
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 03 November 2020
Sec. Livestock Genomics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.562855
This article is part of the Research TopicEpigenetic Variation Influences on Livestock Production and Disease TraitsView all 24 articles
 Xiao-Dong Zheng1,2,3,4,5†
Xiao-Dong Zheng1,2,3,4,5† Jin Cheng1,2†Wen-Juan Qin1,2,6Nyamsuren Balsai1,2Xuan-Jian Shang1,2
Jin Cheng1,2†Wen-Juan Qin1,2,6Nyamsuren Balsai1,2Xuan-Jian Shang1,2 Meng-Ting Zhang1,2
Meng-Ting Zhang1,2 Hong-Quan Chen1,2,6*
Hong-Quan Chen1,2,6*Wandong (WD) cattle has recently been identified as a new Chinese native cattle breed by the National Commission for Livestock and Poultry Genetic Resources. The population size of this breed is less than 10,000. WD cattle and Dabieshan (DB) cattle are sympatric but are raised in different ecological environments, on mountains and plains, respectively, and the body sizes of these two breeds are markedly different. Blood samples were obtained from 8 adult female WD cattle and 7 adult female DB cattle (24 months old). The total RNA was extracted from leukocyte cells, and sequencing experiments were conducted on the Illumina HiSeqTM 4000 platform. After the removal of one outlier sample from the WD cattle breed as determined by principal component analysis (PCA), phylogenetic and population structure analyses indicated that WD and DB cattle formed a distinct Central China cattle group and showed evidence of hybridization between Bos. taurus and Bos. indicus. The immune-regulator CD48 (P = 1.3E-6) was associated with breed-specific traits according to loss-of-function variant enrichment analysis. In addition, 113 differentially expressed genes were identified between the two breeds, many of which are associated with the regulation of body growth, which is the major difference between the two breeds. This study showed that WD cattle belong to the group of hybrids between Bos. Taurus and Bos. indicus, and one novel gene associated with breed traits and multiple differentially expressed genes between these two closely related breeds was identified. The results provide insights into the genetic mechanisms that underlie economically important traits, such as body size, in cattle.
As key providers of milk, meat, leather and labor, cattle are one of the most important livestock species worldwide and have been domesticated since the early Neolithic period (Loftus et al., 1994, 1999; Beja-Pereira et al., 2006). More than 1000 cattle breeds have been identified around the world (Leroy et al., 2016), and 57 indigenous cattle breeds have originated from and been domesticated in China, as described in the State Catalog of Livestock and Poultry Genetic Resource 2008. Extant domesticated cattle have been categorized by many researchers into two major geographic taxa: humpless taurine (Bos. taurus) and humped indicine (Bos. indicus) (Hiendleder et al., 2008; Gibbs et al., 2009; Canavez et al., 2012; Porto-Neto et al., 2013). Recent studies have indicated that cattle breeds from Central China show reliable evidence of having originated from hybridization between Bos. taurus and Bos. indicus (Lai et al., 2006; Lei et al., 2006; Mei et al., 2017; Chen et al., 2018).
Chinese cattle breeds vary in their intrinsic characteristics and are important genetic resources. However, there is pressure on the genetic diversity of cattle, and the worldwide extinction of some (local) cattle breeds is a major concern (Pariset et al., 2010; Biscarini et al., 2015). An interesting example is provided by the Wandong (WD) cattle breed, which was recently identified in Fengyang county, Anhui Province, by the National Commission for Livestock and Poultry Genetic Resources. WD cattle were domesticated in the watershed region between the Yangtze River and Huai River approximately 500 years ago and mainly exhibit two kinds of hair colors, yellow and brown. WD is one of the best breeds reared in China; it is characterized by a medium size and high-quality meat production that can reach the level of “3A plus” identified by the National Committee on Livestock and Poultry Genetic Resources. Additionally, it is one of the breeds that can be raised in the ecological environment in hilly areas with poor land and little rainfall. However, according to the statistics department data, the total breed population of WD cattle has declined alarmingly rapidly from more than 100,000 heads in the 1990s to less than 10,000 heads recently. Therefore, there is an urgent need to protect this precious genetic resource.
Dabieshan (DB) cattle were mainly domesticated in the eastern Dabie Mountains. These animals are small in size and suitable for farming in the mountains. Currently, the total breed population of DB cattle is more than 200,000 heads. The large population and purity of its genetic resources make DB cattle one of the best local livestock breeds for population genetic studies. Companies that raise cattle in this area mainly intend to maintain pure genetic resources. Therefore, it is rare for DB cattle to be in contact with other breeds. In the absence of gene flow, adaptive variants and linked variants through hitchhiking are expected to quickly increase in frequency within isolated populations (Rettelbach et al., 2019) and may evolve into differentiation islands through positive selection. In addition, genetic drift or purifying selection [background selection (BGS)] will reduce the population genomic diversity when a population is not large enough and will increase the number of differentiation islands between different populations (Burri et al., 2015). A gradual increase in the number of differentiation islands may have resulted in the evolution of this unique breed, which is able to adapt to mountainous environments.
In this study, we performed whole-genome RNA sequencing on these two phenotypically diverse domestic Chinese cattle breeds. By combining the obtained whole-genome RNA sequence data and whole-genome sequence data from 11 additional representative taurine and indicine breeds, we explored the taxonomic status of the WD cattle breed in more detail. Since loss-of-function variant association studies are considered extensions of genome wide association studies (Lu et al., 2017; Wen et al., 2018), we also investigated genes and corresponding variants that are associated with agriculturally important traits, such as size, through gene-level loss-of-function (LOF) variant enrichment analysis, which has proven to be efficient in detecting evidence of linkage in whole-genome sequencing data (Artomov et al., 2017; He et al., 2019; Yang et al., 2020). The aims of our analyses were to provide new insights into the population stratification of worldwide domestic cattle breeds and explore the relationship between genetic variants and agricultural traits.
Animal experiments were executed according to the Institutional Animal Care and Use Committee (IACUC) guidelines under current approved protocols at Anhui Agricultural University.
Blood samples were obtained from 8 adult female WD cattle and 7 adult female DB cattle (24 months old) (Figure 1). WD cattle were fed and maintained under the same management conditions at the Daming Agriculture and Animal Technology Development Co., Ltd., which is located in FengYang county (Anhui Province) and has been designated as a WD cattle genetic resource conservation farm, while the DB cattle came from another conservation farm and were fed and maintained at the Anhui Wanjia Modern Agriculture Co., Ltd., Taihu County, Anhui Province.
Figure 1. WD cattle and DB cattle. WD cattle (A) and DB cattle (B) were fed and maintained at the Daming Agriculture and Animal Technology Development Co., Ltd., FengYang County, Anhui Province, and the Anhui Wanjia Modern Agriculture Co., Ltd., Taihu County, Anhui Province, respectively.
