 Jin-Yi Li1,2
Jin-Yi Li1,2 Qi Wang
Qi Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 17 December 2020
Sec. Evolutionary and Genomic Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.564939
Many bacteria belonging to Paenibacillus polymyxa are plant growth-promoting rhizobacteria (PGPR) with the potential to promote plant growth and suppress phytopathogens and have been used as biological control agents (BCAs). However, the growth promotion and biocontrol mechanisms of P. polymyxa have not been thoroughly elucidated thus far. In this investigation, the genome sequences of two P. polymyxa strains, ZF129 and ZF197, with broad anti-pathogen activities and potential for growth promotion were comparatively studied. Comparative and functional analyses of the two sequenced P. polymyxa genomes showed that the ZF129 genome consists of one 5,703,931 bp circular chromosome and two 79,020 bp and 37,602 bp plasmids, designated pAP1 and pAP2, respectively. The complete genome sequence of ZF197 consists of one 5,507,169 bp circular chromosome and one 32,065 bp plasmid, designated pAP197. Phylogenetic analysis revealed that ZF129 is highly similar to two P. polymyxa strains, HY96-2 and SQR-21, while ZF197 is highly similar to P. polymyxa strain J. The genes responsible for secondary metabolite synthesis, plant growth-promoting traits, and systemic resistance inducer production were compared between strains ZF129 and ZF197 as well as other P. polymyxa strains. The results indicated that the variation of the corresponding genes or gene clusters between strains ZF129 and ZF197 may lead to different antagonistic activities of their volatiles or cell-free supernatants against Fusarium oxysporum. This work indicates that plant growth promotion by P. polymyxa is largely mediated by phytohormone production, increased nutrient availability and biocontrol mechanisms. This study provides an in-depth understanding of the genome architecture of P. polymyxa, revealing great potential for the application of this bacterium in the fields of agriculture and horticulture as a PGPR.
Plant growth-promoting rhizobacteria (PGPR) have been identified as environmentally friendly alternatives to traditional agrochemicals for improving crop yield and quality (Kloepper et al., 1980). Paenibacillus polymyxa (formerly Bacillus polymyxa), which is an important beneficial member of the PGPR, has been applied in the fields of agriculture and horticulture in the form of soil inoculants to control a wide array of plant pathogens (Raza et al., 2008). P. polymyxa is a prominent biofertilizer and biocontrol agent that has been reported to suppress a wide variety of fungal and bacterial plant diseases, such as those caused by the fungi Rhizoctonia solani, Fusarium oxysporum, Botrytis cinereal, Phytophthora parasitica var. nicotianae, and Penicillium digitatum (Chen et al., 2011; Lai et al., 2012; Ren et al., 2012; Weselowski et al., 2016; Liu et al., 2018) and the bacteria Pseudomonas syringae pv. tomato, P. syringae pv. lachrymans, Xanthomonas campestris, X. oryzae pv. oryzae, Acidovorax avenae subsp. citrulli, and Ralstonia solanacearum (Li et al., 2010; Shi et al., 2012; Weselowski et al., 2016; Abdallah et al., 2019). Although the suppression of plant diseases by P. polymyxa strains has been continuously reported, further research into their detailed biocontrol mechanisms, especially at the molecular level, is necessary.
Plant growth-promoting rhizobacteria stably colonize the plant rhizosphere and enhance plant growth due to their capacities for phytohormone production, phosphate solubilization, nitrogen fixation, and antibiotic biosynthesis (Xie et al., 2016). P. polymyxa has been reported to produce various potent antimicrobial and volatile compounds that reduce plant disease severity (Eastman et al., 2014), such as antifungal and antibacterial metabolites (Zhao et al., 2011; Mageshwaran et al., 2012; Raza et al., 2015; Liu et al., 2018), thereby promoting growth (Anand et al., 2013; Padda et al., 2016) and inducing plant defenses (Mei et al., 2014; Shi et al., 2017; Luo et al., 2018). The types and amounts of antimicrobial substances generated by a beneficial bacterium affect its antimicrobial spectra and biocontrol efficacy. P. polymyxa can produce several kinds of antibiotic compounds, that can suppress the growth of pathogens under both laboratory and field conditions, including polymyxins and antifungal compounds such as fusaricidin (Choi et al., 2009; Padda et al., 2017; Liu et al., 2018). Moreover, it has been reported that P. polymyxa secretes other types of antibiotics, such as 1-octen-3-ol, benzothiazole, citronellol (Zhao et al., 2011), paenibacillin (Huang and Yousef, 2015), di-n-butyl phthalate (Deng et al., 2011), lipopeptide (Mageshwaran et al., 2012), and phenazine-1-carboxylic acid (Tupinamba et al., 2008), and systemic resistance inducers, including 2,3-butanediol, methanethiol and isoprene (Gao et al., 2010; Lee et al., 2012).
Paenibacillus polymyxa strains are used as soil inoculants in agriculture and horticulture as efficient plant growth promoting rhizobacteria (PGPR). Currently, comparative genomics is recognized as an important tool for identifying and understanding major biocontrol mechanisms and key functional genes among related organisms (Helfrich et al., 2014). The key genes responsible for the production of antimicrobial agents and volatile organic compounds, indoleacetic acid (IAA) synthesis, siderophore secretion, phosphate transporter, and phosphonate cluster biosynthesis in P. polymyxa strains, including P. polymyxa E681 (Kim et al., 2010), P. polymyxa SC2 (Ma et al., 2011), P. polymyxa M1 (Niu et al., 2011), P. polymyxa SQR-21 (Li et al., 2014), P. polymyxa CR1 (Eastman et al., 2014), P. polymyxa Sb3-1 (Rybakova et al., 2015), P. polymyxa YC0136 (Liu et al., 2017a), P. polymyxa YC0573 (Liu et al., 2017b), and P. polymyxa HY96-2 (Luo et al., 2018) were identified by genome sequencing and confirmed by a combination of physiological experiments.
In this study, we demonstrate the sequences and annotations of two strains P. polymyxa (ZF129 and ZF197), and compare the genomes with the thirteen representative P. polymyxa strains that are beneficial to plant growth. Strains ZF129 and ZF197 both exhibit significant broad inhibitory spectra against various plant-pathogenic fungi and bacteria and possess excellent biocontrol characteristics and potential for the biocontrol of vegetable diseases. However, their specific biocontrol mechanisms, especially at the molecular level, are still unclear. By comparing the genomic analysis with the P. polymyxa strains, our aim was to better understand their biocontrol mechanisms at the molecular level. Besides, secondary metabolite biosynthesis, IAA biosynthesis, phosphate solubilization, nitrogen fixation, and systemic resistance inducer production were analyzed via genomic comparison. These data will provide important insights for the study of biocontrol mechanisms and benefit the practical application of strains ZF129 and ZF197 in the field.
Strain ZF129 and ZF197 were isolated from the rhizosphere soil of potatoes grown in Guyuan and Gaomi, respectively, in China. Strains ZF129 and ZF197 were cultivated in LB (Luria broth) medium at 28°C with shaking for 36 h. The morphology of the two strains was observed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM; JEOL 1230 microscope). Genomic DNA was extracted from cultured ZF129 and ZF197 cells (OD600 = 0.8) using a TIANamp Bacteria DNA kit [Tiangen Biotech (Beijing) Co., Ltd]. The antagonistic activities of strains ZF129 and ZF197 against pathogenic bacteria and fungi were assessed through plate bioassays, and all of the experiments were repeated three times.
The P. polymyxa ZF129 and ZF197 strains were cultured in LB medium at 30°C with shaking at 180 rpm in the dark for 48 h. The cell-free culture supernatant was collected by centrifugation at 6,000 × g for 10 min and sequentially filtered through a 0.22 μm organic filter membrane. The filtrate was used for the antimicrobial activity test.
To measure the inhibitory activity against mycelial growth, the prepared cell-free supernatant was added to agar plates (1.5% w/v agar) containing potato dextrose agar (PDA, Merck) to achieve a final concentration of 10% (v/v). LB medium was used as the control. Then, a 6-mm mycelial plug was removed from the margin of the F. oxysporum colony and placed in the center of the PDA plate. The plates were incubated at 28°C for 5 days and examined for fungal growth. The inhibition activity was expressed in terms of the percentage of mycelial growth inhibition and was calculated according to the following formula: inhibition (%) = [(growth in control – growth in treatment)/growth in control] × 100.
A bioassay of volatiles from P. polymyxa ZF129 and ZF197 against F. oxysporum was performed in sealed dishes using a previously described method (Tagg and Mcgiven, 1971) with some modifications. Briefly, 300 mL of bacterial culture was spread on sterile solid LB medium supplemented with 1.5% agar. A 6-mm F. oxysporum mycelial plug taken from the margin of the colony was then placed in the center of a new PDA agar plate. The fungal dish was immediately inverted over the bacterial dish, and the dishes were rapidly sealed with Parafilm. The dishes were incubated at 28°C in the dark until the F. oxysporum mycelium in the controls extended over 3/4 of the plate. Volatiles from LB medium served as controls in place of bacterial volatiles. The diameter (mm) of the fungal colonies were measured, and inhibition activity was calculated in the same way.
The genomes of P. polymyxa ZF129 and ZF197 were sequenced by Igenecode, Beijing, China1. Whole-genome sequencing was performed using the Pacific Biosciences (PacBio) RS II platform, and a 20-kb SMRTbell template was used for library construction. The sequences were assembled de novo using the HGAP v.2.3 program, which uses an overlap-layout-consensus algorithm with the parameter Genome Length of 6000000. The HGAP pipeline uses the longest reads as seeds to recruit all other reads for the construction of highly accurate preassembled reads through a directed acyclic graph-based consensus procedure, which we follow with assembly using off-the-shelf long-read assemblers (Supplementary Table S1; Chin et al., 2013). Graphical views of the genome alignments were generated using CGView (Tatusova et al., 2016). The identification and annotation of the functional genes were performed using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP2) (Tatusova et al., 2016). Transfer RNA (tRNA) and ribosome RNA (rRNA) genes were identified using tRNAscan-SE version 2.0 and RNAmmer version 1.2, respectively (Lagesen et al., 2007; Lowe and Chan, 2016). The functions of the predicted proteins were assigned through comparisons against multiple databases, including the NR (non-redundant) protein databases3 (Li et al., 2002), the RAST (Rapid Annotation using Subsystem Technology) analysis platform (Aziz et al., 2008), Pfam4, SwissProt and the enhanced COG (clusters of orthologous groups of proteins) database5 (Tatusov et al., 2000). In addition, SignalP 4.0 (Bendtsen et al., 2004) and TMHMM 2.0 (Krogh et al., 2001) were used to predict putative signal peptides and transmembrane helices, respectively. PHAST was used for prophage prediction (Arndt et al., 2016), and clustered regularly interspaced short palindromic repeats (CRISPRs) were identified using CRISPR finder (Grissa et al., 2008). Besides genome sequencing and assembly, all of other bioinformatics analysis were also performed by Igenecode, Beijing, China.
The taxonomic positions of strains ZF129 and ZF197 were determined by multilocus gene sequence analysis (MLSA) based on six housekeeping genes (16S rRNA, gapA, gyrA, atpD, rpoA, and rho). The gene sequences were aligned using MUSCLE and trimmed to remove ambiguously aligned regions. Subsequently, the phylogenetic tree was constructed using the maximum likelihood method in MEGA 6.0 (Tamura et al., 2013). Other available gene sequences of the Paenibacillus and Bacillus strains to be used for phylogenetic tree construction were downloaded from the NCBI database (Supplementary Table S2). According to the phylogenetic analysis, two closely related P. polymyxa strains with released complete genome sequences, P. polymyxa HY96-2 (GenBank Accession No. CP025957.1) and P. polymyxa SQR-21 (GenBank Accession No. CP006872.1), were selected for genome comparison with ZF129. Likewise, P. polymyxa J (GenBank Accession No. CP015423.1) was selected for genome comparison with ZF197. Average nucleotide identities (ANIs) (Michael and Ramon, 2009) and in silico DNA-DNA hybridization (DDH) (Meierkolthoff et al., 2013) were calculated using the OrthoANIu algorithm6 and the Genome-to-Genome Distance Calculator (GGDC)7, respectively. Furthermore, complete genome comparisons were conducted with the progressive alignment option of Mauve 2.3.1 comparison software using the ZF129 and ZF197 genomes as the reference genomes (Darling et al., 2004). The Mauve results were also used to create a gene-by-gene orthologous comparison, which was used to create the Veen diagrams using the R package (Venn Diagram) (Chen and Boutros, 2011).
