
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 19 November 2020
Sec. Livestock Genomics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.533611
 Rui Su1,2,3,4,5†
Rui Su1,2,3,4,5† Xian Qiao1†
Xian Qiao1† Yun Gao6
Yun Gao6 Xiaokai Li1Wei Jiang1Wei Chen7,8Yixing Fan1Bingwu Zheng9Yanjun Zhang1,2,3,4Zhihong Liu1,2,3,4Ruijun Wang1,2,3,4Zhiying Wang1,2,3,4Zhixin Wang1,2,3,4
Xiaokai Li1Wei Jiang1Wei Chen7,8Yixing Fan1Bingwu Zheng9Yanjun Zhang1,2,3,4Zhihong Liu1,2,3,4Ruijun Wang1,2,3,4Zhiying Wang1,2,3,4Zhixin Wang1,2,3,4 Wenting Wan10
Wenting Wan10 Yang Dong7,8*Jinquan Li1,2,3,4*
Yang Dong7,8*Jinquan Li1,2,3,4*Mouflon (Ovis orientalis) with its huge and beautiful horns is considered as one of the ancestors of domesticated sheep. The European mouflon (Ovis orientalis musimon) is in the Asiatic mouflon (O. orientalis) clade. In order to provide novel genome information for mouflon, moreover promote genetic analysis of genus Ovis both domestic and wild, we propose to sequence the mouflon genome. We assembled the highly heterozygous mouflon genome based on Illumina HiSeq platform using the next-generation sequencing technology. Finally, the draft genome we accessed approximately 2.69 Gb (42.15% GC), while N50 sizes of contig and scaffold are 110.1 kb and 10.4 Mb, respectively. The contiguity of this assembly is obviously better than earlier versions. Further analyses predicted 20,814 protein-coding genes in the mouflon genome and 12,390 shared gene families among bovine species. It is estimated that the divergence time between O. orientalis musimon and Ovis aries was 7.6 million years ago. The draft mouflon genome assembly will provide data support and theoretical basis for various investigations of the genus Ovis species in future.
Mouflon (Ovis orientalis) is a subspecies of the wild sheep with beautiful curved horns and red-brown coat color (Macdonald, 2001). It is considered to be one of the two ancestors for all modern domesticated sheep breeds (Hiendleder et al., 1998, 2002). This animal has been found in many different countries in Central Asia from Turkey in the west to Pakistan in the east (Hodges, 1997). Its usual habitat is in the mountainous areas up to 3,000 meters above sea level, but mouflon has also been introduced into European forested areas (IUCN Red List of Threatened Species, January, 2008, http://www.iucnredlist.org). Some previous studies considered the European mouflon (Ovis orientalis musimon) as a subspecies of Asiatic mouflon (O. orientalis; Corbet, 1986; Patterson et al., 1993; Rezaei et al., 2009). The survival of wild mouflon is threatened by habitat loss as a result of land reclamation, deforestation, mining activities, and expansion of human settlements (Ptak et al., 2002). It is also threatened by recreational hunters because of the highly prized horns (Garel et al., 2007). In addition, interbreeding with domesticated sheep resulted in the decreased number of genetically pure mouflon in the wild. Due to these factors, certain wild populations of the mouflon are listed as vulnerable by IUCN. This study reports a draft genome assembly of mouflon, and it will lay the ground work for further gene function annotating of the mouflon genome and revealing its evolutionary status, and comparative genomics, as well as promote the conservation of the mouflon and the breeding of genus Ovis in the future.
A total of 780.6 Gb raw data were generated (Supplementary Table 1) from Illumina HiSeq 4000 platform (Illumina, CA, United States). The average GC content is 45.35%. In the quality control step, we filtered out low quality and repetitive data, and finally got 660.7 Gb of clean data for subsequent analysis (Supplementary Tables 1, 2).
The analysis result of all 21-mer sequences from quality-filtered reads was calculated and explained as a frequency graph (Supplementary Figure 1). Two distinct peaks were observed at the depth of 49× and 97×, respectively. The first peak indicated that the mouflon genome may have high heterozygosity. The heterozygosity ratio is 0.42%, belonging to the high heterozygosity ratio genome calculated by GCE (Liu et al., 2013). The repeat ratio is 41.66%. Estimation result of mouflon genome size was 2.87 Gb (Supplementary Table 3). The sequencing depth of clean data is about 230× genome coverage. Based on this result, we used Platanus to assemble this genome in the next step.
