 Vinicius A. C. de Abreu
Vinicius A. C. de Abreu José Perdigão
José Perdigão Sintia Almeida
Sintia Almeida
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet. , 18 January 2021
Sec. Computational Genomics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.575592
This article is part of the Research Topic Computational Genomics Approaches Against Antibacterial Drug Resistance View all 8 articles
Antimicrobial resistance is a major global public health problem, which develops when pathogens acquire antimicrobial resistance genes (ARGs), primarily through genetic recombination between commensal and pathogenic microbes. The resistome is a collection of all ARGs. In microorganisms, the primary method of ARG acquisition is horizontal gene transfer (HGT). Thus, understanding and identifying HGTs, can provide insight into the mechanisms of antimicrobial resistance transmission and dissemination. The use of high-throughput sequencing technologies has made the analysis of ARG sequences feasible and accessible. In particular, the metagenomic approach has facilitated the identification of community-based antimicrobial resistance. This approach is useful, as it allows access to the genomic data in an environmental sample without the need to isolate and culture microorganisms prior to analysis. Here, we aimed to reflect on the challenges of analyzing metagenomic data in the three main approaches for studying antimicrobial resistance: (i) analysis of microbial diversity, (ii) functional gene analysis, and (iii) searching the most complete and pertinent resistome databases.
Bacterial resistance, which is closely associated with the use of antimicrobial agents, is considered one of the most persistent global public health problems (Enne and Bennett, 2010; Giedraitienė et al., 2011). However, it is not a new phenomenon. Resistance to penicillin developed in the 1940s, immediately after the large-scale use of the antibiotic. Healthcare was the first field to face challenges created by the indiscriminate use of antibiotics. However, medicine is not alone, and the fields of agriculture, livestock farming, and aquaculture are also being affected by the increasing, continued use of antibiotics, which drives the selection of resistant bacterial populations in environments and contributes to antimicrobial resistance (Barbosa and Levy, 2000; Van Boeckel et al., 2015; von Wintersdorff et al., 2016).
Antimicrobial resistance (Table 1) develops when pathogens acquire antimicrobial resistance genes (ARGs). The acquisition of ARGs primarily occurs through genetic recombination between commensal and pathogenic microbes and is associated with the conjugation mechanism of horizontal gene transfer (HGT) (Brown and Wright, 2016; Munita and Arias, 2016). Resistance is a mechanism naturally used by bacteria, whether induced or not induced. However, the large-scale use of antibiotics drives the rapid development of highly antimicrobial-resistant strains. Antibiotic resistance spreads through genetic material exchange, primarily between bacteria of the same genus, and, at a minor frequency, between phyla (von Wintersdorff et al., 2016; Wybouw et al., 2016), resulting in the development of potentially harmful bacteria.
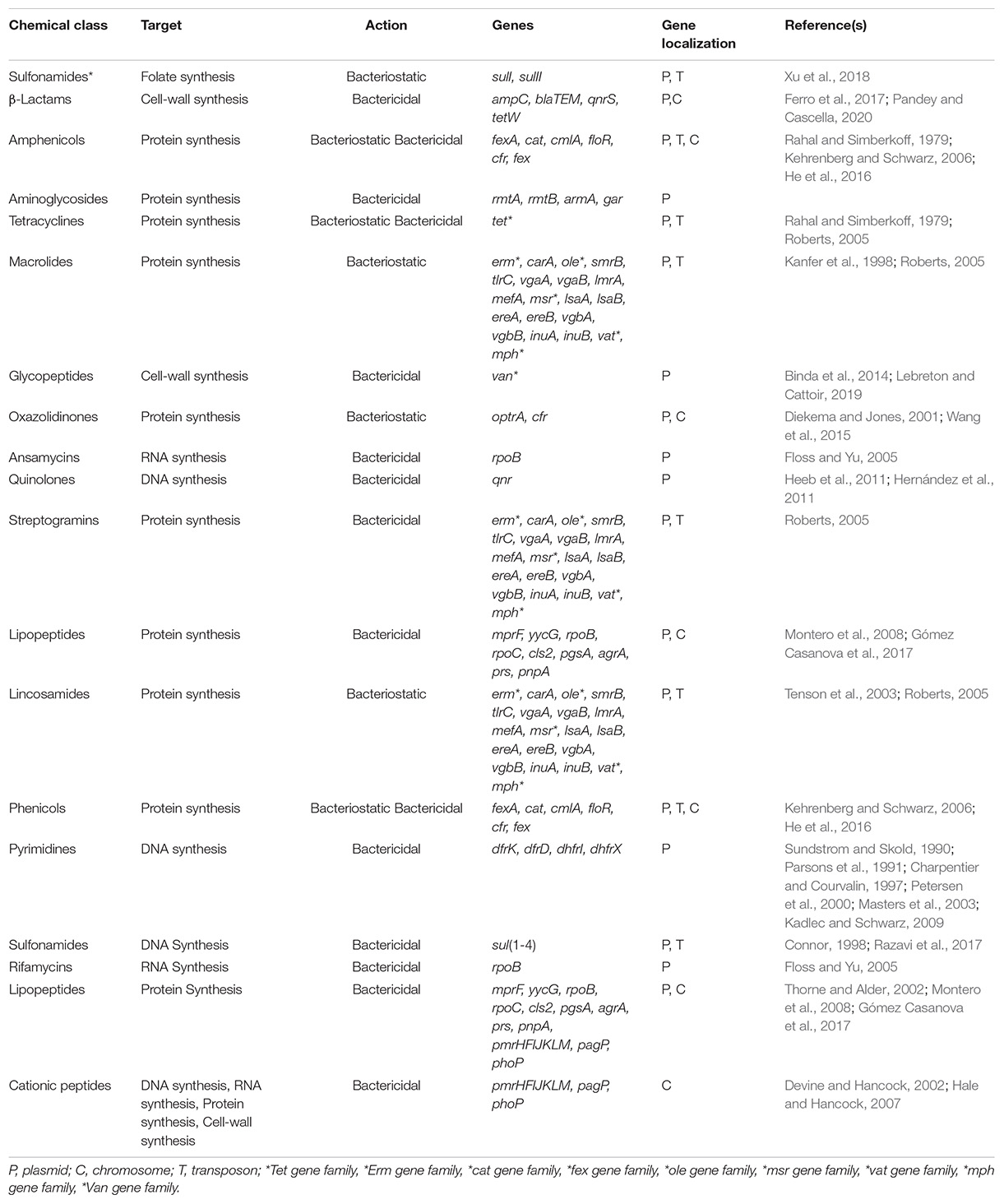
Table 1. Resistance mechanisms, antibiotic resistance genes, and gene localization.
