 Qianyun Guo†
Qianyun Guo† Xunxun Feng
Xunxun Feng Yujie Zhou
Yujie Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 23 September 2020
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.01020
This article is part of the Research Topic Genetics of Familial Hypercholesterolemia: New Insight View all 17 articles
Autosomal dominant familial hypercholesterolemia (FH) affects approximately 1/250, individuals and potentially leads to elevated blood cholesterol and a significantly increased risk of atherosclerosis. Along with improvements in detection and the increased early diagnosis and treatment, the serious burden of FH on families and society has become increasingly apparent. Since FH is strongly associated with proprotein convertase subtilisin/kexin type 9 (PCSK9), increasing numbers of studies have focused on finding effective diagnostic and therapeutic methods based on PCSK9. At present, as PCSK9 is one of the main pathogenic FH genes, its contribution to FH deserves more explorative research.
A hereditary propensity for elevated serum levels of low-density lipoprotein cholesterol (LDL-C) that leading to cardiovascular disease (CVD) is typical FH and affects approximately 1 in 250 individuals. Currently recognized FH-inducing variants that lead to disease occur mostly in the apolipoprotein B (APOB), PCSK9, and LDL receptor (LDLR) genes. While most cases are caused by LDLR variants, they may also be caused by autosomal dominant variants of PCSK9, although less frequently (Defesche et al., 2017; Viigimaa et al., 2018; Sarraju and Knowles, 2019). PCSK9 encodes the proprotein convertase subtilisin/kexin type 9 protein which binds LDL and regulates the numbers of LDLR. As the third gene implicated in FH, PCSK9 has been found to reduce the uptake of LDL-C in the liver by increasing the endosomal and lysosomal degradation of LDLR (Maxwell and Breslow, 2005; Stoekenbroek and Kastelein, 2018). Moreover, experimental studies indicate that PCSK9 might independently accelerate atherosclerosis by enhancing inflammation, endothelial dysfunction, and hypertension (Urban et al., 2013). PCSK9 variants implicated in autosomal dominant hypercholesterolemia (ADH) were first identified in 2003. At the time, they were believed to induce abnormal cholesterol metabolism through undefined mediators. Since then, PCSK9 variants have been extensively investigated. Among these, loss-of-function (LOF) variants are associated with reduced LDL-C levels and coronary artery disease (CAD) risk, while gain-of-function (GOF) variants diminish LDLR levels, thereby inducing hypercholesterolemia (Abifadel et al., 2009). To prevent morbidity and mortality, it is crucial to diagnose FH early. Predominantly, laboratory tests or medical traits including family history are used in diagnosis, however, genetic screening of pathological variants might make definite diagnoses more accessible (Hovingh et al., 2013). To explore PCSK9 variants in FH and their biological roles, we provide a detailed discussion and summary of novel PCSK9 variants. We also discuss the important interplay between PCSK9 and other molecules. Finally, this review sums up novel treatment strategies aimed at PCSK9 in FH to pave the way for future investigative studies.
Seventeen years ago, through the sequencing of 12 exons of the PCSK9 gene, Abifadel et al. (2003) opened the door to researching the role of the PCSK9 gene in FH which suggested a novel mechanism of dyslipidemia. They identified a T to A substitution in exon 2 at nucleotide 625 resulting in a non-synonymous change at codon 127 of arginine for the conserved serine p.(S127R) and the substitution of T to C, resulting in an amino acid substitution of phenylalanine to leucine p.(F216L) among different families. In the following 5 years, many studies have been carried out to discover new variants in PCSK9 (Figure 1). In addition, different populations have different types and frequencies of PCSK9 variants, so we also provide a summary table of PCSK9 variants with their population frequencies and LDL-C levels from previous studies (Tables 1, 2).
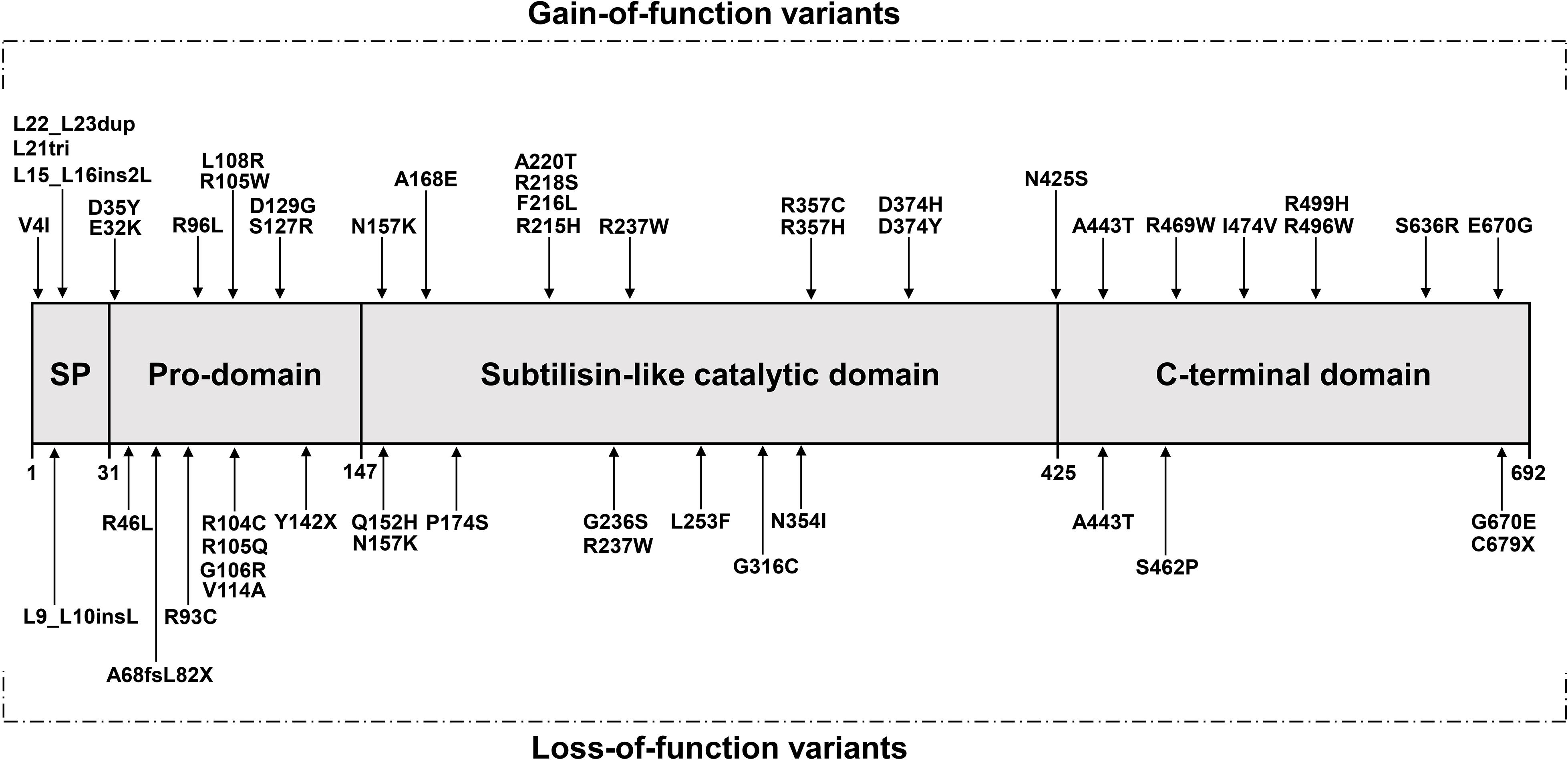
Figure 1. The protein’s domain structure of PCSK9 and location of variants (adopted and redrawn from Maxwell and Breslow, 2005; Soutar and Naoumova, 2007) SP, Signal peptide.
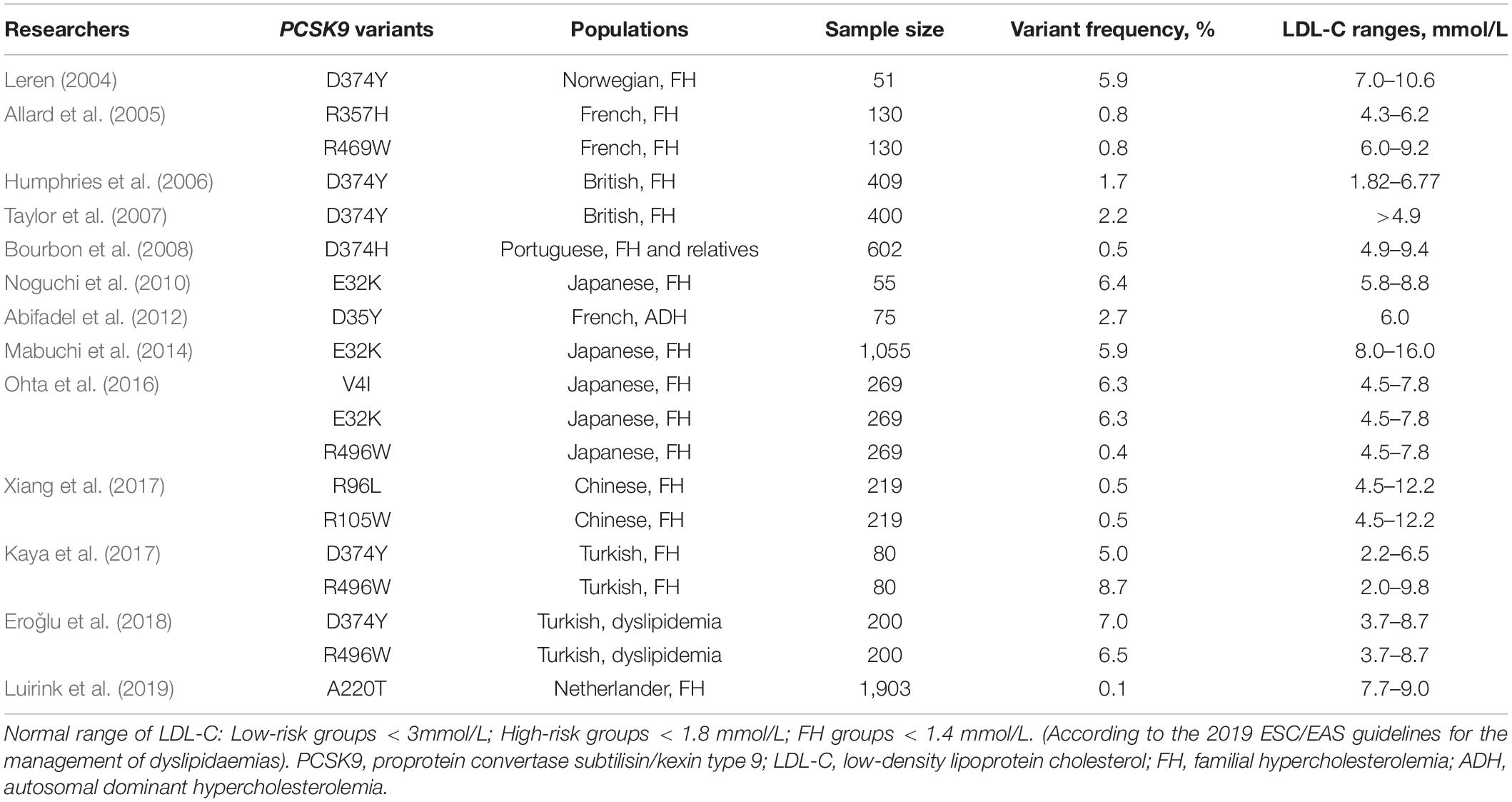
Table 1. Summary of GOF variants among different populations.
