
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 22 January 2021
Sec. Human and Medical Genomics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.512940
 Irina Ponomarenko1
Irina Ponomarenko1 Evgeny Reshetnikov1*
Evgeny Reshetnikov1* Alexey Polonikov2Irina Verzilina1Inna Sorokina3Anna Yermachenko3
Alexey Polonikov2Irina Verzilina1Inna Sorokina3Anna Yermachenko3 Volodymyr Dvornyk4
Volodymyr Dvornyk4 Mikhail Churnosov1
Mikhail Churnosov1Age at menarche (AAM) is an important marker of the pubertal development and function of the hypothalamic–pituitary–ovarian system. It was reported as a possible factor for a risk of uterine leiomyoma (UL). However, while more than 350 loci for AAM have been determined by genome-wide association studies (GWASs) to date, no studies of these loci for their association with UL have been conducted so far. In this study, we analyzed 52 candidate loci for AAM for possible association with UL in a sample of 569 patients and 981 controls. The results of the study suggested that 23 out of the 52 studied polymorphisms had association with UL. Locus rs7759938 LIN28B was individually associated with the disease according to the dominant model. Twenty loci were associated with UL within 11 most significant models of intergenic interactions. Nine loci involved in 16 most significant models of interactions between single-nucleotide polymorphism (SNP), induced abortions, and chronic endometritis were associated with UL. Among the 23 loci associated with UL, 16 manifested association also with either AAM (7 SNPs) or height and/or body mass index (BMI) (13 SNPs). The above 23 SNPs and 514 SNPs linked to them have non-synonymous, regulatory, and expression quantitative trait locus (eQTL) significance for 35 genes, which play roles in the pathways related to development of the female reproductive organs and hormone-mediated signaling [false discovery rate (FDR) ≤ 0.05]. This is the first study reporting associations of candidate genes for AAM with UL.
Uterine leiomyoma (UL), commonly known as fibroids, is a benign tumor of smooth muscle in the uterus (Wise and Laughlin-Tommaso, 2016; Stewart et al., 2017). The symptoms include excessive menstrual bleeding, abdominal pain, and pregnancy complications (Zimmermann et al., 2012; Stewart et al., 2017). About 30 percent of reproductive-age women are diagnosed with UL, which is considered as a major factor of gynecologic morbidity (Wise et al., 2004), one of the primary causes of hospitalizations for gynecological disorders, and the most frequent reason for hysterectomy in the United States (Wise and Laughlin-Tommaso, 2016). ULs bear a substantial economic burden: the annual cost of treating this disorder has been estimated to be about $34 billion in the United States alone, which exceeded the combined cost of treating breast and colon cancers (Cardozo et al., 2012).
The biological mechanisms of UL development are not well understood (Rafnar et al., 2018). Family history is an important risk factor for UL (Commandeur et al., 2015; Wise and Laughlin-Tommaso, 2016). A risk to develop the disease is 2.5-fold greater for first-degree relatives of affected women than the population average (Vikhlyaeva et al., 1995), and the concordance among monozygotic twins is almost twice that of dizygotic twins (Luoto et al., 2000). Heritability estimates of UL from twin studies vary from 26 to 69% in European populations (Snieder et al., 1998; Luoto et al., 2000). More estimates come from recent genome-wide association studies (GWASs). Cha et al. (2011) reported candidate loci for UL in chromosome regions 10q24.33 (OBFC1), 11p15.5 (BET1L), and 22q13.1 (TNRC6B) in a cohort of Japanese patients. One more locus, 17q25.3 (FASN, CCDC57, and SLC16A3), was suggested by the genome-wide linkage and follow-up association studies in a meta-analysis of white women (Eggert et al., 2012). The 11p15.5 (BET1L) locus was later replicated in a sample of Caucasians (Edwards et al., 2013). The 22q13.1 (TNRC6B) locus was replicated in Caucasian, American, and Saudi Arabian cohorts (Edwards et al., 2013; Aissani et al., 2015; Bondagji et al., 2017). However, none of the above loci was replicated in African–American women (Wise et al., 2012). A GWAS of a risk for leiomyoma in African–American women suggested a locus on 22q13.1 (CYTH4) (Hellwege et al., 2017). In 2018, two teams presented results of the first GWAS of UL in European populations (Rafnar et al., 2018; Välimäki et al., 2018) reporting 21 and 22 loci associated with the increased risk of UL.
Age at menarche (AAM) has been suggested as a risk factor for UL (Wise et al., 2004; Terry et al., 2010; Velez Edwards et al., 2013; Wise and Laughlin-Tommaso, 2016). Women with an early AAM have, on average, longer period of menstrual cycling in their life and thus a greater lifelong exposure to estrogens, which may promote growth of UL (Wise et al., 2004). Cell division rate in the myometrium is the highest during the luteal phase of the menstrual cycle; therefore, a longer history of cycling may increase a risk of UL (Wise and Laughlin-Tommaso, 2016). On the other hand, some studies reported no significant association between age of menarche and UL (Samadi et al., 1996; Zimmermann et al., 2012).
The significantly different heritability estimates for UL [from 26 to 69% according to twin studies (Snieder et al., 1998; Luoto et al., 2000) and only 13% according to GWAS data single-nucleotide polymorphism (SNP) heritability (Rafnar et al., 2018)] bring about a problem of so-called “missing heritability.” Studies of candidate genes associated with specific risk factors for UL [e.g., AAM and body mass index (BMI)] may help to solve this problem. For example, more than 350 loci have been reported by GWAS for association with AAM (Day et al., 2017). There are data that candidate genes for AAM (e.g., FTO, LIN28B, MAP2K5, TNNI3K, GPRC5B, and FANCL) may also contribute to various anthropometric traits (e.g., BMI, height, and others) (Ong et al., 2009; Elks et al., 2010; Fernandez-Rhodes et al., 2013; Perry et al., 2014), while some of these traits (e.g., an increased BMI) may be risk factors for UL (Luoto et al., 2000; Wise and Laughlin-Tommaso, 2016; McWilliams and Chennathukuzhi, 2017). However, there have been no studies on the possible association of the candidate genes for AAM with a risk of UL.
The purpose of the present study was to analyze 52 candidate loci for menarcheal age for their possible association with UL. Recently, we studied association of these loci with some phenotypic traits (including AAM, height, and BMI) in the same sample of Caucasian females (Ponomarenko et al., 2019). Using the same sample for the analysis of different traits eliminates between-sample heterogeneity and makes comparison and interpretation of the results more meaningful and robust.
The study protocol was approved by the Regional Ethics Committee of Belgorod State University. All participants signed informed consent documents before entering the study. The participants were recruited through the Perinatal Center of the Belgorod Regional Clinical Hospital of St. Joasaph during 2008–2013. All patients with UL underwent hysterectomy followed by morphological verification of the diagnosis. The control group included women without clinical (asymptomatic women) and ultrasound signs of benign proliferative diseases of the reproductive organs. They were enrolled during regular medical examinations at the above Perinatal Center. A total of 1,620 women were recruited: 600 patients with UL (ICD-10 code D25) and 1,020 controls.
The participants were enrolled under the following exclusion criteria: self-declared non-Russian descent, a birthplace outside of Central Russia (Sorokina et al., 2018), chronic severe disorders of the vital organs (heart, respiratory, or renal failure), cancers of pelvis and breast, and severe autoimmune disorders.
The following data were collected for each participant: physical characteristics (height, weight, and BMI), AAM (determined as described elsewhere; Ponomarenko et al., 2019), characteristics of the menstrual cycle (length and duration of menses), other data about reproductive health (number of pregnancies and childbirths, age at first birth, time since last birth, spontaneous and induced abortions, and infertility), marital status, family history of UL, use of oral contraceptives and age at first oral contraceptive use, and smoking and alcohol use. The participants were also examined for the presence of gynecological disorders (endometrial hyperplasia, endometriosis, adenomyosis, etc.).
The phlebotomy was performed by a certified nurse. DNA extraction from buffy coat was performed according to the protocol used in our previous gene association studies (Ponomarenko et al., 2019).
The SNPs for the study were selected according to the criteria and procedure and using the online tools described elsewhere (Ponomarenko, 2018; Ponomarenko et al., 2019). Briefly, the criteria included the following: (1) associations with AAM or traits, which have common biological pathways with menarche (anthropometric characteristics, obesity, etc.), (2) effect on gene expression (eSNP), (3) regulatory potential (regSNP), (4) tag value (tagSNP), and (5) minor allele fraction (MAF) > 5%.
The regulatory potential of the SNPs and effect on gene expression was estimated using the following online tools: HaploReg (v4.1)1, RegulomeDB (version 1.1)2, rSNPBase3, SNPinfo Web Server–SNP Function Prediction (FuncPred)4, Blood eQTL browser5, and GTEx Portal6.
In total, 52 loci were selected for the present study (Supplementary Tables 1–4). All SNPs appeared to have a significant regulatory potential (Supplementary Table 2), 43 SNPs were eSNPs (Supplementary Table 3), and 29 were tagSNPs.
Fourteen loci among the 52 selected showed association with AAM according to the results of GWAS and 28 loci according to the results of gene association studies (Supplementary Table 4). Seventeen SNPs were associated with anthropometric characteristics (Supplementary Table 4). Ten more polymorphisms were not directly associated with AAM but manifested association or tagged with the traits related to menarche [e.g., polycystic ovary syndrome (PCOS), vitamin D metabolism, and physical characteristics; Supplementary Table 4]. These SNPs included rs1884051 ESR1, rs3020394 ESR1, rs12324955 FTO, rs4633 COMT, rs222020 GC, rs222003 GC, rs1544410 VDR, rs3756261 EGF, rs7766109 F13A1, and rs2252673 INSR. All these SNPs have a significant regulatory potential, nine of them are eSNPs, and eight are tagSNPs.
Several of the selected loci were previously reported to be associated with AAM (14 SNPs), BMI (15 SNPs), and height (16 SNPs) in the studied sample (Ponomarenko et al., 2019).
DNA samples were genotyped using the Sequenom MassARRAY® iPLEX platform at the Centre of Genomic Sciences (University of Hong Kong). The procedure for DNA sample preparation and data quality control are described elsewhere (Ponomarenko et al., 2019). All DNA samples successfully met the following quality control criteria: call rate > 95%, the success rate of duplicate check > 99.5%, and the success rate of the blank check > 90%. Finally, the proportion of the determined genotypes for the 52 SNPs was 98.84%. The samples with a proportion of determined genotypes < 95% were excluded from the analysis (n = 70). Thus, the final study sample included 1,550 participants: 569 women with UL and 981 control subjects.
Association of clinical anamnestic risk factors with UL was assessed using logistic regression analysis as implemented in the epicalc package in the R software environment [version 3.4.0 (2017-04-21)].