Although numerous recent and ongoing research efforts have addressed bacterial virulence and multi-resistance mechanisms, the processes governing bacterial fitness, competition, dissemination, and adaptability remain poorly understood. Little is known about the diversity, distribution, and origin of resistance genes, especially those of most environmental bacteria that cannot be cultured under laboratory conditions (Schmieder and Edwards, 2012). The development, acquisition, and dissemination of ARGs are critical aspects of antimicrobial resistance, and the microbial community as a whole contributes to the generation of the antimicrobial resistome, rather than an individual ARG source organism (Bello-López et al., 2019; De, 2019). Therefore, understanding and identifying HGTs among pathogenic and non-pathogenic species may aid the determination of the mechanisms underlying resistance transmission and dissemination. The use of high-throughput sequencing technologies has made ARG sequence analyses feasible and accessible. Metagenomics, in particular, has facilitated the analysis of antimicrobial resistance in communities.
The term metagenomics, first used by Handelsman et al. (1998), originates from conventional microbial genomics and reflects the fact that pure cultures are not required for sequencing. The metagenomics approach is used to analyze the genomic data of environmental samples without the need to first isolate and culture microorganisms (Roh and Villatte, 2008; Cowan et al., 2015). Metagenomic analysis enables the prediction of new taxa (phyla, orders, genera, and candidate species) and genome reconstruction of organisms that cannot be cultured in vitro. The definition of community structures allows a deeper understanding of the relationships between individual components of a community and their dynamics in response to the selective pressure of a space-time parameter (Alves et al., 2018). Therefore, the metagenomic analysis of taxonomic (structural) assignment facilitates better identification of microbial communities, the discovery of new microbial metabolic capacities, and the inference of microbial functions in microbiomes where they inhabit (Simmons et al., 2014; Eloe-Fadrosh et al., 2016). Thus, sequence-based functional metagenomics is a powerful tool, widely used to discover resistance genes and identify and understand resistance mechanisms (Pehrsson et al., 2013; Xing et al., 2020). The robust structural and functional aspects of metagenomic data aid the study of antibacterial resistance.
A series of pipelines and reviews have focused on describing the best platforms for metagenomic statistical analyses and benchmarking metrics (Bengtsson-Palme et al., 2017; Quince et al., 2017; Boolchandani et al., 2019; Tamames and Puente-Sánchez, 2019; Ye et al., 2019), but this is not our goal. In this review, we have focused on the three main approaches used for metagenomic analysis of antimicrobial resistance: (i) analysis of microbial diversity, (ii) functional gene analysis, and (iii) searching the most complete and relevant resistome databases available. We will also comment on the challenges related to analyzing metagenomic data.
For several years, pathogenic bacteria have been the focus of antibiotic resistance research. This line of research has facilitated the identification of critical mechanisms that mediate bacterial antibiotic resistance. Among the mechanisms of antibiotic resistance, the four most important are (McManus, 1997; Munita and Arias, 2016): (i) enzymatic modification or destruction of the antibiotic, which usually involves the overproduction of enzymes that inactivate the antibiotic (e.g., β-lactamases and aminoglycosides kinases), (ii) alteration of the antibiotic target molecule to reduce its binding capacity, (iii) modification of metabolic pathways and regulatory networks to circumvent the effect of the antibiotic, and (iv) reduction of the intracellular accumulation of the antibiotic by decreasing cellular permeability to it or activating efflux mechanisms to export the harmful molecule.
However, an increasing number of resistance studies have provided new insight into microbial pathogenicity by analyzing the ARGs of both pathogenic and non-pathogenic bacteria (Beceiro et al., 2013; Roberts, 2017). This work raised interest in the genomes of non-pathogenic organisms based on the knowledge that comparative genomic analysis might aid the elucidation of gene associations relevant to antimicrobial resistance and indicate the presence or absence of ARGs. Mass sequencing and complete genome analysis have contributed to important advances in our understanding of bacterial resistance, genes that confer this resistance, and other phenotypes of interest. Moreover, data obtained from genomic analyses have revealed the remarkable genetic plasticity of bacteria, which enables them to respond to a wide variety of threats, including antibiotics. However, to understand the functioning of sets of genes that can acquire antibiotic resistance in resistomes, metagenomic methods are increasingly being used (Ghosh et al., 2013; Costa et al., 2015; Wang et al., 2020; Zhao et al., 2020). Metagenomic approaches can be function- or sequence-based (Schloss and Handelsman, 2003). In sequence-based methods, multiple sequence reads are generated and analyzed using sequence analysis software.
The most comprehensive approach for metagenome sequencing is complete genome sequencing; this approach allows the study of the structural and functional diversities of a microbial community by identifying genes and metabolic pathways and reconstructing almost complete bacterial genomes (Chen and Pachter, 2005; De, 2019). The main advantage of this approach is its sensitivity, as it allows the detection of a greater abundance of species and identification of potential ARGs.
Complete metagenomic sequencing, since it was implemented, has had a tremendous impact on the study of structural and functional microbial diversities in environmental and clinical samples and has been an alternative to rRNA sequencing (Escobar-Zepeda et al., 2018). Alternatively, functional metagenomics employ different approaches to study genes of interest, including gene cloning and sequencing and biochemical analysis (Ngara and Zhang, 2018; Tamames et al., 2019). Functional metagenomics are mostly used for the identification of resistance genes.
However, some challenges affect the quality of metagenomic analysis, with the first being low sensitivity in detecting minority populations that harbor resistance genes, which has proved to be an obstacle at the time of analysis (Lynch and Neufeld, 2015). The second is the low specificity in identifying allelic variants, which can have substantial impact, as different variants can impart different phenotypic susceptibilities (Forslund et al., 2013). To overcome these challenges, metagenomic analyses must employ both sequence- and function-based approaches, including functional gene annotation (Chistoserdovai, 2010; Lam et al., 2015) in the analysis pipeline, and heterologous expression of identified genes (Tripathi and Nailwal, 2020).
