 Firoz Ahmed1,2*
Firoz Ahmed1,2* Monika Sharma3
Monika Sharma3 Abdulsalam Abdullah Al-Ghamdi1Sultan Muhammad Al-Yami1Abdulaziz Musa Al-Salami1
Abdulsalam Abdullah Al-Ghamdi1Sultan Muhammad Al-Yami1Abdulaziz Musa Al-Salami1 Mohammed Y. Refai1,2Mohiuddin Khan Warsi1,2Saad M. Howladar4Mohammed N. Baeshen4
Mohammed Y. Refai1,2Mohiuddin Khan Warsi1,2Saad M. Howladar4Mohammed N. Baeshen4- 1Department of Biochemistry, College of Science, University of Jeddah, Jeddah, Saudi Arabia
- 2University of Jeddah Center for Scientific and Medical Research, University of Jeddah, Jeddah, Saudi Arabia
- 3Department of Chemical Sciences, Indian Institute of Science Education and Research, Mohali, India
- 4Department of Biology, College of Science, University of Jeddah, Jeddah, Saudi Arabia
The emergence of a new coronavirus (CoV), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), responsible for severe respiratory disease in humans termed coronavirus disease of 2019 (COVID-19), became a new global threat for health and the economy. The SARS-CoV-2 genome is about a 29,800-nucleotide-long plus-strand RNA that can form functionally important secondary and higher-order structures called cis-acting RNA elements. These elements can interact with viral proteins, host proteins, or other RNAs and be involved in regulating translation and replication processes of the viral genome and encapsidation of the virus. However, the cis-acting RNA elements and their biological roles in SARS-CoV-2 as well as their comparative analysis in the closely related viral genome have not been well explored, which is very important to understand the molecular mechanism of viral infection and pathogenies. In this study, we used a bioinformatics approach to identify the cis-acting RNA elements in the SARS-CoV-2 genome. Initially, we aligned the full genomic sequence of six different CoVs, and a phylogenetic analysis was performed to understand their evolutionary relationship. Next, we predicted the cis-acting RNA elements in the SARS-CoV-2 genome using the structRNAfinder tool. Then, we annotated the location of these cis-acting RNA elements in different genomic regions of SARS-CoV-2. After that, we analyzed the sequence conservation patterns of each cis-acting RNA element among the six CoVs. Finally, the presence of cis-acting RNA elements across different CoV genomes and their comparative analysis was performed. Our study identified 12 important cis-acting RNA elements in the SARS-CoV-2 genome; among them, Corona_FSE, Corona_pk3, and s2m are highly conserved across most of the studied CoVs, and Thr_leader, MAT2A_D, and MS2 are uniquely present in SARS-CoV-2. These RNA structure elements can be involved in viral translation, replication, and encapsidation and, therefore, can be potential targets for better treatment of COVID-19. It is imperative to further characterize these cis-acting RNA elements experimentally for a better mechanistic understanding of SARS-CoV-2 infection and therapeutic intervention.
Introduction
The emergence of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes severe respiratory disease in humans, is called the coronavirus disease of 2019 (COVID-19). Now COVID-19 has become a threat to global health and the economy. SARS-CoV-2 was initially reported in December 2019 from patients with shortness of breath and severe pneumonia at Wuhan city of Hubei province in China (Ren et al., 2020). Within a very short period, this new CoV infected hundreds of thousands of people across the world. On January 30, 2020, the World Health Organization (WHO) declared the outbreak to be a public health emergency of international concern. Subsequently, on March 11, 2020, WHO announced it as a world pandemic. As of September 11, 2020, 28,328,131 SARS-CoV-2 positive cases, 20,342,740 recovered cases, and 913,919 SARS-CoV-2 related deaths have been reported, and the numbers of COVID-19 positive cases are increasing at an alarming rate.1
Most COVID-19 patients show only mild symptoms of fever and cough. However, severe COVID-19 patients show acute respiratory distress syndrome (ARDS) and organ failure due to a sudden increase of various pro-inflammatory cytokines (Moore and June, 2020; Ren et al., 2020; Yang et al., 2020; Zhang et al., 2020). There is accumulating evidence indicating that host genetic factors play an essential role in inducing pro-inflammatory cytokines in response to SARS-CoV-2 infection (Debnath et al., 2020). The host uses various strategies to restrict viral replication and propagation, including RNA editing of the viral genome and transcriptome. A study analyzed the RNA transcriptomic data and identified two different RNA editing signatures in the SARS-CoV-2 transcriptome: (a) adenines-to-inosines by host deaminases ADARs and (b) cytosines-to-uracils by host deaminases APOBECs (Di Giorgio et al., 2020).
Severe acute respiratory syndrome coronavirus 2 is a zoonotic pathogen that jumps from animals to humans (Woo et al., 2009). The virus is closely related to two previously identified pathogenic CoVs of humans: (a) SARS-CoV emerged from China and was responsible for an epidemic in 2002–2003 of different parts of the world, resulting in 8,096 infections and 774 deaths with a fatality rate 9.6%,2 and (b) MERS-CoV emerged from the Middle East in 2012 and caused 2,494 infections and 858 related deaths globally with a fatality rate of 34.4%3 (Choudhry et al., 2019).
Based upon serological and genotypic features, CoVs are divided into four genera (i) alphacoronavirus (alphaCoVs), (ii) betacoronavirus (betaCoVs), (iii) gammacoronavirus (gammaCoVs), and (iv) deltacoronavirus (deltaCoVs) (Adams et al., 2015, 2016; Lu et al., 2015; Coronaviridae Study Group of the International Committee on Taxonomy of Viruses, 2020).4 Among them, two alphaCoVs (229E/NC_002645 and NL63/NC_005831) and four betaCoVs (OC43, HKU1, MERS-CoV, SARS-CoV, and SARS-CoV-2) are known to infect humans. The new virus, SARS-CoV-2, was isolated from five patients with severe pneumonia admitted December 18–29, 2019, in Jin Yintan Hospital of Wuhan, Hubei province, China (Ren et al., 2020). Scientists used next-generation sequencing technology to identify the novel CoV that was causing COVID-19 in these patients. Briefly, the bronchoalveolar lavage fluid (BAL) sample was isolated from a patient, followed by nucleic acid extraction from the sample. The sequencing library was constructed, and sequencing was performed on the Illumina platform (Illumina, San Diego, CA, United States). The sequencing reads were processed with quality control, including adapter trimming, low-quality read removal, and human and ribosomal read removal. The clean reads were assigned to taxonomic classification with the Kraken 2 software against the reference database containing microorganisms and viruses (Wood et al., 2019). The output result showed that a substantial portion of the sequencing reads was mapped to the betaCoVs, which was further selected for de novo assembly to get a complete genomic sequence of SARS-CoV-2 and confirmed by the Sanger sequencing technique (Ren et al., 2020). In addition, sequence homology and comparative analysis showed that SARS-CoV-2 has 87.7, 87.6, 79.0, and 51.8% nucleotide identity with the sequence of bat-derived SARS CoV (bat SL-CoVZXC21; Genbank MG772934), bat-derived SARS CoV (bat SL-CoVZC45; GenBank MG772933), SARS-CoV (GenBank NC_004718), and MERS-CoV (GenBank NC_019843), respectively (Ren et al., 2020). Consequently, phylogenetic analysis of genome and protein sequences revealed that SARS-CoV-2 has a close evolutionary relationship with bat-derived SARS CoV, followed by SARS-CoV and MERS-CoV (Andersen et al., 2020; Tang et al., 2020). The size of the full-length SARS-CoV-2 genome is about 29,870 nucleotides long plus-strand RNA (Ren et al., 2020). Like a typical mRNA, the SARS-CoV-2 genome has a 5′-cap, a 5′UTR, a 3′-UTR, and a poly-A tail and encodes (i) 16 non-structural proteins (NSP1 to NSP-16) from ORF1a and ORF1b; (ii) 4 structural proteins: spike (S), envelope (E), membrane (M), nucleocapsid (N); and (iii) different accessory proteins: ORF3, ORF6, ORF7a, ORF7b, ORF8, and ORF9b (Perlman and Netland, 2009; Ren et al., 2020).
Severe acute respiratory syndrome coronavirus 2 infection starts when the receptor-binding domain (RBD) of the Spike (S) protein present on the viral surface attaches to its host cell receptor angiotensin-converting enzyme 2 (ACE2; Shang et al., 2020). The Spike (S) protein contains two subunits: (i) the N-terminal S1 subunit responsible for binding with ACE2 and (ii) the C-terminal S2 subunit responsible for membrane fusion between virus and host. After attachment, the host cell protease TMPRSS2 cleaves the S protein into S1 and S2 subunits, promoting receptor association and membrane fusion between the virus and the host. Subsequently, the viral genome passes inside the host cell (Hoffmann et al., 2020). Consequently, the plus-stand RNA of the SARS-CoV-2 genome acts as an mRNA and encodes viral proteins, which hijacks the host protein synthesis machinery for virus assembly and escapes from host immune responses. The most reliable diagnosis for detecting SARS-CoV-2 infection is a quantitative fluorescence-based reverse transcription polymerase chain reaction (RT-qPCR) assay used for quantitative detection of the SARS-CoV-2 virus genome in a patient sample (Bustin and Nolan, 2020). A typical mRNA contains not only the protein-coding region, but also the untranslated region and several cis-acting RNA elements, which interact with trans-factors to regulate translation, localization, and half-life of the mRNA (Ahmed et al., 2009b, 2011, 2020).
The single-stranded RNA of the viral genome can form functionally important secondary and higher-order structures called cis-acting RNA elements, such as internal ribosome entry sites, riboswitches, and many others (Liu et al., 2009). These elements can interact with viral proteins, host proteins, or other RNAs and are involved in regulating translation and replication processes of the viral genome as well as encapsidation of the virus (Liu et al., 2009). For packaging the correct viral genome, the virus employs a broad range of strategies, including using cis-acting RNA elements present in the viral genome called packaging signals, which are selectively recognized by viral capsid proteins and assembled into virions (Masters, 2019; Alhatlani, 2020). The structure of the RNA molecules can be determined through various experimental techniques, including (i) high-resolution methods, such as X-ray crystallography, cryo-electron microscopy, nuclear magnetic resonance (NMR) spectroscopy, and cryo-electron microscopy (Cryo-EM), and (ii) low-resolution methods, such as thermal denaturation, chemical/enzymatic probing, mass spectrometry, RNA engineering, and selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE chemistry) (Felden, 2007). However, determining the RNA structure through experimental techniques is a non-trivial, expensive, and time-consuming process. Complementary computational prediction methods have been developed and successfully implemented to discover new biological insights pertaining to RNA structures (Ahmed et al., 2009a, 2013, 2015; Rehan and Bajouh, 2018; Ahmed, 2019). Many algorithms are currently available to predict the RNA secondary structure for a given RNA sequence. These algorithms use thermodynamics approaches, such as minimizing free energy, maximizing expected accuracy, and sampling-based models (Mathews et al., 2010; Lorenz et al., 2011; Bellaousov et al., 2013; Wu et al., 2015). Previous studies have identified several cis-acting RNA elements in different viral and human genomes (Liu et al., 2009; Wakida et al., 2020). However, cis-acting RNA elements and their precise roles in the SARS-CoV-2 genome have not been well explored, which is very important to understand the molecular mechanism of the viral infection, propagation, and virus encapsidation and to identify potential targets for better treatment of COVID-19 (Robertson et al., 2005; Alhatlani, 2020).
Our study is focused on identifying the cis-acting RNA elements in the SARS-CoV-2 genome using a bioinformatics approach. In this work, we aligned the full genome sequence of six different CoVs, and then, a phylogenetic analysis was performed to understand their evolutionary relationship. Afterward, we predicted the cis-acting RNA elements in the SARS-CoV-2 genome using the structRNAfinder tool. We also modeled these cis-acting RNA elements using a knowledge-based method, RNAComposer, that employs fully automated fragment assembly based on the secondary RNA structure as annotated by the RNAfold algorithm. Then, we annotated the location of these cis-acting RNA elements in different genomic regions of SARS-CoV-2. After that, we analyzed the sequence conservation patterns of each cis-acting RNA element across CoVs. Finally, a comparative analysis of cis-acting RNA elements predicted in different CoVs genomes was performed. Our study identified several cis-acting RNA elements in SARS-CoV-2, which could ultimately be used to understand how these elements interact with host machinery to regulate viral protein translation, genome replication, packaging, and pathogenesis.
