
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 11 November 2020
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.580477
This article is part of the Research TopicGenetic Testing in Pediatric DisordersView all 35 articles
 Ewelina Bukowska-Olech1,2†
Ewelina Bukowska-Olech1,2† Anna Materna-Kiryluk1,3†Joanna Walczak-Sztulpa1†Delfina Popiel3Magdalena Badura-Stronka1,3†
Anna Materna-Kiryluk1,3†Joanna Walczak-Sztulpa1†Delfina Popiel3Magdalena Badura-Stronka1,3† Grzegorz Koczyk3,4†Adam Dawidziuk3
Grzegorz Koczyk3,4†Adam Dawidziuk3 Aleksander Jamsheer1,3*†
Aleksander Jamsheer1,3*†Background: Defects in the development of the first and second pharyngeal arches and their derivatives result in abnormal formation of the craniofacial complex, consequently giving rise to facial dysostoses (FDs). FDs represent a group of rare and highly heterogeneous disease entities that encompass mandibulofacial dysostoses (MFDs) with normal extremities and acrofacial dysostoses (AFDs) with limb anomalies in addition to craniofacial defects.
Methods: We examined 11 families with variable clinical symptoms of FDs, in most of which only one member was affected. We applied two custom gene panels—first comprising 37 genes related to the genetic disorders of craniofacial development such as FDs (On-Demand AmpliSeq Thermo Fisher Scientific gene panel with two primer pools) and second composed of 61 genes and 11 single nucleotide variants (SNVs) known to be involved in the development of skull malformations, mainly in the form of craniosynostoses (SureSelect Agilent Technologies). Targeted next-generation sequencing (NGS) was performed using the Ion Torrent S5 platform. To confirm the presence of each detected variant, we have analyzed a genomic region of interest using Sanger sequencing.
Results: In this paper, we summarized the results of custom targeted gene panel sequencing in the cohort of sixteen patients from 11 consecutive families affected by distinct forms of FDs. We have found three novel pathogenic variants in the TCOF1 gene—c.2145_2148dupAAAG p.(Ser717Lysfs∗42), c.4370delA p.(Lys1457Argfs∗118), c.83G>C p.(Arg28Pro) causing Treacher Collins syndrome type 1, two novel missense variants in the EFTUD2 gene–c.491A>G p.(Asp164Gly) and c.779T>A p.(Ile260Asn) in two female patients affected by acrofacial dysostosis Guion-Almeida type, one previously reported–c.403C>T (p.Arg135Cys), as well as one novel missense variant–c.128C>T p.(Pro43Leu) in the DHODH gene in the male patient with Miller syndrome and finally one known pathogenic variant c.574G>T p.(Glu192∗) in the SF3B4 gene in the patient with Nager syndrome.
Conclusion: Our study confirms the efficiency and clinical utility of the targeted gene panel sequencing and shows that this strategy is suitable and efficient in the molecular screening of variable forms of FDs.
The human skull composes of neuro- and viscerocranium that both develop independently from one another. Neurocranium mainly forms from the first five somites and the unsegmented somitomeres rostral to the first somite, whereas, viscerocranium, namely facial skeleton, forms from pharyngeal arches (Jin et al., 2016).
Defects concerning embryonic development of the first and second pharyngeal arches and their derivatives result in facial dysostoses (FDs), which encompass a group of rare and extremely variable clinical disease entities. FDs have been subdivided into mandibulofacial dysostoses (MFDs) with normal extremities and acrofacial dysostoses (AFDs) with limb anomalies in addition to craniofacial malformations (Table 1; Trainor and Andrews, 2013; Wieczorek, 2013). The characteristic facial features of FDs involve hypoplasia of the maxilla, mandible, zygomatic arches, various anomalies of the external and middle ear, and other facial defects including coloboma of the lower eyelid with partial to total absence of eyelashes or dysmorphic features, such as down-slanting palpebral fissures. Limb defects represent a broad spectrum of post- or preaxial abnormalities, including polydactyly, oligodactyly, and brachydactyly (Ogilvy-Stuart and Parsons, 1991; McDonald and Gorski, 1993; Wieczorek et al., 2009; Ng et al., 2010; Rainger et al., 2012; Czeschik et al., 2013; Trainor and Andrews, 2013; Wieczorek, 2013). Additional clinical signs, which may be present comprise microcephaly, cleft palate, oligodontia, defects of the internal organs, short stature, psychomotor delay, and intellectual disability (Guion-Almeida et al., 2006; Wieczorek, 2013).
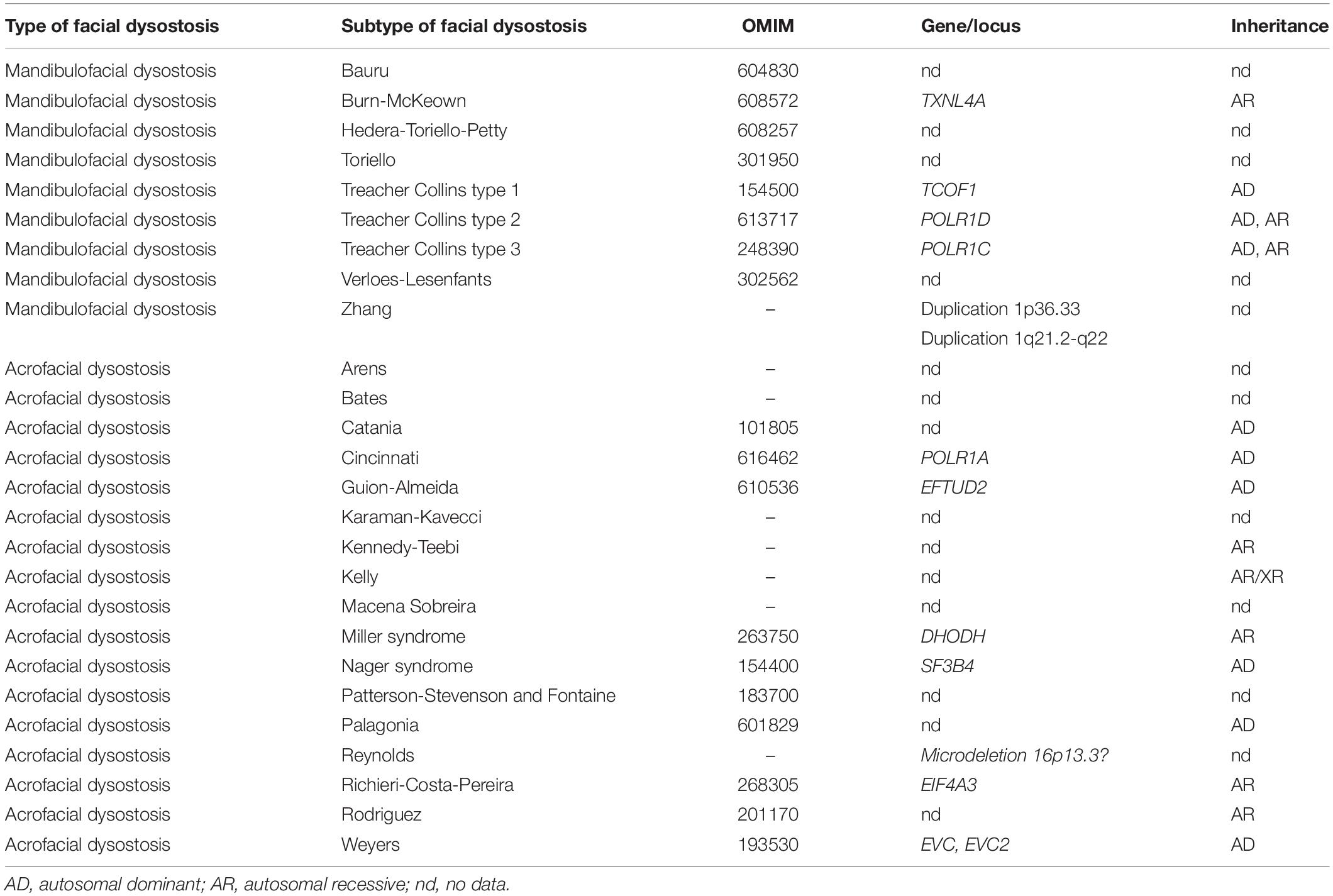
Table 1. Clinical classification of human facial dysostoses.
Importantly, FDs present a high clinical heterogeneity, including a marked inter- and intra-familial variability of their phenotypic expression what in turn frequently impedes reaching an exact clinical diagnosis and its confirmation at a molecular level (Dixon et al., 2004; Vincent et al., 2016). Consequently, many patients affected by FDs remain molecularly undiagnosed and bereft of reliable genetic counseling, prenatal and preimplantation testing, but also of a proper and unequivocal clinical classification (Ng et al., 2010; Bernier et al., 2012; Lines et al., 2012; Fan et al., 2019).