The correspondence of the SNPs to the Hardy–Weinberg equilibrium (HWE) was checked using the chi-square test. The logistic regression method was used to analyze associations of the SNPs with UL assuming additive, recessive, and dominant genetic models. To account for possible confounding effects, the following set of covariates was applied: family history of UL, history of infertility, the presence of induced abortions in the anamnesis, and chronic endometritis as qualitative variables (yes/no), whereas age, BMI, and the number of induced abortions in the anamnesis as quantitative variables (value of the trait) (Table 1). The adaptive permutation test was applied to adjust for multiple comparisons (Che et al., 2014). The significance level was set at pperm < 0.01 (after the Bonferroni correction based on the numbers of genetic models studied).

Table 1. Participant characteristics from case and control groups.
The given sample size (569 patients with UL and 981 controls) was sufficient to detect differences in allelic frequencies between affected subjects and controls at OR = 1.23–1.57 for the additive model, OR = 1.35–1.60 for the dominant model, and OR = 1.40–8.70 for the recessive model (at 80% power, ɑ= 0.05 for two-sided test).
The haplotype blocks were identified using the algorithm implemented in HaploView v.4.2 (Barrett et al., 2005). The association analyses and permutation test were performed using the respective algorithms implemented in the PLINK v. 2.050 software (Purcell et al., 2007)7. The significance level was set at pperm < 0.05.
The interactions between genes were analyzed assuming the two-, three-, and four-locus models and using the model-based multifactor dimensionality reduction (MB-MDR) method (Calle et al., 2008, 2010; Mahachie et al., 2012) in the mbmdr package (version 2.6) in the R software environment [version 3.4.0 (2017-04-21)]. The permutation test was demonstrated to be efficient for analysis of large massifs of GWAS data without reduction of the power (Che et al., 2014). For the permutation test, the following threshold p-values (after the Bonferroni correction based on the numbers of combinations studied for 52 SNPs) were adopted for models of gene–gene interactions: p < 3.8 ∗ 10–5 (<0.05/1,326) for two-locus models, p < 2.3 ∗ 10–6 (<0.05/22,100) for three-locus models, and p < 1.8 ∗ 10–7 (<0.05/270,725) for four-locus models. The significance level was set at pperm ≤ 0.001.
The interactions of the genes with induced abortions and chronic endometritis were analyzed for their possible effect on UL. Induced abortions and chronic endometritis were included in the analysis because (i) they were determined as risk factors for UL in the study sample (Table 1), (ii) induced abortions are a common birth control method of women in Russia as compared with other countries (Sedgh et al., 2007), and (iii) induced abortions are a common cause of post-abortion endometritis and, subsequently, chronic endometritis in Russian women (Douglas et al., 2014). The analysis was performed using MB-MDR with adjustment for covariates (age, BMI, family history of UL, and history of infertility) and multiple comparisons (1,000 permutations) as described above. The permutation test was applied to the selected best models of gene–environment interactions with the significance level of p < 1 ∗ 10–19. The significance level was set at pperm < 0.001.
The most significant models of gene–gene and gene–environment interactions associated with UL were cross-validated using generalized multifactor dimensionality reduction (GMDR) (Lou et al., 2007; Chen et al., 2011)8 and the respective software (Beta 0.9)9. The main parameters of the validation were adjusted for covariates and multiple comparisons using the permutation test (1,000 permutations with 10-fold cross-validation, which provided statistical significance at pperm < 0.001 for a validated model).
The identified interactions and proportion of their contribution to the total variance of the trait were visualized using the MDR method10 and the MDR v. 3.0.2 software11.
The HaploReg v. 4.112 and the data of the European population from the 1000 Genomes Project Phase were utilized to determine SNPs linked to the UL-associated polymorphisms. Then all these loci were analyzed for their functional significance (non-synonymous SNPs, regulatory potential, and eQTLs).
The SIFTonline tool13 was used to analyze non-synonymous SNPs and their functional predictions.
The regulatory potential of the candidate loci for UL was analyzed using SNP Function Prediction (FuncPred)14, rSNPBase15, RegulomeDB (version 1.1)16, and HaploReg (v4.1).
The data from the Blood eQTL browser17 were utilized to assess the eQTL (cis- and trans-eQTL) significance of the studied SNPs in peripheral blood. The eQTL value in other organs and tissues was estimated using the GTEx Portal18 (data release V7 updated on 09/05/2017, dbGaP Accession phs000424.v7.p2). The false discovery rate (FDR) ≤ 0.05 was applied.
The functional significance of the UL-associated genes in metabolic pathways was analyzed using the Gene Ontology Portal tools (PANTHER Overrepresentation Test accessed on 04/13/2017; PANTHER version 12.0 accessed on 07/10/201719) and the FDR test to correct the results for multiple comparisons. GeneMANIA20 (version 3.5.0, accessed on 03/13/2017) and the automatic weighting were used to construct the gene interaction networks. Gene Ontology Portal provides a possibility to evaluate functional significance of genes in various metabolic pathways using seven different data bases (Gene Ontology molecular function, Gene Ontology biological process, PANTHER protein class, PANTHER pathway, PANTHER molecular function, PANTHER biological process, and Reactome pathway) and makes it possible to construct the gene interaction networks based on seven different variants of gene interaction, i.e., physical interactions, predicted interaction, pathways, co-expression, co-localization, genetic interactions, and shared protein domains.
The phases of the study and main results are outlined in Figure 1.
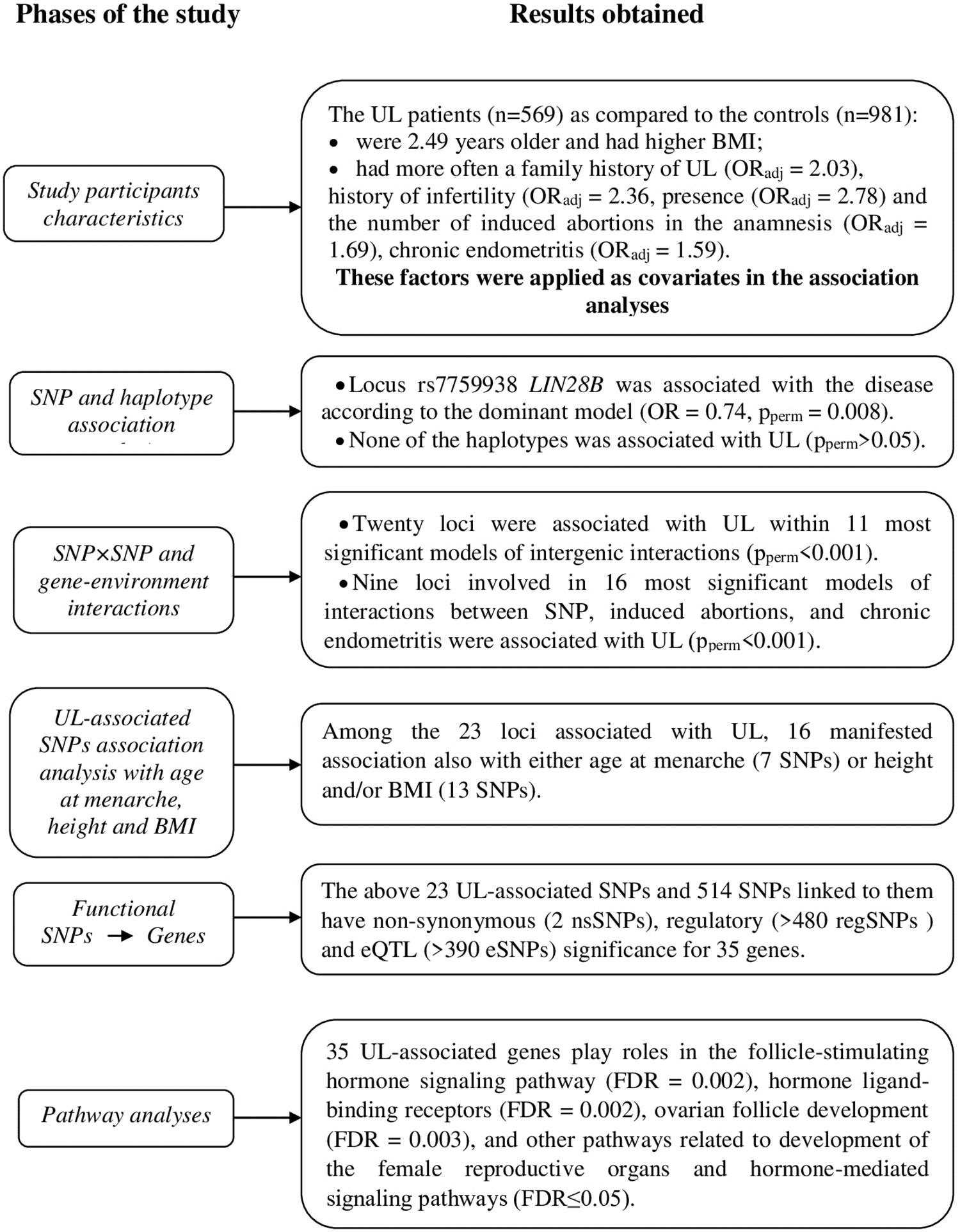
Figure 1. Schematically displayed the phases of the associations study single-nucleotide polymorphism (SNP) candidate genes for age at menarche and uterine leiomyoma (UL) and at each phase at short note the results obtained.
The UL patients (n = 569) were 2.49 years older and had higher BMI as compared with the controls (n = 981) (p < 0.001) (Table 1). They also had more often a family history of UL (adjusted for age and BMI, ORadj = 2.03, 95% CI 1.46–2.98, p < 0.001), history of infertility (ORadj = 2.36, 95% CI 1.75–3.97, p < 0.001), presence (ORadj = 2.78, 95% CI 2.23–3.48, p < 0.001), and the number of induced abortions in the anamnesis (ORadj = 1.69, 95% CI 1.54–1.86, p < 0.001), chronic endometritis (ORadj = 1.59, 95% CI 1.08–2.35, p = 0.02) (Table 1). Therefore, these factors (in addition to age and BMI) were applied as covariates in the association analyses. The higher number of pregnancies in the patients (3.34 vs. 2.45 in cases and controls, respectively) was due to the higher number of the induced abortions (1.59 vs. 0.67 in cases and controls, respectively) in this group as compared with the controls.
The data about the analyzed loci are presented in Supplementary Tables 5, 6. All loci had MAF > 5% and were in correspondence with the HWE (ðbonf > 0.001).
Allele C of the rs7759938 LIN28B locus was associated with UL according to the dominant model (OR = 0.74, 95% CI 0.59–0.92, p = 0.006, pperm = 0.008, power = 82.62%) (Table 2). None of the haplotypes was associated with UL (pperm > 0.05) (Supplementary Table 7).
