 Nivedhitha Mahendran
Nivedhitha Mahendran P. M. Durai Raj Vincent
P. M. Durai Raj Vincent Kathiravan Srinivasan
Kathiravan Srinivasan Chuan-Yu Chang
Chuan-Yu Chang- 1School of Information Technology and Engineering, Vellore Institute of Technology, Vellore, India
- 2Department of Computer Science and Information Engineering, National Yunlin University of Science and Technology, Douliu, Taiwan
Gene Expression is the process of determining the physical characteristics of living beings by generating the necessary proteins. Gene Expression takes place in two steps, translation and transcription. It is the flow of information from DNA to RNA with enzymes’ help, and the end product is proteins and other biochemical molecules. Many technologies can capture Gene Expression from the DNA or RNA. One such technique is Microarray DNA. Other than being expensive, the main issue with Microarray DNA is that it generates high-dimensional data with minimal sample size. The issue in handling such a heavyweight dataset is that the learning model will be over-fitted. This problem should be addressed by reducing the dimension of the data source to a considerable amount. In recent years, Machine Learning has gained popularity in the field of genomic studies. In the literature, many Machine Learning-based Gene Selection approaches have been discussed, which were proposed to improve dimensionality reduction precision. This paper does an extensive review of the various works done on Machine Learning-based gene selection in recent years, along with its performance analysis. The study categorizes various feature selection algorithms under Supervised, Unsupervised, and Semi-supervised learning. The works done in recent years to reduce the features for diagnosing tumors are discussed in detail. Furthermore, the performance of several discussed methods in the literature is analyzed. This study also lists out and briefly discusses the open issues in handling the high-dimension and less sample size data.
Introduction
Deoxy-ribonucleic Acid (DNA) is a hereditary material containing the genetic information, usually found in the cell’s nucleus. The information inside the DNA is made up of a code consisting of four bases, namely, Adenine, Guanine, Cytosine, and Thymine. Adenine pairs with Thymine and Cytosine with Guanine to form base pairs. The base pairs, along with their respective sugar and phosphate molecules, form a Nucleotide. The Nucleotide forms a double helical structure, which looks like a ladder. Gene is the fundamental unit of heredity and is built-up of DNA. Genes are responsible for determining characteristics such as height, color, and many others. Some of the genes manufacture proteins, and some do not. According to the Human Genome Project, there are approximately around 25,000 genes in humans.
There are two copies of genes in every human; one passed on from the parent; almost all the genes are the same, except a few, less than 1% called the Alleles. They determine the unique physical features of a person. Genes manufacture proteins, and proteins, in turn, say what the cell should do (cell functions). The flow starts with DNA, RNA, and then the proteins. The flow of information determines the type of proteins being produced. The process in which the information contained in DNA is transformed into instructions to form proteins and other biochemical molecules is called gene expression. Gene expression assists the cells to react appropriately to the changing environment. The gene expression involves two critical steps in manufacturing the proteins, Transcription and Translation (Raut et al., 2010).
• Transcription: The DNA present in the gene will be copied to form an RNA known as the messenger RNA (mRNA). RNA is similar to DNA; however, it has a single-strand, and instead of Thymine, it has Uracil (U).
• Translation: The messages carried from the transcription by the mRNA will be read by the transfer RNA (tRNA) in the Translation phase. The mRNA can read three letters at a time, which constitutes one Amino acid (Amino acids are the building blocks of proteins).
Proteins play a significant role in cell functioning. Gene expression controls everything, such as when to produce protein, when not to, volume, i.e., increasing or decreasing the amount, etc. It is a kind of on/off switch. When this process does not happen as it is supposed to be, genetic disorders, tumors occur. A detailed study of the gene expression will help find the essential biomarkers that cause genetic disorders and tumors.
There are many techniques available to capture the gene expressions such as Northern blot, RNA protection assay, Reverse Transcription – Polymerase Chain Reaction (RT - PCR), Serial Analysis of Gene Expression (SAGE), Subtractive Hybridization, DNA Microarrays, Second Generation Sequencing (NGS) and many others. Among these, the most widely used these days is DNA Microarray (Raut et al., 2010; Wang and van der Laan, 2011). The DNA microarray technology manages to capture gene expressions of thousands of genes simultaneously. However, the Microarray result is enormous, with a high dimension, which makes the analysis challenging. Thus, it is necessary to perform gene selection to handle the high dimensional problem by removing the redundant and irrelevant genes. There are many computation techniques used in the field of bioinformatics been carried out over the years, such as Pattern Recognition, Data Mining, and many others to manage the high dimensional issue, yet ineffective (Raut et al., 2010).
Hence, in recent years, Machine Learning, which is a part of Artificial Intelligence, has gained the researchers’ attention in genomics and gene expression. Machine Learning is the part of Data Science; its primary purpose is to enable a model to train and learn to make decisions on its own in the future. Machine Learning is commonly categorized as Supervised, Unsupervised, and Semi-supervised or Semi-unsupervised learning. The Supervised involves the labeled data; unsupervised learning involves unlabeled data, and the Semi-supervised or Semi-unsupervised involves handling both labeled and unlabeled data. Machine Learning flows through Pre-processing and Classification or Clustering. In gene expression microarray data, machine learning-based feature selection approaches like gene selection approaches will help to select the required genes from the lot.
Feature selection helps in preserving the informative attributes. Feature selection is primarily applied to the high-dimensional data; in simple terms, feature selection is a dimensionality reduction technique (Kira and Rendell, 1992). Feature selection assists significantly in the fields, which have too many features and relatively scarce samples, for instance, RNA sequencing and DNA Microarray (Ang et al., 2015b).
The primary intent that feature selection got famous in the recent past is to extract the informative subset of features from the original feature space (Ang et al., 2015b). Feature selection techniques aids in overcoming the scare of model overfitting, handling the dimension, better interpretation of the feature space, maximizes prediction accuracy, and maximizes the model training time (Halperin et al., 2005; Sun et al., 2019b). The outcome of Feature selection is the optimal number of features that are relevant to the given class label, which contributes to the process of prediction.
One more technique for dimensionality reduction is Feature Extraction. Feature Selection is part of Feature Extraction (Cárdenas-Ovando et al., 2019). It is the process of transforming the original feature space into a prominent space, which can be a linear or non-linear combination of the original feature space (Anter and Ali, 2020). The major drawback of using Feature Extraction is that it alters the original feature space; eventually, the data interpretability is lost. Also, the transformation is usually expensive (Bermingham et al., 2015).
Gene expression is the flow of genetic information from Deoxy-ribose Nucleic Acid (DNA) to Ribose Nucleic Acid (RNA) to protein or other biomolecule syntheses. Gene expression data is a biological representation of various transcriptions and other chemicals found inside a cell at a given time. As data is recorded directly from DNA, through various experiments, a pertinent computational technique will reveal deep insights about the disease or disorder in the cell, eventually the organism in which the cell belongs (Koul and Manvi, 2020).
On the one hand, the gene expression data is highly dimensional; also, on the other, the sample size is incompetent. The high dimensionality in the data is due to the vast number of values generated for every gene in a genome in the order of thousands. Advanced technologies, for instance, Microarray, assists in analyzing thousands of proteins in a gene in a particular sample. However, the issue with Microarray is that it is expensive (Wahid et al., 2020).
However, the data with vast feature space will have redundant features with unnecessary information that will lead to overfitting, significantly affecting the model’s performance. The primary purpose of implementing the Feature selection or gene selection on gene expression data is to choose the most regulating genes and eliminate the redundant genes that do not contribute to the target class (Pearson et al., 2019).
The gene expression data are usually unlabeled, labeled, or semi-labeled, which leads to the necessity of the concepts of Unsupervised, Supervised, and Semi-supervised feature selection. Unlabeled data has no prior information about the functionalities, whereas it validates the gene selection based on data distribution, variance, and separability. Labeled data consists of meaningful class labels and information about the functionalities. Then gene selection will be performed based on the relevance and importance score of the labeled features. Semi-supervised or Semi-unsupervised combines a small amount of unlabeled data with labeled data and vice versa, which acts as additional information (Yang et al., 2019). This paper discusses the importance of feature selection or gene selection to have an improved result. This paper’s remaining sections discuss the background and development of feature selection, the steps involved in feature selection, a detailed discussion on various works on gene selection in the literature, the open issues, and future research directions concerning the gene expression data and conclusion.
The feature selection methods can be categorized into Supervised, Unsupervised, and Semi-supervised learning models. The survey works in the literature concentrate on either one of the models; for example (Kumar et al., 2017), focuses only on the supervised gene selection methods. Some works also concentrate on one particular feature selection strategy; for example (Lazar et al., 2012), focuses on filter-based techniques. Table 1 shows the comparison of existing reviews with the current survey. Our study categorizes the feature selection strategy into supervised, unsupervised, and semi-supervised methods and discusses the existing approaches in those categories. Also, we have done a detailed discussion of their performances.

Table 1. Comparison of existing reviews with the current survey.
Gene Selection – Background and Development
Gene Selection is the technique applied to the gene expression dataset, such as DNA Microarray, to reduce the number of genes, which are redundant and less expressive or less informative. Gene Selection has its base in the Machine Learning-based Feature Selection technique, which significantly suits the applications that involve thousands of features (Dashtban and Balafar, 2017). Gene Selection techniques are applied mainly for two reasons: finding the informative and expressive genes and removing the original space’s redundant genes. Theoretically, an increase in the number of genes will bring down the model’s performance and compromise the generalization by overfitting. The present works on Gene Selection concentrate mainly on finding the relevant genes, and there is limited research in removing the noise and redundant genes (Wang et al., 2005).
For significant results, it is critical to concentrate on relevancy, redundancy, and complementarity. A gene is considered as relevant when it has necessary information (individually or combined with other genes) about the given class, for example, tumorous or not. According to Yu and Liu (2004), the feature subset can be classified into strongly relevant, weakly relevant, and irrelevant in technical terms. The weakly irrelevant can again be classified into weakly relevant and redundant features and weakly relevant and non-redundant features. Most of the informative features can be found under strongly relevant and weakly relevant, and non-redundant features (Vergara and Estévez, 2014). The same approach is followed in the Gene Selection from the gene expression data. Figure 1 shows the representation of the Gene Selection approach.
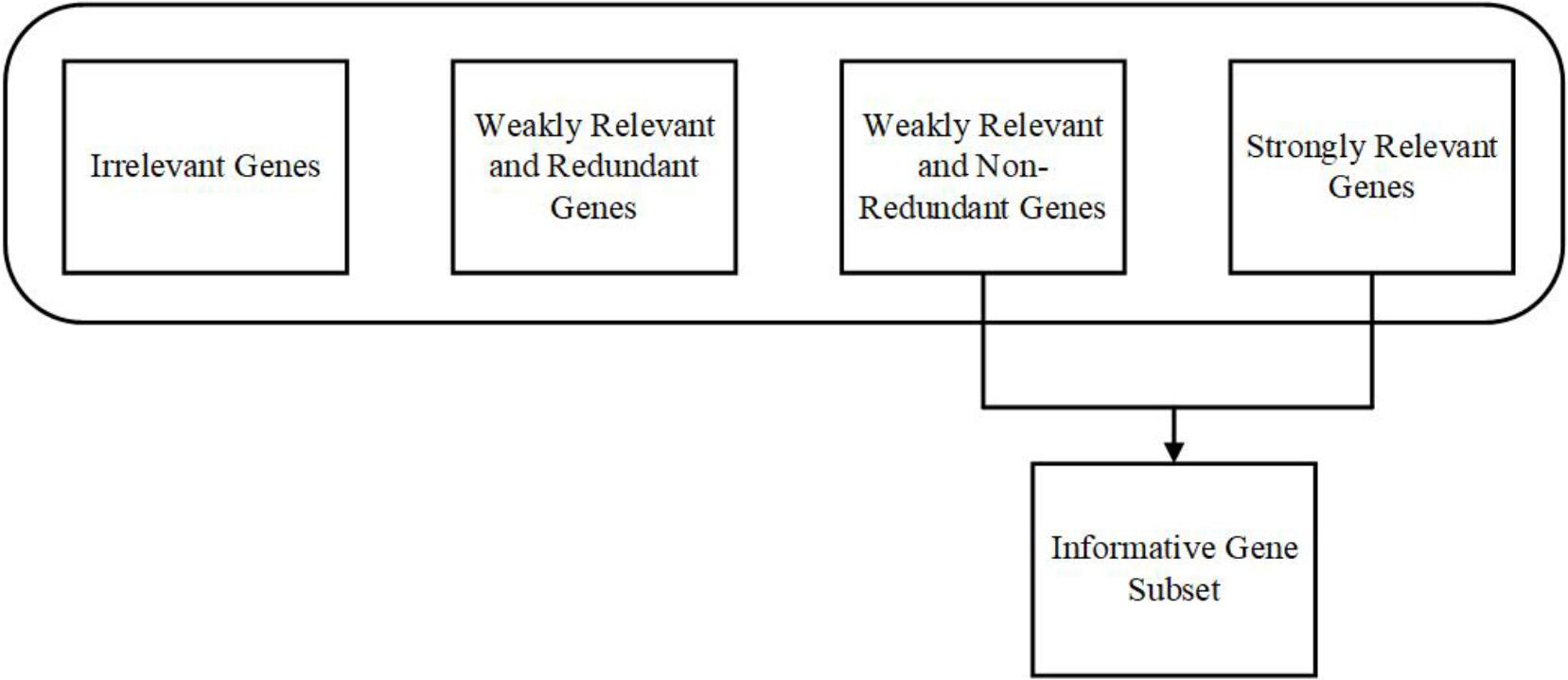
Figure 1. Representation of Gene Selection approach.
Many works in literature (Hu et al., 2010; Hoque et al., 2014; Sun and Xu, 2014) aim to remove redundancy and relevancy from the data with the Mutual Information algorithm’s help in Gene Expression. Many variations in Mutual Information are implemented to tackle these two issues. Along with these two issues, there is one more issue, which many of the existing works fail to address, complementarity. Complementarity is the degree of feature interaction between a gene subset and an individual gene in a given class.
To solve the issues mentioned above, commonly, two approaches are followed in the literature, one is analyzing individual genes, and the other is finding an optimal subset. In analyzing individual genes, the genes are ranked based on their importance scores; genes with a similar score (redundant) and genes with the least score (irrelevant) below a given threshold will be removed. In finding an optimal subset, a search for a minimal subset of genes will be done, satisfying specific criteria and eliminating redundant and irrelevant genes.
In applications such as Text and Genomic Microarray analysis, the central issue is the “Curse of Dimensionality,” where finding the optimal subset of genes is considered an NP-hard problem. Effective learning will be achieved only when the model is trained with relevant and non-redundant genes. However, with an increase in the genes’ dimension, the possible number of optimal gene subsets will also increase exponentially.
In machine learning, feature space is defined as the space associated with a feature vector distributed all over the sample in an n-dimensional space. Moreover, to reduce the dimensionality of such feature space, feature extraction, or feature selection techniques can be used. Feature Selection is a part of the Feature Extraction technique. However, in feature selection, a subset from the original feature space will be formed, whereas, in feature extraction, a new set of feature space will be created that seems to capture the necessary information from the original feature space (Jović et al., 2015). The most commonly used feature extraction techniques are Principle Component Analysis (PCA), Independent Component Analysis (ICA), Expectation-Maximization (EM), and Linear Discriminant Analysis (LDA). Some examples of Feature Selection techniques are RELIEF, Conditional Mutual Information Maximization (CMIM), Correlation Coefficient, Information Gain, and Lasso (Khalid et al., 2014).
The major drawback of using Feature extraction is that the data’s interpretability will be lost in the transformation. Also, the transformation itself will be expensive sometimes (Khalid et al., 2014). Therefore, in this paper, we will discuss various Feature Selection techniques used in Gene Selection, which is less expensive and preserves the data’s interpretability.
The Gene Selection based on machine learning can be classified into three types, Supervised, Unsupervised, and Semi-Supervised. Supervised Gene Selection utilizes the genes that are labeled already (Filippone et al., 2006). The input and output labels are known in advance in this method. However, the data continues to grow and overwhelm the process, leading to data mislabeling, making it unreliable. The main issue in deploying Supervised Gene Selection is overfitting, which can be caused by selecting irrelevant or sometimes eliminating the most relevant gene (Ang et al., 2015b).
Unsupervised Gene Selection, unlike Supervised, will not have any labels to guide the selection process (Filippone et al., 2005). The data used in Unsupervised Gene Selection is unlabelled. That makes it unbiased and serves as an effective way to find the necessary insights into the classification process (Ye and Sakurai, 2017). The main issue in Unsupervised Gene Selection is that it does not consider the interaction among the Genes (correlation), making the resultant gene subset insignificant in the discrimination task (Acharya et al., 2017).
Semi-supervised or Semi-unsupervised Gene Selection is like an add-on to the Supervised and Unsupervised Gene Selection. A Gene Selection is considered semi-supervised when most of the data is labeled, and a Gene Selection is said to be Semi-unsupervised when most of the data is unlabelled. The labeled data in the Semi-supervised or unsupervised is used to increase the distance between the data points that belongs to different classes, whereas the unlabelled data will help identify the geometrical structure of the feature space (Sheikhpour et al., 2017). Figure 2 illustrates the overview of the process involved in Gene Selection.
