 Nicole D. Moss
Nicole D. Moss Lori Sussel
Lori Sussel
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 01 September 2020
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00983
This article is part of the Research Topic Beta-Cell Fate: From Gene Circuits to Disease Mechanisms View all 12 articles
Robust endocrine cell function, particularly β cell function, is required to maintain blood glucose homeostasis. Diabetes can result from the loss or dysfunction of β cells. Despite decades of clinical and basic research, the precise regulation of β cell function and pathogenesis in diabetes remains incompletely understood. In this review, we highlight RNA processing of mRNAs as a rapidly emerging mechanism regulating β cell function and survival. RNA-binding proteins (RBPs) and RNA modifications are primed to be the next frontier to explain many of the poorly understood molecular processes that regulate β cell formation and function, and provide an exciting potential for the development of novel therapeutics. Here we outline the current understanding of β cell specific functions of several characterized RBPs, alternative splicing events, and transcriptome wide changes in RNA methylation. We also highlight several RBPs that are dysregulated in both Type 1 and Type 2 diabetes, and discuss remaining knowledge gaps in the field.
The highly specialized insulin-producing β cell population is located within the pancreatic islet of Langerhans (Figure 1). In humans and other vertebrates, β cells respond to changes in circulating blood glucose levels by secreting insulin. Coupled with the function of the other islet endocrine cell types, β cells help to maintain blood glucose homeostasis; loss or dysfunction of the β cell population results in diabetes. Over the last several decades, substantial research efforts have been directed toward understanding the gene regulatory networks required for the formation and function of the islet cell populations. This has included developmental studies in model organisms that have identified the key transcription factors required to make and maintain functional β cell populations. In addition, translational research approaches in human subjects, including Genome Wide Association Studies (GWAS) and other large sequencing efforts, have identified numerous alleles and mutations associated with increased risk of developing either Type 1 (T1D) or Type 2 (T2D) diabetes, many of which cause β cell dysfunction. Despite these efforts, there remain many gaps in our understanding of the mechanisms that regulate β cells and the pathways that contribute to their pathogenesis in diabetes.