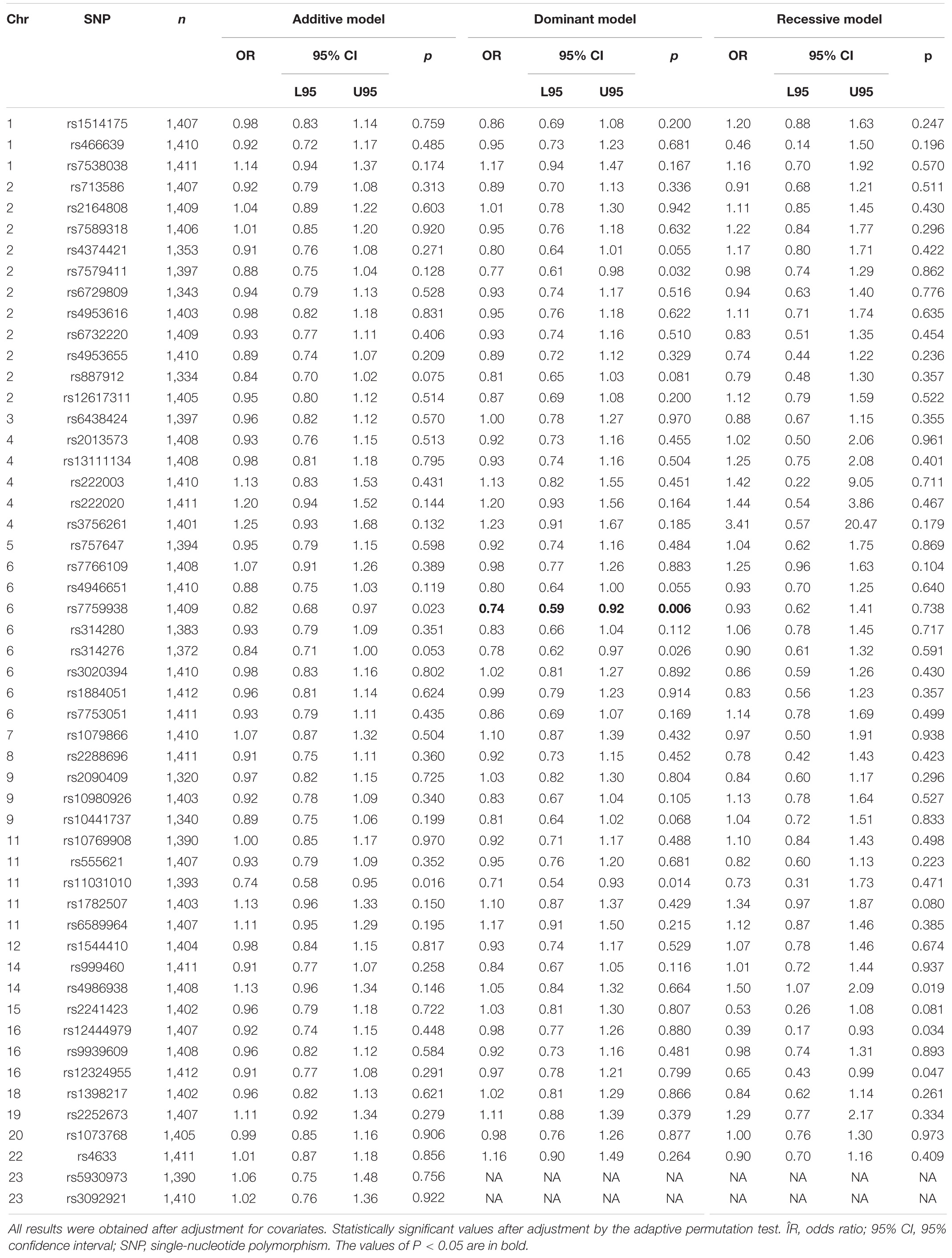
Table 2. Associations of the 52 SNPs with uterine leiomyoma.
In total, 11 most significant two-, three-, and four-locus models of gene–gene interactions associated with UL were determined (Table 3 and Supplementary Table 8). These models included 20 SNPs. Loci of the FSHB, LIN28B, and POMC genes appeared in the largest number of models (seven, six, and five, respectively). Locus rs314276 LIN28B contributed to the most significant gene–gene interaction models at all the levels considered. Among the obtained models of SNP–SNP interactions, the most optimal for prediction was rs314276 LIN28B × rs1782507 FSHB × rs1544410 VDR × rs7589318 POMC (OR = 2.69, Test. Bal. Acc. 54.70, sensitivity 68.72, specificity 55.05, Table 3). It had the highest values of sensitivity and specificity as compared with the others.
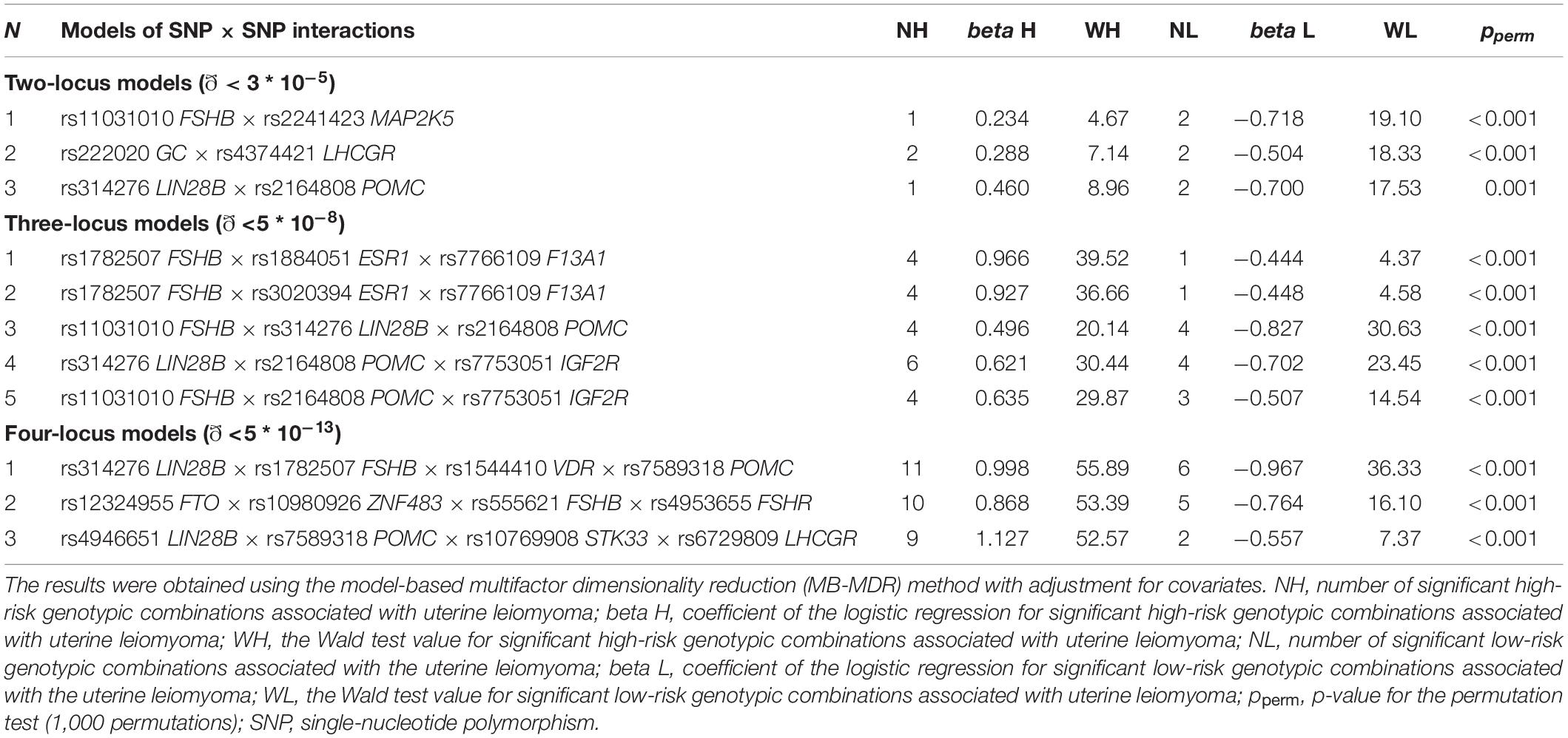
Table 3. The most significant models of SNP–SNP interactions associated with uterine leiomyoma.
The following genotype combinations were determined to have the most significant associations with UL: rs11031010 CA FSHB × rs2241423 GG MAP2K5 (β = −0.66, p = 0.0002), rs222020 TT GC × rs4374421 TC LHCGR (β = -0.44, p = 0.0003), rs1782507 AC FSHB × rs1884051 AG ESR1 × rs7766109 AA F13A1 (β = 0.82, p = 0.0003), and rs11031010 CC FSHB × rs2164808 GA POMC × rs7753051 TT IGF2R (β = 0.49, p = 0.0002) (Supplementary Table 9).
The graph of SNP–SNP interactions of 21 polymorphisms (20 SNPs that make up the 11 best models and rs7759938 LIN28B independently associated with UL) (Figure 2) suggests that these interactions are concerted; the highest contribution to the entropy is made by interactions rs3020394 ESR1 × rs7766109 F13A1 (0.51%), rs2164808 POMC × rs7753051 IGF2R (0.50%), and polymorphism rs7759938 LIN28B (0.51%).
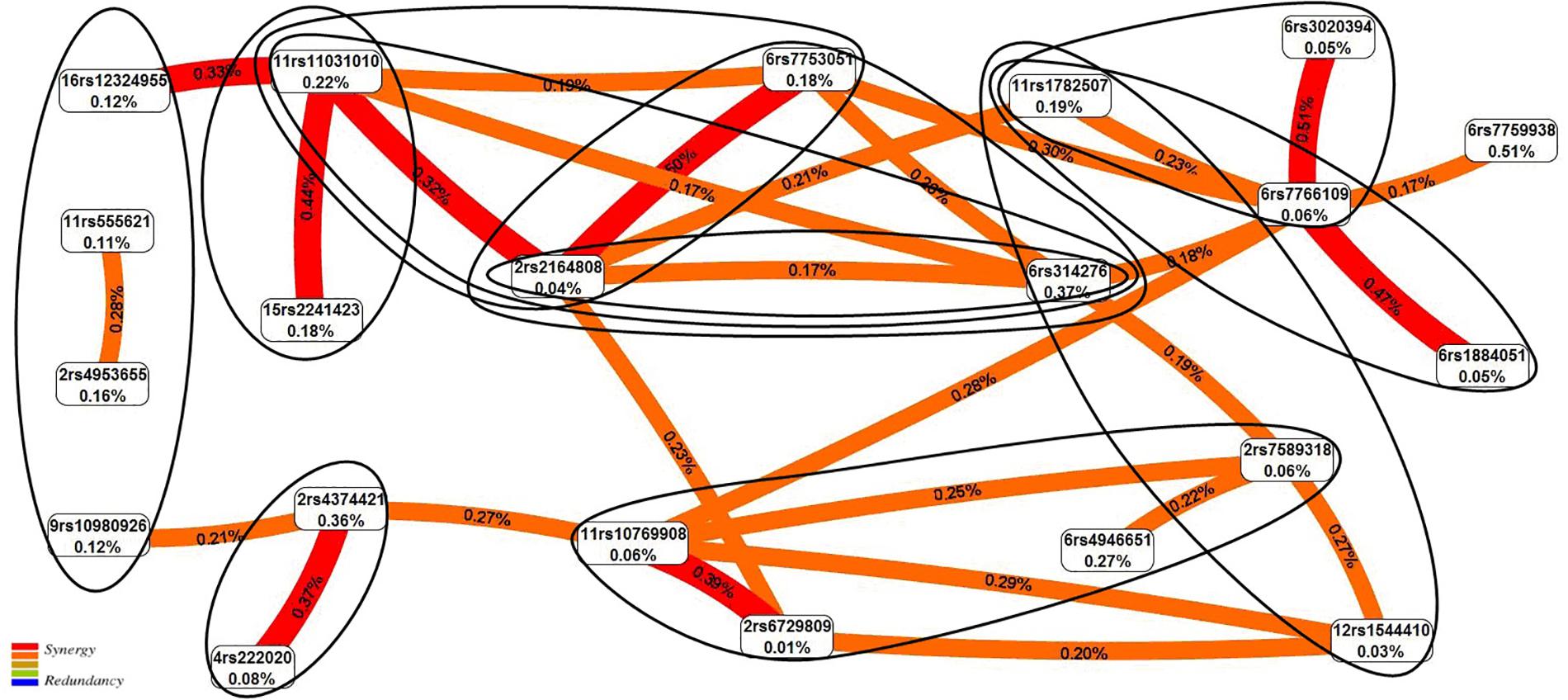
Figure 2. The entropy graph of single-nucleotide polymorphism (SNP)–SNP interactions with uterine leiomyoma based on the multifactor dimensionality reduction (MDR) analysis. Positive values of entropy indicate synergistic interactions, while the negative values indicate redundancy. The polymorphisms are denoted by the chromosome number and rs SNP ID. The red and orange colors denote strong and moderate synergism, respectively. The figures outline the SNP × SNP interactions within the two-, three-, and four-locus models obtained by the MB-MDR method.
