 Shaza Asif
Shaza Asif Nadya M. Morrow
Nadya M. Morrow Erin E. Mulvihill
Erin E. Mulvihill Kyoung-Han Kim
Kyoung-Han Kim
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 15 October 2020
Sec. Epigenomics and Epigenetics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.590369
This article is part of the Research Topic Chromatin Spatial Configuration and Function in Metazoans View all 15 articles
The global prevalence of metabolic disorders, such as obesity, diabetes and fatty liver disease, is dramatically increasing. Both genetic and environmental factors are well-known contributors to the development of these diseases and therefore, the study of epigenetics can provide additional mechanistic insight. Dietary interventions, including caloric restriction, intermittent fasting or time-restricted feeding, have shown promising improvements in patients’ overall metabolic profiles (i.e., reduced body weight, improved glucose homeostasis), and an increasing number of studies have associated these beneficial effects with epigenetic alterations. In this article, we review epigenetic changes involved in both metabolic diseases and dietary interventions in primary metabolic tissues (i.e., adipose, liver, and pancreas) in hopes of elucidating potential biomarkers and therapeutic targets for disease prevention and treatment.
The continuous rise in metabolic diseases, such as obesity, type 2 diabetes (T2D; Table 1) and non-alcoholic fatty liver disease (NAFLD), is one of the leading causes of patient morbidity and mortality worldwide (Saklayen, 2018) and cannot be solely explained by the contribution of genetic and environmental factors. Indeed, epigenetics, which constitutes the reversible and heritable change in gene expression without modification of the underlying nucleotide sequence, serves as a mechanistic bridge. Epigenetic changes influenced by environmental cues can result in altered gene expression associated with metabolic function and dysfunction.
Table 1. Description of commonly used acronyms.
Overnutrition, especially of highly processed foods (Hall et al., 2019), accompanied by erratic diurnal eating patterns, constitute the major environmental contributors to the epidemic state of metabolic diseases today. As such, switching to a regular, nutritious diet can promote processes of maturation and restoration, and protect against the development of chronic metabolic disorders (Di Francesco et al., 2018). Since the applicability of pharmacological interventions in the treatment of metabolic disorders is limited by issues regarding off-target effects, patient compliance and tolerability, as well as lack of sufficiency in disease management (Longo and Panda, 2016); dietary interventions have become a promising, low-risk alternative or supplementary form of therapy. By adjusting meal timing and/or content, dietary interventions have shown continued success in reducing risk factors, inducing beneficial pleiotropic effects and ameliorating disease states (Longo and Panda, 2016).
These dietary interventions involve limiting food intake of entire (i.e., fasting interventions) or selected nutrient compositions (i.e., nutritional interventions), without disturbing energy balance or inducing malnutrition. Specifically, fasting interventions can be categorized into intermittent fasting (IF) and periodic fasting (PF), where food intake is limited either on a daily/weekly basis or on a monthly basis, respectively (Anton et al., 2018; Yong-Quan Ng et al., 2019; Figure 1). IF cycles typically last 24 h and are separated by one or more days, whereas PF cycles last two or more days and are separated by at least a week (Longo and Mattson, 2014). Different forms of IF vary in their timing of meals and include the daily time-restricted feeding (TRF), and the weekly 5:2, 2:1, or 1:1 IF regimens. Moreover, nutritional interventions vary in their meal content and include caloric restriction (CR), dietary restriction (DR), ketogenic diet (KD), and fasting-mimicking diet (FMD).

Figure 1. Classification of dietary interventions. Dietary interventions can be broadly categorized according to varied meal timing (fasting interventions) and meal content (nutritional interventions). Fasting interventions can be further subdivided into periodic fasting (PF) on a monthly basis and intermittent fasting (IF) on a weekly (5:2, 2:1, 1:1 IF) or daily (TRF) basis. ADF, alternate-day fasting; EODF, every-other-day fasting.
Dietary interventions, such as CR and IF, extend lifespan and healthspan in various animal models, including yeast (Lin et al., 2000, 2002; Wu et al., 2013), worms (Wei et al., 2008; Honjoh et al., 2009; Uno et al., 2013), fruit flies (Grandison et al., 2009; Catterson et al., 2018; Villanueva et al., 2019), rodents (Goodrick et al., 1982; Hatori et al., 2012; Chaix et al., 2014; Rusli et al., 2017; Mitchell et al., 2019), and monkeys (Bodkin et al., 2003; Colman et al., 2009; Mattison et al., 2017). Studies done in humans also demonstrate beneficial effects of dietary intervention, specifically regarding overall metabolic improvements in body weight and fat mass (Heilbronn et al., 2005; Johnson et al., 2007; Varady et al., 2009; Harvie et al., 2011; Klempel et al., 2013; Varady et al., 2013; Redman et al., 2018; Anton et al., 2019; Ravussin et al., 2019; Stekovic et al., 2019; Wilkinson et al., 2020), circulating triglyceride (TG) and cholesterol levels (Johnson et al., 2007; Varady et al., 2009; Harvie et al., 2011; Klempel et al., 2013; Varady et al., 2013; Stekovic et al., 2019; Wilkinson et al., 2020), insulin sensitivity and glucose homeostasis (Halberg et al., 2005; Harvie et al., 2011; Sutton et al., 2018; Jamshed et al., 2019), and oxidative stress and inflammation (Johnson et al., 2007; Meydani et al., 2011; Redman et al., 2018; Sutton et al., 2018; Stekovic et al., 2019). Notably, the metabolic benefits of dietary interventions are not completely dependent on total caloric intake. For instance, the 2:1 IF regimen in mice provides comparable metabolic outcomes against obesity and associated metabolic dysfunctions, despite no difference in caloric intake (i.e., isocaloric) in comparison to ad libitum (i.e., normal feeding) (Kim K.H. et al., 2017; Kim R.Y. et al., 2019; Kim Y.H. et al., 2019).
These benefits conferred by dietary interventions involve cellular adaptations within various metabolic tissues, which are mediated by epigenetic modifications. Due to the plasticity of epigenetic factors, environmental changes, such as dietary interventions, which alter food intake and composition, have a significant impact on the epigenome. In this article, we will first review epigenetic changes in metabolic disease with a particular emphasis on adipose tissues, liver, and pancreas. We will primarily focus on DNA methylation and post-translational histone modifications (Figure 2), with the exception of non-coding RNAs reviewed elsewhere (Deiuliis, 2016; Green et al., 2017). Next, we will discuss how fasting as a component of most dietary interventions and caloric restriction modulate epigenetic regulation in these tissues. To conclude, we will also briefly review the epigenetics of gut microbiota and ketone body metabolism in the context of dietary interventions. Overall, the understanding of both metabolic diseases and dietary interventions from an epigenetic perspective will provide new insights for metabolic disease prevention, management and treatment.
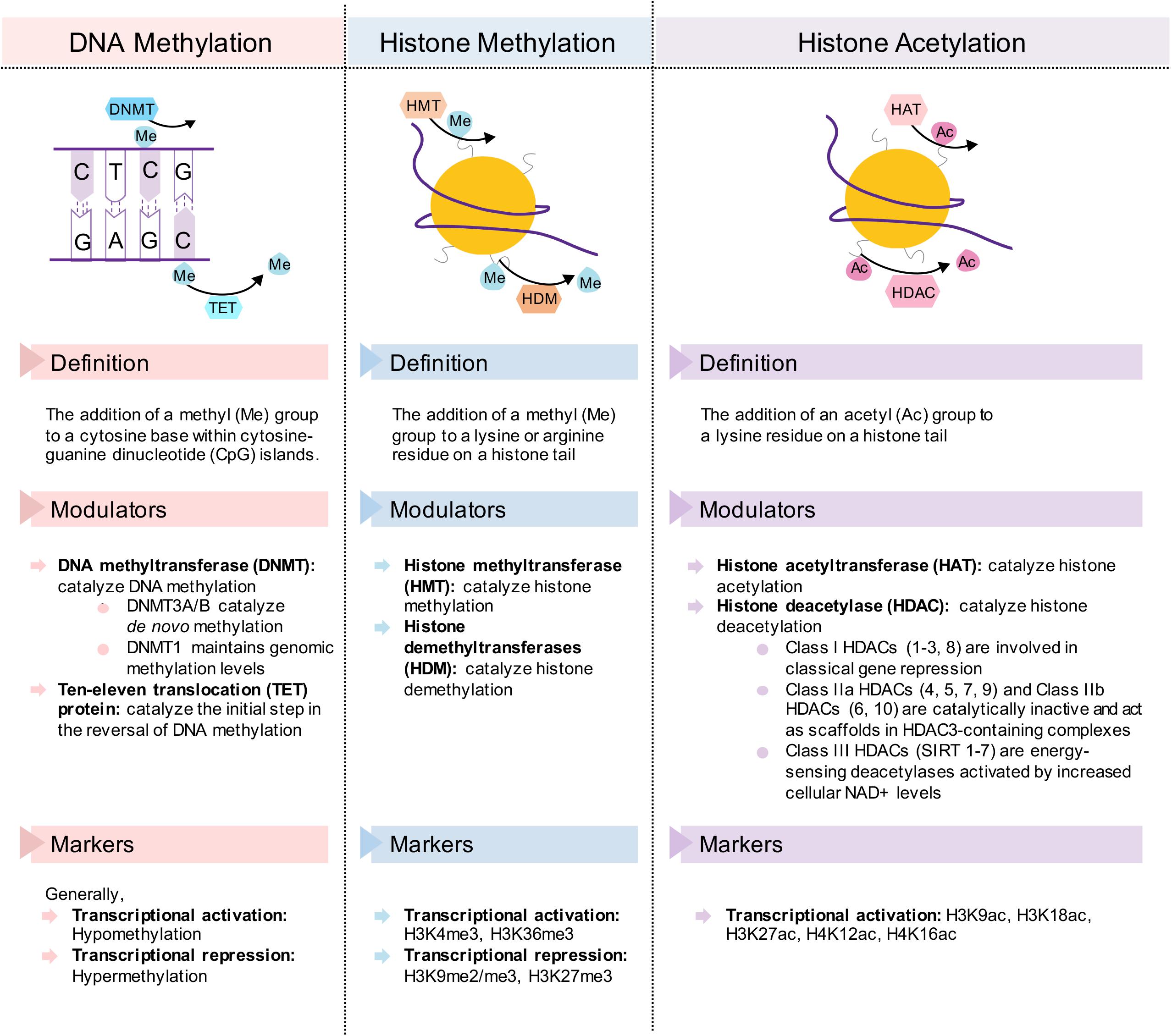
Figure 2. Description of the epigenetic change and its transcriptional modulators and markers in DNA methylation, histone methylation, and histone acetylation. Simplified diagrams show the forward and reverse reactions to each epigenetic mechanism.
Obesity is a serious metabolic disease that has reached worldwide rates of over 27.5% for adults and 47.1% for children (Ng et al., 2014). It has been estimated that obesity is 40−70% inheritable, where genome-wide association studies (GWAS) account for 20% of the variation (Locke et al., 2015). As such, it is becoming increasingly clear that epigenetic modifications serve as a link between environmental and genetic causes of obesity.
Uncontrolled adipose tissue expansion and accompanying dysfunction drive obesity and associated metabolic pathogenesis (Choe et al., 2016). Adipocytes initially expand in size (i.e., hypertrophy) to accommodate increases in energy intake relative to energy expenditure. When adipocytes become lipid-engorged and can no longer store the excess energy, adipogenesis, the process by which pre-adipocytes differentiate into mature adipocytes, expands adipocyte number (i.e., hyperplasia) (Choe et al., 2016; Chait and den Hartigh, 2020).
Adipose tissue is classified into white adipose tissue (WAT) and brown adipose tissue (BAT). WAT stores excess energy in the form of TG and is localized to subcutaneous (i.e., “beneath the skin”) and visceral depots (i.e., “surrounding internal organs”). BAT, by contrast, utilizes stored energy to produce heat in response to stimuli like cold stress, primarily via the uncoupling protein-1 (UCP-1), in a process known as non-shivering thermogenesis (Figure 3). BAT is also distributed subcutaneously (e.g., under the clavicles and in the interscapular region) and viscerally (e.g., perivascular, periviscus and around solid organs) (Sacks and Symonds, 2013; Jung et al., 2019); however, the initial distribution and size of BAT, as found in infants and young children, decreases with age. Moreover, as the “whitening” of brown adipocytes via dysregulated adipogenesis is associated with the development of obesity (Shimizu et al., 2014; Pellegrinelli et al., 2016; Longo et al., 2019), the “browning” of white adipocytes leading to increased thermogenesis may have therapeutic potential in the treatment of obesity (Choe et al., 2016).
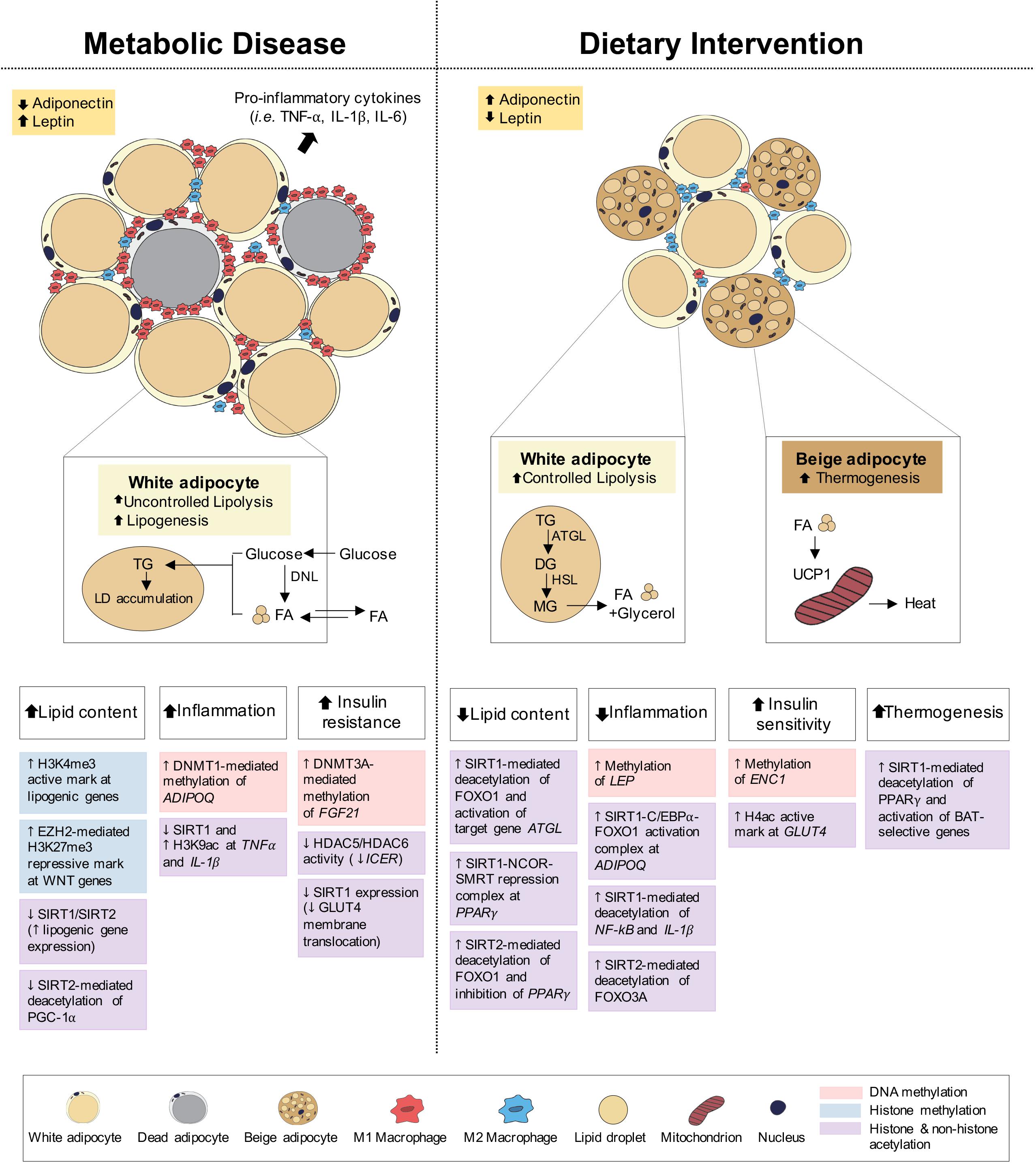
Figure 3. Epigenetic changes of adipose tissue in metabolic disease and dietary intervention. Adipose tissue in metabolic disease (i.e., obesity) predominantly consists of white adipocytes and presents with increased lipid content, inflammation and insulin resistance. Adipose tissue in dietary intervention is interspersed with both white and beige adipocytes and presents with reduced lipid content and inflammation as well as increased insulin sensitivity and thermogenesis. These physiological differences in adipose can be explained by epigenetic changes involving DNA methylation, histone methylation and histone and non-histone acetylation.
Under feeding conditions, adipocytes take up and store circulating glucose and fatty acids (FA) through processes mediated by insulin-stimulated glucose transporter type 4 (GLUT4) translocation to the adipocyte cell surface and lipoprotein lipase (LPL) activity, respectively. Under fasting conditions, adipocyte lipolysis leads to FA release into circulation for use in other metabolic tissues, including muscle, kidney, gut and liver (Ahima and Flier, 2000; Choe et al., 2016; Figure 3). Adipose tissues also function as endocrine organs through secretion of cytokines (e.g., TNFα, IL-1, and IL-6) and hormones (e.g., leptin, adiponectin), to mediate pathways of energy homeostasis, adipocyte differentiation, insulin sensitivity and inflammatory control (Wozniak et al., 2009). The reduction in insulin receptor density within the expanded adipose tissue and the subsequent development of insulin resistance both promote the uncontrollable release of FA, leading to aberrant lipid accumulation and lipotoxicity in peripheral tissues (Choe et al., 2016; Muir et al., 2016; Longo et al., 2019). Lipotoxicity, in combination with reduced anti-inflammatory adiponectin and increased pro-inflammatory cytokine secretion, promotes systemic inflammation associated with obesity, T2D and NAFLD.
DNA methylation is a key epigenetic modification involved in adipose development and function. Changes in DNA methylation in adipocytes have been associated with both the cause and effect of metabolic dysregulation in obesity, where hypomethylation appears to be the dominating change (Sonne et al., 2017). A genome-wide screen has identified 625 significant differentially methylated regions (DMRs) associated with diet-induced obesity phenotypes, of which 232 DMRs correlate with high-fat diet (HFD) alone, and 249 regions are conserved in adipose tissue from obese subjects. Among these, 30 single-nucleotide polymorphisms (SNPs) are associated with T2D (Multhaup et al., 2015). As shown in Table 2, increased and decreased DNA methylations generally correspond to genes involved in positively (i.e., glucose homeostasis) and negatively (i.e., inflammation) regulating adipocyte metabolism, respectively. Additionally, DNA methylation profiles of diet-induced and genetically obese (i.e., ob/ob) mice revealed that methylation changes are more abundant in visceral than subcutaneous adipocytes (Sonne et al., 2017), with visceral fat being the greater contributor to obesity and its associated metabolic dysfunctions (Fox et al., 2007; Neeland et al., 2013).

Table 2. Changes in DNA methylation and gene expression in adipose tissue associated with obesity and high-fat feeding.
DNA methyltransferases (DNMTs) catalyze DNA methylation. In contrast, ten-eleven translocation (TET) proteins catalyze the initial step of the reverse reaction (Figure 2; Jin and Robertson, 2013; Yong-Quan Ng et al., 2019). Altered expression of these DNA methylation modulators in adipose tissue can cause metabolic disease development and/or progression. High DNMT1 expression is found in adipocytes of obese humans (Kim et al., 2015). Similarly, increased Dnmt1 mRNA level is found in WAT of HFD-fed and genetically obese (i.e., db/db) mice compared to chow-fed and wild-type lean mice (Kim et al., 2015). Dnmt1 expression and activity in mouse 3T3-L1 adipocytes are induced by the pro-inflammatory cytokines TNFα and IL-1β (Kim et al., 2015). A known target of DNMT1 is Adipoq, which encodes the key anti-diabetic and anti-inflammatory adipokine, adiponectin (Stern et al., 2016). Over-expression of Dnmt1 in 3T3-L1 adipocytes increases methylation and decreases expression of Adipoq, while its knockdown results in the reverse (Kim et al., 2015), suggesting that direct hypermethylation and heterochromatin formation by DNMT1 at the Adipoq gene promoter is involved in obesity pathogenesis. In addition, the expression of Dnmt3a, not Dnmt3b, is increased in WAT of both diet- and genetically induced obese (i.e., ob/ob) mice (Parrillo et al., 2016; You et al., 2017) and the adipose-specific deletion of Dnmt3a in HFD-fed mice improves insulin sensitivity and glucose tolerance, independent of adiposity (You et al., 2017). Using an unbiased screen, the authors have identified Fgf21 as a key target gene of DNMT3A. Correspondingly, adipose-specific Dnmt3a deletion leads to a decrease in the methylation and an increase in the expression of adipose Fgf21 in both WAT and BAT (You et al., 2017). However, experiments using adipose tissue-specific Dnmt3a and Fgf21 double knockout mice are still required to determine the mechanism of enhanced insulin sensitivity. Additionally, higher DNA methylation levels of FGF21 have been observed in the WAT of T2D patients, which negatively correlated to FGF21 mRNA expression, although DNMT3A levels were not measured (You et al., 2017).
Histone methylation, mediated by histone methyltransferases (HMTs) and reversed by histone demethylases (HDMs) (Figure 2), regulates adipogenesis through the addition and removal of activating and repressing histone methylation marks in adipocytes. Dysregulated adipogenesis through histone methylation impairs adipose tissue development and function and is associated with the maladaptive obesogenic condition.
In adipose tissue of morbidly obese pre-diabetic patients, trimethylation of histone H3 at lysine 4 (H3K4me3), a gene activation mark, is found to be enriched at the promoters of genes associated with adipogenesis and lipid metabolism (e.g., LPL, SREBF2, SCD1, PPARG) (Castellano-Castillo et al., 2019), as well as at the E2F transcription factor 1 (E2F1), a contributor of obesity pathogenesis (Fajas et al., 2002; Haim et al., 2015; Denechaud et al., 2017). This finding suggests that the maintenance of H3K4me3 at promoters of adipogenic genes by HMTs and HDMs can be implicated in the development of obesity. MLL3/MLL4 (KMT2C/KMT2D) are H3K4 mono- and di-methyltransferases that mediate H3K4me3 transcriptional activation of adipogenic genes (e.g., Pparg, Cebpa) in association with the pax transactivation domain interacting protein (PTIP) co-factor (Lee et al., 2008, 2013; Cho et al., 2009). Additionally, LSD1 (KDM1A) catalyzes H3K4 mono- and di-demethylation to activate BAT-selective genes and to repress WAT-selective genes, in association with either nuclear receptor factor 1 (NRF1) or PR domain containing 16 (PRDM16), respectively (Hino et al., 2012; Duteil et al., 2014, 2016; Sambeat et al., 2016; Zeng et al., 2016). LSD1 also promotes BAT thermogenesis by repressing the glucocorticoid-activating enzyme, HSD11B1, thereby preventing the accumulation of excess glucocorticoid in adipose tissue (Zeng et al., 2016). Increased levels and secretion of glucocorticoid in adipose tissues are associated with obesity, insulin resistance and dyslipidemia (Akalestou et al., 2020). Notably, both mice lacking MLL3/MLL4 co-factor PTIP in adipose tissues (aP2-Cre; Paxipflox/flox) and LSD1 in adipose tissues or BAT specifically (Adipoq-Cre; Lsd1flox/flox, Ucp1-Cre; Lsd1flox/flox) exhibit similar obesogenic phenotypes with increased body weight and fat mass as well as dysfunctional BAT, indicated by lipid accumulation and reduced mitochondrial fatty acid oxidation (FAO) (Duteil et al., 2016; Zeng et al., 2016). Altogether, these results suggest that the H3K4 HMT/HDM balance is necessary for maintaining adipocyte function.
Both G9a (EHMT2, euchromatic histone lysine N-methyltransferase 2) and EHMT1 are histone H3 lysine 9 (H3K9) di- and tri-methyltransferases involved in maintaining the H3K9me2/me3 repressive mark. G9a inhibits adipogenesis while promoting the activation of Wnt/β-catenin signaling (Wang L. et al., 2013; Zhang et al., 2014) involved in brown/beige adipocyte development (Chen and Wang, 2018), and Ehmt1 regulates BAT-selective gene programs for BAT development and function in association with Prdm16 (Ohno et al., 2013). G9a (aP2-Cre; Ehmt2flox/flox) and Ehmt1 (Adipoq-Cre; Ehmt1flox/flox) knockout mice develop increased adiposity (Ohno et al., 2013; Wang L. et al., 2013), while adipose tissue-specific Ehmt1 knockout mice also present with reduced BAT thermogenesis and insulin resistance (Ohno et al., 2013). Interestingly, patients with Kleefstra syndrome can have a 9q34 chromosomal deletion containing the EHMT1 gene and display childhood obesity, thus suggesting a plausible role for EHMT1 in obesity development (Cormier-Daire et al., 2003; Willemsen et al., 2012). In contrast, both LSD1 in association with the zinc finger protein 516 (Zfp516) (Sambeat et al., 2016), and Jhdm2a/Jmjd1a (KDM3A) in association with the chromatin remodeling complex SWI/SNF (Tateishi et al., 2009; Abe et al., 2015, 2018) catalyze the demethylation of H3K9me2/me3 to mediate transcriptional activation of BAT-selective genes (e.g., Ucp1, Pgc-1α) and stimulate thermogenic function. Consequently, abrogation of Lsd1 (Ucp1-Cre; Lsd1flox/flox) (Sambeat et al., 2016) or Jhdm2a (Jhdm2a–/–) (Inagaki et al., 2009; Tateishi et al., 2009) in mice results in increased body weight, fat accumulation, and impaired glucose homeostasis, as well as “whitening” of BAT and dysregulated fatty acid metabolism. Taken together, these results suggest an important role for these H3K9 HMTs and HDMs in obesity resistance.
Trimethylation of histone H3 at lysine 27 (H3K27me3), a repressive mark, is increased in the WAT of HFD-fed mice and obese patients (Yi et al., 2016). H3K27me3 is mediated by the Polycomb-Repressive Complex 2 (PRC2), which contains the Enhancer of Zeste Homolog 2 (EZH2) as the enzymatic component, catalyzing di- and tri-methylation of H3K27. EZH2 epigenetically represses Wnt genes (e.g., Wnt1, −6, −10a, −10b) to inhibit Wnt/β-catenin signaling while simultaneously promoting adipogenesis through the upregulation of Pparg and Cebpa (Wang L. et al., 2010; Yi et al., 2016; Jang et al., 2017). GSK126-mediated inhibition of EZH2 in HFD-induced obese mice reduces fat accumulation, improves glucose homeostasis, and increases adipose thermogenesis (Wu et al., 2018).