Secondary metabolite gene clusters prediction was performed using antiSMASH 2.0 on the authors’ Web servers using the default parameters8. Comparative analyses of secondary metabolite gene clusters identified among P. polymyxa ZF129, P. polymyxa ZF197, P. polymyxa HY96-2, P. polymyxa SQR-21, P. polymyxa SC2, and P. polymyxa J were performed based on the Kyoto Encyclopedia of Genes and Genomes database (KEGG9) and the GenBank database.
Functional genes involved in plant growth promotion or plant-bacterial interactions, such as those genes responsible for Indole-3-Acetic Acid (IAA) production, phosphate solubilization, and nitrogen fixation, were searched in the NCBI and KEGG databases. The identities of different functional genes at the amino acid level were compared between P. polymyxa ZF129, P. polymyxa ZF197, P. polymyxa HY96-2, P. polymyxa SQR-21, P. polymyxa SC2, and P. polymyxa J using the BLAST (Basic Local Alignment Search Tool, see text footnote 3).
To screen potential biocontrol agents for use against F. oxysporum, 27 bacteria were isolated from the rhizosphere of potato plants. Among these bacteria, two strains, ZF129 and ZF197, exhibited the highest inhibitory rates of 60.26 and 59.83%, respectively (Supplementary Table S3 and Figure 1). Antagonistic spectrum assays showed that strains ZF129 and ZF197 presented broad, strong antipathogenic activities against various plant-pathogenic fungi and bacteria, including Verticillium dahlia, Corynespora cassiicola, Botrytis cinereal, Fusarium oxysporum, Colletotrichum spp., Rhizoctonia solani, Xanthomonas campestris pv. campestris, Clavibacter michiganensis subsp. sepedonicum, Ralstonia solanacearum, Pseudomonas syringae pv. tomato, and P. syringae pv. lachrymans (Supplementary Table S4 and Supplementary Figure S2).
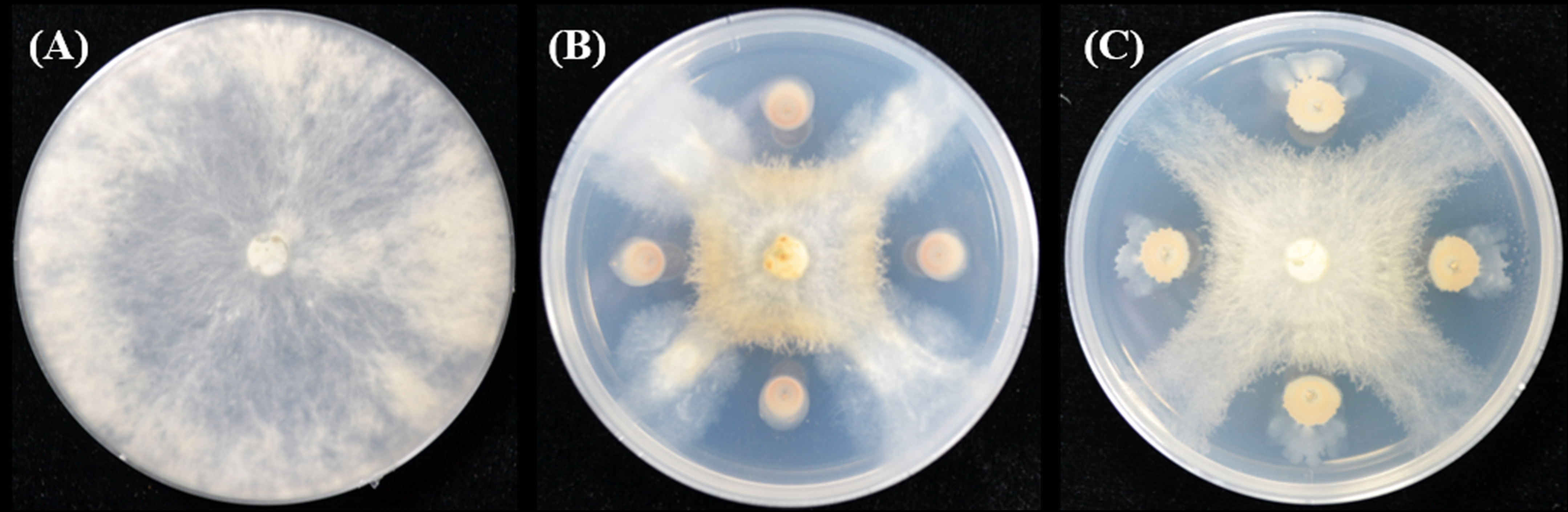
Figure 1. The inhibitory effect of strains ZF129 and ZF197 against F. oxysporum on PDA plates. (A) Control, (B) ZF129 against F. oxysporum, and (C) ZF197 against F. oxysporum.
Strains ZF129 and ZF197 produced antifungal volatile compounds (VOCs) and inhibited the mycelial growth of F. oxysporum with inhibition rates of 21.82 and 26.12%, respectively (Supplementary Figure S3). The cell-free supernatant of strain ZF197 displayed a significant inhibitory effect on the growth of F. oxysporum (56.72%), while the inhibition rate of the cell-free supernatant of ZF129 against F. oxysporum was just 7.73% (Supplementary Table S5). After the antifungal bioassay, mycelial morphology was observed under an optical microscope. The mycelia treated with ZF129 or ZF197 VOCs and the cell-free supernatant of ZF197 exhibited morphological aberrations such as enlargement, distortion and shriveling, whereas no similar changes were noted in the control mycelia.
Paenibacillus polymyxa ZF129 and ZF197 were determined to be motile, Gram-positive, rod-shaped, endospore-forming, facultative anaerobic bacteria belonging to the Paenibacillaceae family (Supplementary Figures S4B,D,F,H). Strains ZF129 and ZF197 grew readily on LB plates at 30°C and produced creamy white or light-yellow sticky colonies with irregular margins after 24 h of incubation (Supplementary Figures S4A,E). The interior of cell sections of ZF129 or ZF197 presented an oval or irregular shape, respectively (Supplementary Figures S4C,G).
The complete genome of P. polymyxa ZF129 comprises a circular 5,703,931 bp chromosome with two additional plasmids, pAP1 and pAP2. The genome of P. polymyxa ZF197 is composed of a circular chromosome of 5,507,169 bp with one plasmid (Supplementary Table S6). The average G + C contents of the ZF129 and ZF197 genomes are 45.34 and 45.60%, respectively, which are similar to those of P. polymyxa HY96-2 (46.50%), P. polymyxa SQR-21 (46.50%), and P. polymyxa J (45.70%), but higher than those of P. polymyxa M1 (44.80%) and P. polymyxa SC2 (44.58%) (Table 1). Graphical circular genomic maps showing the genome structure and functions of P. polymyxa ZF129 and ZF197 are presented in Supplementary Figure S1. In total, 5,149 open reading frames (ORFs) are predicted in the genome of ZF129. In addition to 4,861 protein-coding genes (CDSs), the chromosome contains 156 RNA genes, including 110 tRNA genes, 42 rRNA operons, 4 ncRNAs and 132 pseudogenes (Supplementary Table S6). For the ZF197 strain, 5,054 ORFs are predicted in the genome. In addition to 4,902 CDSs, the chromosome possesses 109 tRNA genes, 39 rRNA genes, 4 sRNA genes, and 244 pseudogenes (Supplementary Table S6). These annotated genes are transcribed in both the positive and negative directions in terms of the direction of DNA replication (Supplementary Figure S1).
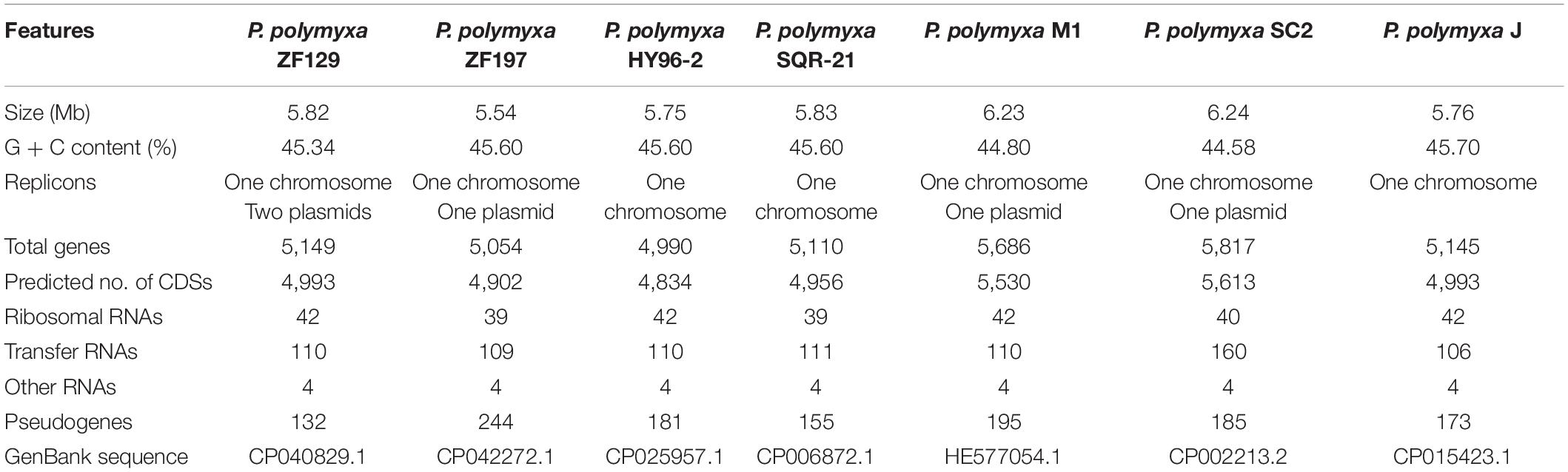
Table 1. Genomic features of P. polymyxa ZF129, ZF197 and other P. polymyxa strains.
The functional categorization of the CDSs of the ZF129 and ZF197 genomes was analyzed using the Clusters of Orthologous Groups of proteins (COG) database (Supplementary Figure S5). The results showed that 3,740 CDSs of ZF129 and 3,717 of the predicted genes of ZF197 were assigned to COG categories, including general function prediction only, transcription, carbohydrate transport and metabolism, amino acid transport and metabolism, signal transduction mechanisms, and ribosomal structure and biogenesis.
For the comparative genomic analysis of P. polymyxa ZF129 and ZF197, five publicly available complete genome sequences of P. polymyxa strains, including HY96-2, SQR-21, SC2, M1, and J, were selected (Table 1). To understand the relationships of P. polymyxa ZF129 and ZF197 with other P. polymyxa and Bacillus strains, a phylogenetic tree was constructed based on the 16S rRNA gene and five housekeeping genes (gapA, gyrA, atpD, rpoA, and rho) (Figure 2 and Supplementary Table S2). As expected, two primary monophyletic clades were corroborated by bootstrap values, which consisted of P. polymyxa and other Bacillus species. Strains ZF129 and ZF197 were clearly classified as P. polymyxa (Figure 2). Based on the observed distance relationships, P. polymyxa ZF129 was closely related to P. polymyxa HY96-2 and SQR-21. However, P. polymyxa ZF197 was closely related to P. polymyxa J. In addition, the taxonomic position of P. polymyxa SC2 and M1 was closer to strain ZF129 than strain ZF197.

Figure 2. Phylogenetic tree highlighting the relative positions of P. polymyxa ZF129 and ZF197 among other P. polymyxa strains and Bacillus species. The phylogenetic tree was constructed based on six housekeeping genes (16S rRNA, gapA, gyrA, atpD, rpoA, and rho) according to the aligned gene sequences using maximum likelihoods derived from MEGA 6.0 software. Bootstrap values (1,000 replicates) are shown at the branch points. The scale bar indicates 0.05 nucleotide substitutions per nucleotide position. The GenBank accession numbers associated with the housekeeping loci of all strains can be found in Supplementary Table S2.