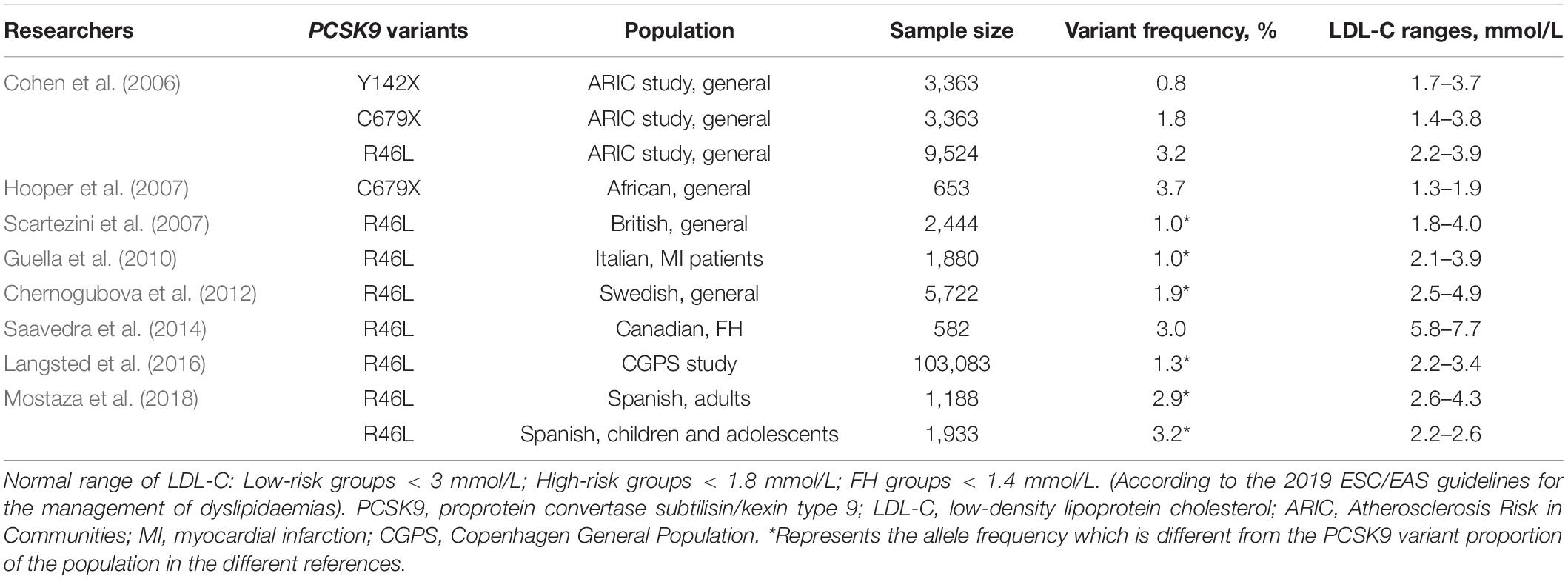
Table 2. Summary of LOF variants among different populations.
In 2004, Timms et al. (2004) recognized a single G→T nucleotide variant present on the K1173 haplotype variant resulting in the non-synonymous p.(D374Y). Leren identified an asparagine to lysine substitution at position 157, p.(N157K) (Leren, 2004). Benjannet et al. (2004) subsequently identified two further PCSK9 variants occurring naturally in the gene pool, namely p.(R218S) and p.(R237W). In addition, Shioji et al. (2004) demonstrated the association between total cholesterol (TC) and LDL-C levels with exon 9/I474V or intron 1/C(-161)T polymorphisms.
In 2005, the amount of plasma LDL-C and the extent of atherosclerosis in coronary arteries were shown by Chen et al. (2005) to depend on a single nucleotide polymorphism resulting in p.(E670G). Subsequently, Allard et al. (2005) found four heterozygous missense variations resulting in changes in PCSK9, namely p.(R218S), p.(R357H), p.(R469W), and p.(A443T) in the coding region and intronic junctions of the PCSK9 gene after analyzing 130 patients with ADH.
From 2006 to 2008, Pisciotta et al. (2006) sequenced multiple genes including PCSK9 in two patients with heterozygous LDLR genes who were diagnosed with homozygous FH (HoFH). They identified one patient with the p.(R496W) variant from her mother and another one with the p.(N425S) variant likely from her deceased father. Patients with mutated LDLR were speculated to suffer worse pathological symptoms if they had an uncommon missense PCSK9 variant. One year later, Bourbon et al. (2008) identified a p.(D374H) variant of PCSK9 in 184 patients and 418 relatives in Portugal, and found that the number of confirmed FH patients increased through cascade screening. They recommended that if patients received appropriate treatment to restrain progress of premature CAD after early identification of FH, their life expectancy and quality of life could be improved. FH patients from New Zealand were observed to have two novel missense variants, namely p.(D129G) and p.(A168E) and together with two established variants from South Africa, namely p.(S127R) and p.(R237W), these gave a PCSK9 variant total of four discovered by Homer et al. (2008) They found that the inhibition of LDLR mediated by PCSK9 occurred independently of PCSK9 release or autocatalytic destruction and speculated that PCSK9 might play a role in cells. In addition, five novel PCSK9 variants were found by Cameron et al. (2008), including p.(R215H), p.(G236S), p.(N354I), p.(A245T), and p.(R272Q), with p.(R215H) resulting in GOF and hypercholesterolemia. Since their effect on the internalization of LDL-C was similar to that of the wild-type (WT) PCSK9, it was demonstrated that p.(R272Q) and p.(A245T) were non-pathological aberrations which maintained normal PCSK9 performance, though both variants were identified in a hypercholesterolemic group. After studying the promoter variant of the PCSK9 gene, Blesa et al. (2008) identified a c.-332C > A variant in the region of the PCSK9 gene promoter that increased transcription of PCSK9. In fact, the variant could result in a 2.5-fold increase in transcription compared to WT, thereby leading to ADH. At the end of the year, Abifadel et al. (2008) found familial combined hyperlipidemia present in two families, with variants of two leucines [designated p.(L15_L16ins2L) and p.(L21tri)] in family members who also had elevated LDL-C, thus suggesting an association.
Cohen et al. (2005) investigated 128 people whose plasma LDL-C was low and identified two nonsense variants including p.(C679X) and p.(Y142X) through determining the coding region sequence of PCSK9. These had been recognized as LOF variants since 2005 as they had opposite effects compared to the GOF PCSK9 variants. Subsequently, in 2006, while examining 38 people with hypocholesterolemia and 25 heterozygotes with hypercholesterolemia, all from different families, Berge et al. (2006) screened for PCSK9 variants. They identified four variants including p.(R46L), p.(G106R), p.(R237W), and p.(N157K) among the two groups. In the same year, Kotowski et al. (2006) employed oligonucleotide hybridization on a chip and deoxyribonucleic acid (DNA) sequencing to identify p.(L253F), p.(A443T), and p.(R46L) missense mutants which were significantly correlated with diminished amounts of LDL-C. Moreover, after sequencing for variations in 403 Caucasians, Yue et al. (2006) identified a c.43_44insCTG mutant that mediated LDL-C level reductions in normal people. In 2007, Fasano et al. (2007) identified a novel variant in exon 1 (c.202delG) of a single nucleotide deletion from one heterozygous patient, that resulted in messenger ribonucleic acid (mRNA) generating peptide frameshift and truncation at p.(A68fsL82X). In the next year, Miyake et al. (2008) found 33 PCSK9 gene sequence variants, with one at p.(R93C) which had a 0.051 R93C allele prevalence in low vs. high LDL-C groups, and was associated with low LDL-C levels. Meanwhile, the LOF variants p.(G236S) and p.(N354I) were described by Cameron et al. (2008). The p.(G236S) variant prevented PCSK9 release from the endoplasmic reticulum (ER) while p.(N354I) resulted in failure of PCSK9 to undergo autocatalytic cleavage.
In the past 10 years, with the emergence of related studies on PCSK9 inhibitors and other treatment of patients with statin-intolerant hyperlipidemia, including patients with FH, less attention has been paid to the analysis of PCSK9 variants than previously (Figure 1).
With respect to LOF variants of PCSK9, Cameron et al. (2009) identified the novel variant p.(S462P) in exon 9 of the PCSK9 gene and suggested that this variant, like the p.(G236S) and p.(N354I) variants, prevented normal C-terminal domain folding precluding release of the protein from the ER. Another LOF variant, the double-mutant p.(R104C)/p.(V114A), which improved the clearance rate of LDL-C (Cariou et al., 2009). Mayne et al. (2011) identified a p.(Q152H) substitution in a French-Canadian family which resulted in a 48% reduction in LDL-C concentration compared with non-carriers. Slimani et al. (2012) investigated PCSK9 and LDLR variants in Tunisian FH families, identifying a new missense variation p.(P174S) that appeared to cause decreased levels of LDL-C with respect to the LDLR genotype in six family members. In Pakistan, FH family members who carried the p.(R105Q) variant had lower levels of total cholesterol suggesting that this variant might cause LOF (Ahmed et al., 2013). Through targeted next-generation sequencing (NGS), the variants p.(R93C) and p.(G670E) were identified by Lee et al. (2017) in nine patients with very low levels of LDL-C. In addition, one carrier of the heterozygous missense variant p.(G316C) that was associated with hypocholesterolemia and steatosis was found (Di Filippo et al., 2017).
Regarding PCSK9 GOF variants, two novel variants were found in 75 patients with ADH and normal APOB and LDLR genes by Abifadel et al. (2012) namely p.(D35Y) and p.(L108R) substitutions. The variants were absent from individuals with normal cholesterol levels and were associated with the presence of ADH in families. The authors also assessed the quantitative and qualitative effects of these PCSK9 variants on lipoprotein granules and their effect on the activity of cholesterol ester transfer protein (CETP). This was the first report of the impact of PCSK9 variants on high density lipoprotein (HDL)-mediated cholesterol release from cells. Ohta et al. (2016) found that the p.(V4I) variant of PCSK9 in 269 clinically diagnosed FH heterozygotes was related to a remarkably increased CAD prevalence accompanied by increasing levels of LDL-C, although the levels of serum lipids and the CAD prevalence between p.(V4I) carriers and non-carriers without LDLR variants remained similar. Di Taranto et al. (2017) demonstrated two GOF variants, p.(S636R) and p.(R357C), considered to cause FH. In their study, the p.(S636R) and p.(R357C) variants showed a lower binding capacity for WT PCSK9 than LDLR. They also found a further two mutants with uncharacterized effects on FH disease progression. Elbitar et al. (2018) sequenced exons from 13 French FH families and discovered a PCSK9 variant at p.(R96C) in a patient with a severe phenotype from a family with the p.(A3396T) APOB variant. They demonstrated that this was the first reported compound heterozygote having both APOB and PCSK9 variants. In addition, Sánchez-Hernández et al. (2019) found a novel PCSK9 variant, p.(R499H) of in two unrelated FH patients from Spain and Italy. This variant resulted in decreased expression of LDLR from in vitro functional assays. There were two related studies on PCSK9 in FH children: DECOPIN and GoTCHA. In the DECOPIN project, Ibarretxe et al. (2018) identified 49 different variants including two in PCSK9 parents and children with FH. In the child-to-parent study, p.(R496W) and p.(L22_L23dup) were found to be pathogenic in families. Luirink et al. (2019) analyzed a total of 1903 FH children with molecular assays in the GoTCHA study. They also conducted candidate gene sequencing in HoFH children whose LDL-C levels were above the lowest level measured in pediatric patients with HoFH. In their study, a GOF variant of PCSK9 p.(A220T) was found in two related patients.
