 Rongxin Zhang
Rongxin Zhang Ke Xiao
Ke Xiao Yu Gu
Yu Gu Hongde Liu
Hongde Liu Xiao Sun
Xiao Sun- State Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing, China
The coronavirus disease 2019 (COVID-19) pandemic caused by SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) has become a global public health emergency. G-quadruplex, one of the non-canonical secondary structures, has shown potential antiviral values. However, little is known about the G-quadruplexes of the emerging SARS-CoV-2. Herein, we characterized the potential G-quadruplexes in both positive and negative-sense viral strands. The identified potential G-quadruplexes exhibited similar features to the G-quadruplexes detected in the human transcriptome. Within some bat- and pangolin-related betacoronaviruses, the G-tracts rather than the loops were under heightened selective constraints. We also found that the amino acid sequence similar to SUD (SARS-unique domain) was retained in SARS-CoV-2 but depleted in some other coronaviruses that can infect humans. Further analysis revealed that the amino acid residues related to the binding affinity of G-quadruplexes were conserved among 16,466 SARS-CoV-2 samples. Moreover, the dimer of the SUD-homology structure in SARS-CoV-2 displayed similar electrostatic potential patterns to the SUD dimer from SARS. Considering the potential value of G-quadruplexes to serve as targets in antiviral strategy, our fundamental research could provide new insights for the SARS-CoV-2 drug discovery.
Introduction
The coronavirus disease 2019 (COVID-19) pandemic, which first broke out in China, has rapidly become a global public health emergency within a few months (Lai et al., 2020). According to the statistics from the Johns Hopkins Coronavirus Resource Center (https://coronavirus.jhu.edu/map.html), as of July 22, 2020, 15 million cases have been confirmed, with a death toll rising to 600,000. Since 2000, humans have suffered at least three coronavirus outbreaks, and they were severe acute respiratory syndrome (SARS) in 2003 (Zhong et al., 2003; Peiris et al., 2004; Zumla et al., 2016; Cui et al., 2019), Middle East respiratory syndrome (MERS) in 2012 (Zumla et al., 2016; Cui et al., 2019), and COVID-19. Scientists identified and sequenced the virus early in this outbreak, and named it SARS-CoV-2 (Gorbalenya et al., 2020). The symptoms of the patients infected with the novel coronavirus vary from person to person, and fever, cough, and fatigue are the most common ones (Guan et al., 2020; Jin et al., 2020; Rothan and Byrareddy, 2020; Zu et al., 2020). The clinical chest CT (computed tomography) and nucleic acid testing are the most typical methods of diagnosing COVID-19 (Jin et al., 2020; Zu et al., 2020). It is worth noting that the recent achievements in AI (artificial intelligence) aid diagnosis technology (Li et al., 2020) and CRISPR-Cas12-based detection methods (Broughton et al., 2020) are expected to expand the diagnosis of COVID-19. Despite the great efforts of the researchers, no specific clinical drugs or vaccines had developed to cope with COVID-19 by the end of September 2020.
SARS-CoV-2 is a betacoronavirus within the Coronaviridae family that is the culprit responsible for the COVID-19 pandemic (Figure 1A) (Guo et al., 2020; Zheng, 2020). Studies have confirmed that SARS-CoV-2 is a positive-sense single-stranded RNA [(+)ssRNA] virus with a total length of approximately 30k. The positive-sense RNA strand of SARS-CoV-2 can serve as a template to produce viral proteins related to replication, structure composition, and other functions or events (Chen et al., 2020; Kim et al., 2020). One of the hotspots is how SARS-CoV-2 entry the host cells. SARS-CoV-2 has shown a great affinity to the angiotensin-converting enzyme 2 (ACE2), which has been proved to be the binding receptor for SARS-CoV-2 (Hoffmann et al., 2020; Walls et al., 2020). After entering the host cells, the viral genomic RNA will be released to the cytoplasm, and the ORF1a/ORF1ab is subsequently translated into replicase polyproteins of pp1a/pp1ab, which will be cleaved into some non-structural proteins (nsps). These non-structure proteins ultimately form the replicase–transcriptase complex for replication and transcription. Along with the full-length positive- and negative-sense RNAs, a nested set of subgenomic RNAs (sgRNAs) are also synthesized, and mainly translated into some structural proteins and accessory proteins. When the assembly is finished, the mature SARS-CoV-2 particles are released from the infected host cells via exocytosis (Shereen et al., 2020). Mounting evidence suggests that bats and pangolins are the suspected natural host and intermediate host of SARS-CoV-2 (Andersen et al., 2020; Lam et al., 2020; Zhang T. et al., 2020; Zhou et al., 2020). Intriguingly, a report from Yongyi Shen et al. showed that SARS-CoV-2 might be the recombination product of Bat-CoV-RaTG13-like virus and Pangolin-CoV-like virus (Xiao et al., 2020).
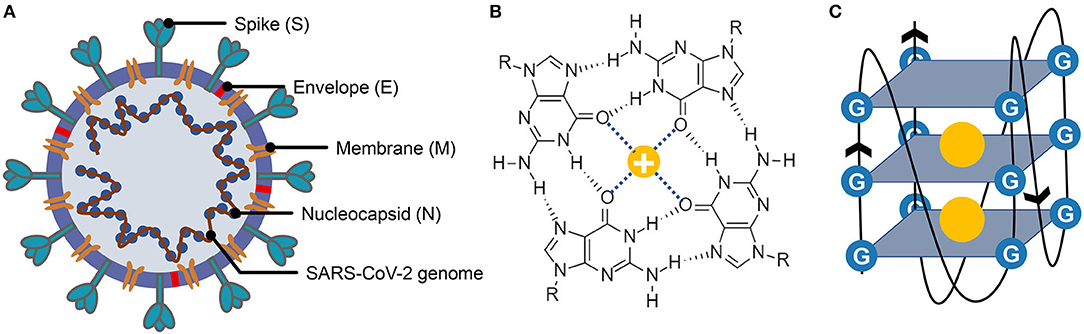
Figure 1. Structure of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), G-quartet, and G-quadruplex. (A) The SARS-CoV-2 particle structure is composed of four structural proteins, which are the spike protein, the envelope protein, the membrane protein, and the nucleocapsid protein. The nucleocapsid proteins are bound to the SARS-CoV-2 genome. (B) Structure of G-quartet; the neighboring guanines are connected via Hoogsteen hydrogen. The cation is indicated by a yellow circle. (C) The G-quadruplex is formed by stacking multiple G-quartets, and the stabilization of the structure is partially determined by the central cations.
G-quadruplexes are the non-canonical nucleic acid structures usually formed in G-rich regions both in DNA and RNA strands (Bochman et al., 2012; Kwok and Merrick, 2017; Varshney et al., 2020). The G-quadruplex is formed by stacking G-quartets (Figure 1B) on top of each other, in which the four guanines making up a G-quartet are connected via Hoogsteen pairs (Figure 1C) (Bochman et al., 2012; Kwok and Merrick, 2017; Spiegel et al., 2020; Varshney et al., 2020). Extensive research indicated that G-quadruplexes were involved in many critical biological processes, including DNA replication (Hoshina et al., 2013; Valton et al., 2014; Valton and Prioleau, 2016; Prorok et al., 2019), telomere regulation (Tang et al., 2007; Wang et al., 2011; Takahama et al., 2013; Moye et al., 2015; Jansson et al., 2019), and RNA translation (Kumari et al., 2007; Gomez et al., 2010; Murat et al., 2018; Jodoin et al., 2019). It has been proven that G-quadruplexes existed in the viral genome and can regulate the viral biological processes, which made it possible to function as potential drug targets for antiviral strategy (Métifiot et al., 2014; Ruggiero and Richter, 2018; Saranathan and Vivekanandan, 2019). A study made by Jinzhi Tan et al. demonstrated that the SARS-unique domain within the nsp3 (nonstructural protein 3) of SARS coronavirus (SARS-CoV) exhibits the binding preference to the G-quadruplex structure in the human transcript and potentially interfere with host cell antiviral response (Tan et al., 2009). They also identified several amino acid residues that were tightly associated with its binding capacity. Yet, whether the SARS-CoV-2 contains a G-quadruplex binding domain and whether the amino acid residues related to the G-quadruplex binding affinity are conserved among SARS-CoV-2 samples need further interpretation. Besides, the G-quadruplexes in some well-known virus, such as HIV-1 (human immunodeficiency viruses type 1) (Perrone et al., 2014; Piekna-Przybylska et al., 2014; Butovskaya et al., 2018, 2019), ZIKV (ZIKA virus) (Fleming et al., 2016), HPV (human papillomavirus) (Tlučková et al., 2013; Marušič et al., 2017) and EBOV (Ebola virus) (Wang et al., 2016a) have been studied. However, in our understanding of the G-quadruplexes, their potential roles in the emerging SARS-CoV-2 are lacking.