In total, nine loci were determined to contribute to gene–environment interactions with induced abortions and chronic endometritis, which were significantly associated with UL. The analysis yielded 16 best two-, three-, and four-order models (Table 4 and Supplementary Table 10). Two loci, rs12324955 FTO and rs11031010 FSHB, were included in the largest number of the best models (11 and 10, respectively) (Table 4). Loci rs4633 COMT and rs314280 LIN28B were significantly associated with UL through the interaction with induced abortions and chronic endometritis. Among the obtained models of gene–environment interactions, the most optimal for prediction was rs12324955 FTO × rs11031010 FSHB × chronic endometritis × aborts (OR = 3.90, Test. Bal. Acc. 64.99, sensitivity 62.57, specificity 70.03, Table 4).
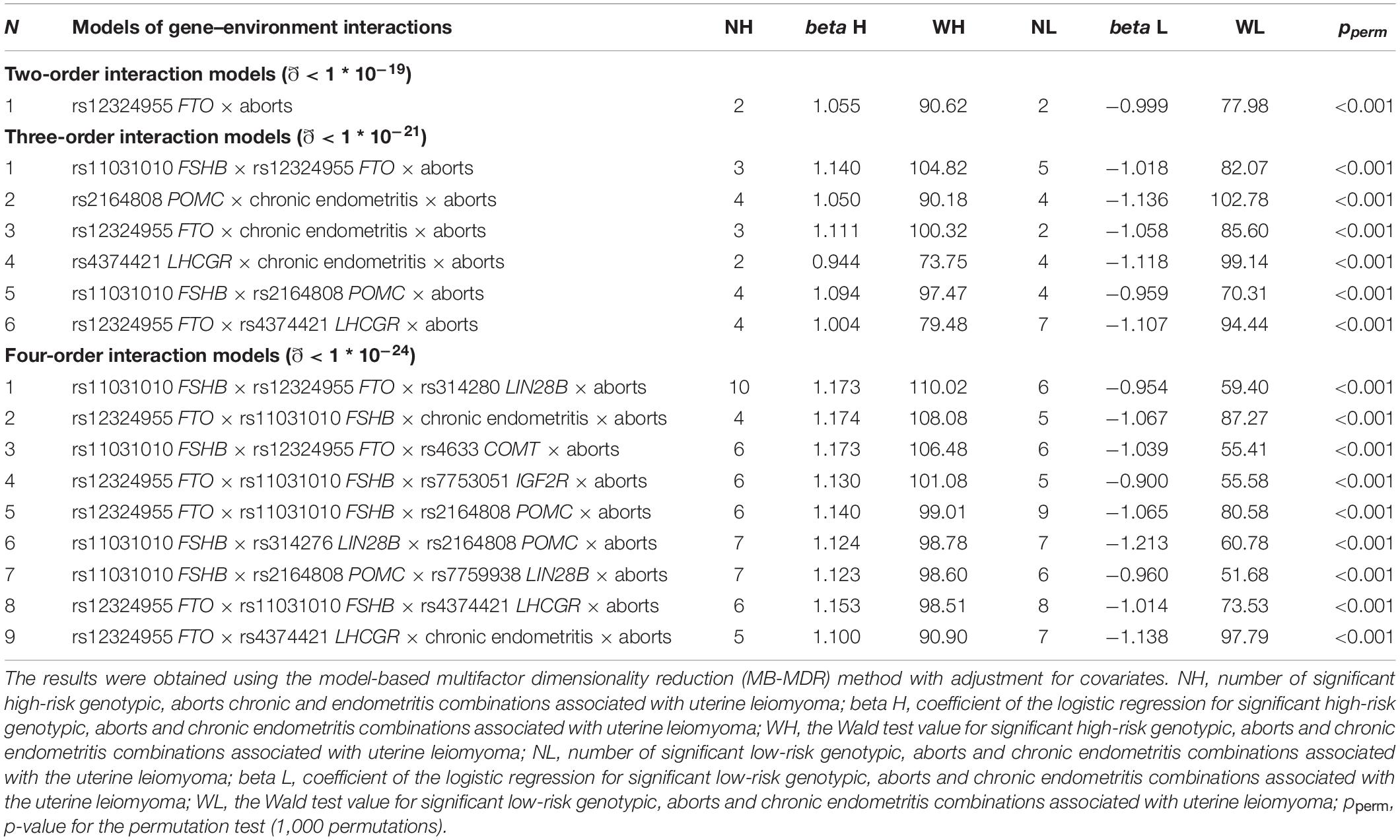
Table 4. The most significant models of gene–environment interactions associated with uterine leiomyoma.
The following combinations of genotypes with chronic endometritis and induced abortions were associated with UL: rs11031010 CC FSHB × rs12324955 GG FTO × abortion (β = 0.92, p = 2.38 ∗ 10–12), rs4374421 TT LHCGR × chronic endometritis × abortion (β = 0.95, p = 2.78 ∗ 10–14), rs12324955 GG FTO × rs11031010 CC FSHB × chronic endometritis × abortion (β = 0.99, p = 5.75 ∗ 10–13) (Supplementary Table 11).
The graph of the interactions between the studied loci, induced abortions, and chronic endometritis suggests that the abortions account for the largest proportion (5.02%) of the trait entropy (Figure 3). In general, the contribution of the most significant gene–environment interactions to UL is 0.31–0.41%, on average.
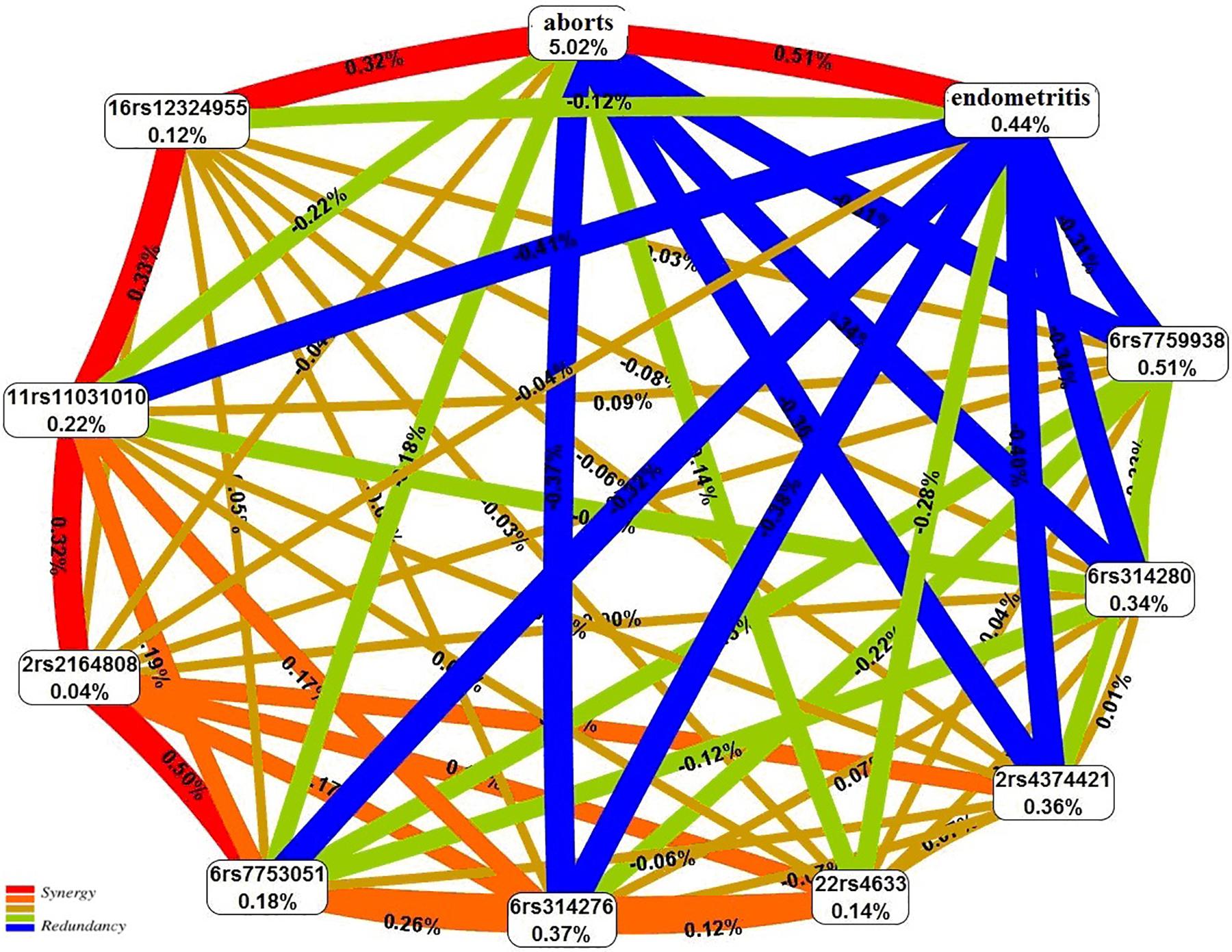
Figure 3. The entropy graph of single-nucleotide polymorphism (SNP) × aborts × chronic endometritis interactions with uterine leiomyoma based on the multifactor dimensionality reduction (MDR) analysis. Positive values of entropy indicate synergistic interactions, while the negative values indicate redundancy. The polymorphisms are denoted by the chromosome number and rs SNP ID. The red and orange colors denote strong and moderate synergism, respectively; brown color denotes the independent effect; and green and blue colors denote moderate and strong antagonism, respectively.