In studies of the PCSK9 variants in Asian populations, Noguchi et al. (2010) identified the p.(E32K) variant in Japanese people. In their study, the frequency of the p.(E32K) variant in clinical FH was significantly higher than that in a control group (6.42 vs. 1.71%). Although this GOF variant might have milder effects than the p.(D374Y) and LDLR variants, it could worsen lipid conditions in true homozygous or double heterozygous probands with LDLR variants. In China, Xiang et al. (2017) identified two PCSK9 variants in FH patients including p.(R96L) and p.(R105W) from the central southern region of China. As GOF variants, these two variants might cause increased LDLR degradation, resulting in a decrease of LDL-C clearance, eventually giving rise to hypercholesterolemia. In their study, they also found another nine novel variants which included seven LDLR variants and two APOB variants.
PCSK9 belongs to the proprotein convertase family of serine proteases. Biological and gene analyses have shown that PCSK9 is a vital regulator of LDLR proteins which in turn, regulate plasma LDL-C. Through indirectly causing the degradation of LDLR, PCSK9 normally downregulates LDLR with LOF PCSK9 variants resulting in low levels of LDL-C in the plasma (Soutar and Naoumova, 2007; Ding and Kullo, 2008; Schmidt et al., 2008; Figure 2).
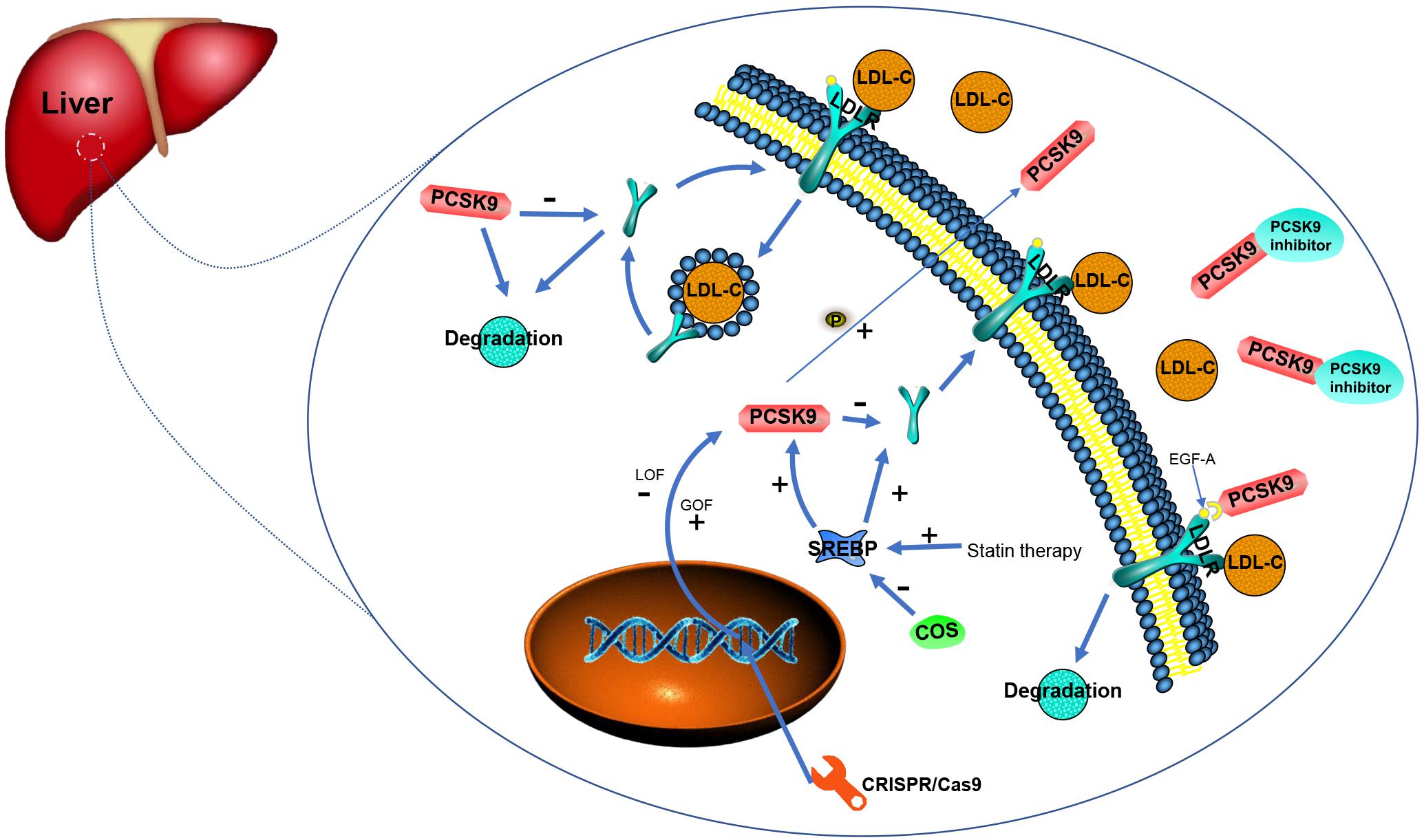
Figure 2. Function and regulation related to PCSK9 and existing/potential therapies for PCSK9 inhibition (adopted and redrawn from Hovingh et al., 2013; German and Shapiro, 2020).
Conversion of PCSK9 is regulated by the sterol regulatory element-binding protein (SREBP) 2, a membrane transcription factor. In addition, transcription to enable PCSK9 production is also regulated by SREBP 2 (Jeong et al., 2008; Wiciński et al., 2017). GOF variants in PCSK9 have been found proven to lead to FH through reducing LDLR protein expression in the liver and decreasing the clearance of circulating cholesterol. Statin therapy is able to inhibit cholesterol biosynthesis followed by the activation of SREBP to increase the expression of LDLR. However, statins also induce the synthesis of PCSK9, leading to the degradation of LDLR (Rashid et al., 2005; Tall, 2006). Transcription of PCSK9, either basal or sterol-regulated, depends on the recognition of a motif in the promoter region by histone nuclear factor P (HINFP). Variants in this region prevent sterol-induced blockade and diminish promoter activity at basal levels in addition to abating promoter activation by SREBP 2 (Li and Liu, 2012). Transcription factors for SREBP depend on binding to the SREBP cleavage-activating protein (SCAP) in the ER membrane. Investigations of SCAP inhibition in a monkey model revealed that when SCAP small interfering RNA (siRNA) encapsulated in lipid nanoparticles is added, both LDL-C levels and PCSK9 are significantly reduced (Murphy et al., 2017). Chitin oligosaccharides (COS) have antioxidant and anti-inflammatory activities and have been recently found to suppress PCSK9 gene expression thereby decreasing the number of LDLRs on the cell surface. Thus, down-regulation of SREBP2 by COS decreases the expression of PCSK9 (Yang et al., 2018).
PCSK9 binds to the epidermal growth factor precursor homology domain A (EGF-A) on extracellular LDLR domains which regulates hepatic LDLRs and leads to their degradation. Asp-374 on the surface of the subtilisin-like catalytic domain of PCSK9 binds the LDLR EGF-A domain. Substitution of the Asp with Tyr (the p.(D374Y) GOF variant enhances PCSK9 affinity for LDLR. This combination of PCSK9 and LDLR may cause a conformational change in LDLR that prevents the recycling of LDLR from the plasma membrane, instead of leading the complex containing PCSK9 and LDLR to degradation (Kwon et al., 2008; Cariou and Dijk, 2020; Martin et al., 2020). Effects on the folding of PCSK9 potentially caused by LOF variants such as p.(Q152H), could trigger increased clearance of LDL-C in the circulation, due to the reduction of LDLR degradation mediated by PCSK9 (Garvie et al., 2016). In tandem, researchers have found that the 314–355 LDLR EGF-A domain and the 153–421 PCSK9 catalytic domain are involved in the interaction between PCSK9 and LDLR that leads to decreased LDLR levels and LDL-C accumulation (Alghamdi et al., 2015). In a strategy using synthetic EGF-A analogs, it has been found that the peptide with the greatest potency enhanced PCSK9 binding affinity compared with WT EGF-A (Schroeder et al., 2014). In addition, a study of the C-terminal domain of PCSK9 created seven de novo mutants of PCSK9 and investigated their affinity toward a calcium-independent mutant of the EGF-A domain. This showed that the p.(G517R), p.(V644R), and p.(V610R) mutants have descending abilities to prevent LDL-C growth in HepG2 cells (Geschwindner et al., 2015). Moreover, ligand-binding (LR) repeats of LDLR have been identified for PCSK9-mediated LDLR degradation and the p.(D203N) variant in the LR5 of full-length LDLR was found to significantly reduce PCSK9 binding (Deng et al., 2019). Interestingly, in studies of post-translational modifications, PCSK9 treatment can also result in ubiquitination of LDLR. Importantly, if the LDLR protein carried variants in its C-terminal ubiquitination sites, it was able to resist PCSK9-mediated degradation (Chen et al., 2011). Recently, it has been observed that phosphorylation may enhance the secretion of PCSK9 from hepatocytes, thus maximizing LDLR degradation through both extracellular and intracellular pathways (Ben Djoudi Ouadda et al., 2019).
The lectin-like ox-LDL receptor-1 (LOX-1) has been shown to play a critical role in inflammatory diseases, including atherosclerosis, where LOX-1 and PCSK9 positively influence each other’s expression and it appears that mitochondria-derived reactive oxygen species (mtROS) may to be important initiators of PCSK9 and LOX-1 expression (Ding et al., 2015). Interestingly, LOX-1 and PCSK9 might be upregulated secondary to induction by ox-LDL in a concentration-dependent manner and ox-LDL-induced human umbilical vein endothelial cell death could be inhibited by PCSK9 siRNA (Wu et al., 2012). Subsequently, in a study of vascular smooth muscle cell mitochondrial DNA (mtDNA) and (SMC)-derived PCSK9 damage, it was found that in the presence of mtROS, there was a positive relationship between mtDNA damage and SMC-derived PCSK9. This interaction leads to cell damage, characterized by apoptosis (Ding et al., 2016). Meanwhile, endothelial cell apoptosis may be repressed through mitogen-activated protein kinase signaling in atherosclerosis by shRNA-PCSK9 targeting of PCSK9 (Li et al., 2017). Investigation of PCSK-induced autophagy mechanisms showed that PCSK9 might be up-regulated in ischemic hearts, thus determining infarct size, cardiac function, and autophagy development through the activation of the ROS-related axis (Ding et al., 2018).
Cascade screening is a vital procedure for identifying people at risk for inherited diseases. For some autosomal dominant diseases, such as FH, relatives can be identified for significant health-affecting interventions, thus significantly increasing life expectancy. Cascade screening is an evidence-based intervention that has been found to reduce cardiovascular morbidity and mortality in the FH population (Knowles et al., 2017). A series of population-based screening and research initiatives, represented by the Dutch Lipid Clinic Network in the Netherlands (DLCNC), have made tremendous progress in raising awareness and treatment of FH (O’Brien et al., 2014). When cascade screening was first introduced in the Netherlands, 2039 relatives of 237 FH cases were found to have FH, 39% of whom had already been receiving treatment. An Australian study also found that among 100 relatives of genetically diagnosed FH patients, 51.4% had pathogenic variants identified by cascade screening. In other recent cases, they have demonstrated other CAD risk factors and have already started using statins without prior diagnosis of FH (Bell et al., 2015; Henderson et al., 2016). In Asia, cascade screening is proving to be an efficient method for the diagnosis of FH in Vietnamese family members. After screening 107 relatives in five FH patients, 56 cases were diagnosed with FH, including three HoFH cases (Truong et al., 2018). Cascade screening is also important in children and adolescents. After analyzing 292 children with FH from 205 parents, researchers found that 20 percent of the parents and 49 percent of the maternal grandparents had an early history of CVD. Similarly, in a Slovenian study, nearly every child diagnosed with FH had a parent who was at high risk for FH. Thus, CVD is still the main cause of FH, and despite the absence of evidence on the long-term safety of drug therapy in childhood, genetic natural history studies confirm the benefits of lifelong low LDL-C levels, so early initiation of cascade screening will facilitate early intervention in the next generation (Galema-Boers et al., 2015; Wiegman et al., 2015; Groselj et al., 2018).