In this study, we depicted the potential G-quadruplexes (PG4s) in SARS-CoV-2 by merging several G-quadruplex prediction tools. The PG4s in SARS-CoV-2 presented similar features to the two-quartet G-quadruplexes in the human transcriptome, which potentially supported the formation and existence of the G-quadruplexes in SARS-CoV-2. Additionally, we investigated the difference in selective constraints between the G-tracts and other nucleotides in the SARS-CoV-2 genome. To further elucidate the possible pathogenic mechanism of SARS-CoV-2, we examined the sequence and structure of the SUD-homology in SARS-CoV-2 that are critical in binding the G-quadruplex structures in host transcripts.
Materials and Methods
Data Collection
We obtained a total of 77 full-length bat-associated betacoronaviruses from the DBatVir (http://www.mgc.ac.cn/DBatVir/) database (Supplementary Table 1) (Chen et al., 2014). We also downloaded the bat coronavirus RaTG13 (MN996532.1) genome from the NCBI virus database (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/), which has shown a high sequence similarity to the SARS-CoV-2 reference genome in previous reports. We acquired the SARS-CoV-2 reference genome from the NCBI virus database under the accession number of NC_045512. In addition to those sequences, nine pangolin coronaviruses were derived from GISAID (https://www.gisaid.org/) database (Shu and McCauley, 2017). To calculate the mutation frequency of each nucleotide in SARS-CoV-2, we downloaded the SARS-CoV-2 alignment sequence file from the GISAID database spanning from December 24, 2019, to April 24, 2020, which contains 16,466 SARS-CoV-2 samples. The ORF1ab amino acid sequences of 24 viruses were retrieved from the NCBI Protein database (https://www.ncbi.nlm.nih.gov/protein/). The detailed accession numbers of the sequence data used in this study are described in Supplementary Tables 1, 2.
Pairwise and Multiple Sequence Alignment, Phylogenetic, and Conservation Analysis
The EMBOSS Needle software, which is based on the Needleman–Wunsch algorithm and is a part of the EMBL-EBI web tools (Madeira et al., 2019), was employed for the pairwise sequence alignment. Clustal Omega (Sievers et al., 2011; Sievers and Higgins, 2018) is a reliable and accurate multiple sequence alignment (MSA) tool that can be performed on large data sets. We utilized this MSA tool to align the viral genomes and the protein sequences under the default paraments, respectively. UGENE (Okonechnikov et al., 2012) is a powerful and user-friendly bioinformatics software, and we choose UGENE to visualize the pairwise and multiple sequence alignment results. We used the MEGA X software (Kumar et al., 2018) to construct the neighbor-joining phylogenetic tree with 1,000 bootstrap replications. To depict the conservation state for each nucleotide site, the GERP++ software (Davydov et al., 2010) was applied to calculate the “Rejected Substitutions” score column by column, which can reflect the strength of constraints for each nucleotide site. The key software or databases are listed in the Supplementary Table 3.
Potential G-Quadruplex Detection
Several open-source G-quadruplex detection software were used to search the PG4s both in the SARS-CoV-2 positive- and negative-sense strands. G4CatchAll (Doluca, 2019), pqsfinder (Hon et al., 2017), and QGRS Mapper (Kikin et al., 2006) were employed to predict the putative G-quadruplexes, respectively; please see ref (Puig Lombardi and Londoño-Vallejo, 2019) for more information about the comparison of those tools mentioned above. The minimum G-tract length was set to two in the three pieces of software, while the max length of the predicted G-quadruplexes was limited to 30. Specifically, the minimum score of the predicted G-quadruplex was set to 10 when using pqsfinder. We utilized BEDTools (Quinlan and Hall, 2010) to sort the PG4s according to their coordinates. Apart from this, we adopted the cG/cC scoring system (Beaudoin et al., 2014) proposed by Jean-Pierre Perreault et al. to delineate the influence of sequence context on PG4s. The PG4s along with 15-nt (nucleotide) upstream and downstream sequence contexts were used to calculate the cG/cC score, and 2.05 was taken as the threshold for the preliminary inference of the G-quadruplex folding capability (Supplementary Table 4) (Beaudoin et al., 2014). Using a customized python script, we implemented the cG/cC scoring system, and the source code of the python script could be found at GitHub.
Homo-Dimer Homology Modeling and Electrostatic Potential Calculation
The homo-dimer of the SUD-homology in SARS-CoV-2 was modeled based on the template of the SARS-CoV SUD structure (PDB ID: 2W2G) through homology modeling. All the modeling process was performed in the Swiss Model (Waterhouse et al., 2018) website (https://swissmodel.expasy.org/) according to the default options. The electrostatic potential was calculated and visualized in the PyMOL software by using the APBS (adaptive Poisson–Boltzmann solver) plugin under the default parameters.
ΔG° Z-score Analysis
The ΔG° z-score for the SARS-CoV-2 genome was retrieved from RNAStructuromeDB (https://structurome.bb.iastate.edu/sars-cov-2) (Andrews et al., 2017). The ΔG° z-score is described as follows (Andrews et al., 2020).
where the MFEnative means the MFE (minimum free energy) ΔG° value predicted by the RNAfold software with a window of 120 nt and step of 1 nt. In addition, the represents the MFE ΔG° value generated by the randomly shuffled sequence with the identical nucleotide composition. The σ is the standard deviation across all the MFE values.
To depict the ΔG° z-score for each nucleotide in the SARS-CoV-2 genome, we utilized the following formula.
where zi is the average ΔG° z-score for nucleotide i, and w denotes the total number of the sliding windows that cover the nucleotide i. indicates the ΔG° z-score for the m-th window. For example, when considering the nucleotide 1,000 under the setting of 120 nt window length and 1-nt step, there are 120 sliding windows covering the nucleotide 1,000. So, the z200, which means the average ΔG° z-score for nucleotide 200, is calculated as the sum of the ΔG° z-score of 120 sliding windows divided by the total number of the sliding windows.
Results
Whole Genome Identification and Annotation of Potential G-Quadruplexes
To get the potential G-quadruplexes in SARS-CoV-2, we took the strategy described as follows (Figure 2A): (i) Predicting the PG4s with three software independently. (ii) Merging the prediction results of the PG4s and evaluating the G-quadruplex folding capabilities by the cG/cC scores. (iii) The PG4s with cG/cC scores higher than the threshold were selected as candidates for further analysis. Here, the threshold for determining whether PG4s can be folded was set to 2.05, as described in the study of Beaudoin et al. (2014). In total, we obtained 24 PG4s (Table 1) in the positive- and negative-sense strands for further analysis.
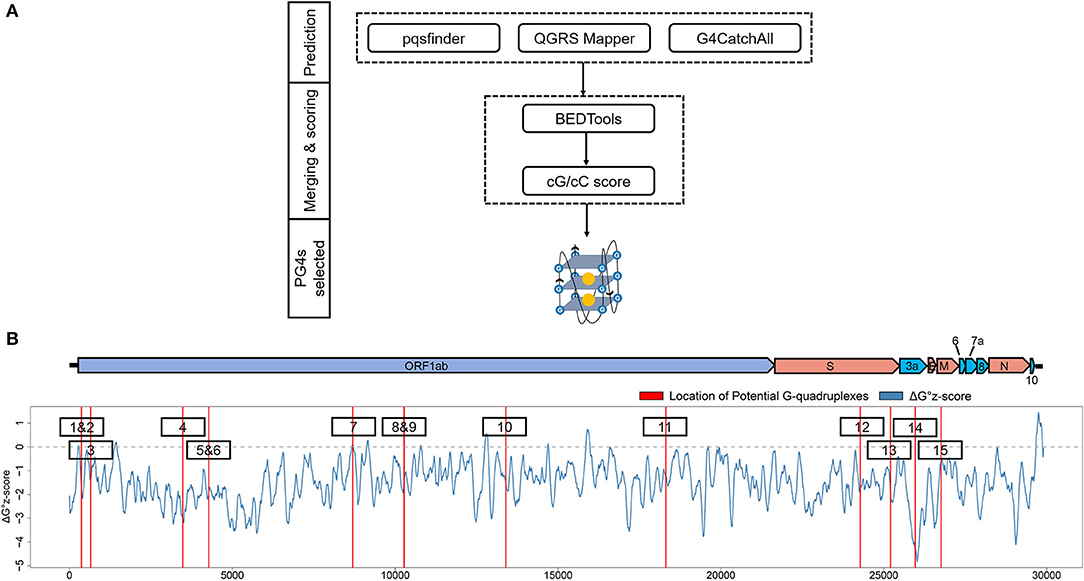
Figure 2. Detection and annotation of the potential G-quadruplexes (PG4s). (A) The schematic flow of PG4s detection. The G-quadruplex prediction tools, pqsfinder, QGRS Mapper, and G4CatchAll were utilized for the prediction of PG4s. BEDTools and the cG/cC scoring system were applied to merge and score the PG4s. After screening, the PG4s that were used in this study were generated. (B) Visualization of the PG4s in the SARS-CoV-2 genome. Top: the genome organization of SARS-CoV-2. Bottom: the average ΔG° z-score for each nucleotide (blue curve) in the SARS-CoV-2 genome; the location of PG4s are plotted with red vertical lines. The order of PG4s is marked with a black box. Please note that only the PG4s in the positive-sense strand are visualized.