The analyses determined 23 loci associated with UL either individually or through gene–gene and gene–environment interactions. We also analyzed whether these SNPs were associated with the other phenotypic characteristics, namely, menarcheal age, height, and BMI in the studied sample (the respective results were reported elsewhere; Ponomarenko et al., 2019). Out of the 23 SNPs, 16 SNPs (69.57%) also manifested association with AAM, height, and/or BMI (Supplementary Table 12). Locus rs4633 COMT manifested association with all the above phenotypes, and three loci (rs2164808 POMC, rs4374421 LHCGR, and rs4946651 LIN28B) were also associated with at least two of the three phenotypes analyzed (i.e., age of menarche and/or height and/or BMI).
None of the 23 polymorphisms associated with UL was missense. However, two of these were in linkage disequilibrium with non-synonymous SNPs. Polymorphism rs4633 was linked to rs4680, which results in an amino acid substitution Val158Met in the COMT protein. This replacement has SIFT Score = 0.02 that corresponds to the predictive value “deleterious.” Another locus, rs2241423 (15q23), was linked to rs7170185, a missense variant (Trp200Arg) in the SKOR1 protein. This amino acid change also has a predictive value of “deleterious” (SIFT Score = 0).
The data on regulatory effects of the UL-associated SNPs are given in Supplementary Table 13. Locus rs4633 COMT appeared to have the most pronounced effect; the significant regulatory potential was also determined for rs314280 LIN28B, rs2164808 POMC, rs12324955 FTO, and rs10769908 STK33 (Supplementary Table 13).
The 514 SNPs linked to the UL-associated loci were analyzed for their regulatory potential (Supplementary Table 14). Among these, more than 460 had regulatory significance. The most pronounced regulatory potential was determined for loci linked to polymorphisms rs314276 and rs7759938 of the LIN28B gene (two SNPs), rs11031010, rs555621, and rs1782507 of the FSHB gene (four SNPs), rs2241423 MAP2K (>10 SNPs), rs10769908 STK33 (two SNPs), and rs4633 COMT (three SNPs) (Supplementary Table 14).
Importantly, the loci that are linked to the UL-associated SNPs showed their regulatory effects in organs and tissues related to pathogenesis of UL, i.e., ovaries, muscle tissue, adipose tissue, various brain regions (hypothalamus, pituitary, etc.), and liver (Commandeur et al., 2015).
Eight of the 23 UL-associated SNPs manifested their association (p < 5 ∗ 10–5, pFDR < 0.05) with the expression level of seven genes [COMT, DNAJC27, F13A1, KIAA0368, SLC22A1, C11orf46 (ARL14EP), and MAP2K5] in peripheral blood (cis-eQTL) (Supplementary Table 15). Five of these SNPs are also linked to the other cis-eQTL loci affecting mRNA transcription level in blood (MAP2K5, SLC22A1, DNAJC27, KIAA0368, and COMT) (Supplementary Table 16). No trans-eQTL SNPs were determined (FDR > 0.05).
According to the data of the Genotype-Tissue Expression (GTEx) project, 16 SNPs of the 23 associated with UL had cis-eQTL significance in various tissues and organs, including those that are related to pathogenesis of UL (p < 8 ∗ 10–5, pFDR ≤ 0.05) (Supplementary Table 17). Allele T (ref) of rs7759938 LIN28B is associated with the lower expression of the gene in hypophysis (β = -0.50, ð = 1.3 ∗ 10–11, pFDR ≤ 0.05), while alternative allele C has a protective effect for UL (OR = 0.74).
Sixteen UL-associated loci manifested strong linkage to more than 380 polymorphisms affecting gene expression (p < 8.5 ∗ 10–5, pFDR ≤ 0.05) in various organs and tissues (Supplementary Table 18). Loci rs1782507, rs11031010, and rs555621 of FSHB appeared to have the most pronounced effect as they were linked to more than 120 SNPs affecting expression of the FSHB, ARL14EP, and RP4-710M3.1 genes in more than 25 organs and tissues.
Overall, 18 of the 23 UL-associated loci have cis-eQTL value: they affect expression of 28 genes. Among these, two SNPs are associated with the mRNA transcription level independently, and 16 SNPs are both individually associated with and linked to other loci affecting gene expression levels.
The roles in biological processes or molecular functions of the UL-associated genes (Supplementary Table 13) and those whose expression is affected by the UL-associated 23 loci according to the eQTL analysis (Supplementary Tables 15–18) were analyzed in silico using annotations from the Gene Ontology databases. We found evidence of enrichment for pathways involved in the follicle-stimulating hormone signaling pathway (FDR = 0.002), hormone ligand-binding receptors (FDR = 0.002), ovarian follicle development (FDR = 0.003), and other pathways related to development of the female reproductive organs and hormone-mediated signaling pathways (FDR ≤ 0.05) (Supplementary Table 19).
The intergenic interactions of the above genes were analyzed using GeneMANIA (see footnote 20). The data on six genes, LINC00577, RP11-509E16.1, RP11-288H12.3, RP4-710M3.1, RP11-34F13.2, and IQCH-AS1, were absent in the database. Therefore, the resulting network included 29 UL-associated genes and 20 other genes significantly interacting with them (Figure 4). The intergenic interactions are executed through pathway (26.52%), co-expression (25.00%), physical (22.76%) and predicted (11.09%) interactions, co-localization (8.47%), shared protein domains (5.87%), and genetic interactions (0.29%) (Supplementary Table 20).
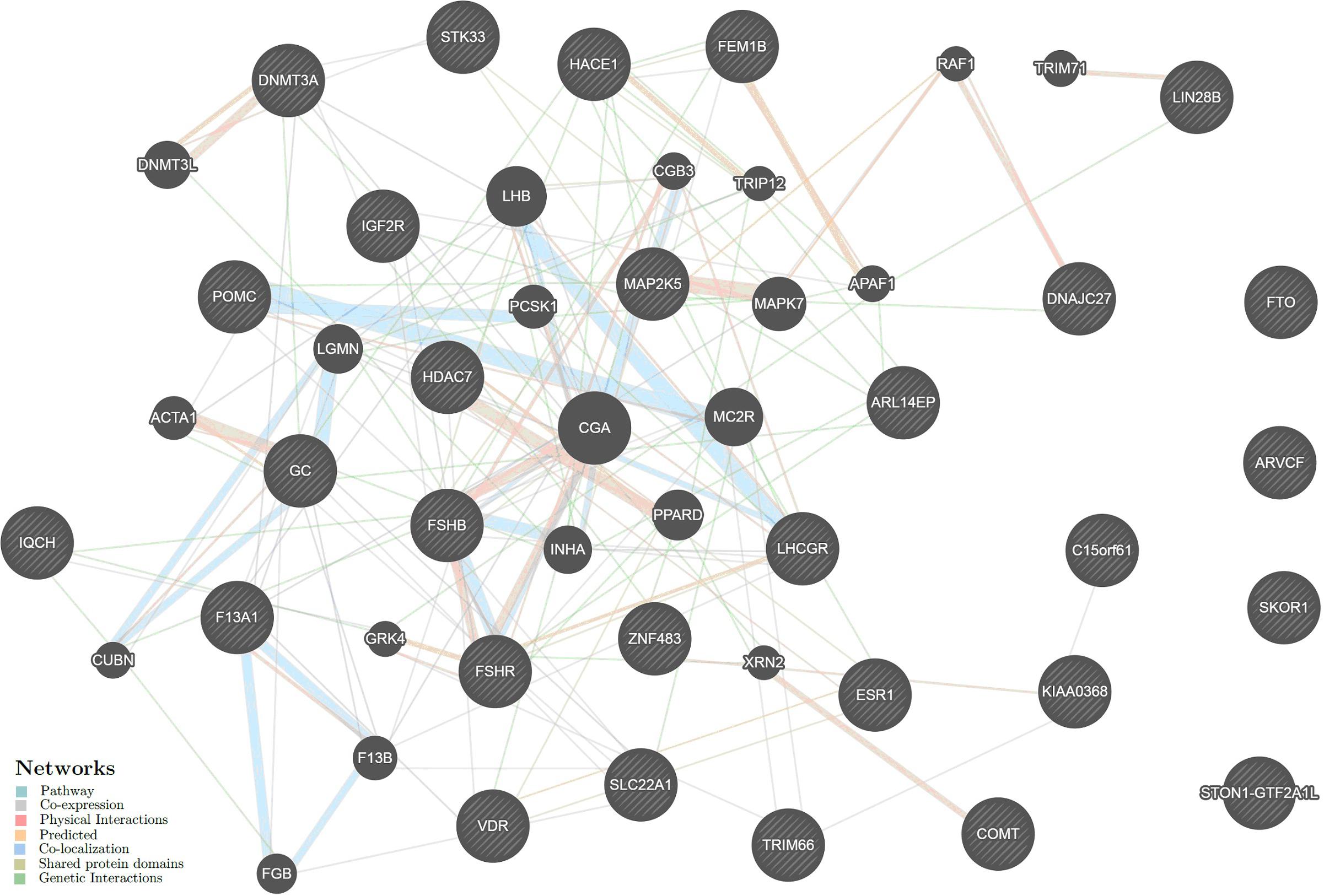
Figure 4. The interaction networks of the candidate genes for uterine leiomyoma inferred using GeneMANIA (http://genemania.org). The candidate genes determined in the present study are cross-shaded.
This study reports for the first time the associations of 23 candidate loci for AAM with UL. Locus rs7759938 of the LIN28B gene demonstrated the most significant associations: its allele Ñ decreased a risk for UL according to the dominant model (OR = 0.74). This locus is also associated with UL within most significant four-order model of polymorphisms interactions with induced abortions.
According to the GTEx Portal database, rs7759938 and 27 SNPs linked to it have the cis-eQTL significance and may affect expression of LIN28B in the pituitary gland (allele Ñ of rs7759938 increases expression of LIN28B). Association of this SNP with menarcheal age was reported previously (Perry et al., 2009; Elks et al., 2010; Delahanty et al., 2013; Perry et al., 2014; Ponomarenko et al., 2019). Several studies suggested that rs7759938 might play a role in the pubertal development and adult height (Elks et al., 2010; Lango Allen et al., 2010; Widén et al., 2010; Leinonen et al., 2012; Cousminer et al., 2013; Perry et al., 2014). Two polymorphisms, rs9391253 and rs314263, which are linked to rs7759938 (r2 ≥ 0.94), were associated with height (Berndt et al., 2013; Wood et al., 2014). Importantly, allele C of rs7759938 was previously determined as associated with later menarche, delayed pubertal growth, and high height (Perry et al., 2009, 2014, Elks et al., 2010; Delahanty et al., 2013). On the other hand, the present study suggested this allele as a protective factor for UL. That means that AAM and a risk for UL may have a shared genetic basis.