Early detection and treatment of FH in individuals and families could help reduce the morbidity and mortality associated with FH. Indeed, the setting up of an FH database and registration system would be a critical measure to enhance the long-term outcomes of FH patients. Currently, in most countries, diagnosis and treatment models are not adequate (Singh and Bittner, 2015; Chen et al., 2019). However, the cost of DNA sequencing in patients has fallen considerably in the last few years and if the rate of progress continues, the current lack of detection and screening might change (Louter et al., 2017). Below, we list the essential and potential ways of FH detection and screening.
Before NGS was widely applied, the genetic diagnosis of FH mainly relied on Sanger sequencing to identify variants in the APOB and PCSK9 genes. After overcoming many of the scalability barriers faced by clinical laboratories using traditional Sanger methods when performing large-scale DNA sequencing according to the guideline and list of genes reported as incidental or secondary findings of the American College of Medical Genetics (ACMG), NGS has gradually been proved to be a reliable and practical molecular screening method for FH pathogenic genes and become one of the main techniques for PCSK9 detection, while Sanger sequencing is mainly used as a verification method to assist the accuracy of NGS detection (Rehm et al., 2013; Rimbert et al., 2016; Kalia et al., 2017; Pek et al., 2018). Moreover, the guidelines for diagnostic NGS of the EuroGentest and European Society of Human Genetics have emphasized that, although NGS testing was still being explored and developed, NGS technology offered potential overall benefits for the diagnosis of patients’ diseases (Matthijs et al., 2016). As NGS are increasingly used for routine FH diagnosis, FH-related variants may be identified exponentially, so detection of disease-related variants in FH patients is critical for early intervention to reduce the risk of CVD. In a study of a British cohort, compared with multiplex polymerase chain reaction and oligonucleotide arrays, the NGS method has shown great analytical performance with approximately 89–100% concordance to other methods (Reiman et al., 2016; Iacocca et al., 2018). What is more, in the first reported NGS test for variants in clinically suspected FH patients in Singapore, the percentage of detected variants was similar to that of western countries, and although no PCSK9 variants were found, it indicated that NGS technology covering all exons of LDLR might be a better strategy (Pek et al., 2018).
In the 2020 technical standard of ACMG, it is recommended that the laboratory must consider the effectiveness of NGS analysis and augment NGS testing with ancillary assays (Bean et al., 2020). In a study of NGS as a potential method for diagnosing FH, NGS-based testing has been shown to involve lower cost and less labor than traditional sequencing genetic testing. This may provide a way to increase the genetic diagnosis of the current low proportion of FH (Norsworthy et al., 2014). With the continuous development of NGS technology, new NGS-based detection is also gradually being applied to FH screening. In a single target NGS panel study, this new NGS is found to be an effective variant detection method, which can better help to understand the phenotype of FH, and is expected to become a personalized diagnosis method for dyslipidemia (Marmontel et al., 2018). In addition, in the first study of capture-based NGS, this NGS method covering the entire LDLR genome region has improved the efficiency of structural variation detection. This method is expected to comprehensively detect the pathogenic variants of LDLR, APOB, and PCSK9 in FH patients (Hsiung et al., 2018).
To relieve the shortage of intensive procedures which have complicated genetic diagnoses, Wright et al. (2008) developed the Multiplex MassARRAY Spectrometry (iPLEX) and identified 56 variants in a number of genes, including one variant in PCKS9, in DNA samples from 92 FH patients. From this study, it is clear that, while the FH iPLEX test is aimed at screening for FH variants in large-scale targeted populations, it is also suitable for population screening. The Agena iPLEX designed by Chiou and Charng (2017). has been found to have higher specificity and sensitivity to FH gene screening, compared with the traditional diagnostic Sanger sequencing procedure, in detecting DNA from 120 FH patients with defined molecular causes.
In contrast to existing detection methods for genetic abnormalities in FH patients, a high-resolution melting (HRM) analysis known as the polymerase chain reaction (PCR) with modifiable melting conditions, has been proven to be more efficient compared to DNA sequencing. Firstly, it is more cost-effective and timesaving. Secondly, as a sensitive and robust technique, it is capable of detecting new sequence changes that would make sense in cascade screening of HF subjects (Liyanage et al., 2008; Whittall et al., 2010; Pećin et al., 2013). In a Turkish FH cohort using HRM analysis on isolated DNA, it was demonstrated that two PCSK9 GOF variants [p.(D374Y) and p.(R496W)] in FH have a higher frequency and different courses of disease compared to other populations around the world (Kaya et al., 2017).
Given the large number of patients with suspected FH, molecular genetic analysis of the entire genome is time-consuming. A new diagnostic tool in the form of a genotyping DNA microarray chip called FH chip based on arrayed primer extension (APEX), has been proposed to accelerate variant screening in Czech FH patients. In this study, researchers found that the validation phase of this FH chip had 100% sensitivity and 99.1% specificity. They suggested that the FH chip could be implemented for genotyping with features of rapidity, reproducibility, specificity, and economy (Dušková et al., 2011). The Belfast Genetics Laboratory aims to keep abreast of evolving technology by simple and economic genetic testing. An FH biochip array protocol is followed by using samples analyzed for FH variants prior to incorporation into cascade screening. Study results estimate that NGS is five times more expensive than the cost of testing and reporting one sample through the FH Biochip technique due to the latter’s high sensitivity and rapid detection ability (Martin et al., 2016).
Appropriate medical treatment is essential to effectively manage FH, including reducing cardiovascular risk and improving the prognosis of affected patients. The current drug treatments for FH mainly include statins, bile acid-binding resins, and cholesterol absorption inhibitors. Considering that the risk of cardiovascular events in FH patients is significantly increased, timely reduction of LDL-C is essential to reduce the risk of CVD. However, traditional drugs may have difficulty in achieving the goal of decreased blood lipid in FH patients, thus, PCSK9 inhibitors with strong lipid-lowering effects have gradually become a new class of drugs for the treatment of FH (Fala, 2016; Papademetriou et al., 2018). In a meta-study, it was found that when evolucomab is given as a 420 mg monthly dose, LDL-C could be reduced by 54.71%, indicating that evolocumab might be a potential drug for FH patients (Eslami et al., 2017). It has also been found that alirocumab treatment is well-tolerated in heterozygous FH (HeFH) patients and could significantly reduce LDL-C by 12 and 24 weeks while evolocumab could effectively reduce the LDL-C levels in HoFH or severe HeFH patients over a median of 4.1 years (Ginsberg et al., 2019; Santos et al., 2020). In summary, PCSK9 inhibitors can significantly reduce LDL-C, even in FH patients who have not yet achieved their LDL-C goals. Therefore, to ensure that FH patients can receive the PCSK9 inhibitor drug therapies, it is necessary to increase the diagnosis rate and conduct family screening in certain populations (Ogura, 2018).
In recent years, with the advent of siRNA, researchers have designed a new generation of drugs to combat PCSK9 named Inclisiran. It can reduce the concentration of PCSK9 in the body by interfering PSCK9 gene expression in hepatocytes with a double-stranded short sequence of RNA, thereby reducing the degradation of LDLR and enhancing the ability of hepatocytes to eliminate LDL-C to reduce its levels (Dyrbuś et al., 2020). In 2017, Ray et al. (2017) found that Inclisiran could significantly reduce LDL-C after the first subcutaneous injection (>50%) and maintained this level for up to 1 year after the first injection. Compared with the placebo group, the Inclisiran group had no serious adverse reactions. In the subsequent phase III clinical study of Inclisiran, compared with placebo, LDL-C in Inclisiran group was significantly reduced, with the efficacy able to last for more than 18 months (Raal et al., 2020; Ray et al., 2020). The introduction of PCSK9 inhibitors is a milestone in the treatment of FH, and Inclisiran provides a new lipid-lowering technology. The clinical trials of the Inclisiran series of drugs are expected to produce a revolutionary new lipid-lowering drug (German and Shapiro, 2020).
In 2003, the characterization of PCSK9’s LDL-C regulatory functions resulted in a landmark paradigm shift in therapies for hypercholesterolemia (Seidah et al., 2019). There is reason to believe that it might soon be possible to achieve effective cholesterol management by permanently and selectively modifying the genome and inactivating the function of target genes with a single injection (Fazio and Tavori, 2014). Among the nine ways to realize PCSK9 inhibition, clustered regularly interspaced short palindromic repeats (CRISPR) technology might have great potential (Mullard, 2017). Though the CRISPR/Cas9 technology offers flexibility for treating hyperlipidemia and is capable of changing the genome to permanently decrease cholesterol levels, more research is needed before their application in human subjects (Banerjee et al., 2016; Chadwick and Musunuru, 2017).
Ding et al. (2014) use CRISPR/CRISPR-associated (Cas) technology to obtain a PCSK9-targeting CRISPR guide RNA and Cas 9 expression in murine livers with an adenovirus to effectively disrupt murine PCSK9 genes, resulting in reduced levels of cholesterol and PCSK9, but elevated amounts of liver LDLR. This momentous finding in genome editing might have potential for the therapy and prevention of CVD in humans. Subsequently, severely diminished levels of blood PCSK9 are seen in mice with humanized livers with almost 50% of highly specific human hepatocyte variants, thus demonstrating the safety and effectiveness of CRISPR-Cas9 in reducing human PCSK9 levels (Wang et al., 2016). In addition, CRISPR/Cas9-mediated genome editing decreases PCSK9 levels in both human and murine hypercholesterolemic models, which could be a valuable tool in the search for novel therapeutic approaches against hypercholesterolemia (Carreras et al., 2019).
It is to get into mammalian cells without a vehicle. Additionally, gene editing using CRISPR/Cas9 technology, which is both sizeable and based mainly on DNA, mRNA, or protein, also poses a significant challenge. Although viral vectors have higher delivery efficiency and adeno-associated viral vectors have recognized efficiency in atherosclerosis research, their biosecurity issues hinder their wide application. Based on the nanocarrier-delivered CRISPR/Cas9 system, Zhang et al. (2019) use a triple targeting strategy to produce a LOF variant in the PCSK9 gene and this strategy might be a potential target therapy for CVD without side effects (Jarrett et al., 2018). In addition, precise knock-in of specific nucleotide changes have proven to be inefficient in non-proliferating cells in vivo. Chadwick et al. (2017) therefore, used PCSK9 base editing, which has the ability to generate alterations in genes without the need for breaks in double-stranded DNA. This demonstrates the ability to precisely introduce therapeutically relevant nucleotide variants into the genome in somatic tissues in adult mammals.
In order to identify ADH disease loci besides PCSK9, LDLR and APOB, on the basis of a genome-wide scan and linkage analysis, Marques-Pinheiro et al. (2010) report a large lineage from France and hypothesized the involvement of a fourth gene, named HCHOLA4, at 16q22.1. In their study, it is shown that other ADH genes do exist, while they also identify nine affected families with no linkage to the HCHOLA4 locus nor the three known genes. Apart from PCSK9, LDLR, and APOB, it has been found that there is a significant relationship between ADH and rs965814 G allele mapping to 8q24.22 through genome-wide analysis on 15 family members (Cenarro et al., 2011).