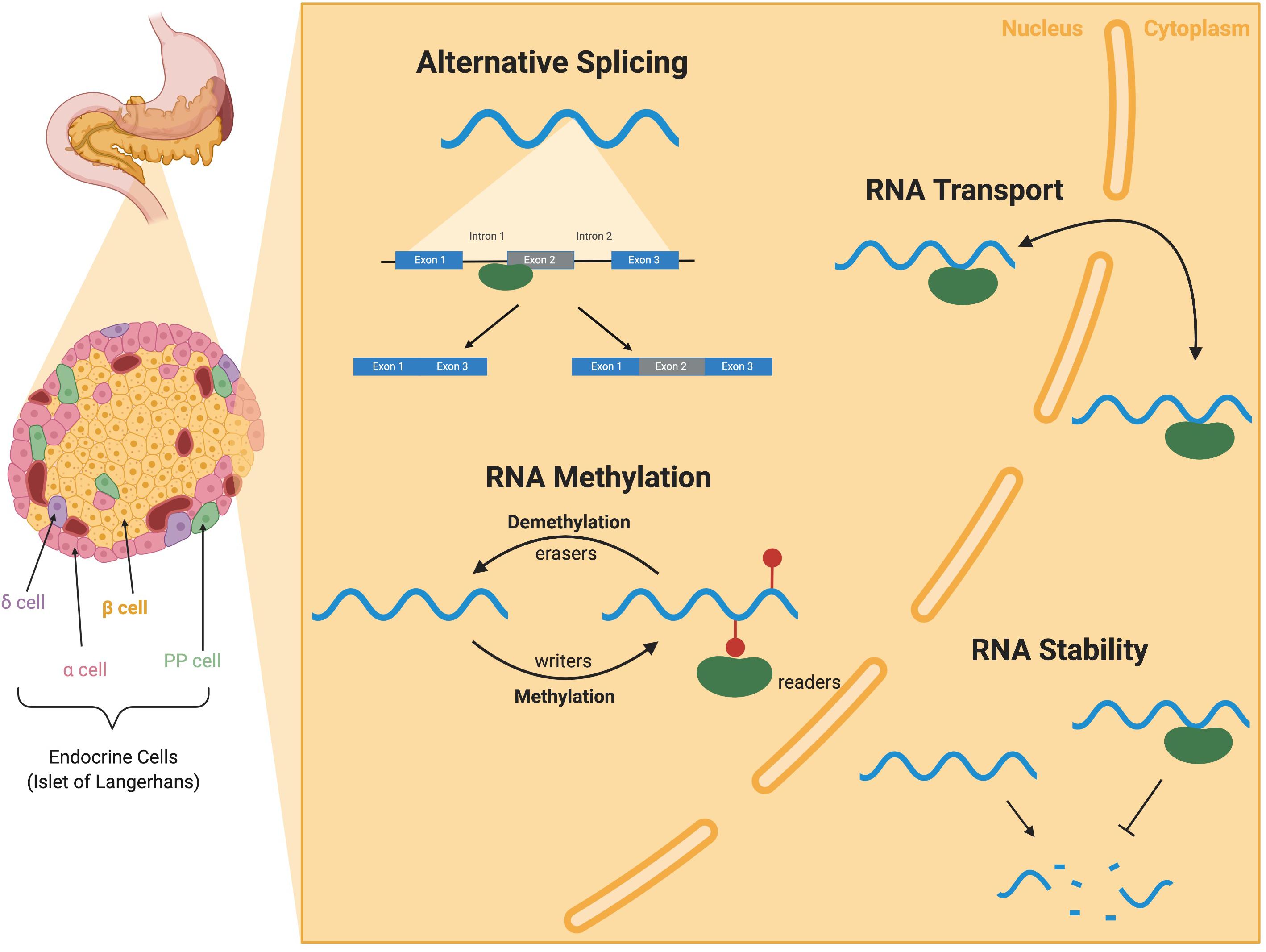
Figure 1. RNA-binding protein (RBP) mediated RNA regulation in the pancreatic β cell. I nsulin-secreting β cells reside in the islets of the pancreas along with several other endocrine cell types (α, δ, and PP cells). RBPs (green) are present in both the nucleus and the cytoplasm of cells and bind to RNA (blue) to perform a variety of functions. RBPs binding to introns and exons of pre-mRNAs contribute to alternative splicing. RBPs can also write, read, and erase methylation modification on mRNAs in RNA methylation. RBPs can also facilitate the transport of RNAs between the nucleus and cytoplasm and throughout the cell. RBP binding to the UTRs can alter mRNA stability and translation. Illustration created with BioRender.
Although much of the research to date has been focused on transcriptional regulation, β cell identity and function are also regulated at the level of mRNA, similar to many other cell types and organ systems. Throughout their life cycle, mRNA molecules undergo extensive processing events to transition from a pre-mRNA molecule to a mature mRNA. These events not only include addition of a 5′-cap and 3′-poly-A tail, but also splicing of introns, nucleotide modification, stability, and subcellular localization (Licatalosi and Darnell, 2010; Figure 1). RNA-binding proteins (RBPs) are responsible for coordinating the events in the lifecycle of an mRNA. Over the past few years, several groups have begun to probe the function of specific RBPs in organogenesis and disease. Many studies have focused on the mis-regulation of mRNAs and RBP function in the context of diabetic complications (adipose, liver, muscle, retina, etc.), rather than specific changes in the β cells (Nutter and Kuyumcu-Martinez, 2018). In the pancreas, only a few groups have delved into the world of RNA regulation, often focusing on a single splicing target or RBP. In this review, we will highlight these studies describing RBP functions, transcriptome wide changes in RBP expression, alternative splicing, and RNA methylation, with a specific focus on regulation of mRNAs in the pancreatic islet population. This is a rapidly emerging field that will undoubtedly provide a unique perspective on a complex disease and will ultimately push the boundaries of therapeutic treatments for diabetes.