Two SNPs of the LIN28B gene (rs314276 and rs4946651) were involved in five out of 11 most significant models of epistatic interactions, and polymorphism rs314280 LIN28B was associated with UL only through interaction with induced abortions and chronic endometritis. All these polymorphisms were reported as associated with AAM (He et al., 2009; Ong et al., 2009; Sulem et al., 2009; Carty et al., 2013). There is evidence that rs314276 may play a role in the pubertal development and height (Ong et al., 2009; Cousminer et al., 2014), BMI, and weight in females (Ong et al., 2011). Previously, we reported the association of the rs314280 and rs4946651 polymorphisms with BMI of adults (Ponomarenko et al., 2019). Two polymorphisms, rs9391253 and rs314263, which are linked to rs314276 (r2 ≥ 0.94), were associated with height (Berndt et al., 2013; Wood et al., 2014). The data of GTEx Portal suggest that the UL-associated SNPs of LIN28B have cis-eQTL significance: rs314276 (with 20 SNPs linked to it) and rs4946651 (with 15 SNPs linked to it) are associated with expression level of LINC00577 in the cortex and basal ganglia of the brain, LIN28B in the pituitary gland, and HACE1 in skeletal muscle; rs314276 and 27 are linked to SNPs that may affect expression of the LIN28B gene in the pituitary gland.
The above four UL-associated SNPs of the LIN28B gene are located in region 6q16.3-q21 about 38 kb from each other and are linked to about 40 SNPs with the regulatory potential and cis-eQTL values (affect transcription of the LIN28B, LINC00577, and HACE1 genes).
LIN28B (lin-28 homolog B) is a member of the LIN28 family of proteins and functions as a repressor of the let-7 family of miRNA (Tsialikas and Romer-Seibert, 2015). These miRNAs contribute to developmental processes in invertebrate animals, while their role in vertebrates is not well understood. Some data suggest that the let-7 miRNAs may contribute to cell cycle control and oncogenesis (Tsialikas and Romer-Seibert, 2015). The target genes of the let-7 miRNA, Kras, Myc, Hmga2, and Igf2bp1 are involved in body size control and metabolism in mammals (Zhu et al., 2011). The function of LINC00577 (LIN28B antisense RNA 1) is unknown; according to the information from the Ensembl database, it belongs to the genes encoding long intergenic non-coding RNAs (lincRNAs), which interact with proteins, DNA, and other RNAs, and thus perform important regulatory functions (Ulitsky and Bartel, 2013). A protein product of the HACE1 (HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1) gene is a potential tumor suppressor and participates in the specific tagging of target proteins that subsequently results in proteasome degradation21.
The rs555621, rs11031010, and rs1782507 polymorphisms of the FSHB gene appeared in the largest number of the most significant models of intergenic interactions (7 out of 11 models); rs11031010 FSHB was presented in 10 out of 16 most significant models gene–environment interactions associated with UL. Recently, we determined association of rs555621 with BMI of adults (Ponomarenko et al., 2019). The above three loci were also associated with AAM (He et al., 2010), and rs11031010 was suggested to be a risk factor of PCOS and affect a level of luteinizing hormone (LH) in patients (Tian et al., 2016).
The FSHB gene encodes a beta-subunit of the follicle-stimulating hormone. This hormone has multiple functions in the female reproductive system, including stimulation proliferation of follicular granulosa cells, rescue of follicles from apoptosis, and synthesis of receptors for the LH on these cells before ovulation.
The recent meta-analysis of 11 GWAS determined the region on chromosome 11 (11p14.1, rs74485684) harboring the FSHB gene as that associated with endometriosis (Sapkota et al., 2017). Importantly, rs11031010 FSHB analyzed in the present study is located just 1.9 kb from the above locus. On the other hand, the first GWAS of UL in European populations (Rafnar et al., 2018) replicated three of the 19 previously reported endometriosis variants (Sapkota et al., 2017). Several loci associated with endometriosis, including rs11031006 located in 11p14.1, were reported for their association with UL in the recent GWAS (Gallagher et al., 2019). Importantly, the rs11031010 polymorphism of the FSHB gene analyzed in the present study is linked to rs11031006 (r2 = 0.64).
An epidemiological meta-analysis of 402,868 women suggested at least twofold higher risk for UL in patients with a history of endometriosis (Gallagher et al., 2019). It was hypothesized that both UL and endometriosis were underlain by hormone-related factors (Rafnar et al., 2018) and thus might have a shared genetic basis (Gallagher et al., 2019). This assumption was supported by the recent findings of syntropic genes for these diseases (Rafnar et al., 2018; Gallagher et al., 2019).
There is evidence that some loci linked to rs11031010 may contribute to various reproductive characters, such as concentration of the luteinizing (rs11031002, r2 = 0.76) and follicle-stimulating (rs11031005, r2 = 0.64) hormones in blood plasma (Ruth et al., 2016), and age at menopause (rs12294104, r2 = 0.61) (Stolk et al., 2012). The SNPs upstream of the FSHB transcription start were implicated into a key role in many reproductive processes (Gajbhiye et al., 2018). It is generally acknowledged that women with late menopause or/and early menarche have, on average, an increased lifetime exposure to estrogen due to the larger number of ovulatory cycles. Since mitotic activity of myometrium is the highest during the luteal phase of the cycle, a longer history of menses may theoretically increase a risk for UL. Likewise, this risk may be higher due to the increasing LH level (Wise and Laughlin-Tommaso, 2016).
The present study determined that the rs2164808 polymorphism of the POMC gene was involved in the largest number of the most significant models of gene–gene (four out of 11 models) and gene–environment (five out of 16 models) interactions associated with UL. Previously, we reported the association of the rs2164808 locus with BMI of adults (Ponomarenko et al., 2019). The association of this locus with AAM was first reported by He et al. (2010). Polymorphism rs1561288, which is linked to rs2164808 (r2 = 0.25), was associated with BMI (Graff et al., 2013). Locus rs2164808 and linked to it rs4665765 are both associated with the transcription level of DNAJC27 (according to the Blood eQTL browser) and RP11-509E16.1 (according to the GTEx Portal database). The data from the SNP Function Prediction and rSNPBase suggest a significant regulatory effect of rs2164808 (proximal regulation and RNA binding protein mediated regulation effects, regulatory potential score 0.334). The POMC (pro-opiomelanocortin-alpha) gene encodes a preproprotein that undergoes tissue-specific posttranslational modifications resulting in up to 10 biologically active peptides, which participate in various cellular processes, including those related to UL (e.g., lipolysis, mobilization of fatty acids, steroidogenesis, and adrenal development) (see footnote 21). The product of the DNAJC27 [DnaJ heat shock protein family (Hsp40) member C27] gene is a GTPase that is associated with the MEK/ERK pathway and may induce cell transformation (see footnote 21). The function of RP11-509E16.1 is unknown; according to the information from the Ensembl database22, it belongs to the genes encoding lincRNAs, which interact with proteins, DNA, and other RNAs and thus perform important regulatory functions (Ulitsky and Bartel, 2013). The polymorphisms located in the regions of the POMC, DNAJC27 (RBJ), and ADCY3 genes were associated with pubertal development, height, BMI, obesity, and type I diabetes mellitus (Gudbjartsson et al., 2008; Lango Allen et al., 2010; Speliotes et al., 2010; Bradfield et al., 2011; Berndt et al., 2013; Cousminer et al., 2013; He et al., 2015).
One more finding of the present study is that associations with UL were determined not only for candidate SNPs for AAM (Supplementary Table 4) but also for seven polymorphisms manifesting association or tagged with the traits related to menarche (e.g., sex steroid hormone and vitamin D metabolism, and physical characteristics; Supplementary Tables 4, 12). These menarche-related traits are important for pathogenesis of UL (Commandeur et al., 2015; Wise and Laughlin-Tommaso, 2016). The latter are involved in the sex steroid hormone pathway (rs3020394 and rs1884051 ESR1, rs4633 COMT, and rs12324955 FTO and rs7766109 F13A1) and vitamin D metabolism (rs1544410 VDR and rs222020 GC) (Supplementary Table 4, see footnote 21), which are important for UL development (Commandeur et al., 2015; Wise and Laughlin-Tommaso, 2016). For example, rs4633 COMT, which is also associated with AAM, height, and BMI in the studied sample (Ponomarenko et al., 2019) and linked to it rs4680 is associated with the COMT expression level in peripheral blood (according to the Blood eQTL browser). The activity of the COMT enzyme is elevated in human leiomyoma tissue as compared with normal myometrium; since this enzyme is essential for estrogen metabolism, the observed association suggests a possible causal role of COMT in UL formation (Commandeur et al., 2015). In addition, the rs4680 polymorphism encodes a replacement substitution Val158Met in COMT: the Met variant has 40% lower activity as compared with the Val one (Chen et al., 2004). Further support of the possible contribution of rs4680 to UL comes from the meta-analysis by Feng et al. (2013), who reported the association of rs4680 with UL.
We did not find a direct association of AAM with UL but determined that 23 candidate loci for AAM might be associated with UL. Among these, several were also associated with age of menarche, height, and/or BMI of adults in the study sample. This may suggest that menarcheal age per se is not a primary risk factor for UL in the studied sample (the population of Russia) and that other factors may be of greater importance. Specifically, we found that the history of induced abortions significantly increased the risk of UL in the study sample (ORadj = 2.78). Our results support the recent findings by other researchers (Shen et al., 2017; Song et al., 2017). Induced abortions are common birth control method in Russia (and generally in the former USSR) (Sedgh et al., 2007; David et al., 2007; Douglas et al., 2014), and Russia (along with Cuba) is a leader by this indicator in the world (Sedgh et al., 2007). Induced abortions in the first trimester of pregnancy may cause multiple health complications, including PCOS, hyperprolactinemia, anovulatory menstruation, disorders of the luteal phase of the menstrual cycle, and post-abortion endometritis (Douglas et al., 2014). Induced recurring abortions result in injuries of the uterus. The patient cohort in the present study included 44.99% individuals who had at least two induced abortions. Uterine injuries may induce the expression of the growth factor that speed ups cellular proliferation, suppresses apoptosis, and increases extracellular matrix production (Wise and Laughlin-Tommaso, 2016). Indeed, our results suggest that the risk for UL directly correlates with the number of the abortions in the anamnesis (ORadj = 1.69): it increases from OR = 3.85 (95% CI 2.79–5.32, p < 0.001) in a case of two abortions up to OR = 13.41 (95% CI 7.22–25.24, p < 0.001) with four and more abortions. Similar results were reported by Song et al. (2017) who showed that the higher number of induced abortions was associated with an increased risk of UL (one induced abortion, OR = 1.32; two induced abortions, OR = 1.45; and ≥ 3 induced abortions, OR = 1.62). PCOS was associated with a 65% increased risk of UL (Wise and Laughlin-Tommaso, 2016). The mechanisms by which PCOS may elevate risk for UL include increased levels of LH or unopposed estrogens and its association with hyperinsulinemia (Wise and Laughlin-Tommaso, 2016). Induced abortions may also be associated with other hormone-related diseases (e.g., breast cancer) (Schneider et al., 2014). Inflammation in the uterus (stress from chronic inflammation) seems a quite possible etiologic factor of UL: studies showed that inflammation increases the production of extracellular matrix and decreases apoptosis in UL (Commandeur et al., 2015; Wise and Laughlin-Tommaso, 2016).