Exome sequencing was performed by Fouchier et al. (2014) along with parametric linkage analysis in a family with FH4 (ADH with unmutated genes for PCSK9, APOB, and LDLR) to identify a fourth ADH relevant locus, leading to the mapping of the ADH locus at 4p13. In addition, ADH associates with four signal-transducing adaptor family member 1 (STAP1) variants, including p.(E97D), p.(L69S), p.(I71T), and p.(D207N). Out of all the STAP1 missense variants analyzed by bioinformatics analysis and available structural information in one man and his two siblings, p.(P176S) which might play a role of affecting cholesterol homeostasis has been associated with FH as the likely most damaging variant (Blanco-Vaca et al., 2018). However, after STAP1 was analyzed in hypercholesterolemic patients from Germany, whose variants are negative in canonical FH genes, it was concluded that the positive predictive value of STAP1 analysis would be comparably small and in order to characterize STAP1 contributions to FH pathogenesis, its molecular interactions should be explored through in vitro functional studies (Danyel et al., 2019). Moreover, despite noting that FH patients carrying STAP1 have lower LDL-C levels than non-carriers, Lamiquiz-Moneo et al. (2020) found no phenotypic penetrance of their genome when exploring potential associations between phenotype and STAP1 variants. What is more, studies of mouse models and carriers of STAP1 variants, indicate that STAP1 might not be the FH or LDL-C modulating gene and should not be considered for FH genetic screening (Loaiza et al., 2020).
The unique mechanism of action of PCSK9 and the identification of its genetic variants have brought a new therapeutic target to FH. Among the numerous variant sites, both GOF and LOF might become new breakthrough points of treatment. Since the use of PCSK9 inhibitors has emerged outside conventional FH treatment strategies, their gradually increasing applications in clinical practice have not only brought good news to FH patients but also new hope for patients with hyperlipidemia that are at high risk of CAD. It is reasonable to believe that, with continuous research progress on PCSK9 in FH, more therapeutic methods and diagnostic methods with superior accuracy, safety, and economy will be applied in FH patients. Interactions of PCSK9 with various cellular components, based on its unique features and influence on the body, are also worthy of further research and discussion. Last, but not least, as a disease related to genetic variant, there are undoubtedly more undiscovered genetic loci related to FH that are worth exploring in addition to the existing FH genes.
QG and XF wrote this review and YZ revised it. All authors read and approved the final manuscript.
This study was supported by grants from the National Key Research and Development Program of China (2017YFC0908800) and Beijing Municipal Administration of Hospitals’ Ascent Plan (DFL20150601) and Mission plan (SML20180601).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ADH, autosomal dominant hypercholesterolemia; APEX, arrayed primer extension; APOB, apolipoprotein B; CAD, coronary artery disease; Cas, CRISPR/CRISPR-associated; CETP, cholesterol ester transfer protein; COS, chitosan oligosaccharides; CRISPR, clustered regularly interspaced short palindromic repeats; CVD, cardiovascular disease; EGF-A, epidermal growth factor precursor homology domain A; ER, endoplasmic reticulum; FH, familial hypercholesterolemia; GOF, gain-of-function; HDL, high density lipoprotein; HeFH, heterozygous FH; HINFP, histone nuclear factor P; HoFH, homozygous FH; HRM, high-resolution melting; LDL-C, low-density lipoprotein cholesterol; LDLR, LDL receptors; LOF, loss-of-function; LOX-1, lectin-like ox-LDL receptor-1; LR, ligand-binding; iPLEX, Multiplex MassARRAY Spectrometry; mRNA, messenger ribonucleic acid; mtDNA, mitochondrial DNA; mtROS, mitochondria-derived reactive oxygen species; NGS, next-generation sequencing; PCR, polymerase chain reaction; PCSK9, proprotein convertase subtilisin/kexin type 9; SCAP, SREBP cleavage-activating protein; SERBP, sterol regulatory element–binding protein; siRNA, small interfering RNA; STAP1, signal-transducing adaptor family member 1; TC, total cholesterol; WT, wild-type.
Abifadel, M., Bernier, L., Dubuc, G., Nuel, G., Rabès, J. P., Bonneau, J., et al. (2008). A PCSK9 variant and familial combined hyperlipidaemia. J. Med. Genet. 45, 780–786. doi: 10.1136/jmg.2008.059980
Abifadel, M., Guerin, M., Benjannet, S., Rabès, J. P., Le Goff, W., Julia, Z., et al. (2012). Identification and characterization of new gain-of-function mutations in the PCSK9 gene responsible for autosomal dominant hypercholesterolemia. Atherosclerosis 223, 394–400. doi: 10.1016/j.atherosclerosis.2012.04.006
Abifadel, M., Rabès, J. P., Devillers, M., Munnich, A., Erlich, D., Junien, C., et al. (2009). Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum. Mutat. 30, 520–529. doi: 10.1002/humu.20882
Abifadel, M., Varret, M., Rabès, J. P., Allard, D., Ouguerram, K., Devillers, M., et al. (2003). Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156. doi: 10.1038/ng1161
Ahmed, W., Whittall, R., Riaz, M., Ajmal, M., Sadeque, A., Ayub, H., et al. (2013). The genetic spectrum of familial hypercholesterolemia in Pakistan. Clin. Chim. Acta. 421, 219–225. doi: 10.1016/j.cca.2013.03.017
Alghamdi, R. H., O’Reilly, P., Lu, C., Gomes, J., Lagace, T. A., and Basak, A. (2015). LDL-R promoting activity of peptides derived from human PCSK9 catalytic domain (153-421): design, synthesis and biochemical evaluation. Eur. J. Med. Chem. 92, 890–907. doi: 10.1016/j.ejmech.2015.01.022
Allard, D., Amsellem, S., Abifadel, M., Trillard, M., Devillers, M., Luc, G., et al. (2005). Novel mutations of the PCSK9 gene cause variable phenotype of autosomal dominant hypercholesterolemia. Hum. Mutat. 26:497. doi: 10.1002/humu.9383
Banerjee, Y., Santos, R. D., Al-Rasadi, K., and Rizzo, M. (2016). Targeting PCSK9 for therapeutic gains: Have we addressed all the concerns? Atherosclerosis 248, 62–75. doi: 10.1016/j.atherosclerosis.2016.02.018
Bean, L. J. H., Funke, B., Carlston, C. M., Gannon, J. L., Kantarci, S., Krock, B. L., et al. (2020). Diagnostic gene sequencing panels: from design to report-a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 22, 453–461. doi: 10.1038/s41436-019-0666-z
Bell, D. A., Pang, J., Burrows, S., Bates, T. R., van Bockxmeer, F. M., Hooper, A. J., et al. (2015). Effectiveness of genetic cascade screening for familial hypercholesterolaemia using a centrally co-ordinated clinical service: an Australian experience. Atherosclerosis 239, 93–100. doi: 10.1016/j.atherosclerosis.2014.12.036
Ben Djoudi Ouadda, A., Gauthier, M. S., Susan-Resiga, D., Girard, E., Essalmani, R., Black, M., et al. (2019). Ser-Phosphorylation of PCSK9 (Proprotein Convertase Subtilisin-Kexin 9) by Fam20C (Family With Sequence Similarity 20, Member C) Kinase Enhances Its Ability to Degrade the LDLR (Low-Density Lipoprotein Receptor). Arterioscler. Thromb. Vasc. Biol. 39, 1996–2013. doi: 10.1161/ATVBAHA.119.313247
Benjannet, S., Rhainds, D., Essalmani, R., Mayne, J., Wickham, L., Jin, W., et al. (2004). NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 279, 48865–48875. doi: 10.1074/jbc.M409699200
Berge, K. E., Ose, L., and Leren, T. P. (2006). Missense mutations in the PCSK9 gene are associated with hypocholesterolemia and possibly increased response to statin therapy. Arterioscler. Thromb. Vasc. Biol. 26, 1094–1100. doi: 10.1161/01.ATV.0000204337.81286.1c
Blanco-Vaca, F., Martín-Campos, J. M., Pérez, A., and Fuentes-Prior, P. (2018). A rare STAP1 mutation incompletely associated with familial hypercholesterolemia. Clin. Chim. Acta. 487, 270–274. doi: 10.1016/j.cca.2018.10.014
Blesa, S., Vernia, S., Garcia-Garcia, A. B., Martinez-Hervas, S., Ivorra, C., Gonzalez-Albert, V., et al. (2008). A new PCSK9 gene promoter variant affects gene expression and causes autosomal dominant hypercholesterolemia. J. Clin. Endocrinol. Metab. 93, 3577–3583. doi: 10.1210/jc.2008-0269
Bourbon, M., Alves, A. C., Medeiros, A. M., Silva, S., and Soutar, A. K. (2008). Investigators of Portuguese FH Study. Familial hypercholesterolaemia in Portugal. Atherosclerosis 196, 633–642. doi: 10.1016/j.atherosclerosis.2007.07.019
Cameron, J., Holla, O. L., Laerdahl, J. K., Kulseth, M. A., Ranheim, T., Rognes, T., et al. (2008). Characterization of novel mutations in the catalytic domain of the PCSK9 gene. J. Intern. Med. 263, 420–431. doi: 10.1111/j.1365-2796.2007.01915.x
Cameron, J., Holla, ØL., Laerdahl, J. K., Kulseth, M. A., Berge, K. E., and Leren, T. P. (2009). Mutation S462P in the PCSK9 gene reduces secretion of mutant PCSK9 without affecting the autocatalytic cleavage. Atherosclerosis 203, 161–165. doi: 10.1016/j.atherosclerosis.2008.10.007
Cariou, B., and Dijk, W. (2020). EGF-A peptides: A promising strategy for PCSK9 inhibition. Atherosclerosis 292, 204–206. doi: 10.1016/j.atherosclerosis.2019.11.010
Cariou, B., Ouguerram, K., Zaïr, Y., Guerois, R., Langhi, C., Kourimate, S., et al. (2009). PCSK9 dominant negative mutant results in increased LDL catabolic rate and familial hypobetalipoproteinemia. Arterioscler. Thromb. Vasc. Biol. 29, 2191–2197. doi: 10.1161/ATVBAHA.109.194191
Carreras, A., Pane, L. S., Nitsch, R., Madeyski-Bengtson, K., Porritt, M., Akcakaya, P., et al. (2019). In vivo genome and base editing of a human PCSK9 knock-in hypercholesterolemic mouse model. BMC Biol. 17:4. doi: 10.1186/s12915-018-0624-2
Cenarro, A., García-Otín, A. L., Tejedor, M. T., Solanas, M., Jarauta, E., Junquera, C., et al. (2011). A presumptive new locus for autosomal dominant hypercholesterolemia mapping to 8q24.22. Clin. Genet. 79, 475–481. doi: 10.1111/j.1399-0004.2010.01485.x