All procedures were performed under the ethical standards of the institutional and national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. We granted ethics approval from the Institutional Review Board of Poznan University of Medical Sciences (no. 741/17 and 742/17 obtained on 22nd June 2017). We examined 11 families with variable clinical symptoms of FDs, in most of which only one member was affected. We extracted genomic DNA from the peripheral blood lymphocytes using the MagCore HF16 Automated Nucleic Acid Extractor and quantified each gDNA using the Agilent Technologies TapeStation 2200 and Qubit.
We designed the custom On-Demand AmpliSeq (Thermo Fisher Scientific) gene panel with two primer pools and performed targeted sequencing of 37 genes related to the genetic disorders of craniofacial development, including FDs (patients 1.4, 1.6, 2–4, 6 and 7; Supplementary Table 1). The gene panel was 145.51 kb in size and comprised 761 amplicons, intervals sequenced for each gene were shown in Supplementary Table 2. The barcoded genomic DNA libraries were constructed according to the ‘manufacturer’s sample preparation protocol (Ion AmpliSeq Library Kit 2.0; On-Demand Panels). Pooled libraries were subjected to emulsion PCR on Ion ChefTM instrument using the Ion 520TM and 530TMKit, according to the standard protocol and sequenced on the Ion Torrent S5 platform.
The custom SureSelect (Agilent Technologies) gene panel comprised of 61 genes and 11 SNVs known to be involved in the development of craniofacial malformations, particularly craniosynostosis (CS), was designed (Supplementary Table 3). Intervals sequenced for each gene were shown in Supplementary Table 4. Targeted sequencing, encompassing 225.709 kb was performed for patient 5. We prepared the NGS library using hybridized capture-based target enrichment approach and sequenced it on the Ion Torrent S5 platform (Bukowska-Olech et al., 2020).
The present analysis follows the approach previously described in Bukowska-Olech et al. (2020). Briefly, the reads were cleaned and aligned to GrCh37 human reference sequence using the TorrentBrowser 5.0.4 software (Thermo Fisher Scientific). The alignments were further processed using IonReporter 5.2 pipeline (Thermo Fisher Scientific), which incorporated variant calling with Torrent Variant Caller 5.4–11. Each variant was examined based on four criteria (read depth− ≥ 20, strandedness− ≤ 4:1, PHRED score ≥ 30 and variant frequency ≥ 0.15). The coverage of individual genes and exons was performed using bedtools 2.27.1 (coverage subcommand) against panel-specific BED files defining coding parts of canonical transcripts for each constituent gene (RefSeq mapped on UCSC hg19 reference; 5 bp padding around each exon included; Supplementary Tables 5–8).
Previous clinical annotation of called variants was obtained from Human Gene Mutation Database (HGMD) Professional (accessed on 14/02/2019), ClinVar (accessed on 27/07/2019) and dbSNP (accessed on 27/07/2019). The predictions for SIFT, PolyPhen, and PhyloP (46-way) tools were retrieved from IonReporter result files. Population allele frequencies were annotated by Ensembl/Variant Effect Predictor (software version 94, database version 94); additionally, gnomAD database was queried for homozygosity/heterozygosity of individual variants (version 2.1.1 of both exome and genome subsets; accessed on 27/07/2019 using tabix 1.5 software). The scores for Combined Annotation Dependent Depletion were obtained from CADD webserver (v1.4, accessed on 27/07/2019). MutationTaster results were obtained using the “query chromosomal position” option of the public webserver (accessed on 27/07/2019).
To confirm the presence of each pathogenic or likely pathogenic variant, as well as of variant of uncertain significance (VUS), we have analyzed the genomic region of interest using Sanger sequencing. The online available Primer3 tool was used to design specific primers for the amplification. The PCR reactions and PCR products purification were carried out following standard protocols. Sanger sequencing was performed using dye-terminator chemistry (kit v.3, ABI 3130XL) and run on automated sequencer Applied Biosystems Prism 3700 DNA Analyzer. Variants were annotated against the reference sequences: DHODH—NG_016271.1, NC_000016.10, NM_001361.4, NP_001352.2; EFTUD2—NG_032674.1, NC_000017.11, NM_004247.3, NP_004238.3; SF3B4—NG_032777.1, NC_000001.11 NM_005850.4, NP_005841.1, TCOF1—NG_011341.1, NC_000005.10, NM_001135243.1, NP_001128715.1 following the Human Genome Variation Society (HGVS) nomenclature guidelines (den Dunnen et al., 2016; Table 2).
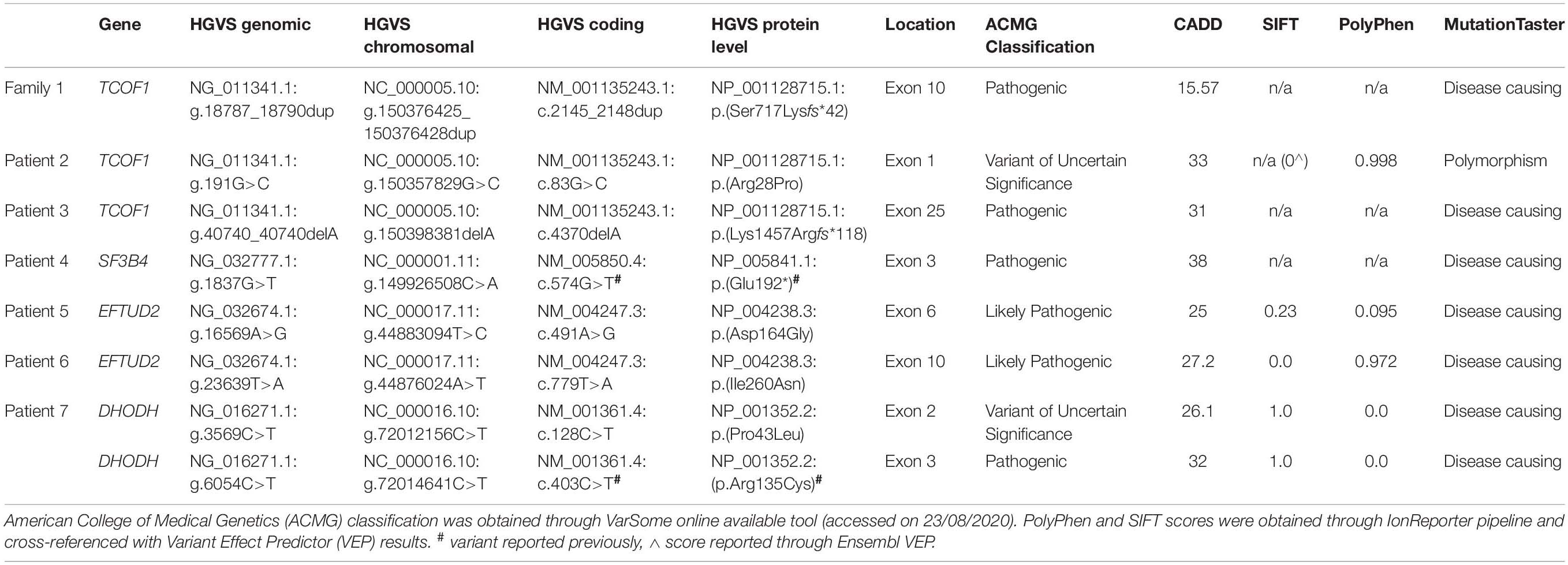
Table 2. Summary of pathogenic variants reported in this paper.
We recruited 11 consecutive families in whom 16 individuals were affected by various forms of FDs. Four families (no. 1–3, 6) received the clinical diagnosis of MFDs defined as hypoplastic maxilla, mandible, and zygomatic arches with down-slanting palpebral fissures. Six families (no. 4, 8–11) were clinically diagnosed with AFDs since the affected individuals showed not only craniofacial but also limb involvement. In one family (family 5), we did not recognize FD based on the dysmorphological assessment. The detailed clinical data of all screened patients is set forth in Supplementary Data Sheet 1.
Targeted NGS allowed us to reach an exact diagnosis of FDs in seven out of eleven families (no. 1–7). Among them, we identified six novel and two previously reported variants in the following genes–TCOF1, SF3B4, EFTUD2, and DHODH (Table 2). The mean coverage for FDs panel ranged from 214 to 430 reads per gene, whereas for CS panel from 272 to 488 reads per gene (Supplementary Tables 5, 7).