Platanus (v. 1.2.1; Kajitani et al., 2014) were used to assemble HiSeq reads. Both paired-end (short-insert 244 bp∼700 bp) and mate pair (long-insert 1 kb∼15 kb) libraries were constructed according to the standard manufacturer’s instructions from Illumina (San Diego, CA, United States). Final draft mouflon genome of 2.69 Gb was assembled (42.15% GC), with contig and scaffold N50 size of 110,147 bp and 10,434,335 bp, respectively, (Table 1). BUSCO analysis identified 95.8% complete and 1.9% fragmented of 2,586 expected genes, respectively, (Supplementary Table 4). To assess base-level accuracy, integrity, and continuity of the genome assembly, Illumina reads from high-quality short-insert libraries were mapped to the assembled mouflon genome (Supplementary Table 5). The result showed that the total mapping ratio of all the 13 short-insert size libraries are up to 99.30%, indicating that our assembly results are of high quality.
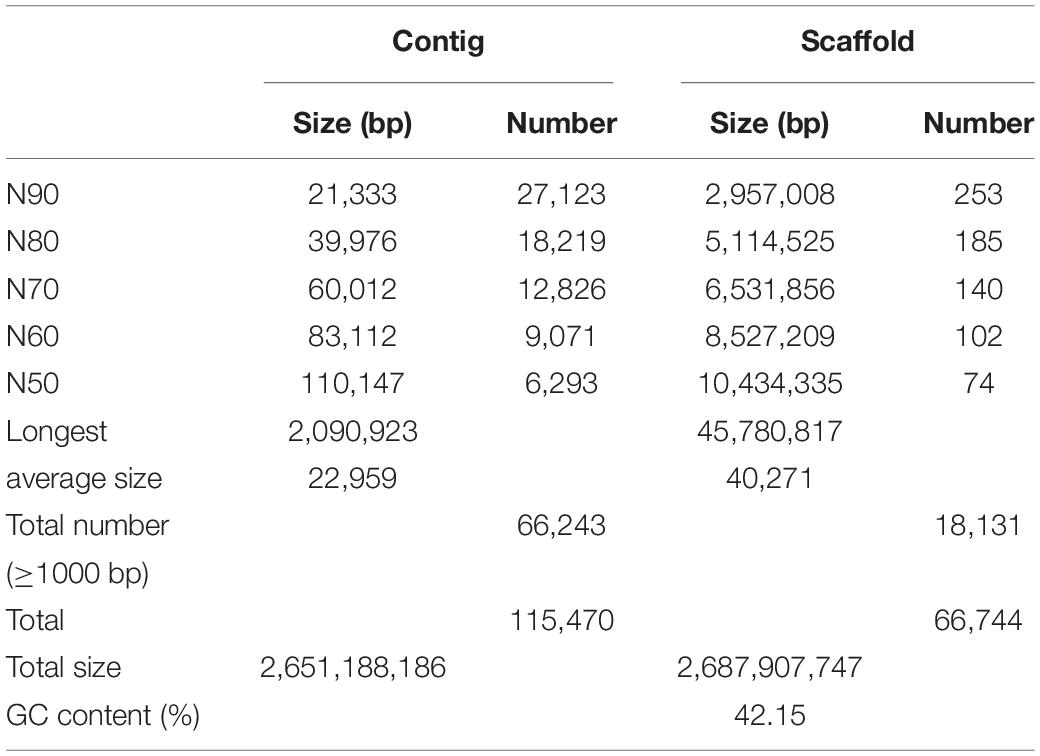
Table 1. Statistics summary of the Platanus assemble results.
Tandem Repeats Finder, de novo prediction approaches and homology to Repbase sequences were combined to annotate repetitive sequences and TEs of mouflon (Supplementary Table 6 and Supplementary Figures 2, 3). Comprehensive analysis results showed that repeated sequences accounted for 45.90% of the mouflon genome assembly and the total length was about 1245.6 Mb (Supplementary Table 7).
Multiple gene prediction methods were used to annotate protein-coding genes in the mouflon genome, including homology-based and de novo predictions. The analyses showed that 20,814 protein-coding genes were annotated in the mouflon genome with an average coding DNA sequence length of 1642 bp (Table 2 and Supplementary Figure 4). The final non-coding RNA annotation results include 482 miRNAs, 535 tRNAs, 114 rRNAs, and 1,428 snRNAs (Supplementary Table 8).