Average nucleotide identity and DNA-DNA hybridization analyses are widely used to calculate whole-genome sequence similarities by comparing genomic data, and strains exhibiting ANI values ≥96% and DDH values ≥70% are typically regarded as the same species (Zhang et al., 2016). In this study, the ANI and DDH values between strains ZF129 and HY96-2 were 98.44 and 93.40%, respectively. Similarly, the ANI and DDH values between strains ZF197 and SC2 were 92.97 and 71.10%, respectively. Notably, the ANI and DDH values between strains ZF129 and ZF197 were 92.49 and 71.90%, respectively (Table 2). Lower ANI and DDH values were obtained when SQR-21, SC2, J, and the other three Bacillus standard strains were used as reference genomes. These findings revealed that strains ZF129, HY96-2, and SQR-21 clustered closely with each other and occupied the same taxonomic position.
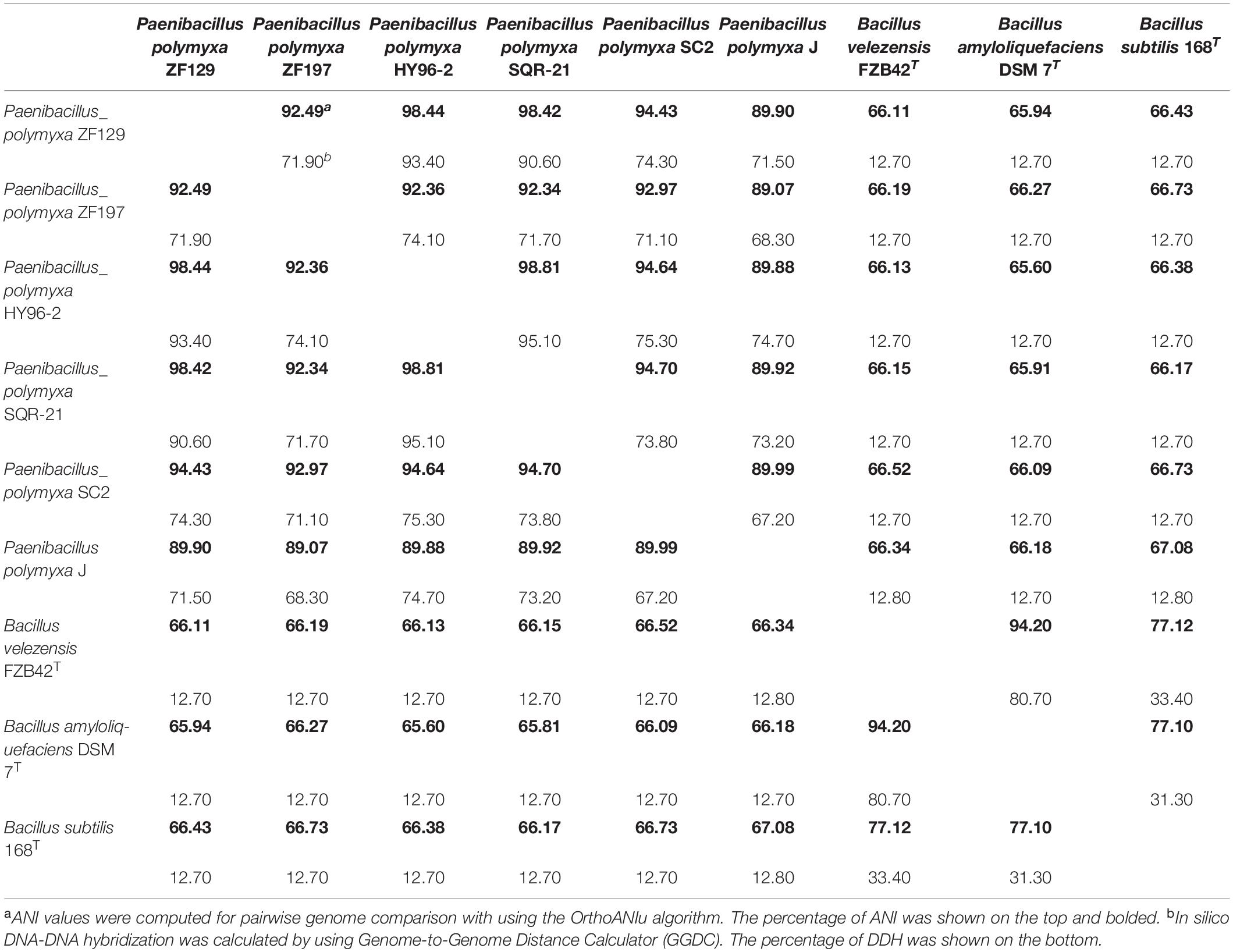
Table 2. Percentage of average nucleotide identities (ANI)a and in silico DNA-DNA hybridization (DDH)b among the selected Paenibacillus polymyxa or Bacillus genomes.
To evaluate the evolutionary distance among these sequenced strains in relation to several P. polymyxa strains, the whole-genome sequences were compared using Mauve. The alignments between ZF129 and ZF197 showed that several gene inversions or deletions were detectable in P. polymyxa ZF129 (Figure 3A). Compared to HY96-2 and SQR-21, a number of gene insertions or deletions and large local collinear block (LCB) inversions were also detected in P. polymyxa ZF129. In comparison to SQR-21, there was no significant deletion of large regions or large LCB inversions in P. polymyxa HY96-2 (Figure 3B). For strain ZF197, the genome sequence was aligned to P. polymyxa J. The results showed that regions with low similarity between the genomes occurred frequently and were distributed randomly between the ZF197 and J strains (Figure 3C). The results of the colinearity analysis were consistent with the synteny plot of the pairwise alignments from all of the above analyses.

Figure 3. Global alignment of the genome sequences of two completely sequenced P. polymyxa strains against three other P. polymyxa genome sequences. (A) Mauve progressive alignment of the ZF129 and ZF197 genomes. (B) Mauve progressive alignment of the ZF129, HY96-2, and SQR-21 genomes. (C) Mauve progressive alignment of the ZF197 and J genomes. The ZF129 genome and the ZF197 genome were used as the reference genomes. Boxes with the same color indicate syntenic regions. Boxes below the horizontal strain line indicate inverted regions. Rearrangements are indicated with colored lines. The scale is in nucleotides. The Venn diagram shows the numbers of shared and unique Clusters of Orthologous Genes at the subspecies and species levels.
To identify the specific genes of P. polymyxa ZF129 and ZF197, we compared their genome sequences with the released complete genome sequences of three P. polymyxa strains, HY96-2, SQR-21, and J (Figure 3). There were 4,127 conserved genes shared between strains ZF129 and ZF197. A total of 1,022 unique genes were present in the genome of ZF129, and 930 unique genes were present in the genome of ZF197. The core genome among ZF129, HY96-2 and SQR-21 was composed of 4,555 orthologous genes. ZF129 shared 106 genes with HY96-2 and 50 genes with SQR-21. Furthermore, 438 unique genes were present in the genome of ZF129, and the functions of most of these genes are still unknown. The analysis also revealed that a core genome consisting of 3,864 genes was common to strains ZF197 and J, while P. polymyxa ZF197 exhibited 1,192 unique genes.
For strain ZF129, 14 gene clusters involved in secondary metabolite production were retrieved, including four clusters encoding NRPSs (non-ribosomal peptide synthetases), three encoding transAT-PKSs (trans-acyl transferase polyketide synthetases) or NRPSs, one encoding an NRPS-like protein, three encoding lanthipeptides, and one each encoding one lassopeptide, one betalactone and one bacteriocin. Ten gene clusters related to secondary metabolite production were retrieved in strain ZF197, including five encoding NRPSs and one each encoding an NRPS-like protein, one lassopeptide, one bacteriocin, one NRPS or beatlactone and one phosphonate (Table 3).
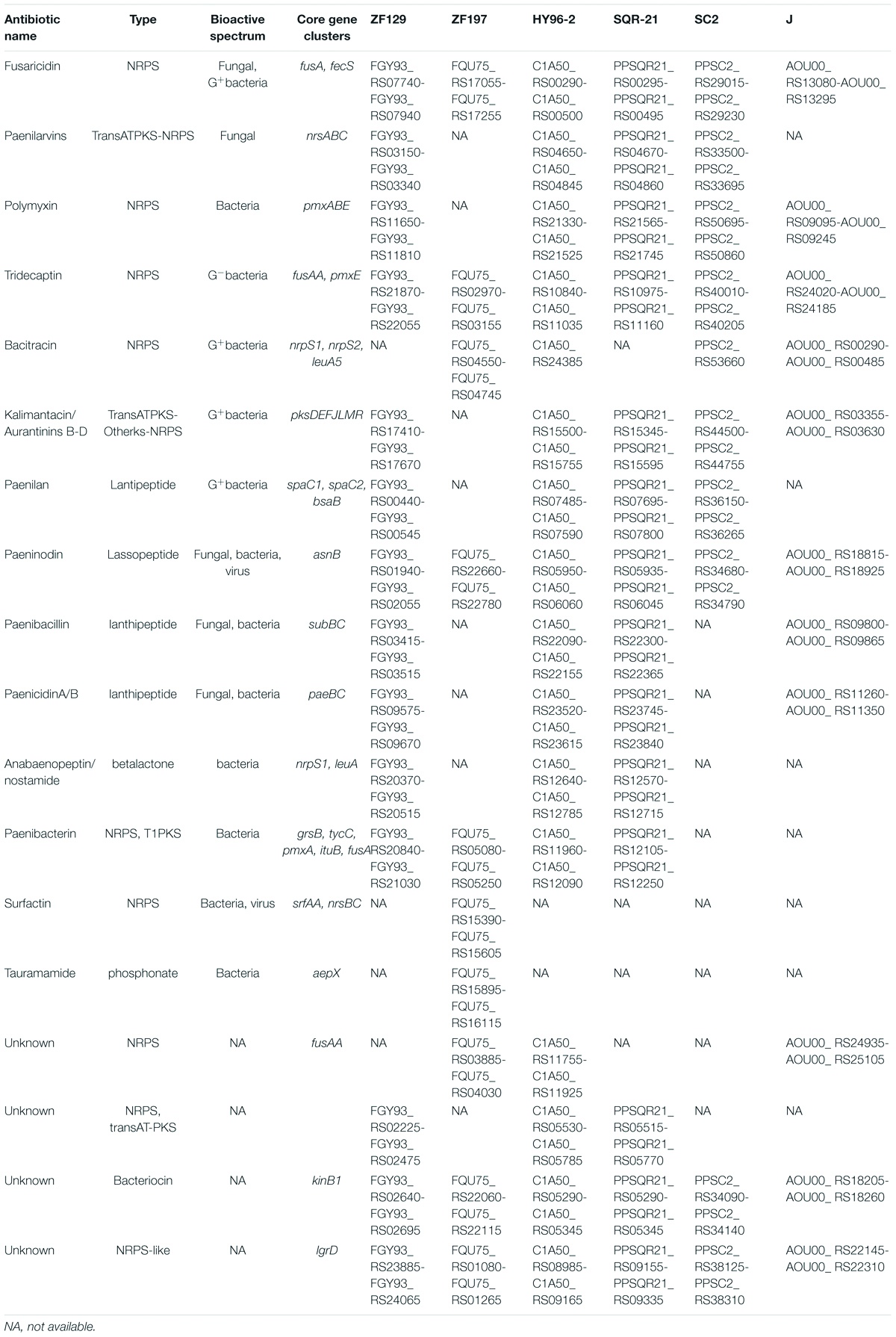
Table 3. Comparison of gene clusters and core genes involved in antibiotic biosynthesis in strains ZF129 and ZF197 as well as strains HY96-2, SQR-21, SC2, and J.
The comparison of genes or gene clusters related to antibiotic synthesis suggested that five secondary metabolites could be synthesized by strains ZF129, HY96-2, SQR-21, ZF197, and J, including fusaricidin, tridecaptin, paeninodin, and two unknown antibiotics. Furthermore, 14 gene clusters related to the biosynthesis of secondary metabolites existed in strains ZF129, HY96-2, and SQR-21 with high identities (Figure 4), and seven gene clusters associated with the biosynthesis of secondary metabolites existed in both strains ZF197 and J with high identities (Figure 5). However, three gene clusters related to the biosynthesis of secondary metabolites, including paenibacterin, surfactin and tauramamide, that were found in strain ZF197 did not exist in strains ZF129, HY96-2, SQR-21 and J. In addition, no gene clusters for the biosynthesis of paenilaryins, polymuxin, kalimantacin, paenilan, paenibacillin, paenicidin, and anabaenopeptin were detected in the ZF197 genome (Figure 5).

