.1534/genetics.109.112466
Tanić, M., and Beck, S. (2017). Epigenome-wide association studies for cancer biomarker discovery in circulating cell-free DNA: technical advances and challenges. Curr. Opin. Genet. Dev. 42, 48–55. doi: 10.1016/j.gde.2017.01.017
Te Pas, M. F., Lebret, B., and Oksbjerg, N. (2017). Measurable biomarkers linked to meat quality from different pig production systems. Arch. Tierz. 60, 271–283. doi: 10.5194/aab-60-271-2017
Thompson, R. P., Nilsson, E., and Skinner, M. K. (2020). Environmental epigenetics and epigenetic inheritance in domestic farm animals. Anim. Reprod. Sci. 220:106316. doi: 10.1016/j.anireprosci.2020.106316
Tian, P., Luo, Y., Li, X., Tian, J., Tao, S., Hua, C., et al. (2017). Negative effects of long-term feeding of high-grain diets to lactating goats on milk fat production and composition by regulating gene expression and DNA methylation in the mammary gland. J. Anim. Sci. Biotechnol. 8:74.
Treppendahl, M. B., Kristensen, L. S., and Grønbæk, K. (2016). Chapter 5 – Biomarkers and methodologies for monitoring epigenetic drug effects in cancer,” in Epigenetic Biomarkers and Diagnostics, ed. J. L. García-Giménez (Boston, MA: Academic Press), 91–118. doi: 10.1016/B978-0-12-801899-6.00005-X
Triantaphyllopoulos, K. A., Ikonomopoulos, I., and Bannister, A. J. (2016). Epigenetics and inheritance of phenotype variation in livestock. Epigenetics Chromatin 9:31.
Usman, T., Yu, Y., and Wang, Y. (2016). CD4 promoter hyper methylation is associated with lower gene expression in clinical mastitis cows and vice versa in the healthy controls. J. Anim. Sci. 94, 38–38. doi: 10.2527/jas2016.94supplement438x
Van Eenennaam, A. L. (2019). Application of genome editing in farm animals: Cattle. Transgenic Res. 28, 93–100. doi: 10.1007/s11248-019-00141-6
Vanselow, J., Yang, W., Herrmann, J., Zerbe, H., Schuberth, H.-J., Petzl, W., et al. (2006). DNA-remethylation around a STAT5-binding enhancer in the αS1-casein promoter is associated with abrupt shutdown of αS1-casein synthesis during acute mastitis. J. Mol. Endocrinol. 37, 463–477. doi: 10.1677/jme.1.02131
Varley, K. E., Gertz, J., Bowling, K. M., Parker, S. L., Reddy, T. E., Pauli-Behn, F., et al. (2013). Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 23, 555–567. doi: 10.1101/gr.147942.112
Veland, N., Lu, Y., Hardikar, S., Gaddis, S., Zeng, Y., Liu, B., et al. (2019). DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic Acids Res. 47, 152–167. doi: 10.1093/nar/gky947
Vinoth, A., Thirunalasundari, T., Shanmugam, M., Uthrakumar, A., Suji, S., and Rajkumar, U. (2018). Evaluation of DNA methylation and mRNA expression of heat shock proteins in thermal manipulated chicken. Cell Stress Chaperones 23, 235–252. doi: 10.1007/s12192-017-0837-2
Vojta, A., Dobrinić, P., Tadić, V., Bočkor, L., Korać, P., Julg, B., et al. (2016). Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 44, 5615–5628. doi: 10.1093/nar/gkw159
Wang, F., Li, J., Li, Q., Liu, R., Zheng, M., Wang, Q., et al. (2017a). Changes of host DNA methylation in domestic chickens infected with Salmonella enterica. J. Genet. 96, 545–550. doi: 10.1007/s12041-017-0818-3
Wang, H., Shi, H., Luo, J., Yi, Y., Yao, D., Zhang, X., et al. (2017b). MiR-145 regulates lipogenesis in goat mammary cells via targeting INSIG1 and epigenetic regulation of lipid-related genes. J. Cell. Physiol. 232, 1030–1040. doi: 10.1002/jcp.25499
Wang, H., Wang, J., Ning, C., Zheng, X., Fu, J., Wang, A., et al. (2017c). Genome-wide DNA methylation and transcriptome analyses reveal genes involved in immune responses of pig peripheral blood mononuclear cells to poly I: C. Sci. Rep. 7:9709.