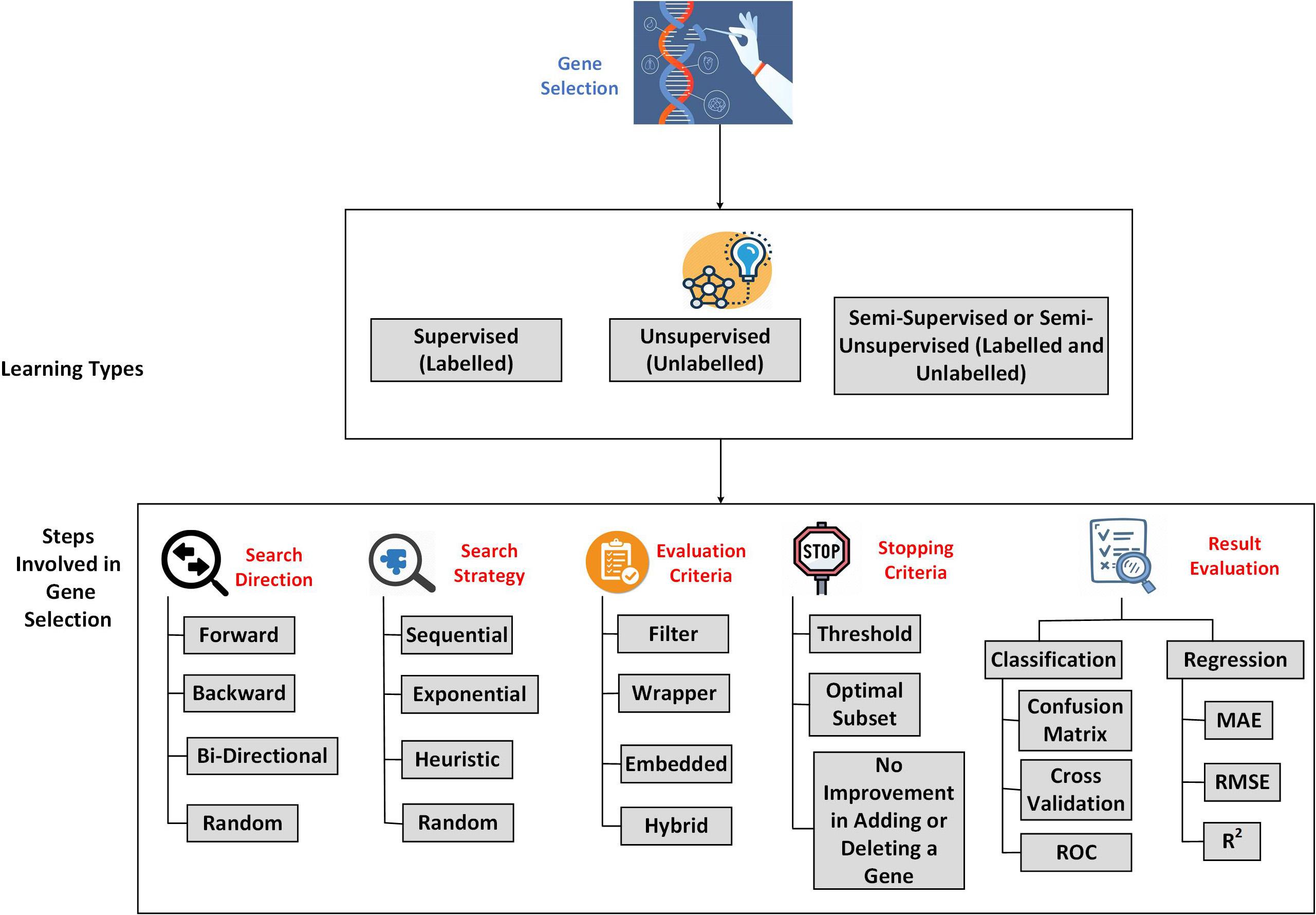
Figure 2. An overview of Gene Selection process.
Steps Involved in Feature Selection
Search Direction
The first stage involved in Feature Selection is to choose a search direction, which serves as a starting point to the process. There are three commonly used search directions:
• Forward Search: In Forward Search, the Search will be started with an empty set, and features are added one by one (Mohapatra et al., 2016).
• Backward Search: Search will be started with the whole set of genes, and the genes will be eliminated one by one with each iteration.
• Bi-directional: Search involves the advantages of Forward Search and Backward Search. The Search starts from both directions by either adding or removing a gene with each iteration (Abinash and Vasudevan, 2018). Other than these, Random Search is also used as a search direction (Wang et al., 2016).
Search Strategy
A good search strategy should attain fast convergence and provide an optimal solution with efficient computational cost and good global search ability (Halperin et al., 2005). There are three most widely used searching strategies:
• Sequential: follows a particular order in finding the best feature subset, for instance, Sequential Forward Search, where the search will be carried out from the start to the end (Chen and Yao, 2017). This strategy is prone to feature interaction and has the risk of attaining local minima (Wang et al., 2016). Examples: Floating Forward or Backward, Linear Forward Search, Beam Search, Greedy Forward Selection, and Backward Elimination.
• Exponential: It is a full-scale search; it guarantees an optimal solution but proves to be expensive. This approach finds all possible feature subsets to choose an optimal subset, which is computationally upscale, especially in high-dimensional datasets such as the Gene Expression Microarray dataset. Some of the examples for Exponential Search are, Exhaustive Search and Branch-and-bound.
• Heuristic Search: It is performed based on a cost measure or a heuristic function, which iteratively improves the solution. Heuristic Search does not always ensure an optimal solution, but it offers an acceptable solution with reasonable time, cost, and memory space (Ruiz et al., 2005). Some examples of Heuristic Search are Best-First Search, Depth-First Search, A∗ Search, Breadth-First Search, and Lowest-Cost-First Search (Russell and Norvig, 2016).
Evaluation Criteria
There are currently four types of evaluation methods used widely; they are Filter, Wrapper, Embedded, and Hybrid. Hybrid and Embedded methods are the recent developments in Gene Selection.
(a) Filter Feature Selection Approach:
Filter helps in identifying the specific abilities of features depending on the inherent properties of the data. The best among the features are identified with relevance score and threshold criteria (Hancer et al., 2018). The features with a low relevance score will be eliminated.
The significant advantages of filter techniques are that they are not dependent on the classifiers, fast and straightforward in terms of computation, and scaled to the immensely dimensioned dataset (Ang et al., 2015b). The common disadvantage is that they consider the data’s univariate features, which means the features are processed individually (Saeys et al., 2007). As a result, there are high chances of ignoring the feature dependencies, which leads to the classifiers’ poor performance compared to other feature selection approaches. Many multivariate filter techniques are introduced to avoid this to some extent (Brumpton and Ferreira, 2016; Djellali et al., 2017; Zhou et al., 2017; Rouhi and Nezamabadi-pour, 2018).
The examples for filter techniques are Pearson Correlation, Fisher Score, Model-based Ranking, and Mutual Information (Lazar et al., 2012) were done in a detailed survey on the filter techniques applied to Gene Expression Microarray data. Figure 3 is the representation of the process involved in the filter approach in gene selection.
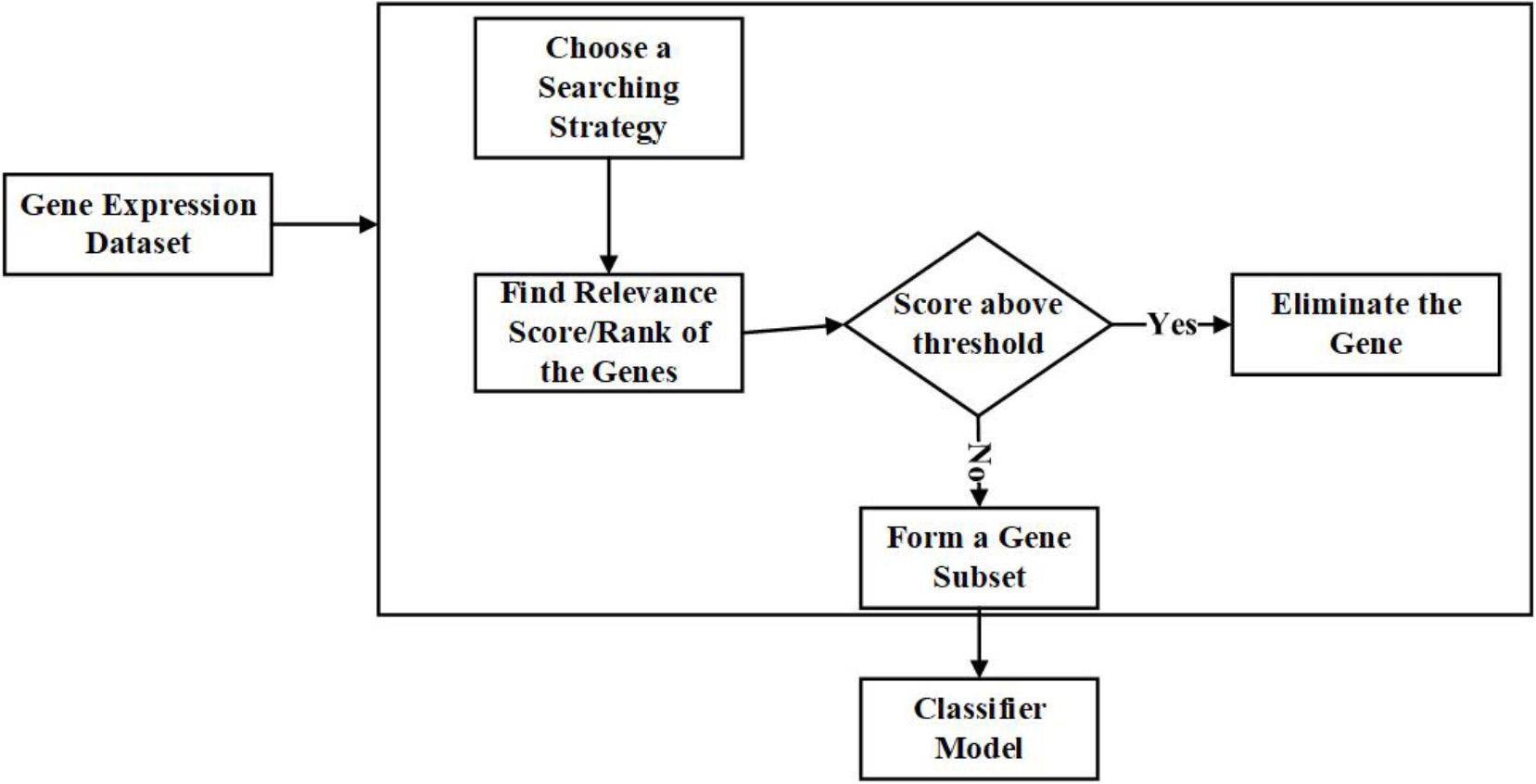
Figure 3. Flow diagram – Filter Feature Selection Approach.
(b) Wrapper Feature Selection Approach:
Unlike the filter approaches, the wrapper approaches wrap the feature subset selection process around the black box’s induction algorithm. Once the search procedure for a feature subspace is defined, various feature subsets will be generated, and the classification algorithm is used to evaluate the selected feature subsets (Blanco et al., 2004). With this approach, it is possible to select features tailored for the induction algorithm (Jadhav et al., 2018). The classification algorithm’s evaluation measures will be optimized while eliminating the features, hence offering better accuracy than the filter approach (Inza et al., 2004; Mohamed et al., 2016).
The significant advantage of using a wrapper approach, as both feature subset generation and the induction algorithm are wrapped together; the model will have the ability to track the feature dependencies (Rodrigues et al., 2014). The common drawback is that it becomes computationally intensive for datasets with high dimensions (Mohamed et al., 2016). Examples of Wrapper techniques are Hill Climbing, Forward Selection, and Backward Elimination. Figure 4 is the representation of the process involved in the wrapper approach.
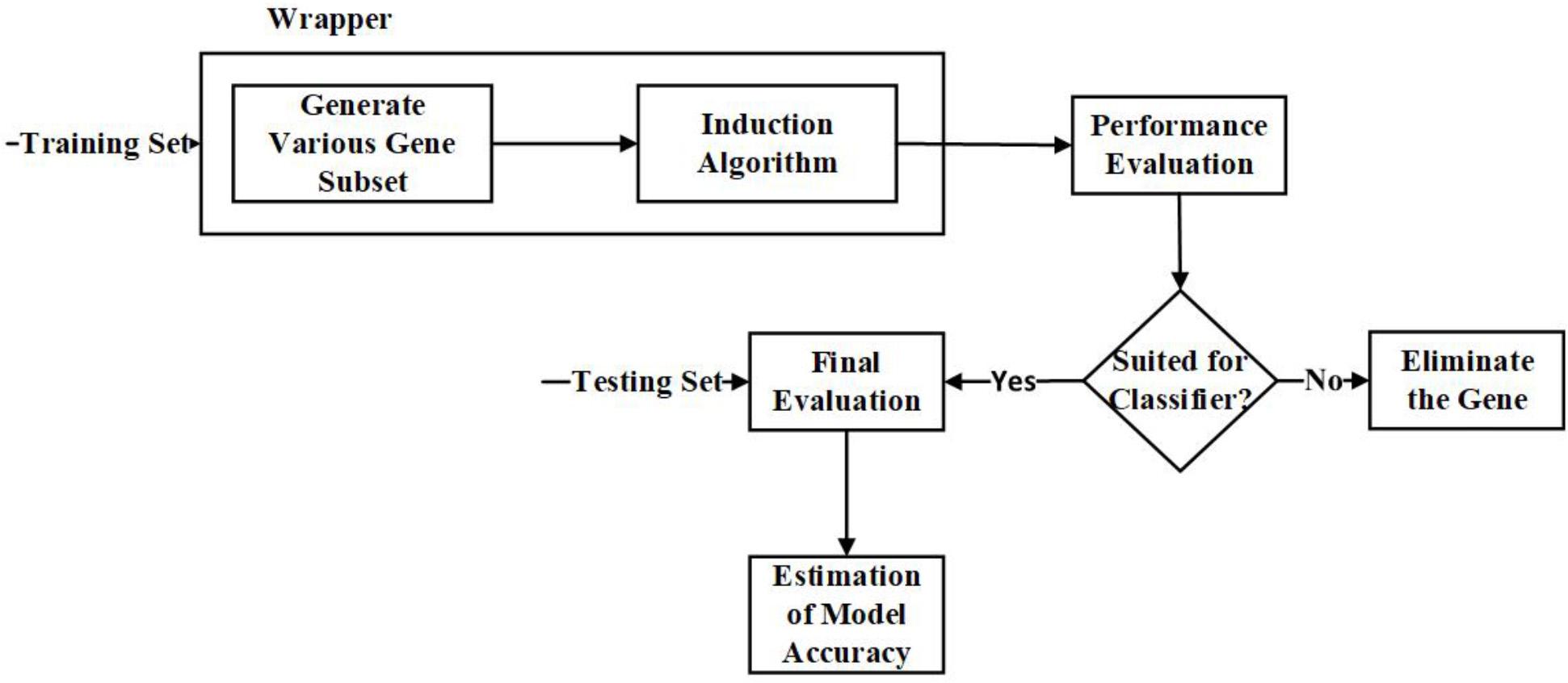
Figure 4. Flow diagram – Wrapper Feature Selection Approach.
(c) Embedded Feature Selection Approach:
In a way, embedded approaches resemble the wrapper approaches, as both depend on the learning algorithm (Hernandez et al., 2007). However, the embedded methods are less computationally intensive than the wrapper methods. The link between the learning algorithm and the feature selection is more robust in embedded methods than the wrapper methods (Huerta et al., 2010). In the embedded methods, the feature selection is made as a part of the classification algorithm; in other terms, the algorithm will have its built-in approaches to select the essential features (Hira and Gillies, 2015).
In the literature, it is mentioned that embedded methods combine the benefits of filter and wrapper methods to improve accuracy. The significant difference between other gene selection approaches and embedded approaches is how the genes are selected and the interaction with the learning algorithm (Chandrashekar and Sahin, 2014; Vanjimalar et al., 2018). Some examples of embedded approaches are ID3, RF, CART, LASSO, L1 Regression, and C4.5. Figure 5 is the representation of the process involved in the embedded approach.
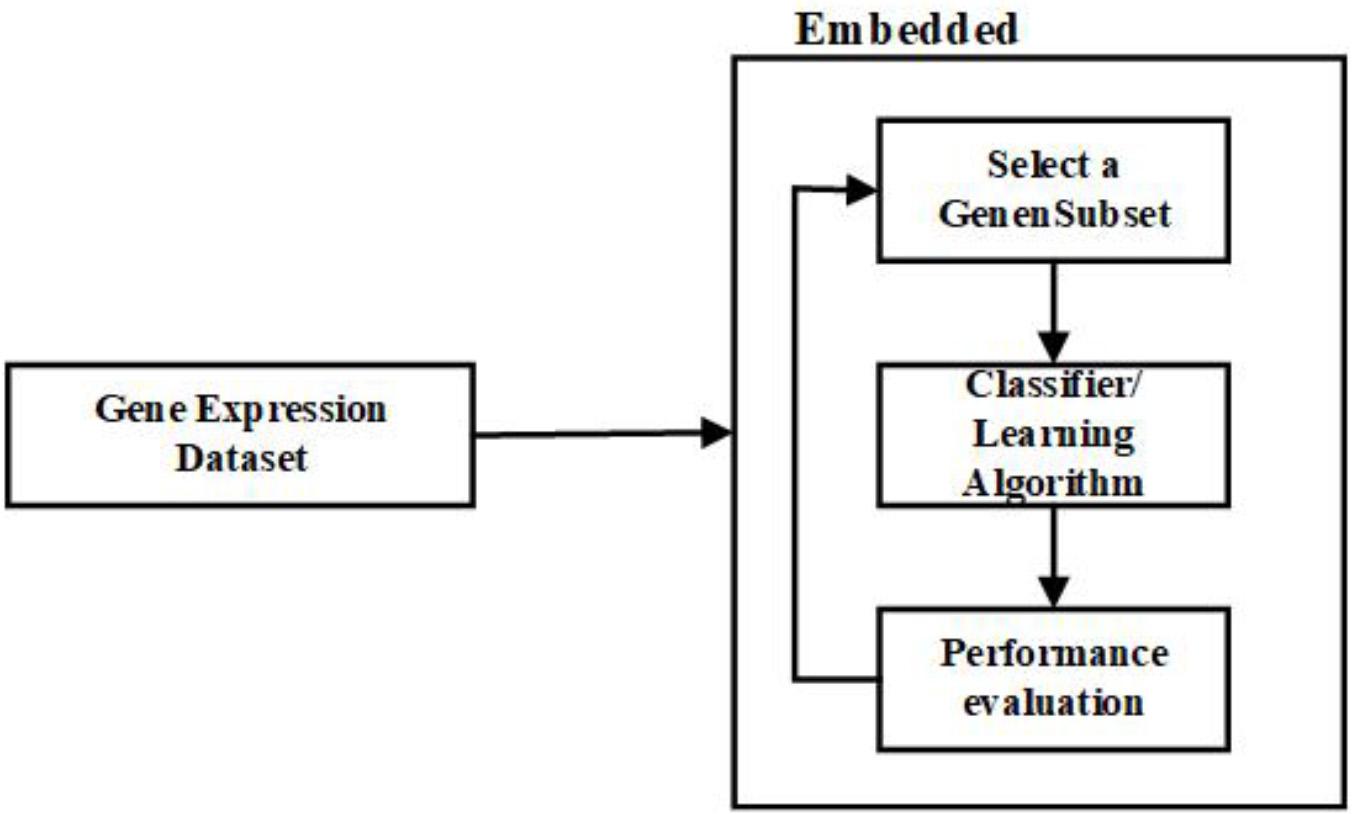
Figure 5. Flow diagram – Embedded Feature Selection Approach.