Dimethylation of histone H3 at lysine 36 (H3K36me2), an activation mark, is found to be protective against impaired adipose tissue function associated with obesity. The nuclear receptor binding SET domain protein 2 (NSD2) HMT mediates dimethylation at H3K36 for the activation of PPARγ-dependent gene programs, critical for mature brown and white adipocyte function. The depletion of the Nsd2-mediated H3K36me2 mark in adipocytes disrupts thermogenic function with “whitening” of BAT and increases insulin resistance of WAT (Zhuang et al., 2018).
Histone acetylation and deacetylation involve the addition and removal of acetyl groups to lysine residues on histone tails, and are mediated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively (Figure 2). Total HDAC activity is decreased in the adipose tissue of obese individuals and HFD-fed mice (Bricambert et al., 2016), and mutations in HDAC4, a class II HDAC, have been associated with obesity (Williams et al., 2010). This reduced HDAC activity is mainly attributed to decreased HDAC5 and HDAC6 (class II HDAC) levels in WAT, which are accompanied by a decrease in inducible cAMP early repressor (Icer) function and an increase in its target activating transcription factor 3 (Atf3), associated with insulin resistance (Bricambert et al., 2016). Additionally, HDAC5 can interact with the GLUT4 enhancer factor (GEF) in adipocytes for the repression of Glut4 promoter activity (Sparling et al., 2008; Weems and Olson, 2011), suggesting a plausible mechanism by which Glut4 expression and insulin-mediated glucose uptake are dysregulated in obesity and T2D. Moreover, the expression of class I HDACs, HDAC1 and HDAC3, is reduced in the adipose tissue of obese female patients (Jannat Ali Pour et al., 2020), however, their role in adipose tissue is not yet well understood.
Sirtuin 1 (SIRT1), the mammalian ortholog of the yeast silent information regulator 2 (Sir2) protein, is a nuclear NAD+-dependent class III HDAC that catalyzes the removal of acetyl groups from protein substrates. SIRT1 is often termed the “master metabolic regulator” due to its ability to modulate the expression of several key metabolic transcription factors and co-factors in response to environmental stimuli (Schug and Li, 2011; Li, 2013). SIRT1 gene and protein expression is significantly reduced in adipose tissue of HFD-fed mice (Yoshizaki et al., 2009, 2010) as well as in chronically obese patients in which it negatively correlates with their body mass index (BMI) (Costa Cdos et al., 2010; Gillum et al., 2011; Perrini et al., 2020). SIRT1 knockdown in human adipose progenitor cells results in a significant increase in cellular lipid content with elevated expression of adipogenic genes (PPARG2, SREBF1C, FASN, ADIPOQ, SLC2A4) (Perrini et al., 2020). Moreover, HFD-fed Sirt1 heterozygous mice (Sirt1+/–) (Xu et al., 2016), adipose-specific Sirt1-KO mice (aP2-Cre; Sirt1flox/flox), and obese patients with decreased adipose SIRT1 expression (Gillum et al., 2011) all exhibit increases in proinflammatory cytokine levels (IL-1β, TNFα, IL-10) and macrophage infiltration (Gillum et al., 2011; Xu et al., 2016) in WAT. Increased inflammation upon Sirt1 deficiency is associated with increased H3K9 acetylation of TNFα and IL-1β promoter sites (Vaquero et al., 2004; Yoshizaki et al., 2009; Gillum et al., 2011; Liu et al., 2011) and reduced adiponectin levels (Qiao and Shao, 2006; Gillum et al., 2011). In addition, the HFD-fed Sirt1 heterozygous mice (Sirt1+/–) present with more severe insulin resistance, compared with wild-type mice (Xu et al., 2016), which may be mediated by reductions in adipose GLUT4 translocation and insulin-stimulated glucose transport (Yoshizaki et al., 2009). These HFD-fed Sirt1+/– mice also exhibit reduced BAT thermogenesis as well as BAT degeneration indicated by mitochondrial dysfunction and loss. Taken together, these studies suggest a protective role of adipose SIRT1 in maintaining lipid and glucose homeostasis and inflammatory control, which is otherwise abrogated in the development of obesity and T2D.
Sirtuin 2 is another NAD+-dependent class III HDAC, which in contrast to the nuclear SIRT1, is primarily cytoplasmic, but can transiently shuttle to the nucleus for deacetylation of transcription factors (de Oliveira et al., 2012; Gomes et al., 2015). SIRT2 expression is found to be decreased in the adipose tissue of HFD-fed mice and obese patients (Krishnan et al., 2012; Perrini et al., 2020) and negatively correlates with their BMI, similar to SIRT1 (Perrini et al., 2020). In obesity, adipose SIRT2 expression is suppressed by adipose hypoxia-induced cellular hypoxia-inducible factor 1-α (HIF1α), which prevents SIRT2-mediated post-translational deacetylation and activation of PGC-1α and its FAO transcriptional gene program (Zhang X. et al., 2010; Krishnan et al., 2012). Similarly, SIRT2 knockdown in isolated human adipose stem cells promotes adipogenesis and lipid accumulation through the induction of PPARG2, SREBF1C, FASN, ADIPOQ, and SLC2A4 gene expression (Perrini et al., 2020), whereas SIRT2 over-expression inhibits this process. Therefore, hypoxia-induced reductions of SIRT2 in obesity may contribute to adipocyte dysregulation by limiting oxidative capacity and increasing lipid mass.
Whether these functions of SIRT2 in adipocytes are mediated by its HDAC activity or cytoplasmic role remains unclear. Previous studies have demonstrated that SIRT2 regulates adipocyte differentiation through direct modulation of FOXO1 acetylation (Jing et al., 2007; Wang and Tong, 2009). On the other hand, SIRT2 controls mitosis by modulating histone H4K16 acetylation (Vaquero et al., 2006) and since mitotic clonal expansion is critical for adipocyte differentiation (Tang and Lane, 2012), this suggests that SIRT2 may regulate adipogenesis through histone modifications. Indeed, it has recently been shown that SIRT6, another class III HDAC, controls mitotic clonal expansion during adipogenesis by repressing kinesin family member 5C (KIF5C) expression with deacetylation of H3K9ac and H3K56ac at its promoter (Chen et al., 2017). Since the loss of Sirt6 blocks adipogenesis and Sirt6 mutant mice are extremely lean and die early with numerous severe metabolic abnormalities (Xiao et al., 2010), these results emphasize that proper development of adipocytes is critical for maintaining metabolic balance. Altogether, the lack of SIRT1-, SIRT2- and SIRT6-dependent deacetylation and activation of specific adipose gene programs can contribute to the development of metabolic conditions, including obesity and T2D.
Modulation of lipid compartmentalization and efficient utilization of excess energy in adipose tissues are critical targets for the treatment of obesity and related metabolic dysfunctions. Dietary interventions, including IF and CR, markedly reduce adipocyte size and depot weights in rodent models of obesity (Wheatley et al., 2011; Kim K.H. et al., 2017; Liu B. et al., 2019; Miyamoto et al., 2019), and confer improvements in adipose tissue inflammation and insulin sensitivity (Anson et al., 2003; Wheatley et al., 2011; Duncan et al., 2016; Gotthardt et al., 2016; Kim K.H. et al., 2017; Li et al., 2017; Kim Y.H. et al., 2019; Liu B. et al., 2019; Figure 3). Additionally, adipose thermogenesis via the induction of WAT “browning”(beige fat) and activation of BAT appear to be predominant pathways (Hatori et al., 2012; Hatting et al., 2017; Kim K.H. et al., 2017; Li et al., 2017), which elevate energy expenditure, mitochondrial biogenesis and energy dissipating capacity (Harms and Seale, 2013; Cypess et al., 2015). Here, we summarize evidence for epigenetic links between dietary interventions and resulting metabolic improvements.
Female obese patients subjected to bariatric surgery with significantly reduced body weight (∼27%) and food intake show reductions in global DNA methylation levels and differentially methylated genes associated with obesity and T2D in adipose tissues (Benton et al., 2015; Dahlman et al., 2015), thus providing context for weight loss and adipocyte reprogramming. These genes are associated with the regulation of body weight (LEPR, FTO), cholesterol homeostasis (CETP, LCAT), blood glucose (IRS1, INSR) (Benton et al., 2015), adipose tissue function (mTOR, RPTOR) (Macartney-Coxson et al., 2017), and epigenetics (FOXP2, HDAC4, DNMT3B) (Benton et al., 2015). Studies investigating DNA methylation changes in adipose tissue upon dietary interventions, however, are limited. In one study, obese women on a 6-month CR diet (1100−1800 kcal/day) who lost >3% of their body fat showed hypermethylation at three genomic loci in their subcutaneous adipose tissue. Genes at these loci were associated with lipid (e.g., PLCL4) and glucose (e.g., ENC1) homeostasis and epigenetic regulation (e.g., PRDM8) (Bouchard et al., 2010). In particular, the ectodermal-neural cortex gene 1 (ENC1), previously associated with obesity (Zhao et al., 2000; Gerlini et al., 2018), was both differentially methylated (increased) and expressed (decreased) after CR treatment (Bouchard et al., 2010). In another study, 36-h of fasting in young, healthy men increased DNA methylation at the promoter site of LEP in subcutaneous adipose tissue, leading to a 3-fold decrease in plasma leptin levels (Hjort et al., 2017). Additionally, rosiglitazone, a PPARγ agonist, and a plausible CR-mimetic, mediates TET2-dependent demethylation of promoter regions of PPARγ target genes, such as ADIPOQ and FABP4, and results in enhanced insulin-stimulated glucose uptake in 3T3-L1 adipocytes (Bian et al., 2018). Altogether, these CR-related DNA methylation changes in adipose tissue can potentially be used as biomarkers of improved adiposity.
Histone methylation changes in adipose tissue as a result of dietary interventions have not yet been studied. Thus, studies showing similar alleviation of the disease state can be used to suggest analogous epigenetic mechanisms. For example, histone demethylases LSD1 of H3K4 (Duteil et al., 2014), JMJD1A of H3K9 (Abe et al., 2018), and UTX (KDM6A) (Zha et al., 2015), and JMJD3 (KDM6B) (Pan et al., 2015) of H3K27 mediate the induction of BAT-selective genes (e.g., Ucp1, Pgc-1α, Ppara, Cidea) in WAT for the development of thermogenically active beige adipocytes. Consequently, whole-body Lsd1 over-expressing mice (Rosa26-Lsd1) present with reduced body weight gain and increased energy expenditure, associated with smaller adipocyte size and greater mitochondrial content in WAT (Duteil et al., 2014). These epigenetic mechanisms of thermogenesis are in response to cold stress. As adipose thermogenesis is a key adaptation seen with the implementation of dietary interventions, such as IF and TRF (Hatori et al., 2012; Hatting et al., 2017; Kim K.H. et al., 2017; Li et al., 2017), these epigenetic modulators may be involved in this process, but require further investigation to establish a causal link.
Although direct evidence of adipose class I/II HDAC participation in dietary interventions is currently lacking, histone acetylation and deacetylation in adipose tissues are associated with the beneficial metabolic effects seen with dietary interventions. For example, 30% CR in HFD-fed mice leads to a significant increase in histone 4 acetylation (H4ac) at the Glut4 promoter, which is associated with increased Glut4 mRNA expression in WAT and decreased plasma glucose levels (Wheatley et al., 2011). Additionally, a number of HATs and HDACs are involved in regulating adipose thermogenesis, which is one of the primary beneficial mechanisms of dietary interventions (Hatori et al., 2012; Hatting et al., 2017; Kim K.H. et al., 2017; Li et al., 2017). The HATs Gcn5/Pcaf (KAT2A) acetylate H3K9 while CBP/p300 acetylate H3K18 and H3K27 for the activation of BAT-selective genes (e.g., Pparg, Prdm16, Angptl4) (Jin et al., 2011, 2014). In contrast, HDAC1 (class I HDAC) (Li et al., 2016), HDAC3 (class IIa HDAC) (Ferrari et al., 2017b; Liao et al., 2018), HDAC9 (class IIa HDAC) (Chatterjee et al., 2014a,b) and HDAC11 (class IV HDAC) (Bagchi et al., 2018) negatively regulate BAT differentiation and thermogenesis. In separate studies, treatment with an HDAC1 inhibitor (i.e., MS-275) (Ferrari et al., 2017a; Rajan et al., 2018) and genetic ablation of HDAC9 (Chatterjee et al., 2014a,b) and HDAC11 (Bagchi et al., 2018) in HFD-fed mice alleviate the obesity phenotype as a result of reduced body weight (Chatterjee et al., 2014a,b; Ferrari et al., 2017a; Rajan et al., 2018), improved glucose tolerance (Ferrari et al., 2017a; Rajan et al., 2018), and increased thermogenesis and “browning” of WAT (Chatterjee et al., 2014a,b; Ferrari et al., 2017a; Rajan et al., 2018). These metabolic benefits were partially mediated by the hyperacetylation and activation of BAT-selective genes (e.g., Ucp1, Pparg, Ppara, Prdm16, Pgc-1α, Cidea) (Chatterjee et al., 2014a,b; Ferrari et al., 2017b; Bagchi et al., 2018). Therefore, as fasting affects the expression and function of class I/II HDACs in the liver (Mihaylova et al., 2011) and hypothalamus (Funato et al., 2011), it would be promising to explore and identify a causal regulator and mechanism of histone acetylation in fasting- and dietary intervention-mediated adipose tissue remodeling and thermogenesis.
Sirtuin 1, a class III HDAC that is upregulated in WAT of mice upon CR (Chen et al., 2008) and fasting (Picard et al., 2004), acts as a negative modulator of adipogenesis. SIRT1 complexes with NCoR/SMRT at the Pparg promoter to co-repress target genes involved in TG storage (Picard et al., 2004) and also post-translationally deacetylates FOXO1 to increase the expression of its target gene Atgl in TG hydrolysis (Chakrabarti et al., 2011).
Another role of SIRT1 is in the attenuation of adipose inflammation, as seen with dietary interventions, such as CR and IF (Wheatley et al., 2011; Kim K.H. et al., 2017; Liu B. et al., 2019). SIRT1, in association with FOXO1 and C/EBPα, forms a transcriptional activator complex at the Adipoq promoter (Qiao and Shao, 2006) for increased stimulation of the anti-inflammatory adiponectin upon CR and IF (Zhu et al., 2004; Kim K.H. et al., 2017). SIRT1-dependent deacetylation of NF-kB and IL-1β promoter sites have also been reported (Yoshizaki et al., 2009; Liu et al., 2011). Similarly, over-expression of SIRT1 (Gillum et al., 2011) and the use of SIRT1 activators (SRT1720, SRT2379, resveratrol) (Yoshizaki et al., 2009; Yoshizaki et al., 2010; Liu et al., 2016) in HFD-fed or genetically obese mice, suppress NF-kB signaling and gene expression (e.g., IL-6, Tnfa, Mcp-1) (Yoshizaki et al., 2009, 2010; Gillum et al., 2011) and reduce macrophage infiltration in WAT (Yoshizaki et al., 2010; Gillum et al., 2011). Altogether, these studies suggest that the anti-inflammatory effects of dietary interventions may be mediated by SIRT1.
Moreover, SIRT1 expression is increased in BAT of mice upon a 48-h fast (Cordeiro et al., 2013) and 40% CR (Wei et al., 2020). Genetic over-expression (Boutant et al., 2015) and activation (SIRT1720) (Feige et al., 2008) of SIRT1 in mice induces BAT thermogenesis and lipid catabolism, which are mediated by increased expression of BAT-selective transcriptional regulators (PPARα, PPARγ, PGC-1α, PGC-1β, FOXO1, FOXO3a), uncoupling and detoxification factors (UCP1, UCP3, SOD1, SOD2) and FAO genes (Mcad, Lcad, Cpt1b, Cpt1a) (Feige et al., 2008; Boutant et al., 2015). Additionally, SIRT1 gain-of-function mice exhibit a greater “browning” phenotype of WAT, indicated by the appearance of smaller adipocytes and elevated brown adipocyte marker genes (Ucp1, Dio2, Cebpb, Cox7a1, Cidea) upon cold exposure, in comparison to wild-type mice. The post-translational SIRT1-dependent lysine deacetylation of PPARγ and its interaction with the browning co-factor Prdm16 allow for this thermogenic remodelling of WAT (Qiang et al., 2012). These studies thereby suggest that the upregulation of SIRT1 may mediate the increased thermogenesis and WAT “browning” seen upon dietary interventions (Hatori et al., 2012; Hatting et al., 2017; Kim K.H. et al., 2017; Li et al., 2017), however further studies are still required to establish a mechanistic link.
Sirtuin 2, another class III HDAC, is also upregulated by CR and fasting in WAT (Wang et al., 2007) and can reduce cellular lipid stores and oxidative stress as seen with dietary interventions (Jing et al., 2007; Wang et al., 2007; Wang and Tong, 2009; Perrini et al., 2020). In adipocytes, the post-translational SIRT2-mediated deacetylation of FOXO1 inhibits the transcriptional activation of PPARγ target genes involved in adipogenesis (Wang and Tong, 2009; Perrini et al., 2020). Additionally, SIRT2 can mediate the post-translational deacetylation of FOXO3a to promote the expression of FOXO target genes involved in the reduction of cellular reactive oxygen species (ROS) (MnSOD), the apoptotic clearance of damaged cells (Bim) and the inhibition of cell proliferation and propagation of mutations (p27kip1) (Wang et al., 2007).
Fatty liver disease is closely associated with both obesity and T2D, with 82% of NAFLD patients presenting with obesity and 48% with T2D, in America (Younossi et al., 2016; Purnell et al., 2017). Intimately linked to systemic energy utilization and storage, the liver functions differently in the fed and fasted states, and its dysregulation can cause NAFLD. During feeding conditions, insulin promotes the storage of glucose into FA and TG or as glycogen through glycogenesis. During fasting, glucagon stimulates the mobilization of TG and glycogen stores for fuel delivery to extra-hepatic tissues, while simultaneously activating hepatic FAO and gluconeogenesis, fueled by adipocyte lipolysis and muscle proteolysis, respectively (Rui, 2014). Under conditions of metabolic dysregulation or disease, insulin resistance of the liver promotes inappropriate upregulation of gluconeogenesis while de novo lipogenesis (DNL) pathways remain insulin sensitive, contributing to hyperglycemia and hepatic lipid accumulation, respectively (Brown and Goldstein, 2008; Figure 4). Impaired FAO and very-low-density lipoprotein (VLDL) secretion as a result of insulin resistance further increase fat deposits in the liver (Bhatt and Smith, 2015). NAFLD develops when hepatic lipid stores exceed 5% of tissue mass, leading to increased inflammation, collagen deposition, fibrosis and cell death. If left untreated, NAFLD can progress to non-alcoholic steatohepatitis (NASH) and may continue to cirrhosis and hepatocellular carcinoma (HCC) (Byrne and Targher, 2015; Friedman et al., 2018). Altogether, the metabolic role of the liver in integrating these endogenous and exogenous fuel sources requires constant transcriptional modulation. Below we highlight some epigenetic changes regulating hepatic gene expression in both disease and dietary interventions.
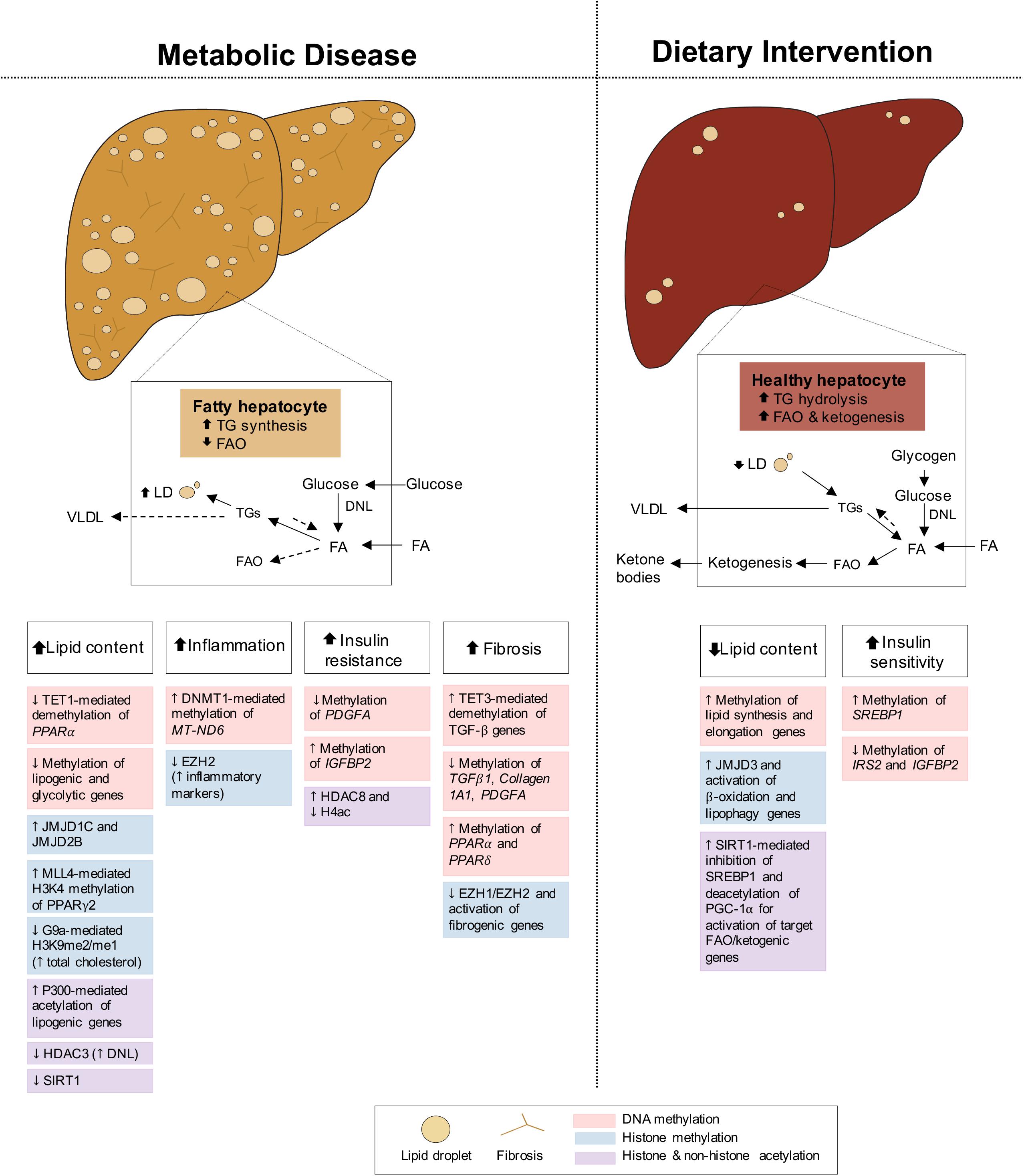
Figure 4. Epigenetic changes of liver in metabolic disease and dietary intervention. Liver in metabolic disease (i.e., NAFLD/NASH) presents with increased lipid content, inflammation, insulin resistance and fibrosis. The liver in dietary intervention has reduced lipid content and improved insulin sensitivity. These physiological differences of the liver can be explained by epigenetic changes involving DNA methylation, histone methylation and histone and non-histone acetylation.
Hepatic methylome and transcriptome studies have identified epigenetic links to the differentially expressed genes underlying
the development of hepatic insulin resistance, T2D, and NAFLD (Ahrens et al., 2013; Nilsson et al., 2015; Kirchner et al., 2016; de Mello et al., 2017; Abderrahmani et al., 2018; Gerhard et al., 2018; Hotta et al., 2018). Notably, in the liver of T2D patients, the majority of significant differentially methylated CpG sites show reduced DNA methylation (Nilsson et al., 2015; Kirchner et al., 2016; Abderrahmani et al., 2018). Hypomethylation of a CpG site within or proximal to the activating transcription factor (ATF)-binding motif of hepatic genes accounts for increased expression of these genes involved in glycolysis (PFKL), DNL (ACACA, FASN), and insulin signaling (PRKCE) in obese and T2D patients (Kirchner et al., 2016). Moreover, the epigenetic induction of PDGFA, which encodes platelet-derived growth factor α (PDGF-AA), appears to be central to hepatic disease progression, as PDGF-AA causes insulin resistance by reducing hepatic insulin receptor density in a protein kinase C (PKC)-dependent manner (Thieringer et al., 2008). In patients with T2D, a CpG site (cg14496282) within PDGFA is found to be hypomethylated, leading to increased hepatic PDGFA expression and PDGF-AA secretion from insulin-resistant human hepatocytes (Abderrahmani et al., 2018). Additionally, PDGFA expression is positively correlated with hepatic fibrosis and NASH risk, and as such, the over-expression of PDGF-AA in mouse liver results in spontaneous liver fibrosis (Thieringer et al., 2008). Since the degree of hepatic fibrosis is an indicator of morbidity and mortality of liver diseases including NAFLD (Dulai et al., 2017), a DNA methylation study conducted with liver samples with NAFLD-related cirrhosis has identified genes enriched in ligand-activated nuclear receptor signaling pathways, involving farnesoid X receptor (FXR), liver X receptor (LXR), and retinoid X receptor (RXR), that play roles in fatty liver disease (Tanaka et al., 2017; Gerhard et al., 2018). Other DNA methylation studies (Ahrens et al., 2013; Nilsson et al., 2015; de Mello et al., 2017; Abderrahmani et al., 2018; Hotta et al., 2018) have also found a number of common differentially methylated sites, including the fibroblast growth factor receptor 2 (FGFR2) involved in liver fibrosis (Thieringer et al., 2008). In addition, hypomethylation of CpG sites within TGFβ1, Collagen 1A1, and PDGFα as well as hypermethylation of CpG sites within PPARα and PPARδ are frequently associated with the increased risk of fibrosis in NAFLD patients (Zeybel et al., 2015; Abderrahmani et al., 2018).
However, the question remains as to why hypomethylation is often found in metabolically dysfunctional livers as well as adipose tissues? Circulating folate levels are reduced in T2D patients compared with non-diabetic subjects (Nilsson et al., 2015). Since folate is a methyl donor in the methylation cycle, hypomethylation in the liver as well as in the pancreas of T2D patients (Dayeh et al., 2014) can be explained by a methyl donor supply consumption imbalance (Zhou et al., 2011; Nilsson et al., 2015). This is supported by a previous study using mice fed with a methyl-deficient diet (Pogribny et al., 2009). Lack of methyl donors accompanied by a loss of genomic cytosine methylation and a change in the expression of hepatic DNA methyltransferases, causes NAFLD and even NASH in mice, highlighting the role of hypomethylation in hepatic steatosis.