Wang, H., Wu, J., Wu, S., Wu, S., and Bao, W. (2017d). DNA methylation differences of the BPI promoter among pig breeds and the regulation of gene expression. RSC Adv. 7, 48025–48030. doi: 10.1039/c7ra05549h
Wang, J., Chen, J., Zhang, J., Gao, B., Bai, X., Lan, Y., et al. (2017e). Castration-induced changes in the expression profiles and promoter methylation of the GHR gene in Huainan male pigs. Anim. Sci. J. 88, 1113–1119. doi: 10.1111/asj.12739
Wang, X., Wang, Z., Wang, Q., Wang, H., Liang, H., and Liu, D. (2017f). Epigenetic modification differences between fetal fibroblast cells and mesenchymal stem cells of the Arbas Cashmere goat. Res. Vet. Sci. 114, 363–369. doi: 10.1016/j.rvsc.2017.07.007
Wang, Y., Luo, M., Jiang, M., Lin, Y., and Zhu, J. (2017g). Comparative analysis of tissue expression and methylation reveals the crucial hypoxia genes in hypoxia-resistant animals. Can. J. Anim. Sci. 98, 204–212.
Wang, H., Liu, Z., Wang, Y., Ma, L., Zhang, W., and Xu, B. (2020a). Genome-wide differential DNA methylation in reproductive, morphological, and visual system differences between queen bee and worker Bee (Apis mellifera). Front. Genet. 11:770. doi: 10.3389/fgene.2020.00770
Wang, M., Feng, S., Ma, G., Miao, Y., Zuo, B., Ruan, J., et al. (2020b). Whole-genome methylation analysis reveals epigenetic variation in cloned and donor pigs. Front. Genet. 11:23. doi: 10.3389/fgene.2020.00023
Wang, M., Liang, Y., Ibeagha-awemu, M. E., Li, M., Zhang, H., Chen, Z., et al. (2020c). Genome-wide DNA methylation analysis of mammary gland tissues from Chinese holstein cows with Staphylococcus aureus induced mastitis. Front. Genet. 19:1295.
Wang, S., Li, F., Liu, J., Zhang, Y., Zheng, Y., Ge, W., et al. (2020d). Integrative analysis of methylome and transcriptome reveals the regulatory mechanisms of hair follicle morphogenesis in cashmere goat. Cells 9:969. doi: 10.3390/cells9040969
Wang, X., Qu, J., Li, J., He, H., Liu, Z., and Huan, Y. (2020e). Epigenetic reprogramming during somatic cell nuclear transfer: recent progress and future directions. Front. Genet. 11:205. doi: 10.3389/fgene.2020.00205
Wang, Y., Zheng, Y., Guo, D., Zhang, X., Guo, S., Hui, T., et al. (2020f). m6A methylation analysis of differentially expressed genes in skin tissues of coarse and fine type liaoning cashmere goats. Front. Genet. 10:1318. doi: 10.3389/fgene.2019.01318
Wang, H., Yang, L., Qu, H., Feng, H., Wu, S., and Bao, W. (2019). Global mapping of H3K4 trimethylation (H3K4me3) and transcriptome analysis reveal genes involved in the response to epidemic diarrhea virus infections in pigs. Animals 9:523. doi: 10.3390/ani9080523
Wang, L., Sun, H., Guan, L., and Liu, J. (2019). Relationship of blood DNA methylation rate and milk performance in dairy cows. J. Dairy Sci. 102, 5208–5211. doi: 10.3168/jds.2018-15869
Wang, J., Bian, Y., Wang, Z., Li, D., Wang, C., Li, Q., et al. (2014). MicroRNA-152 regulates DNA methyltransferase 1 and is involved in the development and lactation of mammary glands in dairy cows. PLoS One 9:e101358. doi: 10.1371/journal.pone.0101358
Wang, J., Yan, X., Nesengani, L. T., Ding, H., Yang, L., and Lu, W. (2018). LPS-induces IL-6 and IL-8 gene expression in bovine endometrial cells “through DNA methylation”. Gene 677, 266–272. doi: 10.1016/j.gene.2018.07.074
Wang, X., Zhang, Y., He, Y. H., Ma, P. P., Fan, L. J., Wang, Y. C., et al. (2013). Aberrant promoter methylation of the CD4 gene in peripheral blood cells of mastitic dairy cows. Genet. Mol. Res. 12, 6228–6239. doi: 10.4238/2013.december.4.10
Wang, X., and Kadarmideen, H. (2019a). Genome-wide DNA methylation analysis using next-generation sequencing to reveal candidate genes responsible for boar taint in pigs. Anim. Genet. 50, 644–659. doi: 10.1111/age.12842
Wang, X., and Kadarmideen, H. (2019b). “Genome-wide DNA methylation profiles of pig testis reveal epigenetic markers/genes for boar taint,” in Proceedings of the 70th Annual Meeting of the European Federation of Animal Science, (Rome: EAAP).