Chadwick, A. C., and Musunuru, K. (2017). Treatment of Dyslipidemia Using CRISPR/Cas9 Genome Editing. Curr. Atheroscler. Rep. 19:32. doi: 10.1007/s11883-017-0668-8
Chadwick, A. C., Wang, X., and Musunuru, K. (2017). In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler. Thromb. Vasc. Biol. 37, 1741–1747. doi: 10.1161/ATVBAHA.117.309881
Chen, P., Chen, X., and Zhang, S. (2019). Current Status of Familial Hypercholesterolemia in China: A Need for Patient FH Registry Systems. Front. Physiol. 10:280. doi: 10.3389/fphys.2019.00280
Chen, S. N., Ballantyne, C. M., Gotto, A. M. Jr., Tan, Y., Willerson, J. T., and Marian, A. J. (2005). A common PCSK9 haplotype, encompassing the E670G coding single nucleotide polymorphism, is a novel genetic marker for plasma low-density lipoprotein cholesterol levels and severity of coronary atherosclerosis. J. Am. Coll Cardiol. 45, 1611–1619. doi: 10.1016/j.jacc.2005.01.051
Chen, Y., Wang, H., Yu, L., Yu, X., Qian, Y. W., Cao, G., et al. (2011). Role of ubiquitination in PCSK9-mediated low-density lipoprotein receptor degradation. Biochem. Biophys. Res. Commun. 415, 515–518. doi: 10.1016/j.bbrc.2011.10.110
Chernogubova, E., Strawbridge, R., Mahdessian, H., Mälarstig, A., Krapivner, S., Gigante, B., et al. (2012). Common and low-frequency genetic variants in the PCSK9 locus influence circulating PCSK9 levels. Arterioscler. Thromb. Vasc. Biol. 32, 1526–1534. doi: 10.1161/ATVBAHA.111.240549
Chiou, K. R., and Charng, M. J. (2017). Detection of common sequence variations of familial hypercholesterolemia in Taiwan using DNA mass spectrometry. J. Clin. Lipidol. 11, 386–393.e6. doi: 10.1016/j.jacl.2016.12.014
Cohen, J., Pertsemlidis, A., Kotowski, I. K., Graham, R., Garcia, C. K., and Hobbs, H. H. (2005). Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165. doi: 10.1038/ng1509
Cohen, J. C., Boerwinkle, E., Mosley, T. H. Jr., and Hobbs, H. H. (2006). Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 354, 1264–1272. doi: 10.1056/NEJMoa054013
Danyel, M., Ott, C. E., Grenkowitz, T., Salewsky, B., Hicks, A. A., Fuchsberger, C., et al. (2019). Evaluation of the role of STAP1 in Familial Hypercholesterolemia. Sci. Rep. 9:11995. doi: 10.1038/s41598-019-48402-y
Defesche, J. C., Gidding, S. S., Harada-Shiba, M., Hegele, R. A., Santos, R. D., and Wierzbicki, A. S. (2017). Familial hypercholesterolaemia. Nat. Rev. Dis. Primers. 3;17093. doi: 10.1038/nrdp.2017.93
Deng, S. J., Alabi, A., Gu, H. M., Adijiang, A., Qin, S., and Zhang, D. W. (2019). Identification of amino acid residues in the ligand binding repeats of LDL receptor important for PCSK9 binding. J. Lipid. Res. 60, 516–527. doi: 10.1194/jlr.M089193
Di Filippo, M., Vokaer, B., and Seidah, N. G. (2017). A case of hypocholesterolemia and steatosis in a carrier of a PCSK9 loss-of-function mutation and polymorphisms predisposing to nonalcoholic fatty liver disease. J. Clin. Lipidol. 11, 1101–1105. doi: 10.1016/j.jacl.2017.06.005
Di Taranto, M. D., Benito-Vicente, A., Giacobbe, C., Uribe, K. B., Rubba, P., Etxebarria, A., et al. (2017). Identification and in vitro characterization of two new PCSK9 Gain of Function variants found in patients with Familial Hypercholesterolemia. Sci. Rep. 7:15282. doi: 10.1038/s41598-017-15543-x
Ding, K., and Kullo, I. J. (2008). Molecular population genetics of PCSK9: a signature of recent positive selection. Pharmacogenet. Genom. 18, 169–179. doi: 10.1097/FPC.0b013e3282f44d99
Ding, Q., Strong, A., Patel, K. M., Ng, S. L., Gosis, B. S., Regan, S. N., et al. (2014). Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 115, 488–492. doi: 10.1161/CIRCRESAHA.115.304351
Ding, Z., Liu, S., Wang, X., Deng, X., Fan, Y., Shahanawaz, J., et al. (2015). Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc. Res. 107, 556–567. doi: 10.1093/cvr/cvv178
Ding, Z., Liu, S., Wang, X., Mathur, P., Dai, Y., Theus, S., et al. (2016). Cross-Talk Between PCSK9 and Damaged mtDNA in Vascular Smooth Muscle Cells: Role in Apoptosis. Antiox. Redox. Sign. 25, 997–1008. doi: 10.1089/ars.2016.6631
Ding, Z., Wang, X., Liu, S., Shahanawaz, J., Theus, S., Fan, Y., et al. (2018). PCSK9 expression in the ischaemic heart and its relationship to infarct size, cardiac function, and development of autophagy. Cardiovasc. Res. 114, 1738–1751. doi: 10.1093/cvr/cvy128
Dušková, L., Kopečková, L., Jansová, E., Tichý, L., Freiberger, T., Zapletalová, P., et al. (2011). An APEX-based genotyping microarray for the screening of 168 mutations associated with familial hypercholesterolemia. Atherosclerosis 216, 139–145. doi: 10.1016/j.atherosclerosis.2011.01.023
Dyrbuś, K., Ga̧sior, M., Penson, P., Ray, K. K., and Banach, M. (2020). Inclisiran-New hope in the management of lipid disorders? J. Clin. Lipidol. 14, 16–27. doi: 10.1016/j.jacl.2019.11.001
Elbitar, S., Susan-Resiga, D., Ghaleb, Y., El Khoury, P., Peloso, G., Stitziel, N., et al. (2018). New Sequencing technologies help revealing unexpected mutations in Autosomal Dominant Hypercholesterolemia. Sci. Rep. 8:1943. doi: 10.1038/s41598-018-20281-9
Eroǧlu, Z., Tetik Vardarlı, A., Düzgün, Z., Gündüz, C., Bozok Çetintaş, V., and Kayıkçıoǧlu, M. (2018). Case-control study on PCSK9 R496W (rs374603772) and D374Y (rs137852912) mutations in Turkish patients with primary dyslipidemia. Anatol. J. Cardiol. 19, 334–340. doi: 10.14744/AnatolJCardiol.2018.86648
Eslami, S. M., Nikfar, S., Ghasemi, M., and Abdollahi, M. (2017). Does Evolocumab, as a PCSK9 Inhibitor, Ameliorate the Lipid Profile in Familial Hypercholesterolemia Patients? A Meta-Analysis of Randomized Controlled Trials. J. Pharm. Pharm. Sci. 20, 81–96. doi: 10.18433/J36C8N
Fala, L. (2016). Repatha (Evolocumab): Second PCSK9 Inhibitor Approved by the FDA for Patients with Familial Hypercholesterolemia. Am. Health Drug Benefits 9, 136–139.
Fasano, T., Cefalù, A. B., Di Leo, E., Noto, D., Pollaccia, D., Bocchi, L., et al. (2007). A novel loss of function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. Arterioscler Thromb Vasc. Biol. 27, 677–681. doi: 10.1161/01.ATV.0000255311.26383.2f
Fazio, S., and Tavori, H. (2014). Peeking into a cool future: genome editing to delete PCSK9 and control hypercholesterolemia in a single shot. Circ. Res. 115, 472–474. doi: 10.1161/CIRCRESAHA.114.304575
Fouchier, S. W., Dallinga-Thie, G. M., Meijers, J. C., Zelcer, N., Kastelein, J. J., Defesche, J. C., et al. (2014). Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ. Res. 115, 552–555. doi: 10.1161/CIRCRESAHA.115.304660
Galema-Boers, J. M., Versmissen, J., Roeters van Lennep, H. W., Dusault-Wijkstra, J. E., Williams, M., Roeters, et al. (2015). Cascade screening of familial hypercholesterolemia must go on. Atherosclerosis 242, 415–417. doi: 10.1016/j.atherosclerosis.2015.07.020
Garvie, C. W., Fraley, C. V., Elowe, N. H., Culyba, E. K., Lemke, C. T., Hubbard, B. K., et al. (2016). Point mutations at the catalytic site of PCSK9 inhibit folding, autoprocessing, and interaction with the LDL receptor. Protein Sci. 25, 2018–2027. doi: 10.1002/pro.3019
German, C. A., and Shapiro, M. D. (2020). Small Interfering RNA Therapeutic Inclisiran: A New Approach to Targeting PCSK9. BioDrugs 34, 1–9. doi: 10.1007/s40259-019-00399-6
Geschwindner, S., Andersson, G. M., Beisel, H. G., Breuer, S., Hallberg, C., Kihlberg, B. M., et al. (2015). Characterisation of de novo mutations in the C-terminal domain of proprotein convertase subtilisin/kexin type 9. Protein Eng. Des. Sel. 28, 117–125. doi: 10.1093/protein/gzv008
Ginsberg, H. N., Tuomilehto, J., Hovingh, G. K., Cariou, B., Santos, R. D., Brown, A. S., et al. (2019). Impact of Age on the Efficacy and Safety of Alirocumab in Patients with Heterozygous Familial Hypercholesterolemia. Cardiovasc. Drugs Ther. 33, 69–76. doi: 10.1007/s10557-019-06852-6
Groselj, U., Kovac, J., Sustar, U., Mlinaric, M., Fras, Z., Podkrajsek, K. T., et al. (2018). Universal screening for familial hypercholesterolemia in children: The Slovenian model and literature review. Atherosclerosis 277, 383–391. doi: 10.1016/j.atherosclerosis.2018.06.858
Guella, I., Asselta, R., Ardissino, D., Merlini, P. A., Peyvandi, F., Kathiresan, S., et al. (2010). Effects of PCSK9 genetic variants on plasma LDL cholesterol levels and risk of premature myocardial infarction in the Italian population. J. Lipid. Res. 51, 3342–3349. doi: 10.1194/jlr.M010009
Henderson, R., O’Kane, M., McGilligan, V., and Watterson, S. (2016). The genetics and screening of familial hypercholesterolaemia. J. Biomed. Sci. 23:39. doi: 10.1186/s12929-016-0256-1
Homer, V. M., Marais, A. D., Charlton, F., Laurie, A. D., Hurndell, N., Scott, R., et al. (2008). Identification and characterization of two non-secreted PCSK9 mutants associated with familial hypercholesterolemia in cohorts from New Zealand and South Africa. Atherosclerosis 196, 659–666. doi: 10.1016/j.atherosclerosis.2007.07.022
Hooper, A. J., Marais, A. D., Tanyanyiwa, D. M., and Burnett, J. R. (2007). The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 193, 445–448. doi: 10.1016/j.atherosclerosis.2006.08.039
Hovingh, G. K., Davidson, M. H., Kastelein, J. J., and O’Connor, A. M. (2013). Diagnosis and treatment of familial hypercholesterolaemia. Eur. Heart J. 34, 962–971. doi: 10.1093/eurheartj/eht015
Hsiung, Y. C., Lin, P. C., Chen, C. S., Tung, Y. C., Yang, W. S., Chen, P. L., et al. (2018). Identification of a novel LDLR disease-causing variant using capture-based next-generation sequencing screening of familial hypercholesterolemia patients in Taiwan. Atherosclerosis 277, 440-447. doi: 10.1016/j.atherosclerosis.2018.08.022