While hypomethylation is more common in hepatic tissues of metabolic disease, patients with NASH exhibit higher hepatic expression of DNMT1, which increases methylation and decreases expression of mitochondrial NADH dehydrogenase 6 (MT-ND6), leading to ultrastructural defects in mitochondrial morphology (Pirola et al., 2013). In addition, increased methylation at a CpG site (cg11669516) and reduced gene expression of insulin-like growth factor binding protein 2 (IGFPB2) are often found in mice and patients with NAFLD and NASH (Ahrens et al., 2013). Consistently, HFD feeding in young mice induces hypermethylation of Igfbp2 and reduces its expression prior to diet-induced obesity and hepatic steatosis development. This epigenetic inhibition of Igfbp2 becomes stable over time in adult mice, suggesting Igfbp2 methylation as a predictable risk indicator of liver disease development (Kammel et al., 2016). Together, genome-wide DNA methylation studies combined with ex vivo and in vitro analyses provide key epigenetic mechanisms underlying NAFLD development and progression in obese and T2D patients, by linking differential methylation states with the regulation of hepatic glucose and lipid metabolism, insulin resistance, and hepatic fibrosis.
In addition to changes in genomic DNA methylation, differential expression of DNA methylation regulators has been associated with the development of hepatic diseases, particularly the TET proteins involved in hydroxymethylation, the initial step in the reversal of DNA methylation. TET1 expression is reduced in both in vitro (HepG2 cells containing FA medium) and in vivo (HFD-fed mice) models of NAFLD (Wang et al., 2020). Loss of Tet1 (Tet–/–) in mice further exacerbates HFD-induced NAFLD, indicated by increased intrahepatic TG levels. This study suggests that hepatic Tet1-mediated hydroxymethylation of the PPARα promoter enhances FAO and thereby prevents NAFLD development.
Furthermore, hepatic fibrosis is a critical pathological process that affects clinical management, as its advancement determines the therapeutic reversibility of NAFLD by leading to irreversible cirrhosis and even HCC (Stal, 2015; Thiele et al., 2017). Abnormal activation of the inflammatory transforming growth factor-beta (TGF-β) signaling pathway along with the transdifferentiation of hepatic stellate cells (HSCs) into proliferative, fibrogenic myofibroblasts, primarily drive hepatic fibrosis through the production of the extracellular matrix (ECM) (Tsuchida and Friedman, 2017). TET3 expression is increased in hepatocytes and HSCs of human fibrotic livers (Xu et al., 2020). In HSCs, TET3 mediates demethylation at specific CpG sites of genes involved in the TGF-β pathway, including TGFB1, to promote profibrotic gene expression and subsequent ECM production. In contrast, siRNA-mediated TET3 knockdown ameliorates liver fibrosis in mice, suggesting its crucial role in the pathological development and progression of hepatic fibrosis. This transdifferentiation of HSCs, largely regulated by a number of epigenetic processes including DNA methylation as described here, and post-translational modification of histones, is reviewed in detail elsewhere (Barcena-Varela et al., 2019).
The development of metabolic dysfunction in liver diseases accompanies global histone modifications including acetylation and methylation (Nie et al., 2017). Notably, alterations in global histone methylation patterns and expression of the regulators, such as HMTs and HDMs, during development and progression of NAFLD have been reported in a number of recent studies.
The hepatic expression of G9a (EHMT2), a H3K9 HMT, is markedly reduced in genetically induced (i.e., db/db) and HFD-fed obese mice (Xue et al., 2018). The liver-specific loss of G9a is associated with a selective decrease in hepatic H3K9me2/me1 and an increase in serum cholesterol levels (Lu et al., 2019). Upon liver injury (e.g., lipopolysaccharide and acetaminophen overdose), G9a mutant mice exhibit severe liver phenotypes associated with increased immune cell infiltration, ROS production and cell death (Zhang et al., 2020), suggesting an epigenetic protective role of G9a in the liver. On the other hand, the H3K9 demethylase, JMJD1C (KDM3), a candidate gene associated with T2D and plasma TG levels (Chasman et al., 2009; Teslovich et al., 2010; Zhang H. et al., 2016), regulates hepatic lipogenic gene expression (e.g., FAS, ACC, SREBF1) by demethylating the H3K9me2/me3 transcriptional repressor marks and leading to increased chromatin accessibility (Viscarra et al., 2020). Over-expression of JMJD1C in the liver increases DNL, whereas liver-specific deletion of Jmjd1c protects mice from diet-induced hepatic steatosis and insulin resistance. Similarly, the H3K9 di- and tri-demethylase JMJD2B (KDM4B), involved in establishing the H3K9me activation mark, is upregulated in livers of diet-induced obese mice, resulting in increased hepatic PPARγ2 expression and induction of hepatic steatosis (Kim et al., 2018). Moreover, Phf2, another H3K9 HDM, specifically demethylates H3K9me2 on the promoter of carbohydrate-responsive element-binding protein (ChREBP) (Bricambert et al., 2018), a major regulator of glycolytic and lipogenic genes (Ortega-Prieto and Postic, 2019). Interestingly, while glucose homeostasis remains preserved, liver-specific Phf2 over-expression results in hepatosteatosis, mediated by increased stearoyl-CoA desaturase (Scd1) expression and accumulation of monounsaturated fatty acids. Conversely, Phf2 silencing leads to liver fibrosis upon a high-fat, high-sucrose diet. With supporting human data, this study suggests Phf2 as a targetable epigenetic checkpoint to prevent NAFLD progression (Bricambert et al., 2018). Together, these studies demonstrate the critical and dynamic implications of H3K9 HMTs and HDMs in the development and progression of liver diseases.
Another HMT EZH2, which catalyzes trimethylation of H3K27 (H3K27me3) for transcriptional repression, also plays a key role in liver diseases. EZH2 expression is reduced in the liver of NAFLD rats and FA-treated HepG2 hepatocytes and is inversely correlated with lipid accumulation and inflammatory marker expression (Vella et al., 2013). The steatosis-related phenotypes are recapitulated when treated with 3-Deazaneplanocin A, an EZH2 inhibitor, suggesting a causal role of EZH2 in NAFLD development. In addition, EZH1, a homolog of EZH2, has H3K27 methyltransferase activity and can partially compensate for the loss of EZH2 (Ezhkova et al., 2011). Notably, when both Ezh1 and Ezh2 are deficient in the liver, the mutant mice develop liver fibrosis with increased fibrogenic gene expression (Fstl1, Fbn1 and Col1a1) (Grindheim et al., 2019).
While PPARγ2 is a master transcriptional factor of adipogenesis in adipocytes, its expression is elevated in the fatty livers of obese animal models and NAFLD patients (Vidal-Puig et al., 1996; Westerbacka et al., 2007). Hepatic PPARγ2 stimulates the uptake and re-esterification of FA into lipid droplets by upregulating Cd36, Fabp4, Mag, Plin2, and Fsp27/Cidec, and thereby promoting steatosis (Kim et al., 2016; Kim K. et al., 2017). This upregulation of hepatic PPARγ2 can be epigenetically achieved by H3K4 methyltransferase MLL4 (KMT2D) (Kim et al., 2016) as well as through defective HDAC3, which normally associates with retinoic acid receptor-related orphan receptor alpha (RORα) to repress PPARγ2 transcription (Kim K. et al., 2017). Together, these studies provide multimodal histone modulatory mechanisms of NAFLD via methyltransferase- and deacetylase-mediated transcriptional regulation of hepatic PPARγ2.
The modulation of hepatic histone acetylation, achieved through HATs and HDACs, contributes to the development of NAFLD. Among HATs, the transcriptional coactivator p300 is activated in mouse models of obesity and T2D, leading to post-translational hyperacetylation of ChREBP, involved in the transcriptional activation of lipogenic (e.g., Acc, Fas) and glycolytic (e.g., Pepck, G6Pase) genes (Bricambert et al., 2010). The activity of hepatic p300 is negatively regulated by salt-inducible kinase 2 (SIK2), thus, liver-specific SIK2 knockdown results in increased transcriptional activity of ChREBP via p300-mediated acetylation. In addition, hepatic p300 over-expression is sufficient to induce NAFLD and insulin resistance. Conversely, inhibition of HAT activity prevents NAFLD: the novel HAT inhibitor, tannic acid, binds to p300 and disrupts its occupancy on lipogenic genes (e.g., Fasn, Acly), leading to hypoacetylation of H3K9ac and H3K36ac (Chung et al., 2019). Together, these findings suggest a role for acetyltransferase p300 in NAFLD development.
Histone deacetylases also have key implications in the regulation and dysregulation of hepatic metabolism. As a significant metabolic hub, the functions of the liver in lipid, carbohydrate and amino acid metabolism as well as detoxification are regulated throughout the day by circadian clocks (Reinke and Asher, 2016). This rhythmic hepatic metabolism is orchestrated by epigenetic modulation, mainly through HDAC3, which is recruited by a key circadian clock component, Rev-ebrα (Feng et al., 2011). Upon hepatic Hdac3 depletion, mice develop hepatosteatosis with increased DNL, suggesting a critical role of histone acetylation-mediated circadian changes in the prevention of NAFLD. Additionally, HDAC8 has been identified as a commonly upregulated gene in dietary and genetic obesity-promoted HCC mouse models as well as in human HCC cells and tissues (Tian et al., 2015). HDAC8 promotes insulin resistance as well as cell proliferation, while its knockdown inhibits NAFLD-HCC tumorigenicity. Specifically, HDAC8, in association with the HMT EZH2, epigenetically regulates the Wnt pathway via decreased histone H4 acetylation and increased H3K27me3 methylation. This finding suggests an epigenetic mechanism involving HDAC8 in the progression of NAFLD-associated HCC.
Sirtuin 1, a class III HDAC, acts on hepatic metabolic regulators in response to hormonal and nutritional signals. Sirt1 levels are reduced in a HFD-induced NAFLD rodent model (Deng et al., 2007) and are significantly lower in obese patients with severe steatosis (Wu et al., 2014; Mariani et al., 2015). Liver-specific deletion or knockdown of Sirt1 in mice leads to fatty liver disease even without a HFD challenge (Rodgers and Puigserver, 2007; Wang R.H. et al., 2010; Xu et al., 2010; Yin et al., 2014). Upon HFD or an alcoholic diet, these Sirt1 mutant mice develop severe liver injury and fibrosis (Purushotham et al., 2009; Yin et al., 2014; Ramirez et al., 2017). Moreover, these mice lacking hepatic Sirt1 exhibit elevated gluconeogenesis, leading to hyperglycemia and insulin resistance, which suggests that hepatic SIRT1 plays a role in not only protecting the liver from steatosis, but also in maintaining whole-body glucose metabolism.
Hepatic lipid accumulation is the primary characteristic and key contributor to NAFLD pathogenesis. Several dietary interventions, such as IF, CR, and KD, have shown protection against and improvement in hepatosteatosis with increased FAO, ketogenesis, and reduced lipogenesis in both obese or diabetic mice (Badman et al., 2009; Baumeier et al., 2015; Kim K.H. et al., 2017; Marinho et al., 2019) and humans (Larson-Meyer et al., 2008; Browning et al., 2011; Sevastianova et al., 2012; Yu et al., 2014; Drinda et al., 2019; Johari et al., 2019; Luukkonen et al., 2020). Additionally, in both rodents and humans, dietary interventions prevent and alleviate the onset of hepatic inflammation (Horrillo et al., 2013; Marinho et al., 2019) and fibrosis (Horrillo et al., 2013; Johari et al., 2019), associated with the more severe NASH and cirrhosis condition of the liver (Figure 4). Taken together, the success of dietary interventions in halting and, in some cases, reversing NAFLD progression, makes it a promising alternative to current therapeutics. Thus, an improved understanding of both accompanying and causal epigenetic changes in the prevention and/or treatment of NAFLD will be necessary for determining novel molecular biomarkers and specific pharmaceutical targets for clinical translation.
Consistent with its anti-aging effect in humans and other model organisms (Fontana and Partridge, 2015; Redman et al., 2018), 40% CR can protect against the age-related increase in global hepatic DNA methylation (Miyamura et al., 1993) and reduce the epigenetic age of the mouse liver by approximately 1.7 years (Maegawa et al., 2017; Wang et al., 2017). In particular, a study comparing young and aged female mice showed that 40% CR delays the epigenetic aging of hepatic lipid metabolism by inducing hypermethylation and down-regulation of key enzymes involved in hepatic insulin resistance (e.g., Srebp1), lipid synthesis (e.g., Acly, Mel, Aacs2, Acac, Pklr, Gpam) and lipid elongation (e.g., Elov15, Elov16). These changes increase insulin sensitivity while reducing lipid content and chain length of TG-associated FA in the livers of old female mice (Hahn et al., 2017). More specifically, the reduction in the level of FA elongases and the subsequent shift in the hepatic TG pool from long-to medium-chain TG (Hahn et al., 2017) is associated with the prevention of diet-induced insulin resistance and liver disease (Ronis et al., 2013; Geng et al., 2016). Sterol regulatory element-binding protein 1 (SREBP1) contributes to insulin resistance by interfering with the binding of FOXO1 to the insulin receptor substrate 2 (Irs2) promoter, thereby repressing Irs2 expression. CR has been shown to improve insulin resistance in mice through hypermethylation and downregulation of Srebp1 as well as through hypomethylation and upregulation of Irs2, the direct target of SREBP1 (Hahn et al., 2017). Moreover, in a separate study, CR-treated mice with a 25% reduction of body weight, exhibit decreased methylation and increased expression of hepatic Igfbp2 – an effect that is abolished by HFD re-feeding (Kammel et al., 2016). Since the reverse, increased methylation and decreased expression of IGFBP2, is found in both mice and humans with NAFLD and NASH (Ahrens et al., 2013), this CR-mediated change in Igfbp2 levels indicates metabolic improvements (Nam et al., 1997; Heald et al., 2006; Carter et al., 2014). CR also regulates the expression of DNA methylation modulators. In both young and old female mice, 40% CR increased Tet3 and Dnmt3a and decreased Tet2, Dnmt1 and Dnmt3b expression in the liver (Hahn et al., 2017). While altered hepatic expression of DNMT and TET enzymes in regulating DNA methylation is not well understood, this finding suggests that CR may modulate hepatic transcripts via dynamic regulation of DNA methylation machinery.
Dietary interventions, such as fasting, can modulate hepatic histone methylation and subsequent gene transcription. In particular, fasting-induced protein expression of JMJD3 (KDM6B), a H3K27me3 HDM (Seok et al., 2018; Byun et al., 2020), stimulates the expression of β-oxidation (e.g., Fgf21, Cpt1a, Mcad) (Seok et al., 2018) and autophagy genes (e.g., Tfeb, Ulkl, Atgl) (Byun et al., 2020) in the liver, thereby promoting the removal of hepatic lipid stores via increased lipolysis and lipophagy. This histone modulation by hepatic JMJD3 in response to fasting is mediated by its association with two transcriptional activating complexes; JMJD3 in complex with PKA-phosphorylated SIRT1 (Ser434) and PPARα (Seok et al., 2018) or PKA phosphorylated JMJD3 (Thr1044) in complex with FGF21 (Byun et al., 2020). The fasting-induced JMJD3-SIRT1-PPARα complex additionally forms a feed-forward regulatory loop, which auto-induces the expression of its genes, including Fgf21, Jmjd3, Sirt1 and Ppara, to amplify the cellular responses under fasting conditions. Importantly, the downregulation of Jmjd3 and its associated factors (Sirt1, Fgf21, Ppara) in the mouse liver results in reduced hepatic β-oxidation and increased steatosis. These data suggest a critical role for histone methylation modulators in mediating the metabolic improvements associated with fasting-related dietary interventions against liver metabolic dysfunction.
Hepatic histone acetylation in the context of dietary interventions mainly involve HDACs. Specifically, under fasting conditions, class IIa HDACs upregulate the hepatic gluconeogenic gene program. Fasting-induced glucagon-secretion in primary mouse hepatocytes promotes protein kinase A (PKA)-mediated phosphorylation and inactivation of SIK2, thereby allowing for the nuclear translocation of unphosphorylated class IIa HDACs (Wang et al., 2011). Nuclear HDAC4 and HDAC5 (class IIa HDACs) in association with HDAC3 (class I HDAC) deacetylate and activate FOXO transcription factors for the induction of gluconeogenic genes (Mihaylova et al., 2011).
Under low nutrient conditions (i.e., IF, CR), an increase in the NAD+/NADH ratio activates hepatic SIRT1, a class III HDAC. The genetic over-expression of SIRT1 or its activation by resveratrol treatment protects against HFD-induced hepatosteatosis and glucose intolerance (Rodgers and Puigserver, 2007; Pfluger et al., 2008) as well as alcoholic diet-induced liver injury and fibrosis (Ajmo et al., 2008; Ramirez et al., 2017). Mechanistically, SIRT1 post-translationally deacetylates and activates PGC-1α, which interacts with its co-factor HNF4α to stimulate the expression of gluconeogenic (e.g., G6pase, Pepck, Fbp1, G6pc) and β-oxidation (e.g., Mcad, Cpt-1a, Dgat2) genes, while repressing glycolytic genes (e.g., Lpk, Gck) (Nemoto et al., 2005; Rodgers et al., 2005; Rodgers and Puigserver, 2007). SIRT1-deacetylated PGC-1α also regulates PPARα target genes involved in hepatic FAO and ketogenesis (Rodgers and Puigserver, 2007; Purushotham et al., 2009; Hayashida et al., 2010). Hepatocyte-specific deletion of Sirt1 results in the hyperacetylation of PGC-1α at PPAR response element (PPRE) sites on target genes, thereby inhibiting PPARα signaling (Purushotham et al., 2009). Additionally, fasting-induced hepatic SIRT1 can post-translationally deacetylate SREBP1 at its DNA-binding domain, leading to its ubiquitin-mediated proteasomal degradation (Hirano et al., 2001; Sundqvist and Ericsson, 2003; Walker et al., 2010). Inhibition of hepatic SREBP activity promotes fat mobilization through the activation of lipolytic and FAO pathways, thereby reducing hepatic fat stores and protecting against hepatic steatosis (Walker et al., 2010). SIRT1 also mediates the acetylation of histones H3 and H4 (i.e., H3K9Ac, H3K56Ac, H3K18Ac and H4K16Ac) (Bosch-Presegue and Vaquero, 2015), for the regulation of chromatin structure and transcriptional activation.
However, the necessity of SIRT1 in IF, TRF, and FMD has not been mechanistically tested yet. Interestingly, a study comparing transcriptomic changes by IF (i.e., ADF) and Sirt1 over-expression in mice concluded that despite functional similarities such as improved insulin sensitivity, Sirt1 gain-of-function does not mimic nor boost the metabolic effects of IF (Boutant et al., 2016). This suggests that Sirt1 may not be the only mediator of fasting-involved dietary interventions. Moreover, the current literature has conflicting results in regard to hepatic SIRT1 expression under fasting and nutritional interventions (i.e., CR). Although most studies show an upregulation of hepatic SIRT1 upon fasting or CR (Cohen et al., 2004; Rodgers et al., 2005; Deng et al., 2007; Rodgers and Puigserver, 2007; Hayashida et al., 2010; Ding et al., 2017), some studies have reported a decrease (Chen et al., 2008) or no change (Barger et al., 2008) in expression. In particular, it was reasoned that the decreased level and activity of Sirt1 in the livers of CR mice was due to low cellular NAD+/NADH levels (Hagopian et al., 2003; Lin et al., 2004; Chen et al., 2008; Kawakami et al., 2012). Furthermore, the liver-specific down-regulation of Sirt1 in these studies resulted in reduced hepatic fat synthesis and improved glucose homeostasis (Chen et al., 2008; Erion et al., 2009) – an effect that contradicts other studies showing increased hepatic FA and cholesterol, impaired glucose tolerance, hepatic inflammation and steatosis (Rodgers and Puigserver, 2007; Purushotham et al., 2009) upon Sirt1 deficiency. Aside from variation in animal species, strain or age used in the studies, differences in hepatic SIRT1 expression may also be attributed to differences in length of fasting or extent of food restriction. Particularly, SIRT1 has been shown to be upregulated in the liver of mice upon long-term fasting (18−24 h), but not short-term fasting (6−8 h), and thus may play a role in the later stages of nutrient depletion (Liu et al., 2008). Some discrepancies might also originate from different experimental settings. Although metabolic phenotyping (e.g., indirect calorimetry) and tissue harvesting are commonly performed after an overnight fast post-feeding day, the metabolic benefits of dietary interventions can take place during fasting and/or refeeding. For example, both the elevation of energy expenditure via adipose thermogenesis by IF (Kim K.H. et al., 2017) and pancreatic β-cell regeneration by FMD (Cheng et al., 2017) occur during the refeeding period after fasting and nutritional interventions. Regardless, these conflicting studies do not undermine the importance of SIRT1 in the liver and the use of SIRT1 agonists or fasting-like mimetics as possible therapeutic options for patients with metabolic syndrome. Overall, the liver-specific epigenetic mechanisms described herein are important in understanding the pathophysiological development of NAFLD and its possible alleviation via dietary interventions.
The pancreas is a secretory organ with both exocrine (acinar) and endocrine (islet) function. Among the pancreatic islet cell types, the glucagon-secreting α-cells and the insulin-secreting β-cells are primarily involved in regulating the metabolic pathways of the fed and fasted states. Under fasting conditions, glucagon is released to increase blood glucose levels through the promotion of hepatic gluconeogenesis and glycogenolysis, whereas under feeding conditions, insulin is secreted to promote the uptake of glucose, amino acids and FA and to stimulate processes of glycogenesis, protein synthesis and DNL in insulin-sensitive metabolic tissues (Piciucchi et al., 2015; Roder et al., 2016; Weisbeck and Jansen, 2017). In the setting of insulin resistance, β-cells increase their insulin secretion (i.e., hyperinsulinemia) to maintain normal glucose levels (Johnson and Alejandro, 2008; Figure 5). However, when β-cells can no longer sustain the increased demand (i.e., hypoinsulinemia), glucose levels rise and initially present as impaired glucose tolerance (Kahn et al., 2014). As β-cell dysfunction progresses, hyperglycaemia and diabetes arise (Weisbeck and Jansen, 2017). Eventually, the hyperactivity of β-cells and high levels of blood glucose and lipids contributing to glucotoxicity and lipotoxicity, can stimulate β-cell apoptosis and further propagate the pathogenesis of T2D (Deng et al., 2010). In addition to the genetic component, T2D is also largely influenced by environmental stressors, such as prolonged physical inactivity and an unhealthy diet (i.e., fatty foods high in dioxins) (Hoyeck et al., 2020; Ibrahim et al., 2020), which result in changes in metabolic gene expression, mediated through epigenetics. Herein we discuss some of the epigenetic changes involved in both T2D development and alleviation via dietary interventions in the pancreas; the key organ regulating both plasma insulin and glucose levels.
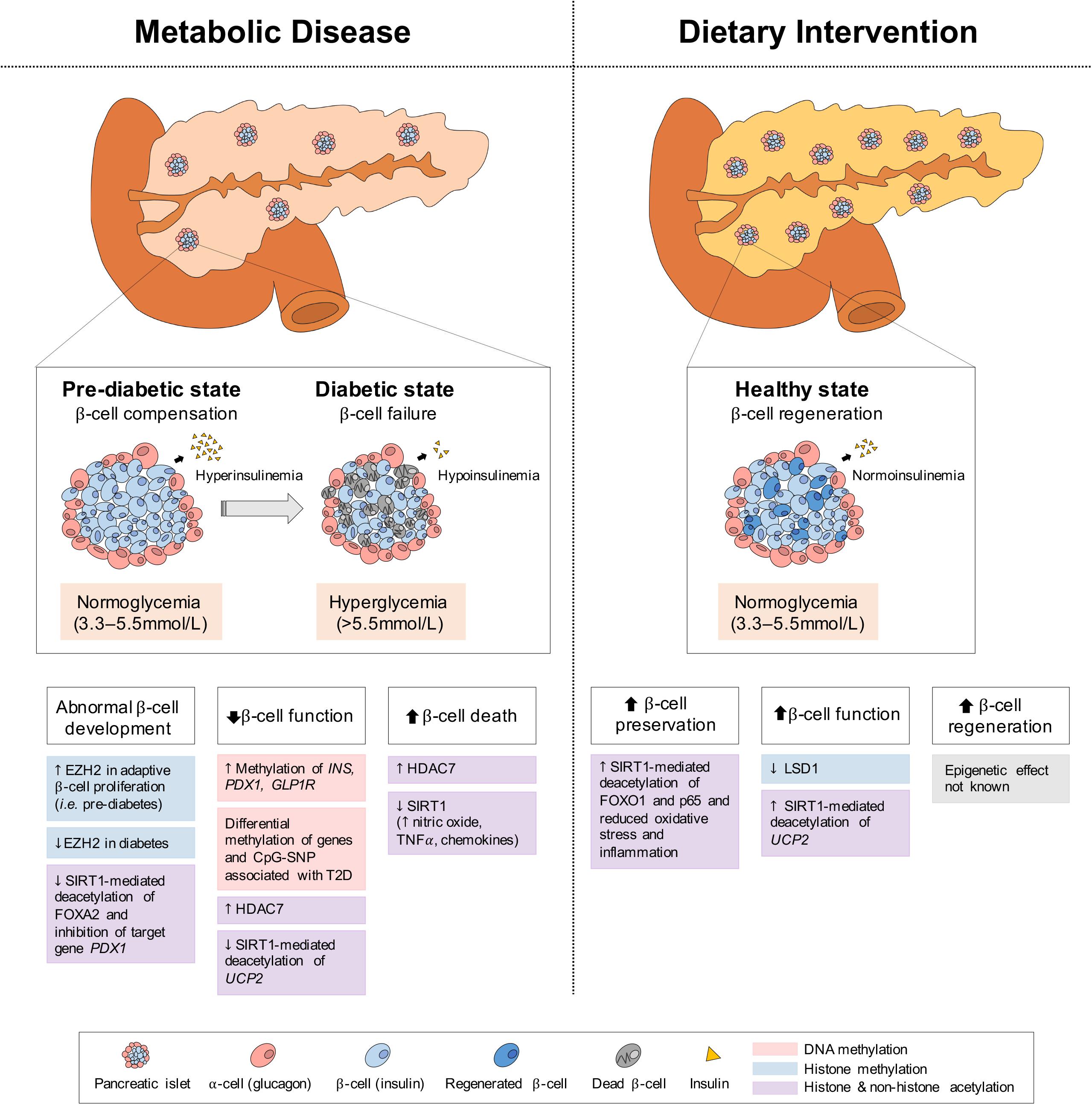
Figure 5. Epigenetic changes of pancreas in metabolic disease and dietary intervention. In metabolic disease (i.e., T2D), the initial proliferation of pancreatic β-cells increase insulin secretion, but eventual β-cell failure leads to hypoinsulinemia and hyperglycemia. In dietary intervention, pancreatic β-cells are preserved through reduced inflammation and oxidative stress and present with increased function (i.e., GSIS) and regeneration. These physiological differences in the pancreas can be explained by epigenetic changes involving DNA methylation, histone methylation and histone and non-histone acetylation.