(d) Hybrid Feature Selection Approach:
Hybrid methods, as the name suggests, is a combination of two different techniques. Here, it can be two different feature selection approaches or different methods with similar criterion or two different strategies. In most cases, the filter and wrapper approaches are combined to form a hybrid approach (Apolloni et al., 2016; Liu et al., 2019). It strives to utilize the benefits of two methods by combining their compatible strengths. Hybrid methods offer better accuracy and computational complexity than the filter and wrapper methods. Also, it is less susceptible to overfitting (Almugren and Alshamlan, 2019). Figure 6 is the representation of the process involved in the hybrid approach.
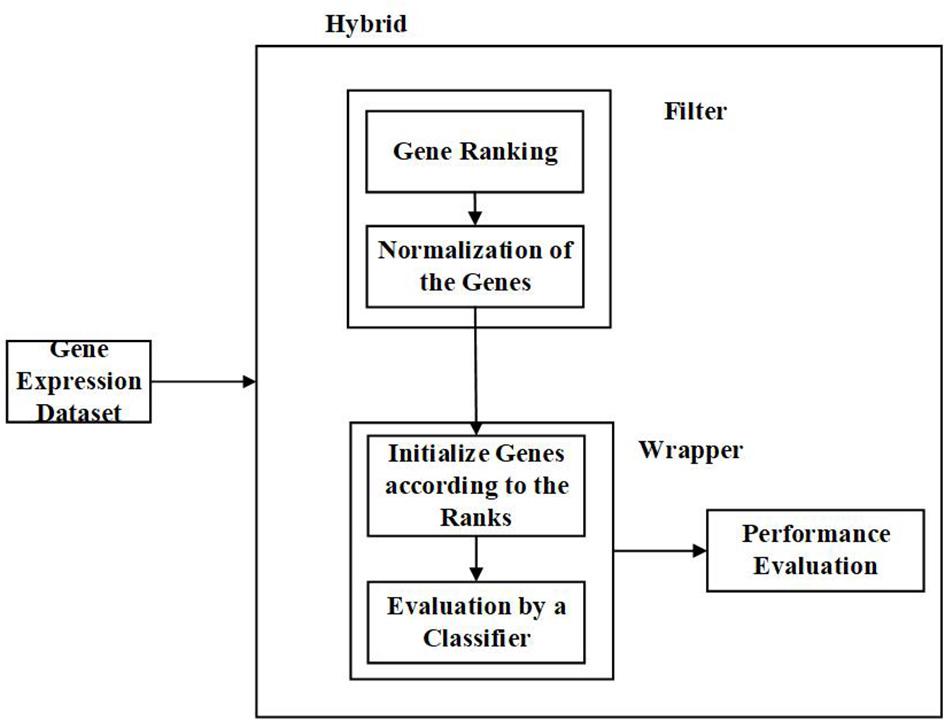
Figure 6. Flow diagram – Hybrid Feature Selection Approach.
Stopping Criteria
The stopping criteria are a kind of threshold used to inform the classifier when to stop selecting the features (Wang et al., 2005). Appropriate stopping criteria will refrain a model from overfitting, thus offer better results, which are computationally cost-effective (Ang et al., 2015b). Some of the commonly used stopping criteria are as follows:
(1) When the search reaches a specific bound, the bound can be several iterations or many features.
(2) The results do not improve with a deletion (or addition) of another feature.
(3) An optimal subset is found. A subset is said to optimal when the classifier’s error rate is less than the preferred threshold.
Evaluating the Results
There are many performance evaluation metrics available in the literature to evaluate and validate the classifier results. In the classification case, i.e., predicting using the categorical attribute, the commonly used error estimation methods are Confusion Matrix, Cross-Validation, and Receiver Optimizer Characteristics (ROC). In the case of regression, i.e., predicting using the continuous attribute, the commonly used error estimation methods are Mean Absolute Error (MAE), Mean Squared Error (MSE), and Coefficient of Determination (R2).
(a) Confusion Matrix: In the case of Multi-class problems, a confusion matrix is the best option to evaluate the classification model (Handelman et al., 2019). For instance, there are four possible results in a binary classification problem with which the model can be evaluated, True Positive, classified correctly, False Positive, erroneous classification, False Negative, erroneously rejected, and True Negative rejected correctly (Braga-Neto et al., 2004). Confusion Matrix offers measures such as Accuracy, Precision, Sensitivity, Specificity, and FMeasure to validate the results of a classifier.
(b) Cross-Validation (CV): It is the process of partitioning the available data into k-sets. Here, k can be any integer depending on the number of folds one needs for the classification or regression task (for instance, k = 10, k = 20, etc.) (Schaffer, 1993; Braga-Neto et al., 2004). CV is most commonly used on the Regression and Classification approaches (Chandrashekar and Sahin, 2014). The main advantage of using CV is that it offers unbiased error estimation, although sometimes it is variable (Bergmeir and Benítez, 2012).
(c) Receiver Optimization Characteristics (ROC): ROC graphs and curves are commonly used for visualizing the performance of the classifiers and select the one showing better performance (Landgrebe and Duin, 2008). As the researches these days are increasingly concentrated on the classification errors and unbalanced class distribution, ROC has gained a lot of attention (Flach, 2016). It is the depiction of the trade-offs between the Sensitivity or benefits (TPR) and the Specificity or costs (FPR) (Fawcett, 2006).
(d) Root Mean Square Error (RMSE): RMSE is a metric commonly used to measure the residuals’ standard deviation or prediction scores. In other words, the deviation in predictions from the regression line. It is given by Elavarasan et al. (2018),
Where, xi – Actual or Observed Values.
– Predicted Values.
n – Total number of sample.
(e) Mean Absolute Error: It is the standard measure of the residuals’ average magnitude (prediction errors), neglecting their directions. It is given by Elavarasan et al. (2018).
Where, xi – Actual or Observed Values,
– Predicted Values.
n – Total number of sample.
(f) Determination Coefficient (R2): It is the measure to estimate how much one variable impacts other variables. It is the change in the percentage of one variable concerning the other. It is given by Elavarasan et al. (2018).
Where, x – first set of values data,
y– the second set of values in the data.
R – Coefficient of determination.
n – Total number of sample.
Machine Learning Based Gene Selection Approaches
Supervised Gene Selection
Supervised Gene Selection involves the data with labeled attributes. Most of the studies done in recent years have concentrated mainly on enhancing and improving the existing supervised gene selection methods.
For instance, Devi Arockia Vanitha et al. (2016) enhanced the Mutual Information (MI) filter method for selecting the informative gene. Also, Joe’s Normalized Mutual Information, an improved version of the standard existing MI approach, was implemented by Maldonado and López (2018). Filter approaches are independent of the classifiers used. Hence, many works are focused on developing filter technologies. For instance, a novel filter approach is mainly based on the Hilbert-Schmidt Independence Criterion (SHS) and motivate by Singular Value Decomposition (SVD). Table 2 shows some of the filter-based gene selection techniques used in the literature to select informative genes.
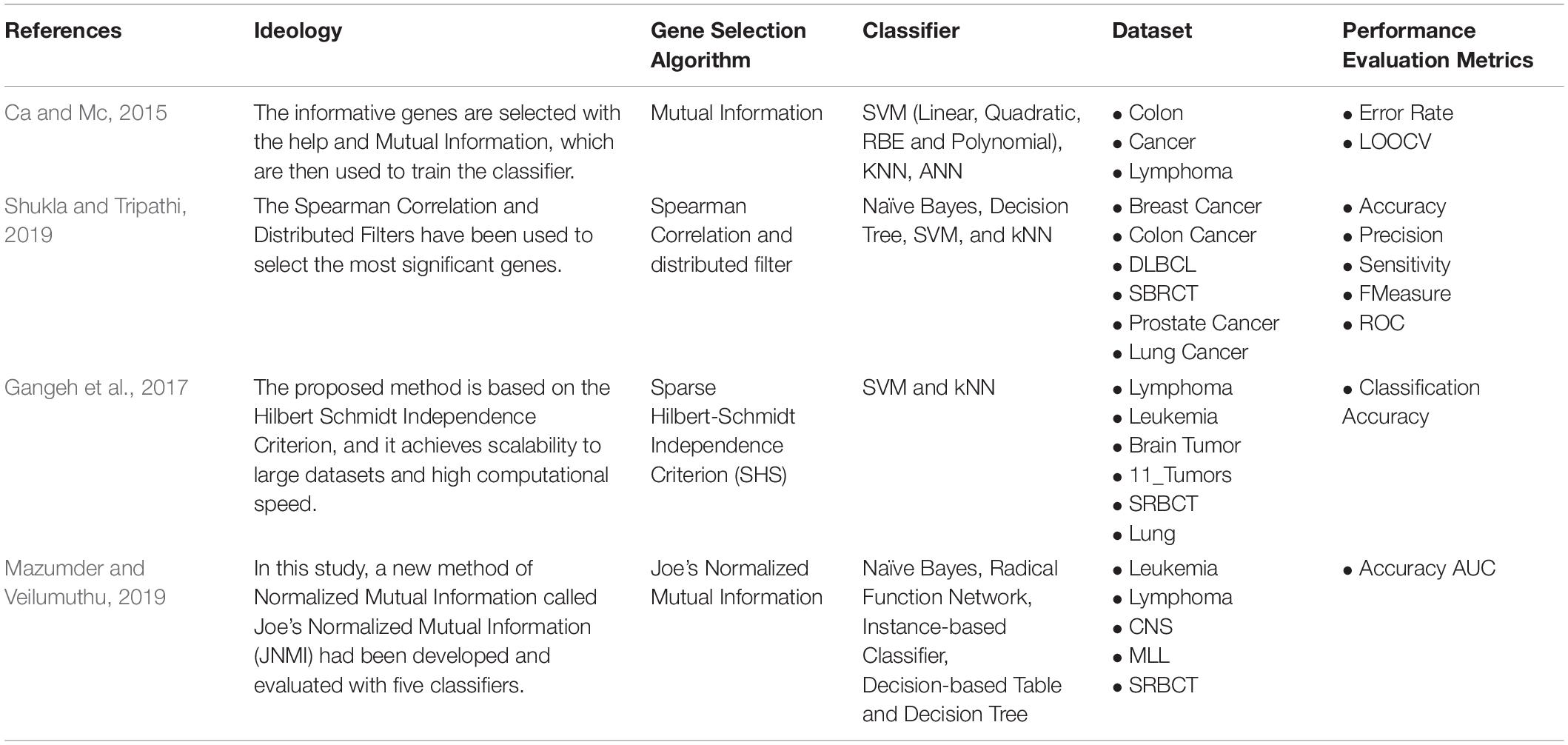
Table 2. Filter-based Supervised Gene Selection.
The wrapper approach is computationally intensive than other feature selection approaches. Works on the wrapper feature selection approach are less because of the issue mentioned above. So, most of the research on the wrapper is focused on improving the computational cost. For instance, Wang A. et al. (2017), Wang H. et al. (2017) implemented a wrapper-based gene selection with Markov Blanket, which reduces the computation time. Many approaches try to enhance the most widely used Support Vector Machine – Recursive Feature Elimination (SVM-RFE), such as Shukla et al. (2018), implemented Support Vector Machine – Bayesian t-test – Recursive Feature Elimination (SVM-BT-RFE), where Bayesian t-test is combined with SVM-RFE to improve the results. Table 3 shows the works done in recent years on Wrapper-based Supervised Gene Selection.
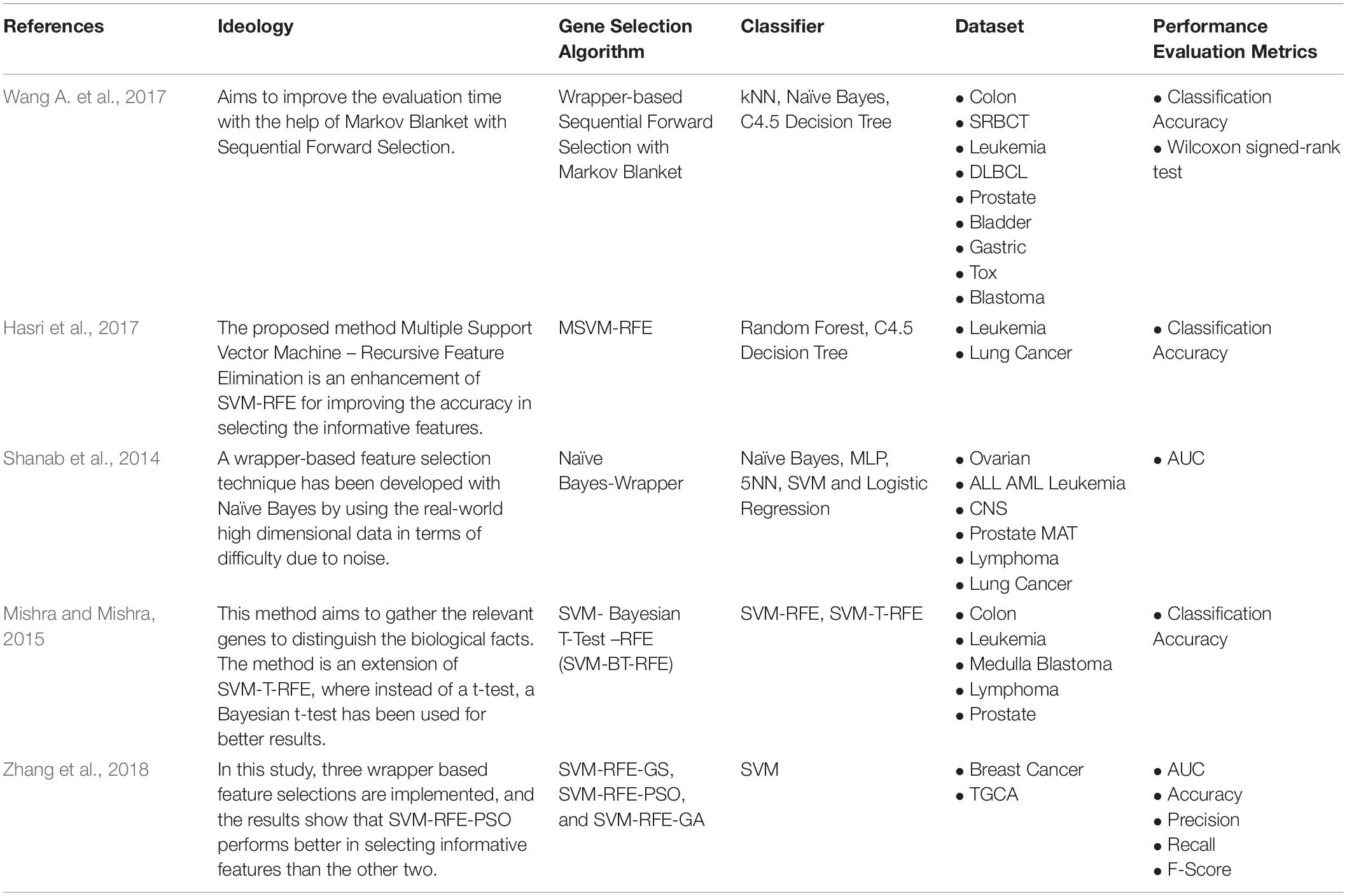
Table 3. Wrapper-based Supervised Gene Selection.
Hybrid Feature Selection is usually the combination of other approaches, mostly filter and wrapper approaches are made into hybrids. For instance, Liao et al. (2014), implemented a filter-wrapper based hybrid approach utilizing the Laplacian score and Sequential Forward and Backward Selection. Also, various works are going on in combining the nature-inspired algorithm. For example, Alshamlan et al. (2015), implemented a Genetic Bee Colony, combining the Genetic Algorithm and Artificial Bee Colony for gene selection. A hybrid of the Salp Swarm Algorithm (SSA) and multi-objective spotted hyena optimizer are implemented in Sharma and Rani (2019). The SSA focuses on diversity, and MOSHO concentrates on convergence. Table 4 consists of the recent works done on Hybrid-based Supervised Gene Selection approaches.