We collected whole-genome RNAseq data from Qinchuan cattle (QC, n = 4, GSE47653), one of the most common local cattle breeds. In addition, whole-genome sequence data from 8 local Iran cattle (PRJEB6119) and 25 local Uganda cattle (PRJEB7061) were downloaded from the European Variation Archive1 website (details in Supplementary Table 5).
The total RNA was extracted from plasma leukocyte cells extracted from samples collected from the jugular vein of each individual using TRIzol reagent (Life Technologies, United States) according to the manufacturer’s instructions. A TruSeq RNA Sample Preparation Kit (Illumina, United States) was used to purify the RNA and synthesize first- and second-strand cDNA. The quality was monitored with a NanoDrop ND-1000 spectrophotometer. The concentration of each sample was more than 50 pg/L, the amount of total RNA was more than 400 pg, and the RNA integrity number (RIN) was higher than 7.0. Then, an Agilent Bioanalyzer 2100 (Agilent Technology, United States) was used to determine the library fragment size distribution and assess whether it was suitable for computational analysis.
Sequence experiments were conducted by the Beijing Genomics Institute (BGI, Shenzhen, China) using the Illumina HiSeqTM 4000 platform. Single reads (50 bp) obtained from sequencing were checked by FastQC (v0.11.5)2, and adapter sequences were removed with Trimmomatic (V0.36) (Bolger et al., 2014). Genome index files were generated by STAR (Dobin et al., 2013) using the gene annotation information GTF file (UCD1.2.95) and the Bos. taurus genome fast file (UMD3.1/bosTau6) (Zimin et al., 2009). Then, whole-transcriptome alignment was performed to obtain all splicing sites the first time. Thereafter, we used the STAR in the two-pass mode to rebuild the genomic index to obtain better alignments around novel splice junctions, and the clean reads were realigned to the Bos. taurus genome with Picard to mark duplicates with the default parameters. Variant calling was performed according to the best practices using the GATK HaplotypeCaller module (Van der Auwera et al., 2013) with the following quality control parameters: QualByDepth (QD) set to less than 2.0 and FisherStrand (FS) set to more than 30. ANNOVAR (Wang et al., 2010) was used to annotate variants relative to RefSeq annotations (UMD3.1).
We obtained the PLINK genotype data format files by using the PLINK 1.9 “–vcf” command with the “–a2-allele” parameter, which will force the VCF (variant call format, generated by GATK program) file reference alleles to be set to A2. After that, the program of “convert” will calculate the number of copies of reference alleles per marker per individual (1 means one copy of the reference allele, 2 means two copies of the reference allele, and 9 means missing data) according to the PLINK SNP file. Principal component analysis was carried out with the overlapping common variants between the local data and the obtained public dataset to identify outlier samples using the smartPCA program of the EIGENSOFT (Patterson et al., 2006) package. A phylogenetic tree was constructed by using the neighbor-joining method in the program PHYLIP v3.6953, and the distance matrices (1 minus the identity-by-state value) were calculated using PLINK 1.07 (Purcell et al., 2007). The population structure was further inferred using ADMIXTURE (Alexander et al., 2009) with the kinship (K) set from 2 to 7.
To identify genetic differentiation between these two typical cattle breeds, we performed single-variant analysis using Fisher’s exact test in PLINK with all variants passing quality control and calculated the genomic inflation based on the P-value distribution by the median chi-squared statistic (Yang et al., 2011). Thereafter, we performed multiple-variant testing with selected variants annotated as LOF variants (such as frame shift insertions, frame shift deletions, no frame shift insertions, no frame shift deletions, non-synonymous SNVs, stop gains, stop losses). Genes with two or more LOF variants were preserved to perform gene-level enrichment analysis. Three independent gene-based analysis methods, the Burden test, the sequence kernel association test (SKAT), and the optimal sequencing kernel association test (SKATO), were applied using the R package SKAT program (Wu et al., 2010, 2011), and one additional chi-squared test was performed with an in-house script.
Clean reads in the RNA-seq data were mapped to the Bos. taurus genome (UMD3.1) using STAR aligner with the TranscriptomeSAM option. The output BAM files were processed using RSEM (Li and Dewey, 2011) to investigate gene expression levels. An in-house script was used to generate an expression matrix with transcripts per kilobase of exon model per million mapped read (TPM) values, which were obtained from the RSEM output gene abundance results. Based on a normal distribution and using log reads count values, a generalized linear model implemented in the R package limma (V 3.37.4) was used to assess differentially expressed genes (DEGs) using the empirical Bayes-moderated t statistics method (eBays). Differentially expressed genes with raw p-values lower than 0.05 and log2-fold changes larger than 334 (a mean value of the absolute value of the log2-fold change plus 2 multiple standard deviations of the absolute value of the log2-fold change) were considered significant. The R package ggplot2 (V3.1.0) was used to construct the volcano plot. To speculate about the potential biological processes related to bovine agricultural traits in which DEGs are involved, we performed the analysis of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with the R package clusterProfiler (Yu et al., 2012).
Fifteen samples from two cattle breeds (WD and DB) were sequenced using RNA-seq technology, generating an average of 24,135,850 raw sequencing reads, from which 23,893,462 clean reads remained after the filtering of low quality reads, and 21,054,824 uniquely mapped reads were finally obtained, with an average genome mapping ratio of 87.23 (Supplementary Table 1). A total of 724,998 single-nucleotide polymorphisms (SNPs) and 44,883 small insertions and deletions (InDels) were detected. A total of 20,819 variants were defined as LOF variants. After the removal of multiallelic variants (MNPs-multiple nucleotide polymorphisms, InDels), 20,575 variants remained. Then, variants (SNPs or InDels) with >90% call rates where each variant was uniquely located in one particular gene were retained. Additionally, the retained genes should have no less than LOF variants. Finally, we obtained 12,670 variants located in 2,720 genes for the subsequent gene-level enrichment analysis.
Whole-genome RNA-seq data from four pooled QC cattle were aligned with data from fifteen local cattle that were subjected to the variant calling procedure described above. More than one million variants were detected. Two additional datasets were obtained, whole-genome sequence data from nine breeds from Uganda (25 individuals; 29,133,488 variants) and one breed from Iran (8 individuals; 19,999,624 variants). A total of 425,728 autosomal variants from these three datasets and were used for the population structure analysis.
