 Meg Whatley1
†
Meg Whatley1
† Abbie Francis1
,2
†
Abbie Francis1
,2
† Zi Ying Ng1
Zi Ying Ng1
 Xin Ee Khoh1
,3
Xin Ee Khoh1
,3
 Marcus D. Atlas1
,4
Marcus D. Atlas1
,4
 Rodney J. Dilley1
,4
,5
Rodney J. Dilley1
,4
,5
 Elaine Y. M. Wong1
,4
,6
*
Elaine Y. M. Wong1
,4
,6
*- 1Ear Science Institute Australia, Nedlands, WA, Australia
- 2Emergency Medicine, The University of Western Australia, Nedlands, WA, Australia
- 3School of Human Sciences, The University of Western Australia, Nedlands, WA, Australia
- 4Ear Sciences Centre, The University of Western Australia, Nedlands, WA, Australia
- 5Centre for Cell Therapy and Regenerative Medicine, The University of Western Australia, Perth, WA, Australia
- 6School of Pharmacy and Biomedical Sciences, Faculty of Health Sciences, Curtin University, Bentley, WA, Australia
Usher syndrome (USH) is an autosomal recessive (AR) disorder that permanently and severely affects the senses of hearing, vision, and balance. Three clinically distinct types of USH have been identified, decreasing in severity from Type 1 to 3, with symptoms of sensorineural hearing loss (SNHL), retinitis pigmentosa (RP), and vestibular dysfunction. There are currently nine confirmed and two suspected USH-causative genes, and a further three candidate loci have been mapped. The proteins encoded by these genes form complexes that play critical roles in the development and maintenance of cellular structures within the inner ear and retina, which have minimal capacity for repair or regeneration. In the cochlea, stereocilia are located on the apical surface of inner ear hair cells (HC) and are responsible for transducing mechanical stimuli from sound pressure waves into chemical signals. These signals are then detected by the auditory nerve fibers, transmitted to the brain and interpreted as sound. Disease-causing mutations in USH genes can destabilize the tip links that bind the stereocilia to each other, and cause defects in protein trafficking and stereocilia bundle morphology, thereby inhibiting mechanosensory transduction. This review summarizes the current knowledge on Usher syndrome with a particular emphasis on mutations in USH genes, USH protein structures, and functional analyses in animal models. Currently, there is no cure for USH. However, the genetic therapies that are rapidly developing will benefit from this compilation of detailed genetic information to identify the most effective strategies for restoring functional USH proteins.
Introduction
Usher Syndrome
Usher syndrome (USH) is an autosomal recessive (AR) disorder characterized by sensorineural hearing loss (SNHL), vision loss due to retinitis pigmentosa (RP), and vestibular dysfunction (Geleoc and Holt, 2014; Mathur and Yang, 2015). USH has an estimated global prevalence of between 4 and 17 cases per 100,000 individuals, and accounts for approximately 50% of all hereditary deaf-blindness cases and 3–6% of all childhood hearing loss (HL) cases (Boughman et al., 1983; Fortnum et al., 2001; Kimberling et al., 2010; Lenarduzzi et al., 2015). The syndrome was first described in 1858, by German ophthalmologist Albrecht Von Graefe, in three siblings with simultaneous congenital deafness and RP (von Graefe, 1858; Boughman et al., 1983). USH was later named after Charles Usher, a Scottish ophthalmologist who established its heritability in 1914 based on 69 cases (Usher, 1914).
Usher syndrome is genetically heterogeneous with nine causative genes confirmed, two suspected, and a further three candidate loci having been mapped. USH has been considered to be a monogenic, genetically heterogeneous disease from the very beginning. However, several studies have shown the digenic inheritance of deafness caused by mutations in USH genes and USH modifier PDZD7 in mice and humans (Zheng et al., 2005, 2012; Ebermann et al., 2011; Bonnet et al., 2016). Zheng et al. (2005, 2012) demonstrated that compound heterozygous Cdh23/Pcdh15; Myo7a/Ush1g; Myo7a/Cdh23; Myo7a/Pcdh15 mice exhibited HL and disorganized hair-cell stereocilia. Inner ear hair cells (HC) have no capacity for regeneration after birth. Once damaged or when these cells begin to deteriorate, which can occur during development, HL progresses and is irreversible (Bermingham-McDonogh and Reh, 2011; Franco and Malgrange, 2017).
An understanding of the USH protein functions and interactions within the inner ear will allow for functional domain analysis and therefore the prediction of mutation pathogenicity within USH genes. This knowledge is critical for the successful development of SNHL treatment in USH patients, since almost all modern therapies, including gene, cell, and drug therapies, rely on the thorough understanding of the molecular basis of the disease. Here, we summarize the current literature on the USH genes and their protein structure, function, and localization, and elucidate the disease mechanisms underlying known USH-causing mutations.
Inner Ear Structure and Function
The organ of Corti is the hearing sensory organ located within the cochlea of the human inner ear and contains approximately 16,000 HC (Schwander et al., 2012; Mathur and Yang, 2015). The HC are arranged as one row of inner HC (IHC) and three rows of outer HC (OHC; Mathur and Yang, 2015; Franco and Malgrange, 2017). All HC have a bundle of approximately 100 actin-rich microvilli, called stereocilia, on their apical surface that are arranged in an inverted V shape, with the length of these projections decreasing stepwise from the tallest stereocilium adjacent to the kinocilium (Pickles et al., 1984; Kachar et al., 2000; Sakaguchi et al., 2009). The single tubulin-filled kinocilium composed of a 9 + 2 microtubule structure is located on the apical surface of HC (Sakaguchi et al., 2009). Upon maturation of the mammalian cochlea, the ankle links and most lateral links are eventually lost and the kinocilium is reabsorbed such that mature mammalian cochlear HC lack kinocilia (Figure 1; Hudspeth and Jacobs, 1979; Verpy et al., 2011). Five different types of supporting cells are organized in rows along the organ of Corti, namely: (1) Hensen’s cells; (2) Deiters’ cells; (3) Pillar cells; (4) Inner phalangeal cells; and (5) Border cells. Supporting cells in mature sensory epithelia preserve the structural integrity of the sensory organs, modulate homeostasis, and maintain the extracellular matrices that enable hair cell mechanotransduction (Slepecky et al., 1995). The sound transduction process occurs at these stereocilia, where mechanical stimuli are converted by a mechanotransduction process into chemical signals, which are then transmitted via auditory nerve fibers through to the brain (Tilney et al., 1980).
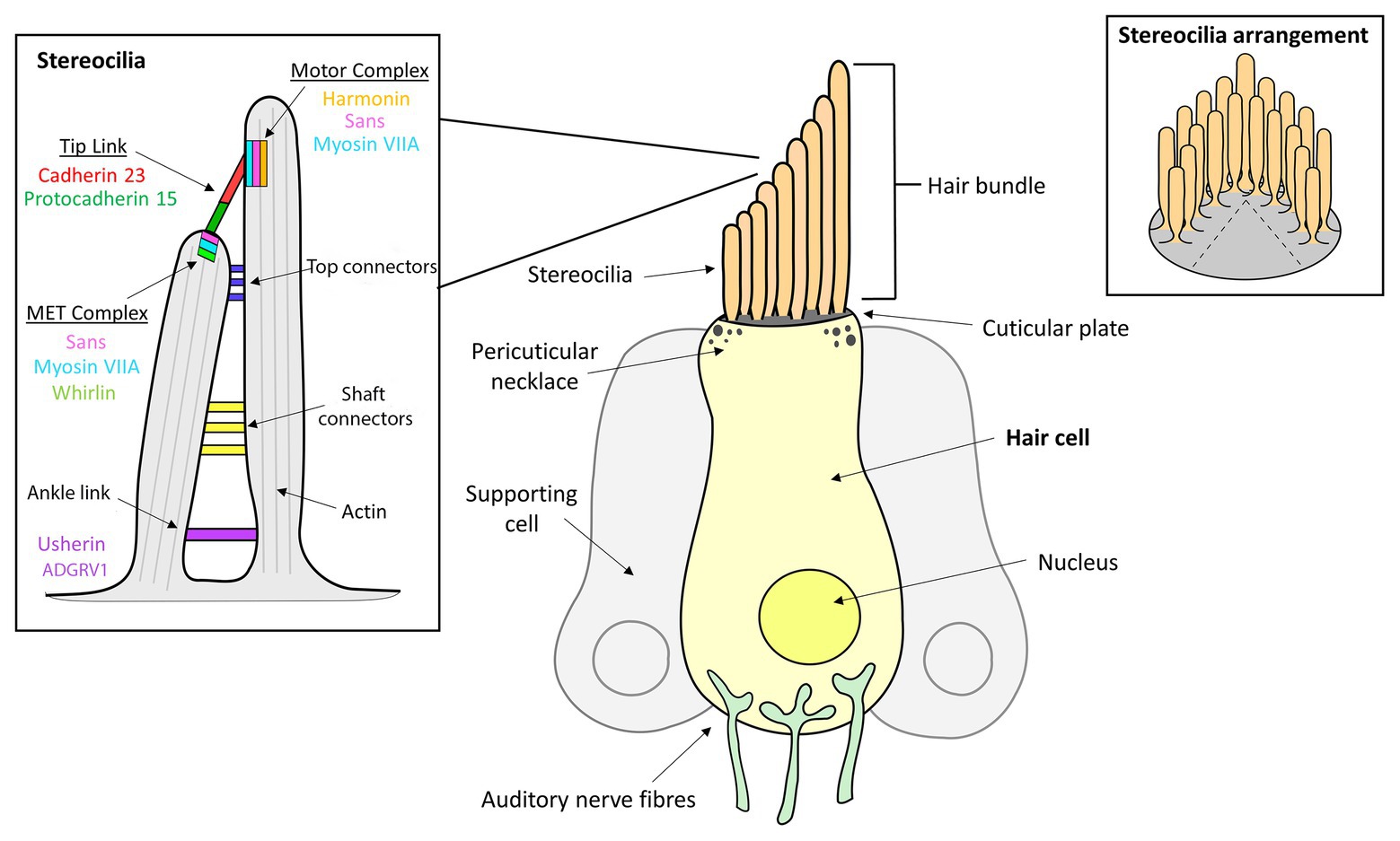
Figure 1. Inner ear hair cell (HC) structure showing the locations of several Usher syndrome (USH) proteins. The apical side of each hair cell expresses a bundle of highly-organized, actin-filled stereocilia, in which the mechanotransduction takes place. The stereocilia are held together by transient ankle links at their base, horizontal shaft links along their length, horizontal tip links near the top and diagonal tip links at their apices.
In the developing cochlea, hair cell development and maturation proceed in two perpendicular gradients: from the base to apex; and from the medial to lateral aspects of the cochlea. During hair bundle development, the kinocilium derives from the primary cilium, migrates from the center to the lateral edge of the hair cell apex. After that, microvilli around the kinocilium elongate to form stereocilia of graded heights. Stereocilia undergo further row-specific, differential outgrowth, eventually forming a hair bundle with a staircase organization (Kelly and Chen, 2009; Wong et al., 2016). The planar polarity and staircase-like pattern of the hair bundle are essential for the mechanoelectrical transduction (MET) function of inner ear sensory cells.
The stereocilia are angled toward the kinocilium and are anchored together through a series of extracellular protein filaments that interconnect and link them, the molecular composition of which change during developmental stages (Ahmed et al., 2006; Sakaguchi et al., 2009; Indzhykulian et al., 2013). Tip links are protein complexes that connect the tip of the shorter stereocilium to their taller neighbor and are a critical component of mechanotransduction since they are involved in transmitting physical force to the mechanotransduction channels located at the tip of the stereocilia (Assad et al., 1991; Sakaguchi et al., 2009; Maeda et al., 2017). Tip links are comprised primarily of protocadherin 15 and cadherin 23, and interact with a number of other USH proteins that both anchor and stiffen the links (Corns et al., 2018). Additionally, stereocilia are joined by horizontal or lateral links that are involved in maintaining the organization and structure of the bundle, as well as the stiffness required for mechanotransduction to occur (Hackney and Furness, 2013). Specifically, there are top and shaft horizontal connectors, which have slightly different composition and function. During early development, the bundles also contain transient ankle links and kinociliary links, involved in interconnecting the base of each of the stereocilia and the kinocilium to the bundle, respectively (Hackney and Furness, 2013).
Mechanoelectrical Transduction
The mechanoelectrical transduction process in the cochlea is essential for the conversion of mechanical signals into chemical responses, to communicate sound signals to the brain (Grati and Kachar, 2011; Blanco-Sanchez et al., 2018). MET relies on the appropriate organization of actin-based stereocilia to open the potassium channels and depolarize the cells (Corns et al., 2018). Mutations causing abnormal development of these HC structures can disrupt the MET process, contributing to SNHL and deafness (Blanco-Sanchez et al., 2018).
The resting potential of HC is approximately −60 mV (Bracho and Budelli, 1978; Gillespie and Muller, 2009). Upon opening of the mechanotransduction channels, the cell depolarizes toward 0 mV, which initiates a signaling cascade (Gillespie and Muller, 2009). Sound waves entering the ear cause displacement of the hair bundle, which results in the tip links “pulling” open the mechanotransduction ion channels, specifically those for potassium and calcium. The ion channels are located at the lower end of the tip link, and their opening causes an inward positive current and depolarization of the cell (Powers et al., 2017). This depolarization activates the release of neurotransmitters at the base of the HC, which excite the auditory nerve fibers and result in sound perception (Gillespie and Muller, 2009; Powers et al., 2017).
Usher Subtypes and Genes
Three clinical subtypes of USH have been defined – Type 1, Type 2, and Type 3 – based upon the presence, severity, and progression of auditory, visual, and vestibular symptoms. Type 1 (USH1) accounts for approximately one third of USH cases and is the most severe form, with profound SNHL and vestibular dysfunction from birth, as well as progressive RP (Smith et al., 1992b, 1994; Weil et al., 1995; Hope et al., 1997). Type 2 (USH2) is the most common subtype, accounting for more than half of all USH cases worldwide (Ng et al., 2019). USH2 patients present with moderate-to-severe SNHL, normal vestibular function, and RP that begins generally during puberty. Type 3 (USH3) is the rarest form, accounting for approximately 2% of all cases (Jouret et al., 2019). Patients with USH3 have normal physiology at birth, with variable levels of hearing, vision, and vestibular deterioration over time (Ng et al., 2019). HL occurs prior to visual symptoms in all three subtypes (Jouret et al., 2019). Fourteen USH loci have been mapped so far, with nine for USH1, three for USH2, two for USH3, as well as one USH modifier and one atypical USH gene (Table 1; Mathur and Yang, 2015). However, some cases are not attributed to these genes and are instead categorized as atypical USH (Otterstedde et al., 2001).
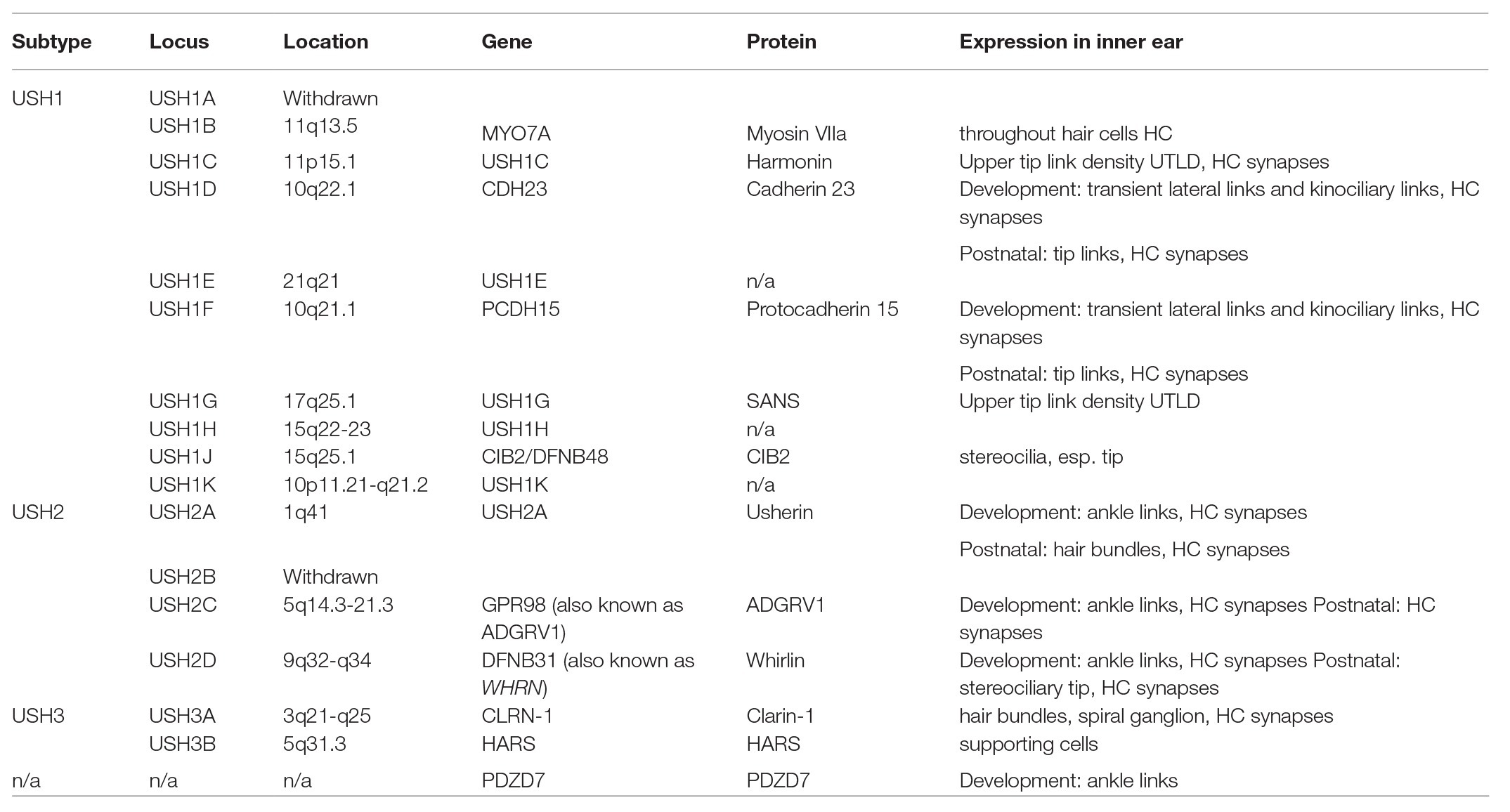
Table 1. The subtypes of USH and their known associated genes and proteins.
USH Type 1
In addition to congenital SNHL and vestibular dysfunction, USH1 patients suffer from tunnel vision within the first decade of life and are often considered legally blind by mid-life due to RP (Lenassi et al., 2014; Ng et al., 2019). USH1 is further subtyped based on the specific causative gene involved. To date, nine causative loci have been mapped for USH1 with mutations in MYO7A mutations being the most common cause of USH1, followed by mutations in CDH23 and PCDH15 (Ahmed et al., 2001; Jouret et al., 2019). USH1A was first described and mapped to the long arm of chromosome 14 at position 32.1 (14q32.1) in France (Kaplan et al., 1992). The USH1A subtype has now been withdrawn as the disease-causing mutations identified by Kaplan et al. (1992) were later found to be located at other USH type 1 loci (Larget-Piet et al., 1994; Gerber et al., 2006). Although the loci of USH1E, USH1H, and USH1K have been mapped, the resulting proteins have not yet been identified (Chaib et al., 1997; Ahmed et al., 2009; Jaworek et al., 2012).
Usher Type 1B: Myosin VIIA
Mutations in MYO7A are associated with USH1B and account for approximately 21% of all USH cases and 50% of USH1 cases (Weil et al., 1995; Roux et al., 2006; Jaijo et al., 2007; Yoshimura et al., 2016; Jouret et al., 2019). Myosin VIIa encodes an actin-binding motor protein that facilitates the movement of intracellular proteins along the actin filaments of HC stereocilia, thereby aiding the migration of proteins into their appropriate positions (Maravillas-Montero and Santos-Argumedo, 2012). Myosin VIIa also participates in the development of actin bundles in HC (Heissler and Manstein, 2012; Sato et al., 2017). Therefore, it contributes to the spatial and temporal organization of proteins in HC, and defects in this mechanism can significantly influence stereocilia formation and affect cell function (Kros et al., 2002; Jaijo et al., 2007). In the cochlea, myosin VIIa is expressed throughout the stereocilia and cytoplasm of both IHC and OHC, particularly at the cuticular plate and peri-cuticular necklace region of stereocilia (Hasson et al., 1995; Heissler and Manstein, 2012; Sato et al., 2017). Abnormal myosin VIIa expression in the inner ear will affect protein trafficking and results in major functional abnormalities of the cochlea HC and their stereocilia, which interferes with sound transduction and therefore leads to SNHL or USH (Kros et al., 2002; Jaijo et al., 2007).
Structure
The MYO7A gene spans 120 kilobases (kb) of genomic DNA, contains 49 exons, and has a 7.4 kb transcribed region (Kimberling and Smith, 1992; Smith et al., 1992a; Guilford et al., 1994; Kelley et al., 1997). MYO7A encodes a 254 kDa, 2215 amino acid (aa) protein comprised of a highly conserved N-terminal motor domain containing actin and ATP binding sites (Figure 2A; Kelley et al., 1997; Cheng et al., 2018). The motor domain binds actin filaments and uses energy generated from ATP hydrolysis to move along the filament, allowing intracellular movement of organelles (Udovichenko et al., 2002; Woolner and Bement, 2009). This domain can be monomeric or dimeric, depending on dimerization with the USH1G protein, SANS (Kiehart et al., 2004; Yu et al., 2017). A recently discovered 56 aa SRC Homology 3 (SH3)-like subdomain has been identified near the motor domain (Maravillas-Montero and Santos-Argumedo, 2012). Next is the neck region, with five isoleucine-glutamine calmodulin-binding (IQ) motifs that interact with calmodulin light chains and act as a lever to allow the motor domain to move (Todorov et al., 2001; Udovichenko et al., 2002; Maravillas-Montero and Santos-Argumedo, 2012). There is a predicted single alpha-helix (SAH) domain that is thought to be a divergent type of SAH as it lacks the dense networks of charged amino acids usually typical of SAH domains (Simm et al., 2017). Lastly is a C-terminal tail region with two sets of Myosin Tail Homology 4 (MyTH4) and Protein-Ezrin-Radixin-Moesin (FERM) domains separated by an SH3 domain (Kelley et al., 1997; Weck et al., 2016). The main function of the tail region is to serve as an anchor to position the motor domain such that it can interact with actin filaments (Inoue and Ikebe, 2003; Li et al., 2017).