In families no 1–3, we found three novel heterozygous variants in the TCOF1 that gave rise to the most well-characterized MFD, i.e., Treacher Collins syndrome (TCS) type 1 (TCS1, MIM:154500). The major clinical features of all affected individuals were summarized in Table 3. The rest of the diagnosed patients was affected by different types of AFDs. In a sporadic female case from family no 4, we identified the heterozygous variant in the SF3B4 gene causing Nager syndrome (MIM:54400). Two other sporadic female cases from families no 5 and 6 harbored de novo heterozygous variants within the EFTUD2 gene and presented with variable symptoms of acrofacial dysostosis Guion-Almeida type (AFDGA, MIM:610536). The comparison of their clinical features was shown in Table 4. Finally, in a male proband from family no 7, we diagnosed Miller syndrome (MIM:263750), as we found two biallelic variants in the DHODH gene. Importantly, none of the detected alterations was present in gnomAD or ExAC databases or found in 200 ethnically matched control alleles from our in-house, population-specific database. Segregation analysis of the causative variants performed in families 1–6 was consistent with the expected autosomal dominant inheritance pattern (Figures 2, 3C,F, 4C, 5D,H), whereas in family 7 with autosomal recessive pattern (Figure 6E). The results of molecular screening and segregation analyses were shown in Figures 1–6 and Supplementary Figures 1A–I.
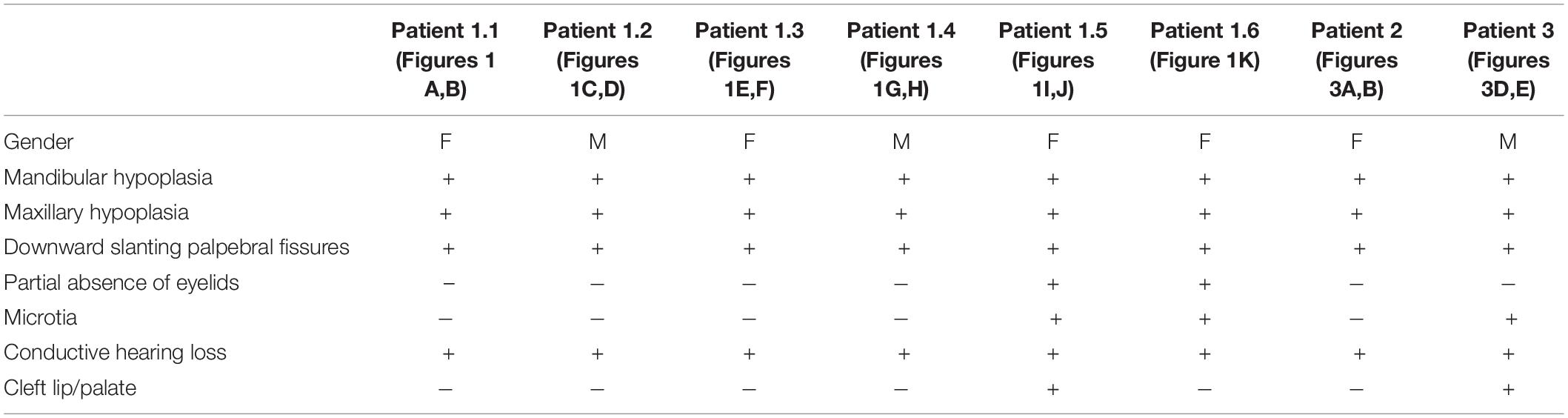
Table 3. Summary of clinical data in reported patients with Treacher Collins syndrome (TCS).
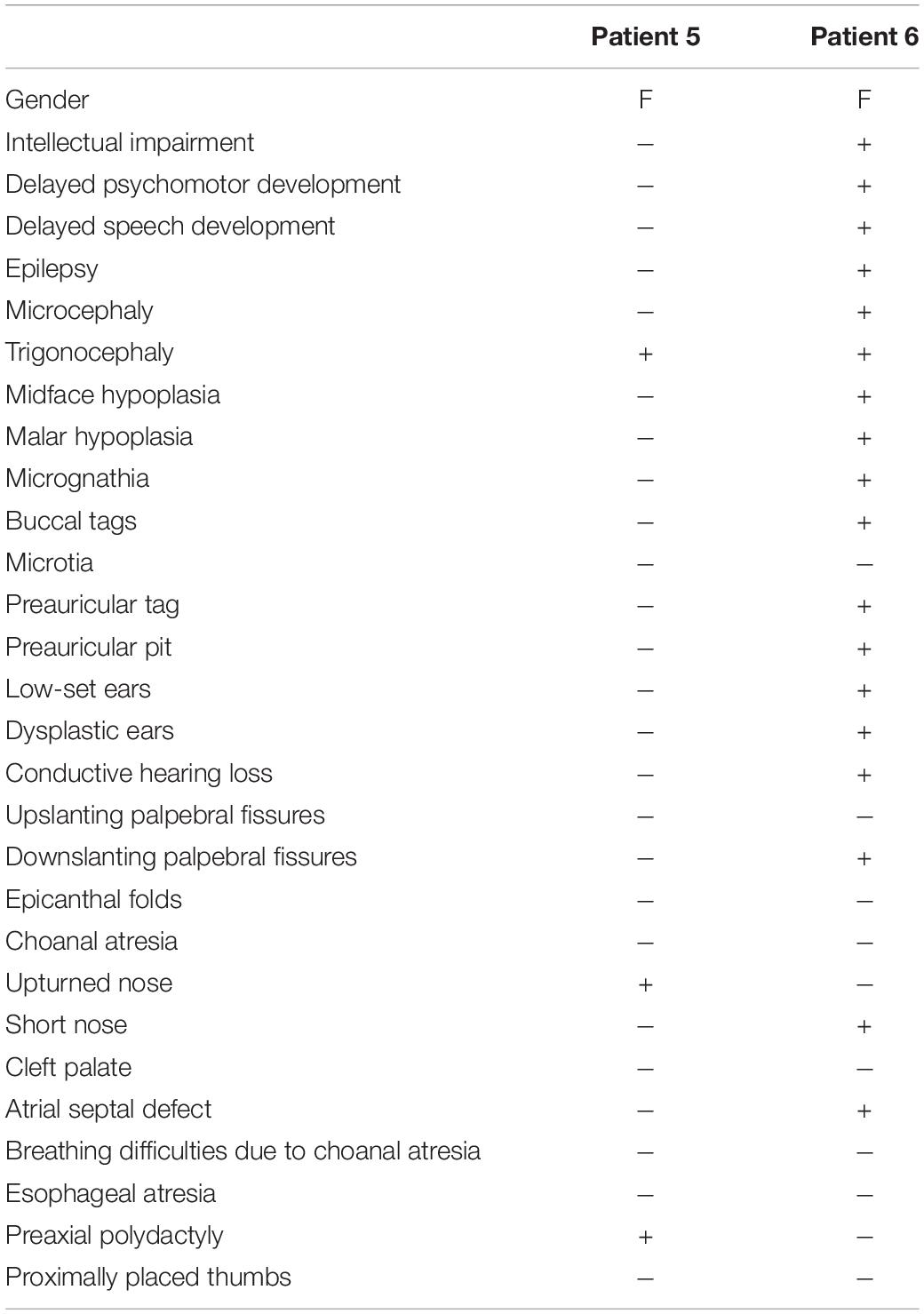
Table 4. Summary of clinical data in reported patients with acrofacial dysostoses Guion-Almeida type (AFDGA).