Principal component analysis (PCA) demonstrated a clear genetic structure, with the samples from local Chinese cattle clustering together except for one sample, WD1, which was removed from the subsequent analysis (Figure 2). The same population affinities were recovered in phylogenetic trees constructed via the neighbor-joining (NJ) method (Figure 3). ADMIXTURE analysis also recapitulated these findings. To choose the best value for K, we set K = 2 to K = 7. When K = 2 or 3, we obtained the smallest cross-validation error (Supplementary Figure 1). Using these parameters we observed three geographically distributed ancestral components, labeled Middle East, Central China and Africa components (Figure 4). We noted that the QC, WD and DB breeds from three separate geographical regions formed a distinct Central China group. All cattle breeds from other regions (the Middle East and Africa) showed evidence of hybridization between Bos. taurus and Bos. indicus.
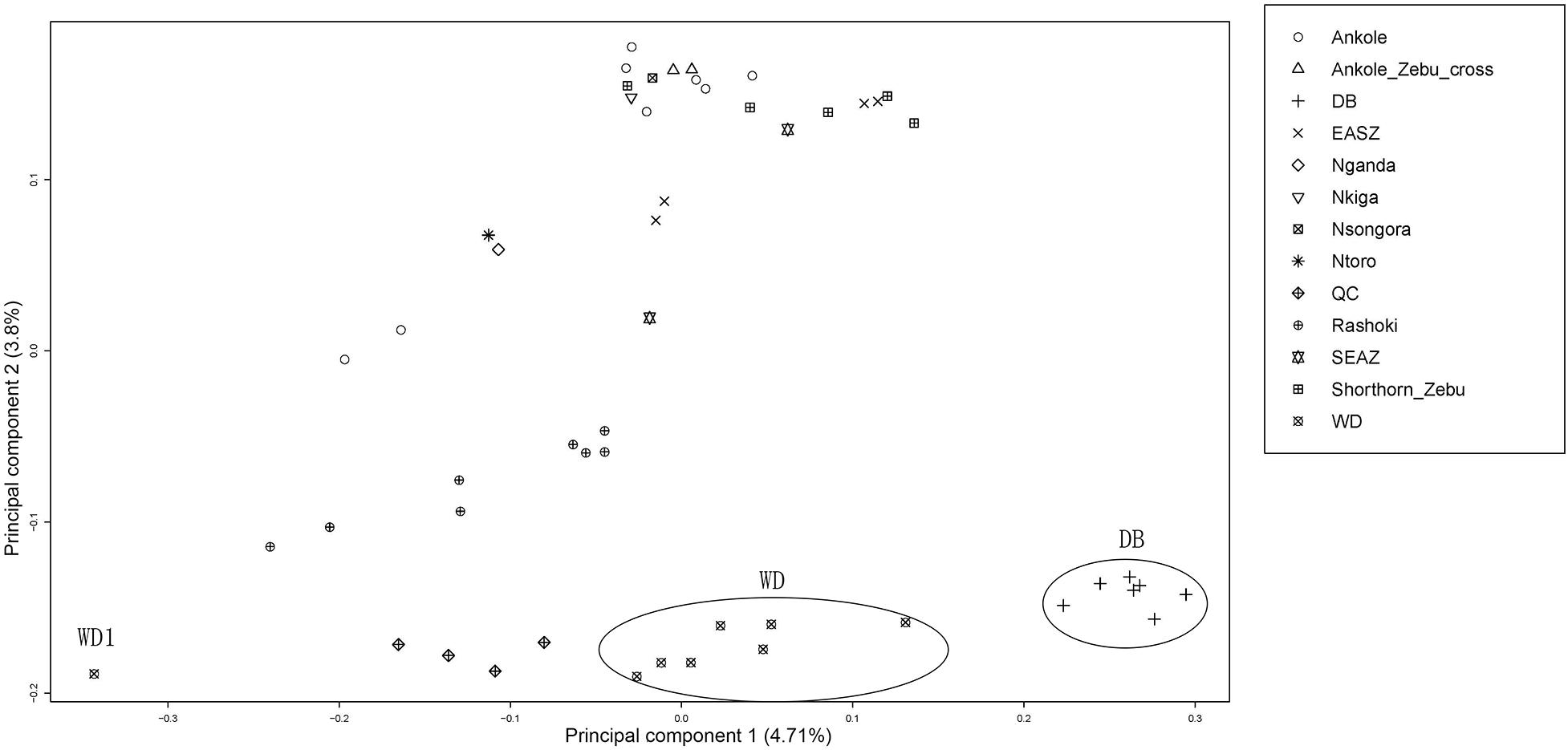
Figure 2. Principal component analysis of 11 cattle breeds. Principal component analysis (PCA) of 11 worldwide distributed cattle breeds.

Figure 3. Phylogenetic tree of 11 cattle breeds. The neighbor-joining (NJ) phylogenetic tree constructed using whole-genome autosome SNP data.
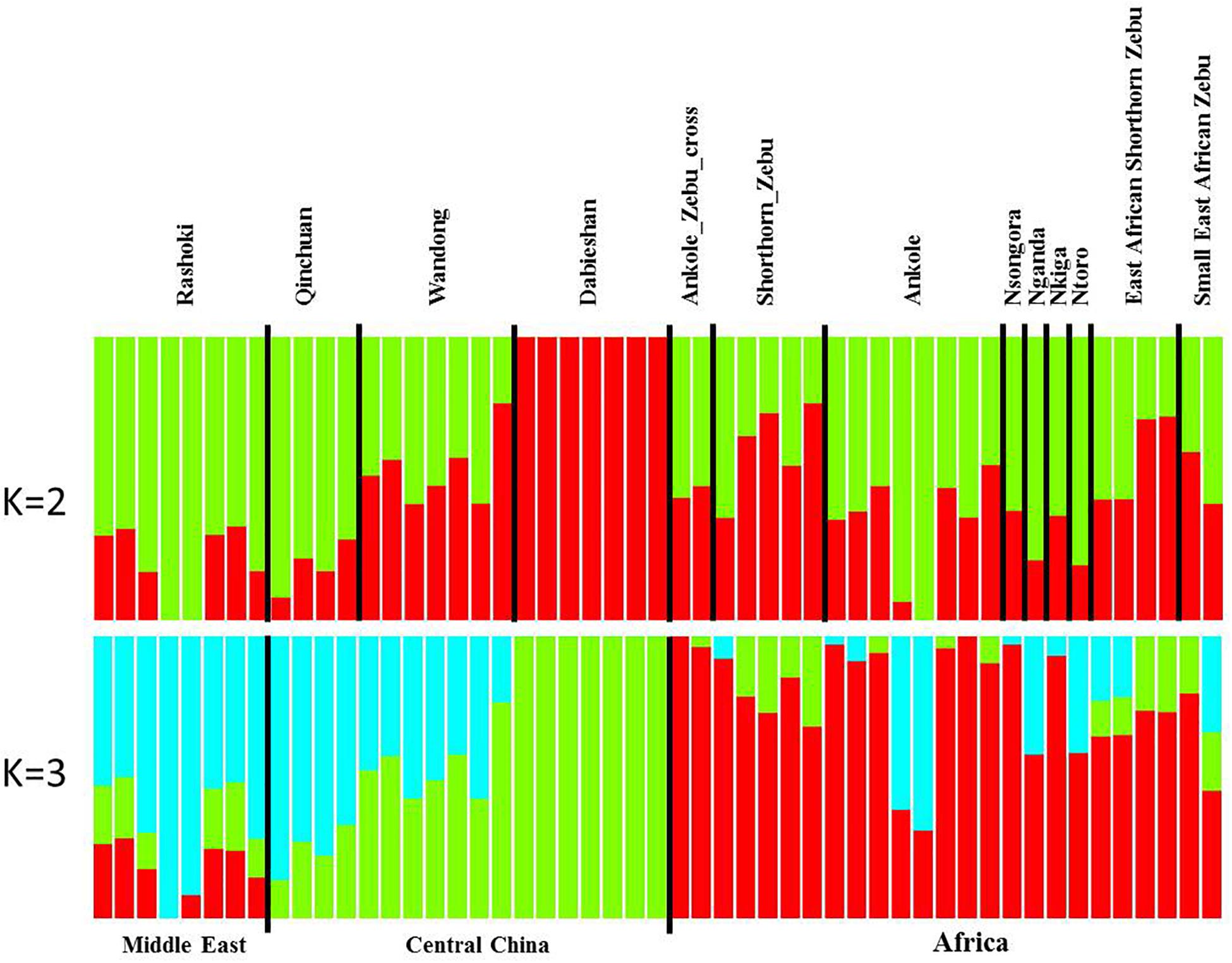
Figure 4. Genetic structure of 11 cattle breeds. Model-based clustering of cattle breeds using ADMIXTURE with K = 2 and K = 3. Breeds are arranged by geographic regions and labeled with the name of each breed.