Horizontal gene transfer is a common method of genetic transfer between species of the same genus or with similar characteristics (Soucy et al., 2015). Thus, studying taxonomic assignments of resistome elements is fundamental for identifying bacteria that shape a resistome. Indeed, the microbial community composition or relative abundance of sampled organisms can be inferred through the taxonomic assignment analysis of resistome elements (Ruppé et al., 2019; Rice et al., 2020). Identifying the bacterial community composition can be accomplished via two distinct approaches: (i) direct measurement of raw data, which does not require the assembly of contigs and (ii) the assembly of contigs for subsequent composition inference. Both strategies have weaknesses and strengths (Mathe, 2002).
Taxonomic classification without the assembly of contigs is a faster approach, with a lower computational cost and no assembly problems (Rodríguez-Brazzarola et al., 2018). However, the quality and length of sequences are important during taxonomic assignment analysis, and poor-quality or short sequences, which are common in the non-assembly based approach, tend to generate matches with low statistical significance (Breitwieser et al., 2019; Ye et al., 2019).
Contrarily, the length of contigs is an advantage for taxonomic classification using contig assembly. Thus, this approach predominantly makes use of databases (Rodríguez-Brazzarola et al., 2018). Moreover, in some cases, contig assembly may enable partial genome reconstruction of a previously unknown organism. However, chimeric contig formation is possible owing to sample heterogeneity, which can be related to sample origin, and sample and sequence quality. All these features are closely linked to assembly quality, which influences classification quality.
In ARG analyses, genome assembly can help differentiate between bacteria in terms of conserved regions like ribosomes, possible HGT regions, and several classes of transposable elements. This is because the reduced size of gene sequences directly impacts gene annotation transfer and studies of biological mechanisms associated with resistance. Thus, taxonomic assignment by contig assembly tends to better facilitate the identification and understanding of resistance mechanisms, such as the understanding of microbiota structural relationship roles in resistome studies. However, it is important to emphasize that researchers must be aware of the type of sample being worked with, if the sample is too heterogeneous and if there is sufficient computational power to analyze the amount of data collected. Even for good-quality, long sequences, taxonomic classification without assembly could be a more appropriate approach from a computational point of view, depending on the dataset and the computational power available (Rodríguez-Brazzarola et al., 2018).
Notably, studying the taxonomic assignments of resistome elements using high-throughput sequencing goes beyond identifying ARGs in host-pathogen relations and can be used to study resistomes in environmental samples, such as those from water reservoirs (Ekwanzala et al., 2020; Yu et al., 2020), hospitals (MetaSUB Consortium et al., 2020), livestock wastewater and feces (Jia S. et al., 2017), human feces (Karkman et al., 2019), soil (Chen et al., 2017), air (Yang et al., 2018; Li et al., 2021), and biogeographical and biogeochemical processes (Quinn et al., 2014; Kuang et al., 2016; Roose-Amsaleg and Laverman, 2016; Liu et al., 2018).
Studying taxonomic signatures enables a better understanding of the relationships between the members of a microbial community. Alternatively, functional metagenomic approach aims to identify functions within the community via the discovery of new enzymes, groups of biosynthetic genes, and ARGs. The functional annotation of a metagenome is similar to its genomic annotation, such that predicted gene sequences are compared to existing sequences in annotated databases (Dong and Strous, 2019). Thus, the high-throughput sequencing of microbial community genomes is a powerful tool to generate information about gene functions, metabolic pathways, and microbial genome evolution (Zhang et al., 2011).
There is a wide range of databases and tools to classify the taxonomic profile of a community and performing functional analyses; thus, the choice of reference database can have important implications for the quality of information obtained. There are three important points regarding sequence- and function-based analyses. First, functional analysis provides an opportunity to perform various sub-analyses, depending on the sequencing depth, including functional category, protein family, gene ontology, protein–protein interaction, pathway, and subsystem analysis. Second, for both types of analysis methods, researchers can work with assembled or non-assembled data. Finally, there are tools, usually open source tools, such as QIIME (Caporaso et al., 2010), Mothur (Schloss et al., 2009), and MEGAN (Huson et al., 2007), that perform both types of analysis.
Genomic annotation employs sequence comparison with similarity-based search tools, such as BLAST+, which was developed by the National Center for Biotechnology Information (NCBI) (Altschul et al., 1997). DIAMOND (Buchfink et al., 2015) performs pairwise sequence alignment for protein and translated DNA searches, which are designed for the high performance analysis of large sequence data; it has the advantage of being fast and is, therefore, attractive for the annotation of huge volumes of metagenomic data. USEARCH (Edgar, 2010) offers search and grouping algorithms that are faster than BLAST. RAPSearch2 (Zhao et al., 2012) is similar to BLAST, in that it uses flexible-length seeds on a reduced amino acid alphabet of ten symbols with the differential. Tools, such as BLAST, offer their own dataset (NR and RefSeq are most used), whereas others offer only alignment options, requiring the use of a third-party dataset (nr/nt, RefSeq, Env_NR, and UniProt). In both cases, it is necessary to download datasets separately or create one’s own local dataset. These tools use their databases for annotation or allow the user to employ a third-party database.
Although there are good database options and tools for comprehensive metagenomic analyses, continuous improvement for the detection and characterization of genetic elements is necessary, as it is important for understanding resistance acquisition over time and evolutionary dynamics. Thus, resistome databases must be constantly updated to include newly identified variant sequences, inserts, and deletions to improve our understanding of these variations in context of resistance (Danko et al., 2019). Moreover, the use of a non-specific or generalist database could generate inherent database bias for the target niche or organism. The choice of an appropriate database for sequence annotation is essential. This choice should be based on the type of data and ecosystem studied. We have highlighted below, the most frequently cited specialist databases for ARGs that allow metagenomic data input (Table 2), including ResFinder, Comprehensive Antibiotic Resistance Database (CARD), MEGARes, ARG-database, and Resfams.