Table 1. The potential G-quadruplexes (PG4s) found in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
To annotate the PG4s, the reference annotation data (in gff3 format) of SARS-CoV-2 were downloaded from the NCBI database with the accession number of NC_045512. First, we focused on the PG4s in the positive-sense strand. Fifteen of the 24 PG4s (67.5%) were located in the positive-sense strand (Table 1); most of them were harbored in non-structural proteins including nsp1, nsp3, nsp4, nsp5, nsp10, and nsp14, with the remaining ones located in the spike protein, orf3a, and the membrane protein. Second, we examined the PG4s in the negative-sense strand, which is an intermediate product of replication. Nine PG4s were scattered in the negative-sense strand (Table 1).
To further characterize the potential canonical secondary structures competitive with G-quadruplexes, the landscape of thermodynamic stability of the SARS-CoV-2 genome was depicted by using ΔG° z-score (Andrews et al., 2020). In general, a positive ΔG° z-score implies that the secondary structure of this region tends to be less stable than the randomly shuffled sequence with the identical nucleotide composition, while a negative ΔG° z-score signifies higher stability than the randomly shuffled sequence. For each nucleotide in the SARS-CoV-2 genome, the ΔG° z-score was calculated for all the 120 nt windows covering the nucleotide, and an average ΔG° z-score was deduced then. Several PG4s are located in positions with a locally higher average ΔG° z-scores (Figure 2B) which implied the relative instability of a canonical secondary structure and the lower possibility to adopt such a competitive structure against the G-quadruplex structure, which may ultimately favor the formation of G-quadruplex structure.
Potential G-Quadruplexes in SARS-CoV-2 Show Analogical Features With the rG4s in the Human Transcriptome
In 2016, Chun Kit Kwok and coworkers profiled the RNA G-quadruplexes in the HeLa transcriptome by using the RNA G-quadruplex sequencing (rG4-seq) technology, and quantified the diversity of these RNA G-quadruplexes (Kwok et al., 2016). We set out to address the question of whether the potential G-quadruplexes in SARS-CoV-2 showed analogical features with the G-quadruplexes found in the human transcriptome and if these PG4s have the ability to form G-quadruplex structures. We noticed that the PG4s in SARS-CoV-2 are all in the two-quartet style. Therefore, we retrieved the two-quartet RNA G-quadruplex sequence data generated in the rG4-seq experiment under the condition of K+ and pyridostatin (PDS). However, for some RTS (reverse transcriptase stalling) sites labeled as two-quartet, there may exist overlapping G-quadruplexes with different loops (e.g., GGCACAGCAGGCATCGGAGGTGAGGCGGGG), and it is difficult to determine which one was formed in the experiment. In order to eliminate the ambiguity, only the RTS sites containing non-overlapping two-quartet G-quadruplex (e.g., GTCATTTTTTGTGTTTGGTTTGGTGGTGGC) were considered.
First, we investigated the loop length distribution pattern of the two-quartet PG4s in both SARS-CoV-2 and the human transcriptome (Figure 3A). As a whole, the two-quartet PG4s in SARS-CoV-2 and the human transcriptome displayed similar loop length distribution patterns, and the loop length of the PG4s in SARS-CoV-2 falls into the scope of the ones from the human transcriptome. The distributions of loop length between the SARS-CoV-2 PG4s and the human two-quartet G-quadruplexes did not show discrepancies (Supplementary Figure 1, Wilcoxon test, p = 0.4552).
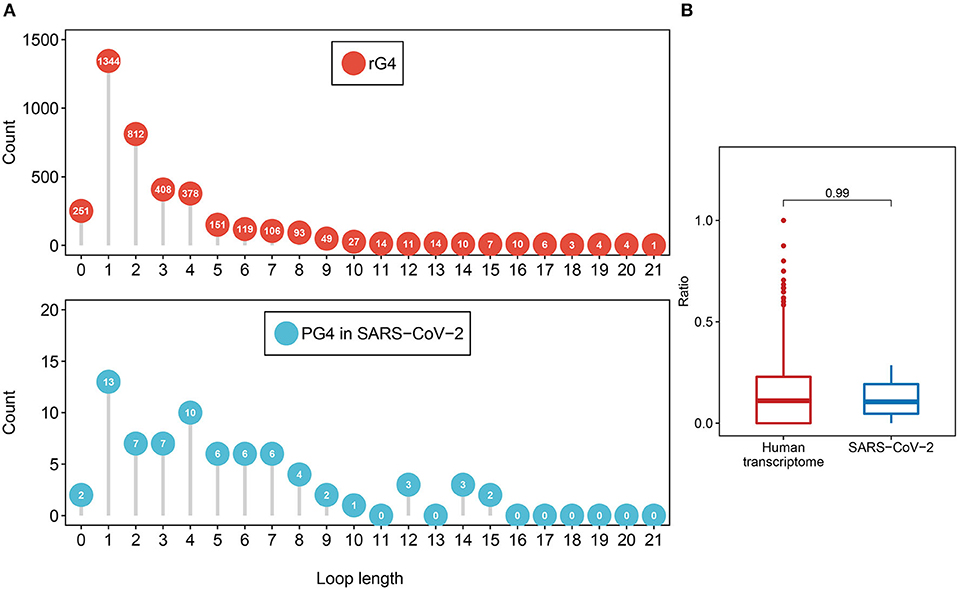
Figure 3. Feature comparison of potential G-quadruplexes found in rG4-seq and SARS-CoV-2. (A) The dot chart represents the number of two-quartet G-quadruplex loops with different lengths in the human transcriptome and SARS-CoV-2, respectively. Top: G-quadruplex in the human transcriptome. Bottom: G-quadruple in SARS-CoV-2. (B) The red boxplot shows the ratio of cytosine in the human transcriptome two-quartet G-quadruplex loops, and the blue boxplot displays the ratio of cytosine in the SARS-CoV-2 PG4 loops.
Considering the fact that the presence of multiple cytosine tracks may hinder the formation of G-quadruplex structures (Beaudoin, 2010; Beaudoin et al., 2014), we examined the cytosine ratio in G-quadruplex loops (Figure 3B). No significant difference in loop cytosine ratios was observed between the SARS-CoV-2 PG4s and the human two-quartet G-quadruplexes (Wilcoxon test, p = 0.9911), which suggested that the loop cytosine ratios between the two types of G-quadruplex were similar.
Taken together, our results suggested that the PG4s in SARS-CoV-2 displayed similar features to the rG4s in the human transcriptome.
Potential G-Quadruplexes Are Under Heightened Selective Constraints in Bat- and Pangolin-Related Betacoronaviruses
Recent research revealed that the G-quadruplexes in human UTRs (untranslated regions) are under selective pressures (Lee et al., 2020), and some coronaviruses on bats and pangolins are closely related to SARS-CoV-2 (Lam et al., 2020; Zhang T. et al., 2020). Consequently, we wondered whether the potential G-quadruplexes in the SARS-CoV-2 genome are under heightened selective constraints. We collected some betacoronavirus genomic sequences of bats and pangolins from several public databases and used the NJ (neighbor-joining) method to construct the phylogenetic tree with 1,000 bootstrap replications (Supplementary Figure 2). The RS (rejected substitutions) score for each site in the SARS-CoV-2 reference genome was evaluated by using the GERP++ software.
We checked the RS score difference between the G-tract (continuous runs of G) nucleotides and other nucleotides. A significant discrepancy was observed, which means that the G-tract nucleotides exhibit heightened selective constraints than other nucleotides in the SARS-CoV-2 genome (Figure 4A, Wilcoxon test, p = 9.254 × 10−8). Considering that the G-tracts are composed of guanines, the conservation of guanines in and outside the G-tracts in the SARS-CoV-2 genome were also compared. We discovered that the guanines in G-tracts are under heightened selective constraints (Figure 4B, Wilcoxon test, p = 3.363 × 10−3). The nucleotides within G-tracts are more relevant to the G-quadruplex structural maintenance than the loops. Then we compared the G-tract and loop RS scores. As a result, the G-tract RS scores were significantly higher than that of the loops (Figure 4C, Wilcoxon test, p = 3.962 × 10−7), which suggests that the G-tracts experienced stronger selective constraints.