Seven of the DB and seven of the WD cattle (one outlying sample was removed on the basis of PCA) were adult females (2 years old). It was obvious that the body size was significantly different between the two breeds (Table 1). To investigate the genetic linkage of this agricultural trait, we used 751,939 autosomal variants obtained from whole-genome RNA-seq of the two breeds, after performing quality control by excluding SNPs with a call rate of <90%, a minor allele frequency (MAF) <0.01, and Hardy–Weinberg equilibrium (HWE) in both cohorts <5e-8. The remaining 318,568 variants were employed to perform the whole-genome association analysis, and the distribution of –log10 P-values across all B. taurus autosomes used for the agricultural trait association of WD and DB cattle are shown in Supplementary Figure 2. After correction for the genomic inflation factor (λgc = 2.2742), there was no single variant showing evidence of a whole-genome significant association.

Table 1. Agricultural traits of two typical cattle breeds.
We performed multiple-variant testing with the rare variants through SKAT analysis in R. Although the quantile-quantile plot of SKAT, SKATO and Burden P-values suggested that there was only moderate genomic inflation or systematic bias in this study (Supplementary Figure 3), no gene could pass the Bonferroni correction P-value threshold (P < 1.84e-5, 0.05/2720). However, there were 87 genes with gene-based association P-values (P_skat, P_skato, and P_burden) of less than 0.05.
We further counted the minor allele numbers of these genes to fill the cross tabulation and performed the chi-squared test. With no evidence of genomic inflation (λgc = 0.1172), we observed a study-wide significant association for the CD48 gene (P_Chi = 1.3E-6, OR = 0.049, 95% CI = 0.011–0.209) and obtained suggestive evidence for an additional three genes (P_Chi < 1e-4) (Figure 5). Finally, there were 30 genes with a P-value of less than 0.05 according to four types of gene-level enrichment analysis methods (Table 2).
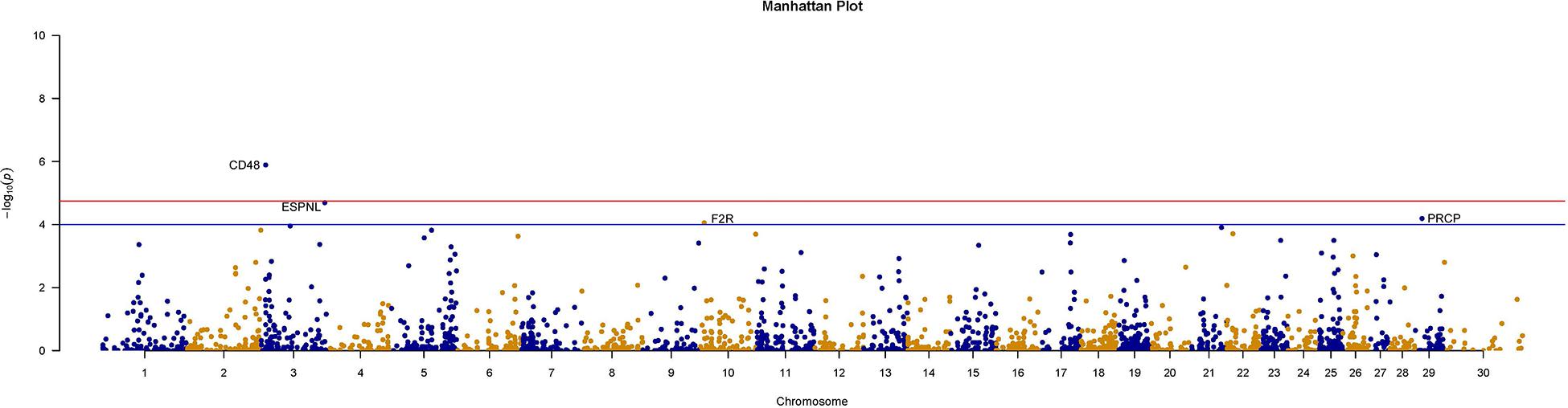
Figure 5. Manhattan plots of size-associated genomic variants based on the chi-squared test. Manhattan plots of -log10 (p-values) for two breeds based on the gene level LOF variant count chi-squared test method. The red horizontal line indicates the study-wide significance level following the Bonferroni correction for the gene-based method [-log10 (0.05/2720)], and the blue horizontal line indicates the suggestive significance level.