Materials and Methods
CoVs Genomic Data
In this study, we took the full genomic sequence of six CoVs: (i) SARS-CoV-2 (accession number: EPI_ISL_402123); two bat-derived SARS CoVs (ii) batCoV batRaTG13 (accession number: EPI_ISL_402131) and (iii) batCoV batZXC21 (MG772934.1); (iv) SARS-CoV (NC_004718.3); (v) MERS-CoV (NC_019843.3); and (vi) Human CoV-NL63 (NC_005831.2). The first two genomic sequences were downloaded from GISAID,5 and the remaining sequences were downloaded from NCBI.6
Phylogenetic Analysis
The genomic sequences in Fasta format of all CoVs were put in a file “All_sequence.fa.” Multiple sequence alignment was then performed on these sequences with the ClustalW-2.1 program using the following commands at default parameters:
After that, a phylogenetic tree was generated by maximum likelihood algorithm using the Akaike information criterion (AIC) as a substitution model and 1,000 bootstrap replications with PhyML 3.0,7 and the best tree was generated on the GTR + G substitution model (Guindon et al., 2010). The phylogenetic tree was visualized using FigTree v1.4.4.8 In order to understand the CoVs similarity, the sequence similarity was plotted with Base-by-Base Version 3 (Tu et al., 2018).
Analysis of cis-Acting RNA Elements
In order to find the potential cis-acting RNA elements, the full genome of CoVs was predicted using the online tool structRNAfinder9 (Arias-Carrasco et al., 2018). Only the “+” strand was analyzed using the “cmsearched” option with e-value 0.01. StructRNAfinder is an automated pipeline, and its back end implements third-party tools, including Infernal (Nawrocki and Eddy, 2013), RNA families (Rfam) database (Nawrocki et al., 2015), RNAfold (Lorenz et al., 2011), and Krona (Ondov et al., 2011) for predicting and annotating RNA families in the genome sequences. The tool uses the nucleotide sequences and/or secondary structure covariance models developed using the Rfam database and identifies the potential novel regulatory RNAs in the genome. The output result displays the list of predicted RNA structures, sequence/structural consensus alignments for each RNA family according to the Rfam database,10 and provides a taxonomic overview of each assigned functional RNA.
Annotations of tRNA in SARS-CoV-2 Genome
To identify the presence of the tRNA gene, the whole genome of SARS-CoV-2 was analyzed using tRNAscan-SE v.2.0.6 (Lowe and Chan, 2016).
Structural Annotations of cis-Acting mRNA Elements
The RNA secondary structures of the viral genomic sequences were predicted using the RNAfold algorithm.11 In order to predict using RNAfold, the genomic regions were broken into fragments consisting of the cis-acting RNA elements, and the secondary structure topology was identified from the larger genome sequence.
The identified potential cis-acting RNA elements were modeled using the RNAComposer algorithm.12 RNAComposer predicts a 3-D structure based on its sequence and secondary structure topology. It utilizes 3-D structure fragments derived from RNA FRABASE (Popenda et al., 2008, 2010), a dictionary of RNA secondary and tertiary structure elements. The secondary structure annotation provided by RNAfold was divided into fragments according to its graph representation, and the best matching 3-D structural fragments were selected to assemble an initial 3-D model. The model was further minimized in torsion angle space and in the Cartesian atom coordinate space.
Results and Discussion
Sequence Similarity of SARS-CoV-2
It was found that SARS-CoV-2 has the highest sequence similarity with batRaTG13 (96.17%), followed by batZXC21 (88.01%), SARS-CoV (79.74%), MERS-CoV (54.13%), and CoV-NL63 (49.92%). The percentage identity score matrix of different CoV is given in Table 1. Furthermore, full-length SARS-CoV-2 sequences were compared with other CoVs to determine their sequence similarity across the genome. A similarity plot using a window size of 500 nucleotides and step size of 50 nucleotides showed that, throughout the genome, SARS-CoV-2 shows the highest similarity with bat CoV batRaTG13 detected in bats from Yunnan Province (Figure 1; Zhou et al., 2020b). Furthermore, we observed the highest sequence similarity between SARS-CoV-2 and bat-derived SARS CoVs (batRaTG13, batZXC21) at the first ∼11,000 nucleotides of the 5′-end and the last ∼5,000 nucleotides of the 3′-end of the genome, and thus, our results show agreement with other studies that SARS-CoV-2 is more closely related with bat-derived SARS-CoV batRaTG13 (Paraskevis et al., 2020). However, a recent study showed a high rate of RNA modification in the host system that could be explained by 87% of the nucleotide synonymous substitution between SARS-CoV-2 and CoV batRaTG13 (Di Giorgio et al., 2020; Li et al., 2020). Thus, the study suggests that previous research might have overestimated the divergence between SARS-CoV-2 and batRaTG13 (Li et al., 2020; Tang et al., 2020).
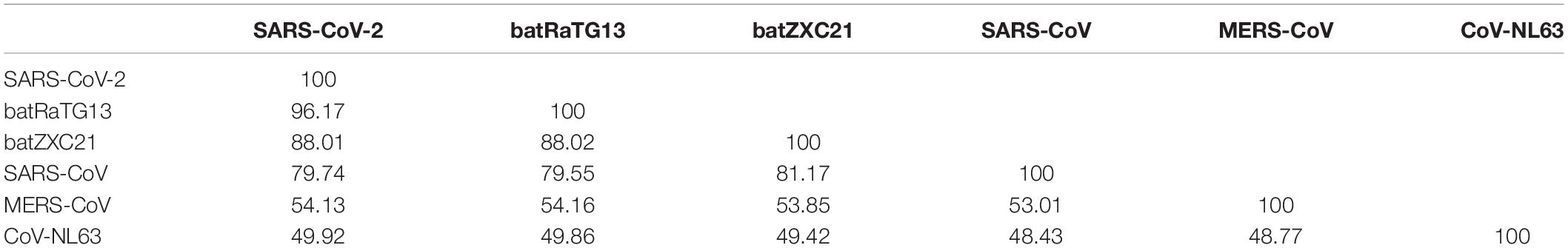
Table 1. Percentage identity score of CoV calculated by CusatalW2.1
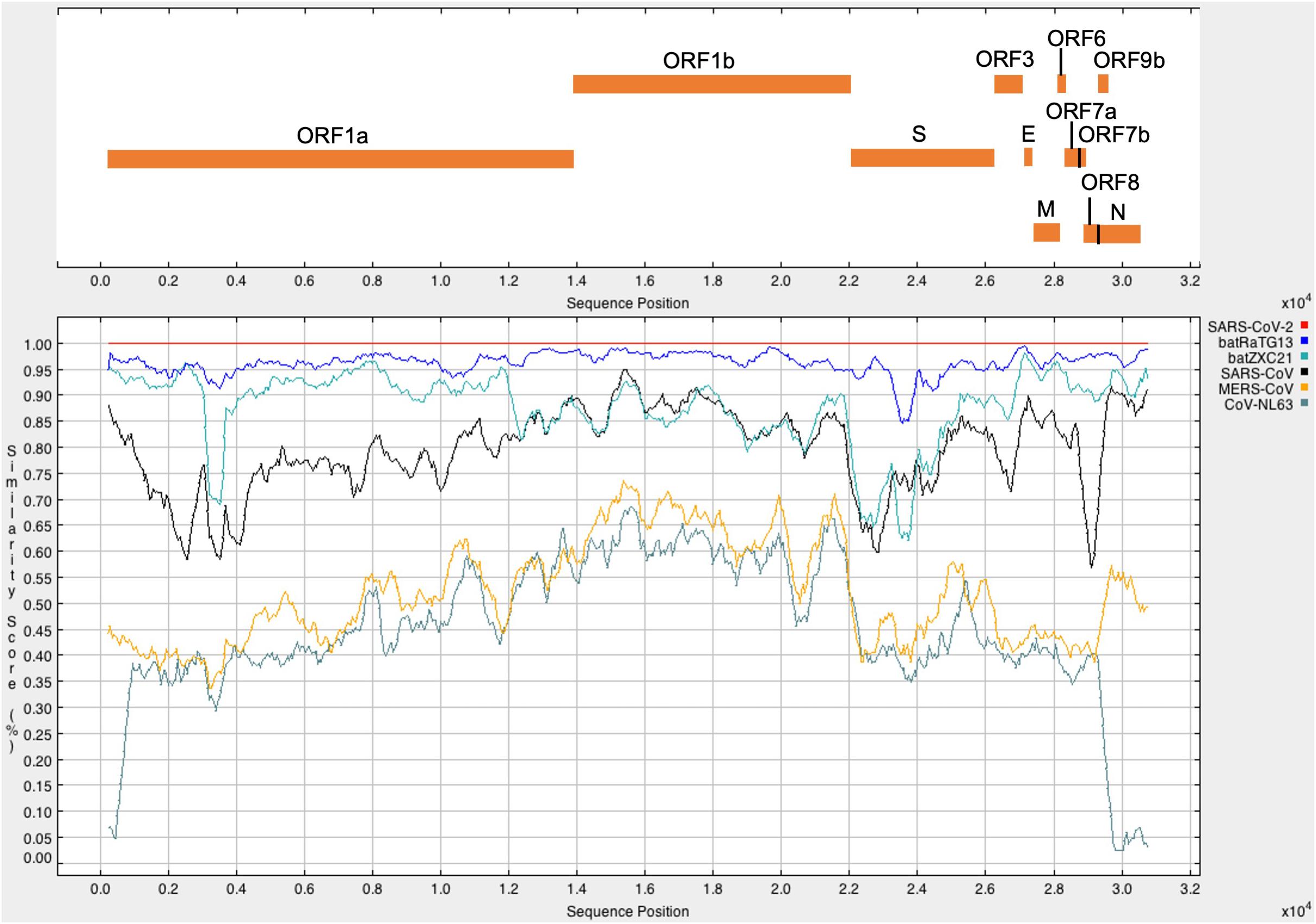
Figure 1. The upper panel represents the genomic organization of SARS-CoV-2 according to positions in the aligned sequences. The lower panel represents the similarity plot of different CoV full-genome sequences with respect to SARS-CoV-2. CoV sequences were aligned using ClustalW2. Each CoV represents in a different color. A single point in the plot shows the percentage identity within a sliding window of 500 nucleotides with a step size of 50 nucleotides. The x-axis represents the nucleotide position of the aligned sequence, and the y-axis represents the similarity score between SARS-CoV-2 and other CoV sequences.
Phylogenetic Relationship of SARS-CoV-2
Phylogenetic analysis of the complete genome sequence revealed that SARS-CoV-2 and batRaTG13 are clustered together in the phylogenetic tree (Figure 2). However, batZXC21 is closely related to SARS-CoV, and the sub-clade that groups MERS-CoV and CoV-NL63 are more distant from a common ancestor (Figure 2). Our study observed that SARS-CoV-2 is genetically closely related to bat-CoV and distantly related to SARS-CoV and, thus, agrees with previous studies suggesting that SARS-CoV-2 may be originated from bats (Lu et al., 2020; Zhou et al., 2020a).