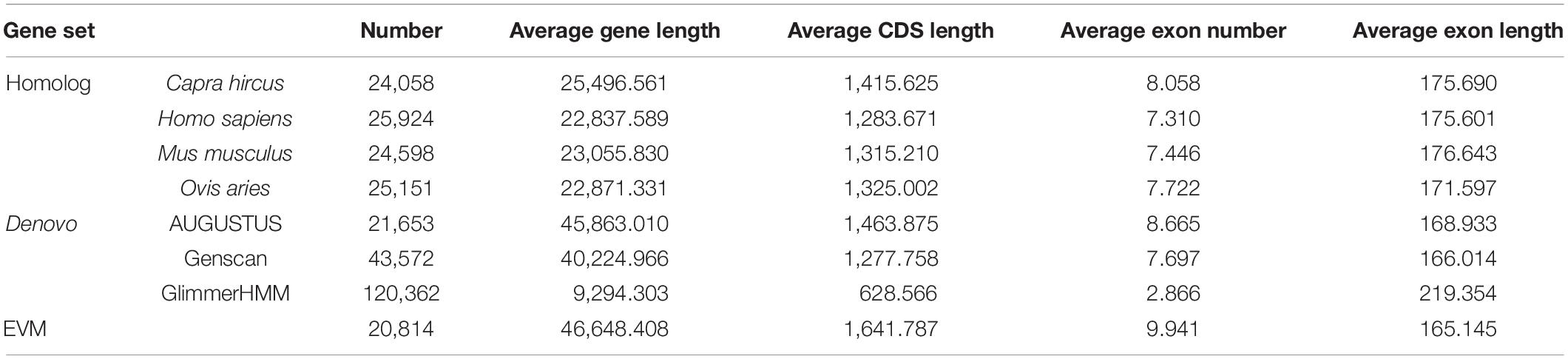
Table 2. General statistics of predicted protein-coding genes.
In order to discern and predict the amount of potential orthologous gene families, gene sequences of O. orientalis musimon, Bison (GCA_000754665.1 Bison_UMD1.0), Bos mutus (GCA_000298355.1 BosGru_v2.0), Bubalus (GCA_000471725.1 UMD_CASPUR_WB_2.0), Camelus ferus (GCA_000311805.1 CB1), Capra hircus (GCA_001704415.1 ARS1), Odocoileus virginianus (GCA_002102435.1 Ovir.te_1.0), Ovis aries (GCA_000298735.2 Oar_v4.0), Pantholops hodgsonii (GCA_000400835.1 PHO1.0) were downloaded from National Center for Biotechnology Information (NCBI)1 and Homo sapiens (GCA_000001405.22 GRCh38.p7) were downloaded from Ensembl Genome Browser2. Among the total of 13,987 mouflon gene families, 140 (1%) appear to be lineage specific. There are 12,390 (88.6%) gene families shared among O. orientalis musimon, Bison, Bubalus, O. aries, and C. hircus. The total of 13,400 gene families were shared between O. orientalis musimon and O. aries as well as 350 gene families were specific to both (Supplementary Table 9 and Supplementary Figures 5, 6). Synteny analysis results show that the relationship of variations between mouflon and sheep genome (Supplementary Figure 7). The left part from outer circle to the inner circle, represent the mouflon scaffold, gene density and repeat sequence density at the corresponding position, respectively, while the right part represents chromosome of sheep, as well as the line between them represents the synteny relationship.
The result of phylogenetic tree showed that O. orientalis musimon was more closely related to C. hircus than P. hodgsonii in the Caprinae family (Supplementary Figure 8a). It is estimated that the divergence time between O. orientalis musimon and O. aries was 7.6 million years ago (Supplementary Figure 8b).
We identified 177 expanded gene families and 2047 contracted gene families in the mouflon genome, which is special among those species in Caprinae (Supplementary Figure 9). The expanded gene families were enriched in 35 GO terms including binding, Cellular Component, etc. (Supplementary Figure 10). We further used the free ratio model to calculate the average Ka/Ks values and the branch-site likelihood ratio test to identify positively selected genes (PSGs). A total of 165 PSGs were identified in the mouflon genome (Supplementary Table 10). The result of KEGG pathway analysis showed that 78 of the PSGs were enriched in 164 pathways, and 7 of these pathways were significantly enriched (P < 0.05; Supplementary Table 11).
According to the results of Pairwise Sequentially Markovian Coalescent (PSMC) analysis, the effective population size (Ne) of the mouflon shows two peak at ∼1 millions of years ago (Mya) and ∼120 thousand years ago (Kya), followed by 3 distinct declines, and 2 distinct increases.