Candidate genes for AAM are associated with UL. Locus rs7759938 LIN28B is associated with UL independently; 20 loci are associated within the 11 most significant intergenic interaction models, and nine loci may contribute to the risk of the disorder through the 16 most significant gene–environment interactions. These associations probably result from multiple effects of more than 510 polymorphisms linked to the above SNPs and implicated in various metabolic processes in organs and tissues that are related to pathogenesis of UL.
The datasets generated for this study can be found in EVA at the following accessions Project: PRJEB40782, Analyses: ERZ1645102.
The studies involving human participants were reviewed and approved by Regional Ethics Committee of Belgorod State University. The patients/participants provided their written informed consent to participate in this study.
IP, MC, and VD made substantial contributions to conception and design, drafted the manuscript, and gave the final approval of the version to be published. ER, AP, IV, IS, and AY analyzed and interpreted of data, drafted the manuscript, and gave the final approval of the version to be published. All the authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.512940/full#supplementary-material
Aissani, B., Zhang, K., and Wiener, H. (2015). Evaluation of GWAS candidate susceptibility loci for uterine leiomyoma in the multi-ethnic NIEHS uterine fibroid study. Front. Genet. 6:241. doi: 10.3389/fgene.2015.00241
Barrett, J. C., Fry, B., Maller, J., and Daly, M. J. (2005). Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265. doi: 10.1093/bioinformatics/bth457
Berndt, S. I., Gustafsson, S., Mägi, R., Ganna, A., Wheeler, E., Feitosa, M. F., et al. (2013). Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat. Genet. 5, 501–512.
Bondagji, N. S., Morad, F. A., Al-Nefaei, A. A., Khan, I. A., Elango, R., Abdullah, L. S., et al. (2017). Replication of GWAS loci revealed the moderate effect of TNRC6B locus on susceptibility of saudi women to develop uterine leiomyomas. J. Obstet. Gynaecol. Res. 43, 330–338. doi: 10.1111/jog.13217
Bradfield, J. P., Qu, H. Q., Wang, K., Zhang, H., Sleiman, P. M., Kim, C. E., et al. (2011). A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 7:e1002293. doi: 10.1371/journal.pgen.1002293
Calle, M. L., Urrea, V., Malats, N., and Van Steen, K. (2010). Mbmdr: an R package for exploring gene-gene interactions associated with binary or quantitative traits. Bioinformatics 17, 2198–2199. doi: 10.1093/bioinformatics/btq352
Calle, M. L., Urrea, V., Vellalta, G., Malats, N., and Steen, K. V. (2008). Improving strategies for detecting genetic patterns of disease susceptibility in association studies. Stat. Med. 27:6532. doi: 10.1002/sim.3431
Cardozo, E. R., Clark, A. D., Banks, N. K., Henne, M. B., Stegmann, B. J., and Segars, J. H. (2012). The estimated annual cost of uterine leiomyomata in the United States. Am. J. Obstet. Gynecol. 206, 211.e1–211.e9.
Carty, C. L., Spencer, K. L., Setiawan, V. W., Fernandez-Rhodes, L., Malinowski, J., Buyske, S., et al. (2013). Replication of genetic loci for ages at menarche and menopause in the multi-ethnic population architecture using genomics and epidemiology (PAGE) study. Hum. Reprod. 6, 1695–1706. doi: 10.1093/humrep/det071
Cha, P. C., Takahashi, A., Hosono, N., Low, S. K., Kamatani, N., Kubo, M., et al. (2011). A genome-wide association study identifies three loci associated with susceptibility to uterine fibroids. Nat. Genet. 43, 447–450. doi: 10.1038/ng.805
Che, R., Jack, J. R., Motsinger-Reif, A. A., and Brown, C. C. (2014). An adaptive permutation approach for genome-wide association study: evaluation and recommendations for use. BioData. Min. 7:9. doi: 10.1186/1756-0381-7-9
Chen, G. B., Xu, Y., Xu, H. M., Li, M. D., Zhu, J., and Lou, X. Y. (2011). Practical and theoretical considerations in study design for detecting gene-gene interactions using MDR and GMDR approaches. PLoS One 6:e16981. doi: 10.1371/journal.pone.0016981
Chen, J., Lipska, B. K., Halim, N., Ma, Q. D., Matsumoto, M., Melhem, S., et al. (2004). Functional analysis of genetic variation in catechol-o-methyltransferase (COMT): effects on mRNA, protein enzyme activity in postmortem human brain. Am. J. Hum. Genet. 2004, 807–821. doi: 10.1086/425589
Commandeur, A. E., Styer, A. K., and Teixeira, J. M. (2015). Epidemiological and genetic clues for molecular mechanisms involved in uterine leiomyoma development and growth. Hum. Reprod. Update 21, 593–615. doi: 10.1093/humupd/dmv030
Cousminer, D. L., Berry, D. J., Timpson, N. J., Ang, W., Thiering, E., Byrne, E. M., et al. (2013). Genome-wide association and longitudinal analyses reveal genetic loci linking pubertal height growth, pubertal timing and childhood adiposity. Hum. Mol. Genet. 13, 2735–2747.
Cousminer, D. L., Stergiakouli, E., Berry, D. J., Ang, W., Groen-Blokhuis, M. M., Körner, A., et al. (2014). Genome-wide association study of sexual maturation in males and females highlights a role for body mass and menarche loci in male puberty. Hum. Mol. Genet. 16, 4452–4464. doi: 10.1093/hmg/ddu150
David, P. H., Reichenbach, L., Savelieva, I., Vartapetova, N., and Potemkina, R. (2007). Women’s reproductive health needs in Russia: what can we learn from an intervention to improvepost-abortion care? Health Policy Plan. 22, 83–94. doi: 10.1093/heapol/czm003
Day, F. R., Thompson, D. J., Helgason, H., Chasman, D. I., Finucane, H., Sulem, P., et al. (2017). Genomic analyses identify hundreds of variants associated with AAM and support a role for puberty timing in cancer risk. Nat. Genet. 49, 834–841.
Delahanty, R. J., Beeghly-Fadiel, A., Long, J. R., Gao, Y. T., Lu, W., Xiang, Y. B., et al. (2013). Evaluation of GWAS-identified genetic variants for age at menarche among Chinese women. Hum. Reprod. 4, 1135–1143. doi: 10.1093/humrep/det011
Douglas, N. I, Pavlova, T. U., Burtseva, T. E., Rad, Y. G., Petrova, P. G., and Odland, J. Ø (2014). Women’s reproductive health in the Sakha Republic (Yakutia). Int. J. Circumpolar Health 30:73. doi: 10.3402/ijch.v73.25872
Edwards, T. L., Michels, K. A., Hartmann, K. E., and Velez Edwards, D. R. (2013). BET1L and TNRC6B associate with uterine fibroid risk among European Americans. Hum. Genet. 132, 943–953. doi: 10.1007/s00439-013-1306-1303
Eggert, S. L., Huyck, K. L., Somasundaram, P., Kavalla, R., Stewart, E. A., Lu, A. T., et al. (2012). Genome-wide linkage and association analyses implicate FASN in predisposition to uterine leiomyomata. Am. J. Hum. Genet. 91, 621–628. doi: 10.1016/j.ajhg.2012.08.009
Elks, C. E., Perry, J. R. B., Sulem, P., Chasman, D. I., Franceschini, N., He, C., et al. (2010). Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat. Genet. 12, 1077–1085.
Feng, Y., Zhao, X., Zhou, C., Yang, L., Liu, Y., and Bian, C. (2013). The associations between the Val158Met in the catechol-O-methyltransferase (COMT) gene and the risk of uterine leiomyoma (ULM). Gene 529, 296–299. doi: 10.1016/j.gene.2013.07.019
Fernandez-Rhodes, L., Demerath, E. W., Cousminer, D. L., Tao, R., Dreyfus, J. G., Esko, T., et al. (2013). Association of adiposity genetic variants with menarche timing in 92,105 women of European descent. Am. J. Epidemiol. 3, 451–460.
Gajbhiye, R., Fung, J. N., and Montgomery, G. W. (2018). Complex genetics of female fertility. NPJ Genom. Med. 3:29. doi: 10.1038/s41525-018-0068-1
Gallagher, C. S., Mäkinen, N., Harris, H. R., Rahmioglu, N., Uimari, O., and Cook, J. P. (2019). Genome-wide association and epidemiological analyses reveal common genetic origins between uterine leiomyomata and endometriosis. Nat. Commun. 10:4857.
Graff, M., Ngwa, J. S., Workalemahu, T., Homuth, G., Schipf, S., Teumer, A., et al. (2013). Genome-wide analysis of BMI in adolescents and young adults reveals additional insight into the effects of genetic loci over the life course. Hum. Mol. Genet. 22, 3597–3607.
Gudbjartsson, D. F., Walters, G. B., Thorleifsson, G., Stefansson, H., Halldorsson, B. V., Zusmanovich, P., et al. (2008). Many sequence variants affecting diversity of adult human height. Nat. Genet. 5, 609–615. doi: 10.1038/ng.122
He, C., Kraft, P., Buring, J. E., Chen, C., Hankison, S. E., Pare, G., et al. (2010). A large-scale candidate-gene association study of age at menarche and age at natural menopause. Hum. Genet. 5, 515–527. doi: 10.1007/s00439-010-0878-4
He, C., Kraft, P., Chen, C., Buring, J. E., Paré, G., Hankinson, S. E., et al. (2009). Genome-wide association studies identify novel loci associated with age at menarche and age at natural menopause. Nat. Genet. 6, 724–728. doi: 10.1038/ng.385
He, M., Xu, M., Zhang, B., Liang, J., Chen, P., Lee, J. Y., et al. (2015). Meta-analysis of genome-wide association studies of adult height in East Asians identifies 17 novel loci. Hum. Mol. Genet. 6, 1791–1800.
Hellwege, J. N., Jeff, J. M., Wise, L. A., Scott, C. G., Melissa, W., and Katherine, E. H. (2017). A multi-stage genome-wide association study of uterine fibroids in African Americans. Hum. Genet. 136, 1363–1373. doi: 10.1007/s00439-017-1836-1831
Lango Allen, H., Estrada, K., Lettre, G., Berndt, S. I., Weedon, M. N., Rivadeneira, F., et al. (2010). Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 7317, 832–838.