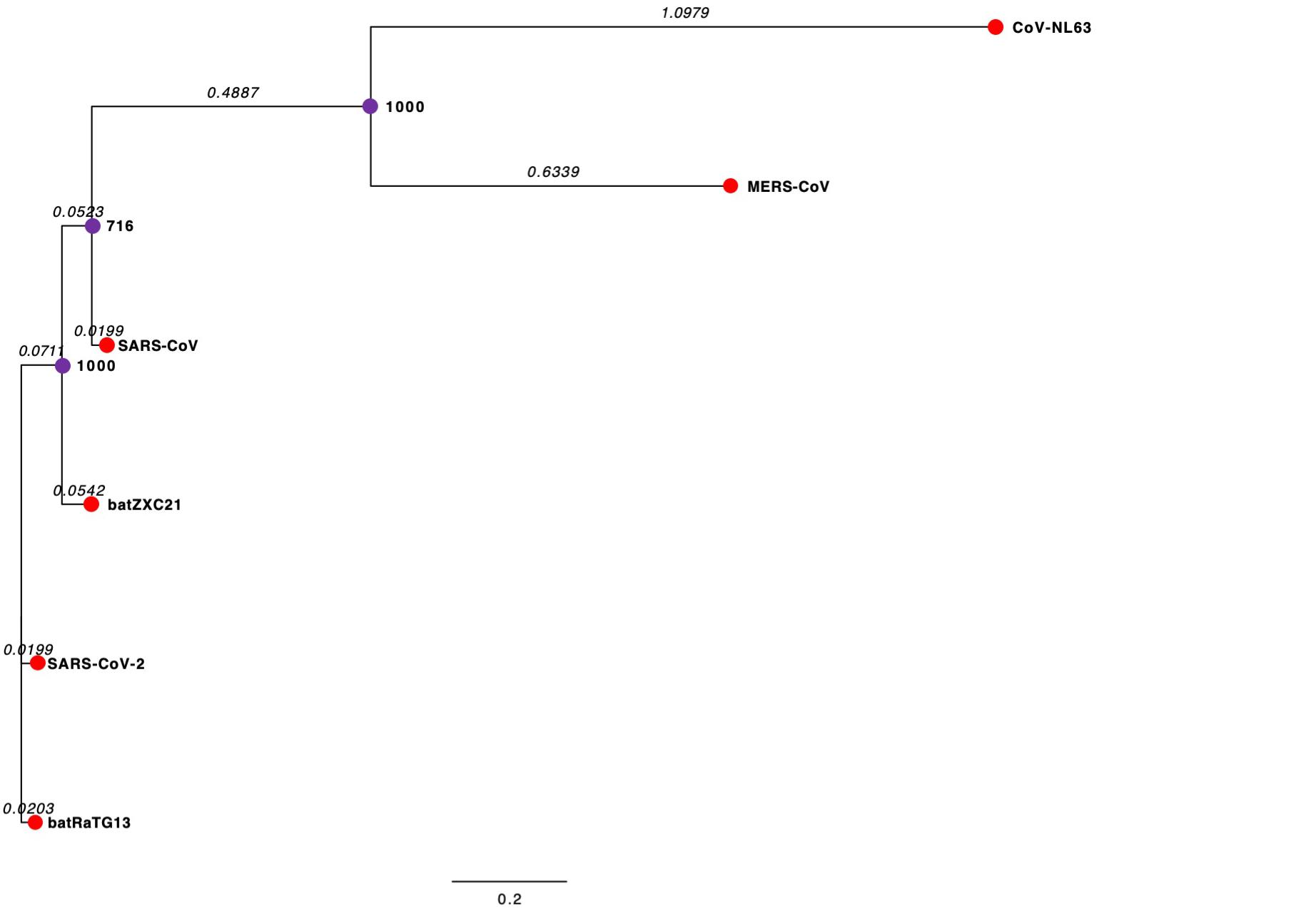
Figure 2. Phylogenetic Tree of CoVs. The external node in the red circle represents each CoV. The internal node in the purple circles represents hypothetical ancestors for the CoVs. Horizontal branch lines represent evolutionary changes over time. The branch length (branch time) value, shown in italics, measures in time or genetic divergence unit. The longer the horizontal line, the larger the number of genetic changes. The bottom of the figure has a bar that provides the scale for the branch lengths. The bootstrap value is presented in bold. The bootstrap value provides confidence for each clade of the observed tree. The higher the bootstrap value, the greater the confidence level of the clade in the phylogenetic tree.
Cis-Acting RNA Elements in SARS-CoV-2 Genome
We analyzed the presence of cis-acting RNA elements in the genome of SARS-CoV-2 (EPI_ISL_402123 from China) using an online tool structRNAfinder9 (Arias-Carrasco et al., 2018). The tool predicted a total of 12 cis-acting RNA elements in the SARS-CoV-2 genome. These elements are broadly divided into three subclasses: (a) Frameshift (Corona_FSE and fiv_FSE), (b) Leader (Thr_leader and S15), and (c) Others-cis (PYLIS_2, MAT2A_D, ClpQY_promoter, Histone3, s2m, Corona_pk3, Corona_pk3, and MS2). Detailed information on these cis-acting RNA elements, including location and structure, is provided in Table 2. The secondary RNA structures spanning the regions consisting of these cis-acting RNA elements were analyzed using the RNAfold algorithm (Supplementary Figure 1), and the secondary annotations were utilized to generate 3-D structures of these elements using RNAComposer (Figures 3–9 and Supplementary Figures 2–6). To better understand the biological role of these cis-acting RNA elements, we compared their location to the protein-coding gene in the SARS-CoV-2 genome (EPI_ISL_402123). A complete result of these cis-acting RNA elements is provided in the following section.
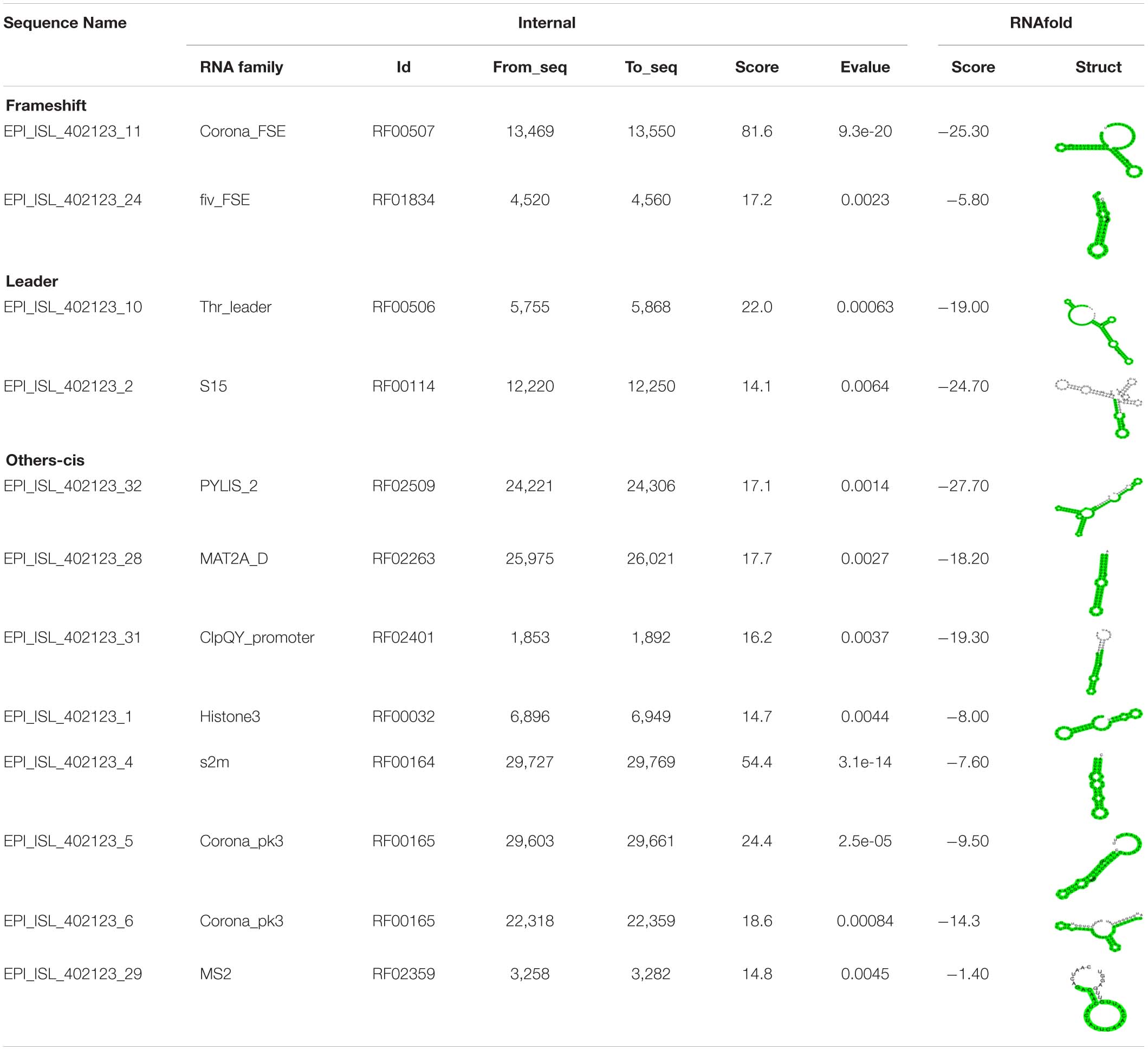
Table 2. Different classes of cis-acting RNA elements and RNA family motifs on SARS-CoV-2 RNA (Accession Number EPI_ISL_402123).
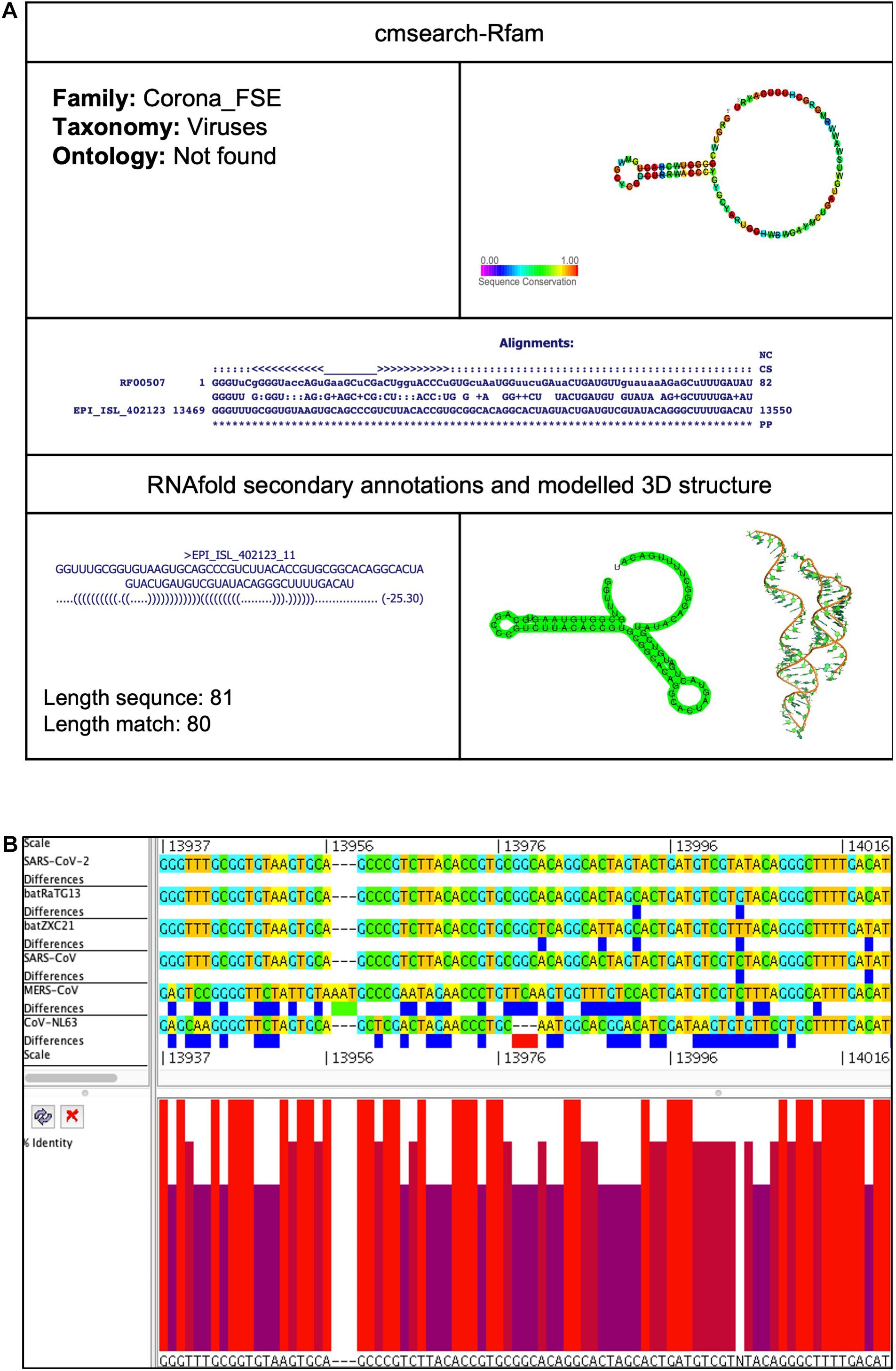
Figure 3. Corona_FSE cis-acting RNA element in the SARS-CoV-2 genome. (A) The sequence and RNA structure of Corona_FSE is located in position 13,469–13,550 of the SARS-CoV-2 genome. In the 3-D structure, the phosphodiester backbone is shown in orange, and the nucleotides are shown in the sticks and filled rings with elemental coloring as C green, O red, and N blue. (B) Conservation of Corona_FSE RNA sequence from various CoVs. The Corona_FSE is located as aligned position 13,937–14,021. The navy blue box indicates mismatched nucleotides between the two sequences, and the red box shows gaps between two sequences. Percentage identity in the histogram in which red indicates a perfect match and magenta shows the low similarity of nucleotide.