Several hundred RBPs have been identified (Hentze et al., 2018), each with the potential of having hundreds of targets within a cell (Keene, 2007; Hogan et al., 2008; Lukong et al., 2008; Blanchette et al., 2009; Li et al., 2014). Some RBPs have ubiquitous expression, while others are transiently expressed during development or restricted to a specific cell type (Gerstberger et al., 2014). Like many other proteins, RBPs are categorized by several modular domains. RBPs recognize RNA targets through a binding domain, in the form of an RNA recognition motif (RRM), K-homology (KH) domain, and RNA-binding zinc-finger (ZnF) domains, or can bind independent of sequence through a double-stranded RNA module (dsRBD) (Lunde et al., 2007). Additionally, RBPs have a variety of enzymatic and/or signaling domains that allow for functional activity (Lunde et al., 2007).
The role of RBPs in the formation and function of pancreatic endocrine cells is only beginning to be appreciated. Only a small number of known RBPs have been studied in the β cell, but as new transcriptomics data becomes available from both healthy and diseased islets, their role in β cell biology will become more apparent. Recently, several studies have identified RBPs that are enriched in pancreatic islet cells and become dysregulated under stress (Juan-Mateu et al., 2017; Jeffery et al., 2019; Ramos-Rodriguez et al., 2019). Stressors including chronic hyperglycemia (Puri and Hebrok, 2012; Brereton et al., 2014), exposure to pro-inflammatory cytokines (Ortis et al., 2010), and palmitate (saturated fatty acid) (Cnop et al., 2014) can result in changes in cellular and molecular identity. In a model of human β cells (EndoC-βH1), treatment with cell stressors (including cytokines, hypoxia, altered lipids, and high and low levels of glucose) also induced dysregulation of many RBPs (Jeffery et al., 2019).
Islet endocrine cells have a specific assemblage of RBPs that perform a variety of functions. Re-analysis of whole transcriptomic RNA-Sequencing (RNA-Seq) data from several human tissues (Eizirik et al., 2012) revealed that human islets share a notable number of RBPs with the brain, and β cells in particular are enriched for many “neuron specific” RBPs (Juan-Mateu et al., 2017; Alvelos et al., 2018). This is not surprising since, despite their disparate developmental origins, neurons and β cells share a large number of transcriptional networks (van Arensbergen et al., 2010). While it is clear that there are a whole host of RBPs expressed in mature insulin-secreting β cells (summarized in Table 1), there remains poor understanding about the role of RBP regulation in the developing pancreas (Baralle and Giudice, 2017). Additionally, the majority of these studies to date are limited to in vitro analysis of RBP requirements and molecular function. RBPs regulate several classes of RNAs, including both coding (mRNA) and non-coding (ncRNA) RNAs. A recent comprehensive review has discussed the critical role of RBPs Dicer and Argonaut in the regulation of miRNAs for β cell function and in T2D (Eliasson and Esguerra, 2020). Here we will provide a brief survey of the expression and function of several prominent RBP families that regulate mRNAs in pancreatic β cells.