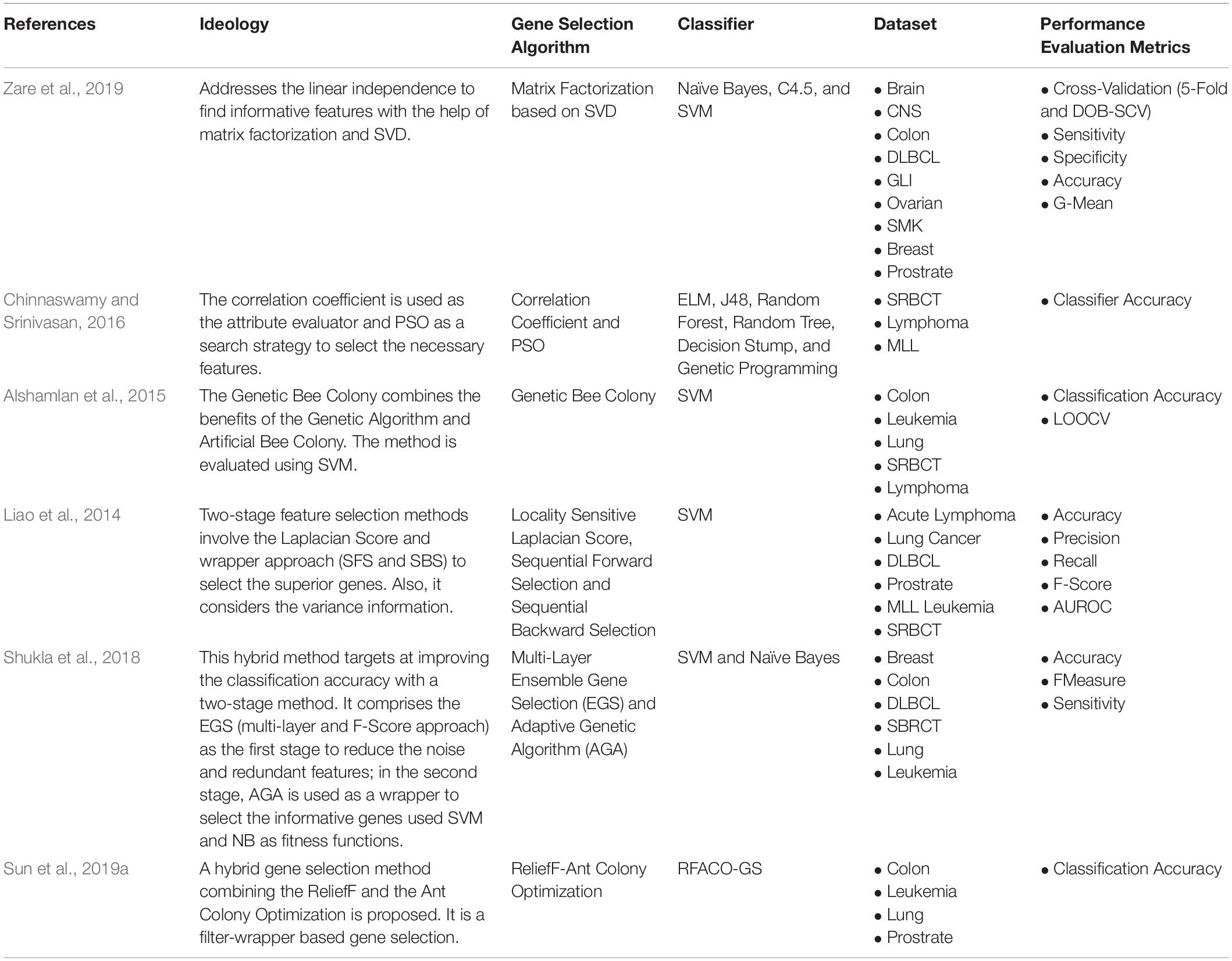
Table 4. Hybrid Supervised Gene Selection.
Ensemble Feature Selection is a combination of the outputs from different expert feature selection approaches. Ghosh et al. (2019a; 2019b), combines the outputs of ReliefF, Chi-square, and Symmetrical Uncertainty (SU) with Union and Intersection of top “n” features. Seijo-Pardo et al. (2016), used a ranking aggregation method to various aggregate ranks from Chi-square, InfoGain, mRmR, and ReliefF. Table 5 shows the different Ensemble-based Supervised Gene Selection approaches used in recent years.
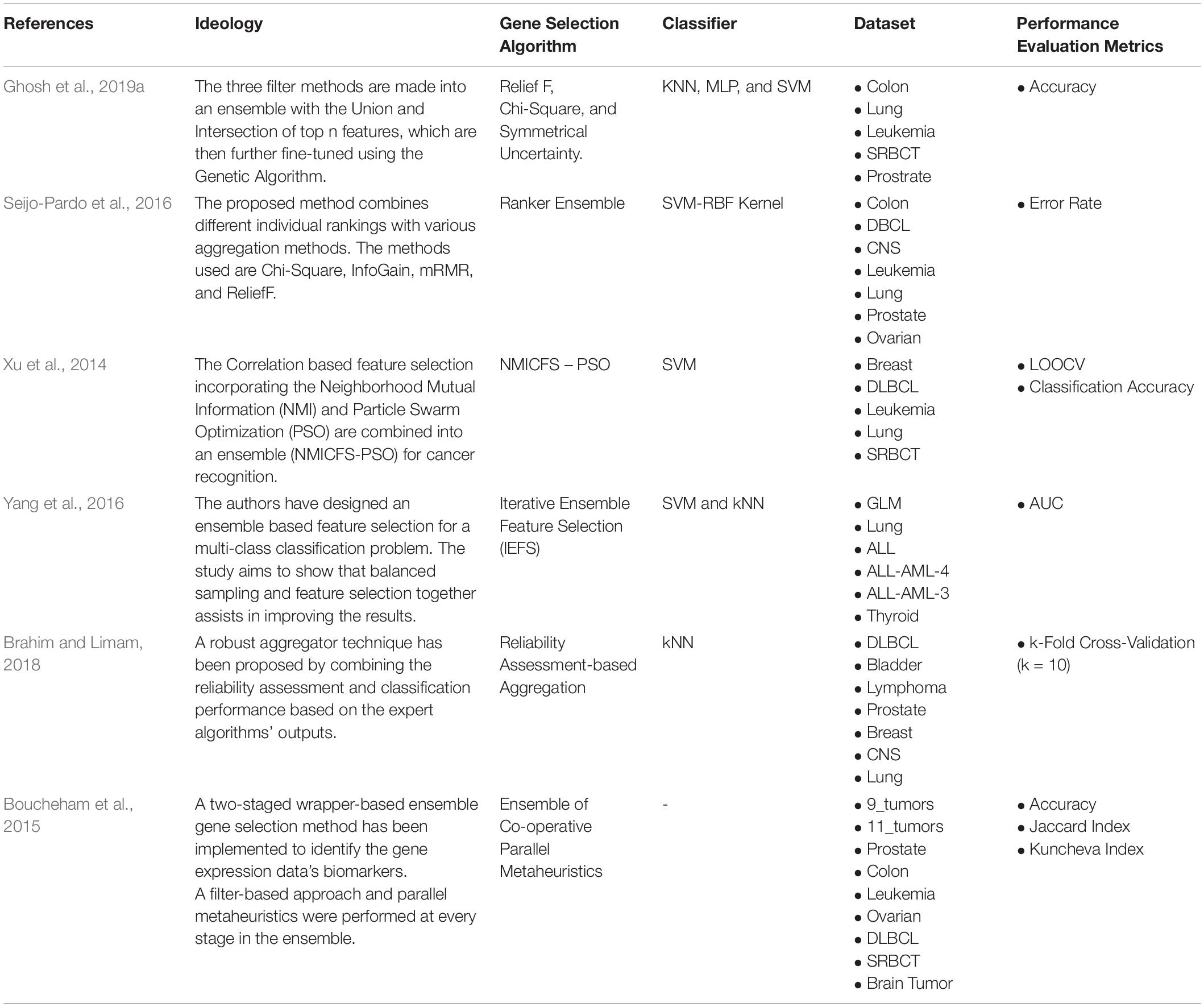
Table 5. Ensemble-based Supervised Gene Selection.
Embedded methods merge the benefits of filter and wrapper methods, where the learning algorithm has a built-in feature selection approach. Ghosh et al. (2019b), implemented a Recursive Memetic Algorithm (RMA) with a wrapper-based approach embedded in it. Also, Guo et al. (2017), used L1 Regularization, along with a feature extraction method for selecting the informative genes. Table 6 shows the various Embedded-based Supervised Gene Selection approaches developed in recent years.
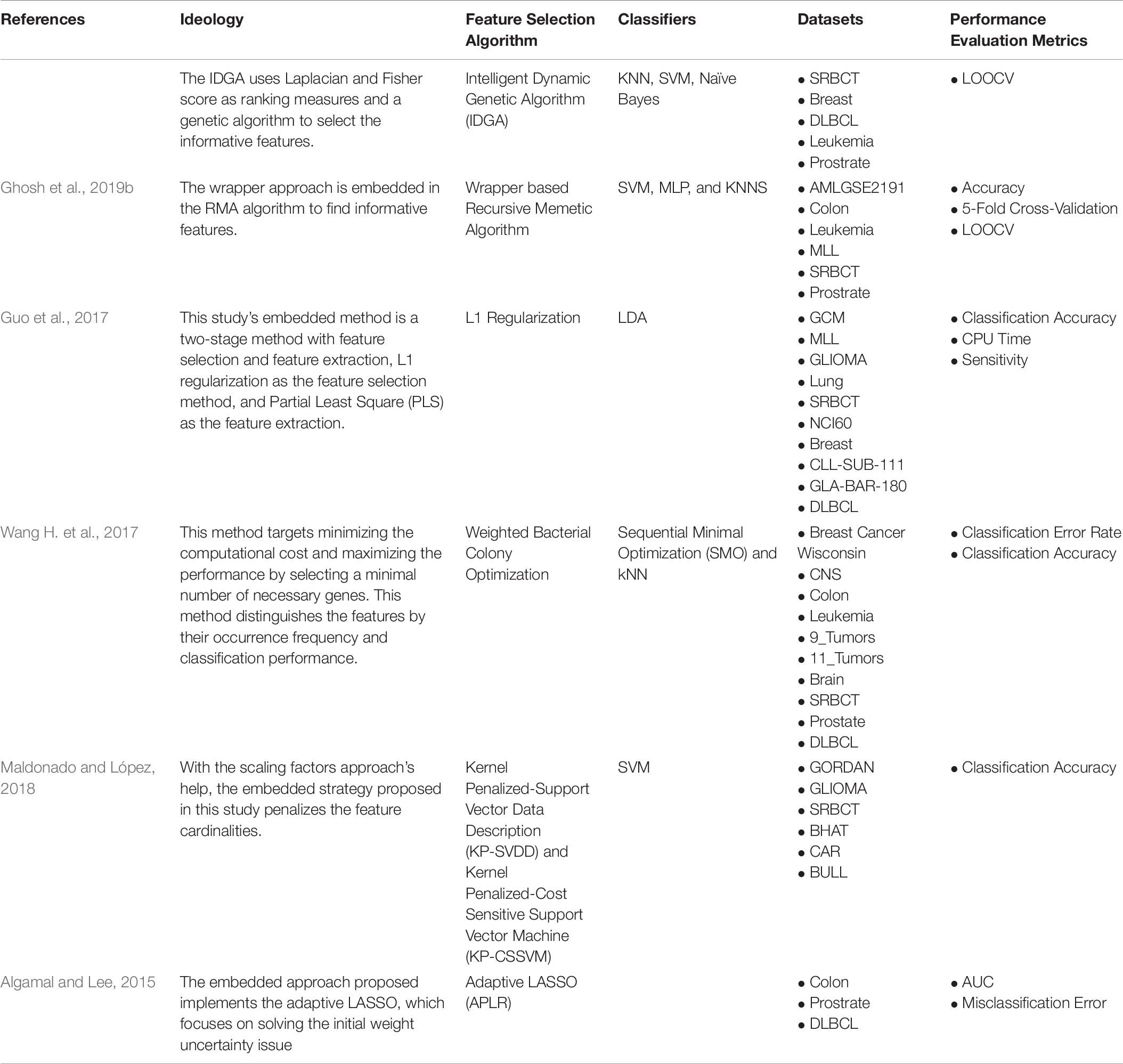
Table 6. Embedded-based Supervised Gene Selection.
Unsupervised Gene Selection
Unsupervised Gene Selection involves data without any labels. Compared to Supervised Gene Selection, works on Unsupervised are less.
There are many novel works done on filter-based unsupervised gene selection, such as Solorio-Fernández et al. (2017), proposed a filter method for both non-numerical and numerical data. It is a combination of kernel approach and spectrum-based feature evaluation. Also, Liu et al. (2018), developed a Deep Sparse Filtering model considering the deep structures, enhancing the results. Many studies on nature-inspired gene selection and the (Guo et al., 2017) implemented the MGSACO to minimize redundancy, thereby increasing the dataset’s relevancy. One another issue with high-dimensional data is dependency maximization. The work in Boucheham et al. (2015) implemented the Hilbert-Schmidt Independence Criterion to eliminate the most dependent genes to handle dependency maximization. Table 7 is the collection of works done in recent years on Filter-based Unsupervised Gene Selection approaches.
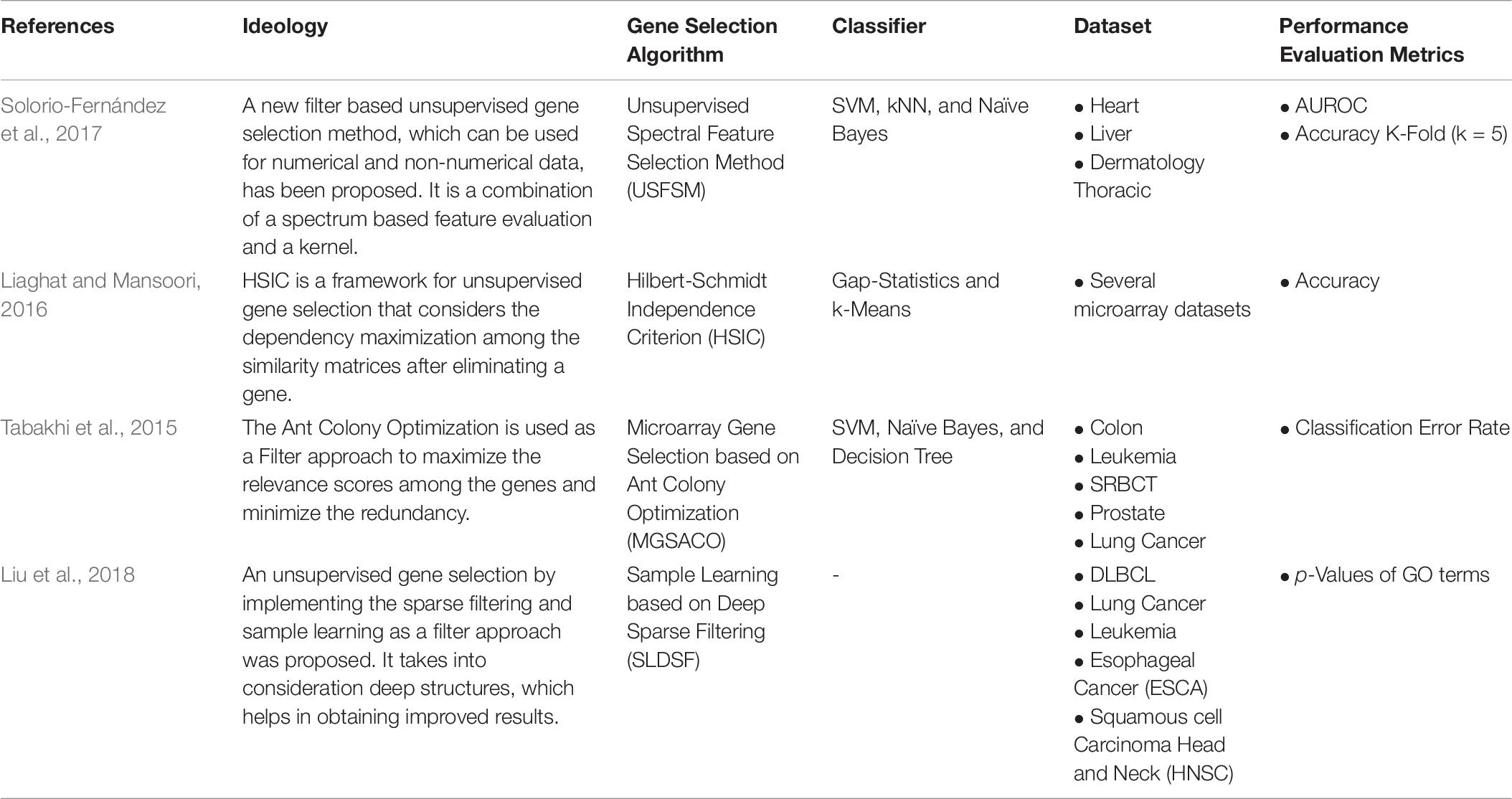
Table 7. Filter-based Unsupervised Gene Selection.
Filter-based gene selection approaches are not dependent on the learning model; on the contrary, wrapper methods are entirely dependent on the learning model. The dependency makes it complicated and has a high computational cost. Hence, the study on wrapper methods is less concentrated. Same with the unsupervised wrapper gene selection, which is less focused. Xu et al. (2017), has implemented SVM-RFE, a wrapper-based gene selection, on unlabeled data to distinguish high-risk and low-risk cancer patients. Table 8 is an example of a wrapper-based Unsupervised Gene Selection approach.

Table 8. Wrapper-based Unsupervised Gene Selection.
Hybrid Unsupervised gene selection is also focused on in the literature as much as the filter approach. Li and Wang (2016), developed a two-stage gene selection approach; it applies the matrix factorization and minimum loss principle. A coarse-fine hybrid gene selection on unlabelled data shows better results than a few other approaches compared to the study. Filter-wrapper hybrid approaches are equally focused on supervised as well as unsupervised gene selection. For instance, Solorio-Fernández et al. (2017), implemented a Laplacian Score Ranking, a filter approach, and Normalised Calinski-Harabasz (LS-WNCH), a wrapper approach as hybrid unsupervised gene selection. It includes the properties of spectral feature selection. Table 9 shows the hybrid-based Unsupervised Gene Selection approaches.
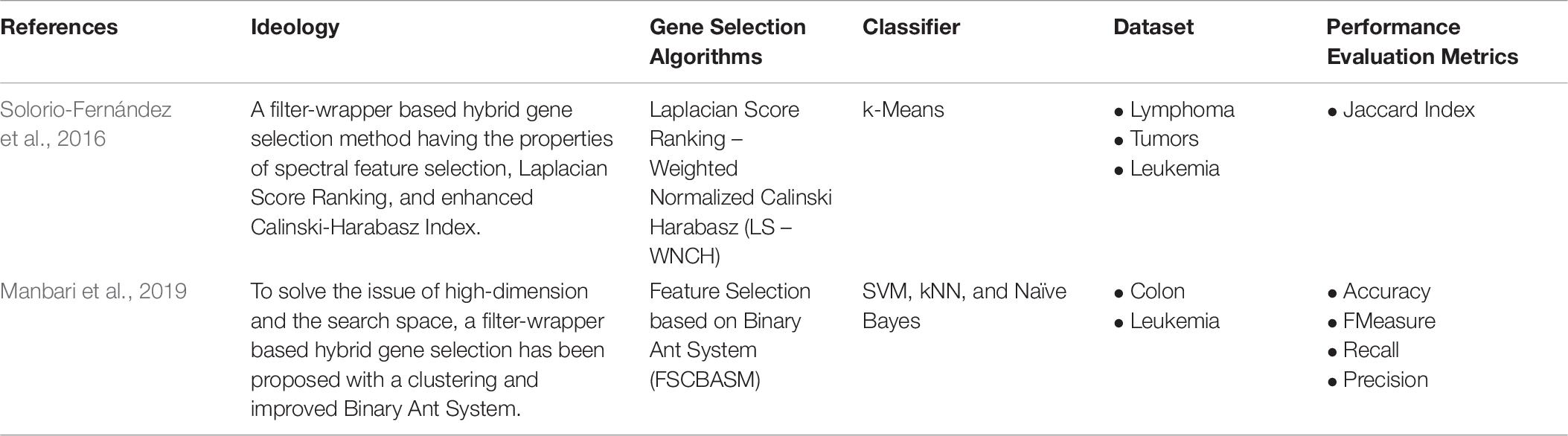
Table 9. Hybrid Unsupervised Gene Selection.