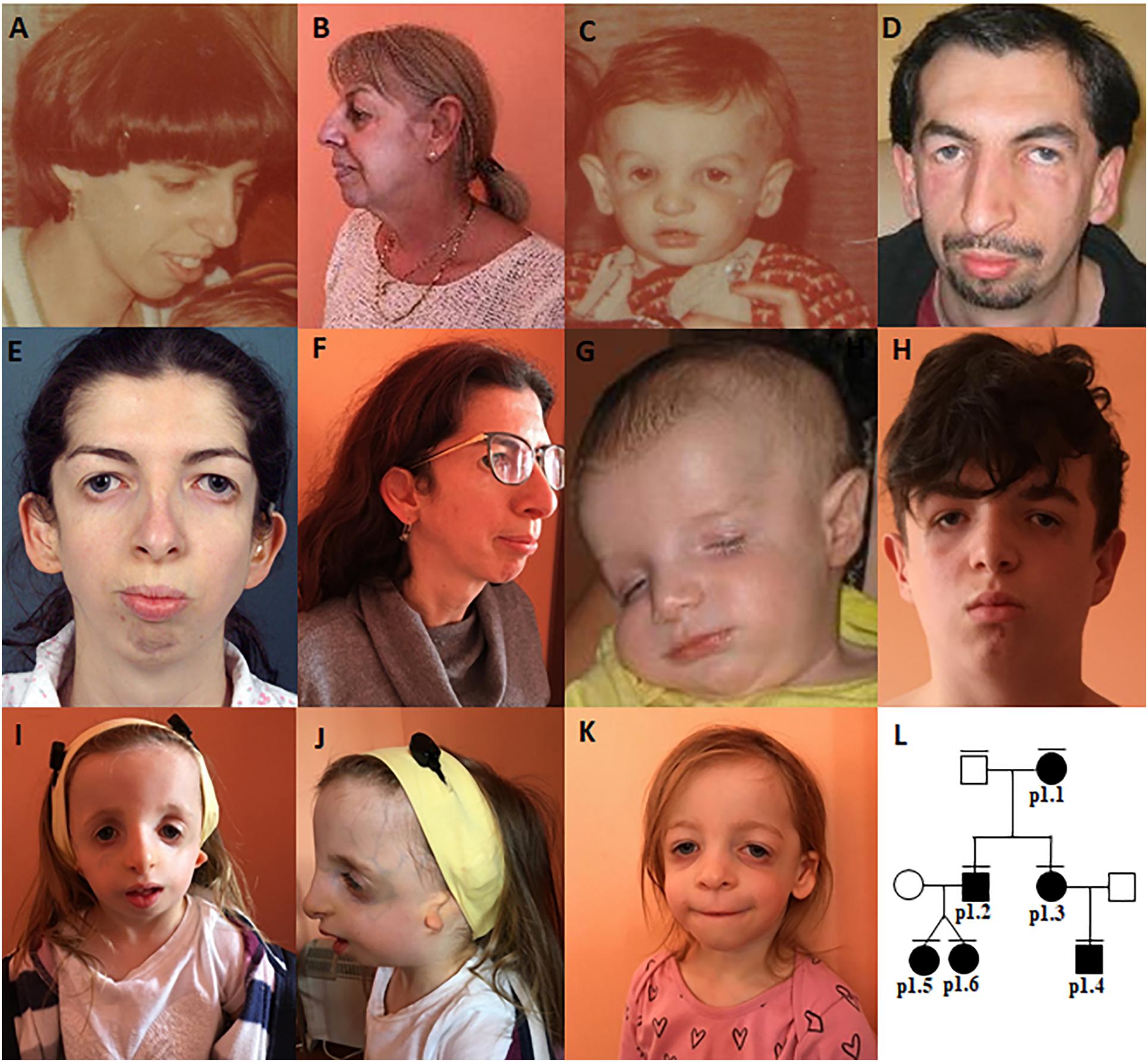
Figure 1. Clinical characteristics of family 1–patient 1.1 (A,B), patient 1.2 (C,D), patient 1.3 (E,F), patient 1.4 (G,H), patient 1.5 (I,J), patient 1.6 (K), The pedigree (L).
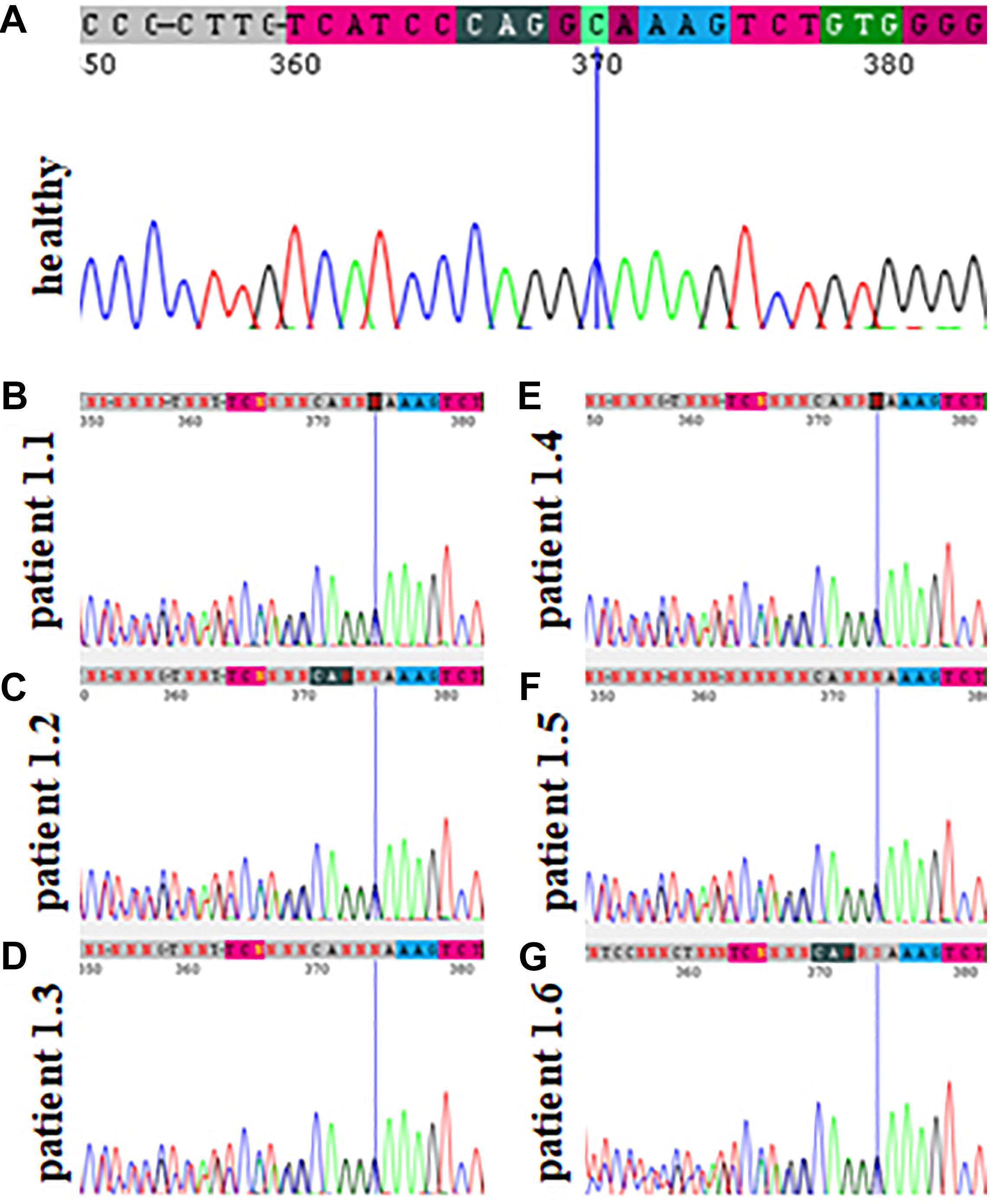
Figure 2. Sanger sequencing results of family 1 showing the absence of the pathogenic variant c.2145_2148dupAAAG p.(Ser717Lysfs*42) within the TCOF1 gene in a healthy family member (A) and its presence in patients affected by Treacher Collins syndrome (B–G).
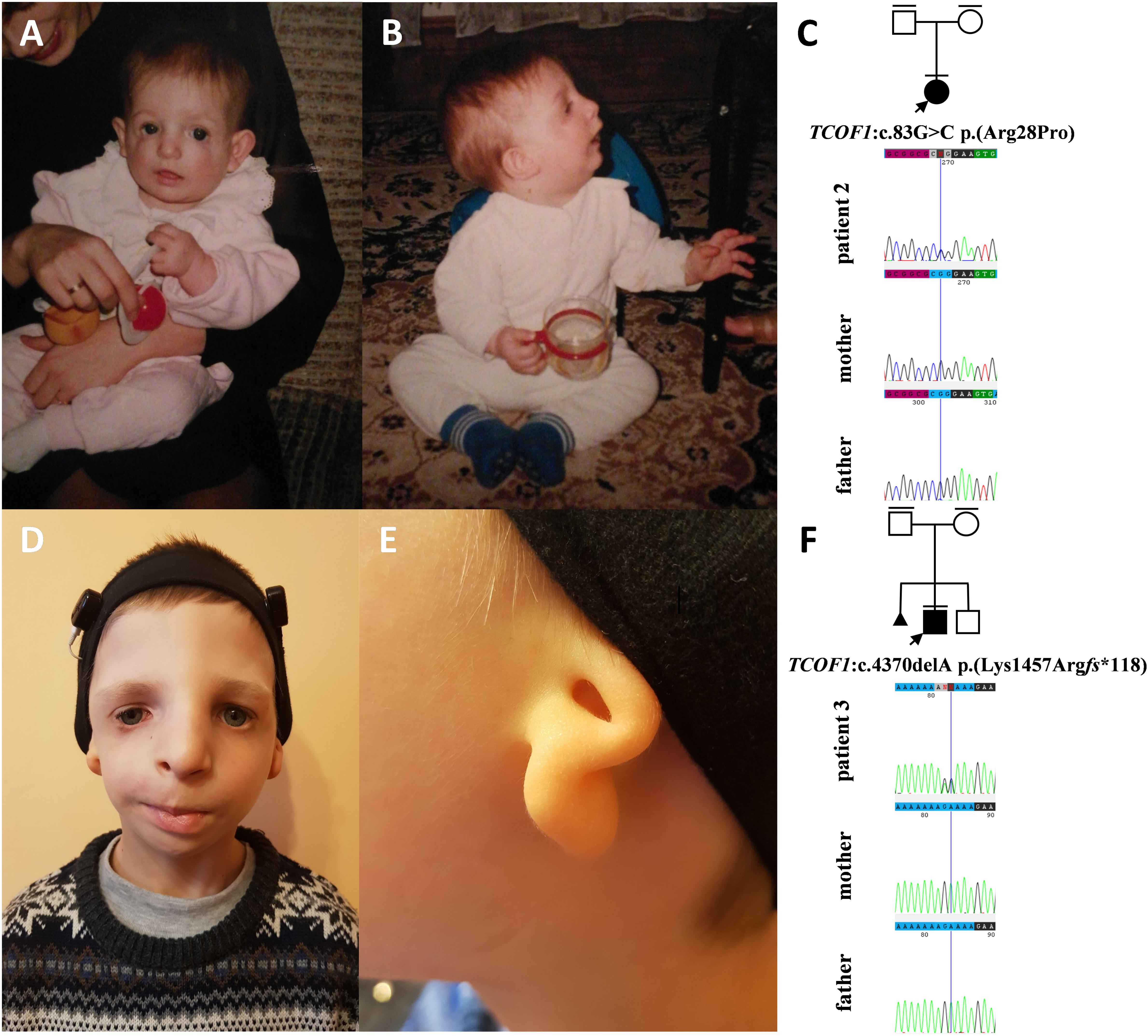
Figure 3. Clinical characteristics and segregation results of patient 2 (A–C) and 3 (D–F) affected by Treacher Collins syndrome. Patient 2 carries a heterozygous variant c.83G>C p.(Arg28Pro) within the TCOF1 gene, which occurred de novo (C) whereas patient 3 carries a heterozygous variant c.4370delA p.(Lys1457Argfs*118) within the TCOF1 gene, which also occurred de novo.