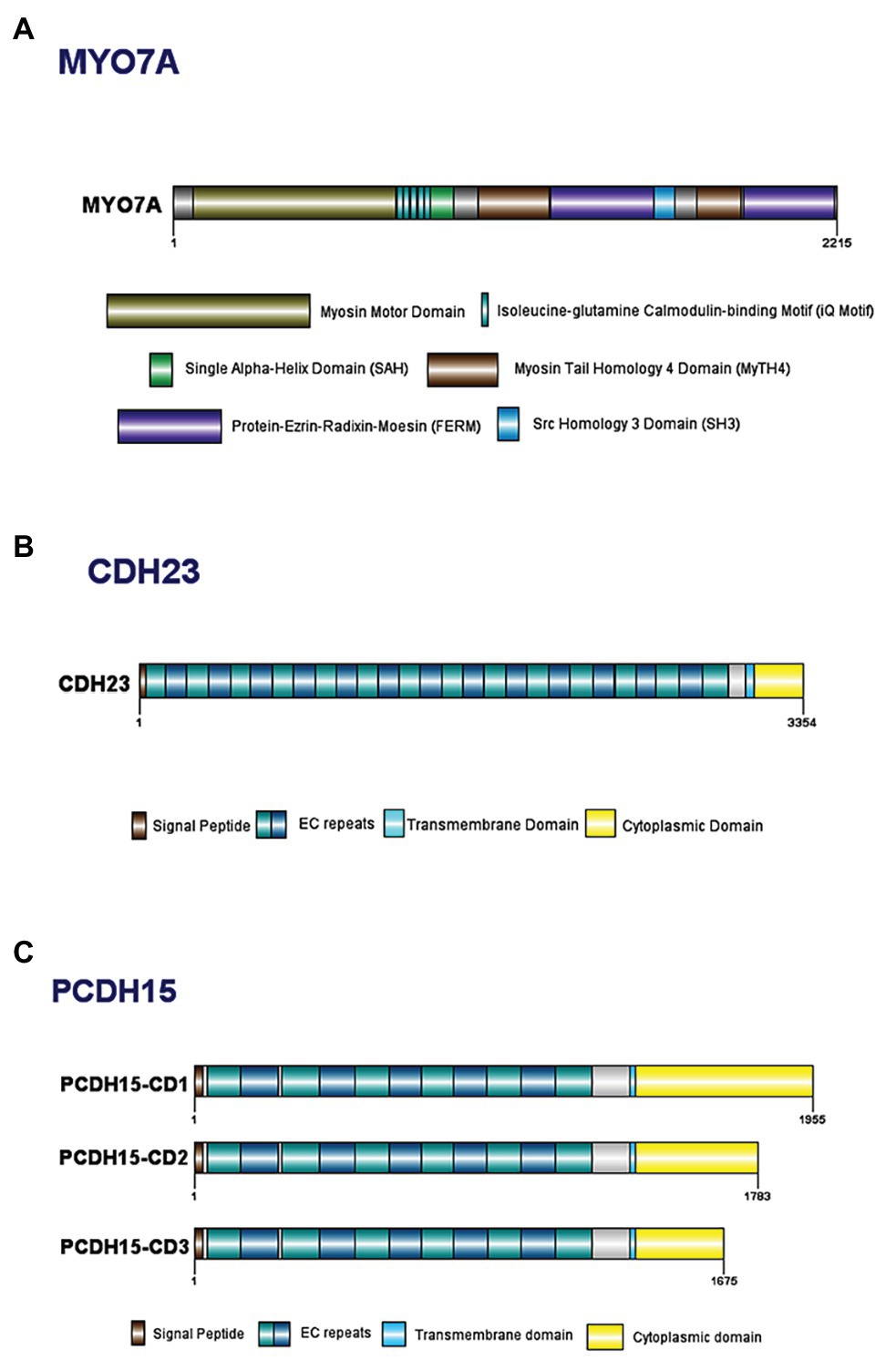
Figure 2. Domain structure of key Usher Type I proteins and their major known isoforms: (A) myosin VIIa; (B) cadherin 23; and (C) protocadherin 15.
Mutations
Various USH mutations are located throughout the MYO7A gene; however, no hotspot mutations have yet been identified (Jaijo et al., 2007). Approximately, 60% of USH1B causative mutations result in a prematurely truncated protein and about 36% are missense mutations (Le Quesne Stabej et al., 2012). As well as USH, two forms of non-syndromic HL and deafness, DFNB2 and DFNA11, are associated with MYO7A mutations (Tamagawa et al., 1996; Weil et al., 1997; Liu et al., 1998). DFNB2 is a rare AR disorder, resulting in severe progressive hearing dysfunction with either congenital or early childhood-onset (Hildebrand et al., 2010). DFNA11 is an autosomal dominant disorder, showing progressive HL with variable vestibular dysfunction (Tamagawa et al., 1996; Venkatesh et al., 2015). The phenotype, either syndromic or isolated HL, is dependent on the location and nature of the mutation within the MYO7A gene, and is the result of allelic heterogeneity (Weil et al., 1997; Venkatesh et al., 2015).
Animal Models
Shaker-1 mice have normal early development, followed by progressive hair bundle disorganization and abnormal auditory responses (Gibson et al., 1995; Mburu et al., 1997; Self et al., 1998). Shaker-1 mice exhibit HL or deafness, circling behavior and a head tossing phenotype (Mburu et al., 1997; Self et al., 1998). Some Shaker-1 models have naturally occurring mutations (e.g., p.R502P and p.R241P), while others have induced mutations within the motor domain or the MyTH/FERM domains of myosin VIIa (Mburu et al., 1997; Self et al., 1998). Headbanger mice have the mutation p.I178F and exhibit HL, abnormal balance and a head tossing phenotype resulting in vestibular and cochlea hair bundle defects (Rhodes et al., 2004). Ewaso mice have the mutation p.I487N and have abnormal vestibular hair cells (Calabro et al., 2019). Dumbo mice have the mutation p.F947I and exhibit disorganized cochlear hair cells during development (Miller et al., 2012). The function of the MYO7A ortholog in zebrafish is similar to that of both humans and mice, and zebrafish with mutations in these genes are known as mariners (Ernest et al., 2000). Mariners are the equivalent to the Shaker-1 mice models and exhibit vestibular dysfunction, as evidenced by circling behaviors, and have disorganized stereocilia and atypical physiological responses to sound (Ernest et al., 2000; Williams, 2008). Mariner zebrafish have mutations affecting the polypeptide domain, motor domain, IQ motifs, and FERM domains (Ernest et al., 2000). Interestingly, 80% of known USH1B-causing missense mutations affect the amino acids that are conserved in the zebrafish myosin VIIa cDNA, further supporting this model to observe specific mutations (Ernest et al., 2000).
Usher Type 1C: Harmonin
The causative gene for the USH type 1C phenotype is USH1C, which codes for the protein harmonin. Harmonin plays a critical role in the auditory, visual, and vestibular systems (Verpy et al., 2000). Mutations in USH1C cause symptoms consistent with USH (Verpy et al., 2000). USH1C accounts for approximately 15% of USH1 cases and 2% of all USH cases (Verpy et al., 2000; Le Quesne Stabej et al., 2012; Jouret et al., 2019).
Structure
Harmonin is a PDZ domain-containing scaffold protein, named after the Greek word for assembly (harmonia) for the role it plays in binding the USH complex (Verpy et al., 2000). There are three PDZ1 domains, two coiled-coil domains, and a Proline/Serine/Threonine (PST) region. PDZ domains bind to specific subcellular domains of other proteins and are involved in the organization and anchoring of protein interactomes (Ponting et al., 1997; Fanning and Anderson, 1999). In the inner ear HC, the three harmonin PDZ domains bind to several USH proteins, including myosin VIIa and cadherin 23, and act as a central scaffold for the bundling and localization of the USH protein complex (Verpy et al., 2000; Boeda et al., 2002; Adato et al., 2005b).
There are at least 10 alternatively spliced isoforms of harmonin, which are divided into three subclasses (a, b, and c), seven of which are alternatively spliced exons only expressed in the inner ear (A-F, G/G; Verpy et al., 2000). This suggests mutations that selectively affect these isoforms would cause non-syndromic recessive deafness rather than USH due to the absence of retinal expression and therefore phenotype (Toriello and Smith, 2013). All isoforms contain the first two PDZ domains and the first coiled-coil domain (Verpy et al., 2000). Subcellular localization of the harmonin isoforms varies during developmental stages (Adato et al., 2005b). Unlike the c isoforms, which are the shortest and only contain the common regions shared by all isoform subclasses, the a and b isoforms all have a third PDZ domain. The b isoforms are the longest and contain a second coiled-coil domain, and a PST region that is responsible for calcium-independent binding of actin. This enables these b isoforms to act as an F-actin-bundling-protein and play a role in stabilization of the stereocilia actin fibers (Verpy et al., 2000; Boeda et al., 2002).
Mutations
The c.496+1G>A mutation in USH1C occurs at a higher frequency than all other USH1 mutations, with more than 9% of USH1 mutant alleles carrying the mutation in one study cohort (Le Quesne Stabej et al., 2012), presenting a possible hotspot. In the Acadian population of Canada, a c.216G>A founder mutation in USH1C accounts for almost all cases of USH1C (Ouyang et al., 2003; Ebermann et al., 2007a). This mutation introduces a cryptic 5' splice site into exon 3, which is used favorably over the original 5' splice site, causing a transcriptional frameshift, premature stop codon, and truncated harmonin protein (Ebermann et al., 2007a). Additionally, mutations in USH1C can also cause non-syndromic recessive deafness type 18 (DFNB18; Ahmed et al., 2002).
Animal Models
Deaf circler (dfcr) mice and deaf circler 2 Jackson (dfcr-2J) mice carry mutations in the Ush1c gene, the mouse ortholog of USH1C, and were developed to improve understanding of the function of harmonin in the inner ear and eye. These mice exhibit morphological abnormalities of the inner ear HC stereocilia and a phenotype of SNHL and vestibular dysfunction, as evidenced by circling behaviors (Johnson et al., 2003). The dfcr mice carry a large 12.8 kb intragenic deletion that removes eight exons from the transcript, resulting in the loss of both coiled-coil domains and the PST region (Johnson et al., 2003; Xu et al., 2009). The dfcr-2J mice carry a single base deletion that causes a frameshift and results in the inappropriate translation of 38 aa and a truncated protein. The mutations of both models alter the subclass b isoforms, which is expressed in both ear and eye, while only the dfcr mutation disrupts the harmonin isoforms a and c (Johnson et al., 2003).
An Ush1c216AA knockin mouse model was generated specifically to study the Acadian USH1C founder mutation in exon 3. These mice are homozygous for a 35 bp deletion that mimics that of USH1C patients (Lentz et al., 2007, 2010). In contrast to the other models, Ush1c216AA knockin mice exhibit SNHL, vestibular dysfunction, and clear retinal degeneration (Lentz et al., 2007, 2010; Vijayakumar et al., 2017).
Usher Type 1D: Cadherin 23
The USH type 1D (USH1D) causative gene, CDH23, encodes cadherin 23 (Bolz et al., 2001; Bork et al., 2001). Cadherin 23 is a member of the cadherin superfamily of calcium-dependent cell adhesion molecules. It is a non-classical cadherin, as characterized by its long extracellular domain that is required for the development and correct morphology of the inner ear HC bundles and, along with protocadherin 15, forms the tip links between stereocilia (Kazmierczak et al., 2007; Colas-Algora and Millan, 2019). CDH23 mRNA is highly expressed in tip links and lateral links of inner ear HC stereocilia (Lagziel et al., 2009).
Structure
The human CDH23 gene spans ~420 kb of genomic DNA and is comprised of 70 exons, that encode a predicted 3354 aa protein. Cadherin 23 has a 5' untranslated region followed by 27 extracellular cadherin (EC) repeat domains, a transmembrane domain, and a unique cytoplasmic domain (CD; Figure 2B; Di Palma et al., 2001b; Oshima et al., 2008). Each EC repeat contains cadherin-specific aa motifs required for cadherin dimerization and calcium binding (Ozawa et al., 1990; Suzuki, 1996; Rowlands et al., 2000). The EC domains form dimers on the cell surface, which bind to dimers on a different cell, thereby linking the cells together. In the absence of calcium, the EC regions are weakened and unable to bind, ultimately resulting in breakage of tip links (Di Palma et al., 2001b). The transmembrane domain consists of single chain glycoprotein repeats and acts as a bridge between the cytoplasmic and extracellular domains and an anchor to fix the cadherin to the cell surface (Shapiro and Weis, 2009). Lastly, the CD contains two PDZ-binding motifs (PBM) that facilitate interactions with other hair bundle proteins that contain PDZ domains, such as harmonin (Boeda et al., 2002; Siemens et al., 2002; Lagziel et al., 2009).
Alternative splicing produces CDH23 isoforms of three different classes (A, B, and C), which are differentially expressed based on tissue location (Lagziel et al., 2009). Each isoform class has two subtypes, which differ based on their inclusion of exon 69 encodes 35 aa, and was frequently referred to as exon 68 by earlier papers that did not classify the exon in the 5'-untranslated region as exon 1 (Takahashi et al., 2016). The A isoforms contain all 27 EC repeats and are the only isoforms expressed in the inner ear and retina (Takahashi et al., 2016). The B and C isoforms lack many key regions, for example, the C isoforms are entirely cytoplasmic, making them unable to serve a cell-cell adhesion role (Takahashi et al., 2016).
Mutations
Mutations in CDH23 are associated with USH1D, accounting for approximately 6% of all USH cases, as well as non-syndromic AR deafness type 12 (DFNB12), and age-related HL (Di Palma et al., 2001b; Jouret et al., 2019). Over 200 pathogenic CDH23 mutations have been identified. Typically, nonsense, frameshift or splice-site mutations that result in truncated non-functional forms of cadherin 23 are associated with USH1D, whereas missense mutations are more often associated with DFNB12 (Bork et al., 2001; Astuto et al., 2002). The spectrum of mutations in the global population shows regional variation, with Japanese HL patients having a particularly high frequency of CDH23 mutations (Mizutari et al., 2015).
Animal Models
The waltzer mouse carries mutations in Cdh23 that results in disorganization of the HC stereocilia (Di Palma et al., 2001a). Waltzer mice are characterized by deafness and a head shaking and circling phenotype indicative of vestibular dysfunction (Wilson et al., 2001). Mutations in the EC repeats can cause disruptions in the ability of calcium to bind, thereby inhibiting cell-cell interactions.
The zebrafish ortholog of CDH23 is expressed in the inner ear HC, brain, olfactory organ, and retina, and the protein has been localized to the distal tips of hair bundles, especially of the tallest stereocilia next to the kinocilium (Sollner et al., 2004). The zebrafish model with cdh23 mutations, sputnik, has reduced or absent MET, likely due to the observed detachment of stereocilia bundles from kinocilium and splayed stereocilia (Nicolson et al., 1998; Seiler and Nicolson, 1999; Sollner et al., 2004).
Usher Type 1F: Protocadherin 15
Mutations in the PCDH15 gene result in one of the most severe forms of Usher syndrome, type 1F (USH1F) which was first described in 1997 (Wayne, 1997; Ahmed et al., 2001). The PCDH15 gene is located on the long arm of chromosome 10 at position 21.1 (10q21.1; Yan and Liu, 2010). PCDH15 contains 1 Mb of DNA and has an open reading frame of 7,021 bp that codes for the cell-cell adhesion protein protocadherin 15 (Ahmed et al., 2001; Le Guédard et al., 2007). Protocadherin 15 is a non-classical cadherin with a long extracellular region (Ahmed et al., 2001, 2003). Within the inner ear HC, protocadherin 15 is expressed throughout the kinociliary link, transient lateral link, tip link, and synapse, as well as the supporting cells and spiral ganglion of the inner ear (Alagramam et al., 2001b; Ahmed et al., 2006; Mathur and Yang, 2015).
Protocadherin 15 is essential for normal function and development of the mammalian inner ear, as it forms the lower portion of the tip link and binds to cadherin 23 (Alagramam et al., 2001b, 2011; Indzhykulian et al., 2013; Perreault-Micale et al., 2014). The structure of the tip link is transient, with two protocadherin 15 proteins binding as the link during development (Indzhykulian et al., 2013). Evidence suggests that individual protocadherin 15 monomers could function as a gating spring, converting displacement of the HC bundle into the mechanical forces required to open the transduction channels. Although the tip link bundle is too stiff to act as a spring, individual protocadherin molecules have low stiffness, are unstable when under high tension, and display a large range of reversible unfolding when Ca2+ levels are reduced. Whether the tip link cadherins are folded during normal hearing remains unknown (Bartsch et al., 2019).
Structure
All protocadherin 15 isoforms are comprised of a N-terminal signal peptide domain, an extracellular domain containing tandemly repeated EC motifs, a transmembrane domain, and one of three C-terminal CDs (Figure 2C; Ahmed et al., 2008). The protocadherin 15 isoforms contain EC repeats of ~100 aa in length, connected to each other via ~10 aa linker regions (Bolz et al., 2002). These repeats are involved in mediating calcium-dependent binding between cadherin proteins (Bolz et al., 2002; Reiners et al., 2005a). For example, the two most N-terminal cadherin repeats bind to the extracellular region of cadherin 23 to form an extracellular link (Ahmed et al., 2006; Kazmierczak et al., 2007; Nie et al., 2016).
There are four alternative isoforms of protocadherin 15 (CD1, CD2, CD3, and secreted) that vary based on their CDs (Ahmed et al., 2008). The largest protocadherin 15 isoform (CD1) contains 32 protein-coding exons and one non-coding exon that translates the longest protein with 1955 aa, CD2 and CC3 contain 1783 aa and 1675 aa, respectively (Figure 2C; Le Guédard et al., 2007). Each of these CDs contains two highly conserved, proline-rich regions that act as binding sites for proteins containing WW and SH3 domains (Sudol, 1998; Alagramam et al., 2001b; Le Guédard et al., 2007). WW and SH3 domains play a role in the construction of signaling complexes and interact with proteins that are often involved in regulating actin filament polymerization; therefore conservation of these amino acid sequences within protocadherin 15 suggest the protein is involved in the regulation of stereocilia planar polarity (Sudol, 1998; Alagramam et al., 2001b). Each of the three isoforms has a different spatiotemporal expression in both developing and mature HC; therefore, it is likely that each of the isoforms plays a specific role in the HC structure or function (Ahmed et al., 2006, 2008; Le Guédard et al., 2007).
Mutations
A large number of disease-causing PCDH15 mutations are splice sites or frameshifts, which result in a truncated or non-functional protein (Le Quesne Stabej et al., 2012). Approximately, 30% of mutations identified within PCDH15 are large deletions, most likely a direct cause of the large size of the gene and low number of protein coding regions (Roux et al., 2006; Le Guédard et al., 2007). Although mutations in the PCDH15 gene account for only 3% of USH cases, an increased incidence of USH1F is observed in the American Ashkenazi Jewish population (Perreault-Micale et al., 2014; Jouret et al., 2019). Mutations in PCDH15 are also associated with non-syndromic deafness type 23 (DFNB23; Ahmed et al., 2003).
Animal Models
The Ames waltzer mouse carries mutations in Pcdh15, and exhibit similar symptoms to human USH1F patients, such as deafness, and show circling behaviors indicative of vestibular dysfunction (Ahmed et al., 2001; Alagramam et al., 2001a; Haywood-Watson et al., 2006). Cochlea and vestibular HC of Ames waltzer mice have disorganized stereocilia bundles and kinocilium, abnormal cell polarization and the OHC lack the V-shaped stereocilia configuration essential for normal MET (Pickles et al., 1984; Alagramam et al., 2001a; Kikkawa et al., 2008). Interestingly, isoform B seems to be the most significant Pcdh15 isoform for hearing. Mice deficient in isoform B are deaf and have abnormal hair bundles, while mice deficient in either isoform A or C have normal hair bundles and hearing (Kikkawa et al., 2008; Webb et al., 2011).
Noddy mice harbor a homozygous p.I108N missense mutation within the first EC repeat and exhibit head-bobbing and circling behaviors, as well as no response to stimulus at any of the frequencies tested, indicative of a complete loss of inner ear function (Geng et al., 2013). Since the two most N-terminal EC repeats of protocadherin 15 interact with those of cadherin 23, the mutation has been shown to block binding of these proteins, resulting in a loss of hair bundle integrity. Specifically, noddy mice lack tip links and the ability to open mechanotransduction channels (Geng et al., 2013).
Zebrafish contain two genes closely related to the human PCDH15 gene, pcdh15a and pcdh15b, which are responsible for the morphology and function of the ear and eye (Seiler et al., 2005; Maeda et al., 2017). Seiler et al. (2005) showed that mutations in pcdh15a result in vestibular dysfunction and deafness, while vision remains unaffected, and vice versa in the pcdh15b gene. It is likely that these genes are differentially expressed in these organs, reflecting the different phenotypes (Seiler et al., 2005). There are two isoforms of pcdh15a in zebrafish, that correspond to the human A and C isoforms, both of which are densely localized at the tip of the HC stereocilia, supporting that protocadherin 15a is a component of the HC tip links (Maeda et al., 2014, 2017).
Usher Type 1G: SANS
The SANS (scaffold protein containing ankyrin repeats and SAM domain) protein plays a critical role in the formation of the USH1 multiprotein complex with harmonin, myosin VIIa, and cadherin 23 (Weil et al., 2003). In mice, SANS expression has been localized to the apical region of HC stereocilia. SANS localizes to the short and middle-row stereocilia of HC and interacts with both of the tip link proteins, cadherin 23, and protocadherin 15. Therefore, SANS is predicted to be involved in a multiprotein complex localized at the lower tip link density (Caberlotto et al., 2011).
Structure
SANS contains 1,380 bp and encodes a 461 aa protein (Weil et al., 2003). The SANS protein contains three N-terminal ankyrin-like domains (ANK 1, 2, and 3), a central region (CENT), followed by a sterile alpha motif (SAM) and a tripeptide, class I PBM at the C-terminal end of the protein (Weil et al., 2003). The class I PBM of SANS interacts with the PDZ1 and PDZ2 domains of the USH2D protein, whirlin (van Wijk et al., 2006). While the SAM and class I PBM of SANS bind the PDZ1 domain of harmonin to form a highly stable complex (Yan et al., 2010).
Mutations
Mutations in SANS cause Usher syndrome type 1G (USH1G), which accounts for approximately 1% of USH cases (Weil et al., 2003; Jouret et al., 2019). Mutations in SANS may also result in non-syndromic HL or in USH with delayed retinal degeneration (Maria Oonk et al., 2015).
Animal Models
Jackson shaker mice that model the USH1G phenotype have been generated by mutating the Sans gene. These mice exhibit deafness caused by degeneration of the sensory cells in the inner ear and vestibular dysfunction characterized by head tossing, circling behaviors and hyperactivity (Kitamura et al., 1992; Kikkawa et al., 2003). Jackson shaker mice do not display retinal abnormalities and cannot be used to study the retinal phenotype of USH1G (Kikkawa et al., 2003). Kitamura et al. (1992) undertook a comparative study on the HC structure of homozygous Jackson shaker mice, and observed the differences in their HC at days P10-P30. The OHC of the homozygous Jackson shaker mutants exhibited incomplete differentiation, and mislocalization and disfiguration of the stereocilia (Kitamura et al., 1992).
Usher Type 1J: CIB2
Calcium and integrin binding protein 2 (CIB2) is a 21.6 kDa protein with 187 aa and encoded by the CIB2 gene, comprised of 6 exons (Huang et al., 2012). CIB2 is categorized as a DNA-dependent protein kinase interacting protein, and is within the CIB protein family. CIB2 is ubiquitously expressed in a wide variety of tissues, specifically within the inner ear, CIB2 is localized to the tips of IHC, OHC, supporting cell cytoplasm, and vestibular HC stereocilia, with more concentrated expression at the tips of the shorter row stereocilia (Riazuddin et al., 2012).
Structure
There are four members of this protein family, CIB1-4, all containing EF-hand domains that change conformation following cation binding (Seki et al., 1999; Gentry et al., 2005; Blazejczyk et al., 2009). CIB2 has three EF-hand motifs, the last two of which mediate the cation binding activity, specifically for binding Ca2+ and Mg2+ (Huang et al., 2012; Michel et al., 2017). In contrast to previous studies, Vallone et al. (2018) showed that CIB2 is unable to function as a calcium sensor under physiological conditions due to a low affinity for Ca2+ ions. However, they demonstrated that CIB2 does have a high affinity for Mg2+ under the same conditions, and is therefore much more likely to function as a magnesium sensor (Vallone et al., 2018).