Ensemble and embedded approaches are studied less than the filter and hybrid methods. Elghazel and Aussem (2013), implemented a Random Cluster Ensemble with k-means as the clustering model. The ECE was constructed with different bootstrap samples at every ensemble partitions. They have also calculated out-of-bag feature importance at every ensemble. Li et al. (2017), developed a Reconstruction-based unsupervised feature selection model, an embedded approach. The model has a filter-based approach embedded in the k-means clustering. Table 10 is the example for Ensemble-based, and Embedded-based Unsupervised Gene Selection approaches.
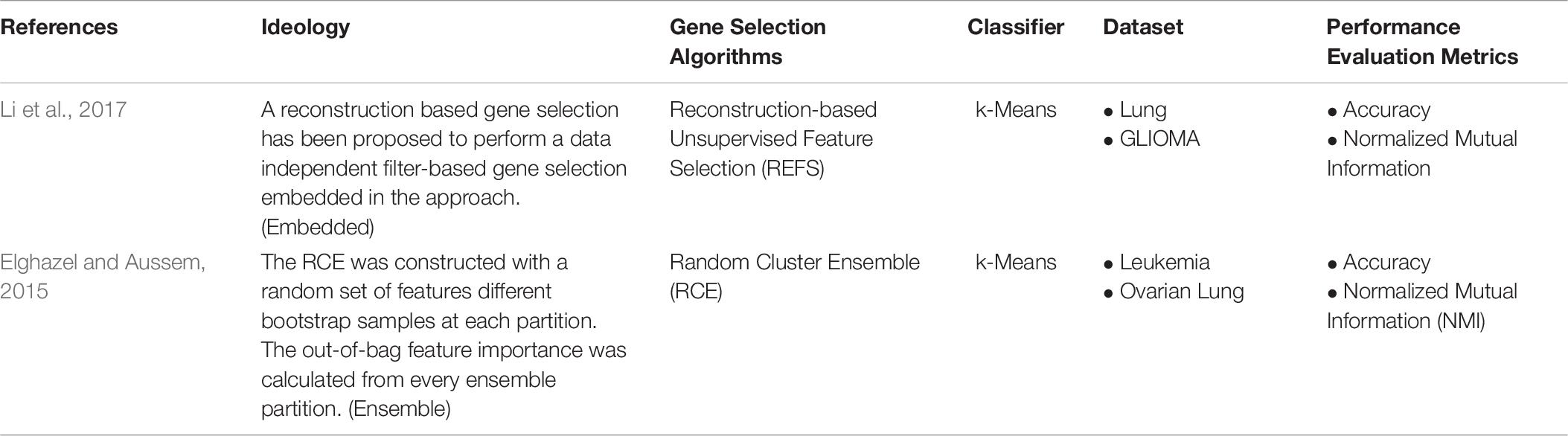
Table 10. Ensemble and embedded Unsupervised Gene Selection.
Semi-Supervised Gene Selection
Semi-supervised gene selection is yet to be explored research area. There are not many works done as much as supervised or unsupervised gene selection. Semi-Supervised or Semi-Unsupervised consists of both labeled and unlabelled data.
Li et al. (2018), combined the benefits of the spectral graph and Mutual Information to develop a Semi-Supervised Maximum Discriminative Local Margin (SemiMM). It takes care of variance, local structure, and MI all at the same time. SVM is used widely in supervised and unsupervised gene selection approaches; in semi-supervised, Ang et al. (2015b), implemented a semi-supervised SVM-RFE (S3VM) for selecting the informative genes, and it proves to be successful. Chakraborty and Maulik (2014), developed a hybrid model; Kernalised Fuzzy Rough Set (KFRS) and S3VM are combined to select the relevant features. The results show that the proposed algorithm is capable of choosing useful biomarkers from the dataset. A semi-supervised embedded approach, Joint Semi-Supervised Feature Selection (JSFS), was developed with a Bayesian approach. The model automatically chooses the informative features and also trains the classifier.
Rajeswari and Gunasekaran (2015), developed an ensemble-based semi-supervised gene selection to improve the quality of the cluster model. Modified Double Selection based Semi-Supervised Cluster Ensemble (MDSVM-SSCE) assists in selecting the most relevant genes. Table 11 shows the Semi-Supervised Gene Selection approaches developed in recent years.
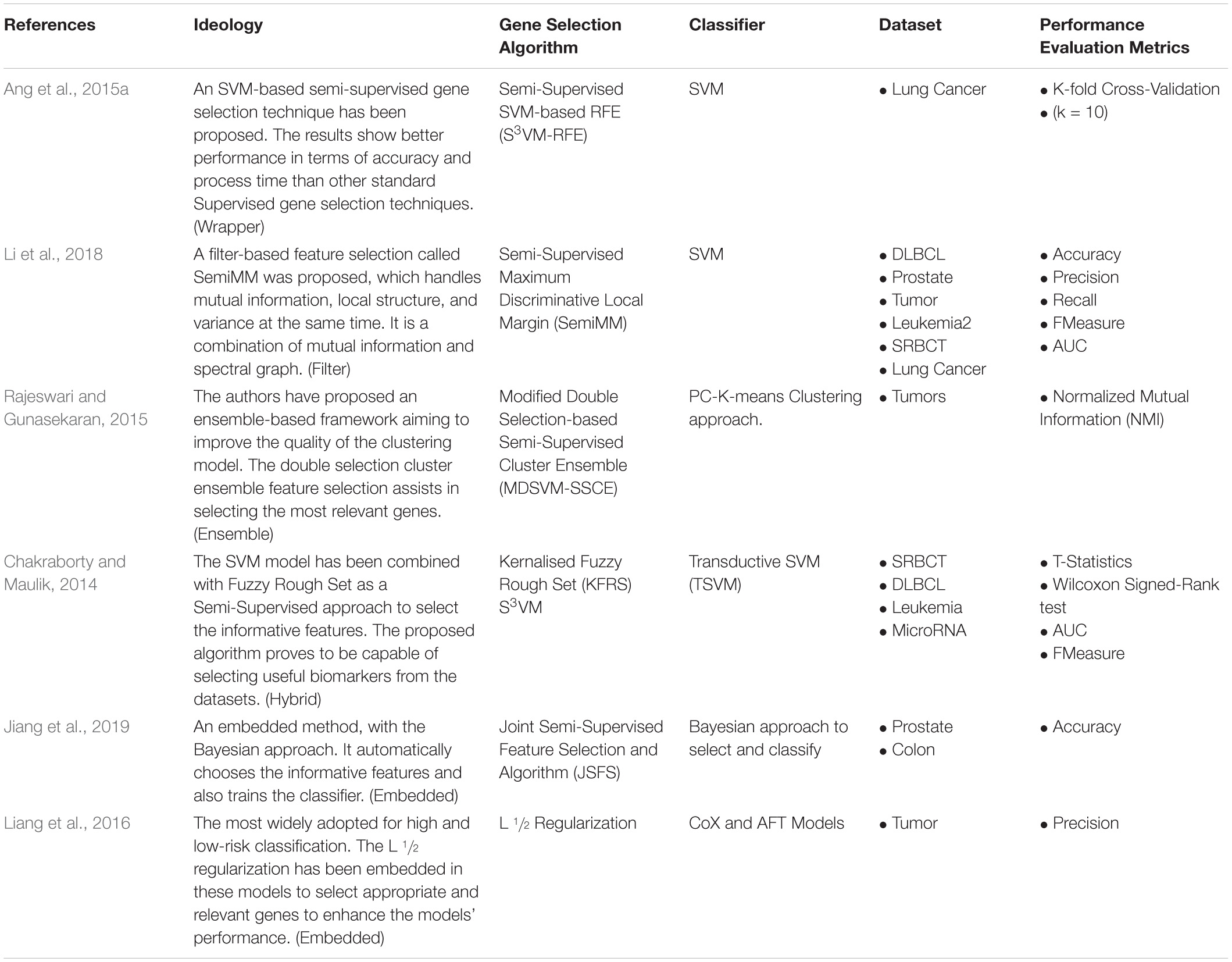
Table 11. Semi-Supervised Gene Selection approaches.
Performance Analysis and Discussion on the Reviewed Literature
In the literature, the top three datasets used widely are Prostate, Leukemia, and Colon. Tables 12–14 shows the respective proposed models’ performance on the datasets mentioned above, along with the number of genes selected.
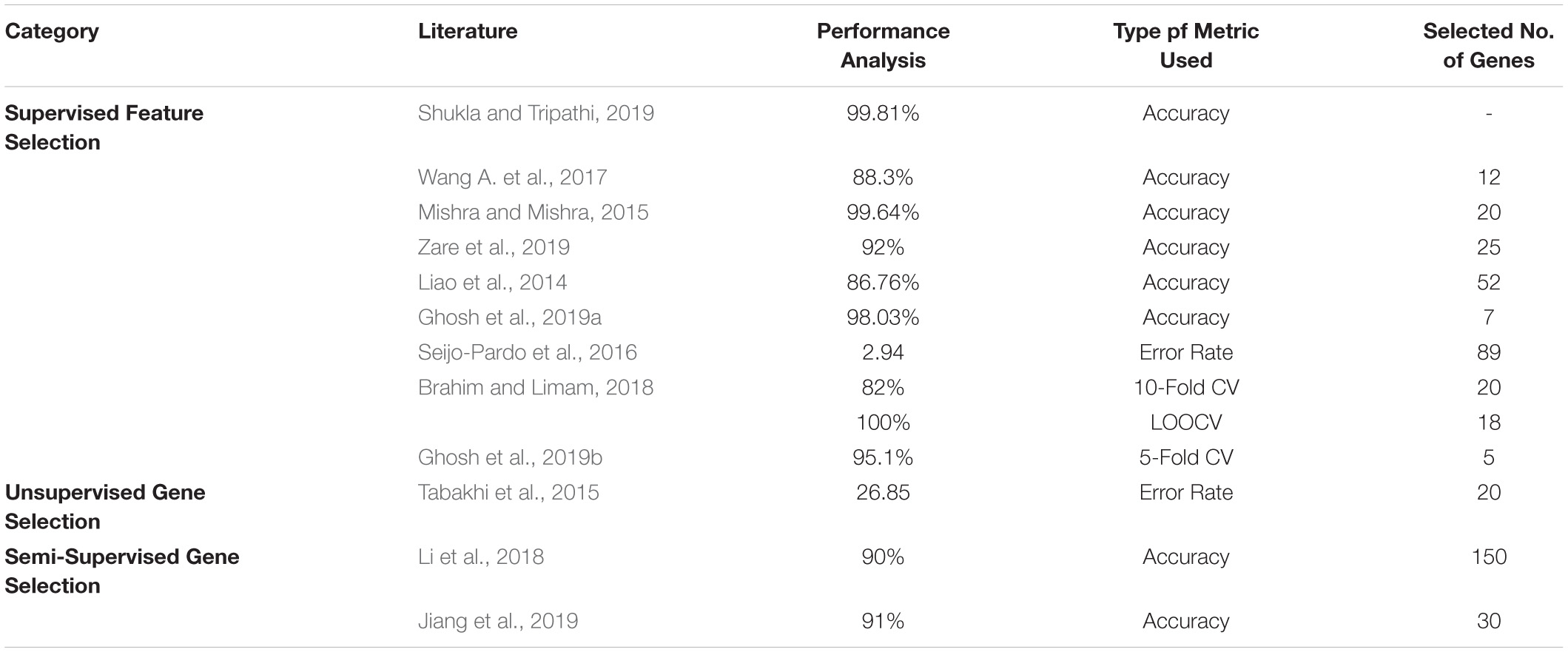
Table 12. Performance analysis of prostate dataset.
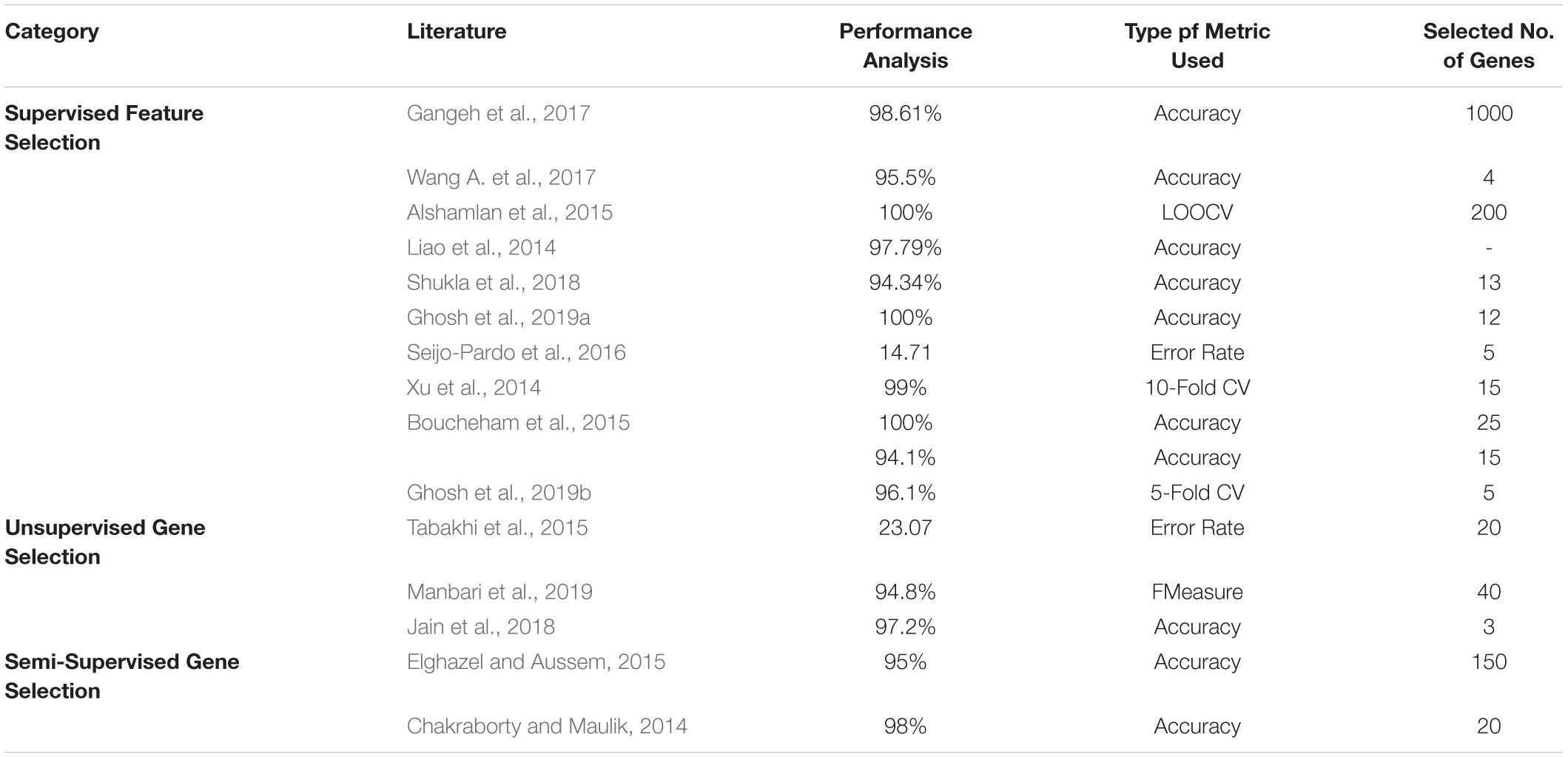
Table 13. Performance analysis on Leukemia dataset.
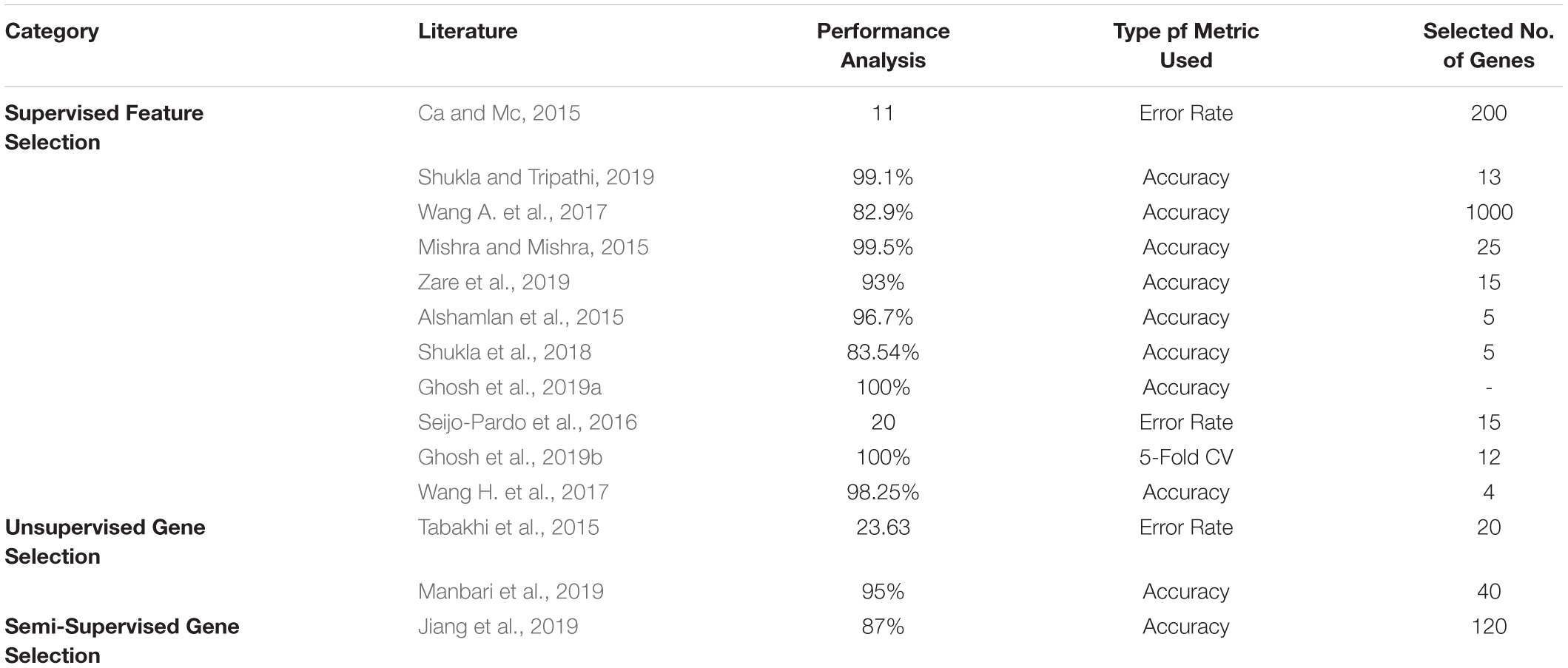
Table 14. Performance analysis of colon dataset.