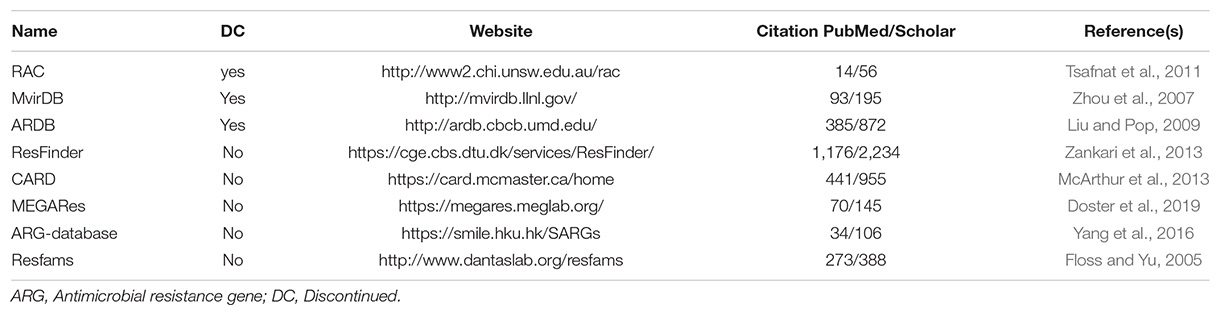
Table 2. Bioinformatic resources for studying ARGs identified using targeted metagenomics.
ResFinder is one of the oldest databases that keeps its sequences up to date. It extracts information from other databases, such as the Lahey1 database and ARDB (both now defunct). ResFinder also sources information from published literature, including reviews (Zankari et al., 2013). It uses the BLAST algorithm to assess sequence similarity. Fully- or draft-assembled sequences from different platforms, genomes or metagenomes, and long or short reads can be used as inputs for ResFinder.
Comprehensive Antibiotic Resistance Database is based on the core components of antimicrobial resistance, including genes and proteins, and utilizes published literature and controlled terminology to robustly investigate data. It is the most commonly used database in metagenomic projects. In addition to having a curated database (Jia B. et al., 2017), it includes resistome data that were computationally predicted in continuation of the ARDB project, which is now defunct.
MEGARes, a database of approximately 8,000 manually curated resistance genes with hierarchical statistical analysis, was published in 2016 and updated in 2019 (Doster et al., 2019). It relies on a specific Galaxy pipeline, although it offers the alternative option of downloading the entire database for integration with custom pipelines. The MEGARes dataset comprises several sources, including the curated CARD database (Doster et al., 2019).
Antimicrobial resistance gene-database is hierarchically structured (ARG type-subtype-reference sequence). Its first version integrated ARGs from ARDB and CARD, and redundant sequences were removed. When it was updated in 2018 (Yin et al., 2018), proteins from the NCBI-NR database were added, thereby tripling the number of sequences in the first version. Based on a specific Galaxy pipeline, the latest version also offers the option to download the database, allowing the integration of the available data with a custom analysis pipeline.
Resfams is organized by ontology with a curated database of protein families and associated profile hidden Markov models (HMMs) and protein sequences from the CARD database, the Lactamase Engineering Database, and Jacoby and Bush’s collection of curated beta-lactamase proteins. It was designed to quantitatively understand the relationship between human and environmental resistomes, with an analysis of over 6000 microbial genomes. It was last updated in 2018 (Gibson et al., 2015).
Although the databases fully complement one another and are often redundant, they continue to be cited as having individual specificities for particular datasets, which hinders recommendations. Given the importance of studies on microbial resistance and the quality of data obtained, it is essential that a platform-independent dataset be available for the antibiotic resistance research community. In one sequence database (DNA/Protein/raw data sequences), INSDC (International Nucleotide Sequence Database), initiatives for the unification and integration have already been implemented. INSDC is a standardization and unification initiative among the main sequence databases (DDBJ, EMBL-EBI, and NCBI), making the data of these databases effectively interchangeable (Karsch-Mizrachi et al., 2018). This type of integration initiative eliminates developer and researcher concerns regarding the “best” dataset for a sample and focuses on the importance and applicability of the analyses and outputs.
Metagenomics is a promising tool for identifying and understanding antibiotic resistance mechanisms, using sequence- and function-based approaches. Notably, however, various analyses of antimicrobial resistance are strongly related to other aspects of the research being carried out, such as mutations, pathogens, metabolic pathways, and gene expression. Reviews analyzing antimicrobial resistance addressing these aspects are strongly recommended.
The most important considerations in a metagenomic resistome study are understanding the nature of the dataset being analyzed and the support that is available for its analysis. If one takes into account the large quantity of data and the complexity of the biological mechanisms involved in antibiotic resistance, it may be preferable to adopt reductionist approaches to decrease bias and increase the objectivity of analyses. It is important to emphasize that the costs of algorithms, computers, and analytical tools are decreasing; in silico predictions based on machine learning are thus becoming more common and have the potential to predict resistance outside databases. This will allow for the development of high-throughput data analysis approaches and the answering more complex questions regarding antimicrobial resistance.
SA and VA wrote the manuscript, as well as guided and reviewed the work. JP revised the writing and formulated the tables. All authors contributed to the article and approved the submitted version.
This work was supported by the Pró-Reitoria de Pesquisa da Universidade Federal do Para – PROPESP/UFPA. JP received grant-aided support by the Brazilian Federal Agency for the scientific research fellowship from FAPESPA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank the Paraense Amazon Foundation for Research Support (FAPESPA) and Pós-graduação em Ciência da Informação for the intermediation of financial support.