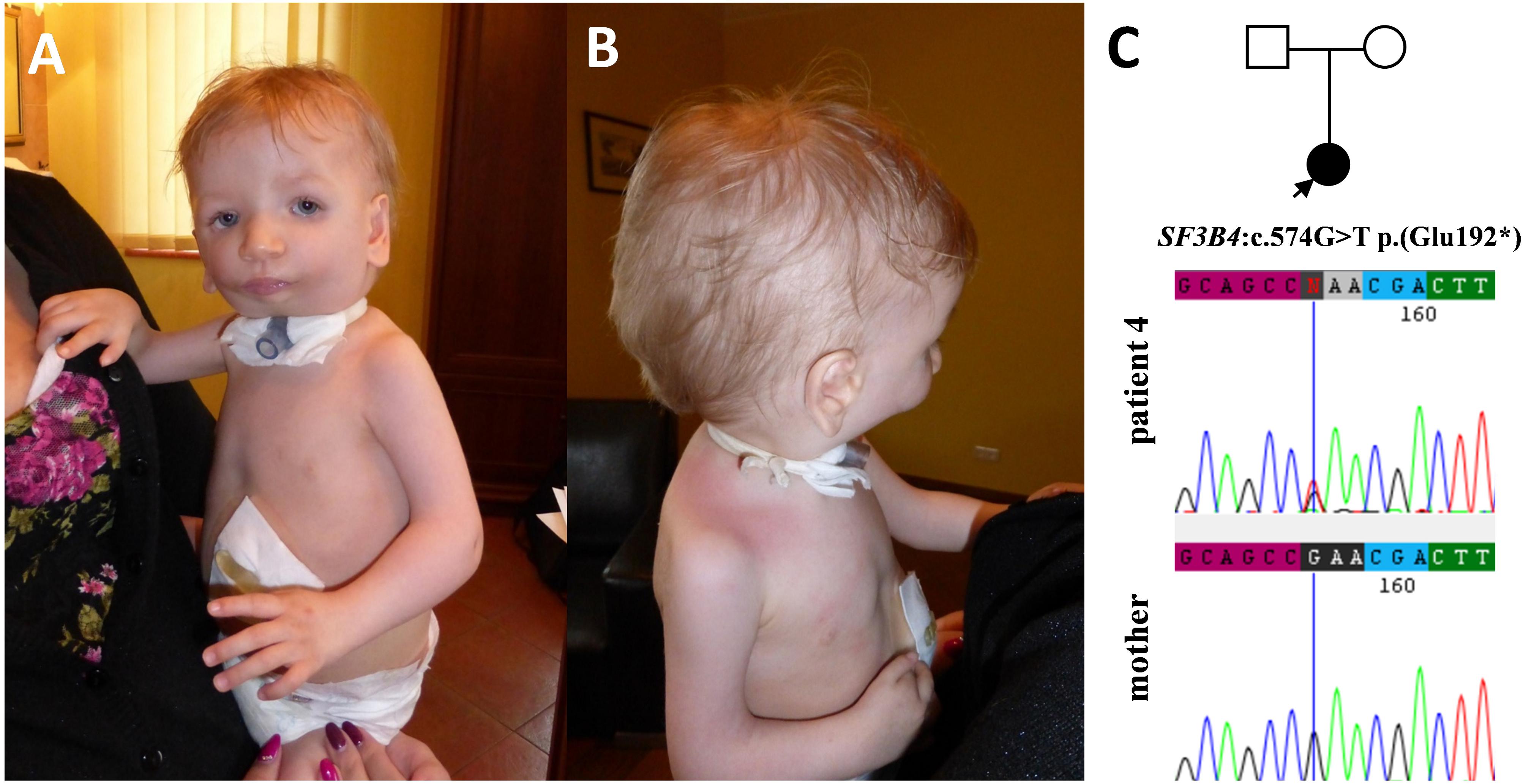
Figure 4. Clinical characteristics of patient 4 affected by Nager syndrome (A,B). Patient 4 carries a heterozygous variant c.574G>T p.(Glu192*) within the SF3B4 gene, which was excluded in her healthy mother (C).
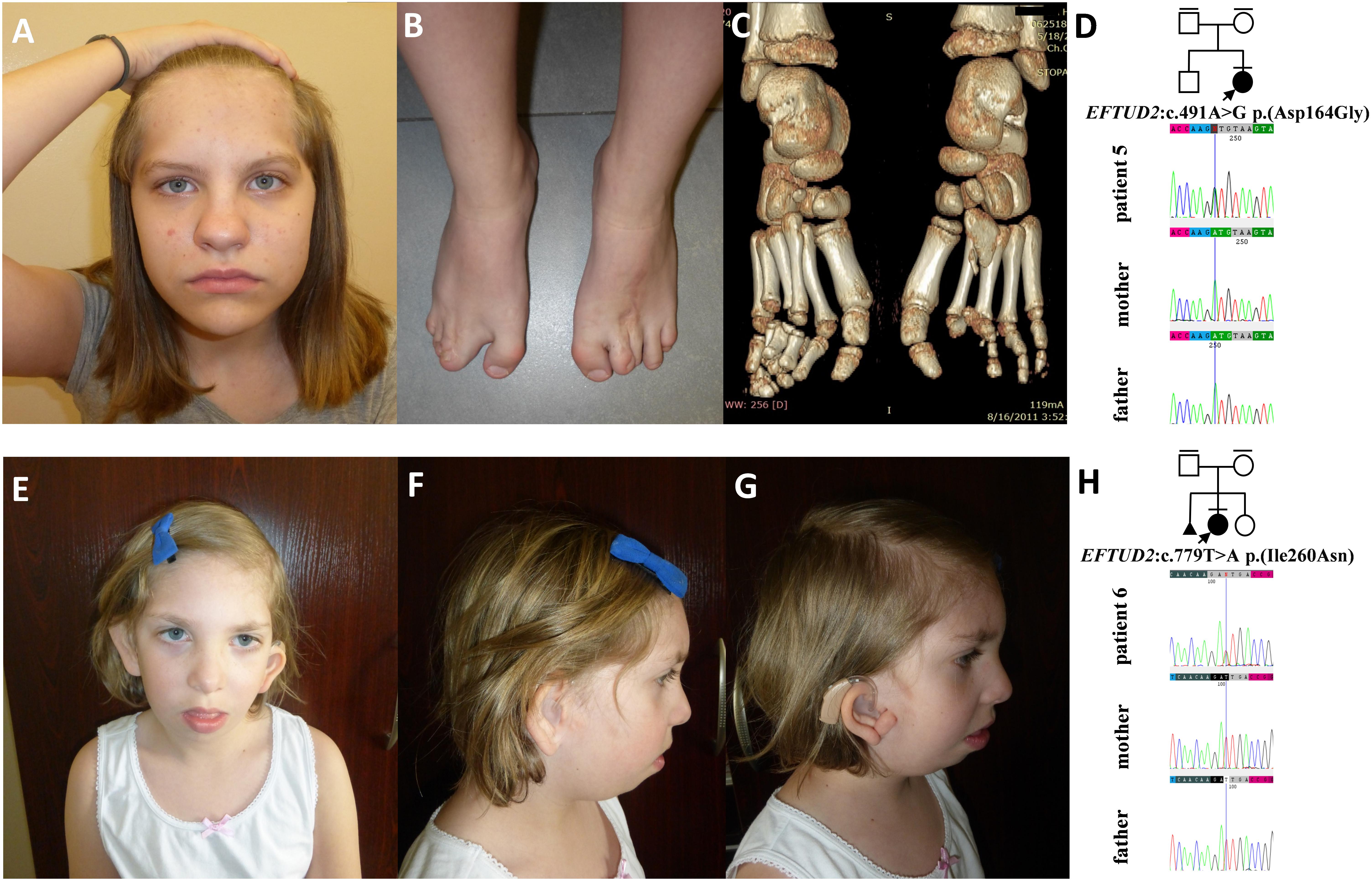
Figure 5. Clinical characteristics and segregation results of patient 5 (A–D) and 6 (E–H) affected by acrofacial dysostosis Guion-Almeida type. Patient 5 carries a heterozygous variant c.491A>G p.(Asp164Gly) within the EFTUD2 gene, which occurred de novo (D) whereas patient 6 carries a heterozygous variant c.779T>A p.(Ile260Asn) within the EFTUD2 gene, which also occurred de novo (H).