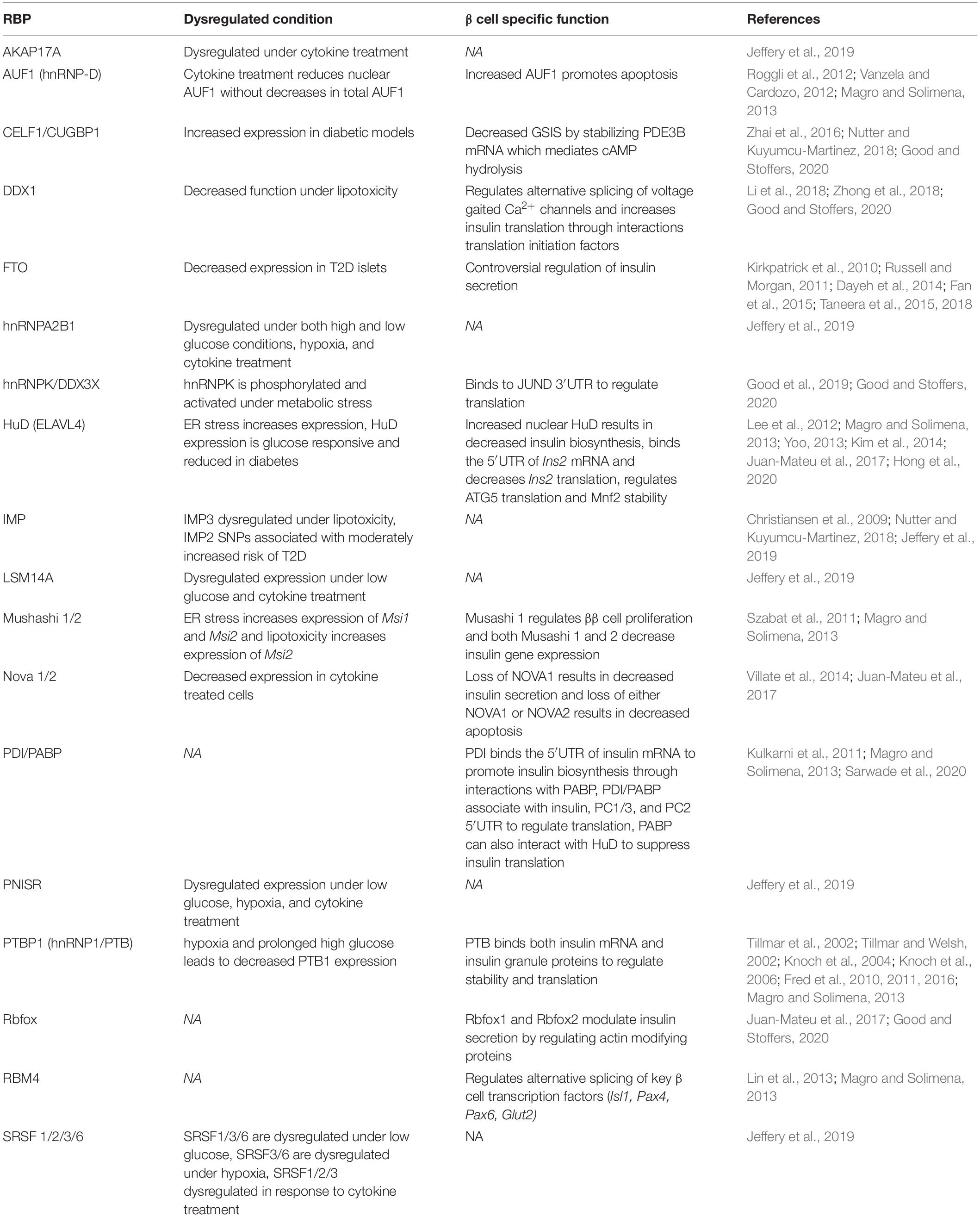
Table 1. RNA-Binding Proteins (RBPs) in the Pancreatic β Cell.