Frameshift
There are two frameshift cis-acting RNA elements predicted in the SARS-CoV-2 genome.
Corona_FSE (Coronavirus frameshifting stimulation element)
This element is located at genomic position 13,469–13,550, overlapping on nsp12 of the SARS-CoV-2 genome (Tables 2, 3). The stem-loop RNA structure of Corona_FSE and its conservation are shown across different CoVs (Figures 3A,B). A previous study showed that Corona_FSE interacts with downstream nucleotides to form a pseudoknot and promote ribosomal frameshifting, which is an important mechanism to express orf1b in CoVs (Baranov et al., 2005).
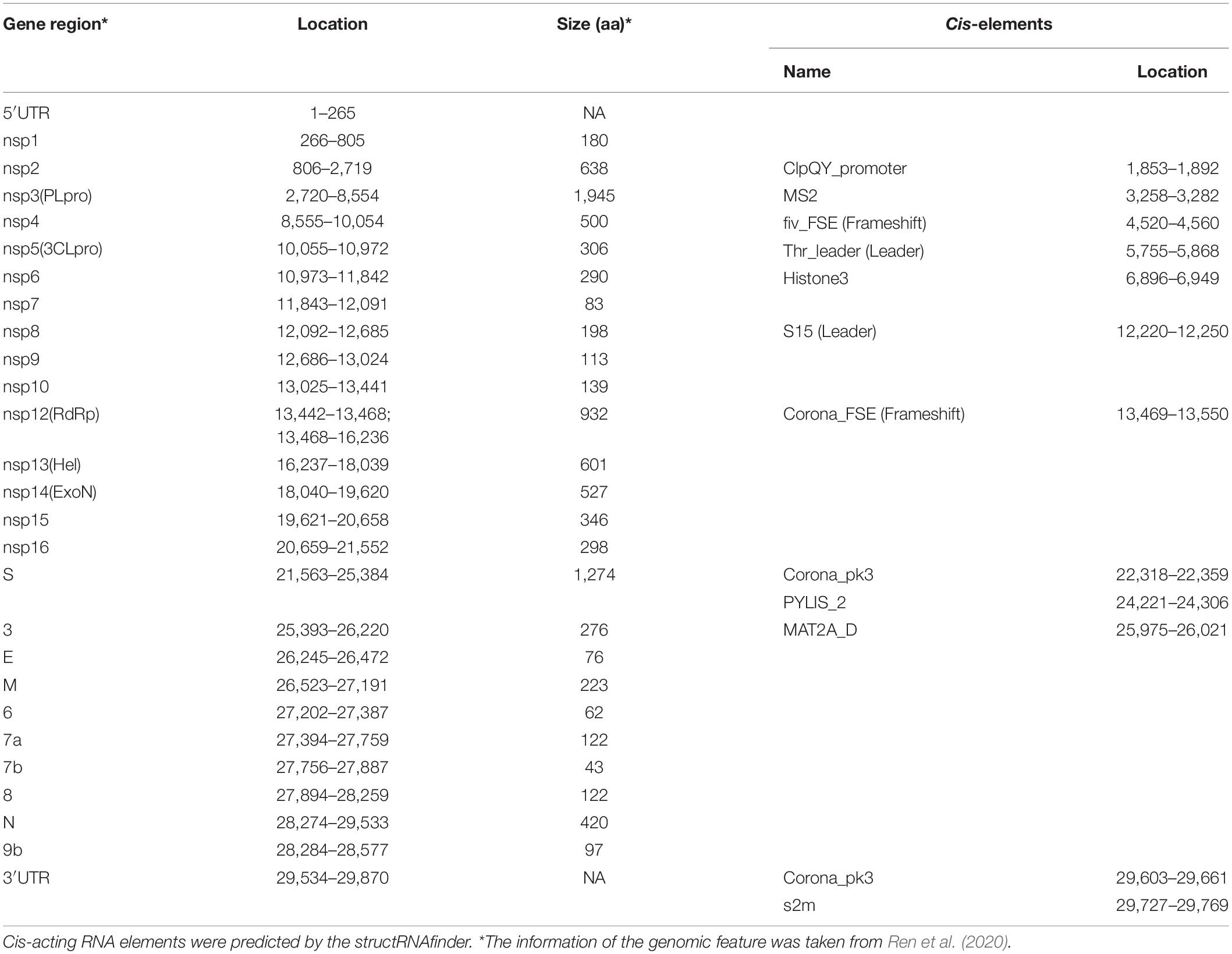
Table 3. Location and size of the putative gene, proteins, cis-elements of SARS-CoV-2 (EPI_ISL_402123).
Fiv_FSE (Feline immunodeficiency virus frameshifting stimulation element)
This element is located at a genomic position 4,520–4,560, overlapping on nsp3 of the SARS-CoV-2 genome (Tables 2, 3). The stem-loop RNA structure of Fiv_FSE and its conservation are shown across different CoVs (Supplementary Figures 2A,B). The Fiv_FSE element is found at a location that uses frameshift during translation in feline immunodeficiency virus (Morikawa and Bishop, 1992; Yu et al., 2005; Gonzalez and Affranchino, 2018). However, we did not observe the Fiv_FSE-associated frameshift in the SARS-CoV-2 genome. FIV is a lentivirus that can infect cats but is not fatal. In contrast, FIV cannot infect humans (Morikawa and Bishop, 1992; Yu et al., 2005; Gonzalez and Affranchino, 2018).
Leader
There are two leader cis-acting RNA elements predicted in the SARS-CoV-2 genome.
Thr_leader (Threonine operon leader)
This element is located at position 5,755–5,868 overlapping on nsp3 of the SARS-CoV-2 genome (Tables 2, 3). The stem-loop RNA structure of Thr_leader and its conservation are shown across different CoVs (Figures 4A,B). The threonine operon leader is an RNA element found in upstream of mRNA that encodes a group of enzymes engaged for the biosynthesis of amino acid threonine. In prokaryotes, transcription and translation occur simultaneously. The transcription mechanism of the operon is regulated by an attenuation mechanism that causes premature termination of the transcript (Kolter and Yanofsky, 1982). The threonine operon is turned “on” to express these enzymes when the threonine level is low and turned “off” to repress the mRNA transcription and translation when the threonine level is high. Thr_leader mRNA’s attenuator sequence adopts two different structures: (a) terminator and (b) antiterminator. In Escherichia coli, RNA polymerase binds and initiates mRNA transcription of the Thr_leader sequence. Subsequently, the ribosome begins to translate the nascent mRNA of the leader sequence into a short leader peptide-rich in threonine. In the presence of excess threonine-charged tRNA, the ribosome translates smoothly to form a leader peptide. Consequently, the attenuator sequence of Thr_leader, located between the mRNA leader sequence and Thr operon gene sequence, forms a terminator structure resulting in release of the RNA polymerase. Thus, the transcription of Thr operon is terminated. If E. coli lacks threonine-charged tRNA, the ribosome stalled at the Thr codons at the mRNA leader sequence; consequently, the attenuator sequence of Thr_leader forms an antiterminator structure, and thus, RNA polymerase continues to transcribe, and ribosome continues to translate the rest of the operon (Kolter and Yanofsky, 1982).
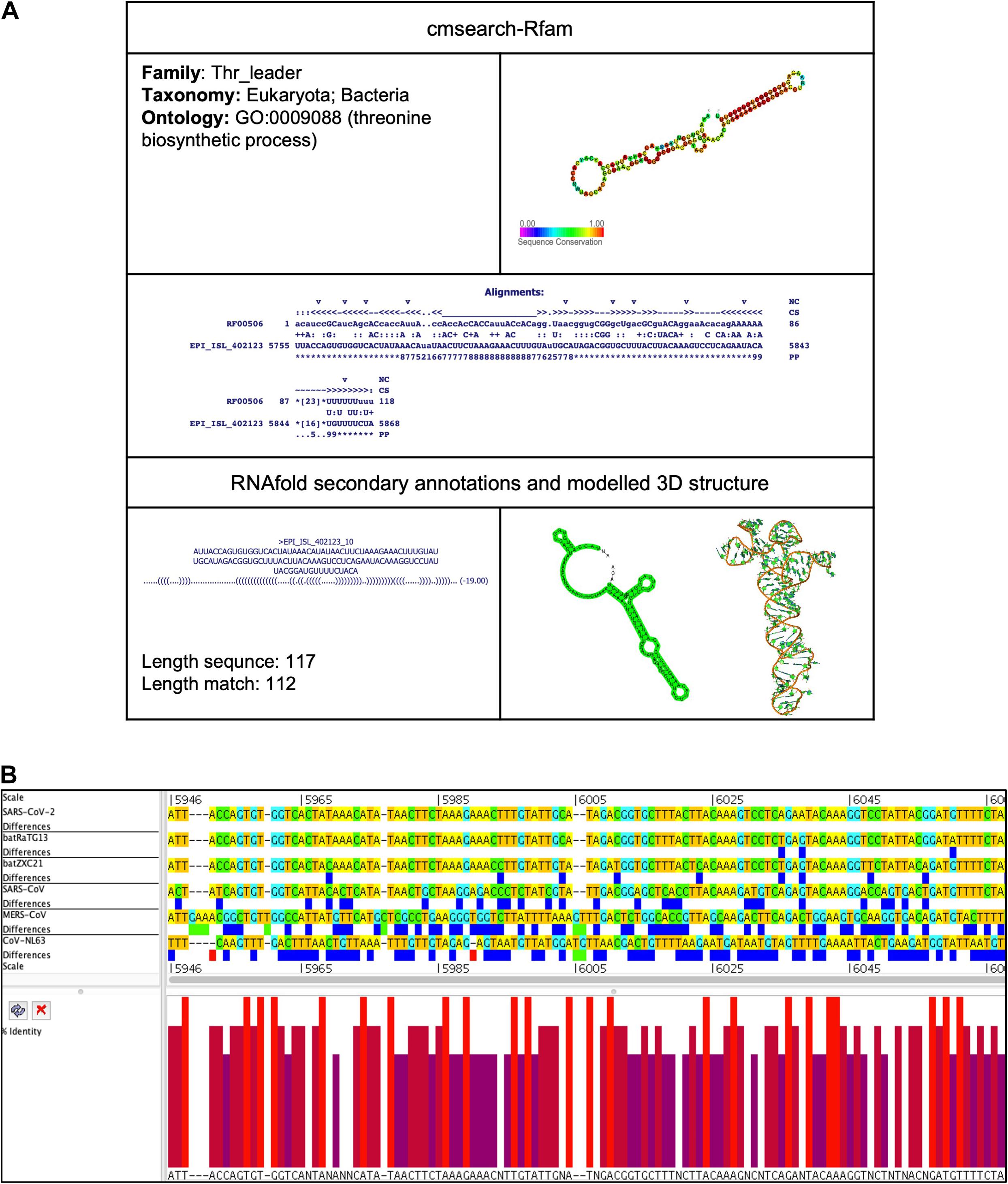
Figure 4. Thr_leader cis-acting RNA element in the SARS-CoV-2 genome. (A) The sequence and RNA structure of Thr_leader is located in te position 5,755–5,868 of the SARS-CoV-2 genome. In the 3-D structure, the phosphodiester backbone is shown in orange, and the nucleotides are shown in the sticks and filled rings with elemental coloring as C green, O red, and N blue. (B) Conservation of Thr_leader RNA sequence from various CoVs. The Thr_leader is located as aligned position 5,946–6,067. The navy blue box indicates mismatched nucleotides between the two sequences, and the red box shows gaps between two sequences. Percentage identity in the histogram in which red indicates a perfect match and magenta indicates the low similarity of nucleotide.