According to the result of KEGG pathway analysis, the PSGs can be divided into 2 groups. The first group is about nutrition and metabolism (Protein digestion and absorption, AGE-RAGE signaling pathway in diabetic complications and Caffeine metabolism). It’s very interesting that we found xanthine dehydrogenase (XDH) enriched in the Caffeine metabolism pathway is related to the milk production of sheep and goat, especially affecting the formation of Lipid droplets (Suárez-Vega et al., 2016; Toral et al., 2016). The second group is about signal transduction (cGMP – PKG signaling pathway, Oxytocin signaling pathway, ECM-receptor interaction and Renin secretion). It is worthwhile to note that some important genes are involved in different pathways at the same time, which need more attention in future studies. For example, nuclear factor of activated T-cells 5 (NFAT5) is important in both immune response and high osmotic stress pathways (Bounedjah et al., 2012). Calcium voltage-gated channel subunit alpha1 F (CACNA1F) and calmodulin 2 (CALM2) were involved in three signal transduction pathways at the same time. CACNA1F encodes a multipass transmembrane protein which acts as an alpha-1 subunit of the voltage-dependent calcium channel, mediating the influx of calcium ions into the cell (Fisher et al., 1997; Strom et al., 1998). Mutations in CACNA1F are reported to be associated with type 2 congenital stationary night blindness (CSNB2; Men et al., 2017), cone-rod dystrophy 3 (CORDX3; Hauke et al., 2013), Aland island eye disease (AIED; Weleber et al., 1989), and X-linked retinitis pigmentosa (XLRP; Zhou et al., 2014). CALM2 is a member of the calmodulin gene family; increased CALM2 mRNA levels may reflect an important role for calmodulin in expansion-induced fetal lung growth of sheep (Gillett et al., 2002). We also found some PSGs related to Lipid metabolism (APOL3, ADARB2, and AP2B1). Apolipoprotein L3 (APOL3) belongs to the apolipoprotein L gene family and has the function of biochemical metabolism by carrying lipids (Duchateau et al., 2001). Adenosine deaminase, RNA specific B2 (ADARB2) related to longevity is associated with metabolic disorders like the abdominal circumference, body mass index, serum triglyceride level, and serum adiponectin level (Oguro et al., 2012). Adaptor related protein complex 2 subunit beta 1 (AP2B1) was reported to be associated with milk yield and fat yield traits (Kolbehdari et al., 2009; Chen et al., 2018), as well as meat quality traits (Piórkowska et al., 2018). To a certain extent, these PSGs reflect the adaptation process of wild sheep domestication to environmental changes.
Population contraction started from 1 Mya, and reached the bottleneck at ∼400 Kya. After 1 Mya, the longer glacial cycle and the cold climate might affect the reduction of the populations (Muller and Macdonald, 1997). This bottleneck of the population may be related to the Penultimate Glaciation (Zheng et al., 2002). As the glaciers receded, the temperature gradually increased and the population began to expand and reached the second flourishing period in 120 Kya which may coincide with warm Eemian interglacial period that genetic diversity of species is enriched (Pilot et al., 2006; Hailer et al., 2012). From ∼120 Kya to the present the Ne has continued to decline, with a short plateau period between ∼400 Kya and ∼900 Kya. The most recent decline involved a decrease of Ne at least a 9-fold, and it happened about ∼40 Kya (Supplementary Figure 11). This process corresponded to the abrupt change of climate after the last glaciations (Cooper et al., 2015).
It is well known that N50 size of contig and scaffold are important indicators of assembly integrity and quality. Compared with the earlier version of mouflon genome assembly Oori1 with contig and scaffold N50 size of 39,721 bp and 2,217,029 bp, respectively3, our result shows better contiguity and quality. Although this assembly vision is only based on scaffold level, the N50 size is also larger than the newly published Marco Polo Sheep (Ovis ammon polii) genome assembly with contig and scaffold N50 size of 30,772 bp and 5,492,388 bp, respectively, (Yang et al., 2017; Supplementary Table 12). The significant improvement of contig has some effects on better gene annotation and the improvement of scaffold is very helpful to the improvement of genome integrity, which revealed that the new version of mouflon genome we assembled was with high quality. In the future we still need to update our sequence technology to upgrade mouflon genome assembly to chromosome level.
In conclusion, the mouflon genome sequencing, assembly, annotation, and evolutionary analysis were reported in this study. This genome assembly will provide valuable resources for the further genetic research of mouflon, and might establish a theoretical basis for the conservation of genetic resources and sheep evolution in the future.
Genomic DNA was extracted from the venous blood of a single (sex unknown) European mouflon (Daqingshan Wild Animal Park, Hohhot, Inner Mongolia, China) with the Tiangen Blood Genomic DNA Extraction Kit (Tiangenbiotech, Beijing). Paired-end libraries with short-insert sizes from 244 bp to 700 bp were constructed with NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, United States), while mate pair libraries with long-insert sizes from 1 kb to 15 kb were constructed with Illumina Nextera Mate Pair Library Preparation Kit (Illumina, United States; Supplementary Table 5). Sample processing was according to the manufacturer’s instructions. Illumina HiSeq 4000 platform (Illumina, CA, United States) were used for sequencing with the PE-150 module (Quail et al., 2012). Then the correction steps were performed with SOAPec_v2.01 (Luo et al., 2012) using the parameters: −k 21, −i 450,000,000.
SOAPec_v2.01 was used for 21-mer frequency distribution analysis with quality-filtered reads from the Illumina platform. All 21-mer sequences were extracted from paired-end reads from short-insert size libraries (<1 kb) that passed quality control, and the frequency of each 21-mer was calculated and plotted. The heterozygosity ratio of genome was calculated by GCE (Liu et al., 2013). The genome size was estimated by the formula Genome_Size = #_Total_Kmer/Expected_Kmer_Depth.