The Hu/ELAV family of RBPs bind to AU-rich elements (AREs) in the 3′UTR of mRNAs and can modulate transcript stability and translation (Hinman and Lou, 2008). Hu/ELAV proteins bind to the AREs through three RRMs (Okano and Darnell, 1997). HuR/ELAVL1 is ubiquitously expressed, while the other three family members (HuB/ELAVL2, HuC/ELAVL3, HuD/ELAVL4) are most highly expressed in neurons (Hinman and Lou, 2008). However, a few recent studies have identified roles for one of these family members, HuD/ELAVL4, in the β cell (Lee et al., 2012; Kim et al., 2014; Hong et al., 2020). Normally, HuD expression is (1) glucose dependent; (2) regulated through insulin receptor (INSR) signaling; and (3) acts as a feedback mechanism that regulates translation of the Preproinsulin2 (Ins2) mRNA (Lee et al., 2012). Rodents encode two prepronsulin (Ins) genes; however, interaction between HuD and the preproinsulin1 (Ins1) transcript was not reported. Insulin is secreted from β cells in response to high levels of glucose. Circulating insulin can then bind the insulin receptor (INSR) on the surface of β cells and, through the PI3K/AKT pathway, the transcriptional repressor FOXO1 is phosphorylated. Phosphorylation of FOXO1 de-represses transcription of HuD. The HuD protein then binds the 5′UTR of Ins2 mRNA and decreases Ins2 translation, maintaining plasma insulin homeostasis. Consistently, HuD–/– mice displayed higher insulin levels and improved glucose tolerance, whereas transgenic mice overexpressing HuD had lower insulin levels and were glucose intolerant, reportedly due to less readily releasable insulin pools (Kim et al., 2014). It has also been demonstrated that nuclear HuD is increased under ER stress resulting in decreased intracellular insulin biosynthesis and decreased plasma insulin homeostasis (Yoo, 2013). In addition to regulating Ins2 translation, HuD also regulates the translation of two genes encoding proteins important for β cell survival in stress conditions. Autophagy-related Gene 5 (ATG5) is a protein that can mediate stress induced β cell death (Fujimoto et al., 2009). HuD binds to AREs in the 3′UTR of Atg5 and enhances the assembly of polysomes to increase ATG5 protein levels. HuD also modulates β cell function through mitochondrial dynamics and stabilizing the mitochondrial gene Mitofusin2 (Mfn2), which encodes a protein which mediates mitochondrial fusion and metabolism, and is an inhibitor of apoptosis in β cells (Baltrusch, 2016; Hong et al., 2020). Taken together, it is clear that RNA regulation by HuD is required for proper function and survival of pancreatic β cells through multiple mechanisms and pathways.
The polypyrimidine-tract-binding proteins (PTBs) are a group of RBPs that function through binding-mediated modifications in target mRNA (Wollerton et al., 2001; Mitchell et al., 2003; Auweter et al., 2007) to either recruit or block other trans-acting factors. PTB has four RRM domains each with specific consensus binding sequences that all bind stretches of pyrimidines (Sawicka et al., 2008). Additionally, PTBs can shuttle between the nucleus and the cytoplasm (Perez et al., 1997; Kamath et al., 2001; Li and Yen, 2002). PTB proteins have been implicated in the regulation of several RNA metabolism events including alternative splicing (Garcia-Blanco et al., 1989), polyadenylation (Lou et al., 1999; Castelo-Branco et al., 2004), mRNA stability (Wollerton et al., 2004), and translation (Jang and Wimmer, 1990). In the pancreas, PTB proteins have been shown to regulate insulin mRNA (human INS and rodent Ins1 and Ins2) (Tillmar et al., 2002; Tillmar and Welsh, 2002; Fred et al., 2010, 2011, 2016) and insulin secretory granule biogenesis (Knoch et al., 2004, 2006). Specifically, the binding of PTB to INS/Ins mRNA increases in response to increased glucose and hypoxia (Tillmar et al., 2002; Tillmar and Welsh, 2002; Fred et al., 2011). Mutations in the INS/Ins 3′UTR or decreases in PTB expression by RNAi both result in decreased insulin and reporter expression respectively (Tillmar et al., 2002; Fred et al., 2016). Furthermore, binding of PTB to the 5′UTR of INS mRNA correspond to cap-independent translation of insulin mRNA (Fred et al., 2011). In addition to changes in PTB binding, T-cell restricted intracellular antigen 1-related protein (TIAR) also increases binding to INS mRNA during glucose stimulation (Fred et al., 2010). These proteins cooperate to regulate INS mRNA stability and biosynthesis (Fred et al., 2010). While cap-independent INS mRNA translation only accounts for a small portion of total translation, it can contribute 40–100% of insulin biosynthesis during stress conditions (Fred et al., 2011). In healthy β cells, transient increase in glucose levels increases PTB binding to 3′UTR promoting mRNA stability and 5′UTR promoting modest levels of cap-independent translation. However, prolonged high glucose exposure results in decreased PTB protein and ultimately decreased insulin biosynthesis (Fred et al., 2010). This is in part due to increased levels of miR-133a which targets PTB mRNA and could explain the mechanism for hyperglycemia-induced β cell dysfunction (Fred et al., 2010).