Wang, X., and Kadarmideen, H. N. (2019c). An epigenome-wide DNA methylation map of testis in pigs for study of complex traits. Front. Genet. 10:405. doi: 10.3389/fgene.2019.00405
Wapinski, O. L., Vierbuchen, T., Qu, K., Lee, Q. Y., Chanda, S., Fuentes, D. R., et al. (2013). Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155, 621–635. doi: 10.1016/j.cell.2013.09.028
Wara, A. B., Chhotaray, S., Shafi, B. U. D., Panda, S., Tarang, M., Yosuf, S., et al. (2019). Role of miRNA signatures in health and productivity of livestock. Int. J. Curr. Microbiol. App. Sci. 8, 727–738. doi: 10.20546/ijcmas.2019.804.079
Waterman, R., Geary, T., Petersen, M., and MacNeil, M. (2017). Effects of reduced in utero and post-weaning nutrition on milk yield and composition in primiparous beef cows. Animal 11, 84–90. doi: 10.1017/s1751731116001257
Weber, M., Hellmann, I., Stadler, M. B., Ramos, L., Pääbo, S., Rebhan, M., et al. (2007). Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39, 457–466. doi: 10.1038/ng1990
Wedd, L., and Maleszka, R. (2016). DNA methylation and gene regulation in honeybees: from genome-wide analyses to obligatory epialleles. Adv. Exp. Med. Biol. 45, 193–211. doi: 10.1007/978-3-319-43624-1_9
Wei, D., Li, A., Zhao, C., Wang, H., Mei, C., Khan, R., et al. (2018). Transcriptional regulation by CpG sites methylation in the core promoter region of the bovine SIX1 gene: roles of histone H4 and E2F2. Int. J. Mol. Sci. 19:213. doi: 10.3390/ijms19010213
Wei, J.-W., Huang, K., Yang, C., and Kang, C.-S. (2017). Non-coding RNAs as regulators in epigenetics. Oncol. Rep. 37, 3–9.
Weksberg, R., Shuman, C., Wilkins-Haug, L., Mann, M., Croughan, M., Stewart, D., et al. (2007). Workshop report: evaluation of genetic and epigenetic risks associated with assisted reproductive technologies and infertility. Fertil. Steril. 88, 27–31. doi: 10.1016/j.fertnstert.2006.11.114
World Health Organization [WHO] (2001). Biomarkers in Risk Assessment: Validity and Vlidation-Environmental Health Criteria 222. Geneva: WHO.
Wu, H., and Zhang, Y. (2014). Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156, 45–68. doi: 10.1016/j.cell.2013.12.019
Wu, J., Zhang, W., and Li, C. (2020a). Recent advances in genetic and epigenetic modulation of animal exposure to high temperature. Front. Genet. 11:e653. doi: 10.3389/fgene.2020.00653
Wu, Y., Chen, J., Sun, Y., Dong, X., Wang, Z., Chen, J., et al. (2020b). PGN and LTA from Staphylococcus aureus induced inflammation and decreased lactation through regulating DNA methylation and histone H3 acetylation in bovine mammary epithelial cells. Toxins 12:238. doi: 10.3390/toxins12040238
Wu, Y., Sun, Y., Dong, X., Chen, J., Wang, Z., Chen, J., et al. (2020c). The synergism of PGN, LTA and LPS in inducing transcriptome changes, inflammatory responses and a decrease in lactation as well as the associated epigenetic mechanisms in bovine mammary epithelial cells. Toxins 12:387. doi: 10.3390/toxins12060387
Wu, S. C., and Zhang, Y. (2010). Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 11, 607–620. doi: 10.1038/nrm2950
Wu, X., and Zhang, Y. (2017). TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 18:517. doi: 10.1038/nrg.2017.33
Wyck, S., Herrera, C., Requena, C. E., Bittner, L., Hajkova, P., Bollwein, H., et al. (2018). Oxidative stress in sperm affects the epigenetic reprogramming in early embryonic development. Epigenetics Chromatin 11:60.
Xiao, P., Zhong, T., Liu, Z., Ding, Y., Guan, W., He, X., et al. (2019). Integrated analysis of methylome and transcriptome changes reveals the underlying regulatory signatures driving the curly wool transformation in Chinese Zhongwei goat. Front. Genet. 10:1263. doi: 10.3389/fgene.2019.01263
Xing, J., and Jiang, Y. (2012). Effect of dietary betaine supplementation on mRNA level of lipogenesis genes and on promoter CpG methylation of fatty acid synthase (FAS) gene in laying hens. Afr. J. Biotechnol. 11, 6633–6640.