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
Alves, L. de F., Westmann, C. A., Lovate, G. L., de Siqueira, G. M. V., Borelli, T. C., and Guazzaroni, M.-E. (2018). Metagenomic Approaches for Understanding New Concepts in Microbial Science. Int. J. Genom. 2018:e2312987. doi: 10.1155/2018/2312987
Barbosa, T. M., and Levy, S. B. (2000). The impact of antibiotic use on resistance development and persistence. Drug Resist. Updates 3, 303–311. doi: 10.1054/drup.2000.0167
Beceiro, A., Tomas, M., and Bou, G. (2013). Antimicrobial Resistance and Virulence: a Successful or Deleterious Association in the Bacterial World? Clin. Microbiol. Rev. 26, 185–230. doi: 10.1128/CMR.00059-12
Bello-López, J. M., Cabrero-Martínez, O. A., Ibáñez-Cervantes, G., Hernández-Cortez, C., Pelcastre-Rodríguez, L. I., Gonzalez-Avila, L. U., et al. (2019). Horizontal Gene Transfer and Its Association with Antibiotic Resistance in the Genus Aeromonas spp. Microorganisms 7:363. doi: 10.3390/microorganisms7090363
Bengtsson-Palme, J., Larsson, D. G. J., and Kristiansson, E. (2017). Using metagenomics to investigate human and environmental resistomes. J. Antimicrob. Chemother. 72, 2690–2703. doi: 10.1093/jac/dkx199
Binda, E., Marinelli, F., and Marcone, G. (2014). Old and New Glycopeptide Antibiotics: Action and Resistance. Antibiotics 3, 572–594. doi: 10.3390/antibiotics3040572
Boolchandani, M., D’Souza, A. W., and Dantas, G. (2019). Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet. 20, 356–370. doi: 10.1038/s41576-019-0108-4
Breitwieser, F. P., Lu, J., and Salzberg, S. L. (2019). A review of methods and databases for metagenomic classification and assembly. Brief. Bioinform. 20, 1125–1136. doi: 10.1093/bib/bbx120
Brown, E. D., and Wright, G. D. (2016). Antibacterial drug discovery in the resistance era. Nature 529, 336–343. doi: 10.1038/nature17042
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Charpentier, E., and Courvalin, P. (1997). Emergence of the trimethoprim resistance gene dfrD in Listeria monocytogenes BM4293. Antimicr. Agents Chemother. 41, 1134–1136. doi: 10.1128/AAC.41.5.1134
Chen, K., and Pachter, L. (2005). Bioinformatics for Whole-Genome Shotgun Sequencing of Microbial Communities. PLoS Comp. Biol. 1:e24. doi: 10.1371/journal.pcbi.0010024
Chen, Q.-L., An, X.-L., Zhu, Y.-G., Su, J.-Q., Gillings, M. R., Ye, Z.-L., et al. (2017). Application of Struvite Alters the Antibiotic Resistome in Soil, Rhizosphere, and Phyllosphere. Environ. Sci. Technol. 51, 8149–8157. doi: 10.1021/acs.est.7b01420
Chistoserdovai, L. (2010). Functional metagenomics: recent advances and future challenges. Biotechnol. Genet. Eng. Rev. 26, 335–352.
Connor, E. E. (1998). Sulfonamide antibiotics. Prim. Care Update OB/GYNS 5, 32–35. doi: 10.1016/S1068-607X(97)00121-2
Costa, P. S., Reis, M. P., Ávila, M. P., Leite, L. R., de Araújo, F. M. G., Salim, A. C. M., et al. (2015). Metagenome of a Microbial Community Inhabiting a Metal-Rich Tropical Stream Sediment. PLoS One 10:e0119465. doi: 10.1371/journal.pone.0119465
Cowan, D., Ramond, J.-B., Makhalanyane, T., and De Maayer, P. (2015). Metagenomics of extreme environments. Curr. Opin. Microbiol. 25, 97–102. doi: 10.1016/j.mib.2015.05.005
Danko, D., Bezdan, D., Afshinnekoo, E., Ahsanuddin, S., Bhattacharya, C., Butler, D. J., et al. (2019). Global Genetic Cartography of Urban Metagenomes and Anti-Microbial Resistance. Microbiology 2019:526. doi: 10.1101/724526
De, R. (2019). Metagenomics: aid to combat antimicrobial resistance in diarrhea. Gut. Pathog. 11, 47. doi: 10.1186/s13099-019-0331-8
Devine, D., and Hancock, R. (2002). Cationic Peptides: Distribution and Mechanisms of Resistance. Curr. Pharmaceut. Design 8, 703–714. doi: 10.2174/1381612023395501
Diekema, D. J., and Jones, R. N. (2001). Oxazolidinone antibiotics. Lancet 358, 1975–1982. doi: 10.1016/S0140-6736(01)06964-1
Dong, X., and Strous, M. (2019). An Integrated Pipeline for Annotation and Visualization of Metagenomic Contigs. Front. Genet. 10:999. doi: 10.3389/fgene.2019.00999
Doster, E., Lakin, S. M., Dean, C. J., Wolfe, C., Young, J. G., Boucher, C., et al. (2019). MEGARes 2.0: a database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Res. 48, D561–D569. doi: 10.1093/nar/gkz1010
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Ekwanzala, M. D., Dewar, J. B., and Momba, M. N. B. (2020). Environmental resistome risks of wastewaters and aquatic environments deciphered by shotgun metagenomic assembly. Ecotoxicol. Environ. Safety 197:110612. doi: 10.1016/j.ecoenv.2020.110612
Eloe-Fadrosh, E. A., Ivanova, N. N., Woyke, T., and Kyrpides, N. C. (2016). Metagenomics uncovers gaps in amplicon-based detection of microbial diversity. Nat. Microbiol. 1, 1–4. doi: 10.1038/nmicrobiol.2015.32
Enne, V. I., and Bennett, P. M. (2010). “Methods to Determine Antibiotic Resistance Gene Silencing,” in Antibiotic Resistance Protocols, eds S. H. Gillespie and T. D. McHugh (Totowa, NJ: Humana Press), 29–44. doi: 10.1007/978-1-60327-279-7_3
Escobar-Zepeda, A., Godoy-Lozano, E. E., Raggi, L., Segovia, L., Merino, E., Gutiérrez-Rios, R. M., et al. (2018). Analysis of sequencing strategies and tools for taxonomic annotation: Defining standards for progressive metagenomics. Sci. Rep. 8:12034. doi: 10.1038/s41598-018-30515-5
Ferro, G., Guarino, F., Cicatelli, A., and Rizzo, L. (2017). β-lactams resistance gene quantification in an antibiotic resistant Escherichia coli water suspension treated by advanced oxidation with UV/H2O2. J. Hazard. Mater. 323, 426–433. doi: 10.1016/j.jhazmat.2016.03.014
Floss, H. G., and Yu, T.-W. (2005). RifamycinMode of Action, Resistance, and Biosynthesis. Chem. Rev. 105, 621–632. doi: 10.1021/cr030112j
Forslund, K., Sunagawa, S., Kultima, J. R., Mende, D. R., Arumugam, M., Typas, A., et al. (2013). Country-specific antibiotic use practices impact the human gut resistome. Genome Res. 23, 1163–1169. doi: 10.1101/gr.155465.113
Ghosh, T. S., Gupta, S. S., Nair, G. B., and Mande, S. S. (2013). In Silico Analysis of Antibiotic Resistance Genes in the Gut Microflora of Individuals from Diverse Geographies and Age-Groups. PLoS One 8:e83823. doi: 10.1371/journal.pone.0083823
Gibson, M. K., Forsberg, K. J., and Dantas, G. (2015). Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 9, 207–216. doi: 10.1038/ismej.2014.106
Giedraitienė, A., Vitkauskienė, A., Naginienė, R., and Pavilonis, A. (2011). Antibiotic Resistance Mechanisms of Clinically Important Bacteria. Medicina 47:19. doi: 10.3390/medicina47030019
Gómez Casanova, N., Siller Ruiz, M., and Muñoz Bellido, J. L. (2017). Mechanisms of resistance to daptomycin in Staphylococcus aureus. Rev. Esp. Quimioter. 30, 391–396.