Both short-insert and long-insert libraries sequenced on Illumina HiSeq 4000 platform were used for hybrid de novo genome assembly. HiSeq reads were assembled using Platanus (v. 1.2.1; Kajitani et al., 2014) with parameters “−k 47 −s 10 −d 0.3 −u 0.1,” for contigs and “−s 47 −v 34 −l 2,” for scaffolds. The completeness of the final assembly were evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO; v.2.0; Simão et al., 2015) with the mammal gene set. To assess the assembly quality, BWA and SAMtools (Li et al., 2009) were used to map Illumina reads for short-insert size libraries with high quality onto the mouflon genome assembly.
Tandem Repeat Finder (v. 4.07b) was used to search the mouflon genome for tandem repeats (Benson, 1999) with default parameters. Transposable elements (TEs) were firstly predicted in the genomes by homology searches to known RepBase TE libraries (Jurka et al., 2005) using RepeatMasker (v. 3.3.0) and RepeatProteinMask (Chen, 2004) with default parameters. A de novo repeat library was constructed using RepeatScout. We obtained consensus sequences and classification information from each repeat family. RepeatScout consensus sequences were used as an input library in order to search for repetitive elements in the assembled genome using RepeatMasker with default parameters.
Protein-coding genes in the mouflon genome were annotate by multiple methods like homology-based predictions and de novo predictions. In homology-based prediction, proteins from C. hircus, H. sapiens, Mus musculus, O. aries were obtained from the NCBI, and mapped to the mouflon genomes using TBLASTN analysis with a cutoff E-value of 1e–5. Then, homologous regions defined by TBLASTN were fed into GeneWise (v. 2.2.0; Birney and Durbin, 2000) to obtain gene models for predicting the gene structure contained in each protein region. BLAST hits corresponding to reference proteins were concatenated by Solar (v. 0.9.6; Beijing Genomics Institute; Li et al., 2016). After removing low-quality records, the genomic sequences of each reference protein were extended upstream and downstream by 2000 bp aimed to represent the protein-coding region. For de novo prediction, AUGUSTUS (v. 2.5.5; Stanke et al., 2006), GENSCAN (v. 1.0; Cai et al., 2014), and glimmerHMM (v. 3.0.2; Majoros et al., 2004) were employed to predict coding genes. Evidence Modeler (released 25 June 2012) was used to combine the homology and de novo predicted gene sets and creat a comprehensive and non-redundant reference gene set.
For tRNA annotation step, tRNAscan-SE (v.1.3.1; Lowe and Eddy, 1997) was used with default parameters for eukaryotes. Homology-based rRNA was annotated by BLASTN which mapping rRNAs to the mouflon genome, with parameters of “E-value = 1e–5.” INFERNAL (v.1.1; Nawrocki et al., 2009) was used to predict miRNA and snRNA genes the with Rfam database (release 11.0; Gardner et al., 2011).
We use standard settings (BLASTP E-value < 1e–5) to apply the OrthoMCL (v.2.0.9) pipeline (Li et al., 2003) for the calculation of all-against-all similarities among O. orientalis musimon, Bison, B. mutus, Bubalus, C. ferus, C. hircus, O. virginianus, O. aries, P. hodgsonii, and H. sapiens, in order to discern and estimate the amount of potential orthologous gene families. Genes were then clustered into gene families using Hcluster_sg with consideration of proteins of out-group species (Homo sapiens). Gene sequences were all downloaded from NCBI (see text footnote 1, respectively). Synteny analysis were carried out to identified the variations between mouflon and sheep genome using the program LAST (LAST,RRID:SCR_006119; Kiełbasa et al., 2011).
Treefam was used in the gene family clustering analysis to identify all 1006 single-copy orthologous genes from the O. orientalis musimon, Bison, B. mutus, Bubalus, C. ferus, C. hircus, O. virginianus, O. aries, P. hodgsonii, and H. sapiens. Mrbayes was used to construct a phylogenetic tree. The species divergence time was estimated based on four-fold degenerate sites via Bayesian relaxed molecular clock (BRMC) approach using the program MULTIDIVTIME as implemented in the Thornian Time Traveler (T3) package4. The divergence time of each node was estimated by the PAML (PAML, RRID:SCR 014932) MCMCtree program v4.5 and calibrated against the timing of the divergence refer to http://www.timetree.org/.