As pancreatic islets are central to T2D development, the methylation status of the promoters of critical genes in islet function and development have been investigated. In islets of T2D patients, the increased DNA methylation of the insulin gene promoter (INS) at 4 CpG sites, correlates negatively with insulin mRNA levels and positively with glycated hemoglobin HbA(1c) levels, which reflect the cumulative blood glucose concentration (Yang et al., 2011). Similarly, in an in vitro model of a rat β-cell line (INS 832/13), 72-h of high glucose exposure (16.7 mmol/L), which recapitulates conditions of hyperglycemia in T2D, increases DNA methylation within the Ins promoter (Yang et al., 2011). Likewise, in another study, 48-h of high glucose exposure (19 mM) in isolated human islets results in differential methylation and expression of genes involved in islet function, including GLRA1, RASD1, VAC14, SLCO5A1, CHRNA5, and PDX1, and a decrease in glucose-stimulated insulin secretion (GSIS) (Hall et al., 2018). A 48-h exposure to palmitate (1 mM), a saturated fatty acid, also reduces GSIS in human islets while inducing methylation changes and differential expression of 290 genes, including the TCF7L2 and GLIS3, markers of T2D risk (Hall et al., 2014). These methylation changes in islets upon high glucose and lipid exposures may be analogous to the effects of glucotoxicity and lipotoxicity in pre-diabetic and diabetic conditions. Moreover, the expression of insulin promoter factor 1 (PDX1), a master transcriptional regulator of β-cell development and function, is reduced with increased methylation in pancreatic islets of T2D patients, which also corresponds to increased HbA1c levels (Yang et al., 2012). Similarly, the expression of the glucagon-like peptide-1 receptor (GLP1R), involved in enhanced GSIS upon GLP-1 peptide binding (Muller et al., 2019), is decreased in pancreatic islets of T2D patients and hyperglycemic rats (Xu et al., 2007; Shu et al., 2009; Taneera et al., 2012). In particular, the methylation level of a CpG site within the GLP1R promoter correlates negatively with its gene expression, but positively with BMI and HbA1c (Hall et al., 2013), suggesting that the obesogenic and diabetic conditions can impact the DNA methylation profile of GLP1R and possibly lead to changes in pancreatic β-cell insulin secretion (Hall et al., 2013).
GWAS have identified multiple loci associated with the T2D risk and accounting for ∼10% of heritable diabetes (Voight et al., 2010). These include loci related to impaired insulin secretion and insulin sensitivity (Ruchat et al., 2009). Additionally, among 40 SNPs previously associated with T2D in human pancreatic islets (McCarthy, 2010), 19 of them (48%) are involved in either introducing or removing a CpG site (Dayeh et al., 2013). Importantly, these CpG-SNP sites are differentially methylated and result in changes in gene expression, alternative splicing events and hormone secretion in human islets. Altered expression of genes associated with T2D risk include TCF7L2, HHEX, CDKN2A, SLC30A8, CDKAL1, ADCY5, and FS1 (Ruchat et al., 2009; McCarthy, 2010). Moreover, in a separate study, genome-wide DNA methylation quantitative trait locus (mQTL) analysis in human pancreatic islets (Olsson et al., 2014) has identified over 67,000 CpG-SNP pairs, with several mQTLs associated with differential expression of T2D- and insulin secretion-related genes (e.g., ADCY5, KCNJ11, INS, PDX1 and GRB10) in human islets. Together, these studies demonstrate that DNA methylation may provide a causal link between SNPs and pancreatic gene function, thereby contributing to T2D development and progression (Dayeh et al., 2013).
Men are at higher risk for T2D than women (Kautzky-Willer et al., 2016). In isolated islets, GSIS is greater in female versus male donors, independent of β-cell number (Sharp et al., 2011). Differences in the DNA methylome also exist in pancreatic islets between men and women; specifically, significant sex-specific differences have been observed in 61 X-chromosome genes and 18 autosomal genes, including NKAP, SPESP1 and APLN, which are expressed at lower levels in female islets. The methylation of NKAP and SPESP1 promoters decreases their expression, and the silencing of Nkap and Apln in clonal cells reduces GSIS (Sharp et al., 2011), suggesting that differential DNA methylation may explain sex differences in insulin secretion and T2D risk.
Compared to the implications of DNA methylation in pancreatic metabolic conditions, our understanding of histone modifications in the onset and progression of diseases is limited. Yet, as many studies focus on the regeneration of pancreatic β-cells for the treatment of T2D, histone methylation via transcriptional programming in pancreatic development and function have been studied. While insulin-secreting β-cells and glucagon-secreting α-cells have different physiological functions, a study revealed that human α, β, and exocrine cells share similar profiles of histone methylation of H3K4me3 and H3K27me3, suggesting an epigenomic plasticity of islet cells and their reprogrammable potentials to treat diabetes (Bramswig et al., 2013). For example, the activation of the S6K1 kinase promotes α to β-cell transition by activating β-cell genes while repressing α-cell genes through histone methylation of the activating H3K4me3 and repressing H3K27me3, respectively (Yi et al., 2018). Human pancreatic α-cells can also be reprogrammed into insulin-producing cells by PDX1, and when transplanted, can treat diabetic mice (Furuyama et al., 2019). Pancreatic β-cell proliferation and expansion are highly active early in life in humans and mice (Meier et al., 2008) and decays with maturation and aging (Teta et al., 2005). Interestingly, expression of EZH2, a HMT of H3K27me3 and a key regulator of cell differentiation and growth, also decreases with aging in mouse pancreatic β-cells, whereas its expression is increased with adaptive β-cell proliferation after streptozotocin-mediated β-cell destruction (Chen et al., 2009). EZH2 can regulate β-cell proliferation by epigenetically repressing the Ink4a/Anf locus, which encodes the cyclin-dependent kinase inhibitor p16INK4a and tumor suppressor p19Arf (Chen et al., 2009). Consequently, mice lacking Ezh2 in pancreatic β-cells exhibit mild diabetes, suggesting a role for pancreatic Ezh2 in β-cell function.
Altered pancreatic histone acetylation plays a part in the development of T2D. Specifically, the expression of HDAC7, a class IIa HDAC, is increased in pancreatic islets from patients with T2D (Daneshpajooh et al., 2017) and over-expression of Hdac7 in rat islets and clonal β-cells reduces insulin content and increases apoptosis, leading to impaired GSIS of β-cells (Daneshpajooh et al., 2017). Conversely, treatment with a class II HDAC inhibitor (MC1568) rescues the dysfunctional insulin release of Hdac7-over-expressed β-cells and human islets from T2D donors (Daneshpajooh et al., 2018). Interestingly, the over-expression of HDAC7 in islets from T2D patients is likely mediated by hypomethylation of HDAC7’s CpG site (Dayeh et al., 2014), suggesting a cooperative epigenetic action.
Pancreatic β-cell SIRT1 (Ramsey et al., 2008) and its genetic polymorphisms are associated with the development of T2D (Dong et al., 2011; Maeda et al., 2011). Additionally, a SIRT1 mutation (L107P), which mildly reduces HDAC activity, has been found in type 1 diabetes (T1D) patients and leads to hyperinflammation with elevated expression of nitric oxide, cytokines (i.e., TNFα), and chemokines in a β-cell line (MIN6) (Biason-Lauber et al., 2013). Pancreas-specific Sirt1-deficient mice (Pdx1-Cre; Sirt1flox/flox) present with glucose intolerance and impaired GSIS of β-cells (Luu et al., 2013; Wang R.H. et al., 2013; Pinho et al., 2015). SIRT1-mediated deacetylation and subsequent repression of Ucp2 normally activates GSIS (Tordjman et al., 2002; Bordone et al., 2006; Chan and Kashemsant, 2006; Brun et al., 2015), however, the absence or reduction of pancreatic SIRT1 results in increased acetylation and expression of Ucp2 (Bordone et al., 2006) and other downstream target genes, such as Pgc-1α, Pparγ (Luu et al., 2013), and Pparα (Maiztegui et al., 2018), leading to decreased GSIS. Similarly, upon high glucose exposure (Brun et al., 2015), or the addition of sucrose (10%) to a normal diet (Maiztegui et al., 2018), pancreatic SIRT1 expression is decreased, while Ucp2 and Pparα expression is increased in human and rodent islets, leading to reductions in insulin content and GSIS of β-cells (Brun et al., 2015; Maiztegui et al., 2018). Moreover, as FOXA2 activation by post-translational SIRT1-mediated deacetylation stimulates the expression of its target gene Pdx1, essential for pancreatic β-cell development and maturation, SIRT1 insufficiency reduces β-cell formation (Wang R.H. et al., 2013). Furthermore, the age-related decline in SIRT1 activity and the accompanying decrease in GSIS from diminished NAD+ biosynthesis suggest an association with age-related metabolic diseases, including T2D (Ramsey et al., 2008). Collectively, these data indicate that genetic and dietary components can have profound effects at the epigenetic level, contributing to β-cell dysfunction and T2D development.
Caloric restriction treatment (30−50%) in rodent models of diabetes (i.e., db/db, aged mice or Zucker diabetic fatty rat) improves glucose tolerance and insulin sensitivity (Colombo et al., 2006; Kanda et al., 2015; Sheng et al., 2016; Rosa et al., 2018), β-cell mass (Ohneda et al., 1995; Bates et al., 2008; He et al., 2012; Kanda et al., 2015) and insulin secretion (Ohneda et al., 1995; Colombo et al., 2006; He et al., 2012; Kanda et al., 2015; Figure 5). These physiological benefits are accompanied by a reduction in the expression of genes related to oxidative and ER stress (i.e., Nox1, Chop10, Tnfa, Sod, Cat, Gpx1) (He et al., 2012; Kanda et al., 2015). In particular, isocaloric 2:1 IF in genetically obese (i.e., ob/ob) mice improves glucose homeostasis with increased postprandial insulin secretion, particularly GSIS (Kim Y.H. et al., 2019). As IF increases plasma GLP-1 levels, this suggests a possibility of an incretin-mediated insulinotropic effect of dietary interventions. Moreover, a FMD in diabetic (i.e., db/db) mice confers improvements in β-cell function as indicated by decreased plasma glucose and increased plasma insulin levels, as well as a reduction in insulin resistance (Cheng et al., 2017). Notably, this study demonstrated that a FMD protects against β-cell failure in late-stage T2D by promoting regeneration of insulin-producing β-cells from Ngn3+ pancreatic progenitor cells, particularly during the re-feeding period. These improvements in β-cell development and function are also seen in T2D patients on CR (Malandrucco et al., 2012; Jackness et al., 2013; Sathananthan et al., 2015).
The limited studies investigating the epigenetic effects of dietary interventions in the pancreas mainly pertain to histone acetylation changes. According to one study, 6 days of TRF (12-h fasting/feeding) in mice enhances GSIS in isolated islets without affecting body weight (Wortham et al., 2019) and is accompanied by histone acetylation of pancreatic islets during the re-feeding period. This epigenetic modulation takes place at sites occupied by the HDM LSD1 (KDM1A), which silences enhancers by removing mono- and dimethyl marks from H3K4 and is implicated in pancreatic endocrine cell development (Vinckier et al., 2020). The β-cell specific loss of Lsd1 results in histone hyperacetylation accompanied by insulin hypersecretion, indicating that the adaptive insulin secretory response to dietary interventions is regulated by the modulation of the epigenome in the pancreas (Rosen et al., 2018).
Another important epigenetic factor mediating the pancreatic response to dietary interventions is SIRT1. CR increases and activates SIRT1 in β-cells, thereby promoting pancreatic insulin secretion and ameliorating the T2D phenotype (Liang et al., 2009; Chen et al., 2010; Deng et al., 2010). Similar to the metabolic benefits conferred by dietary interventions (Chen et al., 2010, 2013), Sirt1 over-expression in mice preserves glucose homeostasis through improvements in insulin secretion and glucose tolerance (Moynihan et al., 2005; Ramsey et al., 2008). Mechanistically, pancreatic SIRT1-mediated repression of Ucp2 increases cellular ATP to promote vesicular exocytosis and release of insulin from β-cells (Moynihan et al., 2005; Bordone et al., 2006; Chan and Kashemsant, 2006; Ramsey et al., 2008). Additionally, to accommodate CR-mediated increases in insulin secretion, β-cells proliferate and increase their mass, resulting in larger pancreas size (Chen et al., 2013) – an effect that is also seen with SIRT1 activation (Wu et al., 2019). CR also lowers pancreatic inflammation and oxidative stress (Deng et al., 2010; Lanza-Jacoby et al., 2013; Harvey et al., 2014), which can otherwise lead to β-cell death and failure associated with T2D pathogenesis. This is likely mediated by SIRT1 activation since SIRT1 over-expression or resveratrol (CR-mimetic) treatment post-translationally deacetylate both p65 for inhibition of the NF-kB inflammatory signaling pathway (Lee et al., 2009) and FOXO1 for defense against oxidative stress (Kitamura et al., 2005; Zhang T. et al., 2016). While limited in number, studies investigating epigenetic changes with dietary interventions in the pancreas highlight key factors and mechanisms involved in ameliorating the T2D condition, and warrant further research on this topic.
The gut is host to trillions of microorganisms, including bacteria, archaea, viruses, and eukaryotes. The combined genome (∼150 times larger than the human genome) and the function of these microorganisms make up the microbiome (Ley et al., 2006a; Qin et al., 2010). While the microbiome influences digestion, gut-hormone secretion, intestinal immunity and inflammation, it is also largely shaped by diet; specifically, diet alterations account for 57% of the changes in gut microbiota populations, whereas genetic mutations only account for 12% (Zhang C. et al., 2010). Cumulating evidence places the gut microbiome and its metabolites at the origin of diet-induced metabolic dysregulation (Ley et al., 2006b), therefore, positive modulations of the gut microbiota by dietary interventions are of therapeutic interest.
Gut microbiota influences the host metabolism through various microbial-derived metabolites, which induce epigenetic alterations of key genes involved in the initiation and progression of diseases (Figure 6). Metabolites, such as folate, choline, vitamin B12, and betaine, can function as methyl donors and participate in DNA methylation processes via the generation of S-adenosylmethionine (SAM). In addition, the gut microbiota ferments complex carbohydrates from the diet to produce small organic acids, most of which are short-chain fatty acids (SCFA) (<95%), such as butyrate, propionate, acetate and lactate (den Besten et al., 2013). These SCFA, particularly butyrate and acetate, inhibit HDACs, leading to transcriptional activation via increased histone acetylation. Additional epigenetic roles of the gut microbiome are well discussed elsewhere (Sharma et al., 2019).
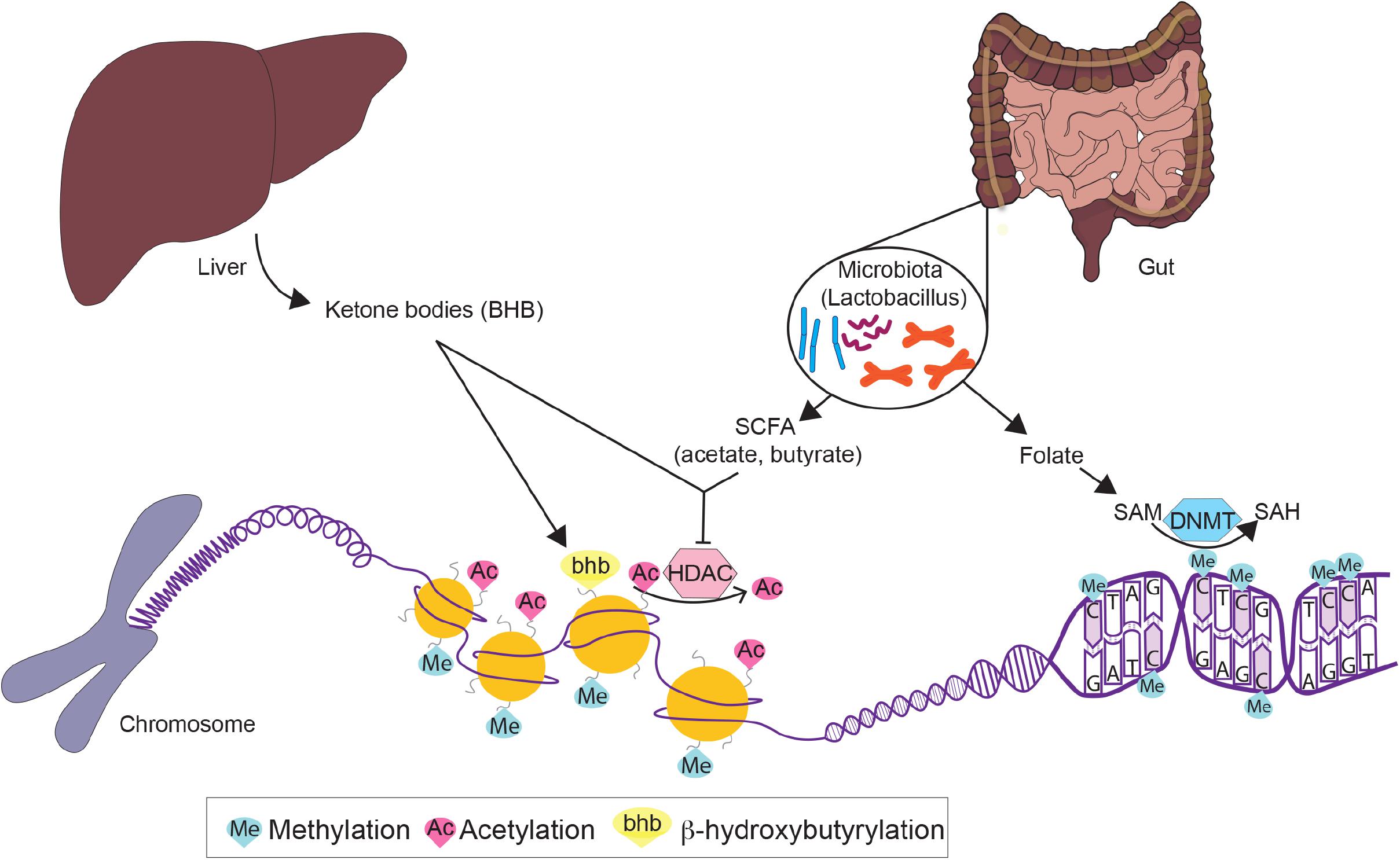
Figure 6. Epigenetic modulation by dietary intervention-induced ketogenesis and gut microbial metabolites. Dietary interventions stimulate ketone body production such as β-hydroxybutyrate, which can modulate gene expression through histone modification (bhb) and inhibition of histone deacetylases (HDACs). Dietary interventions also modulate the gut microbiota, through the release of short-chain fatty acids (SCFA) acetate and butyrate which inhibit HDACs, and folate which provides methyl donors for DNA methyltransferase (DNMT) activity.
Importantly, fasting-feeding cycles can directly impact the gut microbiota (Thaiss et al., 2014). In particular, dietary intervention-mediated (i.e., IF, FMD, CR, KD) remodeling of gut microbial populations confers various health benefits (Li et al., 2017; Beli et al., 2018; Cignarella et al., 2018; Fabbiano et al., 2018; Wang et al., 2018; Rangan et al., 2019; Ang et al., 2020; Liu et al., 2020). The causal and functional roles of these favorable microbes in dietary interventions are supported by microbiota transplantation and elimination (e.g., antibiotic treatment) studies. For example, IF-induced WAT browning and associated metabolic benefits are abolished in microbiota-depleted mice and subsequently restored with IF-microbiota transplantation (Li et al., 2017). This is in part mediated by the expansion of the Lactobacillus bacteria population, resulting in increased levels of serum lactate (Vergnes and Reue, 2014) and acetate (Hernandez et al., 2019), which are taken up by the upregulated monocarboxylate transporter 1 (Mct1) in WAT (Iwanaga et al., 2009; Carriere et al., 2014; Li et al., 2017). Lactobacillus, populated upon IF, CR and FMD, is a probiotic bacterium with health-promoting properties against metabolic diseases (Sharma et al., 2019) and is involved in the production of folate and the fermentation of pyruvate to acetate and lactate. IF can also increase plasma butyrate levels through greater butyrate-producing Odoribacter (Liu et al., 2020). Notably, these health benefits of IF can be achieved through the direct administration of SCFA (Liu et al., 2020), suggesting that metabolites from a re-established microbiome mediate the health benefits of dietary interventions. Although studies of dietary intervention, such as CR and KD, have shown epigenetic-mediated mechanisms of improved gut stem cell homeostasis (Igarashi and Guarente, 2016; Cheng et al., 2019), the association with gut microbiota is still not fully understood and thus warrants further research.
Many of the benefits related to fasting and nutritional interventions, such as IF, CR, FMD and KD, may stem from the activation of the ketogenic pathway and the increased production of ketone bodies (i.e., ketosis). This metabolic switch to ketosis, associated with improvements in lipid and glucose metabolism, contributes to a healthier metabolic state of the tissues discussed in this review. Dysregulation or insufficiency of ketogenesis, by contrast, can be associated with hepatic metabolic abnormalities and may contribute to NAFLD (Cotter et al., 2014; Mannisto et al., 2015; d’Avignon et al., 2018; Fletcher et al., 2019) and liver fibrosis (Puchalska et al., 2019).
Fasting stimulates hepatic ketogenesis to increase systemic ketone body levels. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2) is the rate-limiting enzyme of the ketogenic pathway and catalyzes the conversion of acetoacetyl-CoA to HMG-CoA – the first step in β-hydroxybutyrate (BHB) synthesis. HMGCS2 is a downstream target gene of FOXA2, a key transcription factor of hepatic lipid metabolism. Under feeding conditions, insulin/PI3K/Akt-mediated phosphorylation of FOXA2 reduces its transcriptional activity by nuclear exclusion (Wolfrum et al., 2004; von Meyenn et al., 2013; Newman and Verdin, 2014). Conversely, under fasting conditions, glucagon signaling inhibits SIK2 kinase and allows for p300-mediated post-translational acetylation and activation of FOXA2, resulting in the induction of HMGCS2 transcription (von Meyenn et al., 2013; Newman and Verdin, 2014). In addition to the SIK2-p300-FOXA2 axis, SIRT3, the mitochondrial class III HDAC, directly increases the enzymatic activity of HMGCS2 under fasting conditions through post-translational deacetylation of lysine residues (Shimazu et al., 2010; Newman and Verdin, 2014). Mice lacking Sirt3 exhibit a reduction in fasting-induced BHB production, indicative of hypoketonemia, along with impaired IF-mediated neurological improvements (Liu Y. et al., 2019).
Acetone, acetoacetate (AcAc) and BHB are the three ketone bodies produced by the liver, from which AcAc and BHB are transported primarily to skeletal muscle and the brain as carriers of additional energy, while acetone is mainly released through exhalation. Among the three ketone bodies, BHB is the most abundant in mammals. Notably, recent studies have demonstrated that ketone bodies play a pivotal role as direct or indirect signaling mediators of cellular and metabolic functions, including epigenetic gene regulation (Newman and Verdin, 2014, 2017; Puchalska and Crawford, 2017). Similar to butyrate (Candido et al., 1978; Cousens et al., 1979; Louis and Flint, 2009), BHB inhibits HDACs, particularly HDAC 1, 3, and 4 (class I and IIa) (Shimazu et al., 2013; Figure 6). Both HDAC3 and HDAC4 are responsible for stimulating the expression of gluconeogenic genes (Mihaylova et al., 2011), hence their inhibition by BHB would result in a reduction in plasma glucose levels, as seen in Hdac3-deficient mice (Knutson et al., 2008). In addition, ketosis stimulated by either BHB treatment, 40% CR or overnight fasting in cells and mice all result in histone hyperacetylation, particularly at H3K9 and H3K14, thereby promoting the expression of genes, such as Foxo3a – a core regulator of cellular homeostasis (i.e., cell cycle progression), stress response, and longevity induction (Shimazu et al., 2013). Since both butyrate treatment and HDAC inhibition improve hepatic steatosis (Endo et al., 2013; Zhou et al., 2017, 2018) and glucose homeostasis (Gao et al., 2009; Xiao et al., 2011; Cho et al., 2018; Lee et al., 2018) in both mice and humans, enhancing BHB levels through dietary interventions may provide similar beneficial effects.
Furthermore, elevated BHB levels result in increased histone lysine β-hydroxybutyrylation (kbhb), a novel type of histone post-translational modification (Xie et al., 2016; Figure 6). BHB produced from ketogenesis can be complexed with free molecules of Coenzyme A to form BHB-CoA, the donor for kbhb histone modifications (Sabari et al., 2017). A total of 44 histone kbhb sites have been determined in human embryonic kidney cells (HEK293) and mouse liver. These kbhb marks are found in the promoter sites of target genes and result in transcriptional activation. In particular, kbhb on H3K9 (H3K9bhb) is found in fasted liver (Xie et al., 2016). The H3K9bhb acylation mark targets and leads to the upregulation of genes involved in amino acid catabolism, redox balance and circadian rhythm – mediating these specific processes in the switch from feeding to fasting state (Xie et al., 2016). It is thus tempting to speculate that the metabolic benefits by ketogenic dietary interventions, such as IF, CR, FMD and KD, involve histone kbhb-mediated epigenetic modulation.
Pharmaceutical therapeutics stimulating a state of ketosis similar to that with dietary interventions, such as sodium-glucose co-transporter 2 (SGLT2) inhibitors, are being clinically used in the treatment of NAFLD and T2D (Polidori et al., 2018). SGLT2 inhibitors have shown metabolic improvements in hyperglycemia, adiposity, oxidative stress and inflammation (Komiya et al., 2016; Scheen, 2019). While it has not been clearly understood, increased circulating ketone bodies by SGLT2 inhibitors has been considered one of the mechanisms of action mediating the metabolic benefits (Prattichizzo et al., 2018; Wojcik and Warden, 2019). In addition, in a recent study, loss of the G protein-coupled receptor 43 (Gpr43), activated specifically by AcAc in mice, abolishes IF- and KD-mediated metabolic benefits, including those associated with lipid metabolism (i.e., body weight and fat mass reduction) (Miyamoto et al., 2019). This finding suggests that the improvements seen with dietary interventions are indeed mediated at least in part by ketone body metabolism. Further studies on the ketone body-mediated epigenetic changes in dietary interventions will provide additional mechanistic understanding.