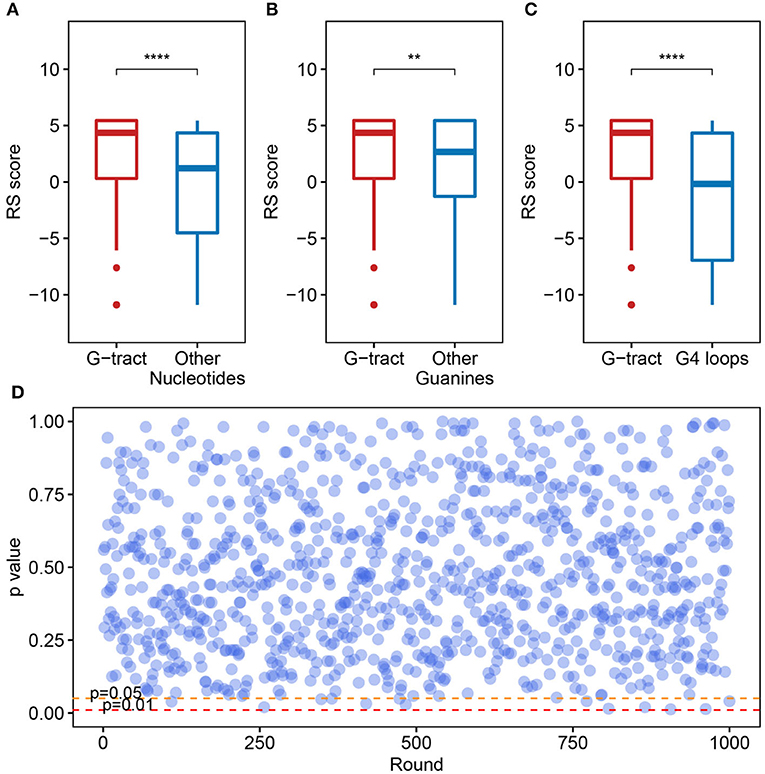
Figure 4. Potential G-quadruplexes exhibit heightened selective constraints in bat and pangolin related betacoronavirus. (A–C) Boxplot showing the difference of nucleotide RS scores in G-tract, other nucleotides, other guanines, and PG4 loops (**p ≤ 0.01, ****p ≤ 0.0001). (D) Test of the RS score difference between the fragments containing PG4s and the randomly selected fragments. The abscissa indicates the round of the test, while the ordinate represents the p-value for each round.
We also checked if the PG4s that are under heightened selective constraints is relevant to its inherent properties rather than the sequence contexts. A random test was performed to check whether the fragments containing PG4s manifested different average RS scores compared with random fragments in the SARS-CoV-2 genome. The fragments containing PG4s were designated as the sequence 100 nt upstream and downstream of the PG4 centers. We conducted 1,000 rounds of tests. In each test, we randomly selected 50 fragments from the SARS-CoV-2 genome with a length of 200 nt and carried out the Wilcoxon test to assess the average RS score difference among the randomly selected fragments and the fragments containing PG4s. The p-value for each round was retained. As a result, no evident difference was observed as few p-values (13/1,000) were < 0.05 (Figure 4D), suggesting that PG4s that are under heightened selective constraints are more likely to be related to its inherent properties rather than sequence contexts.
SARS-CoV-2 Contains Similar SUD to SARS-CoV
SARS-CoV and SARS-CoV-2 share similar nucleic acid sequence compositions, and both can cause acute disease symptoms. It has been confirmed that the SUD in the SARS-CoV could bind to the G-quadruplex structures in human transcripts (Tan et al., 2009), but whether SARS-CoV-2 possesses a similar SUD structure remains unclear. Thus, we explored whether the SARS-CoV-2 genome contains the protein-coding sequence potentially encoding SUD-homology and whether SARS-CoV-2 retains the ability to bind RNA G-quadruplex structures. We collected the ORF1ab amino acid sequences of some coronaviruses belonging to different genera, including seven known coronaviruses that can infect humans. The SUD-homology amino acid sequence was absent in some coronaviruses, especially in alpha, gamma, and delta coronaviruses (Supplementary Figure 3). In contrast, the SUD-homology sequence was retained in several betacoronavirus, particularly in bat- and pangolin-associated ones. Moreover, among the seven coronaviruses that can infect humans, only SARS-CoV and SARS-CoV-2 kept the SUD or SUD-homology sequence, while the sequence in MERS-CoV, HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1 were depleted. Next, we examined the eight key amino acid residues in SUD that are related to G-quadruplex binding affinity according to the previous reports (Figure 5A). Almost all the key amino acid residues are reserved in SARS-CoV-2, with one exception of a conservative replacement of K (Lysine) with R (Arginine). The conservation of the eight amino acid residues within SARS-CoV-2 samples was then investigated, to see if these residues were conserved and the G-quadruplex binding ability of SUD-homology was essential for SARS-CoV-2. We retrieved the nucleotide sequence alignment file of 16,466 SARS-CoV-2 samples from the GISAID database and calculated the mutation frequency for each nucleotide. As a result, limited mutation frequencies were found in the eight amino acid residues compared to the whole genome average mutation frequency (Figure 5B, frequency = 3.96). Although eight nucleotide mutations were detected of glutamate (2,432 E), seven of them were synonymous mutations. Next, we checked the electrostatic potential pattern of the SUD-homology dimer from SARS-CoV-2. The positively charged patches were observed in the core of the SUD-homology dimer, which was surrounded by negatively charged patches (Figure 5C). In contrast, when the dimer was rotated 180°, a slightly inclined narrow cleft with negative potential accompanied by the positively charged patches was discovered (Figure 5D). The above results indicated that the SUD-homology dimer of SARS-CoV-2 and the SUDcore (a shortened version of SUD) dimer of SARS presented analogical electrostatic potential patterns [see ref (Tan et al., 2009) for more details about the electrostatic potential surface of the SUDcore homodimer in SARS]. Similar to the SUD dimer of SARS, we identified the positively charged patches located in the center and back of the SARS-CoV-2 SUD-homology dimer that can potentially bind the G-quadruplex structures (Figures 5C,D).
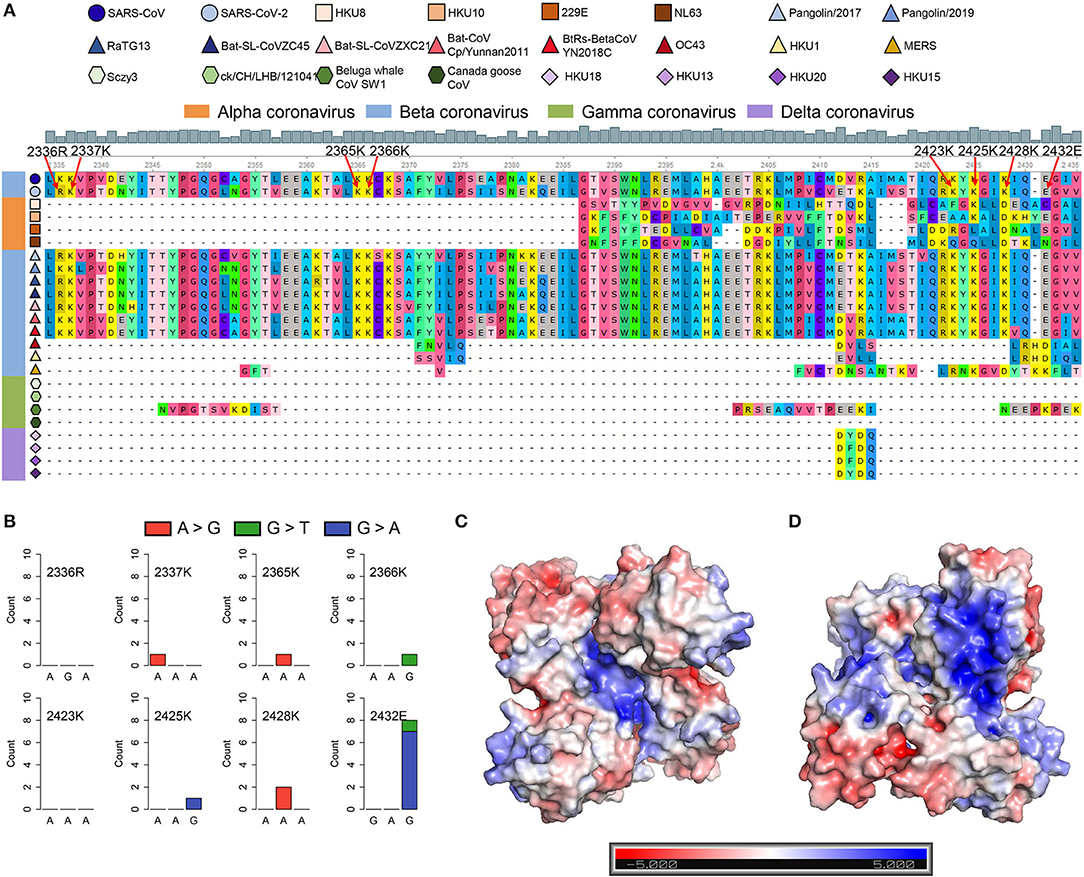
Figure 5. SARS-CoV-2 contains the SARS-unique domain (SUD)-homology sequence and dimer structure. (A) Sequence alignment of four different genera coronaviruses. The shapes in various colors mark different kinds of coronaviruses. The color bar represents different genera of coronaviruses (orange, alphacoronavirus; blue, betacoronavirus; green, gammacoronavirus; purple, deltacoronavirus). The gray histogram shows the consensus of the alignment sites. The eight amino acid residues related to the G-quadruplex binding affinity are labeled by red arrows. (B) Nucleotide mutation count of eight amino acid residues among 16,466 SARS-CoV-2 samples. (C,D) The electrostatic potential surface of the SARS-CoV-2 SUD-homology dimer with different orientations [the front (C) and rotate 180° (D)]. The blue and red showed positive and negative potentials, respectively. The positively charged patches in the center (C) and back (D) of the SUD-homology dimer might be the binding domain for G-quadruplexes.