Xu, T., Seyfert, H., and Shen, X. (2018). Epigenetic mechanisms contribute to decrease stearoyl-CoA desaturase 1 expression in the liver of dairy cows after prolonged feeding of high-concentrate diet. J. Dairy Sci. 101, 2506–2518. doi: 10.3168/jds.2017-12878
Xue, J., Schoenrock, S. A., Valdar, W., Tarantino, L. M., and Ideraabdullah, F. Y. (2016). Maternal vitamin D depletion alters DNA methylation at imprinted loci in multiple generations. Clin. Epigenetics 8:107.
Yagound, B., Smith, N. M., Buchmann, G., Oldroyd, B. P., and Remnant, E. J. (2019). Unique DNA methylation profiles are associated with cis-variation in honey bees. Genome Biol. Evol. 11, 2517–2530. doi: 10.1093/gbe/evz177
Yakovlev, A. (2018). Epigenetic effects in livestock breeding. Russ. J. Genet. 54, 897–909. doi: 10.1134/s1022795418080148
Yang, X., Han, H., De Carvalho, D. D., Lay, F. D., Jones, P. A., and Liang, G. (2014). Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26, 577–590. doi: 10.1016/j.ccr.2014.07.028
Yang, Y., Yamada, T., Hill, K. K., Hemberg, M., Reddy, N. C., Cho, H. Y., et al. (2016a). Chromatin remodeling inactivates activity genes and regulates neural coding. Science 353, 300–305. doi: 10.1126/science.aad4225
Yang, Y., Zhou, R., Mu, Y., Hou, X., Tang, Z., and Li, K. (2016b). Genome-wide analysis of DNA methylation in obese, lean, and miniature pig breeds. Sci. Rep. 6:30160.
Yang, Z., Yang, H., Gong, D., Rose, S., Pirgozliev, V., Chen, X., et al. (2018). Transcriptome analysis of hepatic gene expression and DNA methylation in methionine-and betaine-supplemented geese (Anser cygnoides domesticus). Poult. Sci. 97, 3463–3477. doi: 10.3382/ps/pey242
Ye, J., Wu, W., Li, Y., and Li, L. (2017). Influences of the gut microbiota on DNA methylation and histone modification. Dig. Dis. Sci. 62, 1155–1164. doi: 10.1007/s10620-017-4538-6
Ye, Y., Wu, H., Chen, K., Clapier, C. R., Verma, N., Zhang, W., et al. (2019). Structure of the RSC complex bound to the nucleosome. Science 366, 838–843.
Yearim, A., Gelfman, S., Shayevitch, R., Melcer, S., Glaich, O., Mallm, J.-P., et al. (2015). HP1 is involved in regulating the global impact of DNA methylation on alternative splicing. Cell Rep. 10, 1122–1134. doi: 10.1016/j.celrep.2015.01.038
Yin, Y., Morgunova, E., Jolma, A., Kaasinen, E., Sahu, B., Khund-Sayeed, S., et al. (2017). Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356:eaaj2239. doi: 10.1126/science.aaj2239
Yu, X.-X., Liu, Y.-H., Liu, X.-M., Wang, P.-C., Liu, S., Miao, J.-K., et al. (2018). Ascorbic acid induces global epigenetic reprogramming to promote meiotic maturation and developmental competence of porcine oocytes. Sci. Rep. 8:6132.
Zeng, C., Stroup, E. K., Zhang, Z., Chiu, B. C.-H., and Zhang, W. (2019). Towards precision medicine: advances in 5-hydroxymethylcytosine cancer biomarker discovery in liquid biopsy. Cancer Commun. 39:12. doi: 10.1186/s40880-019-0356-x
Zglejc-Waszak, K., Waszkiewicz, E., and Franczak, A. (2019). Periconceptional undernutrition affects the levels of DNA methylation in the peri-implantation pig endometrium and in embryos. Theriogenology 123, 185–193. doi: 10.1016/j.theriogenology.2018.10.002
Zhang, B., Ban, D., Gou, X., Zhang, Y., Yang, L., Chamba, Y., et al. (2019). Genome-wide DNA methylation profiles in Tibetan and Yorkshire pigs under high-altitude hypoxia. J. Anim. Sci. Biotechnol. 10:25.
Zhang, M., Li, D., Zhai, Y., Wang, Z., Ma, X., Zhang, D., et al. (2020). The landscape of DNA methylation associated with the transcriptomic network of intramuscular adipocytes generates insight into intramuscular fat deposition in chicken. Front. Cell Dev. Biol. 8:206. doi: 10.3389/fcell.2020.00206
Zhang, M., Yan, F.-B., Li, F., Jiang, K.-R., Li, D.-H., Han, R.-L., et al. (2017). Genome-wide DNA methylation profiles reveal novel candidate genes associated with meat quality at different age stages in hens. Sci. Rep. 7:45564.