In this review, we summarized key tissue-specific epigenetic changes implicated in both metabolic diseases and dietary interventions, with a focus on adipose, liver and pancreas. While most of the epigenetic studies have been conducted separately in these metabolic tissues, it is important to recognize that the integrated tissue cross-talks can drive systemic changes in metabolic gene expression and function. For example, secretion of tissue-respective metabolites and hormones, such as adipokines (i.e., leptin, adiponectin), hepatokines (i.e., Fgf21) and pancreatic glucagon and insulin, which as we have discussed are all subject to epigenetic changes, are key mediators of tissue cross-talk and systemic homeostasis (Rosen, 2016; Stern et al., 2016). Therefore, the global metabolic changes seen in several tissues upon disease and dietary intervention make it difficult to not only characterize the adaptive or pathological role of these epigenetic events but, to also pinpoint the primary insult that triggers secondary, systemic aspects of these responses. Temporally identifying the whole-body, tissue-specific epigenetic changes throughout both disease progression and dietary intervention-mediated metabolic improvements will require further investigation. We further discuss some limitations, benefits and potentials to epigenetic modification by dietary intervention.
One of the key limitations of dietary interventions arises from sex differences. Despite the successful outcomes of dietary interventions seen in humans and animal models (Di Francesco et al., 2018; de Cabo and Mattson, 2019), it is not clear whether and to what extent sex differences contribute to the impact of dietary interventions. For example, in the liver, males favor energy utilization by oxidizing FA, whereas females tend to prefer energy storage by converting FA into TG (Tramunt et al., 2020). Furthermore, females primarily store excess energy in subcutaneous fat, which in comparison to visceral fat, allows for greater and longer storage, prevents ectopic fat deposition in other tissues and resists the development of male-predominant metabolic diseases, such as diabetes and NAFLD (Tramunt et al., 2020). Thus, these sex differences in metabolism could lead to different outcomes when subject to dietary interventions. Indeed, unlike male mice showing reduced lipid accumulation, IF increases the hepatic lipid content of female mice (Piotrowska et al., 2016). This result can be further explained by the sex difference in the fasting response of the liver. Upon a short-term fast (6 hr), male mice maintained steady-state metabolism with reductions in anabolic pathways, such as hepatic lipogenesis and gluconeogenesis, whereas females continued to use amino acids for the synthesis of hepatic TG (Della Torre et al., 2018). While it has been suggested that sex differences in the liver are established postnatally via testosterone-mediated DNA methylation (Reizel et al., 2015), this study with transcriptome and metabolomic analyses has demonstrated that the sexual differentiation of the liver exists when mice are born and is largely mediated by the sexually dimorphic hepatic estrogen receptor α (ERα) (Della Torre et al., 2018). Together, this evidence suggests that a comprehensive understanding of sex differences in metabolism is required for safe and efficacious utilization of fasting-involved dietary interventions in both males and females. Particularly, epigenetic changes and their regulatory roles in sexual differentiation in response to dietary interventions remain to be elucidated.
Aging is a key risk factor for metabolic disease development. Extension of lifespan and healthspan by dietary interventions has thus led to an interest in their application for the treatment of metabolic diseases in the elderly (Gensous et al., 2019). At the other extreme, pediatric obesity is also an emerging public health priority; dietary interventions are being considered for this population as well (Vidmar et al., 2019). However, as discussed in this review, most human and animal dietary intervention studies are conducted in young adult and middle-aged individuals, with very limited work in the elderly or pediatric population. Due to the stark differences in the metabolism and physiology among children, young adult and aged individuals, it is necessary to test the safety and efficacy of dietary interventions in these populations. For example, in contrast to young adult or middle-aged individuals, mildly increased body weight and/or BMI in the elderly is often associated with a lower risk of mortality. This finding, known as the “obesity paradox,” suggests a protective role of body fat in the elderly against certain illnesses (i.e., T2D) (Hainer and Aldhoon-Hainerova, 2013). In one study, weight loss (>7.5% body weight) was strongly associated with a reduced survival outcome in T2D elderly patients with a mean age of 62 years (Doehner et al., 2012). Thus, dietary interventions resulting in body weight loss could be detrimental in the elderly (Thorpe and Ferraro, 2004; Locher et al., 2016). Moreover, a study has demonstrated that 4 weeks of TRF in juvenile mice (4-week-old) causes adverse effects including delayed puberty, fatty liver disease and an abnormal gut microbiota shift (Hu et al., 2019). Therefore, the overall risk-to-benefit ratio of dietary interventions in different age populations still remains uncertain and requires further research. Moreover, it would be interesting to see how epigenetic mechanisms govern the differential response to dietary interventions among different age populations with metabolic disease.
Despite the limitations of sex and age, the epigenetic modulation of dietary intervention is still a promising avenue. Due to the reversible nature of epigenetic modifications, the utilization of epigenetic therapies in the treatment of metabolic diseases is encouraging. Both in vitro and in vivo mechanistic studies of current epigenetic therapies, such as inhibitors of DNMT (e.g., 5-aza-2′-deoxycytidine) (Mann et al., 2007, 2010), HDAC (e.g., Trichostatin A) (Zhang et al., 2009) and HAT (e.g., Tannic acid) (Chung et al., 2019) have shown promising preclinical results in targeting hepatic disease processes. However, the low specificity and broad range of outcomes associated with epigenome-targeting agents could lead to side effects (Gius et al., 2004). Interest has also emerged in epigenetic dietary components including the DNMT/HAT inhibitor epigallocatechin-3-gallate (EGCG) found in green tea and the DNMT/HDAC inhibitor resveratrol found in peanuts, grapes and berries (Hardy and Tollefsbol, 2011). Resveratrol, commonly known as a SIRT1 activator, is used as a CR-mimetic in several studies, showing improvements in health and longevity (Baur et al., 2006). Although these natural compounds are associated with several benefits, they do not yet provide the same efficacy in achieving balanced and global effects as with dietary interventions against metabolic diseases. It will, however, be interesting to see whether combining these pharmacological or natural epigenetic modulators with dietary interventions could provide greater efficacy and reduced side effects – a perspective that can be explored in future studies. Overall, the numerous metabolic benefits associated with dietary interventions and their ability to reverse the disease state, makes them encouraging for clinical translation. Although current treatments are mainly associated with risk prevention and disease management, these dietary interventions, in combination with their epigenetic modulators, can serve to effectively improve the prognosis of metabolic diseases.
SA, NM, EM, and K-HK conceived and wrote the manuscript. SA and NM created the figures and table. All authors read and approved the final manuscript.
K-HK was supported by the Heart and Stroke Foundation of Canada Grant-in-Aid (G-18-0022213) and Natural Sciences and Engineering Research Discovery Grant (RGPIN-2018-0870). EM was a Diabetes Canada New Investigator and supported by a grant from the Canadian Institutes of Health Research (PJT156136). SA was supported by the Frederick Banting and Charles Best Canada Graduate Scholarship-Master’s (CGS-M).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Hoon-Ki Sung, Saif Dababneh, Yena Oh, and Cassie Locatelli for their helpful remarks in improving the manuscript.
Abderrahmani, A., Yengo, L., Caiazzo, R., Canouil, M., Cauchi, S., Raverdy, V., et al. (2018). Increased Hepatic PDGF-AA signaling mediates liver insulin resistance in obesity-associated type 2 diabetes. Diabetes 67, 1310–1321. doi: 10.2337/db17-1539
Abe, Y., Fujiwara, Y., Takahashi, H., Matsumura, Y., Sawada, T., Jiang, S., et al. (2018). Histone demethylase JMJD1A coordinates acute and chronic adaptation to cold stress via thermogenic phospho-switch. Nat. Commun. 9:1566.
Abe, Y., Rozqie, R., Matsumura, Y., Kawamura, T., Nakaki, R., Tsurutani, Y., et al. (2015). JMJD1A is a signal-sensing scaffold that regulates acute chromatin dynamics via SWI/SNF association for thermogenesis. Nat. Commun. 6:7052.
Ahima, R. S., and Flier, J. S. (2000). Adipose tissue as an endocrine organ. Trends Endocrinol. Metab. 11, 327–332.
Ahrens, M., Ammerpohl, O., Von Schonfels, W., Kolarova, J., Bens, S., Itzel, T., et al. (2013). DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 18, 296–302. doi: 10.1016/j.cmet.2013.07.004
Ajmo, J. M., Liang, X., Rogers, C. Q., Pennock, B., and You, M. (2008). Resveratrol alleviates alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G833–G842.
Akalestou, E., Genser, L., and Rutter, G. A. (2020). Glucocorticoid metabolism in obesity and following weight loss. Front. Endocrinol. 11:59. doi: 10.3389/fendo.2020.00059
Ang, Q. Y., Alexander, M., Newman, J. C., Tian, Y., Cai, J., Upadhyay, V., et al. (2020). Ketogenic diets alter the gut microbiome resulting in decreased intestinal Th17 cells. Cell 181, 1263–1275.e16.
Anson, R. M., Guo, Z., De Cabo, R., Iyun, T., Rios, M., Hagepanos, A., et al. (2003). Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc. Natl. Acad. Sci. U.S.A. 100, 6216–6220. doi: 10.1073/pnas.1035720100
Anton, S. D., Lee, S. A., Donahoo, W. T., Mclaren, C., Manini, T., Leeuwenburgh, C., et al. (2019). The Effects of time restricted feeding on overweight. Older adults: a pilot study. Nutrients 11:1500. doi: 10.3390/nu11071500
Anton, S. D., Moehl, K., Donahoo, W. T., Marosi, K., Lee, S. A., Mainous, A. G., et al. (2018). Flipping the metabolic switch: understanding and applying the health benefits of fasting. Obesity 26, 254–268. doi: 10.1002/oby.22065
Badman, M. K., Kennedy, A. R., Adams, A. C., Pissios, P., and Maratos-Flier, E. (2009). A very low carbohydrate ketogenic diet improves glucose tolerance in ob/ob mice independently of weight loss. Am. J. Physiol. Endocrinol. Metab. 297, E1197–E1204.
Bagchi, R. A., Ferguson, B. S., Stratton, M. S., Hu, T., Cavasin, M. A., Sun, L., et al. (2018). HDAC11 suppresses the thermogenic program of adipose tissue via BRD2. JCI Insight 3:e120159.
Barcena-Varela, M., Colyn, L., and Fernandez-Barrena, M. G. (2019). Epigenetic mechanisms in hepatic stellate cell activation during liver fibrosis and carcinogenesis. Int. J. Mol. Sci. 20:2507. doi: 10.3390/ijms20102507
Barger, J. L., Kayo, T., Vann, J. M., Arias, E. B., Wang, J., Hacker, T. A., et al. (2008). A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS One 3:e2264. doi: 10.1371/journal.pone.0002264
Bates, H. E., Sirek, A., Kiraly, M. A., Yue, J. T., Riddell, M. C., Matthews, S. G., et al. (2008). Adaptation to intermittent stress promotes maintenance of beta-cell compensation: comparison with food restriction. Am. J. Physiol. Endocrinol. Metab. 295, E947–E958.
Baumeier, C., Kaiser, D., Heeren, J., Scheja, L., John, C., Weise, C., et al. (2015). Caloric restriction and intermittent fasting alter hepatic lipid droplet proteome and diacylglycerol species and prevent diabetes in NZO mice. Biochim. Biophys. Acta 1851, 566–576. doi: 10.1016/j.bbalip.2015.01.013
Baur, J. A., Pearson, K. J., Price, N. L., Jamieson, H. A., Lerin, C., Kalra, A., et al. (2006). Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337–342.
Beli, E., Yan, Y., Moldovan, L., Vieira, C. P., Gao, R., Duan, Y., et al. (2018). Restructuring of the gut microbiome by intermittent fasting prevents retinopathy and prolongs survival in db/db mice. Diabetes 67, 1867–1879.
Benton, M. C., Johnstone, A., Eccles, D., Harmon, B., Hayes, M. T., Lea, R. A., et al. (2015). An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 16:8.
Bhatt, H. B., and Smith, R. J. (2015). Fatty liver disease in diabetes mellitus. Hepatobiliary Surg. Nutr. 4, 101–108.
Bian, F., Ma, X., Villivalam, S. D., You, D., Choy, L. R., Paladugu, A., et al. (2018). TET2 facilitates PPARgamma agonist-mediated gene regulation and insulin sensitization in adipocytes. Metabolism 89, 39–47. doi: 10.1016/j.metabol.2018.08.006
Biason-Lauber, A., Boni-Schnetzler, M., Hubbard, B. P., Bouzakri, K., Brunner, A., Cavelti-Weder, C., et al. (2013). Identification of a SIRT1 mutation in a family with type 1 diabetes. Cell Metab. 17, 448–455.
Bodkin, N. L., Alexander, T. M., Ortmeyer, H. K., Johnson, E., and Hansen, B. C. (2003). Mortality and morbidity in laboratory-maintained Rhesus monkeys and effects of long-term dietary restriction. J. Gerontol. A Biol. Sci. Med. Sci. 58, 212–219.
Bordone, L., Motta, M. C., Picard, F., Robinson, A., Jhala, U. S., Apfeld, J., et al. (2006). Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 4:e31. doi: 10.1371/journal.pbio.0040031
Bosch-Presegue, L., and Vaquero, A. (2015). Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. FEBS J. 282, 1745–1767. doi: 10.1111/febs.13053
Bouchard, L., Rabasa-Lhoret, R., Faraj, M., Lavoie, M. E., Mill, J., Perusse, L., et al. (2010). Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction. Am. J. Clin. Nutr. 91, 309–320. doi: 10.3945/ajcn.2009.28085
Boutant, M., Joffraud, M., Kulkarni, S. S., Garcia-Casarrubios, E., Garcia-Roves, P. M., Ratajczak, J., et al. (2015). SIRT1 enhances glucose tolerance by potentiating brown adipose tissue function. Mol. Metab. 4, 118–131. doi: 10.1016/j.molmet.2014.12.008
Boutant, M., Kulkarni, S. S., Joffraud, M., Raymond, F., Metairon, S., Descombes, P., et al. (2016). SIRT1 gain of function does not mimic or enhance the adaptations to intermittent fasting. Cell Rep. 14, 2068–2075. doi: 10.1016/j.celrep.2016.02.007
Bramswig, N. C., Everett, L. J., Schug, J., Dorrell, C., Liu, C., Luo, Y., et al. (2013). Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. J. Clin. Invest. 123, 1275–1284. doi: 10.1172/jci66514
Bricambert, J., Alves-Guerra, M. C., Esteves, P., Prip-Buus, C., Bertrand-Michel, J., Guillou, H., et al. (2018). The histone demethylase Phf2 acts as a molecular checkpoint to prevent NAFLD progression during obesity. Nat. Commun. 9:2092.
Bricambert, J., Favre, D., Brajkovic, S., Bonnefond, A., Boutry, R., Salvi, R., et al. (2016). Impaired histone deacetylases 5 and 6 expression mimics the effects of obesity and hypoxia on adipocyte function. Mol. Metab. 5, 1200–1207. doi: 10.1016/j.molmet.2016.09.011
Bricambert, J., Miranda, J., Benhamed, F., Girard, J., Postic, C., and Dentin, R. (2010). Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Invest. 120, 4316–4331. doi: 10.1172/jci41624
Brown, M. S., and Goldstein, J. L. (2008). Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 7, 95–96. doi: 10.1016/j.cmet.2007.12.009
Browning, J. D., Baker, J. A., Rogers, T., Davis, J., Satapati, S., and Burgess, S. C. (2011). Short-term weight loss and hepatic triglyceride reduction: evidence of a metabolic advantage with dietary carbohydrate restriction. Am. J. Clin. Nutr. 93, 1048–1052. doi: 10.3945/ajcn.110.007674
Brun, T., Li, N., Jourdain, A. A., Gaudet, P., Duhamel, D., Meyer, J., et al. (2015). Diabetogenic milieus induce specific changes in mitochondrial transcriptome and differentiation of human pancreatic islets. Hum. Mol. Genet. 24, 5270–5284. doi: 10.1093/hmg/ddv247
Byun, S., Seok, S., Kim, Y. C., Zhang, Y., Yau, P., Iwamori, N., et al. (2020). Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 11:807.
Candido, E. P., Reeves, R., and Davie, J. R. (1978). Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 14, 105–113. doi: 10.1016/0092-8674(78)90305-7
Carriere, A., Jeanson, Y., Berger-Muller, S., Andre, M., Chenouard, V., Arnaud, E., et al. (2014). Browning of white adipose cells by intermediate metabolites: an adaptive mechanism to alleviate redox pressure. Diabetes 63, 3253–3265. doi: 10.2337/db13-1885
Carter, S., Li, Z., Lemieux, I., Almeras, N., Tremblay, A., Bergeron, J., et al. (2014). Circulating IGFBP-2 levels are incrementally linked to correlates of the metabolic syndrome and independently associated with VLDL triglycerides. Atherosclerosis 237, 645–651. doi: 10.1016/j.atherosclerosis.2014.09.022
Castellano-Castillo, D., Denechaud, P. D., Fajas, L., Moreno-Indias, I., Oliva-Olivera, W., Tinahones, F., et al. (2019). Human adipose tissue H3K4me3 histone mark in adipogenic, lipid metabolism and inflammatory genes is positively associated with BMI and HOMA-IR. PLoS One 14:e0215083. doi: 10.1371/journal.pone.0215083
Catterson, J. H., Khericha, M., Dyson, M. C., Vincent, A. J., Callard, R., Haveron, S. M., et al. (2018). Short-term, intermittent fasting induces long-lasting gut health and tor-independent lifespan extension. Curr. Biol. 28:1714. doi: 10.1016/j.cub.2018.04.015
Chait, A., and den Hartigh, L. J. (2020). Adipose tissue distribution, inflammation and its metabolic consequences, including diabetes and cardiovascular disease. Front. Cardiovasc. Med. 7:22. doi: 10.3389/fcvm.2020.00022
Chaix, A., Zarrinpar, A., Miu, P., and Panda, S. (2014). Time-restricted feeding is a preventative and therapeutic intervention against diverse nutritional challenges. Cell Metab. 20, 991–1005. doi: 10.1016/j.cmet.2014.11.001
Chakrabarti, P., English, T., Karki, S., Qiang, L., Tao, R., Kim, J., et al. (2011). SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J. Lipid Res. 52, 1693–1701. doi: 10.1194/jlr.m014647
Chan, C. B., and Kashemsant, N. (2006). Regulation of insulin secretion by uncoupling protein. Biochem. Soc. Trans. 34, 802–805. doi: 10.1042/bst0340802
Chasman, D. I., Pare, G., Mora, S., Hopewell, J. C., Peloso, G., Clarke, R., et al. (2009). Forty-three loci associated with plasma lipoprotein size, concentration, and cholesterol content in genome-wide analysis. PLoS Genet. 5:e1000730. doi: 10.1371/journal.pgen.1000730
Chatterjee, T. K., Basford, J. E., Knoll, E., Tong, W. S., Blanco, V., Blomkalns, A. L., et al. (2014a). HDAC9 knockout mice are protected from adipose tissue dysfunction and systemic metabolic disease during high-fat feeding. Diabetes 63, 176–187. doi: 10.2337/db13-1148
Chatterjee, T. K., Basford, J. E., Yiew, K. H., Stepp, D. W., Hui, D. Y., and Weintraub, N. L. (2014b). Role of histone deacetylase 9 in regulating adipogenic differentiation and high fat diet-induced metabolic disease. Adipocyte 3, 333–338. doi: 10.4161/adip.28814
Chen, D., Bruno, J., Easlon, E., Lin, S. J., Cheng, H. L., Alt, F. W., et al. (2008). Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 22, 1753–1757. doi: 10.1101/gad.1650608
Chen, H., Gu, X., Su, I. H., Bottino, R., Contreras, J. L., Tarakhovsky, A., et al. (2009). Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 23, 975–985. doi: 10.1101/gad.1742509
Chen, N., and Wang, J. (2018). Wnt/beta-catenin signaling and obesity. Front. Physiol. 9:792. doi: 10.3389/fphys.2018.00792
Chen, Q., Hao, W., Xiao, C., Wang, R., Xu, X., Lu, H., et al. (2017). SIRT6 is essential for adipocyte differentiation by regulating mitotic clonal expansion. Cell Rep. 18, 3155–3166. doi: 10.1016/j.celrep.2017.03.006
Chen, Y. R., Fang, S. R., Fu, Y. C., Zhou, X. H., Xu, M. Y., and Xu, W. C. (2010). Calorie restriction on insulin resistance and expression of SIRT1 and SIRT4 in rats. Biochem. Cell Biol. 88, 715–722. doi: 10.1139/o10-010
Chen, Y. R., Lai, Y. L., Lin, S. D., Li, X. T., Fu, Y. C., and Xu, W. C. (2013). SIRT1 interacts with metabolic transcriptional factors in the pancreas of insulin-resistant and calorie-restricted rats. Mol. Biol. Rep. 40, 3373–3380. doi: 10.1007/s11033-012-2412-3
Cheng, C. W., Biton, M., Haber, A. L., Gunduz, N., Eng, G., Gaynor, L. T., et al. (2019). Ketone body signaling mediates intestinal stem cell homeostasis and adaptation to diet. Cell 178, 1115–1131.e15.
Cheng, C. W., Villani, V., Buono, R., Wei, M., Kumar, S., Yilmaz, O. H., et al. (2017). Fasting-Mimicking diet promotes Ngn3-driven beta-cell regeneration to reverse diabetes. Cell 168, 775–788.e12.
Cho, H. M., Seok, Y. M., Lee, H. A., Song, M., and Kim, I. (2018). Repression of transcriptional activity of Forkhead Box O1 by histone deacetylase inhibitors ameliorates hyperglycemia in type 2 diabetic rats. Int. J. Mol. Sci. 19:3539. doi: 10.3390/ijms19113539
Cho, Y. W., Hong, S., Jin, Q., Wang, L., Lee, J. E., Gavrilova, O., et al. (2009). Histone methylation regulator PTIP is required for PPARgamma and C/EBPalpha expression and adipogenesis. Cell Metab. 10, 27–39. doi: 10.1016/j.cmet.2009.05.010
Choe, S. S., Huh, J. Y., Hwang, I. J., Kim, J. I., and Kim, J. B. (2016). Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front. Endocrinol. 7:30. doi: 10.3389/fendo.2016.00030
Chung, M. Y., Song, J. H., Lee, J., Shin, E. J., Park, J. H., Lee, S. H., et al. (2019). Tannic acid, a novel histone acetyltransferase inhibitor, prevents non-alcoholic fatty liver disease both in vivo and in vitro model. Mol. Metab. 19, 34–48. doi: 10.1016/j.molmet.2018.11.001
Cignarella, F., Cantoni, C., Ghezzi, L., Salter, A., Dorsett, Y., Chen, L., et al. (2018). Intermittent fasting confers protection in CNS autoimmunity by altering the gut microbiota. Cell Metab. 27, 1222–1235.e6.
Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking, B., Kessler, B., et al. (2004). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392. doi: 10.1126/science.1099196
Colman, R. J., Anderson, R. M., Johnson, S. C., Kastman, E. K., Kosmatka, K. J., Beasley, T. M., et al. (2009). Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325, 201–204. doi: 10.1126/science.1173635
Colombo, M., Kruhoeffer, M., Gregersen, S., Agger, A., Jeppesen, P., Oerntoft, T., et al. (2006). Energy restriction prevents the development of type 2 diabetes in Zucker diabetic fatty rats: coordinated patterns of gene expression for energy metabolism in insulin-sensitive tissues and pancreatic islets determined by oligonucleotide microarray analysis. Metabolism 55, 43–52. doi: 10.1016/j.metabol.2005.07.005
Cordeiro, A., De Souza, L. L., Oliveira, L. S., Faustino, L. C., Santiago, L. A., Bloise, F. F., et al. (2013). Thyroid hormone regulation of Sirtuin 1 expression and implications to integrated responses in fasted mice. J. Endocrinol. 216, 181–193. doi: 10.1530/joe-12-0420
Cormier-Daire, V., Molinari, F., Rio, M., Raoul, O., De Blois, M. C., Romana, S., et al. (2003). Cryptic terminal deletion of chromosome 9q34: a novel cause of syndromic obesity in childhood? J. Med. Genet. 40, 300–303. doi: 10.1136/jmg.40.4.300
Costa Cdos, S., Hammes, T. O., Rohden, F., Margis, R., Bortolotto, J. W., Padoin, A. V., et al. (2010). SIRT1 transcription is decreased in visceral adipose tissue of morbidly obese patients with severe hepatic steatosis. Obes. Surg. 20, 633–639. doi: 10.1007/s11695-009-0052-z
Cotter, D. G., Ercal, B., Huang, X., Leid, J. M., D’avignon, D. A., Graham, M. J., et al. (2014). Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Invest. 124, 5175–5190. doi: 10.1172/jci76388
Cousens, L. S., Gallwitz, D., and Alberts, B. M. (1979). Different accessibilities in chromatin to histone acetylase. J. Biol. Chem. 254, 1716–1723.
Cypess, A. M., Weiner, L. S., Roberts-Toler, C., Franquet Elia, E., Kessler, S. H., Kahn, P. A., et al. (2015). Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab. 21, 33–38. doi: 10.1016/j.cmet.2014.12.009
Dahlman, I., Sinha, I., Gao, H., Brodin, D., Thorell, A., Ryden, M., et al. (2015). The fat cell epigenetic signature in post-obese women is characterized by global hypomethylation and differential DNA methylation of adipogenesis genes. Int. J. Obes. 39, 910–919. doi: 10.1038/ijo.2015.31
Daneshpajooh, M., Bacos, K., Bysani, M., Bagge, A., Ottosson Laakso, E., Vikman, P., et al. (2017). HDAC7 is overexpressed in human diabetic islets and impairs insulin secretion in rat islets and clonal beta cells. Diabetologia 60, 116–125. doi: 10.1007/s00125-016-4113-2
Daneshpajooh, M., Eliasson, L., Bacos, K., and Ling, C. (2018). MC1568 improves insulin secretion in islets from type 2 diabetes patients and rescues beta-cell dysfunction caused by Hdac7 upregulation. Acta Diabetol. 55, 1231–1235. doi: 10.1007/s00592-018-1201-4
d’Avignon, D. A., Puchalska, P., Ercal, B., Chang, Y., Martin, S. E., Graham, M. J., et al. (2018). Hepatic ketogenic insufficiency reprograms hepatic glycogen metabolism and the lipidome. JCI Insight 3:e99762.