Mutations
Mutations in CIB2 have been associated with both non-syndromic deafness type 48 (DFNB48) and USH type 1J (USH1J), as well as congenital muscular dystrophy type 1A (Hager et al., 2008; Riazuddin et al., 2012; Patel et al., 2015a,b). A small number of mutations in CIB2 have been associated with USH1J, including a conservative point mutation, c.192G>C (p.E64D; Riazuddin et al., 2012). The E64 residue is in the N-terminal domain and is thought to communicate with the EF3 cation binding site in the C-terminal domain (Vallone et al., 2018). Therefore, mutations at this site prohibit this inter-domain communication, impairing the cation responsiveness of CIB2. In the presence of physiological Mg2+, the p.E64D mutation causes CIB2 to adopt a less stable partially unfolded conformation that is more prone to aggregation (Vallone et al., 2018). Alternative splicing gives rise to four CIB2 isoforms, and disease-causing mutations in exon 4–6 affect all isoforms (Riazuddin et al., 2012; Michel et al., 2017).
There has been dispute as to whether CIB2 should be considered an USH gene at all. The p.E64D missense mutation that is thought to cause USH1J is only two amino acids away from the p.R66W missense mutation that is exclusively linked to non-syndromic deafness (Seco et al., 2016). In addition, none of the 13 patients in a multi-ethnic cohort (Booth et al., 2018) had an abnormal retinal phenotype, regardless of the mutation type, even null alleles. Lastly, while a retinal phenotype is rarely evident in mouse models of any USH type, many do exhibit some vestibular dysfunction, unlike CIB2 knockout mice (Giese et al., 2017; Michel et al., 2017). Further examination of the p.E64D patients is required to determine whether there may be another cause for their retinal/vestibular phenotypes.
Animal Models
There is currently no specific animal model for USH1J, however, a number of knockout models have been established to investigate the function of CIB2 in different organ systems. Mouse models developed using CRISPR/Cas9 to target exon four with two small deletions (one 9 bp and one 8 bp segment) introducing a premature translational stop and a truncated 109 aa protein (Wang et al., 2017). Loss of CIB2 but not CIB1 affects auditory function and causes profound HL by impacting stereocilia development (Wang et al., 2017). DPOAE thresholds were elevated indicating OHCs function deficits in homozygous knockout mice compared to heterozygous mice, and subsequent experiments have shown IHC are affected to a much lesser extent and only at later timepoints (Wang et al., 2017). Other groups have shown that Cib2 knockout mice are profoundly deaf, but have no retinal or vestibular defects (Giese et al., 2017; Michel et al., 2017; Zou et al., 2017). The deafness is likely due to knockout of CIB2 abolishing MET currents in auditory HC (Giese et al., 2017; Wang et al., 2017). Additionally, knockdown of Cib2 in zebrafish highlighted its role in the correct development, maintenance, and function of IHC (Riazuddin et al., 2012).
Usher Type 2
USH2 patients typically have moderate SNHL from birth and RP, which starts during late puberty or early adulthood (Mathur and Yang, 2015). Although, USH2 was originally characterized as having non-progressive deafness, recent studies have shown that patients may suffer from increased loss of hearing over time, indicating symptoms can be progressive (Hartel et al., 2016). Three responsible genes have been identified so far, as well as USH2B, which was originally mapped to the short arm of chromosome 3, although following molecular analysis this gene is no longer recognized as an USH loci (Hmani et al., 1999; Hmani-Aifa et al., 2009).
Usher Type 2A: Usherin
USH2A was the first USH locus identified, with mutations in this gene causing USH type 2A and accounting for approximately half of all cases of USH (Kimberling et al., 1990; Lewis et al., 1990; Jouret et al., 2019). Approximately, 80% of USH2 cases harbor mutations in the USH2A gene, making it the predominant causative gene for USH2, of the three genes identified (Le Quesne Stabej et al., 2012). USH2A is located on chromosome 1q41 and encodes the Usherin protein, which is involved in developing and maintaining neurosensory cells in both the retina and cochlea (Bhattacharya et al., 2002). Usherin is expressed within the hair bundles during postnatal development in cochlear HC and is localized to the apical inner segment recess that wraps around the connecting cilia in photoreceptor cells (Liu et al., 2007). The cytoplasmic tail of usherin interacts with the PDZ domains of harmonin and whirlin, which are associated with USH types 1C and 2D, respectively (van Wijk et al., 2004; Adato et al., 2005a). An alternatively spliced region predicted to encode 24 additional amino acids has been identified to be specific for cochlea because the sequence is highly expressed in the inner ear (van Wijk et al., 2004; Adato et al., 2005a). This exon was previously not included in the longer USH2A transcript (van Wijk et al., 2004).
Structure
Usherin is a large protein comprised of a number of domains (Figure 3A; Eudy et al., 1998; Weston et al., 2000; Sheng and Sala, 2001; Dona et al., 2018). USH2A has two isoforms, A and B, with isoform B being the predominant form in retina (Figure 3A; Liu et al., 2007; Toms et al., 2015). The short isoform (A) contains 21 exons and codes for an extracellular matrix protein of ~170 kDa and 1546 aa. The long isoform (B) contains 72 exons and codes for a protein of ~580 kDa and 5202 aa, and is crucial for the development of cochlear HC (van Wijk et al., 2004; Adato et al., 2005a).
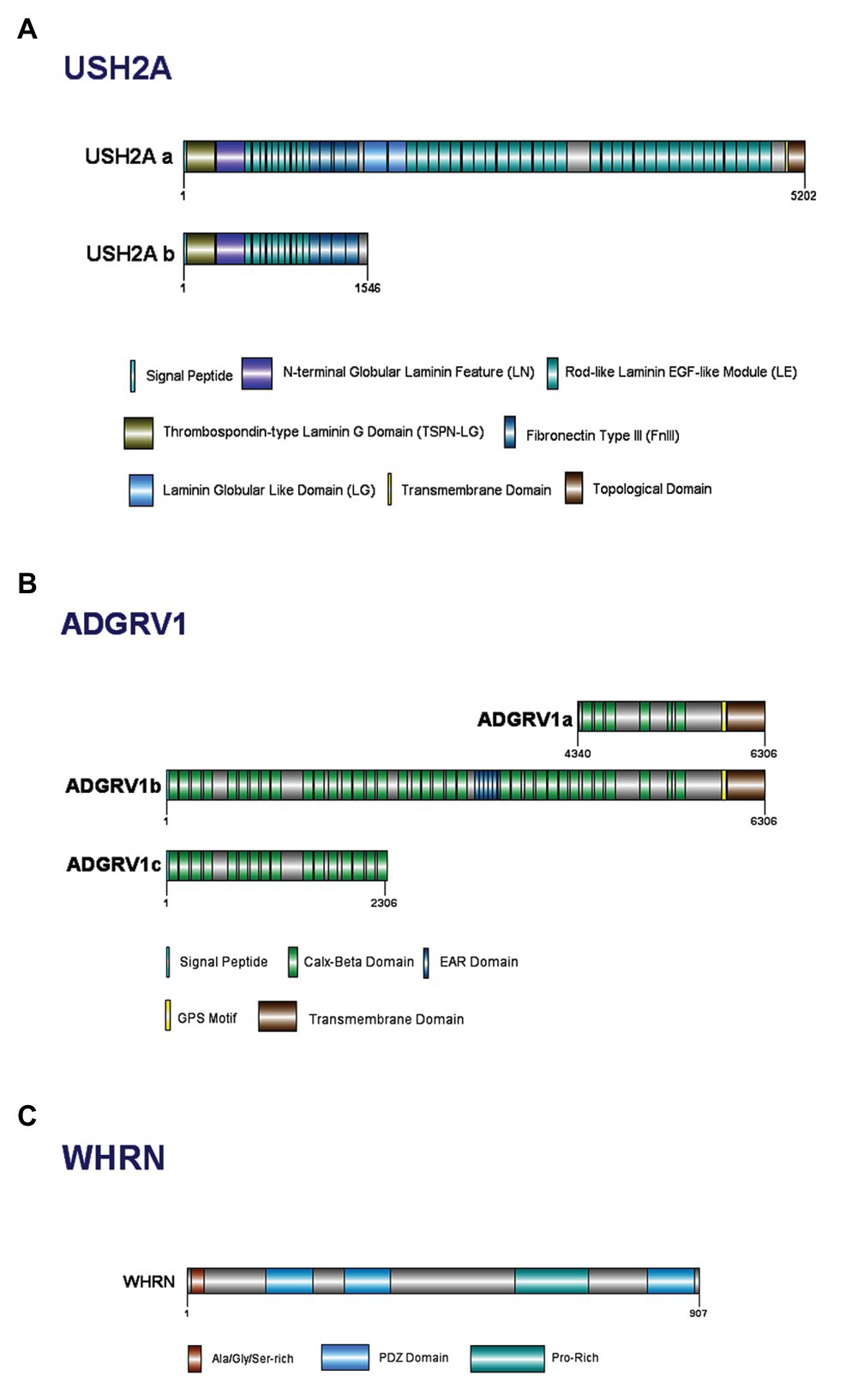
Figure 3. Domain structure of Usher Type II proteins and their major isoforms: (A) usherin; (B) ADGRV1; and (C) whirlin.
The thrombospondin-type LamG domain (TSPN-LG) has homology to the thrombospondin family of extracellular matrix proteins (Taraboletti et al., 1990; Nicosia and Tuszynski, 1994). Thrombospondin-1 has diverse functional properties, including activation of latent TGF-β1, integrin ligation and activation, as well as inhibition of both MMP-2 and MMP-9; the presence of this region in usherin suggests it may play similar roles (Sipes et al., 1999; Bein and Simons, 2000; Murphy-Ullrich and Poczatek, 2000). Following this, is the N-terminal globular laminin feature (LN module), which is required for the polymerization of laminins into the characteristic networks found in basement membranes (Bruch et al., 1989; Beck et al., 1990; Yurchenco and Cheng, 1993; Bork et al., 1996). Next, the 10 rod-like laminin-EGF-like (LE) domains comprise of repeat units of 60 aa containing eight conserved cysteines (Engel, 1989). The arrays of LE domains form rod-like tertiary structures with low flexibility and, as such, may structurally act as a rigid spacer between the amino- and carboxy-terminal domains of usherin (Beck et al., 1990). At the carboxyl terminus of usherin, there are four FN3 repeats (Yan and Liu, 2010). The FN3 repeats tend to form a series of beta-pleated sheet structures, and are associated with integrin binding molecules (Barkalow and Schwarzbauer, 1991; Bowditch et al., 1994).
Mutations
Usherin plays an important role in maintaining the function of the photoreceptor cells and cochlear HC. USH2A has a diverse mutation spectrum, including nonsense, frameshift, missense, splice site-affecting, deletion, and duplication mutations, which can all lead to hearing and vision defects in humans. Two of the most frequent mutations in USH2A occur on exon 13, namely c.2276C>T (p.C759F) and c.2299delG (p.E767fs), with the latter accounting for over 30% of USH2A mutations in an USH cohort (Le Quesne Stabej et al., 2012; Fuster-Garcia et al., 2017). Due to the high frequency of mutations on exon 13, antisense oligonucleotide treatments targeted at skipping this region of the gene are under development (Dona et al., 2018; van Wijk, 2019).
To investigate the possibility of genotype-phenotype correlations, 191 unrelated Spanish patients have been investigated for the c.2299delG and p.C759F (c.2276C>T) mutations in the USH2A gene with USH type II syndrome or non-syndromic retinal diseases (Aller et al., 2004). The c.2299delG mutation was present in patients with clinical signs of USH type II, whereas the p.C759F mutation was only associated with non-syndromic retinitis pigmentosa. Neither of these common mutations in USH2A have been found in patients with non-syndromic HL (Aller et al., 2004). SNHL in patients with retinitis pigmentosa may be determined by alternative splicing and the allelic variants of USH2A present.
Animal Models
Ush2a knockout mice have progressive degeneration of photoreceptor cells and a moderate non-progressive hearing impairment (Adato et al., 2005a). Another Ush2a knockout mice showed progressive photoreceptor degeneration as well as high-frequency threshold elevation (Liu et al., 2007). These Ush2a mutant mice models have provided information on the role of usherin in the auditory system, but limited data on the visual system. Usherin protein appears to be functional in zebrafish despite them lacking a full-length protein (Dona et al., 2018). In addition, ush2a knockout zebrafish have been generated successfully and these exhibit auditory abnormalities and retinal degeneration, replicating the disease phenotypes of human USH2A patients (Han et al., 2018).
Usher Type 2C: ADGRV1
The ADGRV1 gene (also known as VLGR1, GPR98, or MASS1) is located on chromosome 5q14.3-21.3 and has been identified as the causative gene for USH type 2C (USH2C) (Hilgert, 2009a; Hilgert et al., 2009b). It encodes Adhesion G-protein-Coupled Receptor V1 (ADGRV1), a transmembrane receptor protein that is responsible for cochlear development and the formation of stereocilia ankle links (Millan et al., 2011). ADGRV1 is one of the largest human genes, comprising 90 exons and spanning more than 600 kb of genomic DNA (Lentz and Keats, 1999, Updated 2016). ADGRV1 protein is comprised of a large number of distinct domains, and is expressed at high levels within the embryonic nervous system, in particular the ventricular zone (McMillan and White, 2004). It is also present during the development of brain and spinal cord, foetal retina and many other tissue types (Lentz and Keats, 1999, Updated 2016; Hilgert et al., 2009b).
Structure
The ADGRV1 protein belongs to a large N-terminal family B of seven transmembrane segment (7TM) receptors; specifically, to the subfamily with a G-protein-coupled proteolysis site for G-protein signaling (Hilgert et al., 2009b; Yang et al., 2012). ADGRV1 expresses multiple alternatively spliced variants, with three isoforms identified for humans: ADGRV1a, ADGRV1b, and ADGRV1c (Figure 3B; Yang et al., 2012). Isoform ADGRV1a starts in intron 64 of isoform ADGRV1b and contains only the last 26 exons. Containing only 1967 aa, ADGRV1a is the smallest and least abundant within the other isoforms (Lentz and Keats, 1999, Updated 2016; Hilgert et al., 2009b). Isoform ADGRV1b encodes 6306 aa and is the predominant isoform in the retina and inner ear (Lentz and Keats, 1999, Updated 2016; Zou et al., 2011). ADGRV1b has a large extracellular region, consisting of Calx-β domains, epilepsy-associated region (EAR) domains, transmembrane domains, and other exonic fragments in between the Calx-β domains (Myers et al., 2018). Isoform ADGRV1c shares the same start codon as ADGRV1b, but consists of only 31 exons (Hilgert et al., 2009b). The presence of 35 tandem-arranged CalX-β repeat domains mediate the protein-protein interactions between these two domains (McMillan et al., 2002; Yang et al., 2012). The N-terminal 29 aa of ADGRV1b are hydrophobic and may act as a signal sequence for cleavage in the mature protein (Hilgert et al., 2009b). The EAR domain consists of a repeated set of domains that form a seven-bladed propeller structure (Weston et al., 2004).
Mutations
Mutations in ADGRV1 account for 6.6–19% of USH2 cases (Bonnet et al., 2011; Garcia-Garcia et al., 2013). To date, more than 100 variants have been reported in ADGRV1, with about 65 and 25% being missense and small deletions mutations, repectively (Lentz and Keats, 1999, Updated 2016; Bonnet et al., 2011; Garcia-Garcia et al., 2013).
Animal Models
Consistent with the phenotype seen in USH2C patients, Adgrv1/del7TM mutant mice exhibit intact vestibular function but with HL (McMillan and White, 2004; Sadeghi et al., 2004; McGee et al., 2006). These mice have early hair bundle defects leading to a complete loss of HC in the cochlea, demonstrating the degeneration of stereocilia bundles in the OHC; however, they become profoundly deaf by postnatal day 20 (McGee et al., 2006).
Usher Type 2D: Whirlin
The DFNB31 gene is located on chromosome 9q32-q34 and contains 12 exons. It encodes whirlin, a PDZ scaffold protein comprised of 907 aa and five distinct domains (van Wijk et al., 2006). The DFNB31 gene has been reported to be responsible for non-syndromic HL type 31 (DFNB31) and USH type 2D (USH2D; Tlili et al., 2005; Aller et al., 2010). Whirlin is present at the stereociliary tips of mechanosensitive hair bundles and retinal photoreceptor cells (Belyantseva et al., 2005; Ebermann et al., 2007b). The protein isoforms of whirlin play different roles spatially and temporally in the cochlea and retina. It has been shown that in HC stereociliary bases, both full-length and C-terminal whirlin localize to the inner hair cells and vestibular hair cells and participate in the elongation process of the stereocilia in inner ears, while only C-terminal whirlin is present at the outer hair cell stereociliary tip. Full-length whirlin is the only isoform at the stereociliary base in all types of hair cells and critical for the interaction within the USH2 protein network (van Wijk et al., 2006; Mathur and Yang, 2015, 2019).
Structure
Whirlin has two key transcript variants in the inner ear due to alternative transcriptional start sites and/or splicing sites within the gene itself (Yang et al., 2010). The whirlin full-length isoform encodes all 12 DFNB31 exons and contains two N-terminal PDZ domains (PDZ1 and PDZ2), a proline-rich domain and a third C-terminal PDZ domain (Figure 3C; Aller et al., 2010; Yang et al., 2010). The whirlin C-terminal isoform does not have N-terminal PDZ domains and encodes only exons 6–12; however, it retains the proline-rich region and the third PDZ domain near the C-terminus (Aller et al., 2010; Yang et al., 2010). Both isoforms are expressed in the inner ear whereas only the long isoform is expressed in the retina (Yang et al., 2010). It has been postulated that mutations at different positions of the whirlin protein cause USH2D or non-syndromic deafness (DFNB; Mathur and Yang, 2015).
Mutations
DFNB31 mutations appear to be a rare cause of USH, with only two AR deafness families identified from large deafness cohort studies to date (Mburu et al., 2003; Tlili et al., 2005). The mutations reported in USH2D are either located in the C-terminal half or within the coding region specific to the long protein isoform (Mburu et al., 2003; Tlili et al., 2005; Ebermann et al., 2007b). Ebermann et al. (2007a) are the only group to report two patients being compound heterozygous for DFNB31 mutations: one with a nonsense mutation in exon 1, resulting in a truncated protein without PDZ1 and the downstream C-terminal, whereas the other one has a splice site mutation, leading to an in-frame skipping of exon 2 and fusion of PDZ1 and PDZ2 domains. Mutations causing USH are restricted to exons 1–6, which are specific for the long isoform and plays a major role in the retina function. The short isoform expression would remain unaffected if there are mutations in exon 1 or 2 (Audo et al., 2011). Ar non-syndromics severe deafness occurs when mutations are present on both isoforms and affect PDZ3 domain, suggesting the role PDZ3 plays in photoreceptor function and maintenance (Audo et al., 2011).
Animal Models
The whirler mice have a large deletion in the middle of Dfnb31 that causes a frameshift at p.433, leading to a premature termination (Mburu et al., 2003). As a result, the protein is truncated and does not include one of the three whirlin PDZ domains (Mburu et al., 2003). Whirler mice have height-ranked stereocilia and display normal orientation (Mburu et al., 2003); however, their hair bundles are consistently shorter in comparison to wild types and have a U-shaped morphology instead of the usual W-shaped bundle (Mburu et al., 2003).
Usher Type 3
Patients with USH3 typically exhibit symptoms by mid-life including progressive SNHL and RP usually with loss of night vision (nyctalopia), constriction of visual field, as well as variable, progressive vestibular dysfunction (Kimberling et al., 2000; Fields et al., 2002). Symptoms of USH3 are quite variable with vestibular dysfunction, for example, occurring in approximately 50% of cases, and varying rates of hearing and vision symptom progression between patients. Most cases of USH3 do eventually result in profound HL even though early hearing levels are normally good (Millan et al., 2011). Type 3 has the most therapeutic potential due to the late onset of symptoms and relatively slow progression of RP (Geller et al., 2009).
Usher Type 3A: Clarin-1
The CLRN1 gene is located on the long arm of chromosome 3 at position 25.1 (3q25.1), and is the causative gene for USH type 3A (USH3A; Sankila et al., 1995; Joensuu et al., 2001; Isosomppi et al., 2009). CLRN1 is ubiquitously expressed in many tissues throughout the body, and mouse models have identified Clrn1 expression within the HC and the spiral ganglion cells that innervate the sensory epithelium of the cochlea (Adato et al., 2002). Specifically, clarin-1 plays a role in sensory synapses, including HC and photoreceptor cells (Adato et al., 2002). Clarin-1 is also predicted to play a similar role to the USH2 proteins in the development and function of the cochlea HC bundles, but might have a distinct role in the organization of the vestibular system (Mathur and Yang, 2015).
Structure
USH3 is the least common form of USH, accounting for approximately 2% of all cases, although it does occur more frequently in some populations (Jouret et al., 2019). For example, USH3 is responsible for approximately 42% of cases of USH in Finland, all of which show progressive HL (Pakarinen et al., 1995; Sankila et al., 1995). There is also an increased incidence of USH3 in Ashkenazi Jewish populations in comparison to the general population, with some specific mutations being relatively common within individuals of this heritage (Fields et al., 2002; Ben-Yosef et al., 2003; Ness et al., 2003). The gene spans 1,619 bp and encodes the protein clarin-1, consisting of 232 aa and including four transmembrane domains (Adato et al., 2002; Fields et al., 2002). There are 11 alternatively spliced variants identified to date, with the main variant composed of three exons (Fields et al., 2002; Vastinsalo et al., 2011).
Mutations
Mutations in CLRN1 cause USH3A, likely due to the mislocalization and instability of clarin-1 protein and defective intracellular trafficking (Sankila et al., 1995; Joensuu et al., 2001; Isosomppi et al., 2009). The high incidence of USH3 in these two populations is due to a mutation founder effect in each population, specifically the c.300T>C (p.Y176X) mutation known as the Finn mayor mutation in the Finnish population, and the c.143T>C (p.N48K) mutation within Ashkenazi Jews (Joensuu et al., 2001; Ness et al., 2003; Millan et al., 2011).
Animal Models
Clrn1 homozygous knockout mice show a loss of cochlea HC function, as well as a potential ribbon synapse defect in the retina (Geng et al., 2009; Tian et al., 2016). These mice exhibit disorganized HC stereocilia, circling behaviors and deterioration of their organ of Corti within the first 4 months of life (Geller et al., 2009; Geng et al., 2009, 2017). A novel USH3A mouse model, which exhibits delayed-onset progressive HL, similar to that observed in USH3 patients has been developed. The mouse model was created by exploiting the known regulatory element, Atoh1 3' enhancer, to promote transient Clrn1 expression in HC during development. The Clrn1-KO mice were generated harboring a transgene with Clrn1-UTR cDNA (TgAC1), which was regulated by the Atoh1 3' enhancer and a β-globulin basal promoter, to control and down-regulate Clrn1 expression postnatally. In comparison to Clrn1-KO models, the KO-TgAC1 mice experience gradual HL, as shown by slowly decreasing hearing sensitivity with auditory brain stem responses (ABRs), indicating that Clrn1 is critical in postnatal hearing preservation (Geng et al., 2017). Currently, no USH3A mouse models for the retinal phenotype exist, preventing in vivo studies into CLRN1 function within the retina and hindering the development of potential therapies (Geller et al., 2009; Dinculescu et al., 2016).