According to the calculated phylogeny and the divergence time, we used CAFE (v. 2.1; De et al., 2006) to search gene families that under expansion and/or contraction in O. orientalis musimon, Bison, B. mutus, Bubalus, C. ferus, C. hircus, O. virginianus, O. aries, and P. hodgsonii. The parameters were “P = 0.05, number of threads = 4, number of random = 10000, and search for lambda (λ).” CAFE can estimate the global birth and death rate of gene families, infer the most likely gene family size at all internal nodes, identify gene families that have accelerated rates of gain and loss (quantified by a p-value) and identify which branches cause the p-value to be small for significant families. This method estimated the family sizes in the common ancestor, and then defined expansion and contraction by comparing the family size between the current species and the ancestor. We calculated the ratio of non-synonymous substitution rate over synonymous substitution rate (Ka/Ks), which can be used as an indicator of selective pressure acting between all the protein-coding genes. Comparisons of homologous genes with a high Ka/Ks ratio are usually said to be evolving under positive selection, so that we call these genes are PSGs. Ka/Ks were estimated using the software KaKs_Calculator, with the method of model averaging (GMYN). We choose the result with Ka/Ks more than 1(Fisher’ test, P < 0.05), and blast identity more than 80%.
In the final analysis, the demographic history of the mouflon were inferred with the PSMC model (Li and Durbin, 2011). PSMC modeling was done by bootstrapping approach, and the variance of the simulated results were estimated with sampling performed 100 times. We used SAMtools v0.1.19 to obtain Consensus sequences (Li et al., 2009) and the data was divided into non-overlapping 100-bp bins. The analysis parameters were −N25 −t15 −r5 −p “4 + 25 × 2 + 4 + 6.” We removed sites at which the root-mean-square mapping quality of reads covering the site was below 25, the inferred consensus quality was below 20, and read depth was either more than twice or less than one-third of the average read depth across the genome assembly. After filtering, we obtained a diploid consensus genome sequence for the PSMC analysis.
The sequencing reads of each sequencing library have been deposited at NCBI with the Project ID SRP106760. The SRA accession number for the sequenced individual (Sample ID: SAMN06909321) is SRS2175019.
All animal procedures in this paper were approved by the Inner Mongolia Agriculture University Animal Care and Use Committee in accordance with the National Animal Care Standard (GB 14925-2010). All experiments were performed in accordance with relevant guidelines and regulations. All efforts were made to minimize animal suffering.
RS, YD, and JL designed the study. YG assembled the genome and analyzed the data. XQ, RS, YG, and WC participated in manuscript revision. WJ did samples collection. BZ provided the samples. XQ, XL, YF, and WJ extracted DNA. WW constructed libraries. YZ, ZL, RW, ZYW, and ZXW conceived the overall study and revised the manuscript. All authors read and approved the final manuscript.
This work was financially supported by National Natural Science Foundation of China (31860637 and 31660639), Science and technology program of Inner Mongolia Autonomous Region (2019GG243), Agriculture Research System of China (CARS-39-06), Local science and technology development fund projects guided by the central government (2020ZY0007), and Inner Mongolia Agricultural University “Double First-Class” Discipline Innovation Team Construction Talent Cultivation Project (NDSC2018-01).
YG was employed by the company NOWBIO Technology Co. Ltd., Kunming, Yunnan, China.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.533611/full#supplementary-material
IUCN, International Union for Conservation of Nature and Natural Resources; BUSCO, Benchmarking Universal Single-Copy Orthologs; TEs, Transposable elements.
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Birney, E., and Durbin, R. (2000). Using GeneWise in the Drosophila annotation experiment. Genome Res. 10, 547–548. doi: 10.1101/gr.10.4.547
Bounedjah, O., Hamon, L., Savarin, P., Desforges, B., Curmi, P. A., and Pastré, D. (2012). Macromolecular crowding regulates assembly of mRNA stress granules after osmotic stress: new role for compatible osmolytes. J. Biol. Chem. 287, 2446–2458. doi: 10.1074/jbc.m111.292748
Cai, Y., González, J. V., Liu, Z., and Huang, T. (2014). Computational systems biology methods in molecular biology, chemistry biology, molecular biomedicine, and biopharmacy. Biomed. Res. Int. 2014:746814.
Chen, N. (2004). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 4:Unit4.10.
Chen, Z., Yao, Y., Ma, P., Wang, Q., and Pan, Y. (2018). Haplotype-based genome-wide association study identifies loci and candidate genes for milk yield in Holsteins. PLoS One 13:e0192695. doi: 10.1371/journal.pone.0192695
Cooper, A., Turney, C., Hughen, K. A., Brook, B. W., Mcdonald, H. G., and Bradshaw, C. J. (2015). PALEOECOLOGY. Abrupt warming events drove late pleistocene Holarctic megafaunal turnover. Science 349, 602–606. doi: 10.1126/science.aac4315
Corbet, B. G. (1986). The wild sheep of the world Paul Valdez wild sheep and goat International, Mesilla, New Mexico, $40. Oryx 20:58. doi: 10.1017/s0030605300025977
De, B. T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Duchateau, P. N., Pullinger, C. R., Cho, M. H., Eng, C., and Kane, J. P. (2001). Apolipoprotein L gene family: tissue-specific expression, splicing, promoter regions; discovery of a new gene. J. Lipid Res. 42, 620–630.