Hale, J. D., and Hancock, R. E. (2007). Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert. Rev. Anti. Infective Ther. 5, 951–959. doi: 10.1586/14787210.5.6.951
Handelsman, J., Rondon, M. R., Brady, S. F., Clardy, J., and Goodman, R. M. (1998). Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem. Biol. 5, R245–R249. doi: 10.1016/S1074-5521(98)90108-9
He, T., Shen, Y., Schwarz, S., Cai, J., Lv, Y., Li, J., et al. (2016). Genetic environment of the transferable oxazolidinone/phenicol resistance gene optrA in Enterococcus faecalis isolates of human and animal origin. J. Antimicrob. Chemother. 71, 1466–1473. doi: 10.1093/jac/dkw016
Heeb, S., Fletcher, M. P., Chhabra, S. R., Diggle, S. P., Williams, P., and Cámara, M. (2011). Quinolones: from antibiotics to autoinducers. FEMS Microbiol. Rev. 35, 247–274. doi: 10.1111/j.1574-6976.2010.00247.x
Hernández, A., Sánchez, M. B., and Martínez, J. L. (2011). Quinolone Resistance: Much More than Predicted. Front. Microbiol. 2:22. doi: 10.3389/fmicb.2011.00022
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Jia, S., Zhang, X.-X., Miao, Y., Zhao, Y., Ye, L., Li, B., et al. (2017). Fate of antibiotic resistance genes and their associations with bacterial community in livestock breeding wastewater and its receiving river water. Water Res. 124, 259–268. doi: 10.1016/j.watres.2017.07.061
Kadlec, K., and Schwarz, S. (2009). Identification of a Novel Trimethoprim Resistance Gene, dfrK, in a Methicillin-Resistant Staphylococcus aureus ST398 Strain and Its Physical Linkage to the Tetracycline Resistance Gene tet(L). Antimicr. Agents Chemother. 53, 776–778. doi: 10.1128/AAC.01128-08
Kanfer, I., Skinner, M. F., and Walker, R. B. (1998). Analysis of macrolide antibiotics. J. Chromatogr. A 812, 255–286. doi: 10.1016/S0021-9673(98)00276-3
Karkman, A., Pärnänen, K., and Larsson, D. G. J. (2019). Fecal pollution can explain antibiotic resistance gene abundances in anthropogenically impacted environments. Nat. Commun. 10:80. doi: 10.1038/s41467-018-07992-3
Karsch-Mizrachi, I., Takagi, T., and Cochrane, G (2018). The international nucleotide sequence database collaboration. Nucleic Acids Res. 46, D48–D51. doi: 10.1093/nar/gkx1097
Kehrenberg, C., and Schwarz, S. (2006). Distribution of Florfenicol Resistance Genes fexA and cfr among Chloramphenicol-Resistant Staphylococcus Isolates. Antimicrob. Agents Chemother. 50, 1156–1163. doi: 10.1128/AAC.50.4.1156-1163.2006
Kuang, J., Huang, L., He, Z., Chen, L., Hua, Z., Jia, P., et al. (2016). Predicting taxonomic and functional structure of microbial communities in acid mine drainage. ISME J. 10, 1527–1539. doi: 10.1038/ismej.2015.201
Lam, K. N., Cheng, J., Engel, K., Neufeld, J. D., and Charles, T. C. (2015). Current and future resources for functional metagenomics. Front. Microbiol. 6:1196. doi: 10.3389/fmicb.2015.01196
Lebreton, F., and Cattoir, V. (2019). “Resistance to Glycopeptide Antibiotics,” in Bacterial Resistance to Antibiotics – From Molecules to Man, eds B. B. Bonev and N. M. Brown (New York: Wiley), 51–80. doi: 10.1002/9781119593522.ch3
Li, X., Wu, Z., Dang, C., Zhang, M., Zhao, B., Cheng, Z., et al. (2021). A metagenomic-based method to study hospital air dust resistome. Chem. Engin. J. 406:126854. doi: 10.1016/j.cej.2020.126854
Liu, B., and Pop, M. (2009). ARDB–Antibiotic Resistance Genes Database. Nucleic Acids Res. 37, D443–D447. doi: 10.1093/nar/gkn656
Liu, L., Su, J.-Q., Guo, Y., Wilkinson, D. M., Liu, Z., Zhu, Y.-G., et al. (2018). Large-scale biogeographical patterns of bacterial antibiotic resistome in the waterbodies of China. Environ. Int. 117, 292–299. doi: 10.1016/j.envint.2018.05.023
Lynch, M. D. J., and Neufeld, J. D. (2015). Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 13, 217–229. doi: 10.1038/nrmicro3400
Masters, P. A., O’Bryan, T. A., Zurlo, J., Miller, D. Q., and Joshi, N. (2003). Trimethoprim-Sulfamethoxazole Revisited. Arch. Int. Med. 163:402. doi: 10.1001/archinte.163.4.402
Mathe, C. (2002). Current methods of gene prediction, their strengths and weaknesses. Nucleic Acids Res. 30, 4103–4117. doi: 10.1093/nar/gkf543
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The Comprehensive Antibiotic Resistance Database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
McManus, M. C. (1997). Mechanisms of bacterial resistance to antimicrobial agents. Am. J. Health Syst. Pharm. 54, 1420–1433. doi: 10.1093/ajhp/54.12.1420
MetaSUB Consortium, Chng, K. R., Li, C., Bertrand, D., Ng, A. H. Q., Kwah, J. S., et al. (2020). Cartography of opportunistic pathogens and antibiotic resistance genes in a tertiary hospital environment. Nat. Med. 26, 941–951. doi: 10.1038/s41591-020-0894-4
Montero, C. I., Stock, F., and Murray, P. R. (2008). Mechanisms of Resistance to Daptomycin in Enterococcus faecium. Antimicrob. Agents Chemother. 52, 1167–1170. doi: 10.1128/AAC.00774-07
Munita, J. M., and Arias, C. A. (2016). Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 4:15. doi: 10.1128/microbiolspec.VMBF-0016-2015
Ngara, T. R., and Zhang, H. (2018). Recent Advances in Function-based Metagenomic Screening. Genom. Proteom. Bioinform. 16, 405–415. doi: 10.1016/j.gpb.2018.01.002
Pandey, N., and Cascella, M. (2020). Beta Lactam Antibiotics. Treasure Island, FL: StatPearls Publishing. Available online at: http://www.ncbi.nlm.nih.gov/books/NBK545311/ (accessed on June 23, 2020).