Usher Type 3B: HARS
Histidyl-tRNA synthetase (HARS) codes for the protein HARS 1, a member of the class II family of aminoacyl-tRNA synthetases (O’Hanlon and Miller, 2002; Abbott et al., 2017). HARS synthesizes histidyl-transfer RNA (tRNA), a crucial molecule in the incorporation of histidine into proteins (Freist et al., 1999; O’Hanlon and Miller, 2002). Based on the mutation found in two patients, HARS was proposed to be the causative gene of USH3 (Puffenberger et al., 2012). However, these patients show signs of episodic psychosis other than USH symptoms like progressive HL, therefore it is debatable whether it could be considered as the definitive cause of USH type 3B (Puffenberger et al., 2012). To date, the localization of HARS in the inner ear remains unknown and the interactions of HARS within the USH interactome remains unclear and will require further investigations (Nolen et al., 2020).
Structure
The HARS/HARS2 genes span approximately 17.4 kb and contains 13 exons (O’Hanlon and Miller, 2002). They have been mapped to the long arm of chromosome 5 at position 31.3 (5q31.3). Sharing a bi-directional promoter, HARS2 (also known as HARSL) gene is aa synthetase-like gene located in a head-to-head orientation to HARS, with 344 bp intergenic sequence separating their open reading frame (O’Hanlon and Miller, 2002). HARS2 bears striking homology to HARS, and both genes have strong structural and sequence homology over exons 3–12 (O’Hanlon and Miller, 2002). HARS transcripts originate from two distinct promoters: a short transcript maps 15–65 bp upstream of the ORF and a long transcript maps to a distal promoter (O’Hanlon and Miller, 2002).
Mutations
The c.1361A>C (p.Y454S) missense mutation in the gene is the only identified causative variant for USH3B to date. Patients with mutations in HARS not only experience the classical USH symptoms of progressive hearing and vision loss, but also experience episodic hallucinations or psychosis, commonly during periods of acute febrile illness (Puffenberger et al., 2012; Abbott et al., 2017). HARS has been found to be thermally sensitive, in that cells from patients harboring the p.Y454S mutation show reduced levels of protein synthesis at higher temperatures. Additionally, USH3B patients often exhibit delayed motor development, and have a wide-based gait or mild truncal ataxia (Abbott et al., 2017). Due to the additional psychotic and neurological symptoms in patients with this mutation, it is currently undecided whether HARS should be considered an USH gene, or whether it is instead the cause of a different rare syndrome or group of syndromes (Mathur and Yang, 2015).
Animal Models
There was a severe reduction in the number of sensory hair cells in the lateral line at 72 h post fertilization when hars was knocked down in embryos of a transgenic line, Tg(pou4f3:GFP; Waldron et al., 2019). Hars knockdown zebrafish embryos showed a decrease in proliferative cells and an increase in apoptotic cells (Waldron et al., 2019). Stress-related genes such as asns, gpt2, and eif4ebp1 were activated and strongly expressed in the hars knockdown embryos, suggesting that hars knockdown inhibits cell proliferation by inducing the Amino Acid Starvation Response (Waldron et al., 2019).
Interactions Between USH Proteins in the Inner Ear
The proteins implicated in USH are all closely related in their region of expression and function within the inner ear. Many of these proteins interact to form important structures such as stereociliary links and the MET complex. The MET channel consists of different subunits whose genes are involved in hearing impairment, namely they are transmembrane inner ear protein (TMIE), transmembrane channel like protein (TMC1), and tetraspan membrane protein of hair cell stereocilia (TMHS). Interactions of these MET channel subunits with the C-terminal domain of Protocadherin 15 occurs at the tip link in hair cells, and is essential for mechanotransduction MET machinery of hair cells (Xiong et al., 2012; Zhao et al., 2014; Beurg et al., 2015; Kurima et al., 2015).
Each of the USH proteins are known or predicted to interact with several of the other USH protein members (Figure 4). One of the most important pairs of proteins in maintaining stereociliary bundle formation is cadherin 23 and protocadherin 15. These proteins interact with themselves and their extracellular domain homodimers interact in trans with each other at their N-termini (Alagramam et al., 2011). Cadherin 23 and protocadherin 15, localized to the upper and lower parts of the tip link, respectively, form the connections between the HC stereocilia that are critical for stabilizing the stereocilia bundle structure. Mutations affecting either of these proteins can prevent their ability to interact and thereby cause critical defects in tip link formation (Kazmierczak et al., 2007). The tip links are also responsible for transmitting force to the MET complex via interactions with MET complex proteins (Maeda et al., 2014, 2017). Components of the MET (or upper tip link density) complex include myosin VIIa, harmonin, and SANS, which localize to the region on the taller stereocilium where the tip link anchors and interact in a large complex with cadherin 23 to regulate MET and tip link tension (Figure 1; Adato et al., 2005b; Grati and Kachar, 2011; Cunningham and Muller, 2019). Cadherin 23 also frequently complexes with myosin-1c and harmonin, which helps to shape and stabilize the stereocilia of the hair bundles (Liu et al., 2012). These proteins act as a bridge between cadherin 23 and the cytoskeletal actin core of stereocilium through binding of the PDZ domains and anchors cadherin 23 intracellularly to the actin filaments (Boeda et al., 2001, 2002; Adato et al., 2005a).
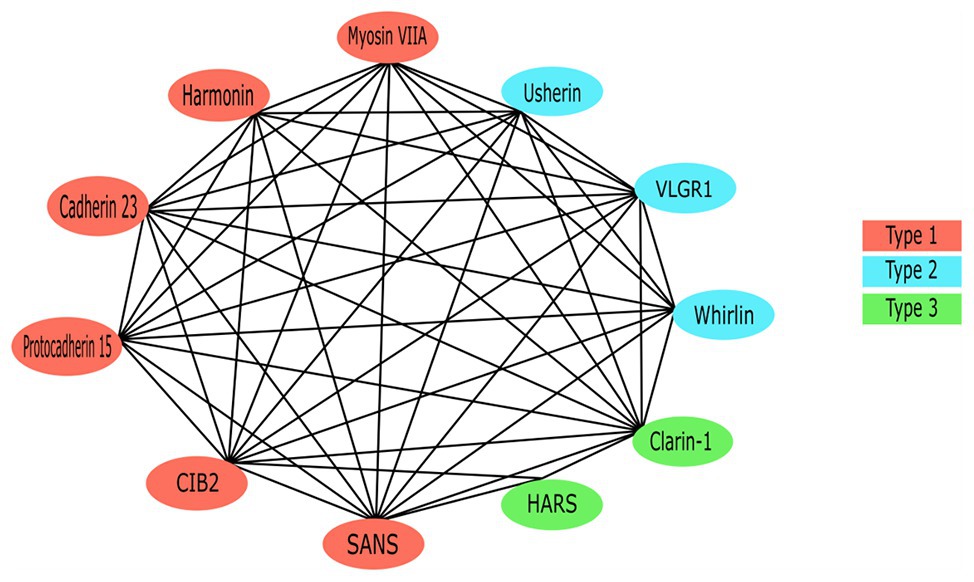
Figure 4. The Usher protein interaction network. All previously reported interactions among Usher proteins have been indicated.
Due to the role it plays as a scaffold protein, harmonin is considered to be at the core of the USH protein interactome (Reiners et al., 2005a). Specifically, the PDZ1 and/or PDZ3 domains, and N-terminal domains of harmonin form a supramodule that binds the SAM domain on SANS with high affinity, forming a stable complex between the two scaffold proteins (Adato et al., 2005b; Yan et al., 2010). The complex binds the tail-region of myosin VIIa to form the upper tip link density or motor complex, which anchors the tip link to the taller stereocilia via binding of the CD of cadherin 23 proteins (Boeda et al., 2002; Adato et al., 2005b; Bahloul et al., 2010; Yan et al., 2010). Known USH-causing missense mutations in both of the genes has been shown to adversely affect the formation of this harmonin/SANS complex and therefore supports that the structure plays a role in normal hearing function (Yan et al., 2010). Additionally, the cytoplasmic tail of protocadherin 15 has been shown to bind the PDZ2 domain of harmonin in vitro (Adato et al., 2005b; Senften et al., 2006). Co-localization of USH2 proteins and harmonin in both the HC and retina may indicate the inclusion of USH2A and ADGRV1 products in the USH-protein supramolecular network (Reiners et al., 2005b). Both usherin and ADGRV1 contain PBM in their C-termini, which bind the PDZ1 domain of harmonin (Reiners et al., 2005b). As well as binding other USH proteins, in vitro assays have identified that the second coiled-coil domain of harmonin isoform b is able to bind the PDZ1 and PDZ2 regions of all other harmonin isoforms (Adato et al., 2005b).
Another central protein is myosin VIIa due to its role in the facilitation of USH protein trafficking via binding of ATP and the actin-core of stereocilia (Udovichenko et al., 2002; Woolner and Bement, 2009). Specifically, the C-terminal MyTH4-FERM domain of myosin VIIa binds to the PDZ domains of harmonin, whirlin as well as the USH modifier, PDZD7 (Li et al., 2017). In the absence of myosin VIIa, harmonin isoform b is not detectable along the stereocilia, implicating a dependence on the motor protein for transport of this isoform to its correct subcellular location, which is not seen in isoforms a or c (Boeda et al., 2002). The CD of protocadherin 15 bind myosin VIIa, suggesting that the two proteins interact in some way to facilitate the development and regulation of HC integrity, roles which both proteins are associated with (Senften et al., 2006). Among the USH proteins, CIB2 only interacts with myosin VIIa and whirlin (Riazuddin et al., 2012). While this interaction is not necessary for localization of CIB2 to the stereocilia tips (Riazuddin et al., 2012), knockout of CIB2 results in mislocalization of whirlin (Michel et al., 2017). Additionally, the PDZ1 and PDZ2 domains of whirlin interact with the class I PBM of SANS (van Wijk et al., 2006). SANS interacts with both tip link proteins, cadherin 23 and protocadherin 15, therefore is predicted to be involved in a multiprotein complex localized at the lower tip link density (Figure 1; Caberlotto et al., 2011).
The USH2 proteins usherin, ADGRV1, and whirlin interact with one another to form a multiprotein complex called the ankle link (or USH2) complex within the cytoplasmic region of ankle links (Mathur and Yang, 2015). The myosin VIIa protein is predicted to transport all three of the USH2 proteins to the base of the stereocilia to form the transient ankle link (Michalski et al., 2007). Ankle links are the thin fibers that connect the bases of neighboring stereocilia and only exist during development (Michalski et al., 2007). Defects in the ankle link complex result in disorganization of the stereocilia bundle. As the correct organization of the stereocilia hair bundle is critical for accurate processing of sound, mutations in the genes coding for these USH2 proteins could cause SNHL through the disruption of the ankle link structure during development (Michalski et al., 2007). The phenotype in USH2A patients with defective usherin is remarkably similar to that seen in USH2C patients with inactivated ADGRV1, therefore suggesting the co-localization of ADGRV1 and usherin in specific sub-cellular compartments (Weston et al., 2004). The long whirlin isoform is responsible for the formation of the USH2 protein complex in vivo; therefore, when whirlin is disrupted the normal cellular localization of usherin and ADGRV1 are abolished, resulting in hearing and visual defects (Yang et al., 2010). The latter, is predicted to be the result of whirlin interacting with usherin, as well as USH1 proteins harmonin and SANS, to stabilize the centrosome-cilium interface region of the photoreceptor cells (Chen et al., 2014; Sorusch et al., 2017).
Clarin-1 interactions are less studied than other USH proteins, although the protein is predicted to interact with myosin VIIa and play a role in regulation of the actin cytoskeleton of the stereocilia (Adato et al., 1999; Tian et al., 2009). Evidence suggests that CLRN1 and MYO7A products may be involved in a synergistic interaction, with symptoms of USH3 being more severe in the presence of a single mutated MYO7A allele (Adato et al., 1999). Therefore, patients harboring a single pathogenic MYO7A mutation, which would not usually be disease causing in isolation, may present with more severe USH symptoms if they also carry two defective CLRN1 alleles (Adato et al., 1999). Additionally, clarin-1 is associated with protocadherin 15, with the USH3A protein predicted to increase the efficiency of assembly and localization of the USH proteins that form the mechanotransduction machinery, through modulation of vesicle recycling (Ogun and Zallocchi, 2014). Specifically, the C-terminal tail of clarin-1 is known to interact with protocadherin 15, since normal localization and function of protocadherin 15 is inhibited in mouse models expressing truncated clarin-1 (Ogun and Zallocchi, 2014).
Pdzd7: an Usher Modifier
PDZD7 encodes a PDZ domain-containing scaffold protein, which is responsible for the organization of the USH2 interactome (Zou et al., 2014; Du et al., 2018). PDZD7 is considered an USH modifier as it interacts with both USH1 and USH2 proteins, including myosin VIIa, usherin, ADGRV1 and whirlin (Ebermann et al., 2010; Zou et al., 2014; Du et al., 2018). As a result, mutations in PDZD7 can cause non-syndromic recessive hearing impairment and also interrupt organization of cells and disrupt the MET process, causing SNHL and deafness as seen in USH (Du et al., 2018). The formation of the USH2 quaternary protein complex relies on the heterodimerization between PDZD7 and whirlin and a subsequent dynamic interplay between USH proteins via their multiple PDZ domains (Chen et al., 2014). Progressive retinal cell death and a reduction in ADGRV1 localization in the connecting cilia in photoreceptor cells were observed in a PDZD7-knockdown zebrafish, which has an USH-like phenotype. In addition, PDZD7-knockout mice exhibit a loss in stereocilia bundle architecture at the ankle-link region, resulting in the attenuation of MET energy (Zou et al., 2014). As such, PDZD7 mutations can cause symptoms of USH via disruptions in its interactions with other proteins rather than through its own specific roles.
Therapeutic Approaches
Unfortunately, despite significant advances in the field, there is still no cure for USH, regardless of the subtype or specific causative gene mutation. Several treatment options are available to help patients manage the symptoms of USH, particularly those of HL. Treatments for the visual and balance symptoms are more limited, with most strategies simply providing support to assist patients in completing basic living tasks. The current treatment for all types of HL, including USH, is auditory rehabilitation using hearing aid devices or cochlear implants. These technologies can significantly aid in communication, yet they are still unable to mimic the quality of natural hearing and, more importantly, they do not treat the underlying cause of the HL. Currently, there are a range of new therapeutic strategies being developed that utilize new technologies such as gene editing and cell based therapies, with some clinical trials currently underway.
Viral-Based Gene Replacement Therapy
One therapeutic approach for genetic disorders including USH is to replace the mutated gene with a wildtype copy of the gene, allowing functional protein to be produced. A popular option for delivering replacement genes is using viral vectors, such as lentivirus and adeno-associated virus (AAV). Local delivery of rAAV2/8 containing a replacement Sans gene into the inner ear completely restored vestibular function and hearing in Sans knockout mice (Emptoz et al., 2017). Although AAVs have a smaller packaging capacity than lentiviral vectors, AAV-mediated delivery of full-length MYO7A cDNA has been effective both in vivo and in vitro, resulting in wildtype expression levels (Allocca et al., 2008; Lopes et al., 2013; Dyka et al., 2014; Trapani et al., 2014). As AAV has limited genetic capacity, many gene sequences are too large to fit into a single vector (Akil, 2020). A full length MYO7A cDNA was successfully reconstituted in an AAV post-infection, however the vectors were in a heterogenous, fragmented genome (“fAAV”) form, with low efficiency (Lopes et al., 2013). To address this, Dyka et al. (2014) used an overlapping approach with dual vectors sharing a central part of the MYO7A cDNA sequence, leading to the expression of full length MYO7A transcript in vitro and in vivo with equal and higher efficiency than fAAV. Several research groups have used dual or multi AAV systems to overcome this limitation in USH studies, including oversized transgene constructs. However, most studies have reported very low levels of protein expression (Jaijo et al., 2007; Allocca et al., 2008; Alagramam et al., 2011; Dyka et al., 2014; Trapani et al., 2014; Maddalena et al., 2018).
Genome Editing-Based Therapy
Zinc-Finger Nucleases and CRISPR/Cas9
Genome editing-based therapy is another promising approach for treating USH as it attempts to correct the mutation in vivo in a site-specific manner, while preserving endogenous regulation of the repaired gene. Zinc-finger nucleases (ZFN) are chimeric proteins generated by fusing a zinc-finger DNA-binding domain (ZF domains) to a DNA-cleavage domain (Durai et al., 2005). ZFN was applied to target a nonsense mutation (p.R31X) in Ush1c cell line, leading to recovered harmonin protein expression, with no apparent off-target effects (Overlack et al., 2012). While ZFN seem like a powerful tool, the clustered regularly interspaced palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) system is now the most advanced and widely accepted methodology due to its simplicity, robustness, and high efficiency (Mishra et al., 2018).
Clustered regularly interspaced palindromic repeats/CRISPR-associated protein 9 was used to successfully correct one of the most prevalent mutations in USH2A (c.2299delG) in patient-derived fibroblasts (Fuster-Garcia et al., 2017). A CRISPR/Cas9 approach has also been used on patient-derived induced pluripotent stem cell (iPSC) harboring mutation in USH2A or MYO7A (Tang et al., 2016; Sanjurjo-Soriano et al., 2019). In these studies, the iPSC were successfully corrected with high efficacy and specificity, with no observed off-target mutagenesis (Tang et al., 2016). Additionally, the corrected iPSC retained their pluripotency and were genetically stable (Sanjurjo-Soriano et al., 2019).
Allergan and Editas Medicine have recently launched the Brilliance clinical trial (NCT03872479) for Leber congenital amaurosis (LCA) 10 patients with an intronic mutation in CEP290 gene. EDIT-101 (AGN-151587) from Editas is designed to locate and remove the mutated CEP290 via based on non-homologous end joining (Maeder et al., 2019). Published results showed sub-retinal delivery of EDIT-101 achieved rapid, productive and sustained CEP290 gene editing in CEP290 mice and somatic primate cells in vivo (Maeder et al., 2019). Concurrently, EDIT-102 is being developed to target USH2A using the same proprietary enzyme, vector, promoters, and route of delivery as EDIT-101. Since USH2A mutations can affect cell receptors in the same way as CEP290 mutations, the USH2A product is expected to benefit from EDIT-101’s therapeutic pipeline.
Drug Therapy
Antisense Oligonucleotides
Antisense oligonucleotides (ASO) are short, synthetic, single-stranded oligodeoxynucleotides that are complementary to the mRNA target (Rinaldi and Wood, 2018). ASO have been used to correct cryptic splicing in Ush1c.216A knock-in mice (Lentz et al., 2013). This partial correction by ASO increases harmonin expression, improves the morphology and structure of stereocilia, leading to a rescue of auditory and vestibular responses in Ush1c mice. This shows that early-treated mice respond better due to an age threshold for effective drug delivery (Lentz et al., 2013; Vijayakumar et al., 2017; Donaldson et al., 2018). Similarly, Ponnath et al. (2018) reported restoration of OHC and IHC in Ush1c mice by ASO-29 treatment.
Two designed ASO successfully targeted a pseudo-exon-causing USH2A mutation, increasing the amount of correctly spliced transcript in USH2A patient-derived fibroblasts (Slijkerman et al., 2016). A novel drug, QR-421a (ProQR Therapeutics) demonstrated a successful “exon skip” in an Ush2a mutant zebrafish model, leading to restored Usherin expression and electroretinogram (ERG) recordings (Dona et al., 2018; van Diepen et al., 2019). QR-421a is currently in clinical trial (Stellar; NCT03780257), with preliminary results reported in April 2020.1
Reducing Endoplasmic Reticulum Stress
The endoplasmic reticulum (ER) is responsible for processing mRNA into protein and appropriately folding those proteins (Szegezdi et al., 2006). Importantly, the ER is also the site at which several USH proteins complex before trafficking as a group into their final position (Blanco-Sanchez et al., 2014). A mutation in a single USH gene can prevent the protein complexes from forming and lead to an accumulation of multiple USH proteins in the ER, which causes ER stress and, ultimately, apoptosis (Blanco-Sanchez et al., 2014). Three proteins in particular – harmonin, cadherin 23 and myosin VIIa – have been shown to co-localize in the ER and preassemble as a complex prior to trafficking to the stereocilia (Blanco-Sanchez et al., 2014). Treatment of erlong (erl) mice, Cdh23 mutant mice carrying a missense mutation with the small molecular compound Salubrinal®, an ER stress inhibitor, caused a reduction in auditory brainstem response thresholds and higher distortion product otoacoustic emission (DPOAE) amplitudes (Han et al., 2012; Hu et al., 2016). Taken together, these findings suggest that inhibition of ER stress may slow or prevent inner ear HC death and subsequent HL in USH patients (Szegezdi et al., 2006).
Improving the Stability of USH Proteins
The most common causative mutation for USH type III in Ashkenazi Jews of European and North American descent is the c.144T>G (p.N48K) mutation in CLRN1 (Fields et al., 2002; Ness et al., 2003). This mutation is thought to adversely affect the folding and stability of the clarin-1 protein, ultimately causing progressive vision and HL (Tian et al., 2009). A novel therapeutic approach is to improve the stability of the mutant clarin-1 protein, thereby allowing it to correctly localize to the plasma membrane and restore function. An intensive high-throughput screening approach identified 48 candidate small molecules that act to stabilize the CLRN1N48K in vitro (Alagramam et al., 2016). A secondary screen of these candidates for proteasome activity found three small molecules that showed clear evidence of specific stabilization of CLRN1N48K. Final validation by immunoblotting narrowed this down to a single candidate, O03, which was effective in the two cell lines tested. Subsequent testing confirmed that the increase in cellular CLRN1N48K following O03 treatment was due to increased stability of the mutant protein (Alagramam et al., 2016). A synthetic modification of O03, BF844, improved the potency and pharmacokinetics of the compound. When applied before or during the early stages of HL, BF844 effectively crossed the blood-labyrinth barrier and mitigated HL in a mouse model (Alagramam et al., 2016). This represents a potential therapeutic candidate for USH3A patients with this mutation that could slow or prevent HL, if applied prior to its onset.
Cell-Based Therapy
Stem Cells and Three-Dimensional Organoids
Given the capacity of pluripotent stem cells (PSCs) to self-renew and differentiate into many cell types, they are now widely used in regenerative medicine (Okano and Kelley, 2012). The stem cells commonly used are embryonic stem cells (ESCs), adult stem cells (ASCs), and iPSC (Dufner-Almeida et al., 2019). Oshima et al. (2010) adapted the protocol and promoted the differentiation of mouse ESCs and iPSC into mechanosensitive HC-like cells.