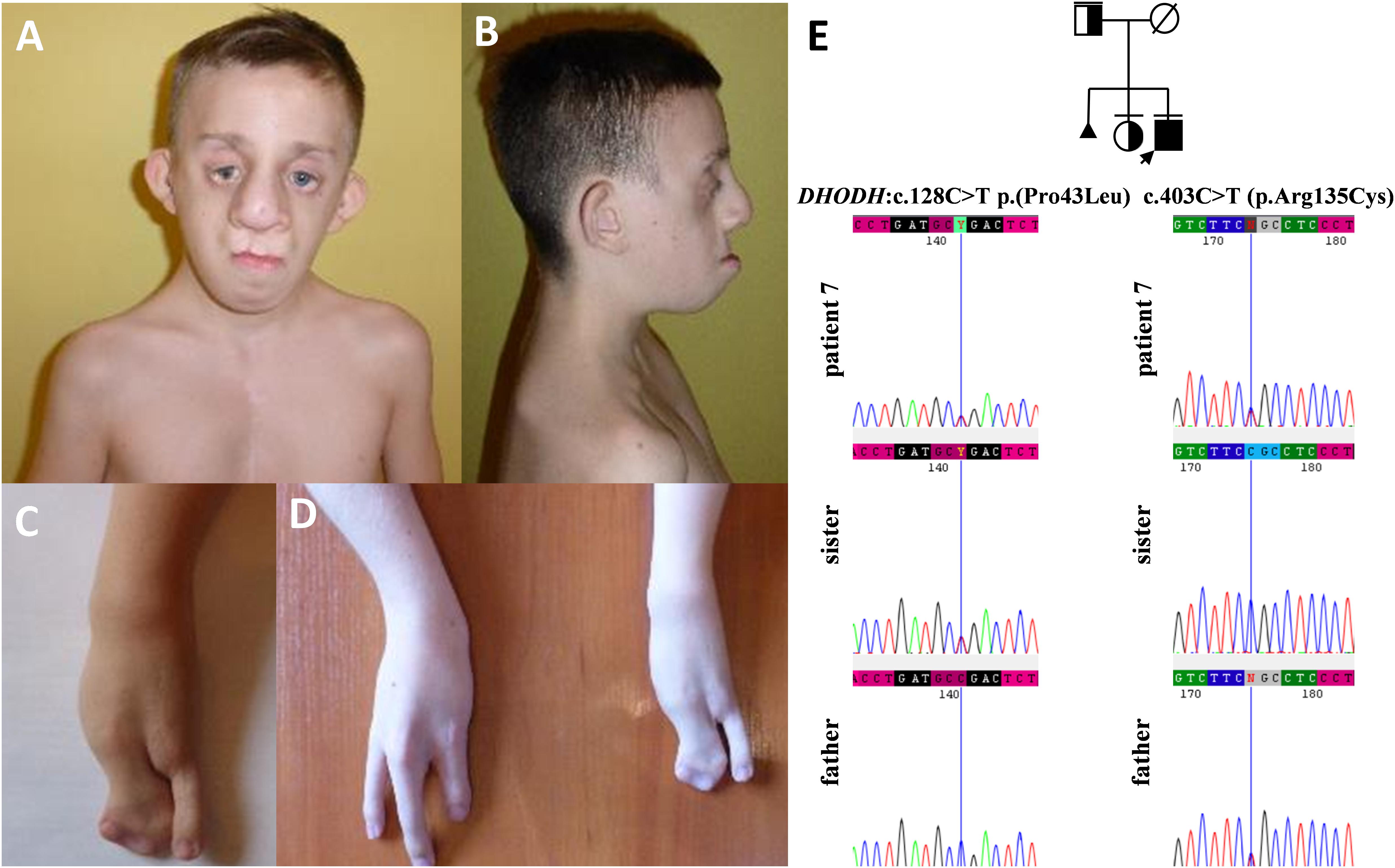
Figure 6. Clinical characteristics (A–D) and segregation results (E) of patient 7 affected by Miller syndrome. Patient 7 carries c.403C>T (p.Arg135Cys) and c.128C>T p.(Pro43Leu) missense variants, both located in trans orientation in the DHODH gene (E).
Before the advent of NGS, identification of the underlying causative genetic lesions in individuals affected by molecularly complex genetic syndromes such as FDs was often difficult or sometimes impossible (Fan et al., 2019). In an excellent review on human facial dysostoses, Wieczorek emphasized that the current classification of FDs bases mostly on the clinical characteristics of the facial features as well as the presence or absence of additional symptoms (Table 1; Wieczorek, 2013). The molecular confirmation of FDs is unquestionably crucial not only for reliable genetic counseling, prenatal and preimplantation testing, but also for a proper and unequivocal clinical classification.
In this paper, we summarized the results of the custom targeted gene panel sequencing in a group of 16 patients from 11 consecutive families affected by distinct forms of FDs that were consulted in our genetic clinic. In this cohort, we detected three novel heterozygous variants within the TCOF1 gene, two of which were frameshifts: c.2145_2148dupAAAG p.(Ser717Lysfs∗42) (family 1; Figure 2), c.4370delA p.(Lys1457Argfs∗118) (family 3; Figure 3F), while one was a missense alteration c.83G>C p.(Arg28Pro) (family 2; Figure 3C). In silico analyses predicted that frameshifts truncating Treacle protein at codons 717 and 1457 were pathogenic, whereas a missense variant p.(Arg28Pro) was initially classified as a VUS (Table 2). The results of segregation analysis performed in family 2 showed that the variant occurred de novo, suggesting its contribution to the patient’s phenotype (Figure 3C). The rest of the diagnosed individuals was affected by various types of AFDs. Among them, we identified one previously reported nonsense heterozygous pathogenic variant in the SF3B4 gene–c.574G>T p.(Glu192∗) (family 4; Figure 4C), two novel missense likely pathogenic heterozygous variants in the EFTUD2 gene, which occurred de novo–c.491A>G p.(Asp164Gly) (family 5; Figure 5D), c.779T>A p.(Ile260Asn) (family 6; Figure 5H), and one previously reported–c.403C>T (p.Arg135Cys) (evaluated as pathogenic), as well as one novel—c.128C>T p.(Pro43Leu) missense variants (evaluated as VUS), both located in trans orientation in the DHODH gene (family 7; Figure 6E).
Pathogenic variants in the TCOF1 but also the POLR1D and POLR1C genes have been previously associated with Treacher Collins syndrome (TCS) types 1–3, respectively (Dixon et al., 1996; Dauwerse et al., 2011; Schaefer et al., 2014). According to the current review of medical literature, in the majority of patients (i.e., 77–89%) causative alterations are found within the TCOF1 gene (encoding Treacle protein). Consistently with these results, we detected all three causative variants in the TCOF1 (families no 1–3), and not in other TCS-related genes (Dauwerse et al., 2011). TCS is characterized by malar hypoplasia, micrognathia, down-slanting palpebral fissures, atresia of the external auditory canals, anomalies of the middle ear ossicles, and bilateral conductive hearing loss (Stovin et al., 1960; Phelps et al., 1981; Trainor et al., 2009; Vincent et al., 2016). Nevertheless, the phenotype often varies not only between, but also within the affected families. We have confirmed that in cases with TCS1 there is no clear genotype-phenotype correlation and that the expressivity of the clinical symptoms shows considerable intra- and interfamilial variability suggesting the influence of additional contributors (Table 3). However, the presence of modifying factors, probably involved in the process of mRNA level regulation, requires further research.