Discussion
The COVID-19 pandemic has caused huge losses to humans and made people pay more attention to public health. A large number of scientists around the world have participated in the fight against the epidemic. The SARS-CoV-2 coronavirus is the key culprit responsible for the outbreak, and no specific inhibitor drugs have been developed yet. G-quadruplexes have shown tremendous potential for the development of anticancer (Han and Hurley, 2000; Balasubramanian et al., 2011; Miller and Rodriguez, 2011; Neidle, 2017) and antiviral drugs (Perrone et al., 2015; Ruggiero and Richter, 2018, 2020), as G-quadruplexes can interfere with many biological processes that are critical to cancer cells and viruses. Therefore, it is necessary to quantify and characterize the PG4s in the SARS-CoV-2 genome to provide a possible novel method for the treatment of COVID-19.
In this study, besides three popular G-quadruplexes prediction tools, the cG/cC scoring system, which is specially designed for the identification of RNA G-quadruplexes, was adopted to determine the PG4s. Indeed, we did not find the G-quadruplexes with three or more G-quartets, which are generally considered to be more stable than the two-quartet G-quadruplexes. One of the controversial issues lies on the stability of the two-quartet G-quadruplexes, especially the folding capability of those G-quadruplexes in vivo. However, it is well-acknowledged that the RNA G-quadruplexes is more stable than their DNA counterparts (Joachimi et al., 2009; Zaccaria and Fonseca Guerra, 2018) and SARS-CoV-2 is a single-strand RNA virus, which may be conducive to its structure formation. Several emerging studies have demonstrated the formation of two-quartet G-quadruplexes in viral sequences (Perrone et al., 2013; Murat et al., 2014; Fleming et al., 2016; Wang et al., 2016a; Zahin et al., 2018; Majee et al., 2020; Zhang Y. et al., 2020), which could serve as antiviral elements under the presence of G-quadruplex ligands. We also did a comprehensive search for viral two-quartet G-quadruplexes, whose formation has been confirmed by experimental methods in vitro, and a total of 16 two-quartet G-quadruplexes were found (Supplementary Table 5). The potential G-quadruplexes in SARS-CoV-2 showed a lower cytosine ratio in loops than that in experimental validated viral G-quadruplexes (Supplementary Figure 4A, Wilcoxon test, p = 0.0018), which is a positive signal for the G-quadruplex formation in SARS-CoV-2. When looking at the loop lengths, the experimental supported G-quadruplexes in other viruses displayed a short and concentrated distribution mode, while the potential G-quadruplexes in SARS-CoV-2 exhibited a relatively uniform distribution mode in <8 nt, which is a hint that maybe part of the sequence could form G-quadruplex structures (Supplementary Figure 4B). The cG/cC scores of the experimental supported G-quadruplexes in other viruses and their 15 nt flanking sequences were calculated as well (Supplementary Table 6). The cG/cC scores of some potential G-quadruplexes in SARS-CoV-2 were relatively high (Supplementary Figure 5); however, the overall cG/cC scores for the potential G-quadruplexes in SARS-CoV-2 were low. When only considering the G-quadruplex sequences itself, the potential G-quadruplexes in SARS-CoV-2 presented higher cG/cC scores (mean = 52.51, standard deviation = 77.29), while the experimental supported G-quadruplexes displayed relatively lower cG/cC scores (mean = 24.39, standard deviation = 37.27). These observations suggest that some of the potential G-quadruplexes in SARS-CoV-2 could be folded into secondary structures. Moreover, the K+ (potassium ion), one of the primary positive ions inside human cells, can strongly support the formation of G-quadruplexes. Nevertheless, whether the SARS-CoV-2 G-quadruplexes could form in vivo requires overwhelming proofs.
Most of the PG4s we detected were located in the positive-sense strand. The G-quadruplex forming sequences in the SARS-CoV genome were presumed to function as the chaperones of SUD, and their interaction was essential for the SARS-CoV genome replication (Kusov et al., 2015). ORF1ab that encodes the replicase proteins is required for the viral replication and transcription. Some PG4s were found to harbor in ORF1ab, and whether these PG4s were related to the replication of the viral genome and interacted with the G-quadruplex binding domain in SARS-CoV-2 is worthy of further investigation. In addition to ORF1ab, there exist several PG4s in the structural and accessory protein-coding sequences as well as the sgRNAs that contain the above protein sequences. Some studies have characterized the impact of G-quadruplex structures on the translation of human transcripts, and an apparent inhibitory effect was observed (Kumari et al., 2007; Beaudoin, 2010; Shahid et al., 2010). The translation of some SARS-CoV-2 proteins requires the involvement of human ribosomes; thus, it is possible to repress the translation of SARS-CoV-2 proteins via stabilizing the G-quadruplex structures. In fact, this inhibition effect has been reported in some other viral studies (Wang et al., 2016b; Majee et al., 2020). The negative-sense strand serves as templates for the synthesis of the positive-sense strand and the subgenomic RNAs. The identified potential G-quadruplexes were broadly distributed in the negative-sense strand of SARS-CoV-2. Notably, we observed one PG4 located at the 3′ end of the negative-sense strand. A previous study confirmed that the stable G-quadruplex structures located at the 3′ end of the hepatitis C virus negative-sense strand could inhibit the RNA synthesis by reducing the activity of the RdRp (RNA-dependent RNA polymerase) (Jaubert et al., 2018). Therefore, it is necessary to further investigate whether the PG4 at the 3′ end of the negative-sense strand of SARS-CoV-2 could inhibit RNA synthesis. In addition, recent studies have detected high-frequency trinucleotide mutations (G28881A, G2882A, and G28883C) in the SARS-CoV-2 genome (Yao et al., 2020; Yin, 2020). G28881A and G28882A always co-occur within the same codon, which means a positive selection of amino acid (Mishra et al., 2020), but the consequence of the trinucleotide mutations was still elusive. We noticed that the trinucleotide mutations were in the G-rich sequence from 28,881 to 28,917 nt (5′ GGGGAACTTCTCCTGCTAGAATGGCTGGCAATGGCGG 3′). The potential G-quadruplex downstream of the trinucleotide mutations was filtered by the cG/cC score system as the presence of cytosine tracks within and flanking of the potential G-quadruplex reduce the cG/cC score; however, in fact, this potential G-quadruplex showed a relative lower MFE (Minimum Free Energy) among all the potential G-quadruplexes we detected. Whether the mutations have an internal causality with the G-rich sequence still needs to be elucidated.
The SUD in SARS, which is thought to be related to its terrible pathogenicity, has displayed binding preference to the G-quadruplexes in human transcripts (Tan et al., 2009). Our analysis revealed that the novel coronavirus SARS-CoV-2 contained a similar domain to SUD as well. Furthermore, several amino acid residues previously reported to be an indispensable part of the G-quadruplexes binding capability are almost retained in SARS-CoV-2. Further exploration indicated that the eight key amino acid residues were conserved in numerous SARS-CoV-2 samples across countries all over the world, suggesting the essentiality of the above residues. It is supposed that the binding of SUD to G-quadruplexes could affect transcript stability and translation, hence, impairing the immune response of host cells (Tan et al., 2009). The expression of host genes in SARS-CoV-2-infected cells is extremely inhibited (Kim et al., 2020); therefore, we speculate that the SARS-CoV-2 may possess a similar mechanism to SARS-CoV that can inhibit the expression of some important genes.