In addition to binding INS/Ins mRNA directly, PTB has been shown to bind and regulate components of the insulin secretory granules in response to changes in blood glucose levels. During glucose stimulated insulin secretion (GSIS), newly synthesized insulin granules preferentially undergo exocytosis (Gold et al., 1982; Halban, 1982) and this 2010process is impaired in T2D. New secretory granules are synthesized in response to glucose stimulation in the β cell, partially through regulation of PTB. Upon β cell stimulation (glucose or GLP-1), PTB is translocated from the nucleus to the cytoplasm (Knoch et al., 2004, 2006), where this process not only promotes stability of insulin mRNA (as described above), but also increases the stability of several insulin secretory granule proteins (Knoch et al., 2004). PTB translocation results from phosphorylation by PKA downstream of GLP-1 receptor and is cAMP-dependent (Knoch et al., 2006). The activated and cytosolic PTB is then able to bind and stabilize mRNAs that code for secretory granule proteins with putative PTB binding sites in the 3′UTR. Furthermore, knockdown of PTB by RNAi results in decreased expression of target mRNAs and secretory granules (Knoch et al., 2004). Taken together, glucose/GPL-1 dependent stimulation of β cells results in cytoplasmic translocation of PTB where it can act to stabilize insulin mRNA and components of the insulin secretory granule. In light of these findings, it is evident that PTB expression and activation represents a critical component in regulating GSIS.
The Neuro-oncological ventral antigens (NOVA) are a family of two RBPs (NOVA1 and NOVA2) that account for approximately 700 alternative splicing events in neurons (Ule et al., 2005, 2006; Licatalosi et al., 2008; Zhang et al., 2010) and have been implicated in regulating alternative polyadenylation (Licatalosi et al., 2008). Both NOVA proteins bind to YCAY consensus sequences in target mRNAs through three hnRNPK-homology (KH)-type RNA binding motifs (Buckanovich and Darnell, 1997; Yang et al., 1998; Jensen et al., 2000). The positions of NOVA binding relative to the alternative splice site determines exon inclusion versus exclusion; exon inclusion is correlated with NOVA downstream binding (Ule et al., 2006; Zhang et al., 2010). Both NOVA1 and NOVA2 are expressed in the pancreatic β cell (Villate et al., 2014; Juan-Mateu et al., 2017) and in vitro studies suggest they contribute to alternative splicing (Eizirik et al., 2012; Villate et al., 2014; Juan-Mateu et al., 2017). Knockdown of Nova1 by RNAi in FACS-purified rat β cells resulted in changes in alternative splicing of 4961 isoforms and impaired GSIS (Villate et al., 2014). In INS-1E cells and MIN6 cells, knockdown of Nova1 disrupts insulin secretion through changes in alternative splicing of key exocytosis factors PLCβ1 and Snap25, and decreases in voltage-dependent Ca2+ current (Villate et al., 2014). NOVA1 has also been shown to regulate alternative splicing of the insulin receptor (INSR), suggesting that NOVA1 is required to promote exon 11 inclusion and expression of the INSR-B form of the receptor (Villate et al., 2014). Additionally, NOVA1 has been implicated in T1D and cytokine-induced apoptosis (Eizirik et al., 2012; Villate et al., 2014). In both cytokine-treated β cells and Nova1 knockdown β cells, apoptosis increases through the upregulation of pro-apoptotic