Figure 4. The predicted NRPS, transATPKS and peptide biosynthetic gene clusters show variation in ZF129 (top), HY96-2 (middle), and SQR-21 (bottom). Dark red indicates the core biosynthetic genes in different gene clusters among the three P. polymyxa strains. The core biosynthetic genes were relabeled in each gene cluster.
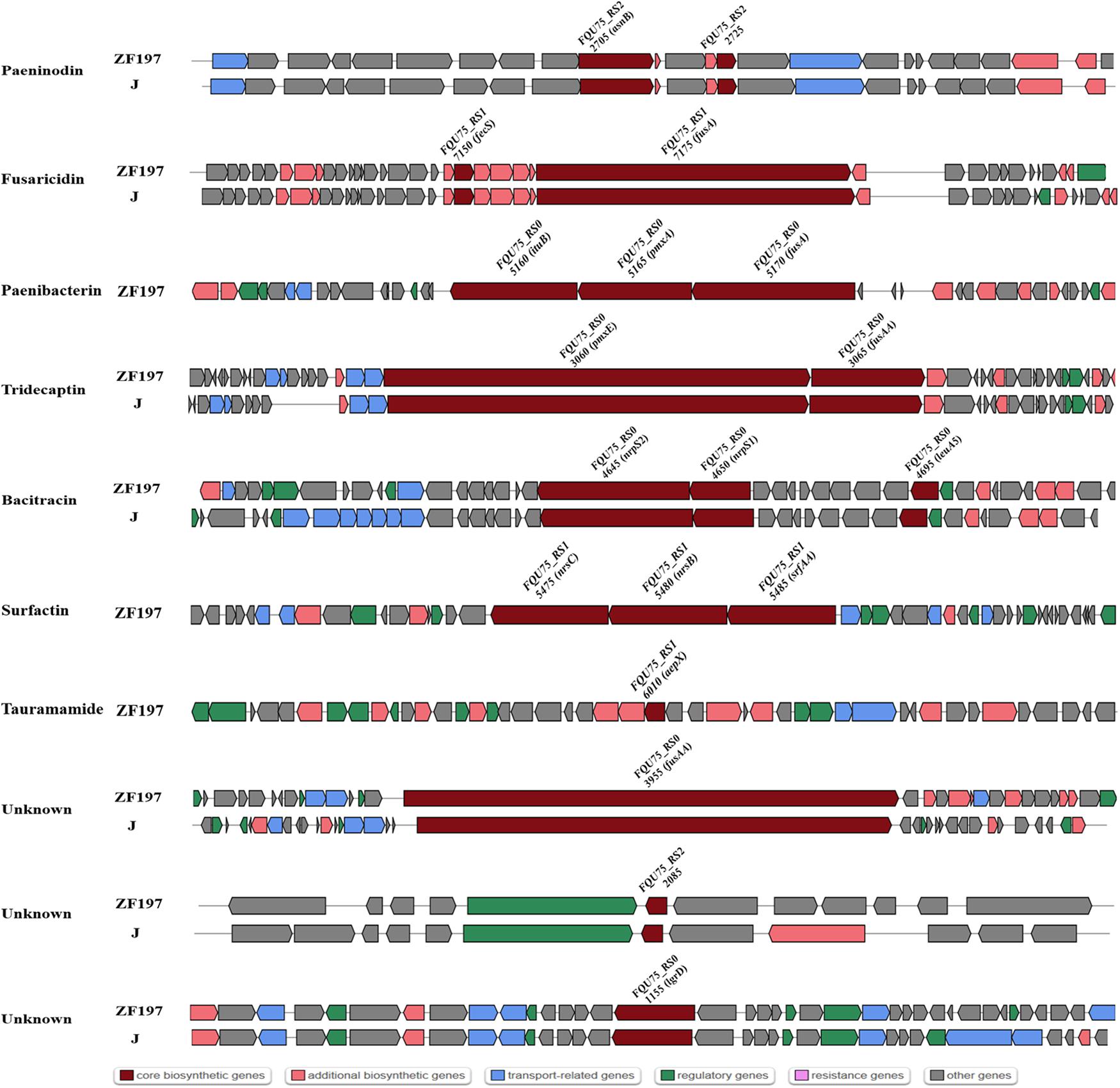
Figure 5. Comparisons of NRPS, transATPKS and peptide clusters in ZF197 (top) and J (bottom). Dark red indicates the core biosynthetic genes in different gene clusters among the three Bacillus strains. The core biosynthetic genes were relabeled in each gene cluster.
The key genes involved in plant growth promotion in P. polymyxa ZF129 and ZF197 were retrieved from the NCBI and KEGG databases, and the identities of these genes among ZF129, ZF197, HY96-2, SQR-21, and J were compared. The results indicated that six key genes (trpC, trpS, trpE, trpD, trpB, and mtrB) responsible for IAA (indole-3-acetic acid production) production were all found in the genomes of ZF129, ZF197, HY96-2, SQR-21 and J, with sequence identities exceeding 94% between P. polymyxa ZF129 and the other strains (Table 4). The results revealed that these strains may be expected to possess similar capacities for plant growth promotion.
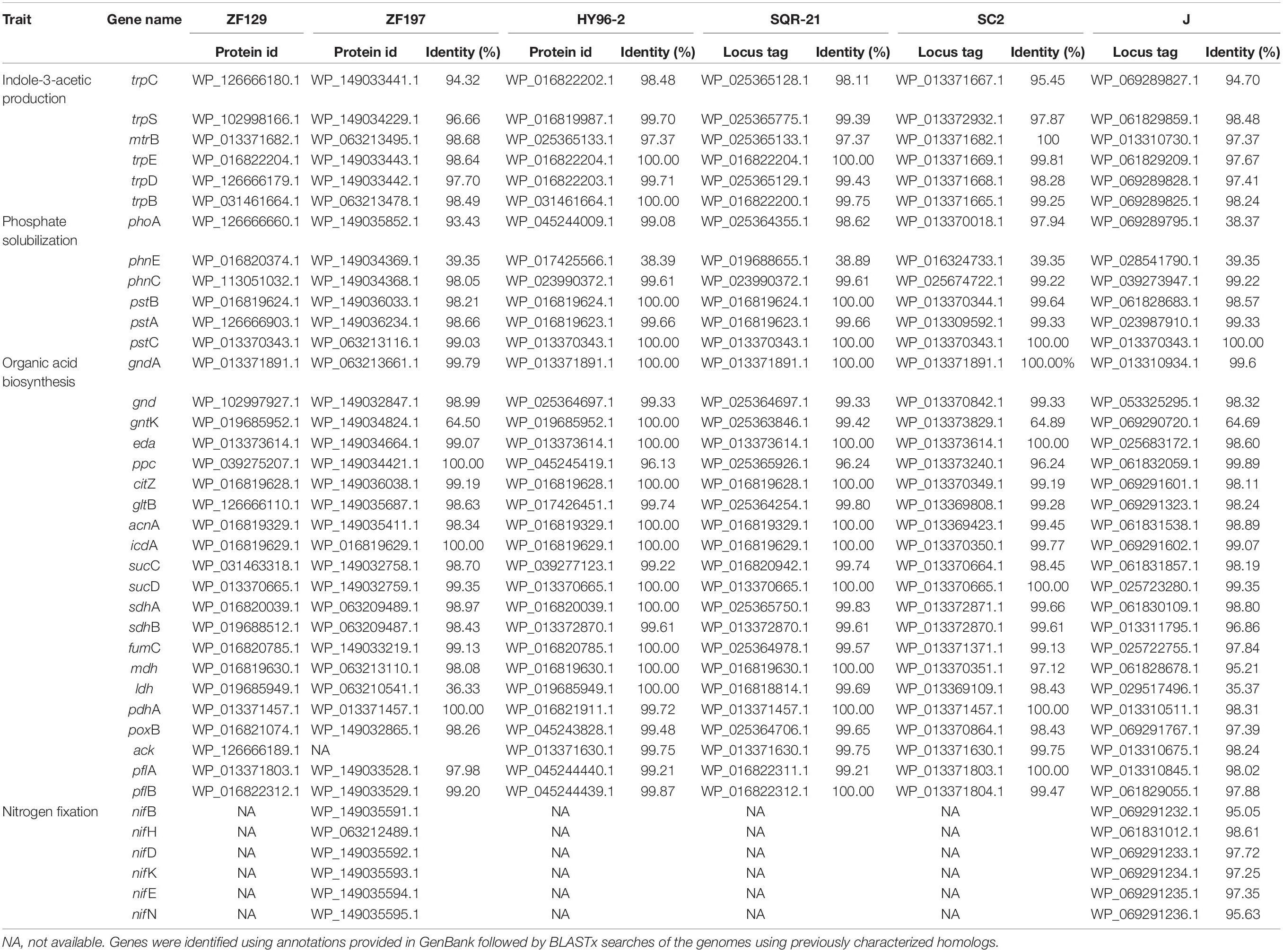
Table 4. Comparison of genes involved in plant growth promotion in strains ZF129 and ZF197 as well as strains HY96-2, SQR-21, SC2, and J (sequence similarity is expressed as a percentage of the amino acid identity).
In addition, six key genes related to phosphate solubilization were all retrieved from the five P. polymyxa strains (Table 4). The sequence identities of the four pstA-C and phnC genes between ZF129 and the other four strains were 98.05–100.00%, while the sequence identities of the phnE gene among the five strains were under 40%. In addition, high consistency of phoA was found between strains ZF129, ZF197, HY96-2, and SQR-21; however, the sequence identity between ZF129 and J was only 38.37%. Furthermore, 20 genes responsible for organic acid biosynthesis were all found in the genomes of ZF129, ZF197, HY96-2, SQR-21 and J, most of which were highly conserved (Table 4). The identities of the corresponding genes between ZF129 and HY96-2 and SQR-21 (96.13–100.00%) were higher than those between the ZF129, ZF197, and J strains (35.37–100.00%), especially for the ldh and gntK genes. The ack gene was found in strains ZF129, HY96-2, SQR-21 and J with sequence identities of 98.24–99.75% but was not detected in strain ZF197. Notably, key genes related to nitrogen fixation (nifB, nifH, nifD, nifK, nifE, and nifN) were only found in the genomes of strains ZF197 and J (Table 4), with sequence identities ranging from 95.05% to 98.61%.
Key genes associated with the synthesis of resistance inducers in P. polymyxa were retrieved from the KEGG database, and the identities of these genes between ZF129, ZF197, HY96-2, SQR-21, and J were compared. The results indicated that key genes for 2,3-butanediol (budA, ilyN), methanethiol (metH, metE), and isoprene (idi, lytB, gcpE, ispF, and ispE) synthesis were found in the genomes of all these strains, with sequence identities exceeding 92% (Table 5). The identities of the corresponding genes between ZF129 and ZF197, HY96-2, and SQR-21 (94.43–100%) were higher than those between ZF129 and J (92.62–99.19%).