Dayeh, T., Volkov, P., Salo, S., Hall, E., Nilsson, E., Olsson, A. H., et al. (2014). Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 10:e1004160. doi: 10.1371/journal.pgen.1004160
Dayeh, T. A., Olsson, A. H., Volkov, P., Almgren, P., Ronn, T., and Ling, C. (2013). Identification of CpG-SNPs associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia 56, 1036–1046. doi: 10.1007/s00125-012-2815-7
de Cabo, R., and Mattson, M. P. (2019). Effects of intermittent fasting on health, aging, and disease. N. Engl. J. Med. 381, 2541–2551. doi: 10.1056/nejmra1905136
de Mello, V. D., Matte, A., Perfilyev, A., Mannisto, V., Ronn, T., Nilsson, E., et al. (2017). Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics 12, 287–295. doi: 10.1080/15592294.2017.1294305
de Oliveira, R. M., Sarkander, J., Kazantsev, A. G., and Outeiro, T. F. (2012). SIRT2 as a therapeutic target for age-related disorders. Front. Pharmacol. 3:82. doi: 10.3389/fphar.2012.00082
Deiuliis, J. A. (2016). MicroRNAs as regulators of metabolic disease: pathophysiologic significance and emerging role as biomarkers and therapeutics. Int. J. Obes. 40, 88–101. doi: 10.1038/ijo.2015.170
Della Torre, S., Mitro, N., Meda, C., Lolli, F., Pedretti, S., Barcella, M., et al. (2018). Short-term fasting reveals amino acid metabolism as a major sex-discriminating factor in the liver. Cell Metab. 28, 256–267.
den Besten, G., Van Eunen, K., Groen, A. K., Venema, K., Reijngoud, D. J., and Bakker, B. M. (2013). The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 54, 2325–2340. doi: 10.1194/jlr.r036012
Denechaud, P. D., Fajas, L., and Giralt, A. (2017). E2F1, a novel regulator of metabolism. Front. Endocrinol. 8:311. doi: 10.3389/fendo.2017.00311
Deng, X., Cheng, J., Zhang, Y., Li, N., and Chen, L. (2010). Effects of caloric restriction on SIRT1 expression and apoptosis of islet beta cells in type 2 diabetic rats. Acta Diabetol. 47(Suppl. 1), 177–185. doi: 10.1007/s00592-009-0159-7
Deng, X. Q., Chen, L. L., and Li, N. X. (2007). The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int. 27, 708–715. doi: 10.1111/j.1478-3231.2007.01497.x
Di Francesco, A., Di Germanio, C., Bernier, M., and De Cabo, R. (2018). A time to fast. Science 362, 770–775.
Ding, S., Jiang, J., Zhang, G., Bu, Y., Zhang, G., and Zhao, X. (2017). Resveratrol and caloric restriction prevent hepatic steatosis by regulating SIRT1-autophagy pathway and alleviating endoplasmic reticulum stress in high-fat diet-fed rats. PLoS One 12:e0183541. doi: 10.1371/journal.pone.0183541
Doehner, W., Erdmann, E., Cairns, R., Clark, A. L., Dormandy, J. A., Ferrannini, E., et al. (2012). Inverse relation of body weight and weight change with mortality and morbidity in patients with type 2 diabetes and cardiovascular co-morbidity: an analysis of the PROactive study population. Int. J. Cardiol. 162, 20–26. doi: 10.1016/j.ijcard.2011.09.039
Dong, Y., Guo, T., Traurig, M., Mason, C. C., Kobes, S., Perez, J., et al. (2011). SIRT1 is associated with a decrease in acute insulin secretion and a sex specific increase in risk for type 2 diabetes in Pima Indians. Mol. Genet. Metab. 104, 661–665. doi: 10.1016/j.ymgme.2011.08.001
Drinda, S., Grundler, F., Neumann, T., Lehmann, T., Steckhan, N., Michalsen, A., et al. (2019). Effects of periodic fasting on fatty liver index-A prospective observational study. Nutrients 11:2601. doi: 10.3390/nu11112601
Dulai, P. S., Singh, S., Patel, J., Soni, M., Prokop, L. J., Younossi, Z., et al. (2017). Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 65, 1557–1565. doi: 10.1002/hep.29085
Duncan, M. J., Smith, J. T., Narbaiza, J., Mueez, F., Bustle, L. B., Qureshi, S., et al. (2016). Restricting feeding to the active phase in middle-aged mice attenuates adverse metabolic effects of a high-fat diet. Physiol. Behav. 167, 1–9. doi: 10.1016/j.physbeh.2016.08.027
Duteil, D., Metzger, E., Willmann, D., Karagianni, P., Friedrichs, N., Greschik, H., et al. (2014). LSD1 promotes oxidative metabolism of white adipose tissue. Nat. Commun. 5:4093.
Duteil, D., Tosic, M., Lausecker, F., Nenseth, H. Z., Muller, J. M., Urban, S., et al. (2016). Lsd1 ablation triggers metabolic reprogramming of brown adipose tissue. Cell Rep. 17, 1008–1021. doi: 10.1016/j.celrep.2016.09.053
Endo, H., Niioka, M., Kobayashi, N., Tanaka, M., and Watanabe, T. (2013). Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: new insight into the probiotics for the gut-liver axis. PLoS One 8:e63388. doi: 10.1371/journal.pone.0063388
Erion, D. M., Yonemitsu, S., Nie, Y., Nagai, Y., Gillum, M. P., Hsiao, J. J., et al. (2009). SirT1 knockdown in liver decreases basal hepatic glucose production and increases hepatic insulin responsiveness in diabetic rats. Proc. Natl. Acad. Sci. U.S.A. 106, 11288–11293. doi: 10.1073/pnas.0812931106
Ezhkova, E., Lien, W. H., Stokes, N., Pasolli, H. A., Silva, J. M., and Fuchs, E. (2011). EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 25, 485–498. doi: 10.1101/gad.2019811
Fabbiano, S., Suarez-Zamorano, N., Chevalier, C., Lazarevic, V., Kieser, S., Rigo, D., et al. (2018). Functional gut microbiota remodeling contributes to the caloric restriction-induced metabolic improvements. Cell Metab. 28:e907.
Fajas, L., Landsberg, R. L., Huss-Garcia, Y., Sardet, C., Lees, J. A., and Auwerx, J. (2002). E2Fs regulate adipocyte differentiation. Dev. Cell 3, 39–49. doi: 10.1016/s1534-5807(02)00190-9
Feige, J. N., Lagouge, M., Canto, C., Strehle, A., Houten, S. M., Milne, J. C., et al. (2008). Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 8, 347–358. doi: 10.1016/j.cmet.2008.08.017
Feng, D., Liu, T., Sun, Z., Bugge, A., Mullican, S. E., Alenghat, T., et al. (2011). A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science 331, 1315–1319. doi: 10.1126/science.1198125
Ferrari, A., Fiorino, E., Longo, R., Barilla, S., Mitro, N., Cermenati, G., et al. (2017a). Attenuation of diet-induced obesity and induction of white fat browning with a chemical inhibitor of histone deacetylases. Int. J. Obes. 41, 289–298. doi: 10.1038/ijo.2016.191
Ferrari, A., Longo, R., Fiorino, E., Silva, R., Mitro, N., Cermenati, G., et al. (2017b). HDAC3 is a molecular brake of the metabolic switch supporting white adipose tissue browning. Nat. Commun. 8:93.
Fletcher, J. A., Deja, S., Satapati, S., Fu, X., Burgess, S. C., and Browning, J. D. (2019). Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 5:e127737.
Fontana, L., and Partridge, L. (2015). Promoting health and longevity through diet: from model organisms to humans. Cell 161, 106–118. doi: 10.1016/j.cell.2015.02.020
Fox, C. S., Massaro, J. M., Hoffmann, U., Pou, K. M., Maurovich-Horvat, P., Liu, C. Y., et al. (2007). Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation 116, 39–48. doi: 10.1161/circulationaha.106.675355
Friedman, S. L., Neuschwander-Tetri, B. A., Rinella, M., and Sanyal, A. J. (2018). Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–922. doi: 10.1038/s41591-018-0104-9
Funato, H., Oda, S., Yokofujita, J., Igarashi, H., and Kuroda, M. (2011). Fasting and high-fat diet alter histone deacetylase expression in the medial hypothalamus. PLoS One 6:e18950. doi: 10.1371/journal.pone.0018950
Furuyama, K., Chera, S., Van Gurp, L., Oropeza, D., Ghila, L., Damond, N., et al. (2019). Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells. Nature 567, 43–48. doi: 10.1038/s41586-019-0942-8
Gao, Z., Yin, J., Zhang, J., Ward, R. E., Martin, R. J., Lefevre, M., et al. (2009). Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58, 1509–1517. doi: 10.2337/db08-1637
Geng, S., Zhu, W., Xie, C., Li, X., Wu, J., Liang, Z., et al. (2016). Medium-chain triglyceride ameliorates insulin resistance and inflammation in high fat diet-induced obese mice. Eur. J. Nutr. 55, 931–940. doi: 10.1007/s00394-015-0907-0
Gensous, N., Franceschi, C., Santoro, A., Milazzo, M., Garagnani, P., and Bacalini, M. G. (2019). The impact of caloric restriction on the epigenetic signatures of aging. Int. J. Mol. Sci. 20:2022. doi: 10.3390/ijms20082022
Gerhard, G. S., Malenica, I., Llaci, L., Chu, X., Petrick, A. T., Still, C. D., et al. (2018). Differentially methylated loci in NAFLD cirrhosis are associated with key signaling pathways. Clin. Epigenetics 10:93.
Gerlini, R., Berti, L., Darr, J., Lassi, M., Brandmaier, S., Fritsche, L., et al. (2018). Glucose tolerance and insulin sensitivity define adipocyte transcriptional programs in human obesity. Mol. Metab. 18, 42–50. doi: 10.1016/j.molmet.2018.09.004
Gillum, M. P., Kotas, M. E., Erion, D. M., Kursawe, R., Chatterjee, P., Nead, K. T., et al. (2011). SirT1 regulates adipose tissue inflammation. Diabetes 60, 3235–3245.
Gius, D., Cui, H., Bradbury, C. M., Cook, J., Smart, D. K., Zhao, S., et al. (2004). Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell 6, 361–371. doi: 10.1016/j.ccr.2004.08.029
Gomes, P., Fleming Outeiro, T., and Cavadas, C. (2015). Emerging role of Sirtuin 2 in the regulation of mammalian metabolism. Trends Pharmacol. Sci. 36, 756–768. doi: 10.1016/j.tips.2015.08.001
Goodrick, C. L., Ingram, D. K., Reynolds, M. A., Freeman, J. R., and Cider, N. L. (1982). Effects of intermittent feeding upon growth and life span in rats. Gerontology 28, 233–241. doi: 10.1159/000212538
Gotthardt, J. D., Verpeut, J. L., Yeomans, B. L., Yang, J. A., Yasrebi, A., Roepke, T. A., et al. (2016). Intermittent fasting promotes fat loss with lean mass retention, increased hypothalamic norepinephrine content, and increased neuropeptide y gene expression in diet-induced obese male mice. Endocrinology 157, 679–691. doi: 10.1210/en.2015-1622
Grandison, R. C., Wong, R., Bass, T. M., Partridge, L., and Piper, M. D. (2009). Effect of a standardised dietary restriction protocol on multiple laboratory strains of Drosophila melanogaster. PLoS One 4:e4067. doi: 10.1371/journal.pone.0004067
Green, C. D., Huang, Y., Dou, X., Yang, L., Liu, Y., and Han, J. J. (2017). Impact of dietary interventions on noncoding RNA networks and mRNAs encoding chromatin-related factors. Cell Rep. 18, 2957–2968. doi: 10.1016/j.celrep.2017.03.001
Grindheim, J. M., Nicetto, D., Donahue, G., and Zaret, K. S. (2019). Polycomb repressive complex 2 proteins EZH1 and EZH2 regulate timing of postnatal hepatocyte maturation and fibrosis by repressing genes with euchromatic promoters in mice. Gastroenterology 156, 1834–1848. doi: 10.1053/j.gastro.2019.01.041
Hagopian, K., Ramsey, J. J., and Weindruch, R. (2003). Influence of age and caloric restriction on liver glycolytic enzyme activities and metabolite concentrations in mice. Exp. Gerontol. 38, 253–266. doi: 10.1016/s0531-5565(02)00203-6
Hahn, O., Gronke, S., Stubbs, T. M., Ficz, G., Hendrich, O., Krueger, F., et al. (2017). Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 18:56.
Haim, Y., Bluher, M., Slutsky, N., Goldstein, N., Kloting, N., Harman-Boehm, I., et al. (2015). Elevated autophagy gene expression in adipose tissue of obese humans: a potential non-cell-cycle-dependent function of E2F1. Autophagy 11, 2074–2088. doi: 10.1080/15548627.2015.1094597
Hainer, V., and Aldhoon-Hainerova, I. (2013). Obesity paradox does exist. Diabetes Care 36(Suppl. 2), S276–S281.
Halberg, N., Henriksen, M., Soderhamn, N., Stallknecht, B., Ploug, T., Schjerling, P., et al. (2005). Effect of intermittent fasting and refeeding on insulin action in healthy men. J. Appl. Physiol. 99, 2128–2136. doi: 10.1152/japplphysiol.00683.2005
Hall, E., Dayeh, T., Kirkpatrick, C. L., Wollheim, C. B., Dekker Nitert, M., and Ling, C. (2013). DNA methylation of the glucagon-like peptide 1 receptor (GLP1R) in human pancreatic islets. BMC Med. Genet. 14:76. doi: 10.1186/1471-2350-14-76
Hall, E., Dekker Nitert, M., Volkov, P., Malmgren, S., Mulder, H., Bacos, K., et al. (2018). The effects of high glucose exposure on global gene expression and DNA methylation in human pancreatic islets. Mol. Cell. Endocrinol. 472, 57–67. doi: 10.1016/j.mce.2017.11.019
Hall, E., Volkov, P., Dayeh, T., Bacos, K., Ronn, T., Nitert, M. D., et al. (2014). Effects of palmitate on genome-wide mRNA expression and DNA methylation patterns in human pancreatic islets. BMC Med. 12:103. doi: 10.1186/1741-7015-12-103
Hall, K. D., Ayuketah, A., Brychta, R., Cai, H., Cassimatis, T., Chen, K. Y., et al. (2019). Ultra-processed diets cause excess calorie intake and weight gain: an inpatient randomized controlled trial of ad libitum food intake. Cell Metab. 30:e63.
Hardy, T. M., and Tollefsbol, T. O. (2011). Epigenetic diet: impact on the epigenome and cancer. Epigenomics 3, 503–518. doi: 10.2217/epi.11.71
Harms, M., and Seale, P. (2013). Brown and beige fat: development, function and therapeutic potential. Nat. Med. 19, 1252–1263. doi: 10.1038/nm.3361
Harvey, A. E., Lashinger, L. M., Hays, D., Harrison, L. M., Lewis, K., Fischer, S. M., et al. (2014). Calorie restriction decreases murine and human pancreatic tumor cell growth, nuclear factor-kappaB activation, and inflammation-related gene expression in an insulin-like growth factor-1-dependent manner. PLoS One 9:e94151. doi: 10.1371/journal.pone.0094151
Harvie, M. N., Pegington, M., Mattson, M. P., Frystyk, J., Dillon, B., Evans, G., et al. (2011). The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: a randomized trial in young overweight women. Int. J. Obes. 35, 714–727. doi: 10.1038/ijo.2010.171
Hatori, M., Vollmers, C., Zarrinpar, A., Ditacchio, L., Bushong, E. A., Gill, S., et al. (2012). Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 15, 848–860. doi: 10.1016/j.cmet.2012.04.019
Hatting, M., Rines, A. K., Luo, C., Tabata, M., Sharabi, K., Hall, J. A., et al. (2017). Adipose tissue CLK2 promotes energy expenditure during high-fat diet intermittent fasting. Cell Metab. 25, 428–437. doi: 10.1016/j.cmet.2016.12.007
Hayashida, S., Arimoto, A., Kuramoto, Y., Kozako, T., Honda, S., Shimeno, H., et al. (2010). Fasting promotes the expression of SIRT1, an NAD+ -dependent protein deacetylase, via activation of PPARalpha in mice. Mol. Cell. Biochem. 339, 285–292. doi: 10.1007/s11010-010-0391-z
He, X. Y., Zhao, X. L., Gu, Q., Shen, J. P., Hu, Y., and Hu, R. M. (2012). Calorie restriction from a young age preserves the functions of pancreatic beta cells in aging rats. Tohoku J. Exp. Med. 227, 245–252. doi: 10.1620/tjem.227.245
Heald, A. H., Kaushal, K., Siddals, K. W., Rudenski, A. S., Anderson, S. G., and Gibson, J. M. (2006). Insulin-like growth factor binding protein-2 (IGFBP-2) is a marker for the metabolic syndrome. Exp. Clin. Endocrinol. Diabetes 114, 371–376.
Heilbronn, L. K., Smith, S. R., Martin, C. K., Anton, S. D., and Ravussin, E. (2005). Alternate-day fasting in nonobese subjects: effects on body weight, body composition, and energy metabolism. Am. J. Clin. Nutr. 81, 69–73. doi: 10.1093/ajcn/81.1.69
Hernandez, M. A. G., Canfora, E. E., Jocken, J. W. E., and Blaak, E. E. (2019). The short-chain fatty acid acetate in body weight control and insulin sensitivity. Nutrients 11:1943. doi: 10.3390/nu11081943
Hino, S., Sakamoto, A., Nagaoka, K., Anan, K., Wang, Y., Mimasu, S., et al. (2012). FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun. 3:758.
Hirano, Y., Yoshida, M., Shimizu, M., and Sato, R. (2001). Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathway. J. Biol. Chem. 276, 36431–36437. doi: 10.1074/jbc.m105200200
Hjort, L., Jorgensen, S. W., Gillberg, L., Hall, E., Brons, C., Frystyk, J., et al. (2017). 36 h fasting of young men influences adipose tissue DNA methylation of LEP and ADIPOQ in a birth weight-dependent manner. Clin. Epigenetics 9:40.
Honjoh, S., Yamamoto, T., Uno, M., and Nishida, E. (2009). Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature 457, 726–730. doi: 10.1038/nature07583
Horrillo, D., Gallardo, N., Lauzurica, N., Barrus, M. T., San Frutos, M. G., Andres, A., et al. (2013). Development of liver fibrosis during aging: effects of caloric restriction. J. Biol. Regul. Homeost. Agents 27, 377–388.
Hotta, K., Kitamoto, T., Kitamoto, A., Ogawa, Y., Honda, Y., Kessoku, T., et al. (2018). Identification of the genomic region under epigenetic regulation during non-alcoholic fatty liver disease progression. Hepatol. Res. 48, E320–E334.
Hoyeck, M. P., Blair, H., Ibrahim, M., Solanki, S., Elsawy, M., Prakash, A., et al. (2020). Long-term metabolic consequences of acute dioxin exposure differ between male and female mice. Sci. Rep. 10:1448.
Hu, D., Mao, Y., Xu, G., Liao, W., Ren, J., Yang, H., et al. (2019). Time-restricted feeding causes irreversible metabolic disorders and gut microbiota shift in pediatric mice. Pediatr. Res. 85, 518–526. doi: 10.1038/s41390-018-0156-z
Ibrahim, M., Macfarlane, E. M., Matteo, G., Hoyeck, M. P., Rick, K. R. C., Farokhi, S., et al. (2020). Functional cytochrome P450 1A enzymes are induced in mouse and human islets following pollutant exposure. Diabetologia 63, 162–178.
Igarashi, M., and Guarente, L. (2016). mTORC1 and SIRT1 cooperate to foster expansion of gut adult stem cells during calorie restriction. Cell 166, 436–450. doi: 10.1016/j.cell.2016.05.044
Inagaki, T., Tachibana, M., Magoori, K., Kudo, H., Tanaka, T., Okamura, M., et al. (2009). Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 14, 991–1001. doi: 10.1111/j.1365-2443.2009.01326.x
Iwanaga, T., Kuchiiwa, T., and Saito, M. (2009). Histochemical demonstration of monocarboxylate transporters in mouse brown adipose tissue. Biomed. Res. 30, 217–225. doi: 10.2220/biomedres.30.217
Jackness, C., Karmally, W., Febres, G., Conwell, I. M., Ahmed, L., Bessler, M., et al. (2013). Very low-calorie diet mimics the early beneficial effect of Roux-en-Y gastric bypass on insulin sensitivity and beta-cell Function in type 2 diabetic patients. Diabetes 62, 3027–3032. doi: 10.2337/db12-1762
Jamshed, H., Beyl, R. A., Della Manna, D. L., Yang, E. S., Ravussin, E., and Peterson, C. M. (2019). Early time-restricted feeding improves 24-hour glucose levels and affects markers of the circadian clock, aging, and autophagy in humans. Nutrients 11:1234. doi: 10.3390/nu11061234
Jang, M. K., Kim, J. H., and Jung, M. H. (2017). Histone H3K9 Demethylase JMJD2B Activates Adipogenesis by Regulating H3K9 Methylation on PPARgamma and C/EBPalpha during Adipogenesis. PLoS One 12:e0168185. doi: 10.1371/journal.pone.0168185
Jannat Ali Pour, N., Meshkani, R., Toolabi, K., Mohassel Azadi, S., Zand, S., and Emamgholipour, S. (2020). Adipose tissue mRNA expression of HDAC1, HDAC3 and HDAC9 in obese women in relation to obesity indices and insulin resistance. Mol. Biol. Rep. 47, 3459–3468. doi: 10.1007/s11033-020-05431-5
Jin, B., and Robertson, K. D. (2013). DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 754, 3–29. doi: 10.1007/978-1-4419-9967-2_1
Jin, Q., Wang, C., Kuang, X., Feng, X., Sartorelli, V., Ying, H., et al. (2014). Gcn5 and PCAF regulate PPARgamma and Prdm16 expression to facilitate brown adipogenesis. Mol. Cell. Biol. 34, 3746–3753. doi: 10.1128/mcb.00622-14
Jin, Q., Yu, L. R., Wang, L., Zhang, Z., Kasper, L. H., Lee, J. E., et al. (2011). Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262. doi: 10.1038/emboj.2010.318
Jing, E., Gesta, S., and Kahn, C. R. (2007). SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 6, 105–114. doi: 10.1016/j.cmet.2007.07.003
Johari, M. I., Yusoff, K., Haron, J., Nadarajan, C., Ibrahim, K. N., Wong, M. S., et al. (2019). A randomised controlled trial on the effectiveness and adherence of modified alternate-day calorie restriction in improving activity of non-alcoholic fatty liver disease. Sci. Rep. 9:11232.
Johnson, J. B., Summer, W., Cutler, R. G., Martin, B., Hyun, D. H., Dixit, V. D., et al. (2007). Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic. Biol. Med. 42, 665–674. doi: 10.1016/j.freeradbiomed.2006.12.005
Johnson, J. D., and Alejandro, E. U. (2008). Control of pancreatic beta-cell fate by insulin signaling: the sweet spot hypothesis. Cell Cycle 7, 1343–1347. doi: 10.4161/cc.7.10.5865
Jung, S. M., Sanchez-Gurmaches, J., and Guertin, D. A. (2019). Brown adipose tissue development and metabolism. Handb. Exp. Pharmacol. 251, 3–36. doi: 10.1007/164_2018_168
Kahn, S. E., Cooper, M. E., and Del Prato, S. (2014). Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383, 1068–1083. doi: 10.1016/s0140-6736(13)62154-6
Kammel, A., Saussenthaler, S., Jahnert, M., Jonas, W., Stirm, L., Hoeflich, A., et al. (2016). Early hypermethylation of hepatic Igfbp2 results in its reduced expression preceding fatty liver in mice. Hum. Mol. Genet. 25, 2588–2599.
Kanda, Y., Hashiramoto, M., Shimoda, M., Hamamoto, S., Tawaramoto, K., Kimura, T., et al. (2015). Dietary restriction preserves the mass and function of pancreatic beta cells via cell kinetic regulation and suppression of oxidative/ER stress in diabetic mice. J. Nutr. Biochem. 26, 219–226. doi: 10.1016/j.jnutbio.2014.10.007
Kautzky-Willer, A., Harreiter, J., and Pacini, G. (2016). Sex and gender differences in risk, pathophysiology and complications of type 2 diabetes mellitus. Endocr. Rev. 37, 278–316. doi: 10.1210/er.2015-1137
Kawakami, K., Nakamura, A., and Goto, S. (2012). Dietary restriction increases site-specific histone H3 acetylation in rat liver: possible modulation by sirtuins. Biochem. Biophys. Res. Commun. 418, 836–840. doi: 10.1016/j.bbrc.2012.01.120
Kim, A. Y., Park, Y. J., Pan, X., Shin, K. C., Kwak, S. H., Bassas, A. F., et al. (2015). Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat. Commun. 6:7585.
Kim, D. H., Kim, J., Kwon, J. S., Sandhu, J., Tontonoz, P., et al. (2016). Critical roles of the histone methyltransferase MLL4/KMT2D in murine hepatic steatosis directed by ABL1 and PPARgamma2. Cell Rep. 17, 1671–1682. doi: 10.1016/j.celrep.2016.10.023
Kim, J. H., Jung, D. Y., Nagappan, A., and Jung, M. H. (2018). Histone H3K9 demethylase JMJD2B induces hepatic steatosis through upregulation of PPARgamma2. Sci. Rep. 8:13734.
Kim, K., Boo, K., Yu, Y. S., Oh, S. K., Kim, H., Jeon, Y., et al. (2017). RORalpha controls hepatic lipid homeostasis via negative regulation of PPARgamma transcriptional network. Nat. Commun. 8:162.
Kim, K. H., Kim, Y. H., Son, J. E., Lee, J. H., Kim, S., Choe, M. S., et al. (2017). Intermittent fasting promotes adipose thermogenesis and metabolic homeostasis via VEGF-mediated alternative activation of macrophage. Cell Res. 27, 1309–1326. doi: 10.1038/cr.2017.126
Kim, R. Y., Lee, J. H., Oh, Y., Sung, H. K., and Kim, K. H. (2019). Assessment of the metabolic effects of isocaloric 2:1 intermittent fasting in mice. J. Vis. Exp. 153. doi: 10.3791/60174
Kim, Y. H., Lee, J. H., Yeung, J. L., Das, E., Kim, R. Y., Jiang, Y., et al. (2019). Thermogenesis-independent metabolic benefits conferred by isocaloric intermittent fasting in ob/ob mice. Sci. Rep. 9:2479.