protein, Bim (Barthson et al., 2011; Villate et al., 2014). Similarly, Nova1 is decreased in β cells treated with cytokines (Villate et al., 2014). Bim is regulated by FOXO3a, however phosphorylation of FOXO3a inhibits it’s function (Sunters et al., 2003; Zhang et al., 2011). In Nova1 knockdown β cells, FoxO3a expression is increased but phosphorylation is decreased, allowing for the subsequent upregulation of Bim. Similarly, NOVA2 has been shown to regulate β cell survival. NOVA2/Nova2 knockdown in INS-1E, EndoC-βH1, and sorted rat β cells resulted in increased apoptosis (Juan-Mateu et al., 2017). Together these studies have identified several roles for NOVA RBPs in the function and survival of pancreatic β cells.
The RBFOX family of RBPs contains three highly conserved members – RBFOX1, RBFOX2, and RBFOX3. RBFOX RBPs all contain an RRM that recognizes the specific (U)GCAUG sequence in target mRNAs to promote alterative splicing and other RNA metabolic functions (Jin et al., 2003; Ponthier et al., 2006). Rbfox2 is nearly ubiquitously expressed across cell types and throughout development, whereas Rbfox1 and Rbfox3 are considerably more cell type specific or only transiently expressed. The functions of the RBFOX proteins have been studied primarily in neurons and muscle tissue, and their activity is often required for development and maturation of these cell types (Gehman et al., 2012; Wei et al., 2015; Jacko et al., 2018). During pancreas development, scRNA-Seq reveals that Rbfox2 is detectable throughout the embryonic (E15.5 and E18.5) mouse pancreas and hESC-derived pancreatic endocrine cells (Krentz et al., 2018). This dataset also shows that Rbfox3 appears to be transiently expressed specifically within the Neurog3+ endocrine progenitor population at E15.5 (Krentz et al., 2018). Rbfox1 is not detectable in embryonic E15.5 or E18.5 mouse pancreas or within the hESC-derived endocrine cells (Krentz et al., 2018). Similarly in the adult pancreas, RNA-Seq on intact islets revealed high expression of Rbfox2; whereas Rbfox1 and Rbfox3 were barely detectable. Further analysis of the individual sorted mouse endocrine cells determined that Rbfox2 is expressed in α, β, and δ cells; with its highest expression in the β cell population (DiGruccio et al., 2016). Within this dataset, Rbfox1 is undetectable in any of the endocrine populations, whereas Rbfox3 expression can be found in δ cells. Consistently, scRNA-Seq in adult islets show that the majority of endocrine cells express Rbfox2, whereas Rbfox3 is predominantly restricted to δ cells and Rbfox1 is undetectable (DiGruccio et al., 2016; The Tabula Muris Consortium et al., 2018). Consistent with the mouse studies, bulk sequencing of human islets and other human tissues identified expression of both RBFOX2 and RBFOX3, but not RBFOX1 in whole islets (Juan-Mateu et al., 2017). Paradoxically, this group proceeded to knockdown Rbfox2 and Rbfox1 in rat INS1-E cells and suggested that both proteins regulate insulin content and insulin secretion through the alternative splicing of genes involved in actin regulation (Juan-Mateu et al., 2017). It is possible that there will be redundant functions of the highly conserved Rbfox family of RBPs in endocrine cells; however, it remains to be determined how their coordinated, transient and/or compensatory expression impacts endocrine cell development and function in disease states.