Fisher, S. E., Ciccodicola, A., Tanaka, K., Curci, A., Desicato, S., D’Urso, M., et al. (1997). Sequence-based exon prediction around the synaptophysin locus reveals a gene-rich area containing novel genes in human proximal Xp. Genomics 45:340. doi: 10.1006/geno.1997.4941
Gardner, P. P., Daub, J., Tate, J., Moore, B. L., Osuch, I. H., Griffiths-Jones, S., et al. (2011). Rfam: wikipedia, clans and the “decimal” release. Nucleic Acids Res. 39, 141–145.
Garel, M., Cugnasse, J. M., Maillard, D., Gaillard, J. M., Hewison, A. J. M., and Dubray, D. (2007). Selective harvesting and habitat loss produce long-term life history changes in a mouflon population. Ecol. Appl. 17, 1607–1618. doi: 10.1890/06-0898.1
Gillett, A. M., Wallace, M. J., Gillespie, M. T., and Hooper, S. B. (2002). Increased expansion of the lung stimulates calmodulin 2 expression in fetal sheep. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, 440–447.
Hailer, F., Kutschera, V. E., Hallström, B. M., Klassert, D., Fain, S. R., Leonard, J. A., et al. (2012). Nuclear genomic sequences reveal that polar bears are an old and distinct bear lineage. Science 336, 344–347. doi: 10.1126/science.1216424
Hauke, J., Schild, A., Neugebauer, A., Lappa, A., Fricke, J., Fauser, S., et al. (2013). A novel large in-frame deletion within the CACNA1F gene associates with a cone-rod dystrophy 3-like phenotype. PLoS One 8:e76414. doi: 10.1371/journal.pone.0076414
Hiendleder, S., Kaupe, B., Wassmuth, R., and Janke, A. (2002). Molecular analysis of wild and domestic sheep questions current nomenclature and provides evidence for domestication from two different subspecies. Proc. Biol. Sci. 269:893. doi: 10.1098/rspb.2002.1975
Hiendleder, S., Mainz, K., Plante, Y., and Lewalski, H. (1998). Analysis of mitochondrial DNA indicates that domestic sheep are derived from two different ancestral maternal sources: no evidence for contributions from urial and argali sheep. J. Hered. 89, 113–120. doi: 10.1093/jhered/89.2.113
Hodges, J. (1997). A world dictionary of livestock breeds, types and varieties. J. Anim. Breed. Genet. 49, 87–88. doi: 10.1016/s0301-6226(97)90045-2
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., and Walichiewicz, J. (2005). Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Kajitani, R., Toshimoto, K., Noguchi, H., Toyoda, A., Ogura, Y., Okuno, M., et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. doi: 10.1101/gr.170720.113
Kiełbasa, S. M., Wan, R., Sato, K., Horton, P., and Frith, M. C. (2011). Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493. doi: 10.1101/gr.113985.110
Kolbehdari, D., Wang, Z., Grant, J. R., Murdoch, B., Prasad, A., Xiu, Z., et al. (2009). A whole genome scan to map QTL for milk production traits and somatic cell score in Canadian Holstein bulls. J. Anim. Breed. Genet. 126, 216–227. doi: 10.1111/j.1439-0388.2008.00793.x
Li, H., and Durbin, R. (2011). Inference of human population history from individual whole-genome sequences. Nature 475:493. doi: 10.1038/nature10231
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, X., Ling, K., Zhang, J., Xie, Y., Wang, L., Yan, Y., et al. (2016). Improved hybridde novogenome assembly of domesticated apple (Malus x domestica). Gigascience 5:35.
Liu, B., Shi, Y., Yuan, J., Hu, X., Zhang, H., Li, N., et al. (2013). Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Q. Biol. 35, 62–67.
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Luo, R., Liu, B., and Xie, Y. (2012). Software and supporting material for SOAPdenovo2: an empirically improved memory-efficient short read de novo assembly. Gigascience 1:18.
Majoros, W. H., Pertea, M., and Salzberg, S. L. (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Men, C. J., Bujakowska, K. M., Comander, J., Place, E., Bedoukian, E. C., Zhu, X., et al. (2017). The importance of genetic testing as demonstrated by two cases ofCACNA1F-associated retinal generation misdiagnosed as LCA. Mol. Vis. 23, 695–706.