Zhang, X., Zhang, S., Ma, L., Jiang, E., Xu, H., Chen, R., et al. (2017). Reduced representation bisulfite sequencing (RRBS) of dairy goat mammary glands reveals DNA methylation profiles of integrated genome-wide and critical milk-related genes. Oncotarget 8, 115326–115344. doi: 10.18632/oncotarget.23260
Zhang, Y., Li, F., Feng, X., Yang, H., Zhu, A., Pang, J., et al. (2017). Genome-wide analysis of DNA Methylation profiles on sheep ovaries associated with prolificacy using whole-genome Bisulfite sequencing. BMC Genomics 18:759. doi: 10.1186/s12864-017-4068-9
Zhang, S., Shen, L., Xia, Y., Yang, Q., Li, X., Tang, G., et al. (2016). DNA methylation landscape of fat deposits and fatty acid composition in obese and lean pigs. Sci. Rep. 6:35063.
Zhang, Y., Meng, J., Gao, Y., Zhang, J., Niu, S., Yu, X., et al. (2016). Changes in methylation of genomic DNA from chicken immune organs in response to H5N1 influenza virus infection. Genet. Mol. Res. 15:e15037382.
Zhang, Y., Wang, X., Jiang, Q., Hao, H., Ju, Z., Yang, C., et al. (2018a). DNA methylation rather than single nucleotide polymorphisms regulates the production of an aberrant splice variant of IL6R in mastitic cows. Cell Stress Chaperones 23, 617–628. doi: 10.1007/s12192-017-0871-0
Zhang, Y., Zhang, X., Shi, J., Tuorto, F., Li, X., Liu, Y., et al. (2018b). Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non-coding RNAs. Nat. Cell Biol. 20, 535–540. doi: 10.1038/s41556-018-0087-2
Zhao, C., Ji, G., Carrillo, J. A., Li, Y., Tian, F., Baldwin, R. L., et al. (2020). The profiling of DNA methylation and its regulation on divergent tenderness in angus beef cattle. Front. Genet. 11:939. doi: 10.3389/fgene.2020.00939
Zhao, Z., and Shilatifard, A. (2019). Epigenetic modifications of histones in cancer. Genome Biol. 20:245.
Zhi, D., Aslibekyan, S., Irvin, M. R., Claas, S. A., Borecki, I. B., Ordovas, J. M., et al. (2013). SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics 8, 802–806. doi: 10.4161/epi.25501
Zhou, C. Y., Johnson, S. L., Gamarra, N. I., and Narlikar, G. J. (2016). Mechanisms of ATP-dependent chromatin remodeling motors. Annu. Rev. Biophys. 45, 153–181. doi: 10.1146/annurev-biophys-051013-022819
Zhou, Y., Xu, L., Bickhart, D. M., Schroeder, S. G., Connor, E. E., Alexander, L. J., et al. (2016). Reduced representation bisulphite sequencing of ten bovine somatic tissues reveals DNA methylation patterns and their impacts on gene expression. BMC Genomics 17:779. doi: 10.1186/s12864-016-3116-1
Zhou, Y., Connor, E. E., Bickhart, D. M., Li, C., Baldwin, R. L., Schroeder, S. G., et al. (2018). Comparative whole genome DNA methylation profiling of cattle sperm and somatic tissues reveals striking hypomethylated patterns in sperm. GigaScience 7:giy039.
Keywords: DNA methylation and histone modification, epigenetics biomarker, application of epigenetics data, growth and development, livestock, productivity, health and disease
Citation: Wang M and Ibeagha-Awemu EM (2021) Impacts of Epigenetic Processes on the Health and Productivity of Livestock. Front. Genet. 11:613636. doi: 10.3389/fgene.2020.613636
Received: 02 October 2020; Accepted: 21 December 2020;
Published: 23 February 2021.
Edited by:
Nick V. L. Serão, Iowa State University, United StatesReviewed by:
Christopher K. Tuggle, Iowa State University, United StatesCopyright © 2021 Her Majesty the Queen in Right of Canada, as represented by the Minister of Agriculture and Agri-Food Canada for the contribution of Mengqi Wang and Eveline M. Ibeagha-Awemu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eveline M. Ibeagha-Awemu, ZXZlbGluZS5pYmVhZ2hhLWF3ZW11QGNhbmFkYS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.