In the vast majority of families affected by AFDs, we have suspected Nager syndrome, which surprisingly was proved genetically only in one individual (patient 4). The families 5 and 7 that were misdiagnosed with Nager syndrome turned out to represent AFDGA (family 6) and Miller syndrome (family 7). Nager syndrome, along with AFDGA and Miller syndrome, share similar facial and skeletal features, hence the unambiguous diagnosis based only on the clinical symptoms may be uncertain (Trainor and Andrews, 2013; Wieczorek, 2013). Variants in the SF3B4 gene that encodes a splicing factor 3B subunit 4 are found in about half of the patients clinically diagnosed with Nager syndrome (Bernier et al., 2012; Czeschik et al., 2013). The main phenotypic features of this condition involve facial characteristics similar to TCS and preaxial limb defects such as hypoplasia or aplasia of the radius and thumbs, radioulnar synostosis or triphalangeal thumbs (Bowen and Harley, 1974; Meyerson et al., 1977; Opitz, 1987; McDonald and Gorski, 1993; Wieczorek, 2013). A nonsense alteration p.(Glu192∗) within splicing factor 3B subunit 4, which occurred in patient 4, was firstly described by Czeschik et al. in a female patient with Nager syndrome, who manifested very similar symptoms to patient 4. Both individuals presented with extreme hypoplasia of the mandible, micrognathia, hypoplastic thumbs, bilateral limited elbow extension, hearing loss and delayed psychomotor development, with absent speech due to severe hypoplasia of the mandibulofacial structures (Czeschik et al., 2013). Additionally, patient 4 had an ectopic left kidney, which was located in the pelvis.
In families no. 5 and 6, we have found novel heterozygous variants in EFTUD2, the gene encoding elongation factor Tu GTP binding domain containing 2, resulting in AFDGA (Guion-Almeida et al., 2006; Lines et al., 2012). This entity was initially classified as mandibulofacial dysostosis Guion-Almeida type (MFDGA). Nevertheless, due to the occurrence of limb anomalies in some patients, it was reclassified to AFDs (Voigt et al., 2013). The clinical spectrum of AFDGA is broad and consists of microcephaly, postnatal-onset growth deficiency, choanal atresia, heart and gastrointestinal anomalies, preaxial polydactyly, slender fingers, and proximally placed thumbs (Guion-Almeida et al., 2006; Wieczorek et al., 2007, 2009; Lines et al., 2012; Gordon et al., 2015). Very recently, new craniofacial phenotypes, i.e., metopic craniosynostosis (CS) and cleft lip and palate, were reported in two individuals carrying heterozygous variants–c.2466+1G>A in intron 24 and c.2333C>A in exon 23 of EFTUD2. These cases were initially suspected of having an alternative craniofacial syndrome and were eventually diagnosed with MFDGA (Lacour et al., 2019). Similarly, patient 5 also presented with very unusual craniofacial features, including metopic synostosis and high forehead that were not suggestive for AFDGA.
Finally, in patient 7, we described compound heterozygous variants in DHODH gene. Biallelic variants in DHODH gene result in defects in pyrimidine biogenesis and give rise to Miller syndrome that has an autosomal recessive inheritance. Limb anomalies include postaxial abnormalities, such as the absence of fifth digits with or without shortening and deviation of forearms with ulnar and radial hypoplasia, and syndactyly (Genee, 1969; Ogilvy-Stuart and Parsons, 1991; Chrzanowska and Fryns, 1993; Ng et al., 2010). The first pathogenic missense variant (p.Arg135Cys) has been reported previously in six individuals affected by Miller syndrome who were compound heterozygous for (p.Arg135Cys) in combination with one of the following alterations–c.155A>G (p.Glu52Gly), c.1036C>T (p.Arg346Trp), and c.1175A>G (p.Asp392Gly). All those patients, including patient 7 (Figures 6A–D), presented with strikingly similar features, encompassing severe micrognathia, cleft lip and/or palate, as well as hypoplasia or aplasia of the postaxial rays of the limbs (Ng et al., 2010; Al Kaissi et al., 2011; Rainger et al., 2012). The second missense variant c.128C>T p.(Pro43Leu) identified by us was novel.
The diagnostic success rate of the presented approach achieved 64%. Undiagnosed cases (families 8–11) may harbor other types of causative alterations that cannot be detected by targeted NGS, such as deep intronic or regulatory mutations affecting genes included in custom gene panels. Alternatively, causative mutations may be located elsewhere in the genome and not in the genes known to be involved in the pathogenesis of FDs. Therefore, in cases without the exact genetic diagnosis, another type of molecular investigation, including whole-genome sequencing (WGS) should be considered. Indeed, the introduction of WGS in the molecular diagnostics of FDs requires advanced in silico and functional analyses, which are both necessary for the interpretation and proving of pathogenicity of genomic alterations (Smedley et al., 2016; Bodea et al., 2018; Posey, 2019). The proportion of molecularly undiagnosed patients, however, clearly suggests that some of the genetic lesions, causative for FDs, lie within non-coding DNA, and could be picked up only by WGS (Kindberg and Bush, 2019).
In conclusion, with this report, we demonstrated the clinical utility of NGS targeted gene panel approach as a valuable and cost-effective first-tier testing in the molecular analysis of patients presenting with FDs. We described the clinical phenotypes of seven families showing distinct forms of FDs and identified six novel and two previously described pathogenic variants. Consequently, we highlighted the need for molecular confirmation of FDs, which is crucial for a reliable genetic counseling, an early prenatal and preimplantation testing, but also a proper and unequivocal clinical classification.
Data generated during this study are included in this published article and its supplementary material. BAMs files were submitted on Sequence Read Archive (SRA) NCBI (SRA submission: PRJNA663125, https://www.ncbi.nlm.nih.gov/bioproject/PRJN A663125.
The studies involving human participants were reviewed and approved by the Bioethics Committee at Poznan University of Medical Sciences (no. 741/17 and 742/17) according to the Good Clinical Practice and Polish law. All patients and their parents agreed to participate in this study. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
AJ, AM-K, and MB-S recruited and clinically diagnosed the patients. EB-O designed the study, performed, analyzed the molecular data, and wrote the manuscript. JW-S participated in data analysis. DP participated in a part of experimental procedures. GK participated in a part of data analysis. AD participated in a part of experimental procedures. AJ designed the study and critically revised and corrected the manuscript. All authors read and approved the final manuscript.
This work was supported by the grant from the Poznan University of Medical Sciences, Poland 502-14-11261860-41259, from the Polish National Science Centre, Poland UMO-2016/23/N/NZ5/02577 to EB-O and UMO-2016/22/E/NZ5/00270 to AJ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to the patients and all family members for participating in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.580477/full#supplementary-material
Al Kaissi, A., Roetzer, K. M., Ulz, P., Heitzer, E., Klaushofer, K., and Grill, F. (2011). Extra phenotypic features in a girl with Miller syndrome. Clin. Dysmorphol. 20, 66–72. doi: 10.1097/MCD.0b013e3283416701
Bernier, F. P., Caluseriu, O., Ng, S., Schwartzentruber, J., Buckingham, K. J., Innes, A. M., et al. (2012). Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am. J. Hum. Genet. 90, 925–933. doi: 10.1016/j.ajhg.2012.04.004
Bodea, C. A., Mitchell, A. A., Bloemendal, A., Day-Williams, A. G., Runz, H., and Sunyaev, S. R. (2018). PINES: phenotype-informed tissue weighting improves prediction of pathogenic noncoding variants. Genome Biol. 19:173. doi: 10.1186/s13059-018-1546-6
Bowen, P., and Harley, F. (1974). Mandibulofacial dysostosis with limb malformations (Nager’s acrofcial dysostosis). Birth Defects Orig. Artic. Ser. 10, 109–115.
Bukowska-Olech, E., Popiel, D., Koczyk, G., Sowiñska-Seidler, A., Socha, M., Wojciechowicz, B., et al. (2020). Adapting SureSelect enrichment protocol to the Ion Torrent S5 platform in molecular diagnostics of craniosynostosis. Sci. Rep. 10:4159. doi: 10.1038/s41598-020-61048-5
Chrzanowska, K., and Fryns, J. P. (1993). Miller postaxial acrofacial dysostosis. The phenotypic changes with age. Genet. Couns. 4, 131–133.