To identify the presence of the tRNA gene, the whole genome of SARS-CoV-2, including nsp3 ORF, was analyzed using tRNAscan-SE v.2.0.6 (Lowe and Chan, 2016). However, we did not find any tRNA gene encoded by the SARS-CoV-2.
S15 (Ribosomal S15 leader)
This element is located at position 12,220–12,250 overlapping on nsp8 of the SARS-CoV-2 genome (Tables 2, 3). The stem-loop RNA structure of S15 and its conservation are shown across different CoVs (Supplementary Figures 3A,B). In E. coli, the S15 structure is present in the ribosomal S15 mRNA. The RNA structure is involved in the translation regulation of S15 protein (Benard et al., 1996). The S15 adopts two alternate structures: (a) a series of 3 hairpins and (b) a pseudoknot.
Others-cis
There are eight other types of cis-acting RNA elements predicted in the SARS-CoV-2 genome.
PYLIS_2 (Pyrrolysine insertion sequence MtmB)
This element is located at position 24,221–24,306 overlapping on S of the SARS-CoV-2 genome (Tables 2, 3). The stem-loop RNA structure of PYLIS_2 and its conservation are shown across different CoVs (Supplementary Figures 4A,B). Some mRNA of methanogenic archaea Methanosarcina barkeri contains this stem-loop structure (Theobald-Dietrich et al., 2005). Previously, this structure was considered to be involved in translating UAG (Stop codon) to unusual amino acid pyrrolysine instead of termination of protein translation (Namy et al., 2004).
MAT2A_D (MAT2A 3′UTR stem loop D)
This element is located at position 25,975–26,021 overlapping on ORF3 of the SARS-CoV-2 genome (Tables 2, 3). The stem-loop RNA structure of MAT2A_D and its conservation are shown across different CoVs (Figures 5A,B). The enzyme methionine adenosyltransferase (MAT) creates S-adenosylmethionine (SAM) by reacting methionine with ATP (Parker et al., 2011). S-adenosylmethionine acts as a methionine donor during DNA methylation and, thus, switches off the gene, and therefore, SAM regulates gene expression. The 3′UTR of MAT II, alpha (MAT2A) mRNA contains six hairpin structures (named A–F) and helps in transcript stability (Parker et al., 2011).
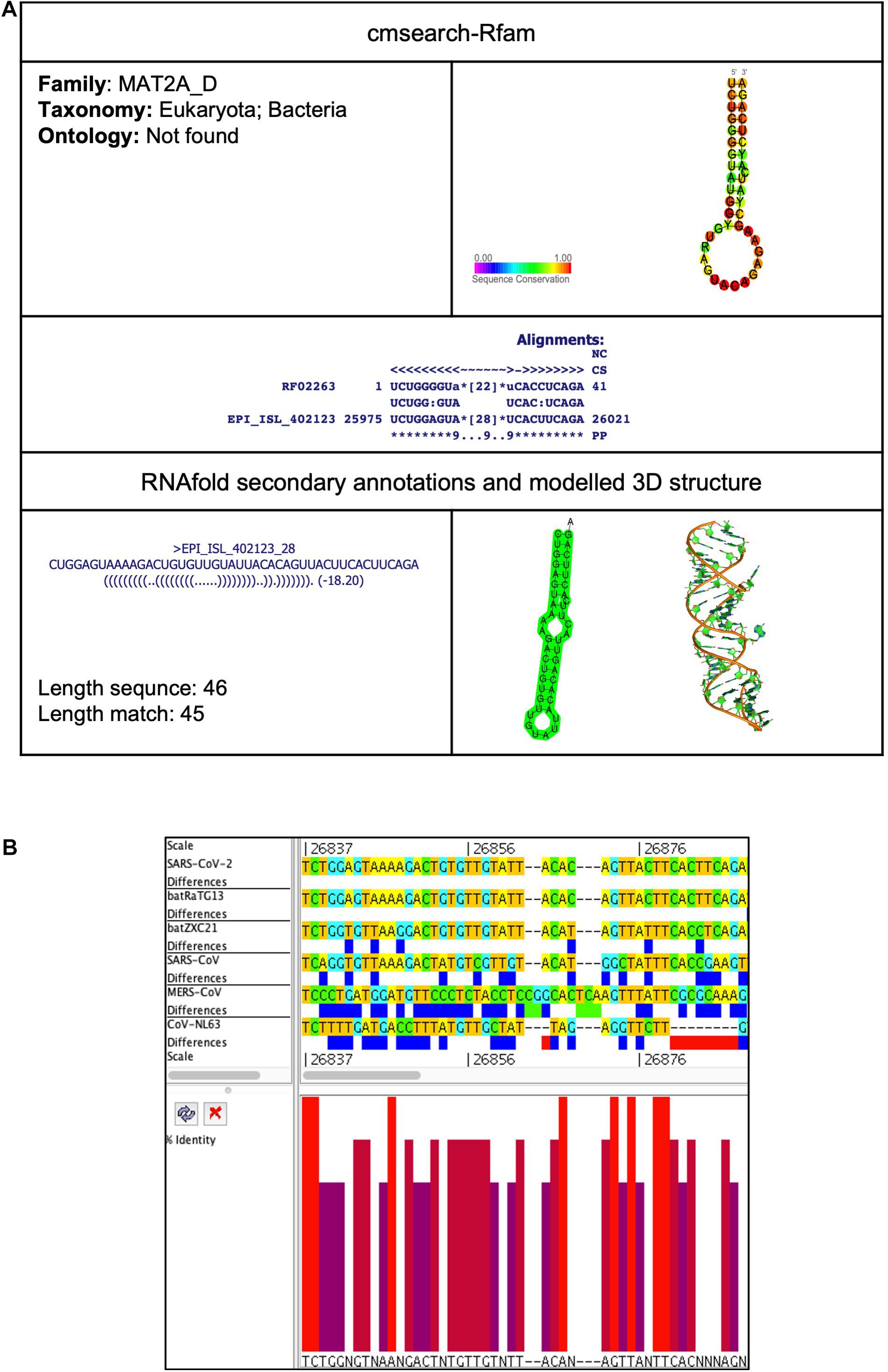
Figure 5. MAT2A_D cis-acting RNA element in the SARS-CoV-2 genome. (A) The sequence and RNA structure of MAT2A_D is located in position 25,975–26,021 of the SARS-CoV-2 genome. In the 3-D structure, the phosphodiester backbone is shown in orange, and the nucleotides are shown in the sticks and filled rings with elemental coloring as C green, O red, and N blue. (B) Conservation of MAT2A_D RNA sequence from various CoVs. The MAT2A_D is located as aligned position 26,837–26,888. The navy blue box indicates mismatched nucleotides between the two sequences, and the red box indicates gaps between two sequences. Percentage identity in the histogram in which red indicates a perfect match and magenta indicates the low similarity of nucleotide.
ClpQY_promoter
This element is located at position 1,853–1,892 overlapping on the nsp2 of the SARS-CoV-2 genome (Tables 2, 3). The stem-loop RNA structure of the ClpQY_promoter and its conservation are shown across different CoVs (Supplementary Figure 5A,B). The heat shock proteins ClpQ (protease) and ClpY (ATPase) are expressed in many bacteria by a single operon under stress conditions to form a complex called ClpQY (also called HslUV) (Nishii and Takahashi, 2003). The ClpQY complex degrades damaged and unwanted proteins. The promoter region of the operon contains a stem-loop structure required for gene expression and mRNA stability.
Histone3 (Histone 3′ UTR stem-loop)
This element is located at position 6,896–6,949 overlapping on nsp3 of the SARS-CoV-2 genome (Tables 2, 3). The stem-loop RNA structure of Histone3 and its conservation are shown across different CoVs (Supplementary Figures 6A,B). The mRNAs of metazoan histone mRNAs lack a poly-A tail at 3′UTR; however, they contain a highly conserved stem-loop region called Histone3 and a purine-rich region about 20 nucleotides downstream (Williams and Marzluff, 1995). In the nucleus, the histone3 element binds with the hairpin-binding protein (HBP) and U7 snRNA to form a processing complex that processes between histone3 and the purine-rich region to generate 3′, which is vital for mature mRNA formation and nucleocytoplasmic transport. In contrast, histone3 in the cytoplasm increases the stability and translation of histone mRNA (Zanier et al., 2002).
s2m (Coronavirus 3′ stem-loop II-like motif)
This element is located at position 29,727–29,769 overlapping on 3′UTR of the SARS-CoV-2 genome (Tables 2, 3). The stem-loop RNA structure of s2m and its conservation are shown across different CoVs (Figures 6A,B). It is a secondary structure RNA motif present in the 3′UTR region of SARS-CoV-2. Studies show that the motif is conserved at the sequence and secondary structure level and found in the genome of other CoVs as well as of astrovirus and equine rhinovirus (Jonassen et al., 1998; Robertson et al., 2005). After viral infection, proteins bind with s2m and help to replace host protein synthesis with viral protein synthesis (Jonassen et al., 1998; Robertson et al., 2005). Although the function of s2m is not very well understood, the study suggests it involves viral replication and packaging. The previous study solved the structure of s2m RNA in SARS-CoV-1 (PDB entry 1XJR), which is highly similar to SARS-CoV-2 and supposed to be a potential target for an antivirus.

Figure 6. s2m cis-acting RNA element in the SARS-CoV-2 genome. (A) The sequence and RNA structure of s2m is located in position 29,727–29,769 of the SARS-CoV-2 genome. In the 3-D structure, the phosphodiester backbone is shown in orange, and the nucleotides are shown in the sticks and filled rings with elemental coloring as C green, O red, and N blue. (B) Conservation of s2m RNA sequence from various CoVs. The s2m is located as aligned position 30,844–30,886. The navy blue box indicates mismatched nucleotides between the two sequences, and the red box indicates gaps between two sequences. Percentage identity in the histogram in which red indicates a perfect match and magenta indicates the low similarity of nucleotide.
Corona_pk3 (Coronavirus 3′ UTR pseudoknot)
This element is located at two positions: one at 22,318–22,359 overlapping on S and another at 29,603–29,661 located at 3′UTR of the SARS-CoV-2 genome (Tables 2, 3). The stem-loop RNA structure of Corona_pk3 and its conservation are shown across different CoVs (Figures 7A,B and 8A,B). It is a ∼55 nucleotide pseudoknot structure found at the 3′ UTR region of the CoV genomes. This cis-acting RNA element is necessary for viral genome replication. However, the mechanism is not fully understood (Williams et al., 1999).