Herein, we briefly depict the possible role of G-quadruplexes in the antiviral mechanism and pathogenicity, and the development of certain G-quadruplex-specific ligands might be a promising antiviral strategy (Figure 6). We call for more researchers to shed light on the relationship between G-quadruplexes and coronaviruses. Only if we have a deeper understanding of coronaviruses can we better cope with the possible novel coronavirus pandemics in the future.
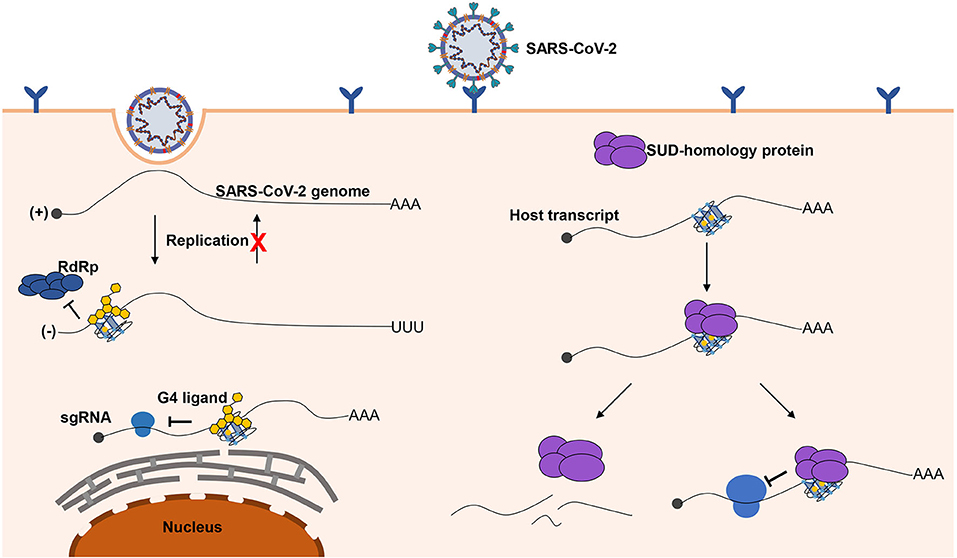
Figure 6. Possible role of G-quadruplexes in the antiviral mechanism and pathogenicity. Left part, G-quadruplexes can function as inhibition elements in the SARS-CoV-2 life cycle. Both the replication and translation could be affected by the G-quadruplex structures. The stable G-quadruplex structures in the 3′ end of the negative-sense strand may interfere with the activity of RdRp; hence, the replication of the negative-sense strands to the positive-sense strands is repressed, so that the SARS-CoV-2 genomes cannot be produced in large quantities. The G-quadruplex structures can suppress the translation process by impairing the elongating of ribosomes, which can hinder the production of proteins required for the virus. The G-quadruplex structures could be stabilized by the specific ligands to enhance the inhibitory effects, which is a promising antiviral strategy. Right part, a possible mechanism for SARS-CoV-2 to impede the expression of human genes. G-quadruplex structures, particularly with longer G-stretches, are the potential binding targets for the G-quadruplex binding domain in SARS-CoV-2, and the interaction of the G-quadruplex binding domain of SARS-CoV-2 with G-quadruplex structures possibly leads to the transcript instability or obstructing of the translation efficiency.
Data Availability Statement
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
This project was under the supervision of XS. RZ, KX, and YG took part in the project. The article was written by RZ and revised by XS and KX. HL participated in the discussion of this project. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 61972084 and 81830053).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Miss Jiaqing Xue for language polishing of this manuscript. This manuscript has been released as a preprint at bioRxiv, https://www.biorxiv.org/content/10.1101/2020.06.05.135749v1.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.587829/full#supplementary-material
References
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., and Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452. doi: 10.1038/s41591-020-0820-9
Andrews, R. J., Baber, L., and Moss, W. N. (2017). RNAStructuromeDB: a genome-wide database for RNA structural inference. Sci. Rep. 7:17269. doi: 10.1038/s41598-017-17510-y
Andrews, R. J., Peterson, J. M., Haniff, H. S., Chen, J., Williams, C., Grefe, M., et al. (2020). An in silico map of the SARS-CoV-2 RNA structurome. bioRxiv [preprint]. doi: 10.1101/2020.04.17.045161
Balasubramanian, S., Hurley, L. H., and Neidle, S. (2011). Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat. Rev. Drug Discov. 10, 261–275. doi: 10.1038/nrd3428
Beaudoin, J. D. (2010). Perreault J-P, 5'-UTR G-quadruplex structures acting as translational repressors. Nucleic Acids Res. 38, 7022–7036. doi: 10.1093/nar/gkq557
Beaudoin, J. D., Jodoin, R., and Perreault, J. P. (2014). New scoring system to identify RNA G-quadruplex folding. Nucleic Acids Res. 42, 1209–1223. doi: 10.1093/nar/gkt904
Bochman, M. L., Paeschke, K., and Zakian, V. A. (2012). DNA secondary structures: stability and function of G-quadruplex structures. Nat. Rev. Genet. 13, 770–780. doi: 10.1038/nrg3296
Broughton, J. P., Deng, X., Yu, G., Fasching, C. L., Servellita, V., Singh, J., et al. (2020). CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38, 870–874. doi: 10.1038/s41587-020-0513-4
Butovskaya, E., Heddi, B., Bakalar, B., Richter, S. N., and Phan, A. T. (2018). Major G-quadruplex form of HIV-1 LTR reveals a (3 + 1) folding topology containing a stem-loop. J. Am. Chem. Soc. 140, 13654–13662. doi: 10.1021/jacs.8b05332
Butovskaya, E., Soldà, P., Scalabrin, M., Nadai, M., and Richter, S. N. (2019). HIV-1 nucleocapsid protein unfolds stable RNA G-quadruplexes in the viral genome and is inhibited by G-quadruplex ligands. ACS Infect. Dis. 5, 2127–2135. doi: 10.1021/acsinfecdis.9b00272
Chen, L., Liu, B., Yang, J., and Jin, Q. (2014). DBatVir: the database of bat-associated viruses. Database 2014:bau021. doi: 10.1093/database/bau021
Chen, Y., Liu, Q., and Guo, D. (2020). Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 92, 418–423. doi: 10.1002/jmv.25681
Cui, J., Li, F., and Shi, Z. L. (2019). Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192. doi: 10.1038/s41579-018-0118-9
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLOS Comp. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
Doluca, O. (2019). G4Catchall: a G-quadruplex prediction approach considering atypical features. J. Theoretic. Biol. 463, 92–98. doi: 10.1016/j.jtbi.2018.12.007
Fleming, A. M., Ding, Y., Alenko, A., and Burrows, C. J. (2016). Zika virus genomic RNA possesses conserved G-quadruplexes characteristic of the flaviviridae family. ACS Infect. Dis. 2, 674–681. doi: 10.1021/acsinfecdis.6b00109
Gomez, D., Guédin, A., Mergny, J. L., Salles, B., Riou, J. F., Teulade-Fichou, M. P., et al. (2010). A G-quadruplex structure within the 5′-UTR of TRF2 mRNA represses translation in human cells. Nucleic Acids Res. 38, 7187–7198. doi: 10.1093/nar/gkq563
Gorbalenya, A. E., Baker, S. C., Baric, R. S., de Groot, R. J., Drosten, C., Gulyaeva, A. A., et al. (2020). The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544. doi: 10.1038/s41564-020-0695-z
Guan, W. J., Ni, Z. Y, Hu, Y., Liang, W. H., Ou, C. Q., He, J. X., et al. (2020). Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 382, 1708–1720. doi: 10.1056/NEJMoa2002032
Guo, Y. R., Cao, Q. D., Hong, Z. S., Tan, Y. Y., Chen, S. D., Jin, H. J., et al. (2020). The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak – an update on the status. Mil. Med. Res. 7:11. doi: 10.1186/s40779-020-00240-0
Han, H., and Hurley, L. H. (2000). G-quadruplex DNA: a potential target for anti-cancer drug design. Trends Pharmacol. Sci. 21, 136–142. doi: 10.1016/S0165-6147(00)01457-7
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8. doi: 10.1016/j.cell.2020.02.052
Hon, J., Martínek, T., Zendulka, J., and Lexa, M. (2017). pqsfinder: an exhaustive and imperfection-tolerant search tool for potential quadruplex-forming sequences in R. Bioinformatics 33, 3373–3379. doi: 10.1093/bioinformatics/btx413