Czeschik, J. C., Voigt, C., Alanay, Y., Albrecht, B., Avci, S., Fitzpatrick, D., et al. (2013). Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Hum. Genet. 132, 885–898. doi: 10.1007/s00439-013-1295-2
Dauwerse, J. G., Dixon, J., Seland, S., Ruivenkamp, C. A. L., van Haeringen, A., Hoefsloot, L. H., et al. (2011). Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat. Genet. 43, 20–22. doi: 10.1038/ng.724
den Dunnen, J. T., Dalgleish, R., Maglott, D. R., Hart, R. K., Greenblatt, M. S., McGowan-Jordan, J., et al. (2016). HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 37, 564–569. doi: 10.1002/humu.22981
Dixon, J., Edvards, S. J., Gladwin, A. J., Dixon, M. J., Loftus, S. K., and Bonner, C. A. (1996). Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. The Treacher Collins Syndrome Collaborative Group. Nat. Genet 12, 130–136. doi: 10.1038/ng0296-130
Dixon, J., Ellis, I., Bottani, A., Temple, K., and Dixon, M. J. (2004). Identification of mutations inTCOF1: Use of molecular analysis in the pre- and postnatal diagnosis of Treacher Collins syndrome. Am. J. Med. Genet. 127A, 244–248. doi: 10.1002/ajmg.a.30010
Fan, X., Wang, Y., Fan, Y., Du, H., Luo, N., Zhang, S., et al. (2019). TCOF1 pathogenic variants identified by Whole-exome sequencing in Chinese Treacher Collins syndrome families and hearing rehabilitation effect. Orphanet J. Rare Dis. 14, 178. doi: 10.1186/s13023-019-1136-z
Gordon, C. T., Weaver, K. N., Zechi-ceide, R. M., Madsen, E. C., Tavares, A. L. P., Oufadem, M., et al. (2015). Mutations in the Endothelin Receptor Type A Cause Mandibulofacial Dysostosis with Alopecia. Am. J. Hum. Genet 96, 519–531. doi: 10.1016/j.ajhg.2015.01.015
Guion-Almeida, M. L., Zechi-Ceide, R. M., Vendramini, S., and Tabith Junior, A. (2006). A new syndrome with growth and mental retardation, mandibulofacial dysostosis, microcephaly, and cleft palate. Clin. Dysmorphol. 15, 171–174. doi: 10.1097/01.mcd.0000220603.09661.7e
Jin, S.-W., Sim, K.-B., and Kim, S.-D. (2016). Development and Growth of the Normal Cranial Vault: An Embryologic Review. J. Korean Neurosurg. Soc. 59, 192–196. doi: 10.3340/jkns.2016.59.3.192
Kindberg, A. A., and Bush, J. O. (2019). Cellular organization and boundary formation in craniofacial development. Genesis 57:e23271. doi: 10.1002/dvg.23271
Lacour, J. C., McBride, L., St Hilaire, H., Mundinger, G. S., Moses, M., Koon, J., et al. (2019). Novel De Novo EFTUD2 Mutations in 2 Cases With MFDM, Initially Suspected to Have Alternative Craniofacial Diagnoses. Cleft Palate. Craniofac. J. 56, 674–678. doi: 10.1177/1055665618806379
Lines, M. A., Huang, L., Schwartzentruber, J., Douglas, S. L., Lynch, D. C., Beaulieu, C., et al. (2012). Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am. J. Hum. Genet. 90, 369–377. doi: 10.1016/j.ajhg.2011.12.023
McDonald, M. T., and Gorski, J. L. (1993). Nager acrofacial dysostosis. J. Med. Genet. 30, 779–782. doi: 10.1136/jmg.30.9.779
Meyerson, M. D., Jensen, K. M., Meyers, J. M., and Hall, B. D. (1977). Nager acrofacial dysostosis: early intervention and long-term planning. Cleft Palate J. 14, 35–40.
Ng, S. B., Buckingham, K. J., Lee, C., Bigham, A. W., Tabor, H. K., Dent, K. M., et al. (2010). Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 42, 30–35. doi: 10.1038/ng.499
Ogilvy-Stuart, A. L., and Parsons, A. C. (1991). Miller syndrome (postaxial acrofacial dysostosis): further evidence for autosomal recessive inheritance and expansion of the phenotype. J. Med. Genet. 28, 695–700. doi: 10.1136/jmg.28.10.695
Opitz, J. M. (1987). Nager “syndrome” versus “anomaly” and its nosology with the postaxial acrofacial dysostosis syndrome of Genee and Wiedemann. Am. J. Med. Genet. 27, 959–963. doi: 10.1002/ajmg.1320270425
Phelps, P. D., Poswillo, D., and Lloyd, G. A. (1981). The ear deformities in mandibulofacial dysostosis (Treacher Collins syndrome). Clin. Otolaryngol. Allied Sci. 6, 15–28.
Posey, J. E. (2019). Genome sequencing and implications for rare disorders. Orphanet J. Rare Dis. 14:153. doi: 10.1186/s13023-019-1127-0
Rainger, J., Bengani, H., Campbell, L., Anderson, E., Sokhi, K., Lam, W., et al. (2012). Miller (genéeacute;wiedemann) syndrome represents a clinically and biochemically distinct subgroup of postaxial acrofacial dysostosis associated with partial deficiency of DHODH. Hum. Mol. Genet. 21, 3969–3983. doi: 10.1093/hmg/dds218
Schaefer, E., Collet, C., Genevieve, D., Vincent, M., Lohmann, D. R., Sanchez, E., et al. (2014). Autosomal recessive POLR1D mutation with decrease of TCOF1 mRNA is responsible for Treacher Collins syndrome. Genet. Med. 16, 720–724. doi: 10.1038/gim.2014.12
Smedley, D., Schubach, M., Jacobsen, J. O. B., Kohler, S., Zemojtel, T., Spielmann, M., et al. (2016). A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am. J. Hum. Genet. 99, 595–606. doi: 10.1016/j.ajhg.2016.07.005
Stovin, J. J., Lyon, J. A. Jr., and Raymond, L. C. (1960). Mandibulofacial dysostosis. Radiology 74, 225–231.
Trainor, P. A., and Andrews, B. T. (2013). Facial dysostoses: Etiology, pathogenesis and management. Am. J. Med. Genet. C. Semin. Med. Genet. 163C, 283–294. doi: 10.1002/ajmg.c.31375
Trainor, P. A., Dixon, J., and Dixon, M. J. (2009). Treacher Collins syndrome: Etiology, pathogenesis and prevention. Eur. J. Hum. Genet. 17, 275–283. doi: 10.1038/ejhg.2008.221
Vincent, M., Genevieve, D., Ostertag, A., Marlin, S., Lacombe, D., Martin-Coignard, D., et al. (2016). Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genet. Med. 18, 49–56. doi: 10.1038/gim.2015.29
Voigt, C., Czeschik, J. C., Albrecht, B., Kuechler, A., Lüdecke, H.-J., Wieczorek, D., et al. (2013). Oto-facial syndrome and esophageal atresia, intellectual disability and zygomatic anomalies - Expanding the phenotypes associated with EFTUD2 mutations. Orphanet J. Rare Dis. 8, 1–12. doi: 10.1186/1750-1172-8-110
Wieczorek, D., Gener, B., Gonzalez, M. J. M., Seland, S., Fischer, S., Hehr, U., et al. (2009). Microcephaly, microtia, preauricular tags, choanal atresia and developmental delay in three unrelated patients: a mandibulofacial dysostosis distinct from Treacher Collins syndrome. Am. J. Med. Genet. A 149A, 837–843. doi: 10.1002/ajmg.a.32747
Wieczorek, D., Shaw-Smith, C., Kohlhase, J., Schmitt, W., Buiting, K., Coffey, A., et al. (2007). Esophageal atresia, hypoplasia of zygomatic complex, microcephaly, cup-shaped ears, congenital heart defect, and mental retardation–new MCA/MR syndrome in two affected sibs and a mildly affected mother? Am. J. Med. Genet. A 143A, 1135–1142. doi: 10.1002/ajmg.a.31752
Keywords: facial dysostoses, mandibulofacial dysostoses, acrofacial dysostoses, pharyngeal arch, rare diseases, craniofacial development, targeted next-generation sequencing
Citation: Bukowska-Olech E, Materna-Kiryluk A, Walczak-Sztulpa J, Popiel D, Badura-Stronka M, Koczyk G, Dawidziuk A and Jamsheer A (2020) Targeted Next-Generation Sequencing in the Diagnosis of Facial Dysostoses. Front. Genet. 11:580477. doi: 10.3389/fgene.2020.580477
Received: 06 July 2020; Accepted: 17 September 2020;
Published: 11 November 2020.
Edited by:
Jessica Duis, Children’s Hospital Colorado, United StatesReviewed by:
Aline L. Petrin, The University of Iowa, United StatesCopyright © 2020 Bukowska-Olech, Materna-Kiryluk, Walczak-Sztulpa, Popiel, Badura-Stronka, Koczyk, Dawidziuk and Jamsheer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleksander Jamsheer, amFtc2hlZXJAd3AucGw=
†ORCID: Aleksander Jamsheer, orcid.org/0000-0003-4058-3901; Ewelina Bukowska-Olech, orcid.org/0000-0003-0509-1696; Anna Materna-Kiryluk, orcid.org/0000-0002-2889-5892; Joanna Walczak-Sztulpa, orcid.org/0000-0002-4662-9336; Grzegorz Koczyk, orcid.org/0000-0002-5414-4689; Magdalena Badura-Stronka, orcid.org/0000-0002-2193-3657
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.