Muller, R. A., and Macdonald, G. J. (1997). Glacial cycles and astronomical forcing. Science 277, 215–218. doi: 10.1126/science.277.5323.215
Nawrocki, E. P., Kolbe, D. L., and Eddy, S. R. (2009). Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337. doi: 10.1093/bioinformatics/btp157
Oguro, R., Kamide, K., Katsuya, T., Akasaka, H., Sugimoto, K., Congrains, A., et al. (2012). A single nucleotide polymorphism of the adenosine deaminase, RNA-specific gene is associated with the serum triglyceride level, abdominal circumference, and serum adiponectin concentration. Exper. Gerontol. 47:183. doi: 10.1016/j.exger.2011.12.004
Patterson, B. D., Wilson, D. E., and Reeder, D. M. (eds) (1993). Mammal species of the world: a taxonomic and geographic reference, 2nd edition. J. Mammal. 75, 236–239. doi: 10.2307/1382262
Pilot, M., Jedrzejewski, W., Branicki, W., Sidorovich, V. E., Jedrzejewska, B., Stachura, K., et al. (2006). Ecological factors influence population genetic structure of European grey wolves. Mol. Ecol. 15, 4533–4553. doi: 10.1111/j.1365-294x.2006.03110.x
Piórkowska, K., Żukowski, K., Ropka-Molik, K., Tyra, M., and Gurgul, A. (2018). A comprehensive transcriptome analysis of skeletal muscles in two Polish pig breeds differing in fat and meat quality traits. Genet. Mol. Biol. 41, 125–136. doi: 10.1590/1678-4685-gmb-2016-0101
Ptak, G., Clinton, M., Barboni, B., Muzzeddu, M., Cappai, P., Tischner, M., et al. (2002). Preservation of the Wild European Mouflon: the first example of genetic management using a complete program of reproductive biotechnologies. Biol. Reprod. 66, 796–801. doi: 10.1095/biolreprod66.3.796
Quail, M. A., Smith, M., Coupland, P., Otto, T. D., Harris, S. R., Connor, T. R., et al. (2012). A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific biosciences and Illumina MiSeq sequencers. BMC Genom. 13:341. doi: 10.1186/1471-2164-13-341
Rezaei, H. R., Naderi, S., Chintauan-Marquier, I. C., Taberlet, P., and Pompanon, F. (2009). Evolution and taxonomy of the wild species of the genus Ovis (Mammalia, Artiodactyla, Bovidae). Mol. Phylogenet. Evol. 54, 315–326. doi: 10.1016/j.ympev.2009.10.037
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO v2: assessing genome assembly and annotation completeness withbenchmarking universal single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, 435–439.
Strom, T. M., Nyakatura, G., Apfelstedtsylla, E., Hellebrand, H., Lorenz, B., Weber, B. H. F., et al. (1998). An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet. 19, 260–263. doi: 10.1038/940
Suárez-Vega, A., Gutiérrez-Gil, B., and Arranz, J. J. (2016). Transcriptome expression analysis of candidate milk genes affecting cheese-related traits in 2 sheep breeds. J. Dairy Sci. 99, 6381–6390. doi: 10.3168/jds.2016-11048
Toral, P. G., Bernard, L., Belenguer, A., Rouel, J., Hervás, G., Chilliard, Y., et al. (2016). Comparison of ruminal lipid metabolism in dairy cows and goats fed diets supplemented with starch, plant oil, or fish oil. J. Dairy Sci. 99, 301–316. doi: 10.3168/jds.2015-10292
Weleber, R. G., Pillers, D. A., Powell, B. R., Hanna, C. E., Magenis, R. E., and Buist, N. R. (1989). Aland Island eye disease (Forsius-Eriksson syndrome) associated with contiguous deletion syndrome at Xp21. Similarity to incomplete congenital stationary night blindness. Arch. Ophthalmol. 107, 1170–1179. doi: 10.1001/archopht.1989.01070020236032
Yang, Y., Wang, Y., Zhao, Y., Zhang, X., Li, R., Chen, L., et al. (2017). Draft genome of the marco polo sheep (Ovis ammon polii). Gigascience 6, 1–7.
Zheng, B., Xu, Q., and Shen, Y. (2002). The relationship between climate change and quaternary glacial cycles on the Qinghai-Tibetan Plateau: review and speculation. Q. Intern. 97–98, 93–101. doi: 10.1016/s1040-6182(02)00054-x
Keywords: mouflon, Ovis orientalis musimon, Illumina sequencing, de novo genome assembly, genus Ovis
Citation: Su R, Qiao X, Gao Y, Li X, Jiang W, Chen W, Fan Y, Zheng B, Zhang Y, Liu Z, Wang R, Wang Z, Wang Z, Wan W, Dong Y and Li J (2020) Draft Genome of the European Mouflon (Ovis orientalis musimon). Front. Genet. 11:533611. doi: 10.3389/fgene.2020.533611
Received: 09 February 2020; Accepted: 26 October 2020;
Published: 19 November 2020.
Edited by:
Göran Andersson, Swedish University of Agricultural Sciences, SwedenReviewed by:
Xiaozhu Wang, Auburn University, United StatesCopyright © 2020 Su, Qiao, Gao, Li, Jiang, Chen, Fan, Zheng, Zhang, Liu, Wang, Wang, Wang, Wan, Dong and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Dong, bG95YWx5YW5nQDE2My5jb20=; Jinquan Li, bGlqaW5xdWFuX25kQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.