Parsons, Y., Hall, R. M., and Stokes, H. W. (1991). A new trimethoprim resistance gene, dhfrX, in the In7 integron of plasmid pDGO100. Antimicrob. Agents Chemother. 35, 2436–2439. doi: 10.1128/AAC.35.11.2436
Pehrsson, E. C., Forsberg, K. J., Gibson, M. K., Ahmadi, S., and Dantas, G. (2013). Novel resistance functions uncovered using functional metagenomic investigations of resistance reservoirs. Front. Microbiol. 7:145. doi: 10.3389/fmicb.2013.00145
Petersen, A., Guardabassi, L., Dalsgaard, A., and Olsen, J. E. (2000). Class I integrons containing a dhfrI trimethoprim resistance gene cassette in aquatic Acinetobacter spp. FEMS Microbiol. Lett. 182, 73–76. doi: 10.1111/j.1574-6968.2000.tb08876.x
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J., and Segata, N. (2017). Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35, 833–844. doi: 10.1038/nbt.3935
Quinn, R. A., Lim, Y. W., Maughan, H., Conrad, D., Rohwer, F., and Whiteson, K. L. (2014). Biogeochemical Forces Shape the Composition and Physiology of Polymicrobial Communities in the Cystic Fibrosis Lung. mBio 5, e00956–13. doi: 10.1128/mBio.00956-13
Rahal, J. J., and Simberkoff, M. S. (1979). Bactericidal and Bacteriostatic Action of Chloramphenicol Against Meningeal Pathogens. Antimicrob. Agents Chemother. 16, 13–18. doi: 10.1128/AAC.16.1.13
Razavi, M., Marathe, N. P., Gillings, M. R., Flach, C.-F., Kristiansson, E., and Joakim Larsson, D. G. (2017). Discovery of the fourth mobile sulfonamide resistance gene. Microbiome 5:160. doi: 10.1186/s40168-017-0379-y
Rice, E. W., Wang, P., Smith, A. L., and Stadler, L. B. (2020). Determining Hosts of Antibiotic Resistance Genes: A Review of Methodological Advances. Environ. Sci. Technol. Lett. 7, 282–291. doi: 10.1021/acs.estlett.0c00202
Roberts, M. C. (2005). Update on acquired tetracycline resistance genes. FEMS Microbiol. Lett. 245, 195–203. doi: 10.1016/j.femsle.2005.02.034
Roberts, M. C. (2017). “Antibiotic-Resistant Environmental Bacteria and Their Role as Reservoirs in Disease,” in Modeling the Transmission and Prevention of Infectious Disease, ed. C. J. Hurst (Cham: Springer International Publishing), 187–212. doi: 10.1007/978-3-319-60616-3_7
Rodríguez-Brazzarola, P., Pérez-Wohlfeil, E., Díaz-del-Pino, S., Holthausen, R., and Trelles, O. (2018). “Analyzing the Differences Between Reads and Contigs When Performing a Taxonomic Assignment Comparison in Metagenomics,” in Bioinformatics and Biomedical Engineering Lecture Notes in Computer Science., eds I. Rojas and F. Ortuño (Cham: Springer International Publishing), 450–460. doi: 10.1007/978-3-319-78723-7_39
Roh, C., and Villatte, F. (2008). Isolation of a low-temperature adapted lipolytic enzyme from uncultivated micro-organism. J. Appl. Microbiol. 105, 116–123. doi: 10.1111/j.1365-2672.2007.03717.x
Roose-Amsaleg, C., and Laverman, A. M. (2016). Do antibiotics have environmental side-effects? Impact of synthetic antibiotics on biogeochemical processes. Environ. Sci. Pollut. Res. 23, 4000–4012. doi: 10.1007/s11356-015-4943-3
Ruppé, E., Ghozlane, A., Tap, J., Pons, N., Alvarez, A.-S., Maziers, N., et al. (2019). Prediction of the intestinal resistome by a three-dimensional structure-based method. Nat. Microbiol. 4, 112–123. doi: 10.1038/s41564-018-0292-6
Schloss, P. D., and Handelsman, J. (2003). Biotechnological prospects from metagenomics. Curr. Opin. Biotechnol. 14, 303–310. doi: 10.1016/S0958-1669(03)00067-3
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. AEM 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schmieder, R., and Edwards, R. (2012). Insights into antibiotic resistance through metagenomic approaches. Fut. Microbiol. 7, 73–89. doi: 10.2217/fmb.11.135
Simmons, C. W., Reddy, A. P., D’haeseleer, P., Khudyakov, J., Billis, K., Pati, A., et al. (2014). Metatranscriptomic analysis of lignocellulolytic microbial communities involved in high-solids decomposition of rice straw. Biotechnol. Biof. 7:495. doi: 10.1186/s13068-014-0180-0
Soucy, S. M., Huang, J., and Gogarten, J. P. (2015). Horizontal gene transfer: building the web of life. Nat. Rev. Genet. 16, 472–482. doi: 10.1038/nrg3962
Sundstrom, L., and Skold, O. (1990). The dhfrI trimethoprim resistance gene of Tn7 can be found at specific sites in other genetic surroundings. Antimicrob. Agents Chemother. 34, 642–650. doi: 10.1128/AAC.34.4.642
Tamames, J., and Puente-Sánchez, F. (2019). SqueezeMeta, A Highly Portable, Fully Automatic Metagenomic Analysis Pipeline. Front. Microbiol. 9:3349. doi: 10.3389/fmicb.2018.03349
Tamames, J., Cobo-Simón, M., and Puente-Sánchez, F. (2019). Assessing the performance of different approaches for functional and taxonomic annotation of metagenomes. BMC Genom. 20:960. doi: 10.1186/s12864-019-6289-6
Tenson, T., Lovmar, M., and Ehrenberg, M. (2003). The Mechanism of Action of Macrolides, Lincosamides and Streptogramin B Reveals the Nascent Peptide Exit Path in the Ribosome. J. Mole. Biol. 330, 1005–1014. doi: 10.1016/S0022-2836(03)00662-4
Thorne, G. M., and Alder, J. (2002). Daptomycin: a novel lipopeptide antibiotic. Clin. Microbiol. Newslett. 24, 33–40. doi: 10.1016/S0196-4399(02)80007-1