Pluripotent stem cells can form embryoid bodies in vitro which are three dimensional (3D) aggregates capable of differentiating into specific lineages (Okano and Kelley, 2012; Omole and Fakoya, 2018), including retinal (Nakano et al., 2012; Phillips et al., 2012; Zhong et al., 2014; Mellough et al., 2015, 2019) and inner ear organoids (Koehler and Hashino, 2014; Liu et al., 2016; Longworth-Mills et al., 2016; Nie et al., 2017). The first inner ear organoid system was developed by Koehler et al. using mouse ESCs, which was later adapted to use human ESCs or iPSC (Koehler et al., 2017). Several groups have published protocols inducing inner ear organoids from aggregates of mouse ESCs (Koehler and Hashino, 2014; Liu et al., 2016; Longworth-Mills et al., 2016; Nie et al., 2017) and human PSCs (Jeong et al., 2018). Single-cell electrophysiology tests identified functional vestibular hair cells in inner ear organoids (Jeong et al., 2018). Numerous improved protocols have been described on including the modulation of signaling pathways, like Wnt, Sonic Hedgehog, and Fgf (Lahlou et al., 2018), addition of extracellular matrix scaffolds (e.g., Matrigel; Koehler et al., 2013, 2017); and addition of bone morphogenetic protein (BMP), transforming growth factor beta (TGFβ), and Wnt antagonists (Zhou et al., 2015). These protocols have been discussed in depth in a recent review (Tang et al., 2020). The PSCs and organoid technology are ideal for creating patient-specific disease models for USH, with these in vitro models possessing an identical genomic profile of the patient, offering opportunities for personalized medicine (Johnson and Hockemeyer, 2015; Yilmaz and Benvenisty, 2019).
Hair Cell Regeneration
Since hearing loss commonly occurs as a result of inner ear hair cell damage and since hair cells are unable to regenerate spontaneously (i.e., quiescent), promoting the regeneration of these hair cells is a key therapeutic goal. Cellular quiescence occurs via inhibition of cyclin/cyclin-dependent kinase (CDK) and hypophosphorylation of retinoblastoma protein (Polyak et al., 1994). CDK inhibitors such as p27Kip1 are expressed in the cochlea at a level sufficient to promote quiescence of the hair cells (Mantela et al., 2005; Laine et al., 2007; Walters et al., 2014). Deletion of p27Kip1 in transgenic mice resulted in proliferation of supporting cells and regeneration of hair cells (Chen and Segil, 1999; Lowenheim et al., 1999; Kil et al., 2011). Hair cell-specific conditional deletion of p27Kip1 in neonatal mice resulted in proliferation and improved survival of hair cells without any adverse effects on hearing (Walters et al., 2014). In response to the knockout of p27Kip1, IHC were more proliferative than OHC (Walters et al., 2014). These findings suggest that p27Kip1 could represent a good therapeutic target to minimize cell death and promote hair cell regeneration in the cochlea. As such, Sound Pharmaceuticals is conducting a phase 1/2 trial (NCT02819856) of SPI-5557, a drug that inhibits p27Kip1, aiming to regenerating cochlea hair cells (Jones, 2018).
Atoh1 (also known as Math1 for “mouse atoh1” and HATH1 for “human atoh1”), a basic helix-loop-helix transcription factor, was first identified to be essential in the generation of inner ear hair cells, and overexpression of Atoh1 enhanced hair cell regeneration (Ben-Arie et al., 1997; Bermingham et al., 1999). Atoh1 has since been in use as a prime candidate in hair cell regeneration research, with varying success rates across published studies. Guinea pigs treated with ATOH1 gene therapy showed a significant increase in the number of hair cells (Atkinson et al., 2014). Overall, these guinea pigs did not have complete recovery of hearing, suggesting that ATOH1 gene therapy alone is unable to convert non-sensory cells into hair cells, nor capable of rescuing the phenotype of surviving hair cells (Atkinson et al., 2014).
Limitations and Considerations for USH Therapies
Given that USH is a syndromic condition affecting multiple senses, it is important to note that current therapeutic trials generally focus on one sense rather than across the entire syndrome. The onset of visual symptoms is later than the onset of audiological symptoms in all USH sybtypes (Mathur and Yang, 2015). Upon identification of HL, genetic testing would indicate if a mutation is present in the known USH-related genes, which provides information on the USH subtype and the disease progression. It follows then that therapeutics could be delivered to a patient before their vision deteriorates and maintaining important protein structures and complexes is a much easier task than restoring them. Delivery of drugs into the inner ear is notoriously difficult. Systemic delivery carries the highest risk for off target effects and, regardless, is hampered by the blood-labyrinth barrier, which is the physiological barrier between the peripheral blood and the inner ear fluids (Hao and Li, 2019; Nyberg et al., 2019). Direct injection into the scala media would result in the highest bioavailability and benefit, however this requires a costly and high risk surgery (Hao and Li, 2019; Nyberg et al., 2019).
There are several barriers to overcome in order to achieve successful gene therapy, including the likelihood of a host immune response being triggered and directed against the vector component or the transgene product, or both, as these vector systems are administered directly to the patient (Williams et al., 2017). Another uncertainty is whether the transgenes have long-term expression and the same functional response as a healthy gene. As for genome editing approaches, there are a number of disadvantageous consequences to be considered – off-target mutagenesis, mosaicism, complex rearrangements, on-site damage, and biallelic modification (Araki et al., 2014). These technical risks can be reduced and addressed by experimental design; for example, designing unique genomic target sequences and evaluate off-target risks with bioinformatics tools, but there are still too many unknowns regarding the mechanisms of DNA repair at this stage (Araki et al., 2014). It is challenging to determine the therapeutic vector dose and the route of administration, as it is not well characterized at this stage. Many strategies have been conducted to overcome human immune responses in a systemic gene transfer, such as delivering at a different site or administering immunosuppressive drugs to prevent or block T-cell responses toward foreign antigens (Sack and Herzog, 2009), much more remains to be done to better understand the efficacy and safety implications of long-term risk of gene therapy administration.
Although stem cell-based therapy is popular, there are several challenges and unanticipated risks to be evaluated before it could be applied clinically. The major concern is the similarities between stem cells and cancer cells, such as the ability to self-renew, the potency of stem cells (pluripotent or multipotent), indefinite growth, and high proliferation rate (Li and Neaves, 2006; Werbowetski-Ogilvie et al., 2009; Herberts et al., 2011). The first point is the survival rate and period of the transplanted cells after their insertion into the patients’ cochlea, without interfering the normal hearing. Alongside the risk of tumorigenesis, potential stem cell migration to inappropriate sites and immune rejection of transplanted stem cells must be considered. Other risk factors include the type of stem cells used, their procurement and culturing history, the level of manipulation and administration site associated with stem cell-based therapy and medicinal products (Herberts et al., 2011).
Future Outlooks
Although USH genes have been mapped and many corresponding pathological mutations identified, the syndrome remains incurable. USH proteins interact in large multiprotein complexes in the inner ear and retina, and are responsible for the trafficking, scaffolding, and signaling of proteins involved in the development and maintenance of the sensory cells of these organs. Within the cochlea, USH proteins play critical roles in the correct organization and function of the HC stereocilia that conduct soundwaves to the brain. A deeper understanding of these complex USH protein networks, including their functions and the downstream effects of protein mutations in the inner ear sensory cells, is critical for understanding USH. Research into the genotype–phenotype associations of USH gene mutations has highlighted the importance of accurate molecular diagnosis for patients, since the nature of the mutation can greatly influence the prognosis (Toms et al., 2015; Bonnet et al., 2016).
Interestingly, all current potential treatments are only targeted at one of the groups of symptoms (HL, vision loss, or balance problems), none have looked at tackling the syndrome as a whole. Currently, a majority of the USH treatments under development are targeted at specific, high-frequency mutations, further emphasizing the need for genetic screening (Toms et al., 2015; Jouret et al., 2019). Meta-analysis of USH-causing mutations have been successful in highlighting the frequency and location of pathological mutations, including hotspot and founder mutations (Jouret et al., 2019).
Significant progress has also been made into the characterization of USH pathogenesis through animal models. Recent studies have begun to elucidate the complex functions and interactions between USH proteins, although there remains a number of unknowns regarding the molecular mechanisms controlling these processes, as well as possible gene interactions. Inner ear organoids generated from human cells may hold the key to the in vitro analysis of human USH genes and proteins. Particularly given that several of the USH genes are responsible for early development, which would be mimicked by the organoids (Liu et al., 2016; Koehler et al., 2017; Jeong et al., 2018). Presently, a number of studies are underway to identify potential USH treatments, including ASO therapy, CRISPR/Cas9 genome editing, translational read-through pharmaceutical drugs, and AAV or lentiviral-mediated therapies. Clinical trials focused on applying these techniques to USH in the hopes of developing a cure, are still underway.
Author Contributions
MA, RD, and EW designed the study. MW, AF, ZN, and XK conducted the research and literature search. MW, AF, ZN, and EW prepared the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Garnett Passe and Rodney Williams Memorial Foundation (Conjoint Grant 2018, Australia) and the Stan Perron Charitable Foundation (Australia).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Abbott, J. A., Guth, E., Kim, C., Regan, C., Siu, V. M., Rupar, C. A., et al. (2017). The usher syndrome type IIIB histidyl-tRNA synthetase mutation confers temperature sensitivity. Biochemistry 56, 3619–3631. doi: 10.1021/acs.biochem.7b00114
Adato, A., Kalinski, H., Weil, D., Chaib, H., Korostishevsky, M., and Bonne-Tamir, B. (1999). Possible interaction between USH1B and USH3 gene products as implied by apparent digenic deafness inheritance. Am. J. Hum. Genet. 65, 261–265. doi: 10.1086/302438
Adato, A., Lefevre, G., Delprat, B., Michel, V., Michalski, N., Chardenoux, S., et al. (2005a). Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum. Mol. Genet. 14, 3921–3932. doi: 10.1093/hmg/ddi416
Adato, A., Michel, V., Kikkawa, Y., Reiners, J., Alagramam, K. N., Weil, D., et al. (2005b). Interactions in the network of Usher syndrome type 1 proteins. Hum. Mol. Genet. 14, 347–356. doi: 10.1093/hmg/ddi031
Adato, A., Vreugde, S., Joensuu, T., Avidan, N., Hamalainen, R., Belenkiy, O., et al. (2002). USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur. J. Hum. Genet. 10, 339–350. doi: 10.1038/sj.ejhg.5200831
Ahmed, Z. M., Goodyear, R., Riazuddin, S., Lagziel, A., Legan, P. K., Behra, M., et al. (2006). The tip-link antigen, a protein associated with the transduction complex of sensory hair cells, is protocadherin-15. J. Neurosci. 26, 7022–7034. doi: 10.1523/JNEUROSCI.1163-06.2006
Ahmed, Z. M., Riazuddin, S., Ahmad, J., Bernstein, S. L., Guo, Y., Sabar, M. F., et al. (2003). PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum. Mol. Genet. 12, 3215–3223. doi: 10.1093/hmg/ddg358
Ahmed, Z. M., Riazuddin, S., Aye, S., Ali, R. A., Venselaar, H., Anwar, S., et al. (2008). Gene structure and mutant alleles of PCDH15: nonsyndromic deafness DFNB23 and type 1 Usher syndrome. Hum. Genet. 124, 215–223. doi: 10.1007/s00439-008-0543-3
Ahmed, Z. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z., Khan, S., Griffith, A. J., et al. (2001). Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am. J. Hum. Genet. 69, 25–34. doi: 10.1086/321277
Ahmed, Z. M., Riazuddin, S., Khan, S. N., Friedman, P. L., Riazuddin, S., and Friedman, T. B. (2009). USH1H, a novel locus for type I Usher syndrome, maps to chromosome 15q22-23. Clin. Genet. 75, 86–91. doi: 10.1111/j.1399-0004.2008.01038.x
Ahmed, Z. M., Smith, T. N., Riazuddin, S., Makishima, T., Ghosh, M., Bokhari, S., et al. (2002). Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum. Genet. 110, 527–531. doi: 10.1007/s00439-002-0732-4
Akil, O. (2020). Dual and triple AAV delivery of large therapeutic gene sequences into the inner ear. Hear. Res. 394:107912. doi: 10.1016/j.heares.2020.107912
Alagramam, K. N., Goodyear, R. J., Geng, R., Furness, D. N., van Aken, A. F., Marcotti, W., et al. (2011). Mutations in protocadherin 15 and cadherin 23 affect tip links and mechanotransduction in mammalian sensory hair cells. PLoS One 6:e19183. doi: 10.1371/journal.pone.0019183
Alagramam, K. N., Gopal, S. R., Geng, R., Chen, D. H., Nemet, I., Lee, R., et al. (2016). A small molecule mitigates hearing loss in a mouse model of Usher syndrome III. Nat. Chem. Biol. 12, 444–451. doi: 10.1038/nchembio.2069
Alagramam, K. N., Murcia, C. L., Kwon, H. Y., Pawlowski, K. S., Wright, C. G., and Woychik, R. P. (2001a). The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat. Genet. 27, 99–102. doi: 10.1038/83837
Alagramam, K. N., Yuan, H., Kuehn, M. H., Murcia, C. L., Wayne, S., Srisailpathy, C. R., et al. (2001b). Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum. Mol. Genet. 10, 1709–1718. doi: 10.1093/hmg/10.16.1709
Aller, E., Jaijo, T., van Wijk, E., Ebermann, I., Kersten, F., Garcia-Garcia, G., et al. (2010). Sequence variants of the DFNB31 gene among Usher syndrome patients of diverse origin. Mol. Vis. 16, 495–500.
Aller, E., Najera, C., Millan, J. M., Oltra, J. S., Perez-Garrigues, H., Vilela, C., et al. (2004). Genetic analysis of 2299delG and C759F mutations (USH2A) in patients with visual and/or auditory impairments. Eur. J. Hum. Genet. 12, 407–410. doi: 10.1038/sj.ejhg.5201138
Allocca, M., Doria, M., Petrillo, M., Colella, P., Garcia-Hoyos, M., Gibbs, D., et al. (2008). Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J. Clin. Invest. 118, 1955–1964. doi: 10.1172/JCI34316
Araki, M., Nojima, K., and Ishii, T. (2014). Caution required for handling genome editing technology. Trends Biotechnol. 32, 234–237. doi: 10.1016/j.tibtech.2014.03.005
Assad, J. A., Shepherd, G. M., and Corey, D. P. (1991). Tip-link integrity and mechanical transduction in vertebrate hair cells. Neuron 7, 985–994. doi: 10.1016/0896-6273(91)90343-x
Astuto, L. M., Bork, J. M., Weston, M. D., Askew, J. W., Fields, R. R., Orten, D. J., et al. (2002). CDH23 mutation and phenotype heterogeneity: a profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am. J. Hum. Genet. 71, 262–275. doi: 10.1086/341558
Atkinson, P. J., Wise, A. K., Flynn, B. O., Nayagam, B. A., and Richardson, R. T. (2014). Hair cell regeneration after ATOH1 gene therapy in the cochlea of profoundly deaf adult Guinea pigs. PLoS One 9:e102077. doi: 10.1371/journal.pone.0102077
Audo, I., Bujakowska, K., Mohand-Said, S., Tronche, S., Lancelot, M. E., Antonio, A., et al. (2011). A novel DFNB31 mutation associated with Usher type 2 syndrome showing variable degrees of auditory loss in a consanguineous Portuguese family. Mol. Vis. 17, 1598–1606.
Bahloul, A., Michel, V., Hardelin, J. P., Nouaille, S., Hoos, S., Houdusse, A., et al. (2010). Cadherin-23, myosin VIIa and harmonin, encoded by Usher syndrome type I genes, form a ternary complex and interact with membrane phospholipids. Hum. Mol. Genet. 19, 3557–3565. doi: 10.1093/hmg/ddq271
Barkalow, F. J., and Schwarzbauer, J. E. (1991). Localization of the major heparin-binding site in fibronectin. J. Biol. Chem. 266, 7812–7818.
Bartsch, T. F., Hengel, F. E., Oswald, A., Dionne, G., Chipendo, I. V., Mangat, S. S., et al. (2019). Elasticity of individual protocadherin 15 molecules implicates tip links as the gating springs for hearing. Proc. Natl. Acad. Sci. U. S. A. 116, 11048–11056. doi: 10.1073/pnas.1902163116
Beck, K., Hunter, I., and Engel, J. (1990). Structure and function of laminin: anatomy of a multidomain glycoprotein. FASEB J. 4, 148–160. doi: 10.1096/fasebj.4.2.2404817
Bein, K., and Simons, M. (2000). Thrombospondin type 1 repeats interact with matrix metalloproteinase 2. Regulation of metalloproteinase activity. J. Biol. Chem. 275, 32167–32173. doi: 10.1074/jbc.M003834200
Belyantseva, I. A., Boger, E. T., Naz, S., Frolenkov, G. I., Sellers, J. R., Ahmed, Z. M., et al. (2005). Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat. Cell Biol. 7, 148–156. doi: 10.1038/ncb1219
Ben-Arie, N., Bellen, H. J., Armstrong, D. L., McCall, A. E., Gordadze, P. R., Guo, Q., et al. (1997). Math1 is essential for genesis of cerebellar granule neurons. Nature 390, 169–172. doi: 10.1038/36579
Ben-Yosef, T., Ness, S. L., Madeo, A. C., Bar-Lev, A., Wolfman, J. H., Ahmed, Z. M., et al. (2003). A mutation of PCDH15 among Ashkenazi Jews with the type 1 Usher syndrome. N. Engl. J. Med. 348, 1664–1670. doi: 10.1056/NEJMoa021502
Bermingham, N. A., Hassan, B. A., Price, S. D., Vollrath, M. A., Ben-Arie, N., Eatock, R. A., et al. (1999). Math1: an essential gene for the generation of inner ear hair cells. Science 284, 1837–1841. doi: 10.1126/science.284.5421.1837
Bermingham-McDonogh, O., and Reh, T. A. (2011). Regulated reprogramming in the regeneration of sensory receptor cells. Neuron 71, 389–405. doi: 10.1016/j.neuron.2011.07.015
Beurg, M., Xiong, W., Zhao, B., Muller, U., and Fettiplace, R. (2015). Subunit determination of the conductance of hair-cell mechanotransducer channels. Proc. Natl. Acad. Sci. U. S. A. 112, 1589–1594. doi: 10.1073/pnas.1420906112
Bhattacharya, G., Miller, C., Kimberling, W. J., Jablonski, M. M., and Cosgrove, D. (2002). Localization and expression of usherin: a novel basement membrane protein defective in people with Usher’s syndrome type IIa. Hear. Res. 163, 1–11. doi: 10.1016/s0378-5955(01)00344-6
Blanco-Sanchez, B., Clement, A., Fierro, J. Jr., Stednitz, S., Phillips, J. B., Wegner, J., et al. (2018). GRXCR1 promotes hair bundle development by destabilizing the physical interaction between Harmonin and sans Usher syndrome proteins. Cell Rep. 25:e1284. doi: 10.1016/j.celrep.2018.10.005
Blanco-Sanchez, B., Clement, A., Fierro, J. Jr., Washbourne, P., and Westerfield, M. (2014). Complexes of Usher proteins preassemble at the endoplasmic reticulum and are required for trafficking and ER homeostasis. Dis. Model. Mech. 7, 547–559. doi: 10.1242/dmm.014068
Blazejczyk, M., Sobczak, A., Debowska, K., Wisniewska, M. B., Kirilenko, A., Pikula, S., et al. (2009). Biochemical characterization and expression analysis of a novel EF-hand Ca2+ binding protein calmyrin2 (Cib2) in brain indicates its function in NMDA receptor mediated Ca2+ signaling. Arch. Biochem. Biophys. 487, 66–78. doi: 10.1016/j.abb.2009.05.002
Boeda, B., El-Amraoui, A., Bahloul, A., Goodyear, R., Daviet, L., Blanchard, S., et al. (2002). Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 21, 6689–6699. doi: 10.1093/emboj/cdf689
Boeda, B., Weil, D., and Petit, C. (2001). A specific promoter of the sensory cells of the inner ear defined by transgenesis. Hum. Mol. Genet. 10, 1581–1589. doi: 10.1093/hmg/10.15.1581
Bolz, H., Reiners, J., Wolfrum, U., and Gal, A. (2002). Role of cadherins in Ca2+-mediated cell adhesion and inherited photoreceptor degeneration. Adv. Exp. Med. Biol. 514, 399–410. doi: 10.1007/978-1-4615-0121-3_24
Bolz, H., von Brederlow, B., Ramirez, A., Bryda, E. C., Kutsche, K., Nothwang, H. G., et al. (2001). Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat. Genet. 27, 108–112. doi: 10.1038/83667
Bonnet, C., Grati, M., Marlin, S., Levilliers, J., Hardelin, J. P., Parodi, M., et al. (2011). Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J. Rare Dis. 6:21. doi: 10.1186/1750-1172-6-21
Bonnet, C., Riahi, Z., Chantot-Bastaraud, S., Smagghe, L., Letexier, M., Marcaillou, C., et al. (2016). An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 24, 1730–1738. doi: 10.1038/ejhg.2016.99
Booth, K. T., Kahrizi, K., Babanejad, M., Daghagh, H., Bademci, G., Arzhangi, S., et al. (2018). Variants in CIB2 cause DFNB48 and not USH1J. Clin. Genet. 93, 812–821. doi: 10.1111/cge.13170
Bork, P., Downing, A. K., Kieffer, B., and Campbell, I. D. (1996). Structure and distribution of modules in extracellular proteins. Q. Rev. Biophys. 29, 119–167. doi: 10.1017/S0033583500005783
Bork, J. M., Peters, L. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z. M., Ness, S. L., et al. (2001). Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 68, 26–37. doi: 10.1086/316954
Boughman, J. A., Vernon, M., and Shaver, K. A. (1983). Usher syndrome: definition and estimate of prevalence from two high-risk populations. J. Chronic Dis. 36, 595–603. doi: 10.1016/0021-9681(83)90147-9
Bowditch, R. D., Hariharan, M., Tominna, E. F., Smith, J. W., Yamada, K. M., Getzoff, E. D., et al. (1994). Identification of a novel integrin binding site in fibronectin. Differential utilization by beta 3 integrins. J. Biol. Chem. 269, 10856–10863.
Bracho, H., and Budelli, R. (1978). The generation of resting membrane potentials in an inner ear hair cell system. J. Physiol. 281, 445–465. doi: 10.1113/jphysiol.1978.sp012432
Bruch, M., Landwehr, R., and Engel, J. (1989). Dissection of laminin by cathepsin G into its long-arm and short-arm structures and localization of regions involved in calcium dependent stabilization and self-association. Eur. J. Biochem. 185, 271–279. doi: 10.1111/j.1432-1033.1989.tb15112.x
Caberlotto, E., Michel, V., Foucher, I., Bahloul, A., Goodyear, R. J., Pepermans, E., et al. (2011). Usher type 1G protein sans is a critical component of the tip-link complex, a structure controlling actin polymerization in stereocilia. Proc. Natl. Acad. Sci. U. S. A. 108, 5825–5830. doi: 10.1073/pnas.1017114108
Calabro, K. R., Boye, S. L., Choudhury, S., Fajardo, D., Peterson, J. J., Li, W., et al. (2019). A novel mouse model of MYO7A USH1B reveals auditory and visual system haploinsufficiencies. Front. Neurosci. 13:1255. doi: 10.3389/fnins.2019.01255
Chaib, H., Kaplan, J., Gerber, S., Vincent, C., Ayadi, H., Slim, R., et al. (1997). A newly identified locus for Usher syndrome type I, USH1E, maps to chromosome 21q21. Hum. Mol. Genet. 6, 27–31. doi: 10.1093/hmg/6.1.27
Chen, P., and Segil, N. (1999). p27(Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development 126, 1581–1590.