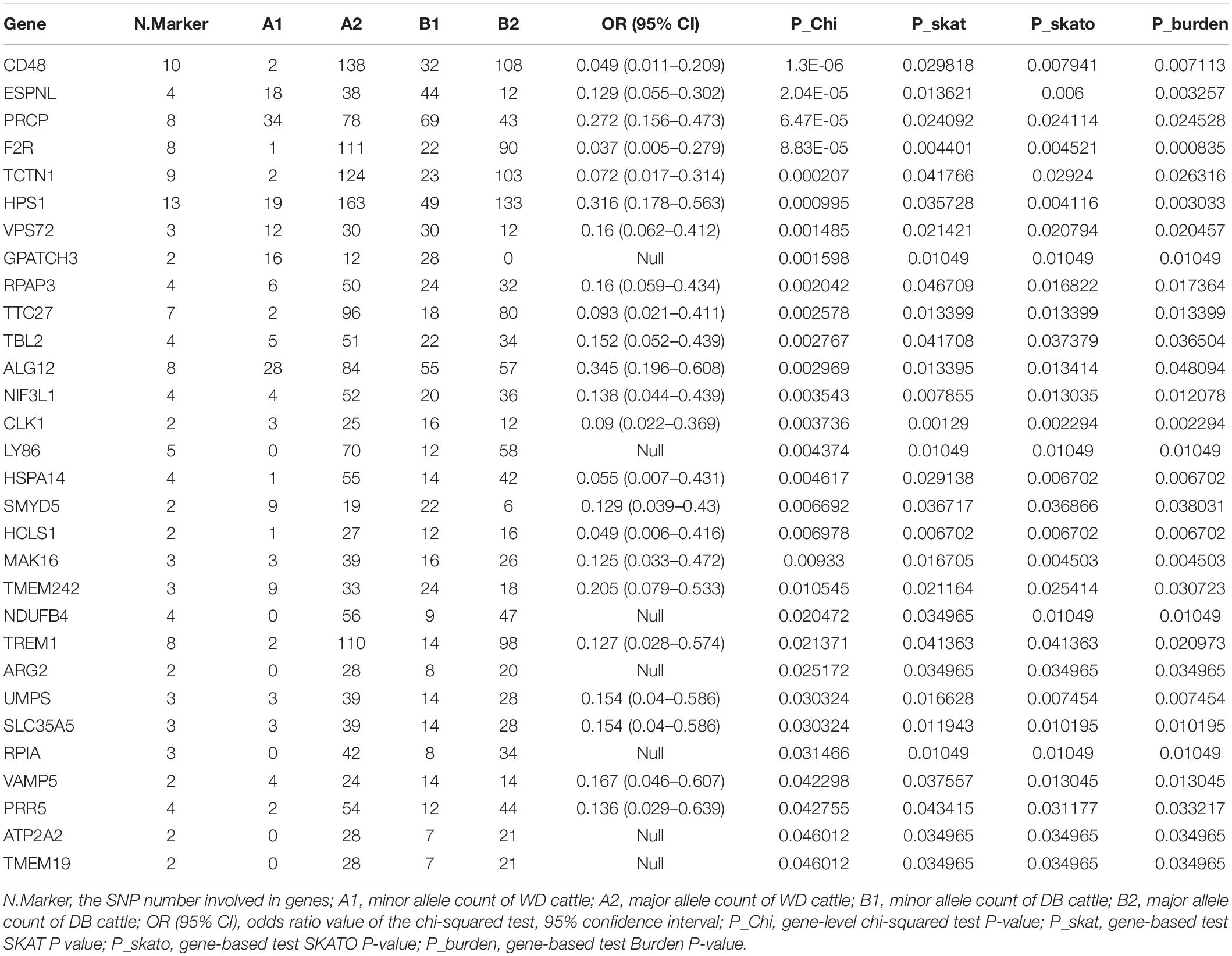
Table 2. 51 genes with P-values of less than 0.05 according to four independent gene-level LOF variant enrichment analysis methods.
Based on the eBays method, genes with associated P-values of less than 0.05 and fold changes greater than 334 were considered as significant. Compared with WD cattle, 86 genes that were upregulated and 27 genes that were downregulated in DB cattle showed significant changes in transcript abundance. After false discovery rate (FDR, Benjamini–Hochberg method) correction, 64 genes were observed to be upregulated, and 21 genes were observed to be downregulated (Supplementary Table 2). A total of 113 DEGs are included in the volcano plot (Supplementary Figure 4).
In total, 151 GO terms were assigned to 130 DEGs, and 134 terms were significantly enriched (Benjamini–Hochberg adjusted P < 0.05). Among these terms, 99 corresponded to biological processes, 28 to cellular components, and 10 to molecular functions (Supplementary Table 3). There were 28 GO terms assigned to the 30 LOF genes, all of which corresponded to cellular components. Only one GO term (GO:0031094) exhibited a P-value of less than 0.05 (Supplementary Table 4) which was involved in the regulation of the platelet dense tubular network (GO:0031094, p_adjust = 9.56e-3). including one suggested F2R gene identified by LOF enrichment analysis.
The findings of this study confirmed and extended the results of previous studies (Mei et al., 2017; Chen et al., 2018). The observed phylogenetic and population structure indicates that these Central Chinese cattle breeds formed an exclusive genetic group. The PCA provided similar results, and PC2 tended to separate populations sampled in Africa and those from the Middle East and Central China. Consistent with the geographic distance, the combination of PC1 and PC2 places the Middle Eastern cattle breed Rashoki between the African and Central Chinese cattle breeds. These data show that WD and DB cattle breeds present extremely low heterozygosity (∼0.2 and 0.18, respectively), while the QC, Rashoki cattle and African cattle breeds all exhibit intermediate heterozygosity (∼0.34–0.38), similar to a previous study of African taurine and indicine breeds (Orozco-terWengel et al., 2015). In contrast to other Central Chinese cattle breeds, WD cattle and DB cattle are raised in isolated environments, on the plains between the Huai and Yangtze rivers and in the mountains, respectively. The relatively less gene flow in these two breeds results in lower heterozygosity and a higher inbreeding coefficient than in other Central Chinese breeds. Additionally, WD cattle recently faced an extinction crisis in which the herd was reduced to six thousand head in 2011.
It remains unclear how genetic changes gradually accumulated in the genome and how these diverging breeds evolved. Reduced gene flow may speed up speciation, leading to large regions around divergently selected trait loci that are protected from recombination between genomic regions containing different locally adapted alleles (Via and West, 2008). The speciation process of restricted recombination caused the retention of these locally adapted variants and linked neutral variants in the distinct population genomes via extensive hitchhiking, probably causing the original ancestor to evolve into these two breeds that are able to adapt to different environments.
The greatest differences in the physical phenotypes of these two breeds are their body size and adaptation to different environments. There are many genes that have been reported to be associated with the economic traits of cattle (Li et al., 2010; Pausch et al., 2017; Raza et al., 2018; Sun et al., 2018; Cheng et al., 2020). In particular, genes such as MYLK4 (Zheng et al., 2019), CRTC3 (Wu et al., 2018), LEPR (Guo et al., 2008), and LHX4 (Ren et al., 2010) have been reported to be associated with the growth traits of Chinese cattle breeds. As body size is one the most important economic/growth traits for the modern breeding industry, it is valuable to investigate the genetic dynamics of this trait.