Hoshina, S., Yura, K., Teranishi, H., Kiyasu, N., Tominaga, A., Kadoma, H., et al. (2013). Human origin recognition complex binds preferentially to G-quadruplex-preferable RNA and single-stranded DNA J. Biol. Chem. 288, 30161–30171. doi: 10.1074/jbc.M113.492504
Jansson, L. I., Hentschel, J., Parks, J. W., Chang, T. R., Lu, C., Baral, R., et al. (2019). Telomere DNA G-quadruplex folding within actively extending human telomerase. Proc. Nat. Acad. Sci. U.S.A. 116, 9350–9359. doi: 10.1073/pnas.1814777116
Jaubert, C., Bedrat, A., Bartolucci, L., Di Primo, C., Ventura, M., Mergny, J. L., et al. (2018). RNA synthesis is modulated by G-quadruplex formation in Hepatitis C virus negative RNA strand. Sci. Rep. 8:8120. doi: 10.1038/s41598-018-26582-3
Jin, Y., Yang, H., Ji, W., Wu, W., Chen, S., Zhang, W., et al. (2020). Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses 12:372. doi: 10.3390/v12040372
Joachimi, A., Benz, A., and Hartig, J. S. (2009). A comparison of DNA and RNA quadruplex structures and stabilities. Bioorgan. Med. Chem. 17, 6811–6815. doi: 10.1016/j.bmc.2009.08.043
Jodoin, R., Carrier, J. C., Rivard, N., Bisaillon, M., and Perreault, J. P. (2019). G-quadruplex located in the 5′UTR of the BAG-1 mRNA affects both its cap-dependent and cap-independent translation through global secondary structure maintenance. Nucleic Acids Res. 47, 10247–10266. doi: 10.1093/nar/gkz777
Kikin, O., D'Antonio, L., and Bagga, P. S. (2006). QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 34(Suppl. 2), W676–W682. doi: 10.1093/nar/gkl253
Kim, D., Lee, J. Y., Yang, J. S., Kim, J. W., Kim, V. N., and Chang, H. (2020). The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921.e10. doi: 10.1016/j.cell.2020.04.011
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kumari, S., Bugaut, A., Huppert, J. L., and Balasubramanian, S. (2007). An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nat. Chem. Biol. 3, 218–221. doi: 10.1038/nchembio864
Kusov, Y., Tan, J., Alvarez, E., Enjuanes, L., and Hilgenfeld, R. (2015). A G-quadruplex-binding macrodomain within the “SARS-unique domain” is essential for the activity of the SARS-coronavirus replication-transcription complex. Virology 484, 313–322. doi: 10.1016/j.virol.2015.06.016
Kwok, C. K., Marsico, G., Sahakyan, A. B., Chambers, V. S., and Balasubramanian, S. (2016). rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat. Methods 13, 841–844. doi: 10.1038/nmeth.3965
Kwok, C. K., and Merrick, C. J. (2017). G-quadruplexes: prediction, characterization, biological application. Trends Biotechnol. 35, 997–1013. doi: 10.1016/j.tibtech.2017.06.012
Lai, C. C., Shih, T. P., Ko, W. C., Tang, H. J., and Hsueh, P. R. (2020). Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): the epidemic and the challenges. Int. J. Antimicrob. Agents. 55:105924. doi: 10.1016/j.ijantimicag.2020.105924
Lam, T. T. Y., Shum, M. H-H., Zhu, H. C., Tong, Y. G., Ni, X. B., Liao, Y-S., et al. (2020). Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature 583, 282–285. doi: 10.1038/s41586-020-2169-0
Lee, D. S. M., Ghanem, L. R., and Barash, Y. (2020). Integrative analysis reveals RNA G-quadruplexes in UTRs are selectively constrained and enriched for functional associations. Nat. Commun. 11:527. doi: 10.1038/s41467-020-14404-y
Li, L., Qin, L., Xu, Z., Yin, Y., Wang, X., Kong, B., et al. (2020). Using artificial intelligence to detect COVID-19 and community-acquired pneumonia based on pulmonary CT: evaluation of the diagnostic accuracy. Radiology 296, E65–E71. doi: 10.1148/radiol.2020200905
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. doi: 10.1093/nar/gkz268
Majee, P., Kumar Mishra, S., Pandya, N., Shankar, U., Pasadi, S., Muniyappa, K., et al. (2020). Identification and characterization of two conserved G-quadruplex forming motifs in the Nipah virus genome and their interaction with G-quadruplex specific ligands. Sci. Rep. 10:1477. doi: 10.1038/s41598-020-58406-8
Marušič, M., Hošnjak, L., Krafčikova, P., Poljak, M., Viglasky, V., and Plavec, J. (2017). The effect of single nucleotide polymorphisms in G-rich regions of high-risk human papillomaviruses on structural diversity of DNA. Biochim. Biophys. Acta Gen. Subj. 1861, 1229–1236. doi: 10.1016/j.bbagen.2016.11.007
Métifiot, M., Amrane, S., Litvak, S., and Andreola, M. L. (2014). G-quadruplexes in viruses: function and potential therapeutic applications. Nucleic Acids Res. 42, 12352–12366. doi: 10.1093/nar/gku999
Miller, K. M., and Rodriguez, R. (2011). G-quadruplexes: selective DNA targeting for cancer therapeutics? Expert Rev. Clin. Pharmacol. 4, 139–142. doi: 10.1586/ecp.11.4
Mishra, A., Pandey, A. K., Gupta, P., Pradhan, P., Dhamija, S., Gomes, J., et al. (2020). Mutation landscape of SARS-CoV-2 reveals three mutually exclusive clusters of leading and trailing single nucleotide substitutions. bioRxiv [preprint]. doi: 10.1101/2020.05.07.082768
Moye, A. L., Porter, K. C., Cohen, S. B., Phan, T., Zyner, K. G., Sasaki, N., et al. (2015). Telomeric G-quadruplexes are a substrate and site of localization for human telomerase. Nat. Commun. 6:7643. doi: 10.1038/ncomms8643
Murat, P., Marsico, G., Herdy, B., Ghanbarian, A., Portella, G., and Balasubramanian, S. (2018). RNA G-quadruplexes at upstream open reading frames cause DHX36- and DHX9-dependent translation of human mRNAs. Genome Biol. 19:229. doi: 10.1186/s13059-018-1602-2
Murat, P., Zhong, J., Lekieffre, L., Cowieson, N. P., Clancy, J. L., Preiss, T., et al. (2014). G-quadruplexes regulate Epstein-Barr virus-encoded nuclear antigen 1 mRNA translation. Nat. Chem. Biol. 10, 358–364. doi: 10.1038/nchembio.1479
Neidle, S. (2017). Quadruplex nucleic acids as targets for anticancer therapeutics. Nat. Rev. Chem. 1:0041. doi: 10.1038/s41570-017-0041
Okonechnikov, K., Golosova, O., and Fursov, M. (2012). the Ut, unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. doi: 10.1093/bioinformatics/bts091
Peiris, J. S. M., Guan, Y., and Yuen, K. Y. (2004). Severe acute respiratory syndrome. Nat. Med. 10, S88–S97. doi: 10.1038/nm1143
Perrone, R., Artusi, S., Butovskaya, E., Nadai, M., Pannecouque, C., and Richter, S. N. (2015). “G-quadruplexes in the human immunodeficiency virus-1 and herpes simplex virus-1: new targets for antiviral activity by small molecules,” in 5th International Conference on Biomedical Engineering in Vietnam, eds V.V. Toi and T.H. Lien Phuong (Cham: Springer International Publishing), 207–210.