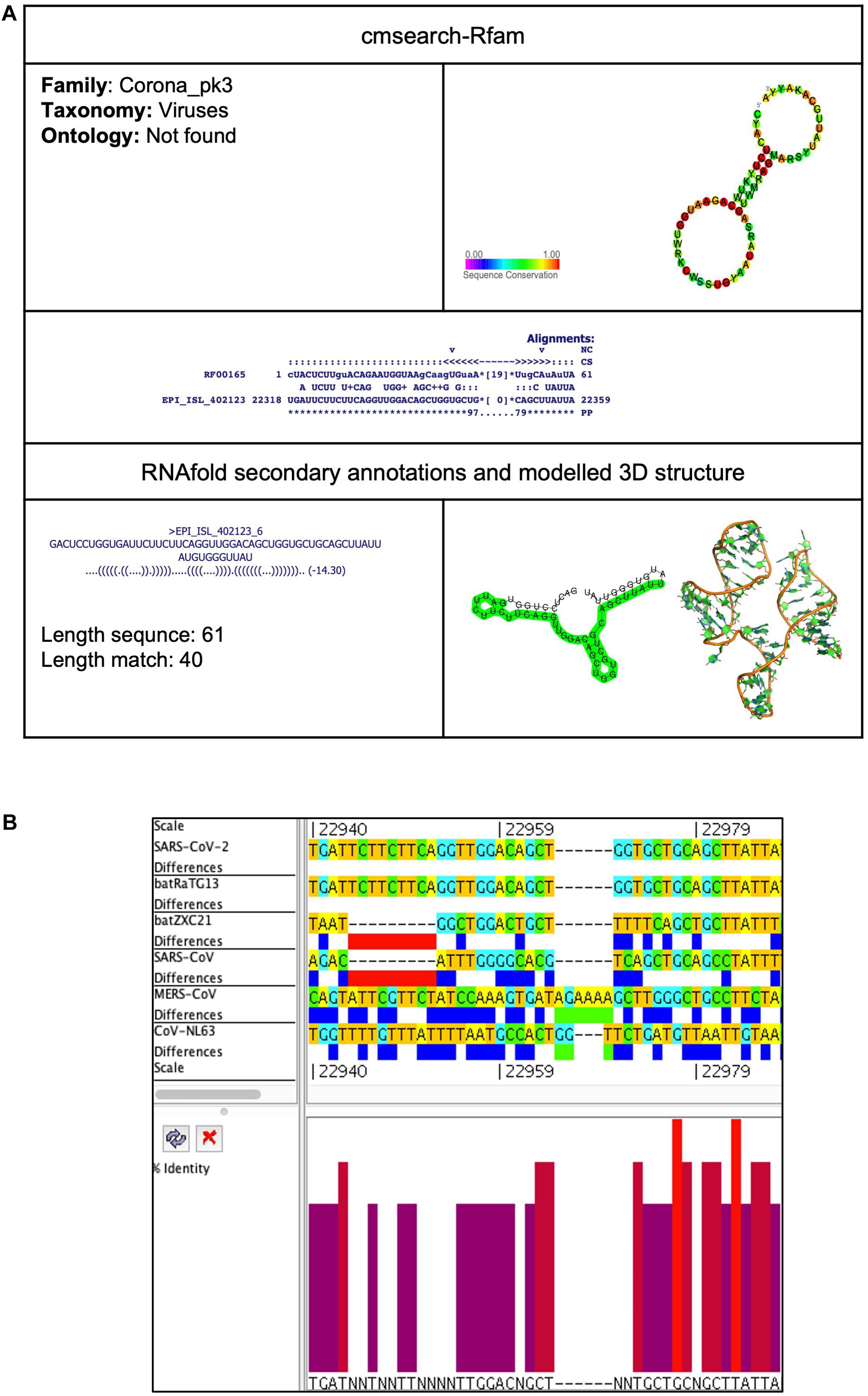
Figure 7. Corona_pk3 cis-acting RNA element in the SARS-CoV-2 genome. (A) The sequence and RNA structure of Corona_pk3 is located in position 22,318–22,359 of the SARS-CoV-2 genome. In the 3-D structure, the phosphodiester backbone is shown in orange, and the nucleotides are shown in the sticks and filled rings with elemental coloring as C green, O red, and N blue. (B) Conservation of Corona_pk3 RNA sequence from various CoVs. The Corona_pk3 is located as aligned position 22,940–22,987. The navy blue box indicates mismatched nucleotides between the two sequences, and the red box indicates gaps between two sequences. Percentage identity in the histogram in which red indicates a perfect match and magenta indicates the low similarity of nucleotide.
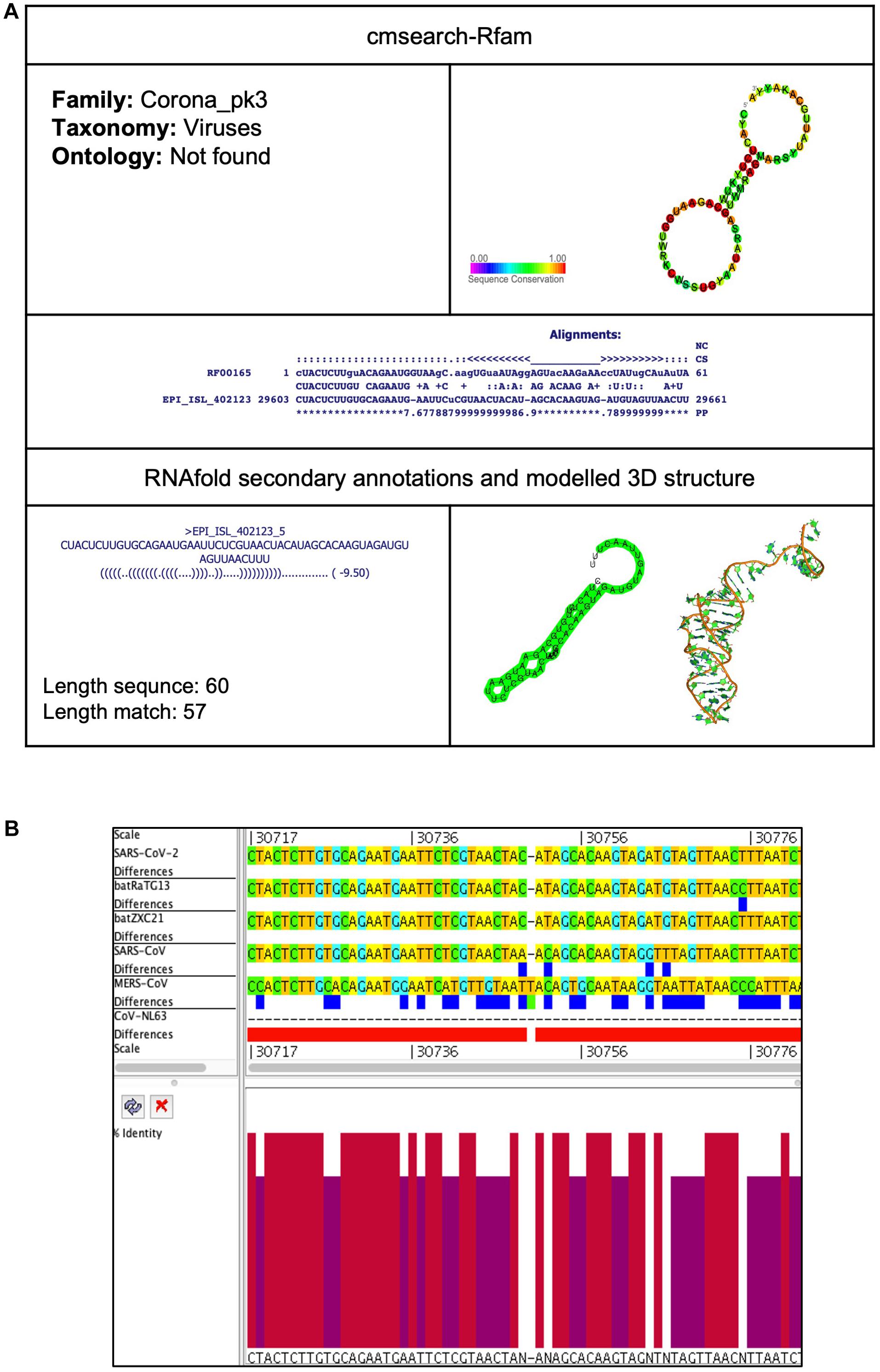
Figure 8. Corona_pk3 cis-acting RNA element in the SARS-CoV-2 genome. (A) The sequence and RNA structure of Corona_pk3 is located in position 29,603–29,661 of the SARS-CoV-2 genome. In the 3-D structure, the phosphodiester backbone is shown in orange, and the nucleotides are shown in the sticks and filled rings with elemental coloring as C green, O red, and N blue. (B) Conservation of Corona_pk3 RNA sequence from various CoVs. The Corona_pk3 is located as aligned position 30,717–30,776. The navy blue box indicates mismatched nucleotides between the two sequences, and the red box indicates gaps between two sequences. Percentae identity in the histogram in which red indicates a perfect match and magenta indicates the low similarity of nucleotide.
MS2 (Bacteriophage MS2 operator hairpin)
This element is located at position 3,258–3,282 overlapping on nsp2 of the SARS-CoV-2 genome (Tables 2, 3). The stem-loop RNA structure of MS2 and its conservation are shown across different CoVs (Figures 9A,B). The Bacteriophage MS2 genome is a 3,569-nucleotides-long positive-stranded RNA and serves as a messenger RNA to encode just four proteins: (a) maturation protein (mat), lysis protein (lys), coat protein (cp), and RNA replicase (rep) (Fiers et al., 1976). These four proteins are expressed by the same mRNA but at different levels regulated by the MS2 RNA hairpin structure. The MS2 RNA hairpin structure is located at –15 to +4 nucleotides relative to the start of the replicase gene (Helgstrand et al., 2002). The coat protein dimer binds to the MS2 RNA hairpin structure and blocks the translation of the viral replicase. Besides this, the binding of the coat protein stimulates (a) self-assembly of phage particles and (b) encapsidation of viral RNA (Ling et al., 1970).
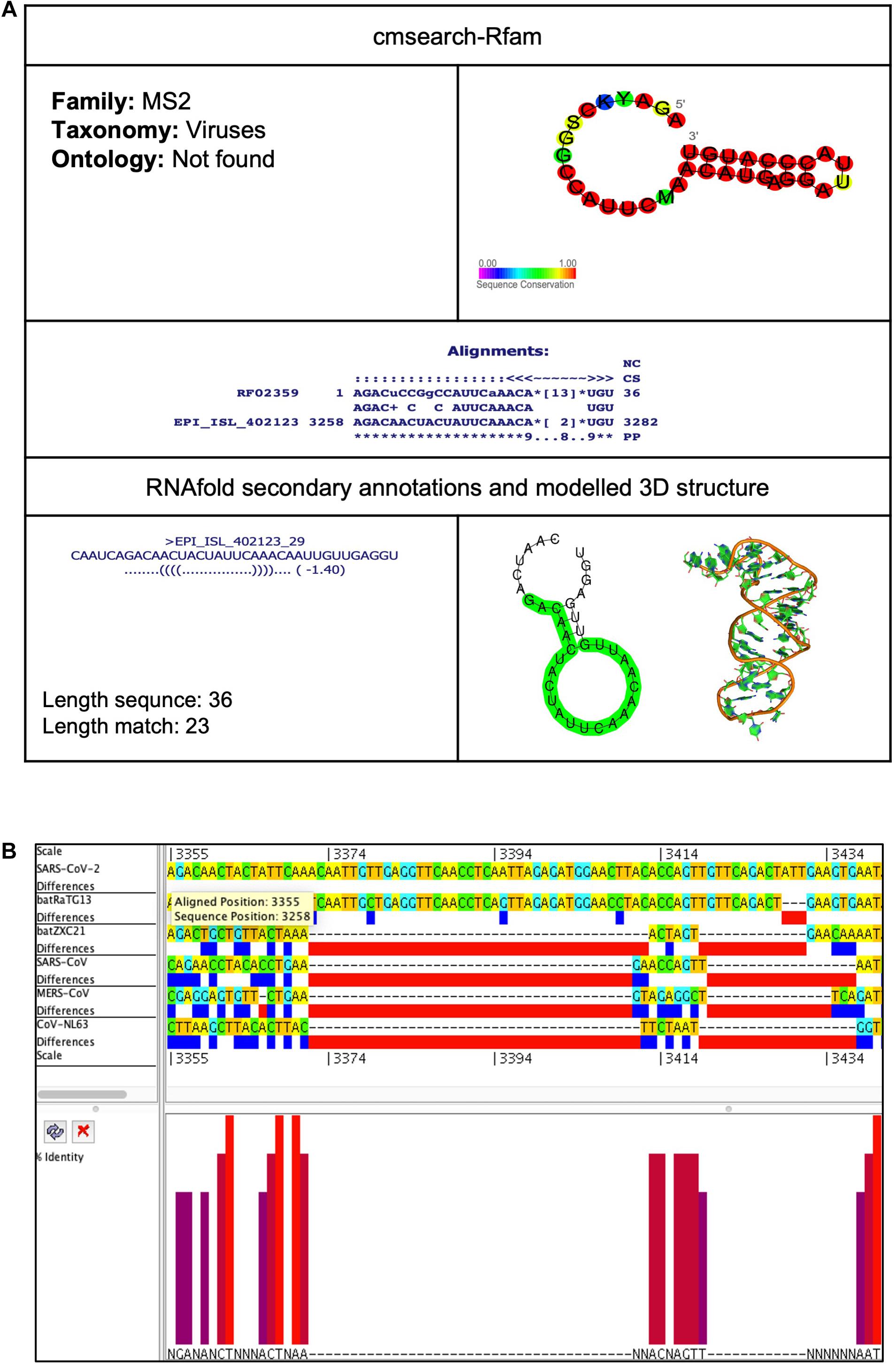
Figure 9. MS2 cis-acting RNA element in the SARS-CoV-2 genome. (A) The sequence and RNA structure of MS2 is located in position 3,258–3,282 of the SARS-CoV-2 genome. In the 3-D structure, the phosphodiester backbone is shown in orange, and the nucleotides are shown in the sticks and filled rings with elemental coloring as C green, O red, and N blue. (B) Conservation of MS2 RNA sequence from various CoVs. The MS2 is located as aligned position 3,355–3,379. The navy blue box indicates mismatched nucleotides between the two sequences, and the red box indicates gaps between two sequences. Percentage identity in the histogram in which red indicates a perfect match and magenta indicates the low similarity of nucleotide.