All three gene selection methods discussed in this paper has its own merits and demerits. From the literature, it is clear that the Supervised Gene Selection is researched the most in recent years, and the Semi-supervised the least. Even though the Semi-Supervised potential is not tapped upon yet, it seems to be the better one among the three. It takes the advantages of Supervised and Unsupervised Gene Selection approaches. It has both labeled and unlabelled data; thus, it combines both the approaches’ benefits, eventually achieving better results. It considers the overlapping genes and handles it with the Unsupervised Gene Selection approach (unlabelled data) and learn and train the learning model with great accuracy and precision with the help of Supervised Gene Selection approaches (labeled data). Figures 7–10 show that the Supervised Gene Selection performs way better than the other two. Still, it might be because there are considerably significantly fewer works in Unsupervised and Semi-Supervised Gene Selection. The abbreviations for the acronyms used in the plot can be found in Table 15. There are several opportunities still untapped in these two areas. We can also notice that many works are concentrated more on Filter approaches as they are simple and computationally effective. However, hybrid approaches are upcoming and promising.

Figure 7. Performance analysis of Supervised Gene Selection Models – Part A.

Figure 8. Performance analysis of Supervised Gene Selection Models – Part B.
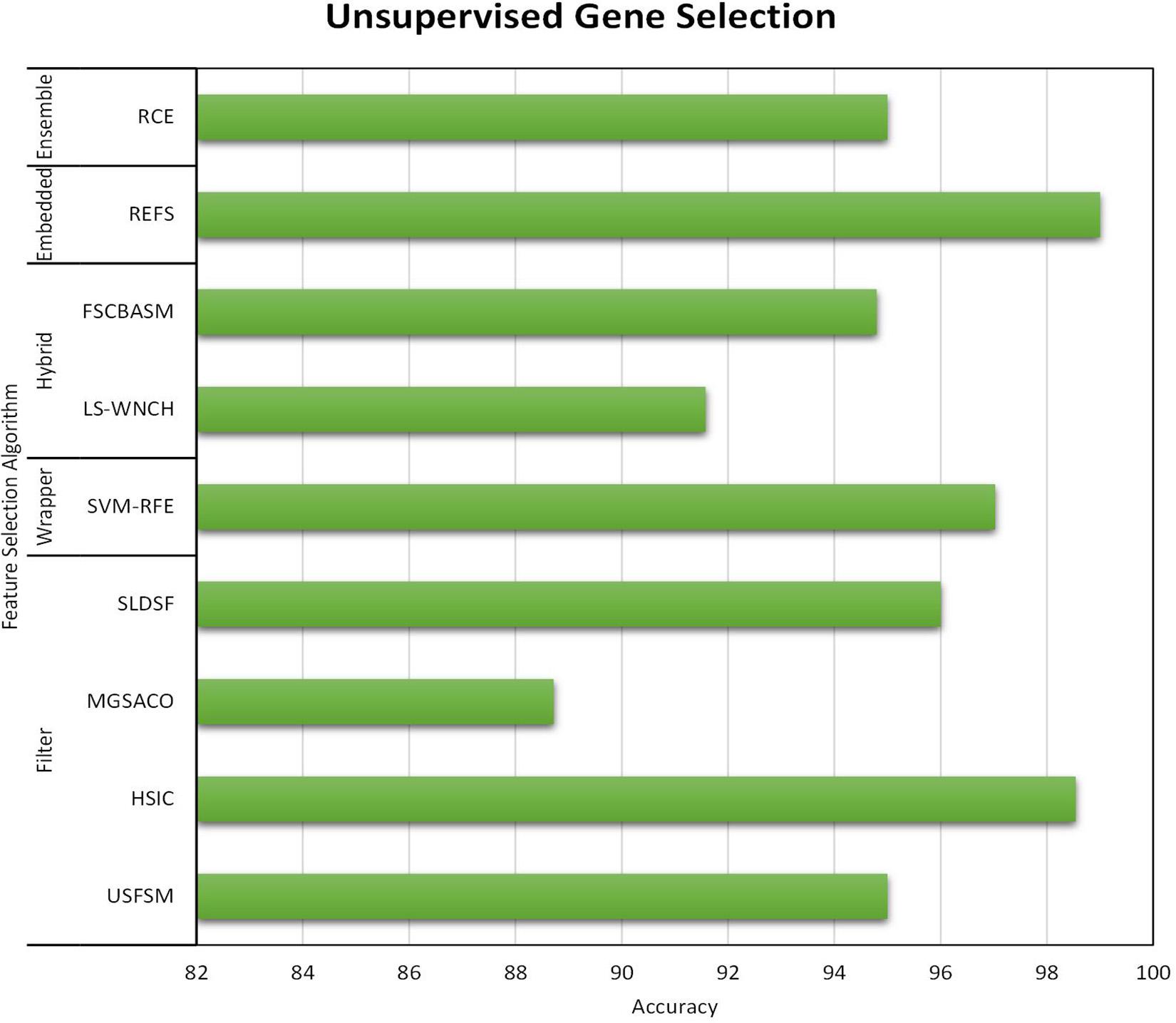
Figure 9. Performance analysis of Unsupervised Gene Selection Models.

Figure 10. Performance analysis of Semi-Supervised Gene Selection Models.

Table 15. Acronyms.
As for the evaluation criteria, in recent years, filter-based approaches are more focused much. Filter methods function independently of the learning model; thus, it is less computationally intensive. As it is less complicated, many researchers target the filter-based approaches in selecting informative genes. Wrapper-based approaches are the least concentrated upon; it is dependent and designed to support the learning model. Wrapper approaches are usually time-consuming and generate high computational overhead. Though other methods are concentrated equally, the hybrid approach proves to be better among the others. Hybrid is a combination of two or more approaches. The most commonly used hybrid method is the Filter-Wrapper combination. In the Hybrid approach, the limitations of the individual approaches are compensated; in other words, it inherits the benefits of two methods. Further, this will minimize computational cost. Hybrid approaches seem to provide better accuracy and reduce over-fitting risks. Apparently, hybrid methods are most suited for high-dimensional datasets such as the gene expression microarray from the literature.
Apart from the discussed literature, many other works focused on nature-inspired and meta-heuristic algorithms in diagnosing cancer. A bio-inspired algorithm is proposed by Dashtban et al. (2018) using the BAT algorithm with more refined and effective multi-objectives. Also, they have proposed a novel local search strategy. Another such BAT inspired algorithm with two-staged gene selection is proposed in Alomari et al. (2017), wherein the first stage is a filter (Minimum Redundancy and Maximum Relevance) and the second stage is the wrapper consisting of BAT and SVM. Other than that, considerable works are done in Particle Swarm Optimization (PSO) by improving and enhancing the existing algorithm. In Jain et al. (2018), the authors implemented a two-phased hybrid gene selection method, combining the improved PSO (iPSO) and Correlation-based Feature Selection (CFS). The proposed method controls the early convergence problem. A recursive PSO is implemented in Prasad et al. (2018); it tries to refine the feature space into more fine-grained. They have also combined existing filter-based feature selection methods with the recursive PSO. KNN and PSO are implemented in Kar et al. (2015) to handle the uncertainty involved in choosing the k-value in KNN. In Han et al. (2015), the authors proposed a Binary PSO (BPSO) to improve the interpretability of the gene selected and improve the prediction accuracy of the model. In Shreem et al. (2014), a nature-inspired algorithm Harmony Search Algorithm (HAS) is embedded with Markov Blanket, which focuses on symmetrical uncertainty Sharbaf et al. (2016) implemented an Ant Colony Optimization based gene selection (ACO) along with Cellular Learning Automata (CLA) as a wrapper method. In another approach (Lai et al., 2016), a hybrid combining filter and wrapper approaches is implemented using Information Gain (IG) and improved Swarm Optimization to find the optimal gene subset. Information Gain (IG) is also implemented along with SVM in Gao et al. (2017) to remove the redundant genes. There are works done in gene selection using the Genetic algorithms with different variations from the existing one. One such work combines the Genetic algorithm and Fuzzy in Nguyen et al. (2015), integrating the two approaches to finding out the optimal gene subset. Genetic Algorithm is also combined with learning automata (GALA) in Motieghader et al. (2017), which improves the time complexity in selecting the gene subset. Statistically, significant models are also implemented, such as the entropy-based measure and rough sets (Chen et al., 2017) and (Xiao et al., 2014; Sun et al., 2019c), testing the statistical significance with p-value and fold change. Decision tree and random forest variances are also worked on, such as the four-state-of art Random forest (Kursa, 2014), decision tree along with PSO (Chen et al., 2014), and a guided regularized Random Forest (Deng and Runger, 2013). Various works are focus on improving the interpretability of the features and reducing the feature space with improvements in the existing models (Zibakhsh and Abadeh, 2013; Cai et al., 2014; García and Sánchez, 2015; Chen et al., 2016; Tang et al., 2018; Cleofas-Sánchez et al., 2019).
Machine Learning techniques are widely used in modern-day research in the field of bioinformatics. The Machine Learning algorithms are available under different criteria, such as the logic-based algorithms (E.g., Decision Trees, Random Forest), perceptron-based algorithms (Neural Network, Multi-layered Perceptron), and Statistical Learning (Naïve Bayes) (Kotsiantis et al., 2007). The classification or prediction models used commonly in the literature discussed in this paper mostly include SVM, KNN, Random Forest, Decision Tree, Naïve Bayes, and Logistic Regression. SVM consists of support vectors that assist in classifying a disease or disorder. The classification depends on the formation of a hyperplane that divides binary classes. The SVM locates the hyperplane with the help of the kernel function. A most important advantage of using SVM is to tackle the outliers (Brown et al., 2000). KNN works on the assumption that the instances within a dataset will be close to one another. Although KNN is easy to understand and implement the algorithm, it lacks the fundamental principle in choosing the value of k. Also, it is sensitive to the distance or similarity function used. Decision Tree is made up of nodes and branches, used mainly because of their effectiveness and speed in calculations. Decision Trees are highly prone to overfitting and underfitting of the data (Czajkowski and Kretowski, 2019). Random Forests are the ensemble of Decision Tree. Naïve Bayes is the statistical classification model. Based on the Bayes Theorem, it works on the assumption that all the features in the dataset are independent and equal.
In general, for continuous and multi-dimensional features, neural networks and SVM show better performance. Whereas, in the case of the categorical or discrete features, the logic-based algorithms, such as the rule learners and decision trees, perform better. SVM and others will need a large sample size to produce high accuracy, but Naïve Bayes works on a small dataset. The training time varies for each algorithm; for example, Naïve Bayes trains quickly because of their single pass of the entries. Also, it does not need much storage space during training and testing. On the contrary, during training, KNN based models require huge storage space and more than that during the testing phase.
In terms of interpretability, the logic-based models are interpreted easily, whereas SVM and neural networks are difficult to interpret. They also have the highest number of parameters, which need optimization and tuning. One algorithm cannot outperform the other. One way to determine the type of algorithm to use is to validate the models and estimate their accuracy and choose the one with better accuracy. Recently, combining the algorithms are proposed to enhance individual algorithm performances. However, the gene expression data has the issue of High Dimension and Low Sample Size (HDLSS), for which machine learning models are less suited. Hence, the Deep Learning and Deep Belief Networks are being researched in recent days and a multi-omics dataset.
In the performance evaluation metrics, the commonly used ones are the Classification Accuracy, Least One Out Cross Validation (LOOCV), k-Fold Cross-Validation, and ROC. Among these, several works use the Classification Accuracy. However, many performance metrics need concentration, such as sensitivity, sensibility, and similarity measures.
Open Issues in Gene Expression Data
The gene expression is a biological process; DNA instructions are transformed into a functional product called the proteins. The cells in a living organism do not need proteins all the time. Certain complex molecular mechanisms must turn the genes on and off. If that does not happen, diseases and disorders will follow.
Deoxy-ribonucleic Acid Microarray is a technology used widely in biomedical research to analyze gene expression to discover the disease or disorder, classify, and predict. The DNA microarray data is also used to predict the responses of a drug or therapies given. There are different types of DNA microarray, such as cDNA (complementary Deoxy-Ribose Nucleic Acid), SNP (Single Nucleotide Polymorphism), and CNV (Copy Number Validation) microarrays (Arevalillo and Navarro, 2013). cDNA is a DNA without introns and formed from a single-stranded RNA. SNP is the variations that can be found only at a single point in a DNA sequence. CNV is a condition where parts of a genome will be repeated, and the repetition will vary from one individual to another. There are many advanced technologies available to analyze gene expression. Most widely used are cDNA bi-color glass slide and Affymetrix GeneChip.
Many challenges and limitations need to be addressed to extract the required knowledge from the gene expression with great precision. The significant difficulties are as follows (Chan et al., 2016; Li and Wang, 2016; Li et al., 2018):
(a) Curse of Dimensionality: The major issue that is researched upon in machine learning is the overfitting of a learning model. The work in García et al. (2017) discusses the curse of dimensionality in detail. Microarray is generally high-dimensional data, ranging from hundreds to thousands and more features. Microarray data prove to be hectic in managing. To handle such huge volumes of data, advanced storage systems are required (Mramor et al., 2005; Abdulla and Khasawneh, 2020).
(b) The gap between the Researchers and Biologists: There is a huge gap among the researchers, biologists and medical practitioners, which led to many unexplored areas in the genomic studies. The opportunity of finding the best techniques and approaches are very less because of the aforementioned gap.
(c) Redundant and Mislabelled Data: Data imbalance and mislabelled data is the most prevailing issue in the Microarray data because of the irregular scanning. The Microarray dataset usually has class imbalance issue, i.e., one class will dominate the entire dataset. When the learning model is trained on a mislabelled and imbalanced data, it will greatly affect the generalization ability of the learning model. Same as the abovementioned issues, redundant and irrelevant data are also the main concern in determining the efficiency of the feature set (Lakshmanan and Jenitha, 2020; Rouhi and Nezamabadi-Pour, 2020).
(d) Difficulty in Retrieving the Biological Information: There are many clinical challenges in retrieving the biological information. The main aim of genomic studies is to discover the significant changes in the gene expression, clinically or biologically. The difficulty is that not everyone will possess high-ended equipment to capture significant changes. Also, in some of the biological processes, the changes in the expression are very subtle and difficult to be identified with analytical methods. Due to the different range of approaches regarding the experimental design, data access, study and batch of reagents used, the data may be erroneous and biased.
Some of the future directions with which the research in this area can be proceeded are as follows:
(a) Enhanced Models for Better Diagnosis of Rare Genetic Disorders:
There are various genetic disorders classified under Monogenic and Polygenic disorders. Monogenic disorders are caused because of modifications in a single gene and inherited genetically. It is rare. Unlike Monogenic, Polygenic are commonly occurring and caused because of modifications in several genes. The genetic illnesses of such types are overwhelming in the recent years. Machine Learning classification and prediction models will diagnose the disorders with great accuracy.
(b) Cancer Prognosis and Prediction:
Cancer is a heterogeneous disease, which is considered to have various subtypes. It is critical to diagnose early to further assist the patients clinically. The importance of grouping high and low risk patients had led to various researches in bioinformatics and machine learning applications. The ability of machine learning models such as Support Vector Machine (SVM), Artificial Neural Networks (ANN) and Bayesian Networks (BN) in the development of classification and predictive models for accurate decisions have to be explored.
(c) Collaborative Platforms in Gene Expressions:
The individual models in Machine Learning will yield better results when applied on gene expression data. However, hybrid methods prove to be successful at many instances. Along with hybrid methods, more research should be done in combining different gene expression data and clinical reports. It is difficult and exhaustive, yet it will offer greater results.
(d) Analyzing Drug Response in Gene Expression Data:
Predicting a drug response to any genetic disorder or disease is an important step. Many recent efforts in analyzing the sensitivity and response to cancer or other diseases are commendable. Still, the main problem in developing a model for drug response is the high dimension and less sample size. The feature selection techniques in Machine Learning assist in reducing the dimensions and improve the accuracy in predicting the drug response.
Conclusion
Gene expression Microarray is a high-dimensional database with less sample size. It needs powerful techniques to handle it and preserve the informative genes by minimizing the redundancy and dependency. This paper discusses the works done in the recent years in the gene expression microarray dataset. The papers are selected from the past six years, the focus is mainly on the supervised, unsupervised and semi-supervised based feature selection in the gene expression data. Further, under those three learning methods, we have chosen papers that concentrate on filter, wrapper, hybrid, embedded and ensemble based gene selection. This study lists out the significant difficulties faced in handling such huge dimensional datasets. To overcome the dimension issues, the gene selection must be made carefully. Although there are a lot of works done in the literature on the gene expression microarray data, there are many open opportunities that need attention. The researches have mainly focused on supervised gene selection with a filter as evaluation methods. The potentials of unsupervised and semi-supervised techniques are yet to be tapped. The semi-supervised technique works with the benefits of supervised and unsupervised techniques combined. Hence, the chances of improved accuracy is high in semi-supervised. The only aim of almost all the works is to achieve higher accuracy the focus on sensitivity, specificity, stability and similarity is scarce. As equally important as the dimensionality issue is the misclassification or mislabelled data. There is a promising future for overcoming these two issues. Another important direction for improvement in gene selection is to develop more ensemble and hybrid evaluation methods. As discussed in the literature, works on hybrid and ensemble are considerably less when compared to filter and wrapper approaches. Hybrid and ensemble methods are capable of providing more accurate results. Apparently, it needs further developments. Research must be done in joint analysis, to combine the clinical reports and the gene expression data. It will help in analyzing various aspects and will offer a different perspective. It would serve as a major breakthrough, yet hectic and exhaustive.