Humphries, S. E., Whittall, R. A., Hubbart, C. S., Maplebeck, S., Cooper, J. A., Soutar, A. K., et al. (2006). Genetic causes of familial hypercholesterolaemia in patients in the UK: relation to plasma lipid levels and coronary heart disease risk. J. Med. Genet. 43, 943–949. doi: 10.1136/jmg.2006.038356
Iacocca, M. A., Chora, J. R., Carrié, A., Freiberger, T., Leigh, S. E., Defesche, J. C., et al. (2018). ClinVar database of global familial hypercholesterolemia-associated DNA variants. Hum. Mutat. 39, 1631–1640. doi: 10.1002/humu.23634
Ibarretxe, D., Rodríguez-Borjabad, C., Feliu, A., Bilbao, J., Masana, L., and Plana, N. (2018). Detecting familial hypercholesterolemia earlier in life by actively searching for affected children: The DECOPIN project. Atherosclerosis 278, 210–216. doi: 10.1016/j.atherosclerosis.2018.09.039
Jarrett, K. E., Lee, C., De Giorgi, M., Hurley, A., Gillard, B. K., Doerfler, A. M., et al. (2018). Somatic Editing of Ldlr With Adeno-Associated Viral-CRISPR Is an Efficient Tool for Atherosclerosis Research. Arterioscler Thromb Vasc. Biol. 38, 1997–2006. doi: 10.1161/ATVBAHA.118.311221
Jeong, H. J., Lee, H. S., Kim, K. S., Kim, Y. K., Yoon, D., and Park, S. W. (2008). Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J. Lipid. Res. 49, 399–409. doi: 10.1194/jlr.M700443-JLR200
Kalia, S. S., Adelman, K., Bale, S. J., Chung, W. K., Eng, C., Evans, J. P., et al. (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 19, 249–255. doi: 10.1038/gim.2016.190
Kaya, E., Kayıkçıoǧlu, M., Tetik Vardarlı, A., Eroǧlu, Z., Payzın, S., and Can, L. (2017). PCSK 9 gain-of-function mutations (R496W and D374Y) and clinical cardiovascular characteristics in a cohort of Turkish patients with familial hypercholesterolemia. Anatol. J. Cardiol. 18, 266–272. doi: 10.14744/AnatolJCardiol.2017.7654
Knowles, J. W., Rader, D. J., and Khoury, M. J. (2017). Cascade Screening for Familial Hypercholesterolemia and the Use of Genetic Testing. JAMA 318, 381–382. doi: 10.1001/jama.2017.8543
Kotowski, I. K., Pertsemlidis, A., Luke, A., Cooper, R. S., Vega, G. L., Cohen, J. C., et al. (2006). A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am. J. Hum. Genet. 78, 410–422. doi: 10.1086/500615
Kwon, H. J., Lagace, T. A., McNutt, M. C., Horton, J. D., and Deisenhofer, J. (2008). Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. U S A. 105, 1820–1825. doi: 10.1073/pnas.0712064105
Lamiquiz-Moneo, I., Restrepo-Córdoba, M. A., Mateo-Gallego, R., Bea, A. M., Del Pino Alberiche-Ruano, M., García-Pavía, P., et al. (2020). Predicted pathogenic mutations in STAP1 are not associated with clinically defined familial hypercholesterolemia. Atherosclerosis 292, 143–151. doi: 10.1016/j.atherosclerosis.2019.11.025
Langsted, A., Nordestgaard, B. G., Benn, M., Tybjærg-Hansen, A., and Kamstrup, P. R. (2016). PCSK9 R46L Loss-of-Function Mutation Reduces Lipoprotein(a), LDL Cholesterol, and Risk of Aortic Valve Stenosis. J. Clin. Endocrinol. Metab. 101, 3281–3287. doi: 10.1210/jc.2016-1206
Lee, C. J., Lee, Y., Park, S., Kang, S. M., Jang, Y., Lee, J. H., et al. (2017). Rare and common variants of APOB and PCSK9 in Korean patients with extremely low low-density lipoprotein-cholesterol levels. PLoS One. 12:e0186446. doi: 10.1371/journal.pone.0186446
Leren, T. P. (2004). Mutations in the PCSK9 gene in Norwegian subjects with autosomal dominant hypercholesterolemia. Clin. Genet. 65, 419–422. doi: 10.1111/j.0009-9163.2004.0238.x
Li, H., and Liu, J. (2012). The novel function of HINFP as a co-activator in sterol-regulated transcription of PCSK9 in HepG2 cells. Biochem. J. 443, 757–768. doi: 10.1042/BJ20111645
Li, J., Liang, X., Wang, Y., Xu, Z., and Li, G. (2017). Investigation of highly expressed PCSK9 in atherosclerotic plaques and ox-LDL-induced endothelial cell apoptosis. Mol. Med. Rep. 16, 1817–1825. doi: 10.3892/mmr.2017.6803
Liyanage, K. E., Hooper, A. J., Defesche, J. C., Burnett, J. R., and van Bockxmeer, F. M. (2008). High-resolution melting analysis for detection of familial ligand-defective apolipoprotein B-100 mutations. Ann. Clin. Biochem. 45(Pt 2), 170–176. doi: 10.1258/acb.2007.007077
Loaiza, N., Hartgers, M. L., Reeskamp, L. F., Balder, J. W., Rimbert, A., Bazioti, V., et al. (2020). Taking One Step Back in Familial Hypercholesterolemia: STAP1 Does Not Alter Plasma LDL (Low-Density Lipoprotein) Cholesterol in Mice and Humans. Arterioscler Thromb Vasc. Biol. 40, 973–985. doi: 10.1161/ATVBAHA.119.313470
Louter, L., Defesche, J., and Roeters van Lennep, J. (2017). Cascade screening for familial hypercholesterolemia: Practical consequences. Atheroscler Suppl. 30, 77–85. doi: 10.1016/j.atherosclerosissup.2017.05.019
Luirink, I. K., Braamskamp, M. J. A. M., Wiegman, A., Hartgers, M. L., Sjouke, B., Defesche, J. C., et al. (2019). The clinical and molecular diversity of homozygous familial hypercholesterolemia in children: Results from the GeneTics of clinical homozygous hypercholesterolemia (GoTCHA) study. J. Clin. Lipidol. 13, 272–278. doi: 10.1016/j.jacl.2018.12.003
Mabuchi, H., Nohara, A., Noguchi, T., Kobayashi, J., Kawashiri, M. A., Inoue, T., et al. (2014). Genotypic and phenotypic features in homozygous familial hypercholesterolemia caused by proprotein convertase subtilisin/kexin type 9 (PCSK9) gain-of-function mutation. Atherosclerosis 236, 54–61. doi: 10.1016/j.atherosclerosis.2014.06.005
Marmontel, O., Charričre, S., Simonet, T., Bonnet, V., Dumont, S., Mahl, M., et al. (2018). Single, short in-del, and copy number variations detection in monogenic dyslipidemia using a next-generation sequencing strategy. Clin. Genet. 94, 132–140. doi: 10.1111/cge.13250
Marques-Pinheiro, A., Marduel, M., Rabčs, J. P., Devillers, M., Villéger, L., Allard, D., et al. (2010). A fourth locus for autosomal dominant hypercholesterolemia maps at 16q22.1. Eur. J. Hum. Genet. 18, 1236–1242. doi: 10.1038/ejhg.2010.94
Martin, R., Latten, M., Hart, P., Murray, H., Bailie, D. A., Crockard, M., et al. (2016). Genetic diagnosis of familial hypercholesterolaemia using a rapid biochip array assay for 40 common LDLR, APOB and PCSK9 mutations. Atherosclerosis 254, 8–13. doi: 10.1016/j.atherosclerosis.2016.09.061
Martin, W. R., Lightstone, F. C., and Cheng, F. (2020). In Silico Insights into Protein-protein Interaction Disruptive Mutations in the PCSK9-LDLR complex. Int. J. Mol. Sci. 21:E1550. doi: 10.3390/ijms21051550
Matthijs, G., Souche, E., Alders, M., Corveleyn, A., Eck, S., Feenstra, I., et al. (2016). Guidelines for diagnostic next-generation sequencing. Eur. J. Hum. Genet. 24, 2–5. doi: 10.1038/ejhg.2015.226
Maxwell, K. N., and Breslow, J. L. (2005). Proprotein convertase subtilisin kexin 9: the third locus implicated in autosomal dominant hypercholesterolemia. Curr. Opin. Lipidol. 16, 167–172. doi: 10.1097/01.mol.0000162321.31925.a3
Mayne, J., Dewpura, T., Raymond, A., Bernier, L., Cousins, M., Ooi, T. C., et al. (2011). Novel loss-of-function PCSK9 variant is associated with low plasma LDL cholesterol in a French-Canadian family and with impaired processing and secretion in cell culture. Clin. Chem. 57, 1415–1423. doi: 10.1373/clinchem.2011.165191
Miyake, Y., Kimura, R., Kokubo, Y., Okayama, A., Tomoike, H., Yamamura, T., et al. (2008). Genetic variants in PCSK9 in the Japanese population: rare genetic variants in PCSK9 might collectively contribute to plasma LDL cholesterol levels in the general population. Atherosclerosis 196, 29–36. doi: 10.1016/j.atherosclerosis.2006.12.035
Mostaza, J. M., Lahoz, C., Salinero-Fort, M. A., de Dios, O., Castillo, E., González-Alegre, T., et al. (2018). R46L polymorphism in the PCSK9 gene: Relationship to lipid levels, subclinical vascular disease, and erectile dysfunction. J. Clin. Lipidol. 12, 1039.e–1046.e. doi: 10.1016/j.jacl.2018.04.004
Mullard, A. (2017). Nine paths to PCSK9 inhibition. Nat. Rev. Drug. Discov. 16, 299–301. doi: 10.1038/nrd.2017.83
Murphy, B. A., Tadin-Strapps, M., Jensen, K., Mogg, R., Liaw, A., Herath, K., et al. (2017). siRNA-mediated inhibition of SREBP cleavage-activating protein reduces dyslipidemia in spontaneously dysmetabolic rhesus monkeys. Metabolism 71, 202–212. doi: 10.1016/j.metabol.2017.02.015
Noguchi, T., Katsuda, S., Kawashiri, M. A., Tada, H., Nohara, A., Inazu, A., et al. (2010). The E32K variant of PCSK9 exacerbates the phenotype of familial hypercholesterolaemia by increasing PCSK9 function and concentration in the circulation. Atherosclerosis 210, 166–172. doi: 10.1016/j.atherosclerosis.2009.11.018
Norsworthy, P. J., Vandrovcova, J., Thomas, E. R., Campbell, A., Kerr, S. M., Biggs, J., et al. (2014). Targeted genetic testing for familial hypercholesterolaemia using next generation sequencing: a population-based study. BMC Med. Genet. 15:70. doi: 10.1186/1471-2350-15-70
O’Brien, E. C., Roe, M. T., Fraulo, E. S., Peterson, E. D., Ballantyne, C. M., Genest, J., et al. (2014). Rationale and design of the familial hypercholesterolemia foundation CAscade SCreening for Awareness and DEtection of Familial Hypercholesterolemia registry. Am. Heart J. 167, 342.e–349.e. doi: 10.1016/j.ahj.2013.12.008
Ogura, M. (2018). PCSK9 inhibition in the management of familial hypercholesterolemia. J. Cardiol. 71, 1–7. doi: 10.1016/j.jjcc.2017.07.002
Ohta, N., Hori, M., Takahashi, A., Ogura, M., Makino, H., Tamanaha, T., et al. (2016). Proprotein convertase subtilisin/kexin 9 V4I variant with LDLR mutations modifies the phenotype of familial hypercholesterolemia. J Clin. Lipidol. 10, 547.e–555.e. doi: 10.1016/j.jacl.2015.12.024
Papademetriou, V., Stavropoulos, K., Papadopoulos, C., Koutsampasopoulos, K., Dimitriadis, K., and Tsioufis, K. (2018). Role of PCSK9 Inhibitors in High Risk Patients with Dyslipidemia: Focus on Familial Hypercholesterolemia. Curr. Pharm. Des. 24, 3647–3653. doi: 10.2174/138161282466618101012465