Comparative Analysis of cis-Acting RNA Elements Across CoVs Genomes
Besides this, we analyzed the cis-acting RNA elements across other CoVs genomes. The batRatG13 genome contains nine cis-acting RNA elements: IRES_HepA, Corona_FSE, fiv_FSE, S15, PYLIS_2, IRE_I, s2m, Corona_pk3, and Corona_pk3 (Supplementary Table 1). The batZXC21 genome contains nine cis-acting RNA elements: Corona_FSE, flavi_FSE, CRISPR-DR61, PYLIS_2, ClpQY_promoter, Histone3, s2m, RSV_RNA, and Corona_pk3 (Supplementary Table 2). The SARS-CoV genome contains only three cis-acting RNA elements: Corona_FSE, s2m, and Corona_pk3 (Supplementary Table 3). The MERS-CoV genome contains only five cis-acting RNA elements: Corona_FSE, rne5, Histone3, Corona_pk3, and Corona_pk3 (Supplementary Table 4). The CoV-NL63 genome contains only two cis-acting RNA elements: Corona_FSE and Corona_pk3 (Supplementary Table 5).
Comparative analysis of cis-acting RNA elements revealed that Corona_FSE and Corona_pk3 are found in all six studied CoVs, indicating their essential function in the CoV (Table 4). The s2m is present in all CoVs except MERS-CoV and CoV-NL63. Furthermore, PYLIS_2 is only present in SARS-CoV-2 and bat CoVs. There are three cis-acting RNA elements (Thr_leader, MAT2A_D, and MS2) that are only present in SARS-CoV-2 (Table 4).

Table 4. Comparative analysis of cis-acting RNA elements across different CoVs.
Mutation or recombination in the genetic material of a human or a virus could alter their proteins’ structure and function (Alzahrani et al., 2020; Korber et al., 2020). An infectious complementary DNA (cDNA) clone of a virus genome is a potent tool to investigate a genomic region’s function and its mutation on viral pathogenesis and transmission (van Dinten et al., 1997). A recent study constructed a full-length infectious clone of SARS-CoV-2 (icSARS-CoV-2) by connecting seven cDNA fragments spanning the SARS-CoV-2 genome (Xie et al., 2020). After transfection into cells, the RNA transcribed from ic-SARS-CoV-2 and produced infectious viruses. Furthermore, the authors developed the reporter virus, icSARS-CoV-2-mNG, by integrating the mNeonGreen reporter gene into the ORF7 region and demonstrated the antiviral activity of IFN-alpha (Xie et al., 2020). The stable clone icSARS-CoV-2-mNG is a powerful tool that is particularly useful for experimental validation and understanding the roles of these cis-acting RNA elements in viral replication, packaging, pathogenesis, and drug screening.
Conclusion
The biological function of cis-acting RNA elements depends upon their proper structure and shape as well as their ability to interact with specific ligands. Therefore, determining the structure of cis-acting RNA elements is not only crucial for understanding the function and mechanism of SARS-CoV-2 infection and replication, but it is also important to reveal their role in the origin and evolution of CoVs.
In this work, we have taken the whole genome sequence of six CoVs. First, we aligned these sequences and then generated a phylogenetic tree to understand their evolutionary relationship. Our analysis found that SARS-CoV-2 is more closely related to bat CoV batRaTG13. After that, we analyzed the SARS-CoV-2 genome to predict the cis-acting RNA elements using bioinformatics approaches. Cis-acting RNA elements interact with trans factors from the host or virus and play an essential role in regulating viral gene expression and replications. Using a bioinformatics approach, we identified 12 significant cis-acting RNA elements located in the SARS-CoV-2 genome. According to the genomic position, the elements appear as ClpQY_promoter, MS2, Fiv_FSE, Thr_leader, Histone3, S15, Corona_FSE, Corona_pk3, PYLIS_2, MAT2A_D, Corona_pk3, and s2m. Among them, Corona_FSE, Corona_pk3, and s2m are highly conserved across most of the study’s CoVs although Thr_leader, MAT2A_D, and MS2 are uniquely present in SARS-CoV-2. These elements are known in the genome of viruses and prokaryotic and eukaryotic organisms; however, specific functions and their molecular mechanisms are still elusive. However, accumulating evidence indicates that these cis-acting RNA elements might regulate viral translation, replication, encapsidation, and pathogenesis. Currently, there is no approved vaccine or treatments available against SARS-CoV-19. Therefore, these cis-acting RNA elements’ role needs to be further characterized experimentally for a better understanding of SARS-CoV-2 infection and to develop therapeutic intervention more rapidly.
Data Availability Statement
This study was conducted on publicly available data. GISAID (https://www.gisaid.org/): SARS-CoV-2 (accession number: EPI_ISL_402123) and batCoV batRaTG13 (accession number: EPI_ISL_402131). NCBI (https://www.ncbi.nlm.nih.gov/): batCoV batZXC21 (MG772934.1), SARS-CoV (NC_004718.3), MERS-CoV (NC_019843.3), and Human CoV-NL63 (NC_005831.2).
Author Contributions
FA conceived the idea, generated the data, analyzed and interpreted the results, wrote, edited and concluded the manuscript, and supervised the project. MS generated the data, examined the results, and wrote and revised the manuscript. AA-G, SA-Y, and AA-S generated the data and wrote the manuscript. MR and MW analyzed the generated data and revised the manuscript. SH and MB verified the generated data and wrote the manuscript. All authors read, revised, and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the technical support provided by the Department of Biochemistry, Department of Biology, and University of Jeddah Center for Scientific and Medical Research (UJCSMR), University of Jeddah. MS acknowledges funding from Science and Engineering Research Board, DST, India for Grant Number: SB/SRS/2019-20/23/CS.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.572702/full#supplementary-material
Footnotes
- ^ https://www.worldometers.info/CoV/
- ^ https://www.who.int/csr/sars/country/table2004_04_21/en/
- ^ https://www.who.int/emergencies/mers-cov/en/
- ^ http://ictv.global/report/
- ^ https://www.gisaid.org/
- ^ https://www.ncbi.nlm.nih.gov/
- ^ http://www.atgc-montpellier.fr/phyml/
- ^ https://github.com/rambaut/figtree/releases
- ^ https://structrnafinder.integrativebioinformatics.me/
- ^ https://rfam.xfam.org/
- ^ http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi
- ^ http://rnacomposer.ibch.poznan.pl
References
Adams, M. J., Lefkowitz, E. J., King, A. M., Bamford, D. H., Breitbart, M., Davison, A. J., et al. (2015). Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2015). Arch. Virol. 160, 1837–1850. doi: 10.1007/s00705-015-2425-z
Adams, M. J., Lefkowitz, E. J., King, A. M., Harrach, B., Harrison, R. L., Knowles, N. J., et al. (2016). Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch. Virol. 161, 2921–2949. doi: 10.1007/s00705-016-2977-6
Ahmed, F. (2019). Integrated network analysis reveals FOXM1 and MYBL2 as key regulators of cell proliferation in non-small cell lung cancer. Front. Oncol. 9:1011. doi: 10.3389/fonc.2019.01011
Ahmed, F., Ansari, H. R., and Raghava, G. P. (2009a). Prediction of guide strand of microRNAs from its sequence and secondary structure. BMC Bioinformatics 10:105. doi: 10.1186/1471-2105-10-105
Ahmed, F., Benedito, V. A., and Zhao, P. X. (2011). Mining functional elements in messenger RNAs: overview, challenges, and perspectives. Front. Plant Sci. 2:84. doi: 10.3389/fpls.2011.00084
Ahmed, F., Dai, X., and Zhao, P. X. (2015). Bioinformatics tools for achieving better gene silencing in plants. Methods Mol. Biol. 1287, 43–60. doi: 10.1007/978-1-4939-2453-0_3
Ahmed, F., Kaundal, R., and Raghava, G. P. (2013). PHDcleav: a SVM based method for predicting human Dicer cleavage sites using sequence and secondary structure of miRNA precursors. BMC Bioinformatics 14(Suppl. 14):S9. doi: 10.1186/1471-2105-14-S14-S9
Ahmed, F., Kumar, M., and Raghava, G. P. (2009b). Prediction of polyadenylation signals in human DNA sequences using nucleotide frequencies. In Silico Biol. 9, 135–148.