Perrone, R., Butovskaya, E., Daelemans, D., Palù, G., Pannecouque, C., and Richter, S. N. (2014). Anti-HIV-1 activity of the G-quadruplex ligand BRACO-19. J. Antimicrob. Chemother. 69, 3248–3258. doi: 10.1093/jac/dku280
Perrone, R., Nadai, M., Poe, J. A., Frasson, I., Palumbo, M., Palù, G., et al. (2013). Formation of a unique cluster of G-quadruplex structures in the HIV-1 nef coding region: implications for antiviral activity. PLoS ONE 8:e73121. doi: 10.1371/journal.pone.0073121
Piekna-Przybylska, D., Sullivan, M. A., Sharma, G., and Bambara, R. A. (2014). U3 region in the HIV-1 genome adopts a G-quadruplex structure in its RNA and DNA sequence. Biochemistry 53, 2581–2593. doi: 10.1021/bi4016692
Prorok, P., Artufel, M., Aze, A., Coulombe, P., Peiffer, I., Lacroix, L., et al. (2019). Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun. 10:3274. doi: 10.1038/s41467-019-11104-0
Puig Lombardi, E., and Londoño-Vallejo, A. (2019). A guide to computational methods for G-quadruplex prediction. Nucleic Acids Res. 48, 1–15. doi: 10.1093/nar/gkz1097
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rothan, H. A., and Byrareddy, S. N. (2020). The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmunity 109:102433. doi: 10.1016/j.jaut.2020.102433
Ruggiero, E., and Richter, S. N. (2018). G-quadruplexes and G-quadruplex ligands: targets and tools in antiviral therapy. Nucleic Acids Res. 46, 3270–3283. doi: 10.1093/nar/gky187
Ruggiero, E., and Richter, S. N. (2020). Viral G-quadruplexes: new frontiers in virus pathogenesis and antiviral therapy. Annu. Rep. Med. Chem. 54, 101–131. doi: 10.1016/bs.armc.2020.04.001
Saranathan, N., and Vivekanandan, P. (2019). G-quadruplexes: more than just a kink in microbial genomes. Trends Microbiol. 27, 148–163. doi: 10.1016/j.tim.2018.08.011
Shahid, R., Bugaut, A., and Balasubramanian, S. (2010). The BCL-2 5′ untranslated region contains an RNA G-quadruplex-forming motif that modulates protein expression. Biochemistry 49, 8300–8306. doi: 10.1021/bi100957h
Shereen, M. A., Khan, S., Kazmi, A., Bashir, N., and Siddique, R. (2020). COVID-19 infection: origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 24, 91–98. doi: 10.1016/j.jare.2020.03.005
Shu, Y., and McCauley, J. (2017). GISAID: Global initiative on sharing all influenza data – from vision to reality. Euro Survelli 22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494
Sievers, F., and Higgins, D. G. (2018). Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 27, 135–145. doi: 10.1002/pro.3290
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Spiegel, J., Adhikari, S., and Balasubramanian, S. (2020). The structure and function of DNA G-quadruplexes. Trends Chem. 2, 123–136. doi: 10.1016/j.trechm.2019.07.002
Takahama, K., Takada, A., Tada, S., Shimizu, M., Sayama, K., Kurokawa, R., et al. (2013). Regulation of telomere length by G-quadruplex telomere DNA- and TERRA-binding protein TLS/FUS. Chem. Biol. 20, 341–350. doi: 10.1016/j.chembiol.2013.02.013
Tan, J., Vonrhein, C., Smart, O. S., Bricogne, G., Bollati, M., Kusov, Y., et al. (2009). The SARS-unique domain (SUD) of SARS coronavirus contains two macrodomains that bind G-quadruplexes. PLoS Pathog. 5:e1000428. doi: 10.1371/journal.ppat.1000428
Tang, J., Kan, Z. Y., Yao, Y., Wang, Q., Hao, Y. H., and Tan, Z. (2007). G-quadruplex preferentially forms at the very 3′ end of vertebrate telomeric DNA. Nucleic Acids Res. 36, 1200–1208. doi: 10.1093/nar/gkm1137
Tlučková, K., Marušič, M., Tóthová, P., Bauer, L., Šket, P., Plavec, J., et al. (2013). Human papillomavirus G-quadruplexes. Biochemistry 52, 7207–7216. doi: 10.1021/bi400897g
Valton, A. L., Hassan-Zadeh, V., Lema, I., Boggetto, N., Alberti, P., Saintomé, C., et al. (2014). G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J. 33, 732–746. doi: 10.1002/embj.201387506
Valton, A. L., and Prioleau, M. N. (2016). G-quadruplexes in DNA replication: a problem or a necessity? Trends Genet. 32, 697–706. doi: 10.1016/j.tig.2016.09.004
Varshney, D., Spiegel, J., Zyner, K., Tannahill, D., and Balasubramanian, S. (2020). The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 21, 459–474. doi: 10.1038/s41580-020-0236-x
Walls, A. C., Park, Y. J., Tortorici, M. A., Wall, A., McGuire, A. T., and Veesler, D. (2020). Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e6. doi: 10.1016/j.cell.2020.02.058
Wang, Q., Liu, J. Q., Chen, Z., Zheng, K. W., Chen, C. Y., Hao, Y. H., et al. (2011). G-quadruplex formation at the 3' end of telomere DNA inhibits its extension by telomerase, polymerase and unwinding by helicase. Nucleic Acids Res. 39, 6229–6237. doi: 10.1093/nar/gkr164
Wang, S. R., Min, Y. Q., Wang, J. Q., Liu, C. X., Fu, B. S., Wu, F., et al. (2016b). A highly conserved G-rich consensus sequence in hepatitis C virus core gene represents a new anti-hepatitis C target. Sci. Adv. 2:e1501535. doi: 10.1126/sciadv.1501535
Wang, S. R., Zhang, Q. Y., Wang, J. Q., Ge, X. Y., Song, Y. Y., Wang, Y. F., et al. (2016a). Chemical targeting of a G-quadruplex RNA in the Ebola Virus L gene. Cell Chem. Biol. 23, 1113–1122. doi: 10.1016/j.chembiol.2016.07.019
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. doi: 10.1093/nar/gky427
Xiao, K., Zhai, J., Feng, Y., Zhou, N., Zhang, X., Zou, J. J., et al. (2020). Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 583, 286–289. doi: 10.1038/s41586-020-2313-x
Yao, H., Lu, X., Chen, Q., Xu, K., Chen, Y., Cheng, L., et al. (2020). Patient-derived mutations impact pathogenicity of SARS-CoV-2. medRxiv [preprint]. doi: 10.1101/2020.04.14.20060160
Yin, C. (2020). Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics 112, 3588–3596. doi: 10.1016/j.ygeno.2020.04.016
Zaccaria, F., and Fonseca Guerra, C. (2018). RNA versus DNA G-quadruplex: the origin of increased stability. Chemistry 24, 16315–16322. doi: 10.1002/chem.201803530
Zahin, M., Dean, W. L., Ghim, S. J., Joh, J., Gray, R. D., Khanal, S., Bossart, G. D., et al. (2018). Identification of G-quadruplex forming sequences in three manatee papillomaviruses. PLoS ONE 13:e0195625. doi: 10.1371/journal.pone.0195625
Zhang, T., Wu, Q., and Zhang, Z. (2020). Probable pangolin origin of sars-cov-2 associated with the COVID-19 outbreak. Curr. Biol. 30, 1346–1351.e2. doi: 10.1016/j.cub.2020.03.022
Zhang, Y., Liu, S., Jiang, H., Deng, H., Dong, C., Shen, W., et al. (2020). G2-quadruplex in the 3'UTR of IE180 regulates pseudorabies virus replication by enhancing gene expression. RNA Biol. 17, 816–827. doi: 10.1080/15476286.2020.1731664
Zheng, J. (2020). SARS-CoV-2: an emerging coronavirus that causes a global threat. Int. J. Biol. Sci. 16, 1678–1685. doi: 10.7150/ijbs.45053
Zhong, N. S., Zheng, B. J., Li, Y. M., Poon, L. L. M., Xie, Z. H., Chan, K. H., et al. (2003). Epidemiology and cause of severe acute respiratory syndrome (SARS) in guangdong, people's Republic of China, in February, 2003. Lancet 362, 1353–1358. doi: 10.1016/S0140-6736(03)14630-2
Zhou, P., Yang, X. L., Wang, X. G., Hu, B., Zhang, L., Zhang, W., et al. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. doi: 10.1038/s41586-020-2012-7
Zu, Z. Y., Jiang, M. D., Xu, P. P., Chen, W., Ni, Q. Q., Lu, G. M., et al. (2020). Coronavirus disease 2019 (COVID-19): a perspective from China. Radiology 2, E15–E25. doi: 10.1148/radiol.2020200490
Keywords: G-quadruplex, SARS-CoV-2, COVID-19, G4, coronavirus, G-quadruplex binding domain, SUD-homology structure
Citation: Zhang R, Xiao K, Gu Y, Liu H and Sun X (2020) Whole Genome Identification of Potential G-Quadruplexes and Analysis of the G-Quadruplex Binding Domain for SARS-CoV-2. Front. Genet. 11:587829. doi: 10.3389/fgene.2020.587829
Received: 27 July 2020; Accepted: 22 October 2020;
Published: 27 November 2020.
Edited by:
Geng Chen, East China Normal University, ChinaReviewed by:
Bernard Fongang, The University of Texas Health Science Center at San Antonio, United StatesCarson Andorf, United States Department of Agriculture (USDA), United States
Copyright © 2020 Zhang, Xiao, Gu, Liu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao Sun, eHN1bkBzZXUuZWR1LmNu