In this study, variant association analysis indicated the existence of moderate genomic inflation between these two cattle breeds, and no associated SNPs were detected. Therefore, based on advantages in detecting associations such as higher efficiency (greater detection power, and lower criteria for multiple testing) (Chen and Wang, 2019), a gene-based association strategy was used to identify physical traits associated with specific genes. Three gene-based methods (SKAT, SKATO, and Burden) showed there was moderate genomic inflation and that no gene could pass the threshold of the Bonferroni correction P-value. However, the chi-squared tests showed no evidence of genomic inflation at the gene level (λgc = 0.1172), and a novel gene, CD48, was found to be significantly associated with the differences in the traits of these two breeds, even after Bonferroni correction. Additionally, three genes with a suggestive association were found: ESPNL (espin-like), PRCP (prolylcarboxypeptidase) and F2R (coagulation factor II thrombin receptor) (P_Chi < 1e-4).
The encoded CD48 protein is located on the surface of lymphocytes and other immune cells and participates in activation and differentiation pathways in these cells. PRCP encodes a member of the peptidase S28 family of serine exopeptidases, which can cleave C-terminal amino acids linked to proline in peptides such as angiotensin II and has been reported to be associated with leanness, cell proliferation and blood pressure regulation. One study showed that PRCP depletion reduces cell proliferation (Adams et al., 2013). F2R is an important member of the GPCR family that is highly expressed in osteoclasts. Another study demonstrated that F2r responds to RANKL (receptor activator of nuclear factor-κB ligand) stimulation to attenuate osteoclastogenesis by inhibiting both the F2r-Akt and F2r-NFκB signaling pathways, which leads to a reduction in the expression of osteoclast genes (Zhang et al., 2020).
Differential gene expression analysis identified 86 genes that were upregulated in DB cattle compared to WD cattle (Supplementary Table 2). Unexpectedly, more than 50 genes were related to the ribosomal unit and endoplasmic reticulum. Ribosomal proteins are essential for protein translation, and specific ribosomal proteins have been shown to play an essential role in tumorigenesis, immune signaling, and development. The targeted deletion of RPL29 (ribosomal protein L29) in mice showed that animals are viable but are up to 50% smaller than their control littermates at weaning age (Kirn-Safran et al., 2007). On the other hand, 27 genes were upregulated in WD cattle compared to DB cattle, most of which were related to immunoreactions, transcription factors, signal transduction and metabolism. Interestingly, one upregulated gene (TLN1, P = 8.13E-6) was involved in the spreading and migration of osteoclasts, which may be correlated with the LOF enrichment results. Among these genes, the transforming growth factor gene (TGFB1) was significantly upregulated in WD cattle (P = 8.65E-7). TGFB1 has been reported to be a key regulator of feed efficiency in beef cattle, and motif discovery analysis showed that most genes co-expressed with TGFB1 were enriched for binding sites related to master regulators of muscle differentiation (Alexandre et al., 2019). Furthermore, a large meta-analysis of human data including 46 genome-wide association (GWA) studies of 133,635 individuals, also highlighted a role of TGFB2 in the TGF-beta signaling pathway in regulating human height (Lango Allen et al., 2010). Searches in the Online Mendelian Inheritance in Man (OMIM) database by using the search keywords “short stature,” “overgrowth,” “skeletal dysplasia,” and “brachydactyly” revealed gene records of TGFBR1 and TGFBR2 (receptors of TGFB1 and TGFB2) and many ribosomal protein genes, consistent with our data.
In summary, we performed a whole-transcriptome study for WD and DB cattle. By using publicly available global cattle data, phylogenetic and PCA analyses indicated that these Central Chinese cattle breeds formed exclusive genetic groups even within breeds of hybrid Bos. taurus × Bos. indicus cattle, and these results are consistent with the results of previous studies (Yu et al., 1999; Mei et al., 2017; Chen et al., 2018), which confirmed that these two local Chinese breeds originated from hybridization between Bos. taurus and Bos. indicus. LOF variant enrichment analysis identified one novel gene, CD48, and three genes with a suggestive association. Among these genes, F2R responds to RANKL stimulation to attenuate osteoclastogenesis, and the PRCP gene has been reported to be involved in cell growth and angiogenesis. Differential gene expression analysis showed that many ribosomal protein genes are upregulated in DB cattle, and many transcription factors and metabolism-related genes are upregulated in WD cattle. All of these genes are connected with the economic trait of body size. However, further large-scale studies are needed to understand the roles of these genes and to identify robust phenotype-associated genes/mutations with a large effect size. Our results indicate that whole-genome RNA_seq experiments are an efficient approach for investigating population genetic diversity and the genetic architecture of complex traits in mammals.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The animal study was reviewed and approved by the Anhui Agricultural University.
X-DZ carried out the data, statistical, and other analyses and wrote the original draft. JC carried out the original investigation and design of the project, sample collection, and project administration. W-JQ, NB, X-JS, and M-TZ carried out the investigation and formal analysis. H-QC carried out the conceptualization, funding acquisition, methodology, project administration, supervision, and writing, review, and editing of the manuscript with input from all other authors. All authors contributed to the article and approved the submitted version.
This study was financially supported by the National Natural Science Foundation of China (NSFC) (grant number 31272402).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Daming Agriculture and Animal Technology Development Co., Ltd., and the Anhui Wanjia Modern Agriculture Co., Ltd., for sample collection from WD and DB cattle. We are also grateful to Prof. Martin F. Bachmann for his editorial help in the preparation of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.562855/full#supplementary-material
Supplementary Figure 1 | ADMIXTURE cross-validation plot. ADMIXTURE cross-validation plot for the whole-genome autosome dataset of 11 worldwide cattle breeds set at K = 2 through 7.
Supplementary Figure 2 | Manhattan plots of P-values based on the single-variant association test. Manhattan plots of -log10 (p-values) for two breeds based on the single-variant association test. The red horizontal line indicates the genome-wide significance level from Bonferroni correction [-log10 (5e-8)] for the single-marker analysis, and the blue horizontal line indicates the suggestive significance level [-log10 (1e-5)].
Supplementary Figure 3 | Quantile-quantile plots of P-values. The observed negative logarithms of the P-values obtained using three types of gene-based methods for the two breeds are plotted against their expected values under the null hypothesis of no association.