Ahmed, F., Senthil-Kumar, M., Dai, X., Ramu, V. S., Lee, S., Mysore, K. S., et al. (2020). pssRNAit-a web server for designing effective and specific plant siRNAs with genome-wide off-target assessment. Plant Physiol. 184, 65–81. doi: 10.1104/pp.20.00293
Alhatlani, B. Y. (2020). In silico identification of conserved cis-acting RNA elements in the SARS-CoV-2 genome. Future Virol. 15, 409–417. doi: 10.2217/fvl-2020-0163
Alzahrani, F. A., Ahmed, F., Sharma, M., Rehan, M., Mahfuz, M., Baeshen, M. N., et al. (2020). Investigating the pathogenic SNPs in BLM helicase and their biological consequences by computational approach. Sci. Rep. 10:12377. doi: 10.1038/s41598-020-69033-8
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., and Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452. doi: 10.1038/s41591-020-0820-9
Arias-Carrasco, R., Vasquez-Moran, Y., Nakaya, H. I., and Maracaja-Coutinho, V. (2018). StructRNAfinder: an automated pipeline and web server for RNA families prediction. BMC Bioinformatics 19:55. doi: 10.1186/s12859-018-2052-2
Baranov, P. V., Henderson, C. M., Anderson, C. B., Gesteland, R. F., Atkins, J. F., and Howard, M. T. (2005). Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology 332, 498–510. doi: 10.1016/j.virol.2004.11.038
Bellaousov, S., Reuter, J. S., Seetin, M. G., and Mathews, D. H. (2013). RNAstructure: web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 41, W471–W474. doi: 10.1093/nar/gkt290
Benard, L., Philippe, C., Ehresmann, B., Ehresmann, C., and Portier, C. (1996). Pseudoknot and translational control in the expression of the S15 ribosomal protein. Biochimie 78, 568–576. doi: 10.1016/s0300-9084(96)80003-4
Bustin, S. A., and Nolan, T. (2020). RT-qPCR testing of SARS-CoV-2: a primer. Int. J. Mol. Sci. 21:3004. doi: 10.3390/ijms21083004
Choudhry, H., Bakhrebah, M. A., Abdulaal, W. H., Zamzami, M. A., Baothman, O. A., Hassan, M. A., et al. (2019). Middle East respiratory syndrome: pathogenesis and therapeutic developments. Future Virol. 14, 237–246. doi: 10.2217/fvl-2018-0201
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses (2020). The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544. doi: 10.1038/s41564-020-0695-z
Debnath, M., Banerjee, M., and Berk, M. (2020). Genetic gateways to COVID-19 infection: implications for risk, severity, and outcomes. FASEB J. 34, 8787–8795. doi: 10.1096/fj.202001115R
Di Giorgio, S., Martignano, F., Torcia, M. G., Mattiuz, G., and Conticello, S. G. (2020). Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 6:eabb5813. doi: 10.1126/sciadv.abb5813
Felden, B. (2007). RNA structure: experimental analysis. Curr. Opin. Microbiol. 10, 286–291. doi: 10.1016/j.mib.2007.05.001
Fiers, W., Contreras, R., Duerinck, F., Haegeman, G., Iserentant, D., Merregaert, J., et al. (1976). Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene. Nature 260, 500–507. doi: 10.1038/260500a0
Gonzalez, S. A., and Affranchino, J. L. (2018). Properties and functions of feline immunodeficiency virus gag domains in virion assembly and budding. Viruses 10:261. doi: 10.3390/v10050261
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Helgstrand, C., Grahn, E., Moss, T., Stonehouse, N. J., Tars, K., Stockley, P. G., et al. (2002). Investigating the structural basis of purine specificity in the structures of MS2 coat protein RNA translational operator hairpins. Nucleic Acids Res. 30, 2678–2685. doi: 10.1093/nar/gkf371
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Kruger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e278. doi: 10.1016/j.cell.2020.02.052
Jonassen, C. M., Jonassen, T. O., and Grinde, B. (1998). A common RNA motif in the 3′ end of the genomes of astroviruses, avian infectious bronchitis virus and an equine rhinovirus. J Gen Virol 79(Pt 4), 715–718. doi: 10.1099/0022-1317-79-4-715
Kolter, R., and Yanofsky, C. (1982). Attenuation in amino acid biosynthetic operons. Annu. Rev. Genet. 16, 113–134. doi: 10.1146/annurev.ge.16.120182.000553
Korber, B., Fischer, W. M., Gnanakaran, S., Yoon, H., Theiler, J., Abfalterer, W., et al. (2020). Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182, 812–827.e819. doi: 10.1016/j.cell.2020.06.043
Li, Y., Yang, X., Wang, N., Wang, H., Yin, B., Yang, X., et al. (2020). The divergence between SARS-CoV-2 and RaTG13 might be overestimated due to the extensive RNA modification. Future Virol. 15, 341–347. doi: 10.2217/fvl-2020-0066
Ling, C. M., Hung, P. P., and Overby, L. R. (1970). Independent assembly of Qbeta and MS2 phages in doubly infected Escherichia coli. Virology 40, 920–929. doi: 10.1016/0042-6822(70)90138-8
Liu, Y., Wimmer, E., and Paul, A. V. (2009). Cis-acting RNA elements in human and animal plus-strand RNA viruses. Biochim. Biophys. Acta 1789, 495–517. doi: 10.1016/j.bbagrm.2009.09.007
Lorenz, R., Bernhart, S. H., Höner Zu Siederdissen, C., Tafer, H., Flamm, C., Stadler, P. F., et al. (2011). ViennaRNA package 2.0. Algorithms Mol. Biol. 6:26. doi: 10.1186/1748-7188-6-26
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Lu, G., Wang, Q., and Gao, G. F. (2015). Bat-to-human: spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 23, 468–478. doi: 10.1016/j.tim.2015.06.003
Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., et al. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574. doi: 10.1016/S0140-6736(20)30251-8
Masters, P. S. (2019). Coronavirus genomic RNA packaging. Virology 537, 198–207. doi: 10.1016/j.virol.2019.08.031
Mathews, D. H., Moss, W. N., and Turner, D. H. (2010). Folding and finding RNA secondary structure. Cold Spring Harb. Perspect. Biol. 2:a003665. doi: 10.1101/cshperspect.a003665
Moore, B. J. B., and June, C. H. (2020). Cytokine release syndrome in severe COVID-19. Science 55:eabb8925. doi: 10.1126/science.abb8925
Morikawa, S., and Bishop, D. H. (1992). Identification and analysis of the gag-pol ribosomal frameshift site of feline immunodeficiency virus. Virology 186, 389–397. doi: 10.1016/0042-6822(92)90004-9
Namy, O., Rousset, J. P., Napthine, S., and Brierley, I. (2004). Reprogrammed genetic decoding in cellular gene expression. Mol. Cell 13, 157–168. doi: 10.1016/s1097-2765(04)00031-0
Nawrocki, E. P., Burge, S. W., Bateman, A., Daub, J., Eberhardt, R. Y., Eddy, S. R., et al. (2015). Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–D137. doi: 10.1093/nar/gku1063
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Nishii, W., and Takahashi, K. (2003). Determination of the cleavage sites in SulA, a cell division inhibitor, by the ATP-dependent HslVU protease from Escherichia coli. FEBS Lett. 553, 351–354. doi: 10.1016/s0014-5793(03)01044-5
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385
Paraskevis, D., Kostaki, E. G., Magiorkinis, G., Panayiotakopoulos, G., Sourvinos, G., and Tsiodras, S. (2020). Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect. Genet. Evol. 79, 104212. doi: 10.1016/j.meegid.2020.104212
Parker, B. J., Moltke, I., Roth, A., Washietl, S., Wen, J., Kellis, M., et al. (2011). New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res. 21, 1929–1943. doi: 10.1101/gr.112516.110
Perlman, S., and Netland, J. (2009). Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7, 439–450. doi: 10.1038/nrmicro2147
Popenda, M., Blazewicz, M., Szachniuk, M., and Adamiak, R. W. (2008). RNA FRABASE version 1.0: an engine with a database to search for the three-dimensional fragments within RNA structures. Nucleic Acids Res. 36, D386–D391. doi: 10.1093/nar/gkm786
Popenda, M., Szachniuk, M., Blazewicz, M., Wasik, S., Burke, E. K., Blazewicz, J., et al. (2010). RNA FRABASE 2.0: an advanced web-accessible database with the capacity to search the three-dimensional fragments within RNA structures. BMC Bioinformatics 11:231. doi: 10.1186/1471-2105-11-231
Rehan, M., and Bajouh, O. S. (2018). Virtual screening of naphthoquinone analogs for potent inhibitors against the cancer-signaling PI3K/AKT/mTOR pathway. J. Cell. Biochem. 120, 1328–1339. doi: 10.1002/jcb.27100
Ren, L. L., Wang, Y. M., Wu, Z. Q., Xiang, Z. C., Guo, L., Xu, T., et al. (2020). Identification of a novel coronavirus causing severe pneumonia in human: a descriptive study. Chin. Med. J. 133, 1015–1024. doi: 10.1097/CM9.0000000000000722
Robertson, M. P., Igel, H., Baertsch, R., Haussler, D., Ares, M. Jr., and Scott, W. G. (2005). The structure of a rigorously conserved RNA element within the SARS virus genome. PLoS Biol. 3:e5. doi: 10.1371/journal.pbio.0030005
Shang, J., Ye, G., Shi, K., Wan, Y., Luo, C., Aihara, H., et al. (2020). Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224. doi: 10.1038/s41586-020-2179-y
Tang, X., Wu, C., Li, X., Song, Y., Yao, X., Wu, X., et al. (2020). On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 7, 1012–1023. doi: 10.1093/nsr/nwaa036
Theobald-Dietrich, A., Giege, R., and Rudinger-Thirion, J. (2005). Evidence for the existence in mRNAs of a hairpin element responsible for ribosome dependent pyrrolysine insertion into proteins. Biochimie 87, 813–817. doi: 10.1016/j.biochi.2005.03.006
Tu, S. L., Staheli, J. P., McClay, C., McLeod, K., Rose, T. M., and Upton, C. (2018). Base-by-base version 3: new comparative tools for large virus genomes. Viruses 10:637. doi: 10.3390/v10110637
van Dinten, L. C., den Boon, J. A., Wassenaar, A. L., Spaan, W. J., and Snijder, E. J. (1997). An infectious arterivirus cDNA clone: identification of a replicase point mutation that abolishes discontinuous mRNA transcription. Proc. Natl. Acad. Sci. U.S.A. 94, 991–996. doi: 10.1073/pnas.94.3.991
Wakida, H., Kawata, K., Yamaji, Y., Hattori, E., Tsuchiya, T., Wada, Y., et al. (2020). Stability of RNA sequences derived from the coronavirus genome in human cells. Biochem. Biophys. Res. Commun. 527, 993–999. doi: 10.1016/j.bbrc.2020.05.008
Williams, A. S., and Marzluff, W. F. (1995). The sequence of the stem and flanking sequences at the 3′ end of histone mRNA are critical determinants for the binding of the stem-loop binding protein. Nucleic Acids Res. 23, 654–662. doi: 10.1093/nar/23.4.654
Williams, G. D., Chang, R. Y., and Brian, D. A. (1999). A phylogenetically conserved hairpin-type 3′ untranslated region pseudoknot functions in coronavirus RNA replication. J. Virol. 73, 8349–8355.
Woo, P. C., Lau, S. K., Huang, Y., and Yuen, K. Y. (2009). Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. 234, 1117–1127. doi: 10.3181/0903-MR-94
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20:257. doi: 10.1186/s13059-019-1891-0
Wu, Y., Shi, B., Ding, X., Liu, T., Hu, X., Yip, K. Y., et al. (2015). Improved prediction of RNA secondary structure by integrating the free energy model with restraints derived from experimental probing data. Nucleic Acids Res. 43, 7247–7259. doi: 10.1093/nar/gkv706
Xie, X., Muruato, A., Lokugamage, K. G., Narayanan, K., Zhang, X., Zou, J., et al. (2020). An infectious cDNA Clone of SARS-CoV-2. Cell Host Microbe 27, 841–848.e843. doi: 10.1016/j.chom.2020.04.004
Yang, X., Yu, Y., Xu, J., Shu, H., Xia, J., Liu, H., et al. (2020). Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir. Med. 8, 475–481. doi: 10.1016/S2213-2600(20)30079-5
Yu, E. T., Zhang, Q., and Fabris, D. (2005). Untying the FIV frameshifting pseudoknot structure by MS3D. J. Mol. Biol. 345, 69–80. doi: 10.1016/j.jmb.2004.10.014
Zanier, K., Luyten, I., Crombie, C., Muller, B., Schumperli, D., Linge, J. P., et al. (2002). Structure of the histone mRNA hairpin required for cell cycle regulation of histone gene expression. RNA 8, 29–46. doi: 10.1017/s1355838202014061
Zhang, C., Wu, Z., Li, J. W., Zhao, H., and Wang, G. Q. (2020). The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int. J. Antimicrob. Agents 55:105954. doi: 10.1016/j.ijantimicag.2020.105954
Zhou, P., Yang, X. L., Wang, X. G., Hu, B., Zhang, L., Zhang, W., et al. (2020a). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. doi: 10.1038/s41586-020-2012-7
Keywords: SARS-CoV-2, COVID-19, cis-acting RNA elements, coronavirus genome organization, coronavirus frameshifting stimulation element, RNA structure alignment
Citation: Ahmed F, Sharma M, Al-Ghamdi AA, Al-Yami SM, Al-Salami AM, Refai MY, Warsi MK, Howladar SM and Baeshen MN (2020) A Comprehensive Analysis of cis-Acting RNA Elements in the SARS-CoV-2 Genome by a Bioinformatics Approach. Front. Genet. 11:572702. doi: 10.3389/fgene.2020.572702
Received: 15 June 2020; Accepted: 14 October 2020;
Published: 23 December 2020.
Edited by:
William C. Cho, QEH, Hong KongReviewed by:
Stefano De Pretis, Center for Genomic Science, Italian Institute of Technology (IIT), ItalySayed Haidar Abbas Raza, Northwest A and F University, China
Copyright © 2020 Ahmed, Sharma, Al-Ghamdi, Al-Yami, Al-Salami, Refai, Warsi, Howladar and Baeshen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Firoz Ahmed, ZmFobWVkMUB1ai5lZHUuc2E=; Zmlyb3ouaW10ZWNoQGdtYWlsLmNvbQ==