Leinonen, J. T., Surakka, I., Havulinna, A. S., Kettunen, J., Luoto, R., Salomaa, V., et al. (2012). Association of LIN28B with adult adiposity-related traits in females. PLoS One 11:e48785. doi: 10.1371/journal.pone.0048785
Lou, X. Y., Chen, G. B., Yan, L., Ma, J. Z., Zhu, J., Elston, R. C., et al. (2007). A generalized combinatorial approach for detecting gene by gene and gene by environment interactions with application to nicotine dependence. Am. J. Hum. Genet. 80, 1125–1137. doi: 10.1086/518312
Luoto, R., Kaprio, J., Rutanen, E. M., Taipale, P., Perola, M., and Koskenvuo, M. (2000). Heritability and risk factors of uterine fibroids-the Finnish Twin Cohort study. Maturitas 37, 15–26. doi: 10.1016/S0378-5122(00)00160-162
Mahachie, J. M., Cattaert, T., Van Lishout, F., Gusareva, E. S., and Van Steen, K. (2012). Lower-order effects adjustment in quantitative traits model-based multifactor dimensionality reduction. PLoS One 7:e29594. doi: 10.1371/journal.pone.0029594
McWilliams, M. M., and Chennathukuzhi, V. M. (2017). Recent advances in uterine fibroid etiology. Semin. Reprod. Med. 35, 181–189. doi: 10.1055/s-0037-1599090
Ong, K. K., Elks, C. E., Li, S., Zhao, J. H., Luan, J., Andersen, B., et al. (2009). Genetic variation in LIN28B is associated with the timing of puberty. Nat. Genet. 6, 729–733. doi: 10.1038/ng.382
Ong, K. K., Elks, C. E., Wills, A. K., Wong, A., Wareham, N. J., Loos, R. J., et al. (2011). Associations between the pubertal timing-related variant in LIN28B and BMI vary across the life course. J. Clin. Endocrinol. Metab. 1, E125–E129.
Perry, J. R., Day, F., Elks, C. E., Sulem, P., Thompson, D. J., Ferreira, T., et al. (2014). Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature 7520, 92–97.
Perry, J. R., Stolk, L., Franceschini, N., Lunetta, K. L., Zhai, G., McArdle, P. F., et al. (2009). Meta-analysis of genome-wide association data identifies two loci influencing age at menarche. Nat. Genet. 6, 648–650.
Ponomarenko, I. V. (2018). Selection of polymorphic loci for association analysis in genetic-epidemiological studies. Res. Result Med. Pharm. 2, 40–54. doi: 10.18413/2313-8955-2018-4-2-0-5
Ponomarenko, I. V., Reshetnikov, E. A., Altuchova, O. B., Polonikov, A. V., Sorokina, I. N., Yermachenko, A., et al. (2019). Association of genetic polymorphisms with age at menarche in Russian women. Gene 686, 228–236. doi: 10.1016/j.gene.2018.11.042
Purcell, S., Neale, B., Todd-Brown, K., Lori, T., Manuel, A. R. F., and David, B. (2007). PLINK: a tool set for wholegenome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Rafnar, T., Gunnarsson, B., Stefansson, O. A., Sulem, P., Ingason, A., Frigge, M. L., et al. (2018). Variants associating with uterine leiomyoma highlight genetic background shared by various cancers and hormone-related traits. Nat. Commun. 9:3636. doi: 10.1038/s41467-018-05428-5426
Ruth, K. S., Campbell, P. J., Chew, S., Lim, E. M., Hadlow, N., Stuckey, B. G., et al. (2016). Genome-wide association study with 1000 genomes imputation identifies signals for nine sex hormone-related phenotypes. Eur. J. Hum. Genet. 24, 284–290. doi: 10.1038/ejhg.2015.102
Samadi, A. R., Lee, N. C., Flanders, W. D., Boring, J. R. III, and Parris, E. B. (1996). Risk factors for self-reported uterine fibroids: a case-control study. Am. J. Public. Health 86, 858–862. doi: 10.2105/ajph.86.6.858
Sapkota, Y., Steinthorsdottir, V., Morris, A. P., Fassbender, A., Rahmioglu, N., De Vivo, I., et al. (2017). Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat. Comunn. 8:15539. doi: 10.1038/ncomms15539
Schneider, A. P. II, Zainer, C. M., Kubat, C. K., Mullen, N. K., and Windisch, A. K. (2014). The breast cancer epidemic: 10 facts. Linacre Q. 81, 244–277. doi: 10.1179/2050854914Y.0000000027
Sedgh, G., Henshaw, S. K., Singh, S., Bankole, A., and Drescher, J. (2007). Legal abortion worldwide: incidence and recent trends. Perspect. Sex. Reprod. Health. 39, 216–225. doi: 10.1363/3921607
Shen, Q., Shu, L., Luo, H., Hu, X., and Zhu, X. (2017). The use of mifepristone in abortion associated with an increased risk of uterine leiomyomas. Medicine 96:e6680. doi: 10.1097/md.0000000000006680
Snieder, H., MacGregor, A. J., and Spector, T. D. (1998). Genes control the cessation of a woman’s reproductive life: a twin study of hysterectomy and age at menopause. J. Clin. Endocrinol. Metab. 83, 1875–1880. doi: 10.1210/jc.83.6.1875
Song, L., Shen, L., Mandiwa, C., Yang, S., Liang, Y., Yuan, J., et al. (2017). Induced and spontaneous abortion and risk of uterine fibroids. J. Womens Health 26, 76–82. doi: 10.1089/jwh.2016.5913
Sorokina, I. N., Rudykh, N. A., Bezmenova, I. N., and Polyakova, I. S. (2018). Population genetic characteristics and genetic epidemiological research of candidate genes associations with multifactorial diseases. Res. Results Biomed. 4, 20–30. doi: 10.18413/2313-8955-2018-4-4-0-3
Speliotes, E. K., Willer, C. J., Berndt, S. I., Monda, K. L., Thorleifsson, G., Jackson, A. U., et al. (2010). Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 11, 937–948.
Stewart, E. A., Cookson, C. L., Gandolfo, R. A., and Schulze-Rath, R. (2017). Epidemiology of uterine fibroids: a systematic review. BJOG 124, 1501–1512. doi: 10.1111/1471-0528.14640
Stolk, L., Perry, J. R., Chasman, D. I., He, C., Mangino, M., Sulem, P., et al. (2012). Meta-analyses identify 13 novel loci associated with age at menopause and highlights DNA repair and immune pathways. Nat. Genet. 44, 260–268.
Sulem, P., Gudbjartsson, D. F., Rafnar, T., Holm, H., Olafsdottir, E. J., Olafsdottir, G. H., et al. (2009). Genome-wide association study identifies sequence variants on 6q21 associated with age at menarche. Nat. Genet. 6, 734–738. doi: 10.1038/ng.383
Terry, K. L., De Vivo, I., Hankinson, S. E., and Missmer, S. A. (2010). Reproductive characteristics and risk of uterine leiomyomata. Fertil. Steril. 94, 2703–2707. doi: 10.1016/j.fertnstert.2010.04.065
Tian, Y., Zhao, H., Chen, H., Peng, Y., Cui, L., Du, Y., et al. (2016). Variants in FSHB are associated with polycystic ovary syndrome and luteinizing hormone level in Han Chinese women. J. Clin. Endocrinol. Metab. 101, 2178–2184. doi: 10.1210/jc.2015-3776
Tsialikas, J., and Romer-Seibert, J. (2015). LIN28: roles and regulation in development and beyond. Development 14, 2397–2404. doi: 10.1242/dev.117580
Ulitsky, I., and Bartel, B. (2013). LincRNAs: genomics, evolution, and mechanisms. Cell 1, 26–46. doi: 10.1016/j.cell.2013.06.020
Välimäki, N., Kuisma, H., Pasanen, A., Heikinheimo, O., Sjöberg, J., Bützow, R., et al. (2018). Genetic predisposition to uterine leiomyoma is determined by loci for genitourinary development and genome stability. eLife 7:e37110. doi: 10.7554/eLife.37110
Velez Edwards, D. R., Baird, D. D., and Hartmann, K. E. (2013). Association of age at menarche with increasing number of fibroids in a cohort of women who underwent standardized ultrasound assessment. Am. J. Epidem. 178, 426–433. doi: 10.1093/aje/kws585
Vikhlyaeva, E. M., Khodzhaeva, Z. S., and Fantschenko, N. D. (1995). Familial predisposition to uterine leiomyomas. Int. J. Gynaecol. Obstet. 51, 127–131. doi: 10.1016/0020-7292(95)02533-I
Widén, E., Ripatti, S., Cousminer, D. L., Surakka, I., Lappalainen, T., Järvelin, M. R., et al. (2010). Distinct variants at LIN28B influence growth in height from birth to adulthood. Am. J. Hum. Genet. 5, 773–782. doi: 10.1016/j.ajhg.2010.03.010
Wise, L. A., and Laughlin-Tommaso, S. K. (2016). Epidemiology of uterine fibroids - from menarche to menopause. Clin. Obstet. Gynecol. 59, 2–24. doi: 10.1097/GRF.0000000000000164
Wise, L. A., Palmer, J. R., Harlow, B. L., Spiegelman, D., Stewart, E. A., Adams-Campbell, L. L., et al. (2004). Reproductive factors, hormonal contraception, and risk of uterine leiomyomata in African-American women: a prospective study. Am. J. Epidemiol. 159, 113–123. doi: 10.1093/aje/kwh016
Wise, L. A., Ruiz-Narvaez, E. A., Palmer, J. R., Cozier, Y. C., Tandon, A., Patterson, N., et al. (2012). African ancestry and genetic risk for uterine leiomyomata. Am. J. Epidemiol. 176, 1159–1168. doi: 10.1093/aje/kws276
Wood, A. R., Esko, T., Yang, J., Vedantam, S., Pers, T. H., Gustafsson, S., et al. (2014). Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 11, 1173–1186.
Zhu, H., Shyh-Chang, N., Segre, A. V., Shinoda, G., Shah, S. P., Einhorn, W. S., et al. (2011). The Lin28/let-7 axis regulates glucose metabolism. Cell 1, 81–94. doi: 10.1016/j.cell.2011.08.033
Keywords: age at menarche, association, gene–environment interactions, gene–gene interaction, single nucleotide polymorphism, uterine leiomyoma
Citation: Ponomarenko I, Reshetnikov E, Polonikov A, Verzilina I, Sorokina I, Yermachenko A, Dvornyk V and Churnosov M (2021) Candidate Genes for Age at Menarche Are Associated With Uterine Leiomyoma. Front. Genet. 11:512940. doi: 10.3389/fgene.2020.512940
Received: 18 November 2019; Accepted: 14 October 2020;
Published: 22 January 2021.
Edited by:
Maxim B. Freidin, King’s College London, United KingdomReviewed by:
Elena Bragina, Research Institute of Medical Genetics, Tomsk National Research Medical Center of the Russian Academy of Sciences, RussiaCopyright © 2021 Ponomarenko, Reshetnikov, Polonikov, Verzilina, Sorokina, Yermachenko, Dvornyk and Churnosov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evgeny Reshetnikov, cmVzaGV0bmlrb3ZAYnN1LmVkdS5ydQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.