Author Contributions
PDRV and C-YC did the conceptualization and supervised the data. C-YC carried out the funding acquisition. NM, PDRV, KS, and C-YC investigated the data and performed the methodology. C-YC and KS carried out the project administration and validated the data. NM, PDRV, and KS wrote, reviewed, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the “Intelligent Recognition Industry Service Research Center” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdulla, M., and Khasawneh, M. T. (2020). G-Forest: an ensemble method for cost-sensitive feature selection in gene expression microarrays. Artif. Intell. Med. 108:101941. doi: 10.1016/j.artmed.2020.101941
Abinash, M. J., and Vasudevan, V. (2018). “A Study on Wrapper-Based Feature Selection Algorithm for Leukemia Dataset,” in Proceedings of the Intelligent Engineering Informatics, (New York, NY: Springer), 311–321. doi: 10.1007/978-981-10-7566-7_31
Acharya, S., Saha, S., and Nikhil, N. (2017). Unsupervised gene selection using biological knowledge: application in sample clustering. BMC Bioinform. 18:513. doi: 10.1186/s12859-017-1933-0
Algamal, Z. Y., and Lee, M. H. (2015). Penalized logistic regression with the adaptive LASSO for gene selection in high-dimensional cancer classification. Exp. Syst. Appl. 42, 9326–9332. doi: 10.1016/j.eswa.2015.08.016
Almugren, N., and Alshamlan, H. (2019). A survey on hybrid feature selection methods in microarray gene expression data for cancer classification. IEEE Access 7, 78533–78548. doi: 10.1109/access.2019.2922987
Alomari, O. A., Khader, A. T., Al-Betar, M. A., and Abualigah, L. M. (2017). Gene selection for cancer classification by combining minimum redundancy maximum relevancy and bat-inspired algorithm. Int. J. Data Min. Bioinform. 9, 32–51. doi: 10.1504/ijdmb.2017.10009480
Alshamlan, H. M., Badr, G. H., and Alohali, Y. A. (2015). Genetic Bee Colony (GBC) algorithm: A new gene selection method for microarray cancer classification. Comput. Biol. Chem. 56, 49–60. doi: 10.1016/j.compbiolchem.2015.03.001
Ang, J. C., Haron, H., and Hamed, H. N. A. (2015a). “Semi-supervised SVM-based feature selection for cancer classification using microarray gene expression data,” in Proceedings of the International Conference on Industrial, Engineering and Other Applications of Applied Intelligent Systems, (Cham: Springer), 468–477. doi: 10.1007/978-3-319-19066-2_45
Ang, J. C., Mirzal, A., Haron, H., and Hamed, H. N. A. (2015b). Supervised, unsupervised, and semi-supervised feature selection: a review on gene selection. IEEE Trans. Comp. Biol. Bioinform. 13, 971–989. doi: 10.1109/tcbb.2015.2478454
Anter, A. M., and Ali, M. (2020). Feature selection strategy based on hybrid crow search optimization algorithm integrated with chaos theory and fuzzy c-means algorithm for medical diagnosis problems. Soft Comp. 24, 1565–1584. doi: 10.1007/s00500-019-03988-3
Apolloni, J., Leguizamón, G., and Alba, E. (2016). Two hybrid wrapper-filter feature selection algorithms applied to high-dimensional microarray experiments. Appl. Soft Comp. 38, 922–932. doi: 10.1016/j.asoc.2015.10.037
Arevalillo, J. M., and Navarro, H. (2013). Exploring correlations in gene expression microarray data for maximum predictive–minimum redundancy biomarker selection and classification. Comput. Biol. Med. 43, 1437–1443. doi: 10.1016/j.compbiomed.2013.07.005
Bergmeir, C., and Benítez, J. M. (2012). On the use of cross-validation for time series predictor evaluation. Inform. Sci. 191, 192–213. doi: 10.1016/j.ins.2011.12.028
Bermingham, M. L., Pong-Wong, R., Spiliopoulou, A., Hayward, C., Rudan, I., Campbell, H., et al. (2015). Application of high-dimensional feature selection: evaluation for genomic prediction in man. Sci. Rep. 5:10312.
Blanco, R., Larrañaga, P., Inza, I., and Sierra, B. (2004). Gene selection for cancer classification using wrapper approaches. Int. J. Patt. Recogn. Artif. Intell. 18, 1373–1390. doi: 10.1142/s0218001404003800
Boucheham, A., Batouche, M., and Meshoul, S. (2015). “An ensemble of cooperative parallel metaheuristics for gene selection in cancer classification,” in Proceedings of the International Conference on Bioinformatics and Biomedical Engineering, (Cham: Springer), 301–312. doi: 10.1007/978-3-319-16480-9_30
Braga-Neto, U., Hashimoto, R., Dougherty, E. R., Nguyen, D. V., and Carroll, R. J. (2004). Is cross-validation better than resubstitution for ranking genes? Bioinformatics 20, 253–258. doi: 10.1093/bioinformatics/btg399
Brahim, A. B., and Limam, M. (2018). Ensemble feature selection for high dimensional data: a new method and a comparative study. Adv. Data Anal. Class. 12, 937–952. doi: 10.1007/s11634-017-0285-y
Brown, M. P., Grundy, W. N., Lin, D., Cristianini, N., Sugnet, C. W., Furey, T. S., et al. (2000). Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl. Acad. Sci. 97, 262–267. doi: 10.1073/pnas.97.1.262
Brumpton, B. M., and Ferreira, M. A. (2016). Multivariate eQTL mapping uncovers functional variation on the X-chromosome associated with complex disease traits. Hum. Genet. 135, 827–839. doi: 10.1007/s00439-016-1674-6
Ca, D. A. V., and Mc, V. (2015). Gene expression data classification using support vector machine and mutual information-based gene selection. Proc. Comp. Sci. 47, 13–21. doi: 10.1016/j.procs.2015.03.178
Cai, H., Ruan, P., Ng, M., and Akutsu, T. (2014). Feature weight estimation for gene selection: a local hyperlinear learning approach. BMC Bioinformatics 15:70. doi: 10.1186/1471-2105-15-70
Cárdenas-Ovando, R. A., Fernández-Figueroa, E. A., Rueda-Zárate, H. A., Noguez, J., and Rangel-Escareño, C. (2019). A feature selection strategy for gene expression time series experiments with hidden Markov models. PLoS One 14:e0223183. doi: 10.1371/journal.pone.0223183
Chakraborty, D., and Maulik, U. (2014). Identifying cancer biomarkers from microarray data using feature selection and semisupervised learning. IEEE J. Transl. Eng. Health Med. 2, 1–11. doi: 10.1109/jtehm.2014.2375820
Chan, W. H., Mohamad, M. S., Deris, S., Zaki, N., Kasim, S., Omatu, S., et al. (2016). Identification of informative genes and pathways using an improved penalized support vector machine with a weighting scheme. Comput. Biol. Med. 77, 102–115. doi: 10.1016/j.compbiomed.2016.08.004
Chandrashekar, G., and Sahin, F. (2014). A survey on feature selection methods. Comp. Electr. Eng. 40, 16–28. doi: 10.1016/j.compeleceng.2013.11.024
Chen, H., Zhang, Y., and Gutman, I. (2016). A kernel-based clustering method for gene selection with gene expression data. J. Biomed. Inform. 62, 12–20. doi: 10.1016/j.jbi.2016.05.007
Chen, K. H., Wang, K. J., Tsai, M. L., Wang, K. M., Adrian, A. M., Cheng, W. C., et al. (2014). Gene selection for cancer identification: a decision tree model empowered by particle swarm optimization algorithm. BMC Bioinform. 15:49. doi: 10.1186/1471-2105-15-8
Chen, Y., and Yao, S. (2017). Sequential search with refinement: model and application with click-stream data. Manag. Sci. 63, 4345–4365. doi: 10.1287/mnsc.2016.2557
Chen, Y., Zhang, Z., Zheng, J., Ma, Y., and Xue, Y. (2017). Gene selection for tumor classification using neighborhood rough sets and entropy measures. J. Biomed. Inform. 67, 59–68. doi: 10.1016/j.jbi.2017.02.007
Chinnaswamy, A., and Srinivasan, R. (2016). “Hybrid feature selection using correlation coefficient and particle swarm optimization on microarray gene expression data,” in Proceedings of the Innovations in bio-inspired computing and applications, (Cham: Springer), 229–239. doi: 10.1007/978-3-319-28031-8_20
Cleofas-Sánchez, L., Sánchez, J. S., and García, V. (2019). Gene selection and disease prediction from gene expression data using a two-stage hetero-associative memory. Prog. Artif. Intell. 8, 63–71. doi: 10.1007/s13748-018-0148-6
Czajkowski, M., and Kretowski, M. (2019). Decision tree underfitting in mining of gene expression data. An evolutionary multi-test tree approach. Exp. Syst. Appl. 137, 392–404. doi: 10.1016/j.eswa.2019.07.019
Dashtban, M., and Balafar, M. (2017). Gene selection for microarray cancer classification using a new evolutionary method employing artificial intelligence concepts. Genomics 109, 91–107. doi: 10.1016/j.ygeno.2017.01.004
Dashtban, M., Balafar, M., and Suravajhala, P. (2018). Gene selection for tumor classification using a novel bio-inspired multi-objective approach. Genomics 110, 10–17. doi: 10.1016/j.ygeno.2017.07.010
Deng, H., and Runger, G. (2013). Gene selection with guided regularized random forest. Pattern Recogn. 46, 3483–3489. doi: 10.1016/j.patcog.2013.05.018
Devi Arockia Vanitha, C., Devaraj, D., and Venkatesulu, M. (2016). Multiclass cancer diagnosis in microarray gene expression profile using mutual information and support vector machine. Intell. Data Anal. 20, 1425–1439. doi: 10.3233/IDA-150203
Djellali, H., Guessoum, S., Ghoualmi-Zine, N., and Layachi, S. (2017). “Fast correlation based filter combined with genetic algorithm and particle swarm on feature selection,” in Proceedings of the 2017 5th International Conference on Electrical Engineering-Boumerdes (ICEE-B), (Piscataway, NJ: IEEE), 1–6.
Elavarasan, D., Vincent, D. R., Sharma, V., Zomaya, A. Y., and Srinivasan, K. (2018). Forecasting yield by integrating agrarian factors and machine learning models: a survey. Comp. Electr. Agricult. 155, 257–282. doi: 10.1016/j.compag.2018.10.024
Elghazel, H., and Aussem, A. (2015). Unsupervised feature selection with ensemble learning. Machine Learn. 98, 157–180. doi: 10.1007/s10994-013-5337-8
Fawcett, T. (2006). An introduction to ROC analysis. Pattern Recogn. Lett. 27, 861–874. doi: 10.1016/j.patrec.2005.10.010
Filippone, M., Masulli, F., and Rovetta, S. (2005). “Unsupervised gene selection and clustering using simulated annealing,” in International Workshop on Fuzzy Logic and Applications, (Berlin: Springer), 229–235. doi: 10.1007/11676935_28
Filippone, M., Masulli, F., and Rovetta, S. (2006). “Supervised classification and gene selection using simulated annealing,” in Proceedings of the 2006 IEEE International Joint Conference on Neural Network Proceedings, (Piscataway, NJ: IEEE), 3566–3571.
Flach, P. A. (2016). ROC analysis. In Encyclopedia of Machine Learning and Data Mining. New York, NY: Springer, 1–8.
Gangeh, M. J., Zarkoob, H., and Ghodsi, A. (2017). Fast and scalable feature selection for gene expression data using hilbert-schmidt independence criterion. IEEE Trans. Comp. Biol. Bioinform. 14, 167–181. doi: 10.1109/tcbb.2016.2631164
Gao, L., Ye, M., Lu, X., and Huang, D. (2017). Hybrid method based on information gain and support vector machine for gene selection in cancer classification. Genom. Prot. Bioinform. 15, 389–395. doi: 10.1016/j.gpb.2017.08.002
García, V., and Sánchez, J. S. (2015). Mapping microarray gene expression data into dissimilarity spaces for tumor classification. Inform. Sci. 294, 362–375. doi: 10.1016/j.ins.2014.09.064
García, V., Sánchez, J. S., Cleofas-Sánchez, L., Ochoa-Domínguez, H. J., and López-Orozco, F. (2017). “An insight on the ‘large G, small n’ problem in gene-expression microarray classification,” in Proceedings of the 8th Iberian Conference on Pattern Recognition and Image Analysis, Faro, 483–490. doi: 10.1007/978-3-319-58838-4_53
Ghosh, M., Adhikary, S., Ghosh, K. K., Sardar, A., Begum, S., and Sarkar, R. (2019a). Genetic algorithm based cancerous gene identification from microarray data using ensemble of filter methods. Med. Biol. Eng. Comp. 57, 159–176. doi: 10.1007/s11517-018-1874-4
Ghosh, M., Begum, S., Sarkar, R., Chakraborty, D., and Maulik, U. (2019b). Recursive memetic algorithm for gene selection in microarray data. Exp. Syst. Appl. 116, 172–185. doi: 10.1016/j.eswa.2018.06.057
Guo, S., Guo, D., Chen, L., and Jiang, Q. (2017). A L1-regularized feature selection method for local dimension reduction on microarray data. Comput. Biol. Chem. 67, 92–101. doi: 10.1016/j.compbiolchem.2016.12.010
Halperin, E., Kimmel, G., and Shamir, R. (2005). Tag SNP selection in genotype data for maximizing SNP prediction accuracy. Bioinformatics 21(Suppl._1), i195–i203.
Han, F., Yang, C., Wu, Y. Q., Zhu, J. S., Ling, Q. H., Song, Y. Q., et al. (2015). A gene selection method for microarray data based on binary PSO encoding gene-to-class sensitivity information. IEEE Trans. Comp. Biol. Bioinform. 14, 85–96. doi: 10.1109/tcbb.2015.2465906
Hancer, E., Xue, B., and Zhang, M. (2018). Differential evolution for filter feature selection based on information theory and feature ranking. Knowl. Based Syst. 140, 103–119. doi: 10.1016/j.knosys.2017.10.028
Handelman, G. S., Kok, H. K., Chandra, R. V., Razavi, A. H., Huang, S., Brooks, M., et al. (2019). Peering into the black box of artificial intelligence: evaluation metrics of machine learning methods. AJR 212, 38–43. doi: 10.2214/ajr.18.20224
Hasri, N. N. M., Wen, N. H., Howe, C. W., Mohamad, M. S., Deris, S., and Kasim, S. (2017). Improved support vector machine using multiple SVM-RFE for cancer classification. Int. J. Adv. Sci. Eng. Inform. Technol. 7, 1589–1594. doi: 10.18517/ijaseit.7.4-2.3394
Hernandez, J. C. H., Duval, B., and Hao, J. K. (2007). “A genetic embedded approach for gene selection and classification of microarray data,” in Proceedings of the European Conference on Evolutionary Computation, Machine Learning and Data Mining in Bioinformatics, (Berlin: Springer), 90–101. doi: 10.1007/978-3-540-71783-6_9
Hira, Z. M., and Gillies, D. F. (2015). A review of feature selection and feature extraction methods applied on microarray data. Adv. Bioinform. 2015:198363.
Hoque, N., Bhattacharyya, D. K., and Kalita, J. K. (2014). MIFS-ND: A mutual information-based feature selection method. Exp.Syst. Appl. 41, 6371–6385. doi: 10.1016/j.eswa.2014.04.019
Hu, Q., Pan, W., An, S., Ma, P., and Wei, J. (2010). An efficient gene selection technique for cancer recognition based on neighborhood mutual information. Int. J. Machine Learn. Cybernet. 1, 63–74. doi: 10.1007/s13042-010-0008-6
Huerta, E. B., Hernández, J. C. H., Caporal, R. M., Cruz, J. F. R., and Montiel, L. A. H. (2010). An efficient embedded gene selection method for microarray gene expression data. Res. Comp. Sci. 50, 289–299.
Inza, I., Larrañaga, P., Blanco, R., and Cerrolaza, A. J. (2004). Filter versus wrapper gene selection approaches in DNA microarray domains. Artif. Intell. Med. 31, 91–103. doi: 10.1016/j.artmed.2004.01.007
Jadhav, S., He, H., and Jenkins, K. (2018). Information gain directed genetic algorithm wrapper feature selection for credit rating. Appl. Soft Comp. 69, 541–553. doi: 10.1016/j.asoc.2018.04.033
Jain, I., Jain, V. K., and Jain, R. (2018). Correlation feature selection based improved-binary particle swarm optimization for gene selection and cancer classification. Appl. Soft Comp. 62, 203–215. doi: 10.1016/j.asoc.2017.09.038
Jiang, B., Wu, X., Yu, K., and Chen, H. (2019). Joint semi-supervised feature selection and classification through Bayesian approach. Proc. AAAI Conf. Artif. Intell. 33, 3983–3990. doi: 10.1609/aaai.v33i01.33013983
Jović, A., Brkić, K., and Bogunović, N. (2015). “A review of feature selection methods with applications,” in Proceedings of the 38th International Convention on Information and Communication Technology, Electronics and Microelectronics (MIPRO), (Piscataway, NJ: IEEE), 1200–1205.
Kar, S., Sharma, K. D., and Maitra, M. (2015). Gene selection from microarray gene expression data for classification of cancer subgroups employing PSO and adaptive K-nearest neighborhood technique. Exp. Syst. Appl. 42, 612–627. doi: 10.1016/j.eswa.2014.08.014
Khalid, S., Khalil, T., and Nasreen, S. (2014). “A survey of feature selection and feature extraction techniques in machine learning,” in Proceedings of the Science and Information Conference, (Piscataway, NJ: IEEE), 372–378.