Supplementary Figure 4 | Volcano plot of DEGs for the two cattle breeds. DEG expression profiling [with transcripts per kilo-base of exon model per million mapped reads (TPM) values] was constructed for the two breeds. Red plots represent genes that are significantly upregulated, and green plots represent genes that are significantly downregulated.
Adams, G. N., Stavrou, E. X., Fang, C., Merkulova, A., Alaiti, M. A., Nakajima, K., et al. (2013). Prolylcarboxypeptidase promotes angiogenesis and vascular repair. Blood 122, 1522–1531. doi: 10.1182/blood-2012-10-460360
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Alexandre, P. A., Naval-Sanchez, M., Porto-Neto, L. R., Ferraz, J. B. S., Reverter, A., and Fukumasu, H. (2019). Systems Biology Reveals NR2F6 and TGFB1 as Key Regulators of Feed Efficiency in Beef Cattle. Front. Genet. 10:230. doi: 10.3389/fgene.2019.00230
Artomov, M., Stratigos, A. J., Kim, I., Kumar, R., Lauss, M., Reddy, B. Y., et al. (2017). Rare Variant, Gene-Based Association Study of Hereditary Melanoma Using Whole-Exome Sequencing. J Natl. Cancer Inst. 109:djx083 doi: 10.1093/jnci/djx083
Beja-Pereira, A., Caramelli, D., Lalueza-Fox, C., Vernesi, C., Ferrand, N., Casoli, A., et al. (2006). The origin of European cattle: evidence from modern and ancient DNA. Proc. Natl. Acad. Sci. U S A. 103, 8113–8118. doi: 10.1073/pnas.0509210103
Biscarini, F., Nicolazzi, E. L., Stella, A., Boettcher, P. J., and Gandini, G. (2015). Challenges and opportunities in genetic improvement of local livestock breeds. Front. Genet. 6:33. doi: 10.3389/fgene.2015.00033
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Burri, R., Nater, A., Kawakami, T., Mugal, C. F., Olason, P. I., Smeds, L., et al. (2015). Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Res. 25, 1656–1665. doi: 10.1101/gr.196485.115
Canavez, F. C., Luche, D. D., Stothard, P., Leite, K. R., Sousa-Canavez, J. M., Plastow, G., et al. (2012). Genome sequence and assembly of Bos indicus. J. Hered. 103, 342–348. doi: 10.1093/jhered/esr153
Chen, N., Cai, Y., Chen, Q., Li, R., Wang, K., Huang, Y., et al. (2018). Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 9:2337. doi: 10.1038/s41467-018-04737-0
Chen, Z., and Wang, K. (2019). Gene-based sequential burden association test. Stat. Med. 38, 2353–2363. doi: 10.1002/sim.8111
Cheng, J., Qin, W. J., Balsaia, N., Shang, X. J., Zhang, M. T., and Chen, H. Q. (2020). Transcriptional activity of FIGLA, NEUROG2, and EGR1 transcription factors associated with polymorphisms in the proximal regulatory region of GPR54 gene in cattle. Anim. Reprod. Sci. 218:106506. doi: 10.1016/j.anireprosci.2020.106506
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Gibbs, R. A., Taylor, J. F., Van Tassell, C. P., Barendse, W., Eversole, K. A., Gill, C. A., et al. (2009). Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324, 528–532. doi: 10.1126/science.1167936
Guo, Y., Chen, H., Lan, X., Zhang, B., Pan, C., Zhang, L., et al. (2008). Novel SNPs of the bovine LEPR gene and their association with growth traits. Biochem. Genet. 46, 828–834. doi: 10.1007/s10528-008-9197-z
He, K. Y., Li, X., Kelly, T. N., Liang, J., Cade, B. E., Assimes, T. L., et al. (2019). Leveraging linkage evidence to identify low-frequency and rare variants on 16p13 associated with blood pressure using TOPMed whole genome sequencing data. Hum. Genet. 138, 199–210. doi: 10.1007/s00439-019-01975-0
Hiendleder, S., Lewalski, H., and Janke, A. (2008). Complete mitochondrial genomes of Bos taurus and Bos indicus provide new insights into intra-species variation, taxonomy and domestication. Cytogenet. Genome Res. 120, 150–156. doi: 10.1159/000118756
Kirn-Safran, C. B., Oristian, D. S., Focht, R. J., Parker, S. G., Vivian, J. L., and Carson, D. D. (2007). Global growth deficiencies in mice lacking the ribosomal protein HIP/RPL29. Dev. Dyn. 236, 447–460. doi: 10.1002/dvdy.21046
Lai, S. J., Liu, Y. P., Liu, Y. X., Li, X. W., and Yao, Y. G. (2006). Genetic diversity and origin of Chinese cattle revealed by mtDNA D-loop sequence variation. Mol. Phylogenet. Evol. 38, 146–154. doi: 10.1016/j.ympev.2005.06.013
Lango Allen, H., Estrada, K., Lettre, G., Berndt, S. I., Weedon, M. N., Rivadeneira, F., et al. (2010). Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467, 832–838. doi: 10.1038/nature09410
Lei, C. Z., Chen, H., Zhang, H. C., Cai, X., Liu, R. Y., Luo, L. Y., et al. (2006). Origin and phylogeographical structure of Chinese cattle. Anim. Genet. 37, 579–582. doi: 10.1111/j.1365-2052.2006.01524.x
Leroy, G., Baumung, R., Boettcher, P., Scherf, B., and Hoffmann, I. (2016). Review: Sustainability of crossbreeding in developing countries; definitely not like crossing a meadow. Animal 10, 262–273. doi: 10.1017/S175173111500213X
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, F., Chen, H., Lei, C. Z., Ren, G., Wang, J., Li, Z. J., et al. (2010). Novel SNPs of the bovine GAD1/gad67 gene and their association with growth traits in three native Chinese cattle breeds. Mol. Biol. Rep. 37, 501–505. doi: 10.1007/s11033-009-9699-8
Loftus, R. T., Ertugrul, O., Harba, A. H., El-Barody, M. A., MacHugh, D. E., Park, S. D., et al. (1999). A microsatellite survey of cattle from a centre of origin: the Near East. Mol. Ecol. 8, 2015–2022. doi: 10.1046/j.1365-294x.1999.00805.x
Loftus, R. T., MacHugh, D. E., Bradley, D. G., Sharp, P. M., and Cunningham, P. (1994). Evidence for two independent domestications of cattle. Proc. Natl. Acad. Sci. U S A. 91, 2757–2761. doi: 10.1073/pnas.91.7.2757
Lu, X., Peloso, G. M., Liu, D. J., Wu, Y., Zhang, H., Zhou, W., et al. (2017). Exome chip meta-analysis identifies novel loci and East Asian-specific coding variants that contribute to lipid levels and coronary artery disease. Nat. Genet. 49, 1722–1730. doi: 10.1038/ng.3978