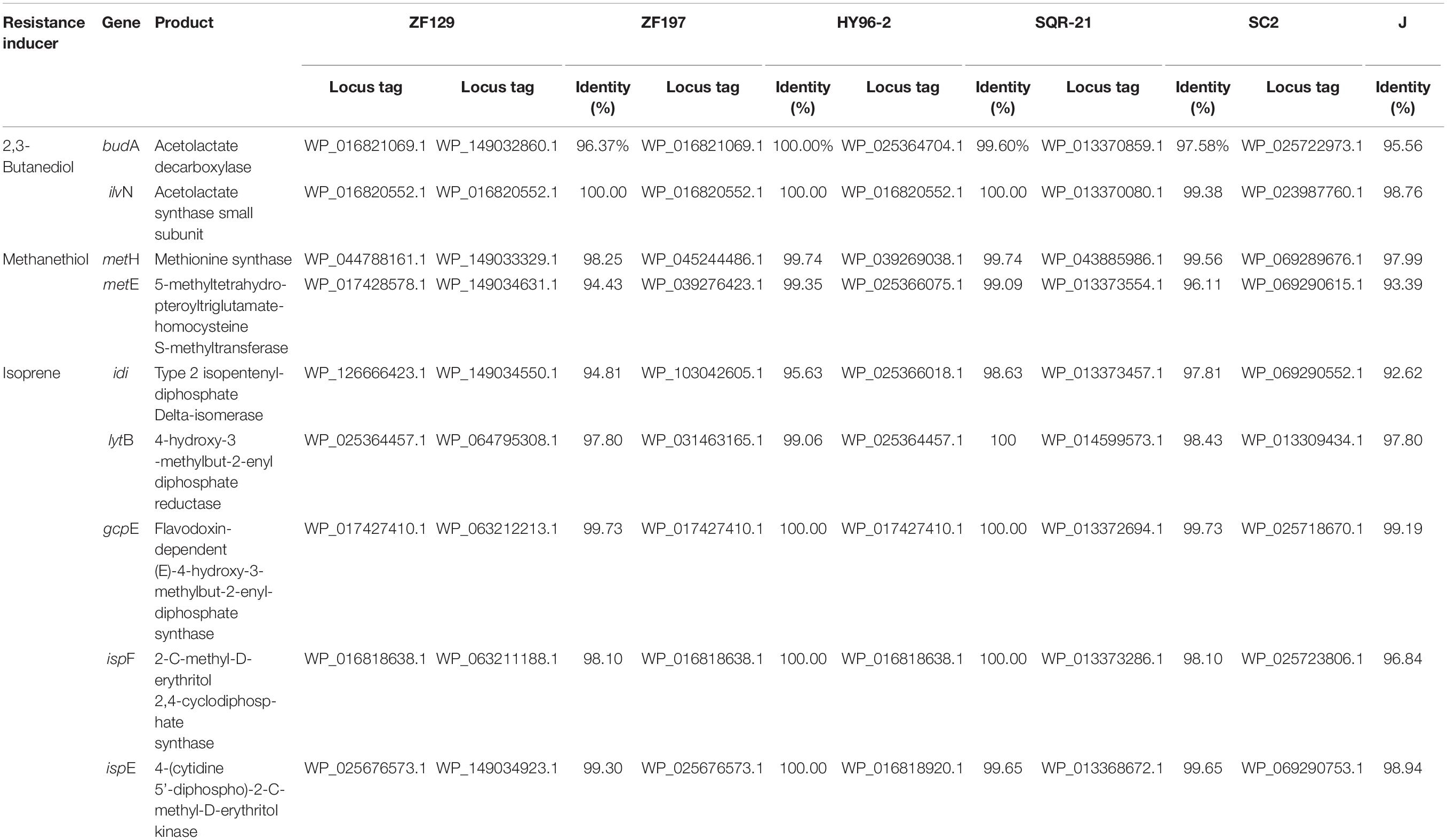
Table 5. Comparison of genes involved in the synthesis of resistance inducers in strains ZF129 and ZF197 as well as strains HY96-2, SQR-21, SC2, and J (sequence similarity is expressed as a percentage of the amino acid identity).
Bacteria of P. polymyxa are agriculturally important microbes and are widely studied for their plant growth-promoting abilities. After more than 100 years of study and analysis of the Paenibacillus genus, it was reclassified into a separate family, Paenibacilliaceae, and was designated as the family’s type genus (Padda et al., 2017). Currently, the previous reports concerning the complete genome of P. polymyxa mainly focused on the general features of the genome and analysis of the effect of this species on promoting growth, but rarely involved the analysis of the biocontrol mechanism (Luo et al., 2018). P. polymyxa ZF129 and ZF197 were isolated from the rhizosphere of potato plants to obtain potential biocontrol agents for use against F. oxysporum. They displayed broad antipathogenic activities and potential for growth promotion. However, their specific biocontrol mechanisms, especially at the molecular level, are still unclear. In this study, the complete genomes of P. polymyxa ZF129 and ZF197 were sequenced and compared with those of other P. polymyxa strains to better understand their biocontrol mechanisms at the molecular level. Phylogenetic analysis were performed to determine the taxonomic position of ZF129 and ZF197 and their relationships with other representative P. polymyxa strains. To clarify the differences in the biocontrol mechanisms between strains ZF129 and ZF197 and other P. polymyxa strains, various genes involved in secondary metabolite biosynthesis, IAA biosynthesis, phosphate solubilization, nitrogen fixation, and systemic resistance inducer production were analyzed via genomic comparison. The results of genome comparison revealed that the genome of strain ZF129 and ZF197 exhibits some degree of variation. And this finding possibly provides an in-depth understanding of the genome architecture of P. polymyxa, revealing great potential for the application of this bacterium in the fields of agriculture and horticulture as a PGPR.
In this study, the evolutionary position of ZF129 and ZF197 relative to other eleven P. polymyxa strains which were sequenced previously were determined by multi-locus sequence analysis (MLSA). Phylogenetic trees, which were constructed based on the 16S rRNA gene and five housekeeping genes (gapA, gyrA, atpD, rpoA, and rho) using maximum likelihood methods, show significant difference with each other. The phylogenetic trees show that the eleven P. polymyxa strains (CR1, YC0573, ATCC15970, YC0136, E681, HY96-2, SQR-21, Sb3-1, M1, SC2, and ZF129) form a monophyletic group. The strain ZF129 was clustered with HY96-2 and SQR-21 in Poly-clade subspecies. However, two P. polymyxa strains (J and ZF197) form the other monophyletic group. Population structure analysis also support that the Poly-clade strains evolved from a common ancestor.
General features of the seven completely sequenced P. polymyxa genomes are presented in Table 1. Immediately evident is the variation in genome size and differences in plasmid content between ZF129 and ZF197, with two plasmids (116,622 bp) or one plasmid (32,065 bp) present in ZF129 or ZF197 (Supplementary Table S6). Genome size varies between 5.70 Mb in ZF129 to 5.51 Mb in ZF197 excluding plasmids, with chromosome coding DNA sequences (CDS) varying between 4,993 and 4,902, respectively. The mean G + C% of seven species is 45.32% and no strain deviates from the mean by >0.6%. The plasmids of SC2 and M1 have a lower G + C% in accordance with previously reported work (Eastman et al., 2014). With the inclusion of plasmids in the calculation, ZF129 has the larger genome and the more CDS (5.82 Mb, 4,993 CDS) with ZF197 in a close second in terms of size, albeit with significantly fewer CDS (5.54 Mb, 4,902 CDS). This difference of 91 CDS between ZF129 and ZF197 may be associated with their different plasmids (Table 1 and Supplementary Table S6). In addition, the remarkable difference of genome annotation and function prediction had been exhibited using different databases, including RAST, Pfam, SwissProt, COG, SignalP, TMHMM, PHAST, and CRISPR Finder (Supplementary Table S6). The differences in CDS between ZF129 and ZF197 is likely the result of different annotation methods employed, which can result in large discrepancies in the total number of genes identified in a genome (Richardson and Watson, 2013).
Average nucleotide identity and DNA-DNA hybridization were used as effective tools for phylogenetic analysis at the genomic level. Strains with ANI values >96% and DDH values ≥70% are considered to be the same species (Zhang et al., 2016). Genome relatedness of the strain ZF129 and ZF197 with P. polymyxa strains from different branches of the phylogenetic tree was analyzed on the basis of ANI and DDH. ANI and DDH values among the representative P. polymyxa strains are shown in Table 2, and according to the ANI and DDH values, the complete genome of ZF129 was most similar (ANI value of 98.44% and DDH value of 93.40%) to that of HY96-2, which is also in the same monophyletic group. However, the ANI and DDH values between ZF197 and four other P. polymyxa genomes were no significant difference except strain J. Lower ANI similarity (<90%) was obtained when P. polymyxa J were used as reference genomes and DDH values were lower than 70%. This genetic difference maybe due to host-specific strain diversity, adaptation and ecological niche. A possible explanation for the result may that strain ZF129 and ZF197 belonged to different subspecies in P. polymyxa subspecies.
Horizontal gene transfer plays an essential role in the diversification of lineages in bacteria, especially with respect to the evolution of defined lineage, species and subspecies (Nowell et al., 2014). A global alignment of ZF129, ZF197, HY96-2, SQR-21, and J chromosomes was performed using Mauve and visualized as local collinear blocks (LCBs) to glean global information into the nucleotide level similarity amongst the sequenced P. polymyxa genomes (Figure 3). The nucleotide level similarity between ZF129 and HY96-2 is markedly higher than the similarity between any other grouping of strains, demonstrating the close relationship of these two strains and supporting our phylogeny showing the ZF129 and HY96-2 strains forming a sub-clade within the P. polymyxa species. Also, readily noticeable is unsimilarity of the ZF197 chromosome compared to any other P. polymyxa strains, with many strain-specific, low similarity regions dispersed throughout the ZF197 genome. Based on the comparative analysis, horizontal gene transfer obviously emerged among the P. polymyxa strains, but the ZF129 genome was highly syntenic with HY96-2, which confirmed the phylogenetic relationship analysis described above (Table 2 and Figure 2).
Comparisons of the genes and gene clusters related to antibiotic synthesis demonstrated more significant differences between the five P. polymyxa strains. In terms of their gene sequences, strains ZF129 and HY96-2 and SQR-21 exhibited relatively high homology, as did strains ZF197 and J. Regarding the control of fungi, fusaricidins are the main antifungal secondary metabolites produced by P. polymyxa, and the corresponding gene clusters were found in all five strains. Fusaricidin is a peptide antibiotic consisting of a group of cyclic depsipeptides with an unusual 15-guanidino-3-hydroxypentadecanoic acid moiety bound to a free amino group (Kajimura and Kaneda, 1996) that has been identified as the most important antifungal compound produced by P. polymyxa (Kajimura and Kaneda, 1997). Many members of the fusaricidin family have been isolated from P. polymyxa, including fusaricidins A-D and LI-F03 to LI-F08 (Han et al., 2012; Liu et al., 2018). The fusaricidin biosynthetic gene cluster (BGC) contains 7 ORFs (fusB, fusC, fusD, fusE, fusF, fusG, and fusTE) totaling 32.4 kb and one large ORF (fusA) of approximately 23.7 kb, encoding a six-module non-ribosomal peptide synthetase involved in fusaricidin production (Li and Jensen, 2008). Indeed, fusaricidins show very high antifungal activities against many plant-pathogenic fungi, especially F. oxysporum (Raza et al., 2009). In addition, fusaricidins from P. polymyxa A21 exhibit excellent antagonistic activity against B. cinerea on tomato (Liu et al., 2018); fusaricidin B is particularly effective against Candida albicans and Saccharomyces cerevisiae (Kajimura and Kaneda, 1996); and the LI-F-type antibiotics LI-F03, LIF04, LI-F05, LI-F06, LI-F07, and LI-F08 produced by P. polymyxa L-1129 and P. polymyxa I exhibit excellent activities against Staphylococcus aureus (Kurusu et al., 1987; Kuroda et al., 2000). Furthermore, gene clusters related to the production of paenilarvins, a class of iturin-like lipopeptide secondary metabolites with broad-spectrum antifungal activities (Sood et al., 2014), were also found in strains ZF129, HY96-2 and SQR-21, but not in ZF197 and J. The different antifungal activities of strains ZF129 and ZF197 identified in this study may have been due to the variation in their gene clusters related to antifungal metabolite synthesis or the production of different antifungal compounds.