Kirchner, H., Sinha, I., Gao, H., Ruby, M. A., Schonke, M., Lindvall, J. M., et al. (2016). Altered DNA methylation of glycolytic and lipogenic genes in liver from obese and type 2 diabetic patients. Mol. Metab. 5, 171–183. doi: 10.1016/j.molmet.2015.12.004
Kitamura, Y. I., Kitamura, T., Kruse, J. P., Raum, J. C., Stein, R., Gu, W., et al. (2005). FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2, 153–163. doi: 10.1016/j.cmet.2005.08.004
Klempel, M. C., Kroeger, C. M., and Varady, K. A. (2013). Alternate day fasting (ADF) with a high-fat diet produces similar weight loss and cardio-protection as ADF with a low-fat diet. Metabolism 62, 137–143. doi: 10.1016/j.metabol.2012.07.002
Knutson, S. K., Chyla, B. J., Amann, J. M., Bhaskara, S., Huppert, S. S., and Hiebert, S. W. (2008). Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 27, 1017–1028. doi: 10.1038/emboj.2008.51
Komiya, C., Tsuchiya, K., Shiba, K., Miyachi, Y., Furuke, S., Shimazu, N., et al. (2016). Ipragliflozin improves hepatic steatosis in obese mice and liver dysfunction in type 2 diabetic patients irrespective of body weight reduction. PLoS One 11:e0151511. doi: 10.1371/journal.pone.0151511
Krishnan, J., Danzer, C., Simka, T., Ukropec, J., Walter, K. M., Kumpf, S., et al. (2012). Dietary obesity-associated Hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 26, 259–270. doi: 10.1101/gad.180406.111
Lanza-Jacoby, S., Yan, G., Radice, G., Lephong, C., Baliff, J., and Hess, R. (2013). Calorie restriction delays the progression of lesions to pancreatic cancer in the LSL-KrasG12D, Pdx-1/Cre mouse model of pancreatic cancer. Exp. Biol. Med. 238, 787–797. doi: 10.1177/1535370213493727
Larson-Meyer, D. E., Newcomer, B. R., Heilbronn, L. K., Volaufova, J., Smith, S. R., Alfonso, A. J., et al. (2008). Effect of 6-month calorie restriction and exercise on serum and liver lipids and markers of liver function. Obesity 16, 1355–1362. doi: 10.1038/oby.2008.201
Lee, H. A., Kang, S. H., Kim, M., Lee, E., Cho, H. M., Moon, E. K., et al. (2018). Histone deacetylase inhibition ameliorates hypertension and hyperglycemia in a model of Cushing’s syndrome. Am. J. Physiol. Endocrinol. Metab. 314, E39–E52.
Lee, J., Saha, P. K., Yang, Q. H., Lee, S., Park, J. Y., Suh, Y., et al. (2008). Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 19229–19234. doi: 10.1073/pnas.0810100105
Lee, J. E., Wang, C., Xu, S., Cho, Y. W., Wang, L., Feng, X., et al. (2013). H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife 2:e01503.
Lee, J. H., Song, M. Y., Song, E. K., Kim, E. K., Moon, W. S., Han, M. K., et al. (2009). Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes 58, 344–351. doi: 10.2337/db07-1795
Ley, R. E., Peterson, D. A., and Gordon, J. I. (2006a). Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848. doi: 10.1016/j.cell.2006.02.017
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006b). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023.
Li, F., Wu, R., Cui, X., Zha, L., Yu, L., Shi, H., et al. (2016). Histone deacetylase 1 (HDAC1) negatively regulates thermogenic program in brown adipocytes via coordinated regulation of histone H3 Lysine 27 (H3K27) deacetylation and methylation. J. Biol. Chem. 291, 4523–4536. doi: 10.1074/jbc.m115.677930
Li, G., Xie, C., Lu, S., Nichols, R. G., Tian, Y., Li, L., et al. (2017). Intermittent fasting promotes white adipose browning and decreases obesity by shaping the gut microbiota. Cell Metab. 26, 672–685.e4.
Li, X. (2013). SIRT1 and energy metabolism. Acta Biochim. Biophys. Sin. 45, 51–60. doi: 10.1093/abbs/gms108
Liang, F., Kume, S., and Koya, D. (2009). SIRT1 and insulin resistance. Nat. Rev. Endocrinol. 5, 367–373. doi: 10.1038/nrendo.2009.101
Liao, J., Jiang, J., Jun, H., Qiao, X., Emont, M. P., Kim, D. I., et al. (2018). HDAC3-selective inhibition activates brown and beige fat through PRDM16. Endocrinology 159, 2520–2527. doi: 10.1210/en.2018-00257
Lin, S. J., Defossez, P. A., and Guarente, L. (2000). Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128. doi: 10.1126/science.289.5487.2126
Lin, S. J., Ford, E., Haigis, M., Liszt, G., and Guarente, L. (2004). Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 18, 12–16. doi: 10.1101/gad.1164804
Lin, S. J., Kaeberlein, M., Andalis, A. A., Sturtz, L. A., Defossez, P. A., Culotta, V. C., et al. (2002). Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418, 344–348. doi: 10.1038/nature00829
Liu, B., Page, A. J., Hatzinikolas, G., Chen, M., Wittert, G. A., and Heilbronn, L. K. (2019). Intermittent fasting improves glucose tolerance and promotes adipose tissue remodeling in male mice fed a high-fat diet. Endocrinology 160, 169–180. doi: 10.1210/en.2018-00701
Liu, T. F., Yoza, B. K., El Gazzar, M., Vachharajani, V. T., and Mccall, C. E. (2011). NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 286, 9856–9864. doi: 10.1074/jbc.m110.196790
Liu, Y., Cheng, A., Li, Y. J., Yang, Y., Kishimoto, Y., Zhang, S., et al. (2019). SIRT3 mediates hippocampal synaptic adaptations to intermittent fasting and ameliorates deficits in APP mutant mice. Nat. Commun. 10:1886.
Liu, Y., Dentin, R., Chen, D., Hedrick, S., Ravnskjaer, K., Schenk, S., et al. (2008). A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 456, 269–273. doi: 10.1038/nature07349
Liu, Z., Dai, X., Zhang, H., Shi, R., Hui, Y., Jin, X., et al. (2020). Gut microbiota mediates intermittent-fasting alleviation of diabetes-induced cognitive impairment. Nat. Commun. 11:855.
Liu, Z., Gan, L., Liu, G., Chen, Y., Wu, T., Feng, F., et al. (2016). Sirt1 decreased adipose inflammation by interacting with Akt2 and inhibiting mTOR/S6K1 pathway in mice. J. Lipid Res. 57, 1373–1381. doi: 10.1194/jlr.m063537
Locher, J. L., Goldsby, T. U., Goss, A. M., Kilgore, M. L., Gower, B., and Ard, J. D. (2016). Calorie restriction in overweight older adults: Do benefits exceed potential risks? Exp. Gerontol. 86, 4–13. doi: 10.1016/j.exger.2016.03.009
Locke, A. E., Kahali, B., Berndt, S. I., Justice, A. E., Pers, T. H., Day, F. R., et al. (2015). Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206.
Longo, M., Zatterale, F., Naderi, J., Parrillo, L., Formisano, P., Raciti, G. A., et al. (2019). Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int. J. Mol. Sci. 20:2358. doi: 10.3390/ijms20092358
Longo, V. D., and Mattson, M. P. (2014). Fasting: molecular mechanisms and clinical applications. Cell Metab. 19, 181–192. doi: 10.1016/j.cmet.2013.12.008
Longo, V. D., and Panda, S. (2016). Fasting, circadian rhythms, and time-restricted feeding in healthy lifespan. Cell Metab. 23, 1048–1059. doi: 10.1016/j.cmet.2016.06.001
Louis, P., and Flint, H. J. (2009). Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 294, 1–8. doi: 10.1111/j.1574-6968.2009.01514.x
Lu, H., Lei, X., and Zhang, Q. (2019). Liver-specific knockout of histone methyltransferase G9a impairs liver maturation and dysregulates inflammatory, cytoprotective, and drug-processing genes. Xenobiotica 49, 740–752. doi: 10.1080/00498254.2018.1490044
Luu, L., Dai, F. F., Prentice, K. J., Huang, X., Hardy, A. B., Hansen, J. B., et al. (2013). The loss of Sirt1 in mouse pancreatic beta cells impairs insulin secretion by disrupting glucose sensing. Diabetologia 56, 2010–2020. doi: 10.1007/s00125-013-2946-5
Luukkonen, P. K., Dufour, S., Lyu, K., Zhang, X. M., Hakkarainen, A., Lehtimaki, T. E., et al. (2020). Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. U.S.A. 117, 7347–7354. doi: 10.1073/pnas.1922344117
Macartney-Coxson, D., Benton, M. C., Blick, R., Stubbs, R. S., Hagan, R. D., and Langston, M. A. (2017). Genome-wide DNA methylation analysis reveals loci that distinguish different types of adipose tissue in obese individuals. Clin. Epigenetics 9:48.
Maeda, S., Koya, D., Araki, S. I., Babazono, T., Umezono, T., Toyoda, M., et al. (2011). Association between single nucleotide polymorphisms within genes encoding sirtuin families and diabetic nephropathy in Japanese subjects with type 2 diabetes. Clin Exp Nephrol 15, 381–390. doi: 10.1007/s10157-011-0418-0
Maegawa, S., Lu, Y., Tahara, T., Lee, J. T., Madzo, J., Liang, S., et al. (2017). Caloric restriction delays age-related methylation drift. Nat. Commun. 8:539.
Maiztegui, B., Roman, C. L., Gagliardino, J. J., and Flores, L. E. (2018). Impaired endocrine-metabolic homeostasis: underlying mechanism of its induction by unbalanced diet. Clin. Sci. 132, 869–881. doi: 10.1042/cs20171616
Malandrucco, I., Pasqualetti, P., Giordani, I., Manfellotto, D., De Marco, F., Alegiani, F., et al. (2012). Very-low-calorie diet: a quick therapeutic tool to improve beta cell function in morbidly obese patients with type 2 diabetes. Am. J. Clin. Nutr. 95, 609–613. doi: 10.3945/ajcn.111.023697
Mann, J., Chu, D. C., Maxwell, A., Oakley, F., Zhu, N. L., Tsukamoto, H., et al. (2010). MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 138:705. doi: 10.1053/j.gastro.2009.10.002
Mann, J., Oakley, F., Akiboye, F., Elsharkawy, A., Thorne, A. W., and Mann, D. A. (2007). Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell Death Differ. 14, 275–285. doi: 10.1038/sj.cdd.4401979
Mannisto, V. T., Simonen, M., Hyysalo, J., Soininen, P., Kangas, A. J., Kaminska, D., et al. (2015). Ketone body production is differentially altered in steatosis and non-alcoholic steatohepatitis in obese humans. Liver Int. 35, 1853–1861. doi: 10.1111/liv.12769
Mariani, S., Fiore, D., Basciani, S., Persichetti, A., Contini, S., Lubrano, C., et al. (2015). Plasma levels of SIRT1 associate with non-alcoholic fatty liver disease in obese patients. Endocrine 49, 711–716. doi: 10.1007/s12020-014-0465-x
Marinho, T. S., Ornellas, F., Barbosa-Da-Silva, S., Mandarim-De-Lacerda, C. A., and Aguila, M. B. (2019). Beneficial effects of intermittent fasting on steatosis and inflammation of the liver in mice fed a high-fat or a high-fructose diet. Nutrition 65, 103–112. doi: 10.1016/j.nut.2019.02.020
Mattison, J. A., Colman, R. J., Beasley, T. M., Allison, D. B., Kemnitz, J. W., Roth, G. S., et al. (2017). Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 8:14063.
Meier, J. J., Butler, A. E., Saisho, Y., Monchamp, T., Galasso, R., Bhushan, A., et al. (2008). Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 57, 1584–1594. doi: 10.2337/db07-1369
Meydani, M., Das, S., Band, M., Epstein, S., and Roberts, S. (2011). The effect of caloric restriction and glycemic load on measures of oxidative stress and antioxidants in humans: results from the CALERIE Trial of Human Caloric Restriction. J. Nutr. Health Aging 15, 456–460. doi: 10.1007/s12603-011-0002-z
Mihaylova, M. M., Vasquez, D. S., Ravnskjaer, K., Denechaud, P. D., Yu, R. T., Alvarez, J. G., et al. (2011). Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145, 607–621. doi: 10.1016/j.cell.2011.03.043
Mitchell, S. J., Bernier, M., Mattison, J. A., Aon, M. A., Kaiser, T. A., Anson, R. M., et al. (2019). Daily fasting improves health and survival in male mice independent of diet composition and calories. Cell Metab. 29, 221–228.e3.
Miyamoto, J., Ohue-Kitano, R., Mukouyama, H., Nishida, A., Watanabe, K., Igarashi, M., et al. (2019). Ketone body receptor GPR43 regulates lipid metabolism under ketogenic conditions. Proc. Natl. Acad. Sci. U.S.A. 116, 23813–23821. doi: 10.1073/pnas.1912573116
Miyamura, Y., Tawa, R., Koizumi, A., Uehara, Y., Kurishita, A., Sakurai, H., et al. (1993). Effects of energy restriction on age-associated changes of DNA methylation in mouse liver. Mutat. Res. 295, 63–69. doi: 10.1016/0921-8734(93)90002-k
Moynihan, K. A., Grimm, A. A., Plueger, M. M., Bernal-Mizrachi, E., Ford, E., Cras-Meneur, C., et al. (2005). Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2, 105–117. doi: 10.1016/j.cmet.2005.07.001
Muir, L. A., Neeley, C. K., Meyer, K. A., Baker, N. A., Brosius, A. M., Washabaugh, A. R., et al. (2016). Adipose tissue fibrosis, hypertrophy, and hyperplasia: Correlations with diabetes in human obesity. Obesity 24, 597–605. doi: 10.1002/oby.21377
Muller, T. D., Finan, B., Bloom, S. R., D’alessio, D., Drucker, D. J., Flatt, P. R., et al. (2019). Glucagon-like peptide 1 (GLP-1). Mol. Metab. 30, 72–130.
Multhaup, M. L., Seldin, M. M., Jaffe, A. E., Lei, X., Kirchner, H., Mondal, P., et al. (2015). Mouse-human experimental epigenetic analysis unmasks dietary targets and genetic liability for diabetic phenotypes. Cell Metab. 21, 138–149. doi: 10.1016/j.cmet.2014.12.014
Nam, S. Y., Lee, E. J., Kim, K. R., Cha, B. S., Song, Y. D., Lim, S. K., et al. (1997). Effect of obesity on total and free insulin-like growth factor (IGF)-1, and their relationship to IGF-binding protein (BP)-1, IGFBP-2, IGFBP-3, insulin, and growth hormone. Int. J. Obes. Relat. Metab. Disord. 21, 355–359. doi: 10.1038/sj.ijo.0800412
Neeland, I. J., Ayers, C. R., Rohatgi, A. K., Turer, A. T., Berry, J. D., Das, S. R., et al. (2013). Associations of visceral and abdominal subcutaneous adipose tissue with markers of cardiac and metabolic risk in obese adults. Obesity 21, E439–E447.
Nemoto, S., Fergusson, M. M., and Finkel, T. (2005). SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 280, 16456–16460. doi: 10.1074/jbc.m501485200
Newman, J. C., and Verdin, E. (2014). Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 25, 42–52. doi: 10.1016/j.tem.2013.09.002
Newman, J. C., and Verdin, E. (2017). beta-hydroxybutyrate: a signaling metabolite. Annu. Rev. Nutr. 37, 51–76. doi: 10.1146/annurev-nutr-071816-064916
Ng, M., Fleming, T., Robinson, M., Thomson, B., Graetz, N., Margono, C., et al. (2014). Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384, 766–781.
Nie, L., Shuai, L., Zhu, M., Liu, P., Xie, Z. F., Jiang, S., et al. (2017). The landscape of histone modifications in a high-fat diet-induced obese (DIO) mouse model. Mol. Cell. Proteomics 16, 1324–1334. doi: 10.1074/mcp.m117.067553
Nilsson, E., Matte, A., Perfilyev, A., De Mello, V. D., Kakela, P., Pihlajamaki, J., et al. (2015). Epigenetic alterations in human liver from subjects with type 2 diabetes in parallel with reduced folate levels. J. Clin. Endocrinol. Metab. 100, E1491–E1501.
Ohneda, M., Inman, L. R., and Unger, R. H. (1995). Caloric restriction in obese pre-diabetic rats prevents beta-cell depletion, loss of beta-cell GLUT 2 and glucose incompetence. Diabetologia 38, 173–179. doi: 10.1007/s001250050267
Ohno, H., Shinoda, K., Ohyama, K., Sharp, L. Z., and Kajimura, S. (2013). EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature 504, 163–167. doi: 10.1038/nature12652
Olsson, A. H., Volkov, P., Bacos, K., Dayeh, T., Hall, E., Nilsson, E. A., et al. (2014). Genome-wide associations between genetic and epigenetic variation influence mRNA expression and insulin secretion in human pancreatic islets. PLoS Genet. 10:e1004886. doi: 10.1371/journal.pgen.1004886
Ortega-Prieto, P., and Postic, C. (2019). Carbohydrate sensing through the transcription factor ChREBP. Front. Genet. 10:472. doi: 10.3389/fgene.2019.00472
Pan, D., Huang, L., Zhu, L. J., Zou, T., Ou, J., Zhou, W., et al. (2015). Jmjd3-mediated H3K27me3 dynamics orchestrate brown fat development and regulate white fat plasticity. Dev. Cell 35, 568–583. doi: 10.1016/j.devcel.2015.11.002
Parrillo, L., Costa, V., Raciti, G. A., Longo, M., Spinelli, R., Esposito, R., et al. (2016). Hoxa5 undergoes dynamic DNA methylation and transcriptional repression in the adipose tissue of mice exposed to high-fat diet. Int. J. Obes. 40, 929–937. doi: 10.1038/ijo.2016.36
Pellegrinelli, V., Carobbio, S., and Vidal-Puig, A. (2016). Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia 59, 1075–1088. doi: 10.1007/s00125-016-3933-4
Perrini, S., Porro, S., Nigro, P., Cignarelli, A., Caccioppoli, C., Genchi, V. A., et al. (2020). Reduced SIRT1 and SIRT2 expression promotes adipogenesis of human visceral adipose stem cells and associates with accumulation of visceral fat in human obesity. Int. J. Obes. 44, 307–319. doi: 10.1038/s41366-019-0436-7
Pfluger, P. T., Herranz, D., Velasco-Miguel, S., Serrano, M., and Tschop, M. H. (2008). Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. U.S.A. 105, 9793–9798. doi: 10.1073/pnas.0802917105
Picard, F., Kurtev, M., Chung, N., Topark-Ngarm, A., Senawong, T., Machado De Oliveira, R., et al. (2004). Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 429, 771–776. doi: 10.1038/nature02583
Piciucchi, M., Capurso, G., Archibugi, L., Delle Fave, M. M., Capasso, M., and Delle Fave, G. (2015). Exocrine pancreatic insufficiency in diabetic patients: prevalence, mechanisms, and treatment. Int. J. Endocrinol. 2015:595649.
Pinho, A. V., Bensellam, M., Wauters, E., Rees, M., Giry-Laterriere, M., Mawson, A., et al. (2015). Pancreas-specific Sirt1-deficiency in mice compromises beta-cell function without development of hyperglycemia. PLoS One 10:e0128012. doi: 10.1371/journal.pone.0128012
Piotrowska, K., Tarnowski, M., Zgutka, K., and Pawlik, A. (2016). Gender differences in response to prolonged every-other-day feeding on the proliferation and apoptosis of hepatocytes in mice. Nutrients 8:176. doi: 10.3390/nu8030176
Pirola, C. J., Gianotti, T. F., Burgueno, A. L., Rey-Funes, M., Loidl, C. F., Mallardi, P., et al. (2013). Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 62, 1356–1363. doi: 10.1136/gutjnl-2012-302962
Pogribny, I. P., Tryndyak, V. P., Bagnyukova, T. V., Melnyk, S., Montgomery, B., Ross, S. A., et al. (2009). Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J. Hepatol. 51, 176–186. doi: 10.1016/j.jhep.2009.03.021
Polidori, D., Iijima, H., Goda, M., Maruyama, N., Inagaki, N., and Crawford, P. A. (2018). Intra- and inter-subject variability for increases in serum ketone bodies in patients with type 2 diabetes treated with the sodium glucose co-transporter 2 inhibitor canagliflozin. Diabetes Obes. Metab. 20, 1321–1326. doi: 10.1111/dom.13224
Prattichizzo, F., De Nigris, V., Micheloni, S., La Sala, L., and Ceriello, A. (2018). Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: Is low-grade inflammation the neglected component? Diabetes Obes. Metab. 20, 2515–2522. doi: 10.1111/dom.13488
Puchalska, P., and Crawford, P. A. (2017). Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 25, 262–284. doi: 10.1016/j.cmet.2016.12.022
Puchalska, P., Martin, S. E., Huang, X., Lengfeld, J. E., Daniel, B., Graham, M. J., et al. (2019). Hepatocyte-macrophage acetoacetate shuttle protects against tissue fibrosis. Cell Metab. 29, 383–398.e7.
Purnell, C. A., Vaca, E. E., and Ellis, M. F. (2017). Conical modification of forearm free flaps for single-stage reconstruction after total orbital exenteration. J. Craniofac. Surg. 28, e767–e769.
Purushotham, A., Schug, T. T., Xu, Q., Surapureddi, S., Guo, X., and Li, X. (2009). Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9, 327–338. doi: 10.1016/j.cmet.2009.02.006
Qiang, L., Wang, L., Kon, N., Zhao, W., Lee, S., Zhang, Y., et al. (2012). Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell 150, 620–632. doi: 10.1016/j.cell.2012.06.027
Qiao, L., and Shao, J. (2006). SIRT1 regulates adiponectin gene expression through Foxo1-C/enhancer-binding protein alpha transcriptional complex. J. Biol. Chem. 281, 39915–39924. doi: 10.1074/jbc.m607215200
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65.
Raciti, G. A., Spinelli, R., Desiderio, A., Longo, M., Parrillo, L., Nigro, C., et al. (2017). Specific CpG hyper-methylation leads to Ankrd26 gene down-regulation in white adipose tissue of a mouse model of diet-induced obesity. Sci. Rep. 7:43526.
Rajan, A., Shi, H., and Xue, B. (2018). Class I and II histone deacetylase inhibitors differentially regulate thermogenic gene expression in brown adipocytes. Sci. Rep. 8:13072.
Ramirez, T., Li, Y. M., Yin, S., Xu, M. J., Feng, D., Zhou, Z., et al. (2017). Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J. Hepatol. 66, 601–609. doi: 10.1016/j.jhep.2016.11.004
Ramsey, K. M., Mills, K. F., Satoh, A., and Imai, S. (2008). Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell 7, 78–88. doi: 10.1111/j.1474-9726.2007.00355.x
Rangan, P., Choi, I., Wei, M., Navarrete, G., Guen, E., Brandhorst, S., et al. (2019). Fasting-mimicking diet modulates microbiota and promotes intestinal regeneration to reduce inflammatory bowel disease pathology. Cell Rep. 26:2704–2719.e6.
Ravussin, E., Beyl, R. A., Poggiogalle, E., Hsia, D. S., and Peterson, C. M. (2019). Early time-restricted feeding reduces appetite and increases fat oxidation but does not affect energy expenditure in humans. Obesity 27, 1244–1254. doi: 10.1002/oby.22518
Redman, L. M., Smith, S. R., Burton, J. H., Martin, C. K., Il’yasova, D., and Ravussin, E. (2018). Metabolic slowing and reduced oxidative damage with sustained caloric restriction support the rate of living and oxidative damage theories of aging. Cell Metab. 27, 805–815.e4.
Reinke, H., and Asher, G. (2016). Circadian clock control of liver metabolic functions. Gastroenterology 150, 574–580. doi: 10.1053/j.gastro.2015.11.043
Reizel, Y., Spiro, A., Sabag, O., Skversky, Y., Hecht, M., Keshet, I., et al. (2015). Gender-specific postnatal demethylation and establishment of epigenetic memory. Genes Dev. 29, 923–933. doi: 10.1101/gad.259309.115
Roder, P. V., Wu, B., Liu, Y., and Han, W. (2016). Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 48:e219. doi: 10.1038/emm.2016.6
Rodgers, J. T., Lerin, C., Haas, W., Gygi, S. P., Spiegelman, B. M., and Puigserver, P. (2005). Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434, 113–118. doi: 10.1038/nature03354
Rodgers, J. T., and Puigserver, P. (2007). Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc. Natl. Acad. Sci. U.S.A. 104, 12861–12866. doi: 10.1073/pnas.0702509104
Ronis, M. J., Baumgardner, J. N., Sharma, N., Vantrease, J., Ferguson, M., Tong, Y., et al. (2013). Medium chain triglycerides dose-dependently prevent liver pathology in a rat model of non-alcoholic fatty liver disease. Exp. Biol. Med. 238, 151–162. doi: 10.1258/ebm.2012.012303
Rosa, C., Campos, J. M., Sa Nakanishi, A. B., Comar, J. F., Martins, I. P., Mathias, P. C. F., et al. (2018). Food restriction promotes damage reduction in rat models of type 2 diabetes mellitus. PLoS One 13:e0199479. doi: 10.1371/journal.pone.0199479
Rosen, E. D. (2016). Epigenomic and transcriptional control of insulin resistance. J. Intern. Med. 280, 443–456. doi: 10.1111/joim.12547
Rosen, E. D., Kaestner, K. H., Natarajan, R., Patti, M. E., Sallari, R., Sander, M., et al. (2018). Epigenetics and epigenomics: implications for diabetes and obesity. Diabetes 67, 1923–1931. doi: 10.2337/db18-0537
Ruchat, S. M., Elks, C. E., Loos, R. J., Vohl, M. C., Weisnagel, S. J., Rankinen, T., et al. (2009). Association between insulin secretion, insulin sensitivity and type 2 diabetes susceptibility variants identified in genome-wide association studies. Acta Diabetol. 46, 217–226. doi: 10.1007/s00592-008-0080-5
Rui, L. (2014). Energy metabolism in the liver. Compr. Physiol. 4, 177–197. doi: 10.1002/cphy.c130024
Rusli, F., Lute, C., Boekschoten, M. V., Van Dijk, M., Van Norren, K., Menke, A. L., et al. (2017). Intermittent calorie restriction largely counteracts the adverse health effects of a moderate-fat diet in aging C57BL/6J mice. Mol. Nutr. Food Res. 61:1600677. doi: 10.1002/mnfr.201600677
Sabari, B. R., Zhang, D., Allis, C. D., and Zhao, Y. (2017). Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 18, 90–101. doi: 10.1038/nrm.2016.140
Sacks, H., and Symonds, M. E. (2013). Anatomical locations of human brown adipose tissue: functional relevance and implications in obesity and type 2 diabetes. Diabetes 62, 1783–1790. doi: 10.2337/db12-1430
Sambeat, A., Gulyaeva, O., Dempersmier, J., Tharp, K. M., Stahl, A., Paul, S. M., et al. (2016). LSD1 interacts with Zfp516 to promote UCP1 transcription and brown fat program. Cell Rep. 15, 2536–2549. doi: 10.1016/j.celrep.2016.05.019
Sathananthan, M., Shah, M., Edens, K. L., Grothe, K. B., Piccinini, F., Farrugia, L. P., et al. (2015). Six and 12 weeks of caloric restriction increases beta cell function and lowers fasting and postprandial glucose concentrations in people with Type 2 diabetes. J. Nutr. 145, 2046–2051. doi: 10.3945/jn.115.210617
Scheen, A. J. (2019). Beneficial effects of SGLT2 inhibitors on fatty liver in type 2 diabetes: a common comorbidity associated with severe complications. Diabetes Metab. 45, 213–223. doi: 10.1016/j.diabet.2019.01.008
Schug, T. T., and Li, X. (2011). Sirtuin 1 in lipid metabolism and obesity. Ann. Med. 43, 198–211. doi: 10.3109/07853890.2010.547211
Seok, S., Kim, Y. C., Byun, S., Choi, S., Xiao, Z., Iwamori, N., et al. (2018). Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid beta-oxidation. J. Clin. Invest. 128, 3144–3159. doi: 10.1172/jci97736
Sevastianova, K., Santos, A., Kotronen, A., Hakkarainen, A., Makkonen, J., Silander, K., et al. (2012). Effect of short-term carbohydrate overfeeding and long-term weight loss on liver fat in overweight humans. Am. J. Clin. Nutr. 96, 727–734. doi: 10.3945/ajcn.112.038695
Sharma, M., Li, Y., Stoll, M. L., and Tollefsbol, T. O. (2019). The epigenetic connection between the gut microbiome in obesity and diabetes. Front. Genet. 10:1329. doi: 10.3389/fgene.2019.01329
Sharp, A. J., Stathaki, E., Migliavacca, E., Brahmachary, M., Montgomery, S. B., Dupre, Y., et al. (2011). DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 21, 1592–1600. doi: 10.1101/gr.112680.110
Sheng, C., Li, F., Lin, Z., Zhang, M., Yang, P., Bu, L., et al. (2016). Reversibility of beta-cell-specific transcript factors expression by long-term caloric restriction in db/db mouse. J. Diabetes Res. 2016:6035046.