Mei, C., Wang, H., Liao, Q., Wang, L., Cheng, G., Zhao, C., et al. (2017). Genetic architecture and selection of Chinese cattle revealed by whole genome resequencing. Mol. Biol. Evol. 35, 688–699. doi: 10.1093/molbev/msx322
Orozco-terWengel, P., Barbato, M., Nicolazzi, E., Biscarini, F., Milanesi, M., Davies, W., et al. (2015). Revisiting demographic processes in cattle with genome-wide population genetic analysis. Front. Genet. 6:191. doi: 10.3389/fgene.2015.00191
Pariset, L., Mariotti, M., Nardone, A., Soysal, M. I., Ozkan, E., Williams, J. L., et al. (2010). Relationships between Podolic cattle breeds assessed by single nucleotide polymorphisms (SNPs) genotyping. J. Anim. Breed Genet. 127, 481–488. doi: 10.1111/j.1439-0388.2010.00868.x
Patterson, N., Price, A. L., and Reich, D. (2006). Population structure and eigenanalysis. PLoS Genet. 2:e190. doi: 10.1371/journal.pgen.0020190
Pausch, H., Emmerling, R., Gredler-Grandl, B., Fries, R., Daetwyler, H. D., and Goddard, M. E. (2017). Meta-analysis of sequence-based association studies across three cattle breeds reveals 25 QTL for fat and protein percentages in milk at nucleotide resolution. BMC Genomics 18:853. doi: 10.1186/s12864-017-4263-8
Porto-Neto, L. R., Sonstegard, T. S., Liu, G. E., Bickhart, D. M., Da Silva, M. V., Machado, M. A., et al. (2013). Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genomics 14:876. doi: 10.1186/1471-2164-14-876
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Raza, S. H. A., Gui, L., Khan, R., Schreurs, N. M., Xiaoyu, W., Wu, S., et al. (2018). Association between FASN gene polymorphisms ultrasound carcass traits and intramuscular fat in Qinchuan cattle. Gene 645, 55–59. doi: 10.1016/j.gene.2017.12.034
Ren, G., Chen, H., Zhang, L. Z., Lan, X. Y., Wei, T. B., Li, M. J., et al. (2010). A coding SNP of LHX4 gene is associated with body weight and body length in bovine. Mol. Biol. Rep. 37, 417–422. doi: 10.1007/s11033-009-9486-6
Rettelbach, A., Nater, A., and Ellegren, H. (2019). How Linked Selection Shapes the Diversity Landscape in Ficedula Flycatchers. Genetics 212, 277–285. doi: 10.1534/genetics.119.301991
Sun, Y., Liu, K., Huang, Y., Lan, X., and Chen, H. (2018). Differential expression of FOXO1 during development and myoblast differentiation of Qinchuan cattle and its association analysis with growth traits. Sci. China Life Sci. 61, 826–835. doi: 10.1007/s11427-017-9205-1
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43
Via, S., and West, J. (2008). The genetic mosaic suggests a new role for hitchhiking in ecological speciation. Mol. Ecol. 17, 4334–4345. doi: 10.1111/j.1365-294X.2008.03921.x
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucl. Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Wen, L., Zhu, C., Zhu, Z., Yang, C., Zheng, X., Liu, L., et al. (2018). Exome-wide association study identifies four novel loci for systemic lupus erythematosus in Han Chinese population. Ann. Rheum. Dis. 77:417. doi: 10.1136/annrheumdis-2017-211823
Wu, M. C., Kraft, P., Epstein, M. P., Taylor, D. M., Chanock, S. J., Hunter, D. J., et al. (2010). Powerful SNP-set analysis for case-control genome-wide association studies. Am. J. Hum. Genet. 86, 929–942. doi: 10.1016/j.ajhg.2010.05.002
Wu, M. C., Lee, S., Cai, T., Li, Y., Boehnke, M., and Lin, X. (2011). Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93. doi: 10.1016/j.ajhg.2011.05.029
Wu, S., Ning, Y., Raza, S. H. A., Zhang, C., Zhang, L., Cheng, G., et al. (2018). Genetic variants and haplotype combination in the bovine CRTC3 affected conformation traits in two Chinese native cattle breeds (Bos Taurus). Genomics 111, 1736–1744. doi: 10.1016/j.ygeno.2018.11.028
Yang, C., Chen, M., Huang, H., Li, X., Qian, D., Hong, X., et al. (2020). Exome-Wide Rare Loss-of-Function Variant Enrichment Study of 21,347 Han Chinese Individuals Identifies Four Susceptibility Genes for Psoriasis. J. Invest. Dermatol. 140, 799–805e791.
Yang, J., Weedon, M. N., Purcell, S., Lettre, G., Estrada, K., Willer, C. J., et al. (2011). Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet. 19, 807–812. doi: 10.1038/ejhg.2011.39
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. doi: 10.1089/omi.2011.0118
Yu, Y., Nie, L., He, Z. Q., Wen, J. K., Jian, C. S., and Zhang, Y. P. (1999). Mitochondrial DNA variation in cattle of south China: origin and introgression. Anim. Genet. 30, 245–250. doi: 10.1046/j.1365-2052.1999.00483.x
Zhang, Y., Wang, H., Zhu, G., Qian, A., and Chen, W. (2020). F2r negatively regulates osteoclastogenesis through inhibiting the Akt and NFkappaB signaling pathways. Int. J. Biol. Sci. 16, 1629–1639. doi: 10.7150/ijbs.41867
Zheng, L., Zhang, G. M., Dong, Y. P., Wen, Y. F., Dong, D., Lei, C. Z., et al. (2019). Genetic Variant of MYLK4 Gene and its Association with Growth Traits in Chinese Cattle. Anim. Biotechnol. 30, 30–35. doi: 10.1080/10495398.2018.1426594
Keywords: whole transcriptome analysis, enrichment analysis, agricultural traits, taxonomic status, native cattle
Citation: Zheng X-D, Cheng J, Qin W-J, Balsai N, Shang X-J, Zhang M-T and Chen H-Q (2020) Whole Transcriptome Analysis Identifies the Taxonomic Status of a New Chinese Native Cattle Breed and Reveals Genes Related to Body Size. Front. Genet. 11:562855. doi: 10.3389/fgene.2020.562855
Received: 16 May 2020; Accepted: 11 September 2020;
Published: 03 November 2020.
Edited by:
George E. Liu, United States Department of Agriculture, United StatesReviewed by:
Lingyang Xu, Chinese Academy of Agricultural Sciences, ChinaCopyright © 2020 Zheng, Cheng, Qin, Balsai, Shang, Zhang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong-Quan Chen, Y2hxY2hxQGFoYXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.