Regarding the control of bacteria, different antibacterial metabolites were predicted to be produced in the five strains using the antiSMASH database, including polymyxin, tridecaptin, bacitracin, kalimantacin, paenilan, and paenibacterin. Polymyxin, which is a kind of non-ribosomal lipopeptide antibiotic, was first isolated from P. polymyxa in 1947, and at least 15 unique polymyxins have been reported (Kim et al., 2010). The gene clusters for polymyxin synthesis span a 40.6 kb region and consist of five ORFs, designated pmxA, pmxB, pmxC, pmxD, and pmxE (Choi et al., 2009). Polymyxin possesses broad-spectrum antibacterical activities, especially against Gram-negative bacteria, and has been recognized as one of the main antibacterial metabolites synthesized by P. polymyxa (Xu et al., 2018). Notably, P. polymyxa ZF197 also exhibited excellent antibacterial activities against many plant-pathogenic bacteria, although no genes or gene clusters involved in the synthesis of polymyxin were found on its chromosome. The strong antibacterial activity of P. polymyxa ZF197 may be associated with other antibacterial metabolites, which will need to be confirmed in further experiments. Tridecaptin is a kind of lipopeptide compound that includes two main members, tridecaptin A and tridecaptin B (Cochrane et al., 2015a,b, 2016). All five P. polymyxa strains were found to contain tridecaptin-related gene clusters, although the identities of the gene clusters between strains ZF129, ZF197, HY96-2, and SQR-21 were clearly higher than those between strains ZF129 and J. In addition, paenibacillin is a recently discovered lantibiotic from P. polymyxa OSY-DF that shows potent activities against bacteria such as Listeria monocytogenes and S. aureus (He et al., 2008). The core gene clusters pae A, paeB, and paeP are reported to be closely related to the biosynthesis of paenibacillin (Huang and Yousef, 2015), but no homologous gene clusters were found in the genome of strain ZF197. Moreover, bacitracin, which belongs to a class of polymyxin antibiotics, is produced at high levels by B. subtilis (Johnson et al., 1945), although the relevant gene clusters were also retrieved in strains ZF197 and J. Bacitractins A, B1, B2, and B3 are regarded as the main active components with potent antimicrobial activities against Gram-positive bacteria, especially against coccus and bacillus bacteria (Suleiman et al., 2017).
It is worth noting that the antibacterial metabolites predicted to be produced by strains ZF129 and ZF197 were significantly different, suggesting that some secondary metabolites may present similar targets in the inhibition of plant pathogens. Interestingly, the study showed that the cell-free supernatant of P. polymyxa ZF197 displayed a more significant inhibitory effect on the growth of F. oxysporum than did that of P. polymyxa ZF129 (Supplementary Figure S3), although the ZF129 and ZF197 strains both harbored the gene cluster involved in the production of fusaricidin, which was the main metabolite responsible for resistance to F. oxysporum. In the antiSMASH database, three secondary metabolites, surfactin, tauramamide and bacitracin, were found in strain ZF197 but not in ZF129. The reason for this unexpected finding is uncertain, but it will be important to determine the relationship between the high efficiency of ZF197 against F. oxysporum and the production of special secondary metabolites. Moreover, four gene clusters encoding unknown secondary metabolites were also found on the genome of strain ZF129 or ZF197; as rare antibiotics, their functions and bioactive spectra require further confirmation. In summary, the genome features of ZF129 and ZF197 showed that they present potential for application in the field to control plant diseases.
Indoleacetic acid is a primary plant hormone synthesized by plant-associated bacteria that has a profound effect on enhancing plant growth and development (Spaepen et al., 2007). P. polymyxa strains have been reported as effective plant growth-promoting bacteria, and one of their beneficial characteristics is the production of auxin-related phytohormones, especially IAA (Timmusk et al., 2005). Five different IAA biosynthetic pathways have been identified in bacteria according to the different intermediates produced during IAA biosynthesis by using tryptophan as a precursor (Patten and Glick, 1996), which mainly include indole-3-acetamide (IAM), indole-3-pyruvate (IPyA), indole-3-ethanol (TOL), indole-3-acetonitrile (IAN), and tryptamine (TAM) (Spaepen et al., 2007). P. polymyxa has been proven to synthesize IAA from the main precursor in a dependent manner and to possess more than one Trp-dependent biosynthetic pathway (Michael et al., 1997). It has been reported that the IPyA pathway is the main IAA production mechanism employed by P. polymyxa (Eastman et al., 2014). The ipdC gene, encoding a key enzyme in the IPyA pathway, has been detected in many P. polymyxa strains, including E681, CR1, and M1 (Phi et al., 2008). In our study, ipdC was not retrieved in the genomes of ZF129 and ZF197, whereas some trp genes were found in the genomes. The results suggested that ZF129 and ZF197 may be able to produce IAA in a tryptophan-independent manner, which needs to be conclusively demonstrated.
Phosphorus (P) is one of the major nutrients in plants, second only to nitrogen in terms of its requirement, and is considered a major essential macronutrient for the growth and development of plants (Passariello et al., 2006). The mineralization of most organic phosphorus compounds is carried out by phosphatases because plants can only utilize P in inorganic form. It has been reported that solubilization of mineral phosphates by bacteria is typically achieved through gluconic acid production. gcd (encoding glucose-1-dehydrogenase) and gad (encoding gluconic acid dehydrogenase) are the main genes responsible for the production of gluconic acid and its conversion (Werra et al., 2009). However, these two genes were not detected in the genomes of ZF129 and ZF197, although gcd and gad have been reported in P. polymyxa strains M1 and E681 (Xie et al., 2016). Another rich source of phosphate in soil is that trapped in the form of phosphonate, and the phosphonate gene cluster (phn) is responsible for the bacterial degradation of phosphonates, which release biologically available phosphate for nearby plants. Many P. polymyxa strains have been reported to carry phn genes (phnABCDEWXM), including strains CR1 and E681 (Xie et al., 2016); however, our comparative genomic analysis revealed that ZF129 and ZF197 do not carry the complete phn cluster, only possessing phnC and phnE. The variations in these genomes might be attributed to gene gain and loss events during evolution.
In addition, the PST (phosphate-specific transport) system, a high-affinity, low-velocity, free-Pi transport system, serves as a major Pi transport system in B. subtilis (Xie et al., 2016). The pst operon of B. subtilis contains pstS, pstC, pstA, pstB1 and pstB2. PstC and PstA are two integral inner membrane proteins, and PstB of Escherichia coli (or PstB1 and PstB2 for B. subtilis) is an ATP-binding protein (Chan and Torriani, 1996). Our comparative genomic analysis revealed that the strains ZF129 and ZF197 carry the pst operon (pstC, pstA, and pstB), and this operon might be associated with phosphonate uptake in response to phosphate deficiency, which needs to be further verified.
Nitrogen is an important limiting element for plant growth and production in agricultural systems since plants only absorb reduced forms of nitrogen, such as ammonia and nitrates. Nitrogenase is an oxygen-sensitive dinitrogen reductase that is produced by certain microorganisms (Bohlool et al., 1992). It can convert inert atmospheric nitrogen (N2) into ammonium (NH4), thereby improving plant growth and crop yields by increasing the concentration of biologically available nitrogen (Oldroyd and Dixon, 2014). Nitrogen fixation is mainly catalyzed by Mo-nitrogenase. The ability to fix nitrogen is found among a limited number of archaeal and bacterial taxa with a wide distribution, including Proteobacteria, Firmicutes, Cyanobacteria, Actinobacteria and Chlorobi (Dos Santos et al., 2012). P. polymyxa has been reported to possess the capacity for nitrogen fixation (Heulin et al., 1994). For instance, P. polymyxa P2b-2R can grow on N-free medium, consistently reduces acetylene in an acetylene reduction assay, and harbors nif gene clusters encoding nitrogenase enzymes (Bal et al., 2012; Anand et al., 2013). However, the contents and organization of nitrogen fixation (nif) genes vary significantly among the different N2-fixing organisms, which raises the question of the origins and evolution of Monitrogenase (Hartmann and Barnum, 2010). For example, 20 nif genes are found within a 24-kb cluster in Klebsiella pneumoniae (Arnold et al., 1988), whereas in Azotobacter vinelandii, the nif genes are dispersed and distributed as two clusters in the genome (Setubal et al., 2009). It has been reported that P. polymyxa and P. terrae likely derived from the same ancestor possessing nif gene clusters, and at least 9 of these genes (nifBHDKENXhesAnifV) have been found to be related to nitrogen fixation (Xie et al., 2014). The nifHDK gene cluster encoding Mo-nitrogenase is responsible for fixing nitrogen, while nifBENX and nifV are responsible for the synthesis and maturation of the FeMo cofactor. The hesA gene encodes an NAD/FAD-binding protein involved in molybdopterin and thiamine biosynthesis (Wang et al., 2013). However, the nif gene cluster is not distributed in all the sequenced P. polymyxa strains, and not all the nif genes are identical (Xie et al., 2014). Our results were consistent with these findings, as a minimal nif cluster (nifBHDKEN) was identified only in P. polymyxa ZF197 and P. polymyxa J and not in three other P. polymyxa strains, ZF129, HY96-2 and SQR-21. Nevertheless, the 7 nif genes identified in ZF197 are of particular interest because of their potential use as a source of transferable genetic elements related to nitrogen fixation to facilitate the development of genetically modified PGPR.
Induced systemic resistance is one of the biocontrol mechanisms whereby beneficial microorganisms control plant diseases (Bargabus et al., 2004). ISR, which is a different form of systemic resistance from systemic acquired resistance (SAR), responds to certain non-pathogenic rhizobacteria and effectively opposes multiple pathogens (Feys and Parker, 2000). It has been reported that biocontrol agents can generate and release systemic resistance inducers such as VOCs (e.g., 2,3-butanediol, methanethiol, isoprene, and butyl acetate) into the surrounding environment, thereby triggering the defensive mechanisms of plants (Lee et al., 2012; Shi et al., 2017). VOCs emitted by Bacillus spp. were shown to be novel determinants of ISR elicitation in Arabidopsis (Ryu et al., 2004). The VOCs released from P. polymyxa have also been proven to induce systemic resistance in Arabidopsis to control the foliar pathogen P. syringae pv. maculicola ES426 (Lee et al., 2012). In this study, all five P. polymyxa strains contained key genes associated with VOC (2,3-butanediol, methanethiol, and isoprene) production, although their sequence identities were different. The results indicated that the ZF129, ZF197, HY96-2, SQR-21, and J strains could induce similar systemic resistance in plants but with varying efficacies owing to the differences in the related genes.
The complete genome sequences of P. polymyxa ZF129 and ZF197 have been deposited in NCBI GenBank under accession numbers CP040829.1, CP040830.1, CP040831.1, CP042272.1, and CP042273.1, respectively. The two strains have also been deposited in the China General Microbiological Culture Collection Center (CGMCC) for Type Culture Collection under accession numbers 17631 and 17632, respectively.
QW and J-YL conceived and designed the experiments. J-YL performed the experiments. J-YL, T-TG, and QW analyzed the data and wrote the manuscript. All authors have read and approved the final manuscript.
This work was supported by funds for team building of talent cultivation and development support plan from China Agricultural University (No. 00109014).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.564939/full#supplementary-material
Supplementary Figure 1 | Graphical circular maps of the P. polymyxa ZF129 chromosome and plasmids pAP1 and pAP2 as well as the P. polymyxa ZF197 chromosome and plasmid pAP197 generated with CGview Server. From the outside to the center, rings 1 and 12 show protein-coding genes oriented in the forward (colored by COG categories) and reverse (colored by COG categories) directions, respectively. Ring 13 shows the G + C% content plot (black), and the innermost ring shows GC skews, where green indicates positive values and purple indicates negative values.
Supplementary Figure 2 | Antagonistic assays of P. polymyxa ZF129 and ZF197 against various plant pathogens. (A,a) Verticillium dahlia, (B,b) Corynespora cassiicola, (C,c) Botrytis cinereal, (D,d) Fusarium oxysporum, (E,e) Colletotrichum spp., (F,f) Rhizoctonia solani, (G,g) Xanthomonas campestris pv. campestris, (H,h) Clavibacter michiganensis subsp. sepedonicum, (I,i) Ralstonia solanacearum, (J,j) Pseudomonas syringae pv. tomato, (K,k) Pseudomonas syringae pv. lachrymans, (L,l) control.
Supplementary Figure 3 | Inhibitory activities of the volatiles or cell-free supernatants of P. polymyxa ZF129 and ZF197. (A) LB plate. (B) Volatiles of ZF129. (C) Volatiles of ZF197. (D) LB medium. (E) Cell-free supernatant of ZF129. (F) Cell-free supernatant of ZF197.
Supplementary Figure 4 | General characteristics of P. polymyxa ZF129 and ZF197. (A,E) Images of ZF129 and ZF197 colony morphology. (B,F) images of ZF129 and ZF197 cells obtained using transmission electron microscopy. (C,G) images of ZF129 and ZF197 cell sections obtained using transmission electron microscopy (Hitachi 7700, Japan), (D,H) images of ZF129 and ZF197 cells obtained using scanning electron microscopy (Hitachi SU8010, Japan).