Tripathi, L. K., and Nailwal, T. K. (2020). Metagenomics: Applications of functional and structural approaches and meta-omics. Rec. Adv. Microbial. Div. 4, 471–505. doi: 10.1016/B978-0-12-821265-3.00020-7
Tsafnat, G., Copty, J., and Partridge, S. R. (2011). RAC: Repository of Antibiotic resistance Cassettes. Database 2011, bar054–bar054. doi: 10.1093/database/bar054
Van Boeckel, T. P., Brower, C., Gilbert, M., Grenfell, B. T., Levin, S. A., Robinson, T. P., et al. (2015). Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sci. U. S. A. 112, 5649–5654. doi: 10.1073/pnas.1503141112
von Wintersdorff, C. J. H., Penders, J., van Niekerk, J. M., Mills, N. D., Majumder, S., van Alphen, L. B., et al. (2016). Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Front. Microbiol. 7:173. doi: 10.3389/fmicb.2016.00173
Wang, S., Yan, Z., Wang, P., Zheng, X., and Fan, J. (2020). Comparative metagenomics reveals the microbial diversity and metabolic potentials in the sediments and surrounding seawaters of Qinhuangdao mariculture area. PLoS One 15:e0234128. doi: 10.1371/journal.pone.0234128
Wang, Y., Lv, Y., Cai, J., Schwarz, S., Cui, L., Hu, Z., et al. (2015). A novel gene, optrA, that confers transferable resistance to oxazolidinones and phenicols and its presence in Enterococcus faecalis and Enterococcus faecium of human and animal origin. J. Antimicr. Chemother. 70, 2182–2190. doi: 10.1093/jac/dkv116
Wybouw, N., Pauchet, Y., Heckel, D. G., and Van Leeuwen, T. (2016). Horizontal Gene Transfer Contributes to the Evolution of Arthropod Herbivory. Genome Biol. Evol. 8, 1785–1801. doi: 10.1093/gbe/evw119
Xing, C., Chen, J., Zheng, X., Chen, L., Chen, M., Wang, L., et al. (2020). Functional metagenomic exploration identifies novel prokaryotic copper resistance genes from the soil microbiome. Metallomics 12, 387–395. doi: 10.1039/C9MT00273A
Xu, R., Yang, Z.-H., Wang, Q.-P., Bai, Y., Liu, J.-B., Zheng, Y., et al. (2018). Rapid startup of thermophilic anaerobic digester to remove tetracycline and sulfonamides resistance genes from sewage sludge. Sci. Tot. Environ. 612, 788–798. doi: 10.1016/j.scitotenv.2017.08.295
Yang, Y., Jiang, X., Chai, B., Ma, L., Li, B., Zhang, A., et al. (2016). ARGs-OAP: online analysis pipeline for antibiotic resistance genes detection from metagenomic data using an integrated structured ARG-database. Bioinformatics 32, 2346–2351. doi: 10.1093/bioinformatics/btw136
Yang, Y., Zhou, R., Chen, B., Zhang, T., Hu, L., and Zou, S. (2018). Characterization of airborne antibiotic resistance genes from typical bioaerosol emission sources in the urban environment using metagenomic approach. Chemosphere 213, 463–471. doi: 10.1016/j.chemosphere.2018.09.066
Ye, S. H., Siddle, K. J., Park, D. J., and Sabeti, P. C. (2019). Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 178, 779–794. doi: 10.1016/j.cell.2019.07.010
Yin, X., Jiang, X.-T., Chai, B., Li, L., Yang, Y., Cole, J. R., et al. (2018). ARGs-OAP v2.0 with an expanded SARG database and Hidden Markov Models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics 34, 2263–2270. doi: 10.1093/bioinformatics/bty053
Yu, K., Li, P., Chen, Y., Zhang, B., Huang, Y., Huang, F.-Y., et al. (2020). Antibiotic resistome associated with microbial communities in an integrated wastewater reclamation system. Water Res. 173:115541. doi: 10.1016/j.watres.2020.115541
Zankari, E., Hasman, H., Kaas, R. S., Seyfarth, A. M., Agerso, Y., Lund, O., et al. (2013). Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. J. Antimicrob. Chemother. 68, 771–777. doi: 10.1093/jac/dks496
Zhang, T., Zhang, X.-X., and Ye, L. (2011). Plasmid metagenome reveals high levels of antibiotic resistance genes and mobile genetic elements in activated sludge. PLoS One 6:e26041. doi: 10.1371/journal.pone.0026041
Zhao, R., Yu, K., Zhang, J., Zhang, G., Huang, J., Ma, L., et al. (2020). Deciphering the mobility and bacterial hosts of antibiotic resistance genes under antibiotic selection pressure by metagenomic assembly and binning approaches. Water Res. 186:116318. doi: 10.1016/j.watres.2020.116318
Zhao, Y., Tang, H., and Ye, Y. (2012). RAPSearch2: a fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 28, 125–126. doi: 10.1093/bioinformatics/btr595
Keywords: antimicrobial resistance genes, horizontal gene transfer, metagenomic analysis, resistome, Shotgun metagenome sequencing, database
Citation: de Abreu VAC, Perdigão J and Almeida S (2021) Metagenomic Approaches to Analyze Antimicrobial Resistance: An Overview. Front. Genet. 11:575592. doi: 10.3389/fgene.2020.575592
Received: 23 June 2020; Accepted: 04 December 2020;
Published: 18 January 2021.
Edited by:
Debmalya Barh, Institute of Integrative Omics and Applied Biotechnology (IIOAB), IndiaReviewed by:
João Marcelo Pereira Alves, University of São Paulo, BrazilCopyright © 2021 de Abreu, Perdigão and Almeida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinicius A. C. de Abreu, dmFicmV1QHVmcGEuYnI=; Sintia Almeida, c2ludGlhYWxtZWlkYUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.