Chen, Q., Zou, J., Shen, Z., Zhang, W., and Yang, J. (2014). Whirlin and PDZ domain-containing 7 (PDZD7) proteins are both required to form the quaternary protein complex associated with Usher syndrome type 2. J. Biol. Chem. 289, 36070–36088. doi: 10.1074/jbc.M114.610535
Cheng, L., Yu, H., Jiang, Y., He, J., Pu, S., Li, X., et al. (2018). Identification of a novel MYO7A mutation in Usher syndrome type 1. Oncotarget 9, 2295–2303. doi: 10.18632/oncotarget.23408
Colas-Algora, N., and Millan, J. (2019). How many cadherins do human endothelial cells express? Cell. Mol. Life Sci. 76, 1299–1317. doi: 10.1007/s00018-018-2991-9
Corns, L. F., Johnson, S. L., Roberts, T., Ranatunga, K. M., Hendry, A., Ceriani, F., et al. (2018). Mechanotransduction is required for establishing and maintaining mature inner hair cells and regulating efferent innervation. Nat. Commun. 9:4015. doi: 10.1038/s41467-018-06307-w
Cunningham, C. L., and Muller, U. (2019). Molecular structure of the hair cell mechanoelectrical transduction complex. Cold Spring Harb. Perspect. Med. 9:a033167. doi: 10.1101/cshperspect.a033167
Di Palma, F., Holme, R. H., Bryda, E. C., Belyantseva, I. A., Pellegrino, R., Kachar, B., et al. (2001a). Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat. Genet. 27, 103–107. doi: 10.1038/83660
Di Palma, F., Pellegrino, R., and Noben-Trauth, K. (2001b). Genomic structure, alternative splice forms and normal and mutant alleles of cadherin 23 (Cdh23). Gene 281, 31–41. doi: 10.1016/s0378-1119(01)00761-2
Dinculescu, A., Stupay, R. M., Deng, W. T., Dyka, F. M., Min, S. H., Boye, S. L., et al. (2016). AAV-mediated Clarin-1 expression in the mouse retina: implications for USH3A gene therapy. PLoS One 11:e0148874. doi: 10.1371/journal.pone.0148874
Dona, M., Slijkerman, R., Lerner, K., Broekman, S., Wegner, J., Howat, T., et al. (2018). Usherin defects lead to early-onset retinal dysfunction in zebrafish. Exp. Eye Res. 173, 148–159. doi: 10.1016/j.exer.2018.05.015
Donaldson, T. N., Jennings, K. T., Cherep, L. A., McNeela, A. M., Depreux, F. F., Jodelka, F. M., et al. (2018). Antisense oligonucleotide therapy rescues disruptions in organization of exploratory movements associated with Usher syndrome type 1C in mice. Behav. Brain Res. 338, 76–87. doi: 10.1016/j.bbr.2017.10.012
Du, H., Ren, R., Chen, P., Xu, Z., and Wang, Y. (2018). Identification of binding partners of deafness-related protein PDZD7. Neural Plast. 2018: 2062346. doi: 10.1155/2018/2062346
Dufner-Almeida, L. G., Cruz, D. B. D., Mingroni Netto, R. C., Batissoco, A. C., Oiticica, J., and Salazar-Silva, R. (2019). Stem-cell therapy for hearing loss: are we there yet? Braz. J. Otorhinolaryngol. 85, 520–529. doi: 10.1016/j.bjorl.2019.04.006
Durai, S., Mani, M., Kandavelou, K., Wu, J., Porteus, M. H., and Chandrasegaran, S. (2005). Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 33, 5978–5990. doi: 10.1093/nar/gki912
Dyka, F. M., Boye, S. L., Chiodo, V. A., Hauswirth, W. W., and Boye, S. E. (2014). Dual adeno-associated virus vectors result in efficient in vitro and in vivo expression of an oversized gene, MYO7A. Hum. Gene Ther. Methods 25, 166–177. doi: 10.1089/hgtb.2013.212
Ebermann, I., Lopez, I., Bitner-Glindzicz, M., Brown, C., Koenekoop, R. K., and Bolz, H. J. (2007a). Deafblindness in French Canadians from Quebec: a predominant founder mutation in the USH1C gene provides the first genetic link with the Acadian population. Genome Biol. 8:R47. doi: 10.1186/gb-2007-8-4-r47
Ebermann, I., Phillips, J. B., Liebau, M. C., Koenekoop, R. K., Schermer, B., Lopez, I., et al. (2010). PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Invest. 120, 1812–1823. doi: 10.1172/JCI39715
Ebermann, I., Phillips, J. B., Liebau, M. C., Koenekoop, R. K., Schermer, B., Lopez, I., et al. (2011). PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Invest. 121:821. doi: 10.1172/JCI46312
Ebermann, I., Scholl, H. P., Charbel Issa, P., Becirovic, E., Lamprecht, J., Jurklies, B., et al. (2007b). A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum. Genet. 121, 203–211. doi: 10.1007/s00439-006-0304-0
Emptoz, A., Michel, V., Lelli, A., Akil, O., Boutet de Monvel, J., Lahlou, G., et al. (2017). Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G. Proc. Natl. Acad. Sci. U. S. A. 114, 9695–9700. doi: 10.1073/pnas.1708894114
Engel, J. (1989). EGF-like domains in extracellular matrix proteins: localized signals for growth and differentiation? FEBS Lett. 251, 1–7. doi: 10.1016/0014-5793(89)81417-6
Ernest, S., Rauch, G. -J., Haffter, P., Geisler, R., Petit, C., and Nicolson, T. (2000). Mariner is defective in myosin VIIA: a zebrafish model for human hereditary deafness. Hum. Mol. Genet. 9, 2189–2196. doi: 10.1093/hmg/9.14.2189
Eudy, J. D., Weston, M. D., Yao, S., Hoover, D. M., Rehm, H. L., Ma-Edmonds, M., et al. (1998). Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 280, 1753–1757. doi: 10.1126/science.280.5370.1753
Fanning, A. S., and Anderson, J. M. (1999). PDZ domains: fundamental building blocks in the organization of protein complexes at the plasma membrane. J. Clin. Invest. 103, 767–772. doi: 10.1172/JCI6509
Fields, R. R., Zhou, G., Huang, D., Davis, J. R., Moller, C., Jacobson, S. G., et al. (2002). Usher syndrome type III: revised genomic structure of the USH3 gene and identification of novel mutations. Am. J. Hum. Genet. 71, 607–617. doi: 10.1086/342098
Fortnum, H. M., Summerfield, A. Q., Marshall, D. H., Davis, A. C., and Bamford, J. M. (2001). Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. BMJ 323, 536–540. doi: 10.1136/bmj.323.7312.536
Franco, B., and Malgrange, B. (2017). Concise review: regeneration in mammalian cochlea hair cells: help from supporting cells transdifferentiation. Stem Cells 35, 551–556. doi: 10.1002/stem.2554
Freist, W., Verhey, J. F., Ruhlmann, A., Gauss, D. H., and Arnez, J. G. (1999). Histidyl-tRNA synthetase. Biol. Chem. 380, 623–646. doi: 10.1515/BC.1999.079
Fuster-Garcia, C., Garcia-Garcia, G., Gonzalez-Romero, E., Jaijo, T., Sequedo, M. D., Ayuso, C., et al. (2017). USH2A gene editing using the CRISPR system. Mol. Ther. Nucleic Acids 15, 529–541. doi: 10.1016/j.omtn.2017.08.003
Garcia-Garcia, G., Besnard, T., Baux, D., Vache, C., Aller, E., Malcolm, S., et al. (2013). The contribution of GPR98 and DFNB31 genes to a Spanish Usher syndrome type 2 cohort. Mol. Vis. 19, 367–373.
Geleoc, G. S., and Holt, J. R. (2014). Sound strategies for hearing restoration. Science 344:1241062. doi: 10.1126/science.1241062
Geller, S. F., Guerin, K. I., Visel, M., Pham, A., Lee, E. S., Dror, A. A., et al. (2009). CLRN1 is nonessential in the mouse retina but is required for cochlear hair cell development. PLoS Genet. 5:e1000607. doi: 10.1371/journal.pgen.1000607
Geng, R., Geller, S. F., Hayashi, T., Ray, C. A., Reh, T. A., Bermingham-McDonogh, O., et al. (2009). Usher syndrome IIIA gene clarin-1 is essential for hair cell function and associated neural activation. Hum. Mol. Genet. 18, 2748–2760. doi: 10.1093/hmg/ddp210
Geng, R., Omar, A., Gopal, S. R., Chen, D. H., Stepanyan, R., Basch, M. L., et al. (2017). Modeling and preventing progressive hearing loss in Usher syndrome III. Sci. Rep. 7:13480. doi: 10.1038/s41598-017-13620-9
Geng, R., Sotomayor, M., Kinder, K. J., Gopal, S. R., Gerka-Stuyt, J., Chen, D. H., et al. (2013). Noddy, a mouse harboring a missense mutation in protocadherin-15, reveals the impact of disrupting a critical interaction site between tip-link cadherins in inner ear hair cells. J. Neurosci. 33, 4395–4404. doi: 10.1523/JNEUROSCI.4514-12.2013
Gentry, H. R., Singer, A. U., Betts, L., Yang, C., Ferrara, J. D., Sondek, J., et al. (2005). Structural and biochemical characterization of CIB1 delineates a new family of EF-hand-containing proteins. J. Biol. Chem. 280, 8407–8415. doi: 10.1074/jbc.M411515200
Gerber, S., Bonneau, D., Gilbert, B., Munnich, A., Dufier, J. L., Rozet, J. M., et al. (2006). USH1A: chronicle of a slow death. Am. J. Hum. Genet. 78, 357–359. doi: 10.1086/500275
Gibson, F., Walsh, J., Mburu, P., Varela, A., Brown, K. A., Antonio, M., et al. (1995). A type VII myosin encoded by the mouse deafness gene shaker-1. Nature 374, 62–64. doi: 10.1038/374062a0
Giese, A. P. J., Tang, Y. Q., Sinha, G. P., Bowl, M. R., Goldring, A. C., Parker, A., et al. (2017). CIB2 interacts with TMC1 and TMC2 and is essential for mechanotransduction in auditory hair cells. Nat. Commun. 8:43. doi: 10.1038/s41467-017-00061-1
Gillespie, P. G., and Muller, U. (2009). Mechanotransduction by hair cells: models, molecules, and mechanisms. Cell 139, 33–44. doi: 10.1016/j.cell.2009.09.010
Grati, M., and Kachar, B. (2011). Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc. Natl. Acad. Sci. U. S. A. 108, 11476–11481. doi: 10.1073/pnas.1104161108
Guilford, P., Ben Arab, S., Blanchard, S., Levilliers, J., Weissenbach, J., Belkahia, A., et al. (1994). A non-syndrome form of neurosensory, recessive deafness maps to the pericentromeric region of chromosome 13q. Nat. Genet. 6, 24–28. doi: 10.1038/ng0194-24
Hackney, C. M., and Furness, D. N. (2013). The composition and role of cross links in mechanoelectrical transduction in vertebrate sensory hair cells. J. Cell Sci. 126, 1721–1731. doi: 10.1242/jcs.106120
Hager, M., Bigotti, M. G., Meszaros, R., Carmignac, V., Holmberg, J., Allamand, V., et al. (2008). Cib2 binds integrin alpha7Bbeta1D and is reduced in laminin alpha2 chain-deficient muscular dystrophy. J. Biol. Chem. 283, 24760–24769. doi: 10.1074/jbc.M801166200
Han, S., Liu, X., Xie, S., Gao, M., Liu, F., Yu, S., et al. (2018). Knockout of ush2a gene in zebrafish causes hearing impairment and late onset rod-cone dystrophy. Hum. Genet. 137, 779–794. doi: 10.1007/s00439-018-1936-6
Han, F., Yu, H., Tian, C., Chen, H. E., Benedict-Alderfer, C., Zheng, Y., et al. (2012). A new mouse mutant of the Cdh23 gene with early-onset hearing loss facilitates evaluation of otoprotection drugs. Pharmacogenomics J. 12, 30–44. doi: 10.1038/tpj.2010.60
Hao, J., and Li, S. K. (2019). Inner ear drug delivery: recent advances, challenges, and perspective. Eur. J. Pharm. Sci. 126, 82–92. doi: 10.1016/j.ejps.2018.05.020
Hartel, B. P., Lofgren, M., Huygen, P. L., Guchelaar, I., Lo-A-Njoe Kort, N., Sadeghi, A. M., et al. (2016). A combination of two truncating mutations in USH2A causes more severe and progressive hearing impairment in Usher syndrome type IIa. Hear. Res. 339, 60–68. doi: 10.1016/j.heares.2016.06.008
Hasson, T., Heintzelman, M. B., Santos-Sacchi, J., Corey, D. P., and Mooseker, M. S. (1995). Expression in cochlea and retina of myosin VIIa, the gene product defective in Usher syndrome type 1B. Proc. Natl. Acad. Sci. U. S. A. 92, 9815–9819. doi: 10.1073/pnas.92.21.9815
Haywood-Watson, R. J. II, Ahmed, Z. M., Kjellstrom, S., Bush, R. A., Takada, Y., Hampton, L. L., et al. (2006). Ames Waltzer deaf mice have reduced electroretinogram amplitudes and complex alternative splicing of Pcdh15 transcripts. Invest. Ophthalmol. Vis. Sci. 47, 3074–3084. doi: 10.1167/iovs.06-0108
Heissler, S. M., and Manstein, D. J. (2012). Functional characterization of the human myosin-7a motor domain. Cell. Mol. Life Sci. 69, 299–311. doi: 10.1007/s00018-011-0749-8
Herberts, C. A., Kwa, M. S., and Hermsen, H. P. (2011). Risk factors in the development of stem cell therapy. J. Transl. Med. 9:29. doi: 10.1186/1479-5876-9-29
Hildebrand, M. S., Thorne, N. P., Bromhead, C. J., Kahrizi, K., Webster, J. A., Fattahi, Z., et al. (2010). Variable hearing impairment in a DFNB2 family with a novel MYO7A missense mutation. Clin. Genet. 77, 563–571. doi: 10.1111/j.1399-0004.2009.01344.x
Hilgert, N. (2009a). Novel human pathological mutations. Gene symbol: GPR98. Disease: Usher syndrome 2C. Hum. Genet. 125:342.
Hilgert, N., Kahrizi, K., Dieltjens, N., Bazazzadegan, N., Najmabadi, H., Smith, R. J., et al. (2009b). A large deletion in GPR98 causes type IIC Usher syndrome in male and female members of an Iranian family. J. Med. Genet. 46, 272–276. doi: 10.1136/jmg.2008.060947
Hmani, M., Ghorbel, A., Boulila-Elgaied, A., Ben Zina, Z., Kammoun, W., Drira, M., et al. (1999). A novel locus for Usher syndrome type II, USH2B, maps to chromosome 3 at p23-24.2. Eur. J. Hum. Genet. 7, 363–367. doi: 10.1038/sj.ejhg.5200307
Hmani-Aifa, M., Benzina, Z., Zulfiqar, F., Dhouib, H., Shahzadi, A., Ghorbel, A., et al. (2009). Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family. Eur. J. Hum. Genet. 17, 474–482. doi: 10.1038/ejhg.2008.167
Hope, C. I., Bundey, S., Proops, D., and Fielder, A. R. (1997). Usher syndrome in the city of Birmingham—prevalence and clinical classification. Br. J. Ophthalmol. 81, 46–53. doi: 10.1136/bjo.81.1.46
Hu, J., Li, B., Apisa, L., Yu, H., Entenman, S., Xu, M., et al. (2016). ER stress inhibitor attenuates hearing loss and hair cell death in Cdh23(erl/erl) mutant mice. Cell Death Dis. 7:e2485. doi: 10.1038/cddis.2016.386
Huang, H., Bogstie, J. N., and Vogel, H. J. (2012). Biophysical and structural studies of the human calcium- and integrin-binding protein family: understanding their functional similarities and differences. Biochem. Cell Biol. 90, 646–656. doi: 10.1139/o2012-021
Hudspeth, A. J., and Jacobs, R. (1979). Stereocilia mediate transduction in vertebrate hair cells (auditory system/cilium/vestibular system). Proc. Natl. Acad. Sci. U. S. A. 76, 1506–1509. doi: 10.1073/pnas.76.3.1506
Indzhykulian, A. A., Stepanyan, R., Nelina, A., Spinelli, K. J., Ahmed, Z. M., Belyantseva, I. A., et al. (2013). Molecular remodeling of tip links underlies mechanosensory regeneration in auditory hair cells. PLoS Biol. 11:e1001583. doi: 10.1371/journal.pbio.1001583
Inoue, A., and Ikebe, M. (2003). Characterization of the motor activity of mammalian myosin VIIA. J. Biol. Chem. 278, 5478–5487. doi: 10.1074/jbc.M210489200
Isosomppi, J., Vastinsalo, H., Geller, S. F., Heon, E., Flannery, J. G., and Sankila, E. M. (2009). Disease-causing mutations in the CLRN1 gene alter normal CLRN1 protein trafficking to the plasma membrane. Mol. Vis. 15, 1806–1818.
Jaijo, T., Aller, E., Beneyto, M., Najera, C., Graziano, C., Turchetti, D., et al. (2007). MYO7A mutation screening in Usher syndrome type I patients from diverse origins. J. Med. Genet. 44:e71. doi: 10.1136/jmg.2006.045377
Jaworek, T. J., Bhatti, R., Latief, N., Khan, S. N., Riazuddin, S., and Ahmed, Z. M. (2012). USH1K, a novel locus for type I Usher syndrome, maps to chromosome 10p11.21-q21.1. J. Hum. Genet. 57, 633–637. doi: 10.1038/jhg.2012.79
Jeong, M., O’Reilly, M., Kirkwood, N. K., Al-Aama, J., Lako, M., Kros, C. J., et al. (2018). Generating inner ear organoids containing putative cochlear hair cells from human pluripotent stem cells. Cell Death Dis. 9:922. doi: 10.1038/s41419-018-0967-1
Joensuu, T., Hamalainen, R., Yuan, B., Johnson, C., Tegelberg, S., Gasparini, P., et al. (2001). Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am. J. Hum. Genet. 69, 673–684. doi: 10.1086/323610
Johnson, K. R., Gagnon, L. H., Webb, L. S., Peters, L. L., Hawes, N. L., Chang, B., et al. (2003). Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum. Mol. Genet. 12, 3075–3086. doi: 10.1093/hmg/ddg332
Johnson, J. Z., and Hockemeyer, D. (2015). Human stem cell-based disease modeling: prospects and challenges. Curr. Opin. Cell Biol. 37, 84–90. doi: 10.1016/j.ceb.2015.10.007
Jones, D. (2018). To hear a whisper: biotechs chase new thinking to restore hearing. Nat. Biotechnol. 36, 1128–1129. doi: 10.1038/nbt1218-1128
Jouret, G., Poirsier, C., Spodenkiewicz, M., Jaquin, C., Gouy, E., Arndt, C., et al. (2019). Genetics of Usher syndrome: new insights from a meta-analysis. Otol. Neurotol. 40, 121–129. doi: 10.1097/MAO.0000000000002054
Kachar, B., Parakkal, M., Kurc, M., Zhao, Y., and Gillespie, P. G. (2000). High-resolution structure of hair-cell tip links. Proc. Natl. Acad. Sci. U. S. A. 97, 13336–13341. doi: 10.1073/pnas.97.24.13336
Kaplan, J., Gerber, S., Bonneau, D., Rozet, J. M., Delrieu, O., Briard, M. L., et al. (1992). A gene for Usher syndrome type I (USH1A) maps to chromosome 14q. Genomics 14, 979–987. doi: 10.1016/s0888-7543(05)80120-x
Kazmierczak, P., Sakaguchi, H., Tokita, J., Wilson-Kubalek, E. M., Milligan, R. A., Muller, U., et al. (2007). Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 449, 87–91. doi: 10.1038/nature06091
Kelley, P. M., Weston, M. D., Chen, Z. Y., Orten, D. J., Hasson, T., Overbeck, L. D., et al. (1997). The genomic structure of the gene defective in Usher syndrome type Ib (MYO7A). Genomics 40, 73–79. doi: 10.1006/geno.1996.4545
Kelly, M. C., and Chen, P. (2009). Development of form and function in the mammalian cochlea. Curr. Opin. Neurobiol. 19, 395–401. doi: 10.1016/j.conb.2009.07.010
Kiehart, D. P., Franke, J. D., Chee, M. K., Montague, R. A., Chen, T. L., Roote, J., et al. (2004). Drosophila crinkled, mutations of which disrupt morphogenesis and cause lethality, encodes fly myosin VIIA. Genetics 168, 1337–1352. doi: 10.1534/genetics.104.026369
Kikkawa, Y. S., Pawlowski, K. S., Wright, C. G., and Alagramam, K. N. (2008). Development of outer hair cells in Ames waltzer mice: mutation in protocadherin 15 affects development of cuticular plate and associated structures. Anat. Rec. 291, 224–232. doi: 10.1002/ar.20632
Kikkawa, Y., Shitara, H., Wakana, S., Kohara, Y., Takada, T., Okamoto, M., et al. (2003). Mutations in a new scaffold protein sans cause deafness in Jackson shaker mice. Hum. Mol. Genet. 12, 453–461. doi: 10.1093/hmg/ddg042
Kil, J., Gu, R., Fero, M. L., and Lynch, E. (2011). The cyclin dependent kinase inhibitor p27kip1 maintains terminal differentiation in the mouse organ of corti. Open Otorhinolaryngol. J. 5, 25–34. doi: 10.2174/1874428101105010025
Kimberling, W. J., Hildebrand, M. S., Shearer, A. E., Jensen, M. L., Halder, J. A., Trzupek, K., et al. (2010). Frequency of Usher syndrome in two pediatric populations: implications for genetic screening of deaf and hard of hearing children. Genet. Med. 12, 512–516. doi: 10.1097/GIM.0b013e3181e5afb8
Kimberling, W. J., Orten, D., and Pieke-Dahl, S. (2000). Genetic heterogeneity of Usher syndrome. Adv. Otorhinolaryngol. 56, 11–18. doi: 10.1159/000059077
Kimberling, W., and Smith, R. J. (1992). Gene mapping of the Usher syndromes. Otolaryngol. Clin. North Am. 25, 923–934. doi: 10.1016/S0030-6665(20)30915-4
Kimberling, W. J., Weston, M. D., Moller, C., Davenport, S. L., Shugart, Y. Y., Priluck, I. A., et al. (1990). Localization of Usher syndrome type II to chromosome 1q. Genomics 7, 245–249. doi: 10.1016/0888-7543(90)90546-7
Kitamura, K., Kakoi, H., Yoshikawa, Y., and Ochikubo, F. (1992). Ultrastructural findings in the inner ear of Jackson shaker mice. Acta Otolaryngol. 112, 622–627. doi: 10.3109/00016489209137451
Koehler, K. R., and Hashino, E. (2014). 3D mouse embryonic stem cell culture for generating inner ear organoids. Nat. Protoc. 9, 1229–1244. doi: 10.1038/nprot.2014.100
Koehler, K. R., Mikosz, A. M., Molosh, A. I., Patel, D., and Hashino, E. (2013). Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature 500, 217–221. doi: 10.1038/nature12298
Koehler, K. R., Nie, J., Longworth-Mills, E., Liu, X.-P., Lee, J., Holt, J. R., et al. (2017). Generation of inner ear organoids containing functional hair cells from human pluripotent stem cells. Nat. Biotechnol. 35, 583–589. doi: 10.1038/nbt.3840
Kros, C. J., Marcotti, W., van Netten, S. M., Self, T. J., Libby, R. T., Brown, S. D., et al. (2002). Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat. Neurosci. 5, 41–47. doi: 10.1038/nn784
Kurima, K., Ebrahim, S., Pan, B., Sedlacek, M., Sengupta, P., Millis, B. A., et al. (2015). TMC1 and TMC2 localize at the site of mechanotransduction in mammalian inner ear hair cell stereocilia. Cell Rep. 12, 1606–1617. doi: 10.1016/j.celrep.2015.07.058
Lagziel, A., Overlack, N., Bernstein, S. L., Morell, R. J., Wolfrum, U., and Friedman, T. B. (2009). Expression of cadherin 23 isoforms is not conserved: implications for a mouse model of Usher syndrome type 1D. Mol. Vis. 15, 1843–1857.