Supplementary Figure 5 | COG functional categorization of two sequenced P. polymyxa genomes. COG functional categorization was performed using tools available in the Clusters of Orthologous Groups of proteins database. The left panel indicates the COG functional classification of ZF129, and the right panel indicates the COG functional classification of ZF197.
Supplementary Table 1 | The genome was assembled using the HGAP assembly pipeline in this study.
Supplementary Table 2 | The locus tag information of 16S rRNA genes and five housekeeping genes used for phylogenetic tree construction in this study.
Supplementary Table 3 | Inhibition rates of 27 biocontrol bacteria against F. oxysporum.
Supplementary Table 4 | The effects of strains ZF129 and ZF197 against various plant pathogens.
Supplementary Table 5 | Inhibitory activities of the volatiles or cell-free supernatants of P. polymyxa ZF129 and ZF197.
Supplementary Table 6 | Genome statistics.
Abdallah, Y., Yang, M., Zhang, M., Masum, M. M. I., Ogunyemi, S. O., Hossain, A., et al. (2019). Plant growth promotion and suppression of bacterial leaf blight in rice by Paenibacillus polymyxa Sx3. Lett. Appl. Microbiol. 68, 423–429. doi: 10.1111/lam.13117
Anand, R., Grayston, S., and Chanway, C. (2013). N2-fixation and seedling growth promotion of lodgepole pine by endophytic Paenibacillus polymyxa. Microb. Ecol. 66, 369–374. doi: 10.1007/s00248-013-0196-1
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Arnold, W., Rump, A., Klipp, W., Priefer, U. B., and Pühler, A. (1988). Nucleotide sequence of a 24,206-base-pair DNA fragment carrying the entire nitrogen fixation gene cluster of Klebsiella pneumoniae. J. Mol. Biol. 203, 715–738. doi: 10.1016/0022-2836(88)90205-7
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bal, A., Anand, R., Berge, O., and Chanway, C. P. (2012). Isolation and identification of diazotrophic bacteria from internal tissues of Pinus contorta and Thuja plicata. Can. J. For. Res. 42, 807–813.
Bargabus, R. L., Zidack, N. K., Sherwood, J. E., and Jacobsen, B. J. (2004). Screening for the identification of potential biological control agents that induce systemic acquired resistance in sugar beet. Biol. Control 30, 342–350. doi: 10.1016/j.biocontrol.2003.11.005
Bendtsen, J. D., Nielsen, H., Heijne, G., and Brunak, S. (2004). Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783–795. doi: 10.1016/j.jmb.2004.05.028
Bohlool, B. B., Ladha, J. K., Garrity, D. P., and George, T. (1992). Biological nitrogen fixation for sustainable agriculture: a perspective. Plant Soil 141, 1–11. doi: 10.1007/BF00011307
Chan, F. Y., and Torriani, A. J. (1996). PstB protein of the phosphate-specific transport system of Escherichia coli is an ATPase. J. Bacteriol. 178, 3974–3977. doi: 10.1128/jb.178.13.3974-3977.1996
Chen, H., and Boutros, P. C. (2011). VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. doi: 10.1186/1471-2105-12-35
Chen, X., Wang, G., Xu, M., Jin, J., and Liu, X. (2011). Antifungal peptide produced by Paenibacillus polymyxa brf-1 isolated from soybean rhizosphere. Afr. J. Microbiol. Res. 4, 2692–2698. doi: 10.1186/1471-2164-11-713
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Choi, S. K., Park, S. Y., Kim, R., Kim, S. B., Lee, C. H., Kim, J. F., et al. (2009). Identification of a polymyxin synthetase gene cluster of Paenibacillus polymyxa and heterologous expression of the gene in Bacillus subtilis. J. Bacteriol. 191, 3350–3358. doi: 10.1128/JB.01728-08
Cochrane, S. A., Findlay, B., Bakhtiary, A., Acedo, J. Z., and Vederas, J. C. (2016). Antimicrobial lipopeptide tridecaptin A 1 selectively binds to Gram-negative lipid II. Proc. Natl. Acad. Sci. U.S.A. 113, 11561–11566. doi: 10.1073/pnas.1608623113
Cochrane, S. A., Li, X., He, S., Yu, M., Wu, M., and Vederas, J. C. (2015a). Synthesis of tridecaptin-antibiotic conjugates with in vivo activity against gram-negative bacteria. J. Med. Chem. 58, 9779–9785. doi: 10.1021/acs.jmedchem.5b01578
Cochrane, S. A., Lohans, C. T., Belkum, M. J., Bels, M. A., and Vederas, J. C. (2015b). Studies on tridecaptin B1, a lipopeptide with activity against multidrug resistant Gram-negative bacteria. Org. Biomol. Chem. 13, 6073–6081. doi: 10.1039/c5ob00780a
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403.
Deng, Y., Lu, Z., Lu, F., Zhang, C., Wang, Y., Zhan, H., et al. (2011). Identification of li-f type antibiotics and di-n-butyl phthalate produced by Paenibacillus polymyxa. J. Microbiol. Methods 85, 175–182. doi: 10.1016/j.mimet.2011.02.013
Dos Santos, P. C., Fang, Z., Mason, S. W., Setubal, J. C., and Dixon, R. (2012). Distribution of nitrogen fixation and nitrogenase-like sequences amongst microbial genomes. BMC Genomics 13:162. doi: 10.1186/1471-2164-13-162
Eastman, A. W., Heinrichs, D. E., and Yuan, Z. C. (2014). Comparative and genetic analysis of the four sequenced Paenibacillus polymyxa genomes reveals a diverse metabolism and conservation of genes relevant to plant-growth promotion and competitiveness. BMC Genomics 15:851. doi: 10.1186/1471-2164-15-851
Feys, B. J., and Parker, J. E. (2000). Interplay of signaling pathways in plant disease resistance. Trends Genet. 16, 449–455. doi: 10.1016/s0168-9525(00)02107-7
Gao, J., Xu, H., Li, Q. J., Feng, X. H., and Li, S. (2010). Optimization of medium for one-step fermentation of inulin extract from jerusalem artichoke tubers using Paenibacillus polymyxa ZJ-9 to produce R,R-2,3-butanediol. Bioresour. Technol. 101, 7076–7082. doi: 10.1016/j.biortech.2010.03.143
Grissa, I., Vergnaud, G., and Pourcel, C. (2008). CRISPRcompar: a website to compare clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 36, W145–W148.
Han, J. W., Kim, E. Y., Lee, J. M., Kim, Y. S., Bang, E., and Kim, B. S. (2012). Site-directed modification of the adenylation domain of the fusaricidin nonribosomal peptide synthetase for enhanced production of fusaricidin analogs. Biotechnol. Lett. 34, 1327–1334. doi: 10.1007/s10529-012-0913-8
Hartmann, L. S., and Barnum, S. R. (2010). Inferring the evolutionary history of mo-dependent nitrogen fixation from phylogenetic studies of nifK and nifDK. J. Mol. Evol. 71, 70–85. doi: 10.1007/s00239-010-9365-8
He, Z., Yuan, C., Zhang, L., and Yousef, A. E. (2008). N-terminal acetylation in paenibacillin, a novel lantibiotic. FEBS Lett. 582, 2782–2792. doi: 10.1016/j.febslet.2008.07.008
Helfrich, E. J., Reiter, S., and Piel, J. (2014). Recent advances in genome-based polyketide discovery. Curr. Opin. Biotechnol. 29, 107–115. doi: 10.1016/j.copbio.2014.03.004
Heulin, T., Berge, O., Mavingui, P., Gouzou, L., Hebbar, K. P., and Hebbar, K. J. (1994). Bacillus polymyxa and Rahnella aquatilis, the dominant N2-fixing bacteria associated with wheat rhizosphere in French soils. Eur. J. Soil Biol. 30, 35–42.
Huang, E., and Yousef, A. E. (2015). Biosynthesis of paenibacillin, a lantibiotic with n-terminal acetylation, by Paenibacillus polymyxa. Microbiol. Res. 181, 15–21. doi: 10.1016/j.micres.2015.08.001
Johnson, B. A., Anker, H., and Meleney, F. L. (1945). Bacitracin: a new antibiotic produced by a member of the B. subtilis group. Science 102, 376–377. doi: 10.1126/science.102.2650.376
Kajimura, Y., and Kaneda, M. (1996). Fusaricidin a, a new depsipeptide antibiotic produced by Bacillus polymyxa KT-8 taxonomy, fermentation, isolation, structure elucidation and biological activity. J. Antibiot. 49, 129–135. doi: 10.7164/antibiotics.49.129
Kajimura, Y., and Kaneda, M. (1997). Fusaricidins B, C and D, new depsipeptide antibuotics produced by Bacillus polymyxa KT-8: isolation, structure elucidation and biological activity. J. Antibiot. 50, 220–228. doi: 10.7164/antibiotics.50.220
Kim, J. F., Jeong, H., Park, S. Y., Kim, S. B., Park, Y. K., Choi, S. K., et al. (2010). Genome sequence of the polymyxin-producing plant-probiotic rhizobacterium Paenibacillus polymyxa E681. J. Bacteriol. 192, 6103–6104. doi: 10.1128/JB.00983-10
Kloepper, J. W., Leong, J., Teintze, M., and Schroth, M. N. (1980). Enhanced plant growth by siderophores produced by plant growth-promoting rhizobacteria. Nature 286, 885–886. doi: 10.1038/286885a0
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kuroda, J., Fukai, T., Konishi, M., Uno, J., and Nomura, T. (2000). ChemInform abstract: LIF antibiotics, a family of antifungal cyclic depsipeptides produced by Bacillus polymyxa L-1129. Chemlnform 31. doi: 10.1002/chin.200042217
Kurusu, K., Ohba, K., Arai, T., and Fukushima, K. (1987). New peptide antibiotics LI-F03, F04, F05, F07, and F08, produced by Bacillus polymyxa. I. Isolation and characterization. J. Antibiot. 40, 1506–1514. doi: 10.7164/antibiotics.40.1506
Lagesen, K., Hallin, P., Rodland, E., Staerfeldt, H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lai, K., Chen, S., Hu, M., Hu, Q., Geng, P., Weng, Q., et al. (2012). Control of postharvest green mold of citrus fruit by application of endophytic Paenibacillus polymyxa strain SG-6. Postharvest Biol. Technol. 69, 40–48. doi: 10.1016/j.postharvbio.2012.03.001
Lee, B., Farag, M. A., Park, H. B., Kloepper, J. W., Lee, S. H., and Ryu, C. M. (2012). Induced resistance by a long-chain bacterial volatile: elicitation of plant systemic defense by a C13 volatile produced by Paenibacillus polymyxa. PLoS One 7:e48744. doi: 10.1371/journal.pone.0048744
Li, B., Su, T., Yu, R., Tao, R., Wu, Z., Soad, A. E., et al. (2010). Inhibitory activity of Paenibacillus macerans and Paenibacillus polymyxa against Ralstonia solanacearum. Afr. J. Microbiol. Res. 4, 2048–2054. doi: 10.1186/1471-2164-11-537
Li, J., and Jensen, S. E. (2008). Nonribosomal biosynthesis of fusaricidins by Paenibacillus polymyxa PKB1 involves direct activation of a d-amino acid. Chem. Biol. 15, 118–127. doi: 10.1016/j.chembiol.2007.12.014
Li, S., Yang, D., Qiu, M., Shao, J., Guo, R., Shen, B., et al. (2014). Complete genome sequence of Paenibacillus polymyxa SQR-21, a plant growth-promoting rhizobacterium with antifungal activity and rhizosphere colonization ability. Genome Announc. 2:e00281-14. doi: 10.1128/genomeA.00281-14
Li, W., Jaroszewski, L., and Godzik, A. (2002). Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18, 77–82. doi: 10.1093/bioinformatics/18.1.77
Liu, H., Liu, K., Li, Y., Wang, C., Hou, Q., Xu, W., et al. (2017a). Complete genome sequence of\r, Paenibacillus polymyxa\r, YC0136, a plant growth-promoting rhizobacterium isolated from tobacco rhizosphere. Genome Announc. 5:e01635-16. doi: 10.1128/genomeA.01635-16
Liu, H., Wang, C., Li, Y., Liu, K., Hou, Q., Xu, W., et al. (2017b). Complete genome sequence of Paenibacillus polymyxa YC0573, a plant growth-promoting rhizobacterium with antimicrobial activity. Genome Announc. 5:e01636-16. doi: 10.1128/genomeA.01636-16
Liu, W., Xiaoli, W. U., Bai, X., Zhang, H., Dong, D., Zhang, T., et al. (2018). Fusaricidins in Paenibacillus polymyxa A21 and their antagonistic activity against Botrytis cinerea on tomato. Front. Agric. Sci. Eng. 5, 262–270.