Pećin, I., Whittall, R., Futema, M., Sertić, J., Reiner, Z., Leigh, S. E., et al. (2013). Mutation detection in Croatian patients with familial hypercholesterolemia. Ann. Hum. Genet. 77, 22–30. doi: 10.1111/j.1469-1809.2012.00735.x
Pek, S. L. T., Dissanayake, S., Fong, J. C. W., Lin, M. X., Chan, E. Z. L., Tang, J. I., et al. (2018). Spectrum of mutations in index patients with familial hypercholesterolemia in Singapore: Single center study. Atherosclerosis 269, 106-116. doi: 10.1016/j.atherosclerosis.2017.12.028
Pisciotta, L., Priore Oliva, C., Cefalù, A. B., Noto, D., Bellocchio, A., Fresa, R., et al. (2006). Additive effect of mutations in LDLR and PCSK9 genes on the phenotype of familial hypercholesterolemia. Atherosclerosis 186, 433–440. doi: 10.1016/j.atherosclerosis.2005.08.015
Raal, F. J., Kallend, D., Ray, K. K., Turner, T., Koenig, W., Wright, R. S., et al. (2020). Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 382, 1520–1530. doi: 10.1056/NEJMoa1913805
Rashid, S., Curtis, D. E., Garuti, R., Anderson, N. N., Bashmakov, Y., Ho, Y. K., et al. (2005). Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc. Natl. Acad. Sci. U S A. 102, 5374–5379. doi: 10.1073/pnas.0501652102
Ray, K. K., Landmesser, U., Leiter, L. A., Kallend, D., Dufour, R., Karakas, M., et al. (2017). Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 376, 1430–1440. doi: 10.1056/NEJMoa1615758
Ray, K. K., Wright, R. S., Kallend, D., Koenig, W., Leiter, L. A., Raal, F. J., et al. (2020). Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 382, 1507–1519. doi: 10.1056/NEJMoa1912387
Rehm, H. L., Bale, S. J., Bayrak-Toydemir, P., Berg, J. S., Brown, K. K., Deignan, J. L., et al. (2013). ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 15, 733–747. doi: 10.1038/gim.2013.92
Reiman, A., Pandey, S., Lloyd, K. L., Dyer, N., Khan, M., Crockard, M., et al. (2016). Molecular testing for familial hypercholesterolaemia-associated mutations in a UK-based cohort: development of an NGS-based method and comparison with multiplex polymerase chain reaction and oligonucleotide arrays. Ann. Clin. Biochem. 53, 654–662. doi: 10.1177/0004563216629170
Rimbert, A., Pichelin, M., Lecointe, S., Marrec, M., Le Scouarnec, S., Barrak, E., et al. (2016). Identification of novel APOB mutations by targeted next-generation sequencing for the molecular diagnosis of familial hypobetalipoproteinemia. Atherosclerosis 250, 52-56. doi: 10.1016/j.atherosclerosis.2016.04.010
Saavedra, Y. G., Dufour, R., Davignon, J., and Baass, A. (2014). PCSK9 R46L, lower LDL, and cardiovascular disease risk in familial hypercholesterolemia: a cross-sectional cohort study. Arterioscler Thromb Vasc. Biol. 34, 2700–2705. doi: 10.1161/ATVBAHA.114.304406
Sánchez-Hernández, R. M., Di Taranto, M. D., Benito-Vicente, A., Uribe, K. B., Lamiquiz-Moneo, I., Larrea-Sebal, A., et al. (2019). The Arg499His gain-of-function mutation in the C-terminal domain of PCSK9. Atherosclerosis 289, 162–172. doi: 10.1016/j.atherosclerosis.2019.08.020
Santos, R. D., Stein, E. A., Hovingh, G. K., Blom, D. J., Soran, H., Watts, G. F., et al. (2020). Long-Term Evolocumab in Patients With Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 75, 565–574. doi: 10.1016/j.jacc.2019.12.020
Sarraju, A., and Knowles, J. W. (2019). Genetic Testing and Risk Scores: Impact on Familial Hypercholesterolemia. Front. Cardiovasc. Med. 6:5. doi: 10.3389/fcvm.2019.00005
Scartezini, M., Hubbart, C., Whittall, R. A., Cooper, J. A., Neil, A. H., and Humphries, S. E. (2007). The PCSK9 gene R46L variant is associated with lower plasma lipid levels and cardiovascular risk in healthy U.K. men. Clin. Sci. 113, 435–441. doi: 10.1042/CS20070150
Schmidt, R. J., Zhang, Y., Zhao, Y., Qian, Y. W., Wang, H., Lin, A., et al. (2008). A novel splicing variant of proprotein convertase subtilisin/kexin type 9. DNA Cell. Biol. 27, 183–189. doi: 10.1089/dna.2007.0667
Schroeder, C. I., Swedberg, J. E., Withka, J. M., Rosengren, K. J., Akcan, M., Clayton, D. J., et al. (2014). Design and synthesis of truncated EGF-A peptides that restore LDL-R recycling in the presence of PCSK9 in vitro. Chem. Biol. 21, 284–294. doi: 10.1016/j.chembiol.2013.11.014
Seidah, N. G., Prat, A., Pirillo, A., Catapano, A. L., and Norata, G. D. (2019). Novel strategies to target proprotein convertase subtilisin kexin 9: beyond monoclonal antibodies. Cardiovasc. Res. 115, 510–518. doi: 10.1093/cvr/cvz003
Shioji, K., Mannami, T., Kokubo, Y., Inamoto, N., Takagi, S., Goto, Y., et al. (2004). Genetic variants in PCSK9 affect the cholesterol level in Japanese. J. Hum. Genet. 49, 109–114. doi: 10.1007/s10038-003-0114-3
Singh, S., and Bittner, V. (2015). Familial hypercholesterolemia–epidemiology, diagnosis, and screening. Curr. Atheroscler. Rep. 17:482. doi: 10.1007/s11883-014-0482-5
Slimani, A., Jelassi, A., Jguirim, I., Najah, M., Rebhi, L., Omezzine, A., et al. (2012). Effect of mutations in LDLR and PCSK9 genes on phenotypic variability in Tunisian familial hypercholesterolemia patients. Atherosclerosis 222, 158–166. doi: 10.1016/j.atherosclerosis.2012.02.018
Soutar, A. K., and Naoumova, R. P. (2007). Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 4, 214–225. doi: 10.1038/ncpcardio0836
Stoekenbroek, R. M., and Kastelein, J. J. P. (2018). Proprotein convertase subtilisin/kexin type 9: from genetics to clinical trials. Curr. Opin. Cardiol. 33, 269–275. doi: 10.1097/HCO.0000000000000517
Tall, A. R. (2006). Protease variants, LDL, and coronary heart disease. N. Engl. J. Med. 354, 1310–1312. doi: 10.1056/NEJMe068026
Taylor, A., Tabrah, S., Wang, D., Sozen, M., Duxbury, N., Whittall, R., et al. (2007). Multiplex ARMS analysis to detect 13 common mutations in familial hypercholesterolaemia. Clin. Genet. 71, 561–568. doi: 10.1111/j.1399-0004.2007.00807.x
Timms, K. M., Wagner, S., Samuels, M. E., Forbey, K., Goldfine, H., Jammulapati, S., et al. (2004). A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum. Genet. 114, 349–353. doi: 10.1007/s00439-003-1071-9
Truong, T. H., Kim, N. T., Nguyen, M. N. T., Pang, J., Hooper, A. J., Watts, G. F., et al. (2018). Homozygous familial hypercholesterolaemia in Vietnam: Case series, genetics and cascade testing of families. Atherosclerosis 277, 392–398. doi: 10.1016/j.atherosclerosis.2018.06.013
Urban, D., Pöss, J., Böhm, M., and Laufs, U. (2013). Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J. Am. Coll. Cardiol. 62, 1401–1408. doi: 10.1016/j.jacc.2013.07.056
Viigimaa, M., Heinsar, S., Lovic, D., Katsimardou, A., Piperidou, A., and Duishvili, D. (2018). New Horizons in the Pathogenesis, Pathophysiology and Treatment of Familial Hypercholesterolaemia. Curr. Pharm. Des. 24, 3599–3604. doi: 10.2174/1381612824666181009105305
Wang, X., Raghavan, A., Chen, T., Qiao, L., Zhang, Y., Ding, Q., et al. (2016). CRISPR-Cas9 Targeting of PCSK9 in Human Hepatocytes In Vivo-Brief Report. Arterioscler Thromb. Vasc. Biol. 36, 783–786. doi: 10.1161/ATVBAHA.116.307227
Whittall, R. A., Scartezini, M., Li, K., Hubbart, C., Reiner, Z., Abraha, A., et al. (2010). Development of a high-resolution melting method for mutation detection in familial hypercholesterolaemia patients. Ann. Clin. Biochem. 47(Pt 1), 44–55. doi: 10.1258/acb.2009.009076
Wiciński, M., Żak, J., Malinowski, B., Popek, G., and Grześk, G. (2017). PCSK9 signaling pathways and their potential importance in clinical practice. EPMA J. 8, 391–402. doi: 10.1007/s13167-017-0106-6
Wiegman, A., Gidding, S. S., Watts, G. F., Chapman, M. J., Ginsberg, H. N., Cuchel, M., et al. (2015). Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur. Heart J. 36, 2425–2437. doi: 10.1093/eurheartj/ehv157
Wright, W. T., Heggarty, S. V., Young, I. S., Nicholls, D. P., Whittall, R., Humphries, S. E., et al. (2008). Multiplex MassARRAY spectrometry (iPLEX) produces a fast and economical test for 56 familial hypercholesterolaemia-causing mutations. Clin. Genet. 74, 463–468. doi: 10.1111/j.1399-0004.2008.01071.x
Wu, C. Y., Tang, Z. H., Jiang, L., Li, X. F., Jiang, Z. S., and Liu, L. S. (2012). PCSK9 siRNA inhibits HUVEC apoptosis induced by ox-LDL via Bcl/Bax-caspase9-caspase3 pathway. Mol. Cell Biochem. 359, 347–358. doi: 10.1007/s11010-011-1028-6
Xiang, R., Fan, L. L., Lin, M. J., Li, J. J., Shi, X. Y., Jin, J. Y., et al. (2017). The genetic spectrum of familial hypercholesterolemia in the central south region of China. Atherosclerosis 258, 84–88. doi: 10.1016/j.atherosclerosis.2017.02.007
Yang, X., Zhang, J., Chen, L., Wu, Q., and Yu, C. (2018). Chitosan oligosaccharides enhance lipid droplets via down-regulation of PCSK9 gene expression in HepG2 cells. Exp. Cell Res. 366, 152–160. doi: 10.1016/j.yexcr.2018.03.013
Yue, P., Averna, M., Lin, X., and Schonfeld, G. (2006). The c.43_44insCTG variation in PCSK9 is associated with low plasma LDL-cholesterol in a Caucasian population. Hum. Mutat. 27, 460–466. doi: 10.1002/humu.20316
Keywords: gene, genetics, proprotein convertase subtilisin/kexin type 9, familial hypercholesterolemia, variant
Citation: Guo Q, Feng X and Zhou Y (2020) PCSK9 Variants in Familial Hypercholesterolemia: A Comprehensive Synopsis. Front. Genet. 11:1020. doi: 10.3389/fgene.2020.01020
Received: 30 March 2020; Accepted: 10 August 2020;
Published: 23 September 2020.
Edited by:
Uma Ramaswami, Royal Free London NHS Foundation Trust, United KingdomReviewed by:
Jernej Kovac, University Medical Centre Ljubljana, SloveniaCopyright © 2020 Guo, Feng and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yujie Zhou, YXp6eWoxMkAxNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.