Lahlou, H., Nivet, E., Lopez-Juarez, A., Fontbonne, A., Assou, S., and Zine, A. (2018). Enriched differentiation of human otic sensory progenitor cells derived from induced pluripotent stem cells. Front. Mol. Neurosci. 11:452. doi: 10.3389/fnmol.2018.00452
Laine, H., Doetzlhofer, A., Mantela, J., Ylikoski, J., Laiho, M., Roussel, M. F., et al. (2007). p19(Ink4d) and p21(Cip1) collaborate to maintain the postmitotic state of auditory hair cells, their codeletion leading to DNA damage and p53-mediated apoptosis. J. Neurosci. 27, 1434–1444. doi: 10.1523/JNEUROSCI.4956-06.2007
Larget-Piet, D., Gerber, S., Bonneau, D., Rozet, J. M., Marc, S., Ghazi, I., et al. (1994). Genetic heterogeneity of Usher syndrome type 1 in French families. Genomics 21, 138–143. doi: 10.1006/geno.1994.1235
Le Guédard, S., Faugère, V., Malcolm, S., Claustres, M., and Roux, A. -F. (2007). Large genomic rearrangements within the PCDH15 gene are a significant cause of USH1F syndrome. Mol. Vis. 13, 102–107.
Le Quesne Stabej, P., Saihan, Z., Rangesh, N., Steele-Stallard, H. B., Ambrose, J., Coffey, A., et al. (2012). Comprehensive sequence analysis of nine Usher syndrome genes in the UK national collaborative Usher study. J. Med. Genet. 49, 27–36. doi: 10.1136/jmedgenet-2011-100468
Lenarduzzi, S., Vozzi, D., Morgan, A., Rubinato, E., D’Eustacchio, A., Osland, T. M., et al. (2015). Usher syndrome: an effective sequencing approach to establish a genetic and clinical diagnosis. Hear. Res. 320, 18–23. doi: 10.1016/j.heares.2014.12.006
Lenassi, E., Saihan, Z., Cipriani, V., Le Quesne Stabej, P., Moore, A. T., Luxon, L. M., et al. (2014). Natural history and retinal structure in patients with Usher syndrome type 1 owing to MYO7A mutation. Ophthalmology 121, 580–587. doi: 10.1016/j.ophtha.2013.09.017
Lentz, J. J., Gordon, W. C., Farris, H. E., MacDonald, G. H., Cunningham, D. E., Robbins, C. A., et al. (2010). Deafness and retinal degeneration in a novel USH1C knock-in mouse model. Dev. Neurobiol. 70, 253–267. doi: 10.1002/dneu.20771
Lentz, J. J., Jodelka, F. M., Hinrich, A. J., McCaffrey, K. E., Farris, H. E., Spalitta, M. J., et al. (2013). Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med. 19, 345–350. doi: 10.1038/nm.3106
Lentz, J., and Keats, B. (1999). “Usher syndrome type II” in GeneReviews. eds. M. Adam, H. Ardinger, and P. RA (Seattle (WA): University of Washington, Seattle).
Lentz, J., Pan, F., Ng, S. S., Deininger, P., and Keats, B. (2007). Ush1c216A knock-in mouse survives Katrina. Mutat. Res. 616, 139–144. doi: 10.1016/j.mrfmmm.2006.11.006
Lewis, R. A., Otterud, B., Stauffer, D., Lalouel, J. M., and Leppert, M. (1990). Mapping recessive ophthalmic diseases: linkage of the locus for Usher syndrome type II to a DNA marker on chromosome 1q. Genomics 7, 250–256. doi: 10.1016/0888-7543(90)90547-8
Li, J., He, Y., Weck, M. L., Lu, Q., Tyska, M. J., and Zhang, M. (2017). Structure of Myo7b/USH1C complex suggests a general PDZ domain binding mode by MyTH4-FERM myosins. Proc. Natl. Acad. Sci. U. S. A. 114, E3776–E3785. doi: 10.1073/pnas.1702251114
Li, L., and Neaves, W. B. (2006). Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 66, 4553–4557. doi: 10.1158/0008-5472.CAN-05-3986
Liu, X., Bulgakov, O. V., Darrow, K. N., Pawlyk, B., Adamian, M., Liberman, M. C., et al. (2007). Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. U. S. A. 104, 4413–4418. doi: 10.1073/pnas.0610950104
Liu, X. Z., Hope, C., Walsh, J., Newton, V., Ke, X. M., Liang, C. Y., et al. (1998). Mutations in the myosin VIIA gene cause a wide phenotypic spectrum, including atypical Usher syndrome. Am. J. Hum. Genet. 63, 909–912. doi: 10.1086/302026
Liu, X. P., Koehler, K. R., Mikosz, A. M., Hashino, E., and Holt, J. R. (2016). Functional development of mechanosensitive hair cells in stem cell-derived organoids parallels native vestibular hair cells. Nat. Commun. 7:11508. doi: 10.1038/ncomms11508
Liu, S., Li, S., Zhu, H., Cheng, S., and Zheng, Q. Y. (2012). A mutation in the cdh23 gene causes age-related hearing loss in Cdh23(nmf308/nmf308) mice. Gene 499, 309–317. doi: 10.1016/j.gene.2012.01.084
Longworth-Mills, E., Koehler, K. R., and Hashino, E. (2016). Generating inner ear organoids from mouse embryonic stem cells. Methods Mol. Biol. 1341, 391–406. doi: 10.1007/7651_2015_215
Lopes, V. S., Boye, S. E., Louie, C. M., Boye, S., Dyka, F., Chiodo, V., et al. (2013). Retinal gene therapy with a large MYO7A cDNA using adeno-associated virus. Gene Ther. 20, 824–833. doi: 10.1038/gt.2013.3
Lowenheim, H., Furness, D. N., Kil, J., Zinn, C., Gultig, K., Fero, M. L., et al. (1999). Gene disruption of p27(Kip1) allows cell proliferation in the postnatal and adult organ of corti. Proc. Natl. Acad. Sci. U. S. A. 96, 4084–4088. doi: 10.1073/pnas.96.7.4084
Maddalena, A., Tornabene, P., Tiberi, P., Minopoli, R., Manfredi, A., Mutarelli, M., et al. (2018). Triple vectors expand aav transfer capacity in the retina. Mol. Ther. 26, 524–541. doi: 10.1016/j.ymthe.2017.11.019
Maeda, R., Kindt, K. S., Mo, W., Morgan, C. P., Erickson, T., Zhao, H., et al. (2014). Tip-link protein protocadherin 15 interacts with transmembrane channel-like proteins TMC1 and TMC2. Proc. Natl. Acad. Sci. U. S. A. 111, 12907–12912. doi: 10.1073/pnas.1402152111
Maeda, R., Pacentine, I. V., Erickson, T., and Nicolson, T. (2017). Functional analysis of the transmembrane and cytoplasmic domains of pcdh15a in zebrafish hair cells. J. Neurosci. 37, 3231–3245. doi: 10.1523/JNEUROSCI.2216-16.2017
Maeder, M. L., Stefanidakis, M., Wilson, C. J., Baral, R., Barrera, L. A., Bounoutas, G. S., et al. (2019). Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 25, 229–233. doi: 10.1038/s41591-018-0327-9
Mantela, J., Jiang, Z., Ylikoski, J., Fritzsch, B., Zacksenhaus, E., and Pirvola, U. (2005). The retinoblastoma gene pathway regulates the postmitotic state of hair cells of the mouse inner ear. Development 132, 2377–2388. doi: 10.1242/dev.01834
Maravillas-Montero, J. L., and Santos-Argumedo, L. (2012). The myosin family: unconventional roles of actin-dependent molecular motors in immune cells. J. Leukoc. Biol. 91, 35–46. doi: 10.1189/jlb.0711335
Maria Oonk, A. M., van Huet, R. A., Leijendeckers, J. M., Oostrik, J., Venselaar, H., van Wijk, E., et al. (2015). Nonsyndromic hearing loss caused by USH1G mutations: widening the USH1G disease spectrum. Ear Hear. 36, 205–211. doi: 10.1097/AUD.0000000000000095
Mathur, P., and Yang, J. (2015). Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 1852, 406–420. doi: 10.1016/j.bbadis.2014.11.020
Mathur, P. D., and Yang, J. (2019). Usher syndrome and non-syndromic deafness: functions of different whirlin isoforms in the cochlea, vestibular organs, and retina. Hear. Res. 375, 14–24. doi: 10.1016/j.heares.2019.02.007
Mburu, P., Liu, X. Z., Walsh, J. Jr., Cope, M. J., Gibson, F., Kendrick-Jones, J., et al. (1997). Mutation analysis of the mouse myosin VIIA deafness gene. Genes Funct. 1, 191–203. doi: 10.1046/j.1365-4624.1997.00020.x
Mburu, P., Mustapha, M., Varela, A., Weil, D., El-Amraoui, A., Holme, R. H., et al. (2003). Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat. Genet. 34, 421–428. doi: 10.1038/ng1208
McGee, J., Goodyear, R. J., McMillan, D. R., Stauffer, E. A., Holt, J. R., Locke, K. G., et al. (2006). The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J. Neurosci. 26, 6543–6553. doi: 10.1523/JNEUROSCI.0693-06.2006
McMillan, D. R., Kayes-Wandover, K. M., Richardson, J. A., and White, P. C. (2002). Very large G protein-coupled receptor-1, the largest known cell surface protein, is highly expressed in the developing central nervous system. J. Biol. Chem. 277, 785–792. doi: 10.1074/jbc.M108929200
McMillan, D. R., and White, P. C. (2004). Loss of the transmembrane and cytoplasmic domains of the very large G-protein-coupled receptor-1 (VLGR1 or Mass1) causes audiogenic seizures in mice. Mol. Cell. Neurosci. 26, 322–329. doi: 10.1016/j.mcn.2004.02.005
Mellough, C. B., Bauer, R., Collin, J., Dorgau, B., Zerti, D., Dolan, D. W. P., et al. (2019). An integrated transcriptional analysis of the developing human retina. Development 146:dev169474. doi: 10.1242/dev.169474
Mellough, C. B., Collin, J., Khazim, M., White, K., Sernagor, E., Steel, D. H., et al. (2015). IGF-1 signaling plays an important role in the formation of three-dimensional laminated neural retina and other ocular structures from human embryonic stem cells. Stem Cells 33, 2416–2430. doi: 10.1002/stem.2023
Michalski, N., Michel, V., Bahloul, A., Lefevre, G., Barral, J., Yagi, H., et al. (2007). Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J. Neurosci. 27, 6478–6488. doi: 10.1523/JNEUROSCI.0342-07.2007
Michel, V., Booth, K. T., Patni, P., Cortese, M., Azaiez, H., Bahloul, A., et al. (2017). CIB2, defective in isolated deafness, is key for auditory hair cell mechanotransduction and survival. EMBO Mol. Med. 9, 1711–1731. doi: 10.15252/emmm.201708087
Millan, J. M., Aller, E., Jaijo, T., Blanco-Kelly, F., Gimenez-Pardo, A., and Ayuso, C. (2011). An update on the genetics of usher syndrome. J. Ophthalmol. 2011:417217. doi: 10.1155/2011/417217
Miller, K. A., Williams, L. H., Rose, E., Kuiper, M., Dahl, H. H., and Manji, S. S. (2012). Inner ear morphology is perturbed in two novel mouse models of recessive deafness. PLoS One 7:e51284. doi: 10.1371/journal.pone.0051284
Mishra, R., Joshi, R. K., and Zhao, K. (2018). Genome editing in rice: recent advances, challenges, and future implications. Front. Plant Sci. 9:1361. doi: 10.3389/fpls.2018.01361
Mizutari, K., Mutai, H., Namba, K., Miyanaga, Y., Nakano, A., Arimoto, Y., et al. (2015). High prevalence of CDH23 mutations in patients with congenital high-frequency sporadic or recessively inherited hearing loss. Orphanet. J. Rare Dis. 10:60. doi: 10.1186/s13023-015-0276-z
Murphy-Ullrich, J. E., and Poczatek, M. (2000). Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 11, 59–69. doi: 10.1016/S1359-6101(99)00029-5
Myers, K. A., Nasioulas, S., Boys, A., McMahon, J. M., Slater, H., Lockhart, P., et al. (2018). ADGRV1 is implicated in myoclonic epilepsy. Epilepsia 59, 381–388. doi: 10.1111/epi.13980
Nakano, T., Ando, S., Takata, N., Kawada, M., Muguruma, K., Sekiguchi, K., et al. (2012). Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10, 771–785. doi: 10.1016/j.stem.2012.05.009
Ness, S. L., Ben-Yosef, T., Bar-Lev, A., Madeo, A. C., Brewer, C. C., Avraham, K. B., et al. (2003). Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J. Med. Genet. 40, 767–772. doi: 10.1136/jmg.40.10.767
Ng, T. K., Tang, W., Cao, Y., Chen, S., Zheng, Y., Xiao, X., et al. (2019). Whole exome sequencing identifies novel USH2A mutations and confirms Usher syndrome 2 diagnosis in Chinese retinitis pigmentosa patients. Sci. Rep. 9:5628. doi: 10.1038/s41598-019-42105-0
Nicolson, T., Rusch, A., Friedrich, R. W., Granato, M., Ruppersberg, J. P., and Nusslein-Volhard, C. (1998). Genetic analysis of vertebrate sensory hair cell mechanosensation: the zebrafish circler mutants. Neuron 20, 271–283. doi: 10.1016/s0896-6273(00)80455-9
Nicosia, R. F., and Tuszynski, G. P. (1994). Matrix-bound thrombospondin promotes angiogenesis in vitro. J. Cell Biol. 124, 183–193. doi: 10.1083/jcb.124.1.183
Nie, J., Koehler, K. R., and Hashino, E. (2017). Directed differentiation of mouse embryonic stem cells into inner ear sensory epithelia in 3D culture. Methods Mol. Biol. 1597, 67–83. doi: 10.1007/978-1-4939-6949-4_6
Nie, H., Liu, Y., Yin, X., Cao, H., Wang, Y., Xiong, W., et al. (2016). Plasma membrane targeting of protocadherin 15 is regulated by the golgi-associated chaperone protein PIST. Neural Plast. 2016:8580675. doi: 10.1155/2016/8580675
Nolen, R. M., Hufnagel, R. B., Friedman, T. B., Turriff, A. E., Brewer, C. C., Zalewski, C. K., et al. (2020). Atypical and ultra-rare Usher syndrome: a review. Ophthalmic Genet. 41, 1–12. doi: 10.1080/13816810.2020.1747090
Nyberg, S., Abbott, N. J., Shi, X., Steyger, P. S., and Dabdoub, A. (2019). Delivery of therapeutics to the inner ear: the challenge of the blood-labyrinth barrier. Sci. Transl. Med. 11:eaao0935. doi: 10.1126/scitranslmed.aao0935
Ogun, O., and Zallocchi, M. (2014). Clarin-1 acts as a modulator of mechanotransduction activity and presynaptic ribbon assembly. J. Cell Biol. 207, 375–391. doi: 10.1083/jcb.201404016
O’Hanlon, T. P., and Miller, F. W. (2002). Genomic organization, transcriptional mapping, and evolutionary implications of the human bi-directional histidyl-tRNA synthetase locus (HARS/HARSL). Biochem. Biophys. Res. Commun. 294, 609–614. doi: 10.1016/S0006-291X(02)00525-9
Okano, T., and Kelley, M. W. (2012). Stem cell therapy for the inner ear: recent advances and future directions. Trends Amplif. 16, 4–18. doi: 10.1177/1084713812440336
Omole, A. E., and Fakoya, A. O. J. (2018). Ten years of progress and promise of induced pluripotent stem cells: historical origins, characteristics, mechanisms, limitations, and potential applications. PeerJ 6:e4370. doi: 10.7717/peerj.4370
Oshima, A., Jaijo, T., Aller, E., Millan, J., Carney, C., Usami, S., et al. (2008). Mutation profile of the CDH23 gene in 56 probands with Usher syndrome type I. Hum. Mutat. 29, E37–E46. doi: 10.1002/humu.20761
Oshima, K., Shin, K., Diensthuber, M., Peng, A. W., Ricci, A. J., and Heller, S. (2010). Mechanosensitive hair cell-like cells from embryonic and induced pluripotent stem cells. Cell 141, 704–716. doi: 10.1016/j.cell.2010.03.035
Otterstedde, C. R., Spandau, U., Blankenagel, A., Kimberling, W. J., and Reisser, C. (2001). A new clinical classification for Usher’s syndrome based on a new subtype of Usher’s syndrome type I. Laryngoscope 111, 84–86. doi: 10.1097/00005537-200101000-00014
Ouyang, X. M., Hejtmancik, J. F., Jacobson, S. G., Xia, X. J., Li, A., Du, L. L., et al. (2003). USH1C: a rare cause of USH1 in a non-Acadian population and a founder effect of the Acadian allele. Clin. Genet. 63, 150–153. doi: 10.1046/j.0009-9163.2002.00004.x
Overlack, N., Goldmann, T., Wolfrum, U., and Nagel-Wolfrum, K. (2012). Gene repair of an Usher syndrome causing mutation by zinc-finger nuclease mediated homologous recombination. Invest. Ophthalmol. Vis. Sci. 53, 4140–4146. doi: 10.1167/iovs.12-9812
Ozawa, M., Hoschutzky, H., Herrenknecht, K., and Kemler, R. (1990). A possible new adhesive site in the cell-adhesion molecule uvomorulin. Mech. Dev. 33, 49–56. doi: 10.1016/0925-4773(90)90134-8
Pakarinen, L., Karjalainen, S., Simola, K. O., Laippala, P., and Kaitalo, H. (1995). Usher’s syndrome type 3 in Finland. Laryngoscope 105, 613–617. doi: 10.1288/00005537-199506000-00010
Patel, K., Giese, A. P., Grossheim, J. M., Hegde, R. S., Delio, M., Samanich, J., et al. (2015a). Correction: a novel C-terminal CIB2 (calcium and integrin binding protein 2) mutation associated with non-syndromic hearing loss in a hispanic family. PLoS One 10:e0141259. doi: 10.1371/journal.pone.0141259
Patel, K., Giese, A. P., Grossheim, J. M., Hegde, R. S., Delio, M., Samanich, J., et al. (2015b). A novel C-terminal CIB2 (calcium and integrin binding protein 2) mutation associated with non-syndromic hearing loss in a hispanic family. PLoS One 10:e0133082. doi: 10.1371/journal.pone.0133082
Perreault-Micale, C., Frieden, A., Kennedy, C. J., Neitzel, D., Sullivan, J., Faulkner, N., et al. (2014). Truncating variants in the majority of the cytoplasmic domain of PCDH15 are unlikely to cause Usher syndrome 1F. J. Mol. Diagn. 16, 673–678. doi: 10.1016/j.jmoldx.2014.07.001
Phillips, M. J., Wallace, K. A., Dickerson, S. J., Miller, M. J., Verhoeven, A. D., Martin, J. M., et al. (2012). Blood-derived human iPS cells generate optic vesicle-like structures with the capacity to form retinal laminae and develop synapses. Invest. Ophthalmol. Vis. Sci. 53, 2007–2019. doi: 10.1167/iovs.11-9313
Pickles, J. O., Comis, S. D., and Osborne, M. P. (1984). Cross-links between stereocilia in the Guinea pig organ of Corti, and their possible relation to sensory transduction. Hear. Res. 15, 103–112. doi: 10.1016/0378-5955(84)90041-8
Polyak, K., Kato, J. Y., Solomon, M. J., Sherr, C. J., Massague, J., Roberts, J. M., et al. (1994). p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 8, 9–22. doi: 10.1101/gad.8.1.9
Ponnath, A., Depreux, F. F., Jodelka, F. M., Rigo, F., Farris, H. E., Hastings, M. L., et al. (2018). Rescue of outer hair cells with antisense oligonucleotides in usher mice is dependent on age of treatment. J. Assoc. Res. Otolaryngol. 19, 1–16. doi: 10.1007/s10162-017-0640-x
Ponting, C. P., Phillips, C., Davies, K. E., and Blake, D. J. (1997). PDZ domains: targeting signalling molecules to sub-membranous sites. BioEssays 19, 469–479. doi: 10.1002/bies.950190606
Powers, R. E., Gaudet, R., and Sotomayor, M. (2017). A partial calcium-free linker confers flexibility to inner-ear protocadherin-15. Structure 25, 482–495. doi: 10.1016/j.str.2017.01.014
Puffenberger, E. G., Jinks, R. N., Sougnez, C., Cibulskis, K., Willert, R. A., Achilly, N. P., et al. (2012). Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS One 7:e28936. doi: 10.1371/journal.pone.0028936
Reiners, J., Marker, T., Jurgens, K., Reidel, B., and Wolfrum, U. (2005a). Photoreceptor expression of the Usher syndrome type 1 protein protocadherin 15 (USH1F) and its interaction with the scaffold protein harmonin (USH1C). Mol. Vis. 11, 347–355.
Reiners, J., van Wijk, E., Marker, T., Zimmermann, U., Jurgens, K., te Brinke, H., et al. (2005b). Scaffold protein harmonin (USH1C) provides molecular links between Usher syndrome type 1 and type 2. Hum. Mol. Genet. 14, 3933–3943. doi: 10.1093/hmg/ddi417
Rhodes, C. R., Hertzano, R., Fuchs, H., Bell, R. E., de Angelis, M. H., Steel, K. P., et al. (2004). A Myo7a mutation cosegregates with stereocilia defects and low-frequency hearing impairment. Mamm. Genome 15, 686–697. doi: 10.1007/s00335-004-2344-x
Riazuddin, S., Belyantseva, I. A., Giese, A. P., Lee, K., Indzhykulian, A. A., Nandamuri, S. P., et al. (2012). Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat. Genet. 44, 1265–1271. doi: 10.1038/ng.2426
Rinaldi, C., and Wood, M. J. A. (2018). Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14, 9–21. doi: 10.1038/nrneurol.2017.148
Roux, A. F., Faugere, V., Le Guedard, S., Pallares-Ruiz, N., Vielle, A., Chambert, S., et al. (2006). Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%. J. Med. Genet. 43, 763–768. doi: 10.1136/jmg.2006.041954
Rowlands, T. M., Symonds, J. M., Farookhi, R., and Blaschuk, O. W. (2000). Cadherins: crucial regulators of structure and function in reproductive tissues. Rev. Reprod. 5, 53–61. doi: 10.1530/ror.0.0050053
Sack, B. K., and Herzog, R. W. (2009). Evading the immune response upon in vivo gene therapy with viral vectors. Curr. Opin. Mol. Ther. 11, 493–503.