Kira, K., and Rendell, L. A. (1992). “A practical approach to feature selection,” in Proceedings of the Machine Learning, (Burlington, MA: Morgan Kaufmann), 249–256. doi: 10.1016/b978-1-55860-247-2.50037-1
Kotsiantis, S. B., Zaharakis, I., and Pintelas, P. (2007). Supervised machine learning: A review of classification techniques. Emerg. Artif. Intell. Appl. Comp. Eng. 160, 3–24.
Koul, N., and Manvi, S. S. (2020). “Machine-Learning Algorithms for Feature Selection from Gene Expression Data,” in Statistical Modelling and Machine Learning Principles for Bioinformatics Techniques, Tools, and Applications, eds K. G. Srinivasa, G. M. Siddesh, and S. R. Manisekhar (Singapore: Springer), 151–161. doi: 10.1007/978-981-15-2445-5_10
Kumar, C. A., Sooraj, M. P., and Ramakrishnan, S. (2017). A comparative performance evaluation of supervised feature selection algorithms on microarray datasets. Proc. Comp. Sci. 115, 209–217. doi: 10.1016/j.procs.2017.09.127
Lai, C. M., Yeh, W. C., and Chang, C. Y. (2016). Gene selection using information gain and improved simplified swarm optimization. Neurocomputing 218, 331–338. doi: 10.1016/j.neucom.2016.08.089
Lakshmanan, B., and Jenitha, T. (2020). Optimized feature selection and classification in microarray gene expression cancer data. Ind. J. Public Health Res. Dev. 11, 347–352. doi: 10.37506/v11/i1/2020/ijphrd/193842
Landgrebe, T. C., and Duin, R. P. (2008). Efficient multiclass ROC approximation by decomposition via confusion matrix perturbation analysis. IEEE Trans. Patt. Anal. Machine Intell. 30, 810–822. doi: 10.1109/tpami.2007.70740
Lazar, C., Taminau, J., Meganck, S., Steenhoff, D., Coletta, A., Molter, C., et al. (2012). A survey on filter techniques for feature selection in gene expression microarray analysis. IEEE Trans. Comp. Biol. Bioinform. 9, 1106–1119. doi: 10.1109/tcbb.2012.33
Li, J., Tang, J., and Liu, H. (2017). “Reconstruction-based Unsupervised Feature Selection: An Embedded Approach,” in Proceedings of the International Joint Conference on Artificial Intelligence, IJCAI, Macao2159–2165.
Li, J., and Wang, F. (2016). Towards unsupervised gene selection: a matrix factorization framework. IEEE Trans. Comp. Biol. Bioinform. 14, 514–521. doi: 10.1109/tcbb.2016.2591545
Li, Z., Liao, B., Cai, L., Chen, M., and Liu, W. (2018). Semi-supervised maximum discriminative local margin for gene selection. Sci. Rep. 8, 1–11. doi: 10.15373/22778179/oct2013/41
Liaghat, S., and Mansoori, E. G. (2016). Unsupervised selection of informative genes in microarray gene expression data. Int. J. Appl. Pattern Recogn. 3, 351–367. doi: 10.1504/ijapr.2016.082237
Liang, Y., Chai, H., Liu, X. Y., Xu, Z. B., Zhang, H., and Leung, K. S. (2016). Cancer survival analysis using semi-supervised learning method based on cox and aft models with l 1/2 regularization. BMC Med. Genom. 9:11. doi: 10.1201/b16589
Liao, B., Jiang, Y., Liang, W., Zhu, W., Cai, L., and Cao, Z. (2014). Gene selection using locality sensitive Laplacian score. IEEE Trans. Comp. Biol. Bioinform. 11, 1146–1156. doi: 10.1109/tcbb.2014.2328334
Liu, H., Zhou, M., and Liu, Q. (2019). An embedded feature selection method for imbalanced data classification. IEEE J. Autom. Sin. 6, 703–715. doi: 10.1109/jas.2019.1911447
Liu, J., Cheng, Y., Wang, X., Zhang, L., and Wang, Z. J. (2018). Cancer characteristic gene selection via sample learning based on deep sparse filtering. Sci. Rep. 8, 1–13.
Maldonado, S., and López, J. (2018). Dealing with high-dimensional class-imbalanced datasets: embedded feature selection for SVM classification. Appl. Soft Comp. 67, 94–105. doi: 10.1016/j.asoc.2018.02.051
Manbari, Z., AkhlaghianTab, F., and Salavati, C. (2019). Hybrid fast unsupervised feature selection for high-dimensional data. Exp. Syst. Appl. 124, 97–118. doi: 10.1016/j.eswa.2019.01.016
Mazumder, D. H., and Veilumuthu, R. (2019). An enhanced feature selection filter for classification of microarray cancer data. ETRI J. 41, 358–370. doi: 10.4218/etrij.2018-0522
Mishra, S., and Mishra, D. (2015). SVM-BT-RFE: An improved gene selection framework using Bayesian T-test embedded in support vector machine (recursive feature elimination) algorithm. Karbala Int. J. Modern Sci. 1, 86–96. doi: 10.1016/j.kijoms.2015.10.002
Mohamed, E., El Houby, E. M., Wassif, K. T., and Salah, A. I. (2016). Survey on different methods for classifying gene expression using microarray approach. Int. J. Comp. Appl. 975:8887.
Mohapatra, P., Chakravarty, S., and Dash, P. K. (2016). Microarray medical data classification using kernel ridge regression and modified cat swarm optimization based gene selection system. Swarm Evol. Comp. 28, 144–160. doi: 10.1016/j.swevo.2016.02.002
Motieghader, H., Najafi, A., Sadeghi, B., and Masoudi-Nejad, A. (2017). A hybrid gene selection algorithm for microarray cancer classification using genetic algorithm and learning automata. Inform. Med. Unlocked 9, 246–254. doi: 10.1016/j.imu.2017.10.004
Mramor, M., Leban, G., Demšar, J., and Zupan, B. (2005). “Conquering the curse of dimensionality in gene expression cancer diagnosis: tough problem, simple models,” in Proceedings of the Conference on Artificial Intelligence in Medicine in Europe (Berlin: Springer), 514–523. doi: 10.1007/11527770_68
Nguyen, T., Khosravi, A., Creighton, D., and Nahavandi, S. (2015). Hierarchical gene selection and genetic fuzzy system for cancer microarray data classification. PLoS One 10:e012036. doi: 10.1371/journal.pone.0120364
Pearson, W., Tran, C. T., Zhang, M., and Xue, B. (2019). “Multi-Round Random Subspace Feature Selection for Incomplete Gene Expression Data,” in Proceedings of the 2019 IEEE Congress on Evolutionary Computation (CEC), (Piscataway, NJ: IEEE), 2544–2551.
Prasad, Y., Biswas, K. K., and Hanmandlu, M. (2018). A recursive PSO scheme for gene selection in microarray data. Appl. Soft Comp. 71, 213–225. doi: 10.1016/j.asoc.2018.06.019
Rajeswari, R., and Gunasekaran, G. (2015). Semi-Supervised Tumor Data Clustering via Spectral Biased Normalized Cuts. (Salem: IJERT).
Raut, S. A., Sathe, S. R., and Raut, A. (2010). “Bioinformatics: Trends in gene expression analysis,” in Proceedings of the 2010 International Conference on Bioinformatics and Biomedical Technology, (Chengdu: IEEE), 97–100.
Rodrigues, D., Pereira, L. A., Nakamura, R. Y., Costa, K. A., Yang, X. S., Souza, A. N., et al. (2014). A wrapper approach for feature selection based on bat algorithm and optimum-path forest. Exp. Syst. Appl. 41, 2250–2258. doi: 10.1016/j.eswa.2013.09.023
Rouhi, A., and Nezamabadi-pour, H. (2018). “Filter-based feature selection for microarray data using improved binary gravitational search algorithm,” in Proceedings of the 2018 3rd Conference on Swarm Intelligence and Evolutionary Computation (CSIEC), (Piscataway, NJ: IEEE), 1–6.
Rouhi, A., and Nezamabadi-Pour, H. (2020). Feature Selection in High-Dimensional Data. In Optimization, Learning, and Control for Interdependent Complex Networks. Cham: Springer, 85–128.
Ruiz, R., Riquelme, J. C., and Aguilar-Ruiz, J. S. (2005). Heuristic search over a ranking for feature selection. In International Work-Conference on Artificial Neural Networks. Berlin: Springer, 742–749.
Russell, S. J., and Norvig, P. (2016). Artificial Intelligence: A Modern Approach. London: Pearson Education Limited.
Saeys, Y., Inza, I., and Larrañaga, P. (2007). A review of feature selection techniques in bioinformatics. Bioinformatics 23, 2507–2517. doi: 10.1093/bioinformatics/btm344
Schaffer, C. (1993). Selecting a classification method by cross-validation. Machine Learn. 13, 135–143. doi: 10.1007/bf00993106
Seijo-Pardo, B., Bolón-Canedo, V., and Alonso-Betanzos, A. (2016). “. Using a feature selection ensemble on DNA microarray datasets,” in Proceedings of the ESANN 2016 proceedings, European Symposium on Artificial Neural Networks, Computational Intelligence and Machine Learning, Bruges.
Shanab, A. A., Khoshgoftaar, T. M., and Wald, R. (2014). “Evaluation of wrapper-based feature selection using hard, moderate, and easy bioinformatics data,” in Proceedings of the 2014 IEEE International Conference on Bioinformatics and Bioengineering, (Piscataway, NJ: IEEE), 149–155.
Sharbaf, F. V., Mosafer, S., and Moattar, M. H. (2016). A hybrid gene selection approach for microarray data classification using cellular learning automata and ant colony optimization. Genomics 107, 231–238. doi: 10.1016/j.ygeno.2016.05.001
Sharma, A., and Rani, R. (2019). C-HMOSHSSA: Gene selection for cancer classification using multi-objective meta-heuristic and machine learning methods. Comput. Methods Prog. 178, 219–235. doi: 10.1016/j.cmpb.2019.06.029
Sheikhpour, R., Sarram, M. A., Gharaghani, S., and Chahooki, M. A. Z. (2017). A survey on semi-supervised feature selection methods. Pattern Recogn. 64, 141–158. doi: 10.1016/j.patcog.2016.11.003
Shreem, S. S., Abdullah, S., and Nazri, M. Z. A. (2014). Hybridising harmony search with a Markov blanket for gene selection problems. Inform. Sci. 258, 108–121. doi: 10.1016/j.ins.2013.10.012
Shukla, A. K., Singh, P., and Vardhan, M. (2018). A hybrid gene selection method for microarray recognition. Biocybernet. Biomed. Eng. 38, 975–991. doi: 10.1016/j.bbe.2018.08.004
Shukla, A. K., and Tripathi, D. (2019). Identification of potential biomarkers on microarray data using distributed gene selection approach. Math. Biosci. 315:108230. doi: 10.1016/j.mbs.2019.108230
Solorio-Fernández, S., Carrasco-Ochoa, J. A., and Martínez-Trinidad, J. F. (2016). A new hybrid filter–wrapper feature selection method for clustering based on ranking. Neurocomputing 214, 866–880. doi: 10.1016/j.neucom.2016.07.026
Solorio-Fernández, S., Martínez-Trinidad, J. F., and Carrasco-Ochoa, J. A. (2017). A new unsupervised spectral feature selection method for mixed data: a filter approach. Pattern Recogn. 72, 314–326. doi: 10.1016/j.patcog.2017.07.020
Sun, L., Kong, X., Xu, J., Zhai, R., and Zhang, S. (2019a). A hybrid gene selection method based on ReliefF and ant colony optimization algorithm for tumor classification. Sci. Rep. 9:8978.
Sun, L., and Xu, J. (2014). Feature selection using mutual information based uncertainty measures for tumor classification. Biomed. Mater. Eng. 24, 763–770. doi: 10.3233/bme-130865
Sun, L., Zhang, X., Qian, Y., Xu, J., and Zhang, S. (2019b). Feature selection using neighborhood entropy-based uncertainty measures for gene expression data classification. Inform. Sci. 502, 18–41. doi: 10.1016/j.ins.2019.05.072
Sun, L., Zhang, X. Y., Qian, Y. H., Xu, J. C., Zhang, S. G., and Tian, Y. (2019c). Joint neighborhood entropy-based gene selection method with fisher score for tumor classification. Appl. Intell. 49, 1245–1259. doi: 10.1007/s10489-018-1320-1
Tabakhi, S., Najafi, A., Ranjbar, R., and Moradi, P. (2015). Gene selection for microarray data classification using a novel ant colony optimization. Neurocomputing 168, 1024–1036. doi: 10.1016/j.neucom.2015.05.022
Tang, C., Cao, L., Zheng, X., and Wang, M. (2018). Gene selection for microarray data classification via subspace learning and manifold regularization. Med. Biol. Eng. Comp. 56, 1271–1284. doi: 10.1007/s11517-017-1751-6
Vanjimalar, S., Ramyachitra, D., and Manikandan, P. (2018). “A Review on Feature Selection Techniques for Gene Expression Data,” in Proceedings of the 2018 IEEE International Conference on Computational Intelligence and Computing Research (ICCIC), (Piscataway, NJ: IEEE), 1–4.
Vergara, J. R., and Estévez, P. A. (2014). A review of feature selection methods based on mutual information. Neural Comp. Appl. 24, 175–186. doi: 10.1007/s00521-013-1368-0
Wahid, A., Khan, D. M., Iqbal, N., Khan, S. A., Ali, A., Khan, M., et al. (2020). Feature selection and classification for gene expression data using novel correlation based overlapping score method via Chou’s 5-steps rule. Chemometr. Intell. Lab. Syst. 199:103958. doi: 10.1016/j.chemolab.2020.103958
Wang, A., An, N., Yang, J., Chen, G., Li, L., and Alterovitz, G. (2017). Wrapper-based gene selection with Markov blanket. Comput. Biol. Med. 81, 11–23. doi: 10.1016/j.compbiomed.2016.12.002
Wang, H., Jing, X., and Niu, B. (2017). A discrete bacterial algorithm for feature selection in classification of microarray gene expression cancer data. Knowl. Based Syst. 126, 8–19. doi: 10.1016/j.knosys.2017.04.004
Wang, H., and van der Laan, M. J. (2011). Dimension reduction with gene expression data using targeted variable importance measurement. BMC Bioinformatics 12:312.
Wang, L., Wang, Y., and Chang, Q. (2016). Feature selection methods for big data bioinformatics: A survey from the search perspective. Methods 111, 21–31. doi: 10.1016/j.ymeth.2016.08.014
Wang, Y., Tetko, I. V., Hall, M. A., Frank, E., Facius, A., Mayer, K. F., et al. (2005). Gene selection from microarray data for cancer classification—a machine learning approach. Comput. Biol. Chem. 29, 37–46. doi: 10.1016/j.compbiolchem.2004.11.001
Xiao, Y., Hsiao, T. H., Suresh, U., Chen, H. I. H., Wu, X., Wolf, S. E., et al. (2014). A novel significance score for gene selection and ranking. Bioinformatics 30, 801–807. doi: 10.1093/bioinformatics/btr671
Xu, G., Zhang, M., Zhu, H., and Xu, J. (2017). A 15-gene signature for prediction of colon cancer recurrence and prognosis based on SVM. Gene 604, 33–40. doi: 10.1016/j.gene.2016.12.016
Xu, J., Sun, L., Gao, Y., and Xu, T. (2014). An ensemble feature selection technique for cancer recognition. Biomed. Mater. Eng. 24, 1001–1008. doi: 10.3233/bme-130897
Yang, J., Zhou, J., Zhu, Z., Ma, X., and Ji, Z. (2016). Iterative ensemble feature selection for multiclass classification of imbalanced microarray data. J. Biol. Res. Thessaloniki 23:13.
Yang, Y., Yin, P., Luo, Z., Gu, W., Chen, R., and Wu, Q. (2019). Informative Feature Clustering and Selection for Gene Expression Data. IEEE Access 7, 169174–169184. doi: 10.1109/access.2019.2952548
Ye, X., and Sakurai, T. (2017). “Unsupervised Feature Learning for Gene Selection in Microarray Data Analysis,” in Proceedings of the 1st International Conference on Medical and Health Informatics 2017, ed. Y.-C. Ho (New York, NY: Association for Computing Machinery), 101–106.
Yu, L., and Liu, H. (2004). Efficient feature selection via analysis of relevance and redundancy. J. Mach. Learn. Res. 5, 1205–1224.
Zare, M., Eftekhari, M., and Aghamollaei, G. (2019). Supervised feature selection via matrix factorization based on singular value decomposition. Chemometr. Intell. Lab. Syst. 185, 105–113. doi: 10.1016/j.chemolab.2019.01.003
Zhang, Y., Deng, Q., Liang, W., and Zou, X. (2018). An efficient feature selection strategy based on multiple support vector machine technology with gene expression data. Biomed. Res. Int. 2018:7538204.
Zhou, Y., Wang, P., Wang, X., Zhu, J., and Song, P. X. K. (2017). Sparse multivariate factor analysis regression models and its applications to integrative genomics analysis. Genet. Epidemiol. 41, 70–80. doi: 10.1002/gepi.22018
Keywords: gene selection, machine learning, microarray gene expression, supervised gene selection, unsupervised gene selection
Citation: Mahendran N, Durai Raj Vincent PM, Srinivasan K and Chang C-Y (2020) Machine Learning Based Computational Gene Selection Models: A Survey, Performance Evaluation, Open Issues, and Future Research Directions. Front. Genet. 11:603808. doi: 10.3389/fgene.2020.603808
Received: 08 September 2020; Accepted: 29 October 2020;
Published: 10 December 2020.
Edited by:
Hua Zhong, Wuhan University, ChinaReviewed by:
José Salvador Sánchez, University of Jaume I, SpainYongjin Lu, Virginia State University, United States
Copyright © 2020 Mahendran, Durai Raj Vincent, Srinivasan and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: P. M. Durai Raj Vincent, cG12aW5jZW50QHZpdC5hYy5pbg==; Chuan-Yu Chang, Y2h1YW55dUB5dW50ZWNoLmVkdS50dw==