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Luo, Y. C., Cheng, Y. J., Yi, J. C., Zhang, Z. J., Luo, Q., Zhang, D. J., et al. (2018). Complete genome sequence of industrial biocontrol strain Paenibacillus polymyxa HY96-2 and further analysis of its biocontrol mechanism. Front. Microbiol. 9:1520. doi: 10.3389/fmicb.2018.01520
Ma, M., Wang, C., Ding, Y., Li, L., Shen, D., Jiang, X., et al. (2011). Complete genome sequence of Paenibacillus polymyxa SC2, a strain of plant growth-promoting rhizobacterium with broad-spectrum antimicrobial activity. J. Bacteriol. 193, 311–312. doi: 10.1128/JB.01234-10
Mageshwaran, V., Walia, S., and Annapurna, K. (2012). Isolation and partial characterization of antibacterial lipopeptide produced by Paenibacillus polymyxa HKA-15 against phytopathogen Xanthomonas campestris pv. phaseoli M-5. World J. Microbiol. Biotechnol. 28, 909–917. doi: 10.1007/s11274-011-0888-y
Mei, L., Liang, Y., Zhang, L., Wang, Y., and Guo, Y. (2014). Induced systemic resistance and growth promotion in tomato by an indole-3-acetic acid-producing strain of Paenibacillus polymyxa. Ann. Appl. Biol. 165, 270–279. doi: 10.1111/aab.12135
Meierkolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Michael, R., and Ramon, R. M. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 106, 19126–19131. doi: 10.1073/pnas.0906412106
Michael, L., Thierry, H., and Anton, H. (1997). Production of auxin and other indolic and phenolic compounds by Paenibacillus polymyxa strains isolated from different proximity to plant roots. FEMS Microbiol. Ecol. 22, 325–334. doi: 10.1111/j.1574-6941.1997.tb00384.x
Niu, B., Rueckert, C., Blom, J., Wang, Q., and Borriss, R. (2011). The genome of the plant growth-promoting rhizobacterium Paenibacillus polymyxa M-1 contains nine sites dedicated to nonribosomal synthesis of lipopeptides and polyketides. J. Bacteriol. 193, 5862–5863. doi: 10.1128/JB.05806-11
Nowell, R. W., Green, S., Laue, B. E., and Sharp, P. M. (2014). The extent of genome flux and its role in the differentiation of bacterial lineages. Genome Biol. Evol. 6, 1514–1529. doi: 10.1093/gbe/evu123
Oldroyd, G. E., and Dixon, R. (2014). Biotechnological solutions to the nitrogen problem. Curr. Opin. Biotechnol. 26, 19–24. doi: 10.1016/j.copbio.2013.08.006
Padda, K. P., Puri, A., and Chanway, C. P. (2016). Effect of GFP tagging of Paenibacillus polymyxa P2b-2r on its ability to promote growth of canola and tomato seedlings. Biol. Fertil. Soils 52, 377–387. doi: 10.1007/s00374-015-1083-3
Padda, K. P., Puri, A., and Chanway, C. P. (2017). “Paenibacillus polymyxa: a prominent biofertilizer and biocontrol agent for sustainable agriculture,” in Agriculturally Important Microbes for Sustainable Agriculture, eds V. Meena, P. Mishra, J. Bisht, and A. Pattanayak (Singapore: Springer), 165–191. doi: 10.1007/978-981-10-5343-6_6
Passariello, C., Forleo, C., Micheli, V., Schippa, S., Leone, R., Mangani, S., et al. (2006). Biochemical characterization of the class B acid phosphatase (AphA) of Escherichia coli MG1655. Biochim. Biophys. Acta 1764, 13–19. doi: 10.1016/j.bbapap.2005.08.028
Patten, C. L., and Glick, B. R. (1996). Bacterial biosynthesis of indole-3-acetic acid. Can. J. Microbiol. 42, 207–220. doi: 10.1139/m96-032
Phi, Q. T., Park, Y. M., Ryu, C. M., Park, S. H., and Ghim, S. Y. (2008). Functional identification and expression of indole-3-pyruvate decarboxylase from Paenibacillus polymyxa E681. J. Microbiol. Biotechnol. 18, 1235–1244. doi: 10.1016/j.mimet.2008.02.015
Raza, W., Yang, W., and Shen, Q. R. (2008). Paenibacillus polymyxa: antibiotics, hydrolytic enzymes and hazard assessment. J. Plant Pathol. 90, 419–430. doi: 10.2307/41998534
Raza, W., Yang, X., Wu, H., Wang, Y., Xu, Y., and Shen, Q. R. (2009). Isolation and characterisation of fusaricidin-type compound-producing strain of Paenibacillus polymyxa SQR-21 active against Fusarium oxysporum f.sp. nevium. Eur. J. Plant Pathol. 125, 471–483. doi: 10.1007/s10658-009-9496-1
Raza, W., Yuan, J., Ling, N., Huang, Q. W., and Shen, Q. R. (2015). Production of volatile organic compounds by an antagonistic strain Paenibacillus polymyxa WR-2 in the presence of root exudates and organic fertilizer and their antifungal activity against Fusarium oxysporum f. sp. niveum. Biol. Control 80, 89–95. doi: 10.1016/j.biocontrol.2014.09.004
Ren, X., Zhang, N., Cao, M., Wu, K., Shen, Q. R., and Huang, Q. W. (2012). Biological control of tobacco black shank and colonization of tobacco roots by a Paenibacillus polymyxa strain C5. Biol. Fertil. Soils 48, 613–620. doi: 10.1007/s00374-011-0651-4
Richardson, E. J., and Watson, M. (2013). The automatic annotation of bacterial genomes. Brief. Bioinform. 14, 1–12. doi: 10.1093/bib/bbs007
Rybakova, D., Wetzlinger, U., Müller, H., and Berg, G. (2015). Complete genome sequence of Paenibacillus polymyxa strain SB3-1, a soilborne bacterium with antagonistic activity toward plant pathogens. Genome Announc. 3:e00052-15. doi: 10.1128/genomeA.00052-15
Ryu, C. M., Farag, M. A., Hu, C. H., Reddy, M. S., Kloepper, J. W., and Paré, P. W. (2004). Bacterial Volatiles Induce Systemic Resistance in Arabidopsis. Plant Physiol. 134, 1017–1026. doi: 10.1104/pp.103.026583
Setubal, J. C., Dos Santos, P., Goldman, B. S., Ertesvåg, H., Espin, G., Rubio, L. M., et al. (2009). Genome sequence of Azotobacter vinelandii, an obligate aerobe specialized to support diverse anaerobic metabolic processes. J. Bacteriol. 191, 4534–4545. doi: 10.1128/JB.00504-09
Shi, Y., Niu, K., Huang, B., Liu, W., and Ma, H. (2017). Transcriptional responses of creeping bentgrass to 2,3-butanediol, a bacterial volatile compound (bvc) analogue. Molecules 22:1318. doi: 10.3390/molecules22081318
Shi, Y., Yang, L., Wang, X., Gao, Y., Liu, W., and Lou, K. (2012). Biocontrol of bacterial spot diseases of muskmelon using Paenibacillus polymyxa G-14. Afr. J. Biotechnol. 11, 16845–16851. doi: 10.5897/AJB12.1435
Sood, S., Steinmetz, H., Beims, H., Mohr, K. I., Stadler, M., Djukic, M., et al. (2014). Paenilarvins: iturin family lipopeptides from the honey bee pathogen Paenibacillus larvae. Cheminform 15, 1947–1955. doi: 10.1002/cbic.201402139
Spaepen, S., Vanderleyden, J., and Remans, R. (2007). Indole-3-acetic acid in microbial and microorganism-plant signaling. FEMS Microbiol. Rev. 31, 425–448. doi: 10.1111/j.1574-6976.2007.00072.x
Suleiman, S. A., Song, F., Su, M., Hang, T., and Song, M. (2017). Analysis of bacitracin and its related substances by liquid chromatography tandem mass spectrometry. J. Pharm. Anal. 7, 48–55. doi: 10.1016/j.jpha.2016.06.001
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tatusov, R. L., Galperin, M. Y., Natale, D. A., and Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. doi: 10.1093/nar/28.1.33
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Timmusk, S., Grantcharova, N., and Wagner, E. G. (2005). Paenibacillus polymyxa invades plant roots and forms biofilms. Appl. Environ. Microbiol. 71, 7292–7300. doi: 10.1128/AEM.71.11.7292-7300.2005
Tupinamba, G. S., Silva, A. J. R. D., Alviano, C. S., Souto-Padron, T., and Alviano, D. S. (2008). Antimicrobial activity of Paenibacillus polymyxa SCE2 against some mycotoxin-producing fungi. J. Appl. Microbiol. 105, 1044–1053. doi: 10.1111/j.1365-2672.2008.03844.x
Wang, L., Zhang, L., Liu, Z., Zhao, D., Liu, X., Zhang, B., et al. (2013). A minimal nitrogen fixation gene cluster from Paenibacillus sp. WLY78 enables expression of active nitrogenase in Escherichia coli. PLoS Genet. 9:e1003865. doi: 10.1371/journal.pgen.1003865
Werra, P. D., Péchy-Tarr, M., Keel, C., and Maurhofer, M. (2009). Role of gluconic acid production in the regulation of biocontrol traits of Pseudomonas fluorescens CHA0. Appl. Environ. Microbiol. 75, 4162–4174. doi: 10.1128/AEM.00295-09
Weselowski, B., Nathoo, N., Eastman, A. W., Macdonald, J., and Yuan, Z. C. (2016). Isolation, identification and characterization of Paenibacillus polymyxa CR1 with potentials for biopesticide, biofertilization, biomass degradation and biofuel production. BMC Microbiol. 16:244. doi: 10.1186/s12866-016-0860-y
Xie, J., Shi, H., Du, Z., Wang, T., Liu, X., and Chen, S. (2016). Comparative genomic and functional analysis reveal conservation of plant growth promoting traits in Paenibacillus polymyxa and its closely related species. Sci. Rep. 6:21329. doi: 10.1038/srep21329
Xie, J. B., Du, Z., Bai, L., Tian, C., Zhang, Y., Xie, J. Y., et al. (2014). Comparative genomic analysis of N2-fixing and Non-N2-fixing Paenibacillus spp.: organization, evolution and expression of the nitrogen fixation genes. PLoS Genet. 10:e1004231. doi: 10.1371/journal.pgen.1004231
Xu, Y., Lin, J., Cui, T., Srinivas, S., and Feng, Y. J. (2018). Mechanistic insights into transferable polymyxin resistance among gut bacteria. J. Biol. Chem. 293, 4350–4365. doi: 10.1074/jbc.RA117.000924
Zhang, Y. C., Fan, Q. R., and Loria, R. (2016). A re-evaluation of the taxonomy of phytopathogenic genera Dickeya and Pectobacterium using whole-genome sequencing data. Syst. Appl. Microbiol. 39, 252–259.
Keywords: genome sequencing, Paenibacillus polymyxa, plant growth-promoting, secondary metabolites, taxonomic position, inhibitory activity
Citation: Li J-Y, Gao T-T and Wang Q (2020) Comparative and Functional Analyses of Two Sequenced Paenibacillus polymyxa Genomes Provides Insights Into Their Potential Genes Related to Plant Growth-Promoting Features and Biocontrol Mechanisms. Front. Genet. 11:564939. doi: 10.3389/fgene.2020.564939
Received: 22 May 2020; Accepted: 13 October 2020;
Published: 17 December 2020.
Edited by:
Qunfeng Dong, Loyola University Chicago, United StatesReviewed by:
Felipe Luiz Pereira, Instituto Federal Catarinense, BrazilCopyright © 2020 Li, Gao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qi Wang, d2FuZ3FpQGNhdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.