Shimazu, T., Hirschey, M. D., Hua, L., Dittenhafer-Reed, K. E., Schwer, B., Lombard, D. B., et al. (2010). SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 12, 654–661. doi: 10.1016/j.cmet.2010.11.003
Shimazu, T., Hirschey, M. D., Newman, J., He, W., Shirakawa, K., Le Moan, N., et al. (2013). Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. doi: 10.1126/science.1227166
Shimizu, I., Aprahamian, T., Kikuchi, R., Shimizu, A., Papanicolaou, K. N., Maclauchlan, S., et al. (2014). Vascular rarefaction mediates whitening of brown fat in obesity. J. Clin. Invest. 124, 2099–2112. doi: 10.1172/jci71643
Shu, L., Matveyenko, A. V., Kerr-Conte, J., Cho, J. H., Mcintosh, C. H., and Maedler, K. (2009). Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum. Mol. Genet. 18, 2388–2399. doi: 10.1093/hmg/ddp178
Sonne, S. B., Yadav, R., Yin, G., Dalgaard, M. D., Myrmel, L. S., Gupta, R., et al. (2017). Obesity is associated with depot-specific alterations in adipocyte DNA methylation and gene expression. Adipocyte 6, 124–133. doi: 10.1080/21623945.2017.1320002
Sparling, D. P., Griesel, B. A., Weems, J., and Olson, A. L. (2008). GLUT4 enhancer factor (GEF) interacts with MEF2A and HDAC5 to regulate the GLUT4 promoter in adipocytes. J. Biol. Chem. 283, 7429–7437. doi: 10.1074/jbc.m800481200
Stal, P. (2015). Liver fibrosis in non-alcoholic fatty liver disease - diagnostic challenge with prognostic significance. World J. Gastroenterol. 21, 11077–11087. doi: 10.3748/wjg.v21.i39.11077
Stekovic, S., Hofer, S. J., Tripolt, N., Aon, M. A., Royer, P., Pein, L., et al. (2019). Alternate day fasting improves physiological and molecular markers of aging in healthy, non-obese humans. Cell Metab. 30, 462–476.e6.
Stern, J. H., Rutkowski, J. M., and Scherer, P. E. (2016). Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 23, 770–784. doi: 10.1016/j.cmet.2016.04.011
Sundqvist, A., and Ericsson, J. (2003). Transcription-dependent degradation controls the stability of the SREBP family of transcription factors. Proc. Natl. Acad. Sci. U.S.A. 100, 13833–13838. doi: 10.1073/pnas.2335135100
Sutton, E. F., Beyl, R., Early, K. S., Cefalu, W. T., Ravussin, E., and Peterson, C. M. (2018). Early time-restricted feeding improves insulin sensitivity, blood pressure, and oxidative stress even without weight loss in men with prediabetes. Cell Metab. 27, 1212–1221.e3.
Tanaka, N., Aoyama, T., Kimura, S., and Gonzalez, F. J. (2017). Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol. Ther. 179, 142–157. doi: 10.1016/j.pharmthera.2017.05.011
Taneera, J., Lang, S., Sharma, A., Fadista, J., Zhou, Y., Ahlqvist, E., et al. (2012). A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 16, 122–134. doi: 10.1016/j.cmet.2012.06.006
Tang, Q. Q., and Lane, M. D. (2012). Adipogenesis: from stem cell to adipocyte. Annu. Rev. Biochem. 81, 715–736. doi: 10.1146/annurev-biochem-052110-115718
Tateishi, K., Okada, Y., Kallin, E. M., and Zhang, Y. (2009). Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 458, 757–761. doi: 10.1038/nature07777
Teslovich, T. M., Musunuru, K., Smith, A. V., Edmondson, A. C., Stylianou, I. M., Koseki, M., et al. (2010). Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713.
Teta, M., Long, S. Y., Wartschow, L. M., Rankin, M. M., and Kushner, J. A. (2005). Very slow turnover of beta-cells in aged adult mice. Diabetes 54, 2557–2567. doi: 10.2337/diabetes.54.9.2557
Thaiss, C. A., Zeevi, D., Levy, M., Zilberman-Schapira, G., Suez, J., Tengeler, A. C., et al. (2014). Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell 159, 514–529. doi: 10.1016/j.cell.2014.09.048
Thiele, M., Madsen, B. S., and Krag, A. (2017). Is liver stiffness equal to liver fibrosis? Hepatology 65:749. doi: 10.1002/hep.28791
Thieringer, F., Maass, T., Czochra, P., Klopcic, B., Conrad, I., Friebe, D., et al. (2008). Spontaneous hepatic fibrosis in transgenic mice overexpressing PDGF-A. Gene 423, 23–28. doi: 10.1016/j.gene.2008.05.022
Thorpe, R. J. Jr., and Ferraro, K. F. (2004). Aging, obesity, and mortality: misplaced concern about obese older people? Res. Aging 26, 108–129. doi: 10.1177/0164027503258738
Tian, Y., Wong, V. W., Wong, G. L., Yang, W., Sun, H., Shen, J., et al. (2015). Histone deacetylase HDAC8 promotes insulin resistance and beta-catenin activation in NAFLD-associated hepatocellular carcinoma. Cancer Res. 75, 4803–4816. doi: 10.1158/0008-5472.can-14-3786
Tordjman, K., Standley, K. N., Bernal-Mizrachi, C., Leone, T. C., Coleman, T., Kelly, D. P., et al. (2002). PPARalpha suppresses insulin secretion and induces UCP2 in insulinoma cells. J. Lipid. Res. 43, 936–943.
Tramunt, B., Smati, S., Grandgeorge, N., Lenfant, F., Arnal, J. F., Montagner, A., et al. (2020). Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 63, 453–461. doi: 10.1007/s00125-019-05040-3
Tsuchida, T., and Friedman, S. L. (2017). Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411.
Uno, M., Honjoh, S., Matsuda, M., Hoshikawa, H., Kishimoto, S., Yamamoto, T., et al. (2013). A fasting-responsive signaling pathway that extends life span in C. elegans. Cell Rep. 3, 79–91. doi: 10.1016/j.celrep.2012.12.018
Vaquero, A., Scher, M., Lee, D., Erdjument-Bromage, H., Tempst, P., and Reinberg, D. (2004). Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 16, 93–105. doi: 10.1016/j.molcel.2004.08.031
Vaquero, A., Scher, M. B., Lee, D. H., Sutton, A., Cheng, H. L., Alt, F. W., et al. (2006). SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 20, 1256–1261. doi: 10.1101/gad.1412706
Varady, K. A., Bhutani, S., Church, E. C., and Klempel, M. C. (2009). Short-term modified alternate-day fasting: a novel dietary strategy for weight loss and cardioprotection in obese adults. Am. J. Clin. Nutr. 90, 1138–1143. doi: 10.3945/ajcn.2009.28380
Varady, K. A., Bhutani, S., Klempel, M. C., Kroeger, C. M., Trepanowski, J. F., Haus, J. M., et al. (2013). Alternate day fasting for weight loss in normal weight and overweight subjects: a randomized controlled trial. Nutr. J. 12:146.
Vella, S., Gnani, D., Crudele, A., Ceccarelli, S., De Stefanis, C., Gaspari, S., et al. (2013). EZH2 down-regulation exacerbates lipid accumulation and inflammation in in vitro and in vivo NAFLD. Int. J. Mol. Sci. 14, 24154–24168. doi: 10.3390/ijms141224154
Vergnes, L., and Reue, K. (2014). Adaptive thermogenesis in white adipose tissue: is lactate the new brown(ing)? Diabetes 63, 3175–3176. doi: 10.2337/db14-0815
Vidal-Puig, A., Jimenez-Linan, M., Lowell, B. B., Hamann, A., Hu, E., Spiegelman, B., et al. (1996). Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J. Clin. Invest. 97, 2553–2561. doi: 10.1172/jci118703
Vidmar, A. P., Goran, M. I., and Raymond, J. K. (2019). Time-limited eating in pediatric patients with obesity: a case series. J. Food Sci. Nutr. Res. 2, 236–244.
Villanueva, J. E., Livelo, C., Trujillo, A. S., Chandran, S., Woodworth, B., Andrade, L., et al. (2019). Time-restricted feeding restores muscle function in Drosophila models of obesity and circadian-rhythm disruption. Nat. Commun. 10:2700.
Vinckier, N. K., Patel, N. A., Geusz, R. J., Wang, A., Wang, J., Matta, I., et al. (2020). LSD1-mediated enhancer silencing attenuates retinoic acid signalling during pancreatic endocrine cell development. Nat. Commun. 11:2082.
Viscarra, J. A., Wang, Y., Nguyen, H. P., Choi, Y. G., and Sul, H. S. (2020). Histone demethylase JMJD1C is phosphorylated by mTOR to activate de novo lipogenesis. Nat. Commun. 11:796.
Voight, B. F., Scott, L. J., Steinthorsdottir, V., Morris, A. P., Dina, C., Welch, R. P., et al. (2010). Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet. 42, 579–589.
von Meyenn, F., Porstmann, T., Gasser, E., Selevsek, N., Schmidt, A., Aebersold, R., et al. (2013). Glucagon-induced acetylation of Foxa2 regulates hepatic lipid metabolism. Cell Metab. 17, 436–447. doi: 10.1016/j.cmet.2013.01.014
Walker, A. K., Yang, F., Jiang, K., Ji, J. Y., Watts, J. L., Purushotham, A., et al. (2010). Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 24, 1403–1417. doi: 10.1101/gad.1901210
Wang, B., Moya, N., Niessen, S., Hoover, H., Mihaylova, M. M., Shaw, R. J., et al. (2011). A hormone-dependent module regulating energy balance. Cell 145, 596–606. doi: 10.1016/j.cell.2011.04.013
Wang, F., Nguyen, M., Qin, F. X., and Tong, Q. (2007). SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6, 505–514. doi: 10.1111/j.1474-9726.2007.00304.x
Wang, F., and Tong, Q. (2009). SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1’s repressive interaction with PPARgamma. Mol. Biol. Cell 20, 801–808. doi: 10.1091/mbc.e08-06-0647
Wang, J., Zhang, Y., Zhuo, Q., Tseng, Y., Wang, J., Ma, Y., et al. (2020). TET1 promotes fatty acid oxidation and inhibits NAFLD progression by hydroxymethylation of PPARalpha promoter. Nutr. Metab. 17:46.
Wang, L., Jin, Q., Lee, J. E., Su, I. H., and Ge, K. (2010). Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 7317–7322. doi: 10.1073/pnas.1000031107
Wang, L., Xu, S., Lee, J. E., Baldridge, A., Grullon, S., Peng, W., et al. (2013). Histone H3K9 methyltransferase G9a represses PPARgamma expression and adipogenesis. EMBO J. 32, 45–59. doi: 10.1038/emboj.2012.306
Wang, R. H., Li, C., and Deng, C. X. (2010). Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int. J. Biol. Sci. 6, 682–690. doi: 10.7150/ijbs.6.682
Wang, R. H., Xu, X., Kim, H. S., Xiao, Z., and Deng, C. X. (2013). SIRT1 deacetylates FOXA2 and is critical for Pdx1 transcription and beta-cell formation. Int. J. Biol. Sci. 9, 934–946. doi: 10.7150/ijbs.7529
Wang, S., Huang, M., You, X., Zhao, J., Chen, L., Wang, L., et al. (2018). Gut microbiota mediates the anti-obesity effect of calorie restriction in mice. Sci. Rep. 8:13037.
Wang, T., Tsui, B., Kreisberg, J. F., Robertson, N. A., Gross, A. M., Yu, M. K., et al. (2017). Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 18:57.
Weems, J., and Olson, A. L. (2011). Class II histone deacetylases limit GLUT4 gene expression during adipocyte differentiation. J. Biol. Chem. 286, 460–468. doi: 10.1074/jbc.m110.157107
Wei, M., Fabrizio, P., Hu, J., Ge, H., Cheng, C., Li, L., et al. (2008). Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 4:e13. doi: 10.1371/journal.pgen.0040013
Wei, X., Jia, R., Wang, G., Hong, S., Song, L., Sun, B., et al. (2020). Depot-specific regulation of NAD(+)/SIRTs metabolism identified in adipose tissue of mice in response to high-fat diet feeding or calorie restriction. J. Nutr. Biochem. 80:108377. doi: 10.1016/j.jnutbio.2020.108377
Weisbeck, A., and Jansen, R. J. (2017). Nutrients and the pancreas: an epigenetic perspective. Nutrients 9:283. doi: 10.3390/nu9030283
Westerbacka, J., Kolak, M., Kiviluoto, T., Arkkila, P., Siren, J., Hamsten, A., et al. (2007). Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 56, 2759–2765. doi: 10.2337/db07-0156
Wheatley, K. E., Nogueira, L. M., Perkins, S. N., and Hursting, S. D. (2011). Differential effects of calorie restriction and exercise on the adipose transcriptome in diet-induced obese mice. J. Obes. 2011:265417.
Wilkinson, M. J., Manoogian, E. N. C., Zadourian, A., Lo, H., Fakhouri, S., Shoghi, A., et al. (2020). Ten-hour time-restricted eating reduces weight, blood pressure, and atherogenic lipids in patients with metabolic syndrome. Cell Metab. 31, 92–104.e5.
Willemsen, M. H., Vulto-Van Silfhout, A. T., Nillesen, W. M., Wissink-Lindhout, W. M., Van Bokhoven, H., Philip, N., et al. (2012). Update on Kleefstra Syndrome. Mol. Syndromol. 2, 202–212.
Williams, S. R., Aldred, M. A., Der Kaloustian, V. M., Halal, F., Gowans, G., Mcleod, D. R., et al. (2010). Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am. J. Hum. Genet. 87, 219–228. doi: 10.1016/j.ajhg.2010.07.011
Wojcik, C., and Warden, B. A. (2019). Mechanisms and evidence for heart failure benefits from SGLT2 inhibitors. Curr. Cardiol. Rep. 21:130.
Wolfrum, C., Asilmaz, E., Luca, E., Friedman, J. M., and Stoffel, M. (2004). Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature 432, 1027–1032. doi: 10.1038/nature03047
Wortham, M., Liu, F., Fleischman, J. Y., Wallace, M., Mulas, F., Vinckier, N. K., et al. (2019). Nutrient regulation of the islet epigenome controls adaptive insulin secretion. bioRxiv [Preprint]. doi: 10.1101/742403
Wozniak, S. E., Gee, L. L., Wachtel, M. S., and Frezza, E. E. (2009). Adipose tissue: the new endocrine organ? A review article. Dig. Dis. Sci. 54, 1847–1856. doi: 10.1007/s10620-008-0585-3
Wu, S. Y., Liang, J., Yang, B. C., and Leung, P. S. (2019). SIRT1 activation promotes beta-cell regeneration by activating endocrine progenitor cells via AMPK signaling-mediated fatty acid oxidation. Stem Cells 37, 1416–1428. doi: 10.1002/stem.3073
Wu, T., Liu, Y. H., Fu, Y. C., Liu, X. M., and Zhou, X. H. (2014). Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 44, 410–418.
Wu, X., Wang, Y., Wang, Y., Wang, X., Li, J., Chang, K., et al. (2018). GSK126 alleviates the obesity phenotype by promoting the differentiation of thermogenic beige adipocytes in diet-induced obese mice. Biochem. Biophys. Res. Commun. 501, 9–15. doi: 10.1016/j.bbrc.2018.04.073
Wu, Z., Liu, S. Q., and Huang, D. (2013). Dietary restriction depends on nutrient composition to extend chronological lifespan in budding yeast Saccharomyces cerevisiae. PLoS One 8:e64448. doi: 10.1371/journal.pone.0064448
Xiao, C., Giacca, A., and Lewis, G. F. (2011). Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes 60, 918–924. doi: 10.2337/db10-1433
Xiao, C., Kim, H. S., Lahusen, T., Wang, R. H., Xu, X., Gavrilova, O., et al. (2010). SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J. Biol. Chem. 285, 36776–36784. doi: 10.1074/jbc.m110.168039
Xie, Z., Zhang, D., Chung, D., Tang, Z., Huang, H., Dai, L., et al. (2016). Metabolic regulation of gene expression by histone lysine beta-hydroxybutyrylation. Mol. Cell 62, 194–206. doi: 10.1016/j.molcel.2016.03.036
Xu, F., Gao, Z., Zhang, J., Rivera, C. A., Yin, J., Weng, J., et al. (2010). Lack of SIRT1 (Mammalian Sirtuin 1) activity leads to liver steatosis in the SIRT1+/- mice: a role of lipid mobilization and inflammation. Endocrinology 151, 2504–2514. doi: 10.1210/en.2009-1013
Xu, F., Zheng, X., Lin, B., Liang, H., Cai, M., Cao, H., et al. (2016). Diet-induced obesity and insulin resistance are associated with brown fat degeneration in SIRT1-deficient mice. Obesity 24, 634–642. doi: 10.1002/oby.21393
Xu, G., Kaneto, H., Laybutt, D. R., Duvivier-Kali, V. F., Trivedi, N., Suzuma, K., et al. (2007). Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: possible contribution to impaired incretin effects in diabetes. Diabetes 56, 1551–1558. doi: 10.2337/db06-1033
Xu, Y., Sun, X., Zhang, R., Cao, T., Cai, S. Y., Boyer, J. L., et al. (2020). A positive feedback loop of TET3 and TGF-beta1 promotes liver fibrosis. Cell Rep. 30, 1310–1318.e5.
Xue, W., Huang, J., Chen, H., Zhang, Y., Zhu, X., Li, J., et al. (2018). Histone methyltransferase G9a modulates hepatic insulin signaling via regulating HMGA1. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 338–346. doi: 10.1016/j.bbadis.2017.10.037
Yang, B. T., Dayeh, T. A., Kirkpatrick, C. L., Taneera, J., Kumar, R., Groop, L., et al. (2011). Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA(1c) levels in human pancreatic islets. Diabetologia 54, 360–367. doi: 10.1007/s00125-010-1967-6
Yang, B. T., Dayeh, T. A., Volkov, P. A., Kirkpatrick, C. L., Malmgren, S., Jing, X., et al. (2012). Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol. Endocrinol. 26, 1203–1212. doi: 10.1210/me.2012-1004
Yi, S. A., Lee, J., Park, J. W., Han, J., Lee, M. G., Nam, K. H., et al. (2018). S6K1 controls epigenetic plasticity for the expression of pancreatic alpha/beta cell marker genes. J. Cell. Biochem. 119, 6674–6683. doi: 10.1002/jcb.26853
Yi, S. A., Um, S. H., Lee, J., Yoo, J. H., Bang, S. Y., Park, E. K., et al. (2016). S6K1 phosphorylation of H2B mediates EZH2 trimethylation of H3: a determinant of early adipogenesis. Mol. Cell 62, 443–452. doi: 10.1016/j.molcel.2016.03.011
Yin, H., Hu, M., Liang, X., Ajmo, J. M., Li, X., Bataller, R., et al. (2014). Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 146, 801–811. doi: 10.1053/j.gastro.2013.11.008
Yong-Quan Ng, G., Yang-Wei Fann, D., Jo, D. G., Sobey, C. G., and Arumugam, T. V. (2019). Dietary restriction and epigenetics: part I. Cond. Med. 2, 284–299.
Yoshizaki, T., Milne, J. C., Imamura, T., Schenk, S., Sonoda, N., Babendure, J. L., et al. (2009). SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol. Cell. Biol. 29, 1363–1374. doi: 10.1128/mcb.00705-08
Yoshizaki, T., Schenk, S., Imamura, T., Babendure, J. L., Sonoda, N., Bae, E. J., et al. (2010). SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 298, E419–E428.
You, D., Nilsson, E., Tenen, D. E., Lyubetskaya, A., Lo, J. C., Jiang, R., et al. (2017). Dnmt3a is an epigenetic mediator of adipose insulin resistance. eLife 6:e30766.
Younossi, Z. M., Koenig, A. B., Abdelatif, D., Fazel, Y., Henry, L., and Wymer, M. (2016). Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. doi: 10.1002/hep.28431
Yu, H., Jia, W., and Guo, Z. (2014). Reducing liver fat by low carbohydrate caloric restriction targets hepatic glucose production in non-diabetic obese adults with non-alcoholic fatty liver disease. J. Clin. Med. 3, 1050–1063. doi: 10.3390/jcm3031050
Zeng, X., Jedrychowski, M. P., Chen, Y., Serag, S., Lavery, G. G., Gygi, S. P., et al. (2016). Lysine-specific demethylase 1 promotes brown adipose tissue thermogenesis via repressing glucocorticoid activation. Genes Dev. 30, 1822–1836. doi: 10.1101/gad.285312.116
Zeybel, M., Hardy, T., Robinson, S. M., Fox, C., Anstee, Q. M., Ness, T., et al. (2015). Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin. Epigenetics 7:25.
Zha, L., Li, F., Wu, R., Artinian, L., Rehder, V., Yu, L., et al. (2015). The histone demethylase UTX promotes brown adipocyte thermogenic program via coordinated regulation of H3K27 demethylation and acetylation. J. Biol. Chem. 290, 25151–25163. doi: 10.1074/jbc.m115.662650
Zhang, C., Zhang, M., Wang, S., Han, R., Cao, Y., Hua, W., et al. (2010). Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 4, 232–241. doi: 10.1038/ismej.2009.112
Zhang, H., Wheeler, W., Hyland, P. L., Yang, Y., Shi, J., Chatterjee, N., et al. (2016). A powerful procedure for pathway-based meta-analysis using summary statistics identifies 43 pathways associated with Type II diabetes in European populations. PLoS Genet. 12:e1006122. doi: 10.1371/journal.pgen.1006122
Zhang, L., Wan, J., Jiang, R., Wang, W., Deng, H., Shen, Y., et al. (2009). Protective effects of trichostatin A on liver injury in septic mice. Hepatol. Res. 39, 931–938. doi: 10.1111/j.1872-034x.2009.00521.x
Zhang, T., Kim, D. H., Xiao, X., Lee, S., Gong, Z., Muzumdar, R., et al. (2016). FoxO1 plays an important role in regulating beta-cell compensation for insulin resistance in male mice. Endocrinology 157, 1055–1070. doi: 10.1210/en.2015-1852
Zhang, X., Lam, K. S., Ye, H., Chung, S. K., Zhou, M., Wang, Y., et al. (2010). Adipose tissue-specific inhibition of hypoxia-inducible factor 1{alpha} induces obesity and glucose intolerance by impeding energy expenditure in mice. J. Biol. Chem. 285, 32869–32877. doi: 10.1074/jbc.m110.135509
Zhang, Y., Xue, W., Zhang, W., Yuan, Y., Zhu, X., Wang, Q., et al. (2020). Histone methyltransferase G9a protects against acute liver injury through GSTP1. Cell Death Differ. 27, 1243–1258. doi: 10.1038/s41418-019-0412-8
Zhang, Z. C., Liu, Y., Li, S. F., Guo, L., Zhao, Y., Qian, S. W., et al. (2014). Suv39h1 mediates AP-2alpha-dependent inhibition of C/EBPalpha expression during adipogenesis. Mol. Cell. Biol. 34, 2330–2338. doi: 10.1128/mcb.00070-14
Zhao, L., Gregoire, F., and Sul, H. S. (2000). Transient induction of ENC-1, a Kelch-related actin-binding protein, is required for adipocyte differentiation. J. Biol. Chem. 275, 16845–16850. doi: 10.1074/jbc.275.22.16845
Zhou, D., Chen, Y. W., Zhao, Z. H., Yang, R. X., Xin, F. Z., Liu, X. L., et al. (2018). Sodium butyrate reduces high-fat diet-induced non-alcoholic steatohepatitis through upregulation of hepatic GLP-1R expression. Exp. Mol. Med. 50, 1–12. doi: 10.1038/s12276-018-0183-1
Zhou, D., Pan, Q., Xin, F. Z., Zhang, R. N., He, C. X., Chen, G. Y., et al. (2017). Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J. Gastroenterol. 23, 60–75. doi: 10.3748/wjg.v23.i1.60
Zhou, S. S., Zhou, Y. M., Li, D., and Lun, Y. Z. (2011). Dietary methyl-consuming compounds and metabolic syndrome. Hypertens. Res. 34, 1239–1245. doi: 10.1038/hr.2011.133
Zhu, M., Miura, J., Lu, L. X., Bernier, M., Decabo, R., Lane, M. A., et al. (2004). Circulating adiponectin levels increase in rats on caloric restriction: the potential for insulin sensitization. Exp. Gerontol. 39, 1049–1059. doi: 10.1016/j.exger.2004.03.024
Keywords: non-alcoholic fatty liver disease, type 2 diabetes, obesity, dietary interventions, intermittent fasting, caloric restriction, DNA methylation, histone modification
Citation: Asif S, Morrow NM, Mulvihill EE and Kim K-H (2020) Understanding Dietary Intervention-Mediated Epigenetic Modifications in Metabolic Diseases. Front. Genet. 11:590369. doi: 10.3389/fgene.2020.590369
Received: 01 August 2020; Accepted: 21 September 2020;
Published: 15 October 2020.
Edited by:
Mayra Furlan-Magaril, National Autonomous University of Mexico, MexicoReviewed by:
Narendra Singh, Stowers Institute for Medical Research, United StatesCopyright © 2020 Asif, Morrow, Mulvihill and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erin E. Mulvihill, ZW11bHZpaGlAdW90dGF3YS5jYQ==; Kyoung-Han Kim, aGFua2ltQHVvdHRhd2EuY2E=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.