Sadeghi, M., Cohn, E. S., Kelly, W. J., Kimberling, W. J., Tranebjoerg, L., and Moller, C. (2004). Audiological findings in Usher syndrome types IIa and II (non-IIa). Int. J. Audiol. 43, 136–143. doi: 10.1080/14992020400050019
Sakaguchi, H., Tokita, J., Muller, U., and Kachar, B. (2009). Tip links in hair cells: molecular composition and role in hearing loss. Curr. Opin. Otolaryngol. Head Neck Surg. 17, 388–393. doi: 10.1097/MOO.0b013e3283303472
Sanjurjo-Soriano, C., Erkilic, N., Baux, D., Mamaeva, D., Hamel, C., Meunier, I., et al. (2019). Genome editing in patient iPSC efficiently corrects the most prevalent USH2A mutations and reveals intriguing mutant mRNA expression profiles. Mol. Ther. Methods Clin. Dev. 17, 156–173. doi: 10.1016/j.omtm.2019.11.016
Sankila, E. M., Pakarinen, L., Kaariainen, H., Aittomaki, K., Karjalainen, S., Sistonen, P., et al. (1995). Assignment of an Usher syndrome type III (USH3) gene to chromosome 3q. Hum. Mol. Genet. 4, 93–98. doi: 10.1093/hmg/4.1.93
Sato, O., Komatsu, S., Sakai, T., Tsukasaki, Y., Tanaka, R., Mizutani, T., et al. (2017). Human myosin VIIa is a very slow processive motor protein on various cellular actin structures. J. Biol. Chem. 292, 10950–10960. doi: 10.1074/jbc.M116.765966
Schwander, M., Kachar, B., and Muller, U. (2012). The cell biology of hearing. J. Cell Biol. 190, 9–20. doi: 10.1083/jcb.201001138
Seco, C. Z., Giese, A. P., Shafique, S., Schraders, M., Oonk, A. M., Grossheim, M., et al. (2016). Novel and recurrent CIB2 variants, associated with nonsyndromic deafness, do not affect calcium buffering and localization in hair cells. Eur. J. Hum. Genet. 24, 542–549. doi: 10.1038/ejhg.2015.157
Seiler, C., Finger-Baier, K. C., Rinner, O., Makhankov, Y. V., Schwarz, H., Neuhauss, S. C., et al. (2005). Duplicated genes with split functions: independent roles of protocadherin15 orthologues in zebrafish hearing and vision. Development 132, 615–623. doi: 10.1242/dev.01591
Seiler, C., and Nicolson, T. (1999). Defective calmodulin-dependent rapid apical endocytosis in zebrafish sensory hair cell mutants. J. Neurobiol. 41, 424–434. doi: 10.1002/(SICI)1097-4695(19991115)41:3<424::AID-NEU10>3.0.CO;2-G
Seki, N., Hattori, A., Hayashi, A., Kozuma, S., Ohira, M., Hori, T., et al. (1999). Structure, expression profile and chromosomal location of an isolog of DNA-PKcs interacting protein (KIP) gene. Biochim. Biophys. Acta 1444, 143–147. doi: 10.1016/s0167-4781(98)00253-x
Self, T., Mahony, M., Fleming, J., Walsh, J., Brown, S. D. M., and Steel, K. P. (1998). Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 125, 557–566.
Senften, M., Schwander, M., Kazmierczak, P., Lillo, C., Shin, J. B., Hasson, T., et al. (2006). Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J. Neurosci. 26, 2060–2071. doi: 10.1523/JNEUROSCI.4251-05.2006
Shapiro, L., and Weis, W. I. (2009). Structure and biochemistry of cadherins and catenins. Cold Spring Harb. Perspect. Biol. 1:a003053. doi: 10.1101/cshperspect.a003053
Sheng, M., and Sala, C. (2001). PDZ domains and the organization of supramolecular complexes. Annu. Rev. Neurosci. 24, 1–29. doi: 10.1146/annurev.neuro.24.1.1
Siemens, J., Kazmierczak, P., Reynolds, A., Sticker, M., Littlewood-Evans, A., and Muller, U. (2002). The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions. Proc. Natl. Acad. Sci. U. S. A. 99, 14946–14951. doi: 10.1073/pnas.232579599
Simm, D., Hatje, K., and Kollmar, M. (2017). Distribution and evolution of stable single alpha-helices (SAH domains) in myosin motor proteins. PLoS One 12:e0174639. doi: 10.1371/journal.pone.0174639
Sipes, J. M., Krutzsch, H. C., Lawler, J., and Roberts, D. D. (1999). Cooperation between thrombospondin-1 type 1 repeat peptides and alpha(v)beta(3) integrin ligands to promote melanoma cell spreading and focal adhesion kinase phosphorylation. J. Biol. Chem. 274, 22755–22762. doi: 10.1074/jbc.274.32.22755
Slepecky, N. B., Henderson, C. G., and Saha, S. (1995). Post-translational modifications of tubulin suggest that dynamic microtubules are present in sensory cells and stable microtubules are present in supporting cells of the mammalian cochlea. Hear. Res. 91, 136–147. doi: 10.1016/0378-5955(95)00184-0
Slijkerman, R. W., Vache, C., Dona, M., Garcia-Garcia, G., Claustres, M., Hetterschijt, L., et al. (2016). Antisense oligonucleotide-based splice correction for USH2A-associated retinal degeneration caused by a frequent deep-intronic mutation. Mol. Ther. Nucleic Acids 5:e381. doi: 10.1038/mtna.2016.89
Smith, R. J., Berlin, C. I., Hejtmancik, J. F., Keats, B. J., Kimberling, W. J., Lewis, R. A., et al. (1994). Clinical diagnosis of the Usher syndromes. Usher syndrome consortium. Am. J. Med. Genet. 50, 32–38. doi: 10.1002/ajmg.1320500107
Smith, R. J., Lee, E. C., Kimberling, W. J., Daiger, S. P., Pelias, M. Z., Keats, B. J., et al. (1992a). Localization of two genes for Usher syndrome type I to chromosome 11. Genomics 14, 995–1002. doi: 10.1016/s0888-7543(05)80122-3
Smith, R. J., Pelias, M. Z., Daiger, S. P., Keats, B., Kimberling, W., and Hejtmancik, J. F. (1992b). Clinical variability and genetic heterogeneity within the Acadian Usher population. Am. J. Med. Genet. 43, 964–969. doi: 10.1002/ajmg.1320430612
Sollner, C., Rauch, G. J., Siemens, J., Geisler, R., Schuster, S. C., Muller, U., et al. (2004). Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature 428, 955–959. doi: 10.1038/nature02484
Sorusch, N., Bauss, K., Plutniok, J., Samanta, A., Knapp, B., Nagel-Wolfrum, K., et al. (2017). Characterization of the ternary Usher syndrome SANS/ush2a/whirlin protein complex. Hum. Mol. Genet. 26, 1157–1172. doi: 10.1093/hmg/ddx027
Sudol, M. (1998). From Src homology domains to other signaling modules: proposal of the ‘protein recognition code’. Oncogene 17, 1469–1474. doi: 10.1038/sj.onc.1202182
Suzuki, S. T. (1996). Structural and functional diversity of cadherin superfamily: are new members of cadherin superfamily involved in signal transduction pathway? J. Cell. Biochem. 61, 531–542. doi: 10.1002/(SICI)1097-4644(19960616)61:4%3C531::AID-JCB6%3E3.0.CO;2-P
Szegezdi, E., Logue, S. E., Gorman, A. M., and Samali, A. (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885. doi: 10.1038/sj.embor.7400779
Takahashi, S., Mui, V. J., Rosenberg, S. K., Homma, K., Cheatham, M. A., and Zheng, J. (2016). Cadherin 23-C regulates microtubule networks by modifying CAMSAP3’s function. Sci. Rep. 6:28706. doi: 10.1038/srep28706
Tamagawa, Y., Kitamura, K., Ishida, T., Ishikawa, K., Tanaka, H., Tsuji, S., et al. (1996). A gene for a dominant form of non-syndromic sensorineural deafness (DFNA11) maps within the region containing the DFNB2 recessive deafness gene. Hum. Mol. Genet. 5, 849–852. doi: 10.1093/hmg/5.6.849
Tang, Z. H., Chen, J. R., Zheng, J., Shi, H. S., Ding, J., Qian, X. D., et al. (2016). Genetic correction of induced pluripotent stem cells from a deaf patient with MYO7A mutation results in morphologic and functional recovery of the derived hair cell-like cells. Stem Cells Transl. Med. 5, 561–571. doi: 10.5966/sctm.2015-0252
Tang, P. C., Hashino, E., and Nelson, R. F. (2020). Progress in modeling and targeting inner ear disorders with pluripotent stem cells. Stem Cell Rep. 14, 996–1008. doi: 10.1016/j.stemcr.2020.04.008
Taraboletti, G., Roberts, D., Liotta, L. A., and Giavazzi, R. (1990). Platelet thrombospondin modulates endothelial cell adhesion, motility, and growth: a potential angiogenesis regulatory factor. J. Cell Biol. 111, 765–772. doi: 10.1083/jcb.111.2.765
Tian, G., Lee, R., Ropelewski, P., and Imanishi, Y. (2016). Impairment of vision in a mouse model of Usher syndrome type III. Invest. Ophthalmol. Vis. Sci. 57, 866–875. doi: 10.1167/iovs.15-16946
Tian, G., Zhou, Y., Hajkova, D., Miyagi, M., Dinculescu, A., Hauswirth, W. W., et al. (2009). Clarin-1, encoded by the Usher syndrome III causative gene, forms a membranous microdomain: possible role of clarin-1 in organizing the actin cytoskeleton. J. Biol. Chem. 284, 18980–18993. doi: 10.1074/jbc.M109.003160
Tilney, L. G., Derosier, D. J., and Mulroy, M. J. (1980). The organization of actin filaments in the stereocilia of cochlear hair cells. J. Cell Biol. 86, 244–259. doi: 10.1083/jcb.86.1.244
Tlili, A., Charfedine, I., Lahmar, I., Benzina, Z., Mohamed, B. A., Weil, D., et al. (2005). Identification of a novel frameshift mutation in the DFNB31/WHRN gene in a Tunisian consanguineous family with hereditary non-syndromic recessive hearing loss. Hum. Mutat. 25:503. doi: 10.1002/humu.9333
Todorov, P. T., Hardisty, R. E., and Brown, S. D. (2001). Myosin VIIA is specifically associated with calmodulin and microtubule-associated protein-2B (MAP-2B). Biochem. J. 354, 267–274. doi: 10.1042/0264-6021:3540267
Toms, M., Bitner-Glindzicz, M., Webster, A., and Moosajee, M. (2015). Usher syndrome: a review of the clinical phenotype, genes and therapeutic strategies. Expert Rev. Ophthalmol. 10, 241–256. doi: 10.1586/17469899.2015.1033403
Toriello, H. V., and Smith, S. D. (2013). Hereditary hearing loss and its syndromes. Oxford, England, United Kingdom: Oxford University Press.
Trapani, I., Colella, P., Sommella, A., Iodice, C., Cesi, G., de Simone, S., et al. (2014). Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 6, 194–211. doi: 10.1002/emmm.201302948
Udovichenko, I. P., Gibbs, D., and Williams, D. S. (2002). Actin-based motor properties of native myosin VIIa. J. Cell Sci. 115, 445–450.
Usher, C. H. (1914). On the inheritance of retinitis pigmentosa; with note of cases. Royal London Ophthalmic Hospital reports. Vol. 19. London, United Kingdom: Royal Society of London in Ophthalmology and Hospital, 130–236.
Vallone, R., Dal Cortivo, G., D’Onofrio, M., and Dell’Orco, D. (2018). Preferential binding of mg2+ over Ca2+ to CIB2 triggers an allosteric switch impaired in Usher syndrome type 1J. Front. Mol. Neurosci. 11:274. doi: 10.3389/fnmol.2018.00274
van Diepen, H., Dulla, K., Chan, H. L., Schulkens, I., Beumer, W., Vorthoren, L., et al. (2019). QR-421a, an antisense oligonucleotide, for the treatment of retinitis pigmentosa due to USH2A exon 13 mutations. Invest. Ophthalmol. Vis. Sci. 60, 3250–3251.
van Wijk, E. (2019). Antisense oligonucleotide-induced splice modulation for the treatment of Usher syndrome type 2A. Acta Ophthalmol. 97. doi: 10.1111/j.1755-3768.2019.8174
van Wijk, E., Pennings, R. J., te Brinke, H., Claassen, A., Yntema, H. G., Hoefsloot, L. H., et al. (2004). Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am. J. Hum. Genet. 74, 738–744. doi: 10.1086/383096
van Wijk, E., van der Zwaag, B., Peters, T., Zimmermann, U., Te Brinke, H., Kersten, F., et al. (2006). The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum. Mol. Genet. 15, 751–765. doi: 10.1093/hmg/ddi490
Vastinsalo, H., Jalkanen, R., Dinculescu, A., Isosomppi, J., Geller, S., Flannery, J. G., et al. (2011). Alternative splice variants of the USH3A gene Clarin 1 (CLRN1). Eur. J. Hum. Genet. 19, 30–35. doi: 10.1038/ejhg.2010.140
Venkatesh, M. D., Moorchung, N., and Puri, B. (2015). Genetics of non syndromic hearing loss. Med. J. Armed Forces India 71, 363–368. doi: 10.1016/j.mjafi.2015.07.003
Verpy, E., Leibovici, M., Michalski, N., Goodyear, R. J., Houdon, C., Weil, D., et al. (2011). Stereocilin connects outer hair cell stereocilia to one another and to the tectorial membrane. J. Comp. Neurol. 519, 194–210. doi: 10.1002/cne.22509
Verpy, E., Leibovici, M., Zwaenepoel, I., Liu, X. Z., Gal, A., Salem, N., et al. (2000). A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat. Genet. 26, 51–55. doi: 10.1038/79171
Vijayakumar, S., Depreux, F. F., Jodelka, F. M., Lentz, J. J., Rigo, F., Jones, T. A., et al. (2017). Rescue of peripheral vestibular function in Usher syndrome mice using a splice-switching antisense oligonucleotide. Hum. Mol. Genet. 26, 3482–3494. doi: 10.1093/hmg/ddx234
von Graefe, A. (1858). Vereinzelte Beobachtungen und Bermerkungen. Exceptionelles verhalten des Gesichtsfeldes bei Pigmentenartung der Netzhaut. Albrecht von Graefes. Arch. Klin. Ophthalmol. 4, 250–253.
Waldron, A., Wilcox, C., Francklyn, C., and Ebert, A. (2019). Knock-down of histidyl-tRNA synthetase causes cell cycle arrest and apoptosis of neuronal progenitor cells in vivo. Front. Cell Dev. Biol. 7:67. doi: 10.3389/fcell.2019.00067
Walters, B. J., Liu, Z., Crabtree, M., Coak, E., Cox, B. C., and Zuo, J. (2014). Auditory hair cell-specific deletion of p27Kip1 in postnatal mice promotes cell-autonomous generation of new hair cells and normal hearing. J. Neurosci. 34, 15751–15763. doi: 10.1523/JNEUROSCI.3200-14.2014
Wang, Y., Li, J., Yao, X., Li, W., Du, H., Tang, M., et al. (2017). Loss of CIB2 causes profound hearing loss and abolishes mechanoelectrical transduction in mice. Front. Mol. Neurosci. 10:401. doi: 10.3389/fnmol.2017.00401
Wayne, S. (1997). Localization of the Usher syndrome type 1F gene (Ush1F) to chromosome 10. Am. J. Hum. Mol. Genet. 61:A300.
Webb, S. W., Grillet, N., Andrade, L. R., Xiong, W., Swarthout, L., Della Santina, C. C., et al. (2011). Regulation of PCDH15 function in mechanosensory hair cells by alternative splicing of the cytoplasmic domain. Development 138, 1607–1617. doi: 10.1242/dev.060061
Weck, M. L., Crawley, S. W., Stone, C. R., and Tyska, M. J. (2016). Myosin-7b promotes distal tip localization of the intermicrovillar adhesion complex. Curr. Biol. 26, 2717–2728. doi: 10.1016/j.cub.2016.08.014
Weil, D., Blanchard, S., Kaplan, J., Guilford, P., Gibson, F., Walsh, J., et al. (1995). Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 374, 60–61. doi: 10.1038/374060a0
Weil, D., El-Amraoui, A., Masmoudi, S., Mustapha, M., Kikkawa, Y., Laine, S., et al. (2003). Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum. Mol. Genet. 12, 463–471. doi: 10.1093/hmg/ddg051
Weil, D., Kussel, P., Blanchard, S., Levy, G., Levi-Acobas, F., Drira, M., et al. (1997). The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat. Genet. 16, 191–193. doi: 10.1038/ng0697-191
Werbowetski-Ogilvie, T. E., Bosse, M., Stewart, M., Schnerch, A., Ramos-Mejia, V., Rouleau, A., et al. (2009). Characterization of human embryonic stem cells with features of neoplastic progression. Nat. Biotechnol. 27, 91–97. doi: 10.1038/nbt.1516
Weston, M. D., Eudy, J. D., Fujita, S., Yao, S., Usami, S., Cremers, C., et al. (2000). Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am. J. Hum. Genet. 66, 1199–1210. doi: 10.1086/302855
Weston, M. D., Luijendijk, M. W., Humphrey, K. D., Moller, C., and Kimberling, W. J. (2004). Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am. J. Hum. Genet. 74, 357–366. doi: 10.1086/381685
Williams, D. S. (2008). Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. Vis. Res. 48, 433–441. doi: 10.1016/j.visres.2007.08.015
Williams, D. S., Chadha, A., Hazim, R., and Gibbs, D. (2017). Gene therapy approaches for prevention of retinal degeneration in Usher syndrome. Gene Ther. 24, 68–71. doi: 10.1038/gt.2016.81
Wilson, S. M., Householder, D. B., Coppola, V., Tessarollo, L., Fritzsch, B., Lee, E. -C., et al. (2001). Mutations in Cdh23 cause nonsyndromic hearing loss in waltzer mice. Genomics 74, 228–233. doi: 10.1006/geno.2001.6554
Wong, E. Y., Xu, C. Y., Brahmachary, M., and Xu, P. X. (2016). A novel ENU-induced mutation in Myo6 causes vestibular dysfunction and deafness. PLoS One 11:e0154984. doi: 10.1371/journal.pone.0154984
Woolner, S., and Bement, W. M. (2009). Unconventional myosins acting unconventionally. Trends Cell Biol. 19, 245–252. doi: 10.1016/j.tcb.2009.03.003
Xiong, W., Grillet, N., Elledge, H. M., Wagner, T. F. J., Zhao, B., Johnson, K. R., et al. (2012). TMHS is an integral component of the mechanotransduction machinery of cochlear hair cells. Cell 151, 1283–1295. doi: 10.1016/j.cell.2012.10.041
Xu, Z., Ricci Aj Fau-Heller, S., and Heller, S. (2009). Rethinking how hearing happens. Neuron 62, 305–307. doi: 10.1016/j.neuron.2009.04.019
Yan, D., and Liu, X. Z. (2010). Genetics and pathological mechanisms of Usher syndrome. J. Hum. Genet. 55, 327–335. doi: 10.1038/jhg.2010.29
Yan, J., Pan, L., Chen, X., Wu, L., and Zhang, M. (2010). The structure of the harmonin/sans complex reveals an unexpected interaction mode of the two Usher syndrome proteins. Proc. Natl. Acad. Sci. U. S. A. 107, 4040–4045. doi: 10.1073/pnas.0911385107
Yang, J., Liu, X., Zhao, Y., Adamian, M., Pawlyk, B., Sun, X., et al. (2010). Ablation of whirlin long isoform disrupts the USH2 protein complex and causes vision and hearing loss. PLoS Genet. 6:e1000955. doi: 10.1371/journal.pgen.1000955
Yang, J., Wang, L., Song, H., and Sokolov, M. (2012). Current understanding of usher syndrome type II. Front. Biosci. 17, 1165–1183. doi: 10.2741/3979
Yilmaz, A., and Benvenisty, N. (2019). Defining human pluripotency. Cell Stem Cell 25, 9–22. doi: 10.1016/j.stem.2019.06.010
Yoshimura, H., Miyagawa, M., Kumakawa, K., Nishio, S. Y., and Usami, S. (2016). Frequency of Usher syndrome type 1 in deaf children by massively parallel DNA sequencing. J. Hum. Genet. 61, 419–422. doi: 10.1038/jhg.2015.168
Yu, I. M., Planelles-Herrero, V. J., Sourigues, Y., Moussaoui, D., Sirkia, H., Kikuti, C., et al. (2017). Myosin 7 and its adaptors link cadherins to actin. Nat. Commun. 8:15864. doi: 10.1038/ncomms15864
Yurchenco, P. D., and Cheng, Y. S. (1993). Self-assembly and calcium-binding sites in laminin. A three-arm interaction model. J. Biol. Chem. 268, 17286–17299.
Zhao, B., Wu, Z., Grillet, N., Yan, L., Xiong, W., Harkins-Perry, S., et al. (2014). TMIE is an essential component of the mechanotransduction machinery of cochlear hair cells. Neuron 84, 954–967. doi: 10.1016/j.neuron.2014.10.041
Zheng, Q. Y., Scarborough, J. D., Zheng, Y., Yu, H., Choi, D., and Gillespie, P. G. (2012). Digenic inheritance of deafness caused by 8J allele of myosin-VIIA and mutations in other Usher I genes. Hum. Mol. Genet. 21, 2588–2598. doi: 10.1093/hmg/dds084
Zheng, Q. Y., Yan, D., Ouyang, X. M., Du, L. L., Yu, H., Chang, B., et al. (2005). Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Hum. Mol. Genet. 14, 103–111. doi: 10.1093/hmg/ddi010
Zhong, X., Gutierrez, C., Xue, T., Hampton, C., Vergara, M. N., Cao, L. H., et al. (2014). Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 5:4047. doi: 10.1038/ncomms5047
Zhou, S., Flamier, A., Abdouh, M., Tetreault, N., Barabino, A., Wadhwa, S., et al. (2015). Differentiation of human embryonic stem cells into cone photoreceptors through simultaneous inhibition of BMP, TGFbeta and Wnt signaling. Development 142, 3294–3306. doi: 10.1242/dev.125385
Zou, J., Chen, Q., Almishaal, A., Mathur, P. D., Zheng, T., Tian, C., et al. (2017). The roles of USH1 proteins and PDZ domain-containing USH proteins in USH2 complex integrity in cochlear hair cells. Hum. Mol. Genet. 26, 624–636. doi: 10.1093/hmg/ddw421
Zou, J., Luo, L., Shen, Z., Chiodo, V. A., Ambati, B. K., Hauswirth, W. W., et al. (2011). Whirlin replacement restores the formation of the USH2 protein complex in whirlin knockout photoreceptors. Invest. Ophthalmol. Vis. Sci. 52, 2343–2351. doi: 10.1167/iovs.10-6141
Keywords: hearing loss, Usher syndrome, hair cell, stereocilia, retinitis pigmentosa, photoreceptor, inner ear
Citation: Whatley M, Francis A, Ng ZY, Khoh XE, Atlas MD, Dilley RJ and Wong EYM (2020) Usher Syndrome: Genetics and Molecular Links of Hearing Loss and Directions for Therapy. Front. Genet. 11:565216. doi: 10.3389/fgene.2020.565216
Edited by:
Jordi Pérez-Tur, Superior Council of Scientific Investigations (CSIC), SpainReviewed by:
Hiroshi Nakanishi, Hamamatsu University School of Medicine, JapanIgnacio del Castillo, Ramón y Cajal University Hospital, Spain
Copyright © 2020 Whatley, Francis, Ng, Khoh, Atlas, Dilley and Wong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elaine Y. M. Wong, ZWxhaW5lLndvbmdAZWFyc2NpZW5jZS5vcmcuYXU=
†These authors have contributed equally to this work