The serine/arginine (SR)-rich proteins are a large family of RBPs characterized by their serine/arginine rich domain and an RRM (Shepard and Hertel, 2009). SR proteins are involved in several aspects of RNA metabolism including both constitutive and alternative splicing events (Zhou and Fu, 2013). SR proteins can interact with core components of the spliceosome (U1 and U2 snRNPs) to promote or inhibit splice site usage. Additionally, SR proteins function in regulating mRNA transport and translation (Zhong et al., 2009). While the function of SR proteins in other cell types and systems have been reviewed extensively (Shepard and Hertel, 2009; Zhong et al., 2009; Zhou and Fu, 2013), relatively little is known about their role in β cells.
Nearly all members of the SR protein family are expressed in the mouse pancreas (The Tabula Muris Consortium et al., 2018) and several of these SR proteins become dysregulated in diabetes (Jeffery et al., 2019). In pancreatic endocrine cells, several SRSF proteins interact with the long non-coding RNA Paupar to influence the alternative splicing of Pax6 to confer differential genomic binding of the PAX6 transcription factor (Kiselev et al., 2012; Singer et al., 2019). β cells express a higher ratio of the shorter PAX6 isoform lacking the 5a exon while α cells predominantly express the longer PAX6 5a isoform (Singer et al., 2019). Together, the differential expression in healthy vs. diabetic endocrine cells and function of SR proteins in mediating transcription factor function make SR proteins an interesting candidate for evaluating the role of RBPs in the onset of diabetes. Of note, the only functional studies of this family of RBPs have been on SRSF3 and many of the SR proteins themselves undergo cell type specific and stress induced alternative splicing, leaving extensive opportunity to evaluate their β cell specific functions in healthy and diabetic states.
Until recently, attempts to identify the genetic causes of T1D and T2D have predominantly relied on GWAS to identify single nucleotide polymorphisms (SNPs) and associated gene expression changes that could contribute to disease. Although these approaches have successfully identified a number of causative candidate alleles, they overlook altered splicing events that may affect gene function rather than expression levels. Furthermore, it is now apparent that differences in co- and posttranscriptional processing such as alternative splicing and N6-methyladenosine (m6A) modifications, can more effectively differentiate between T2D β cells than transcriptomics alone (De Jesus et al., 2019).
Messenger RNA splicing occurs co-transcriptionally to remove introns from the pre-mRNA. This process is carried out by the spliceosome and is coordinated by a series of RBPs. In addition to removing introns, splicing machinery can also vary mRNAs through alternative exon and splice site usage, referred to as alternative splicing. Over 90% of human genes undergo alternative splicing, which more than quadruples the number of potential gene products (Fairbrother et al., 2002; Johnson et al., 2003). Recent work comparing transcriptomes of T2D diabetic and healthy donors identified dysregulation of 26% of alternative splicing events (Jeffery et al., 2019). The highest proportion of alternatively spliced genes in this study function in gene regulation. For example, the authors observed dysregulation of the alternative splicing regulators such as SRSF RBPs, which could each regulate hundreds of splicing events within the β cell. Stress induction of the EndoC-βH1 β cell line supported findings from human diabetic islets, showing a decrease in splicing regulators. Moreover, removal of the stress restored splicing factor expression and changes in transcriptome wide splicing events. Additionally, in a model of T1D, cytokine exposure of FAC sorted rat β cells resulted in differential expression of more than 20 RBPs involved in alternative splicing and changes in alternative splicing of cytokine regulated genes (Ortis et al., 2010). These groups all suggest that changes in the splicing landscape as well as changes in β cell differentiation markers, may be a mechanism of stress response to avoid apoptosis during the onset of diabetes, making the study of alternative splicing regulation in the β cell critical for not only understanding the pathogenesis of the disease but also in designing innovative treatment plans.
The onset of T2D and accelerated dysfunction of β cells has also been attributed to many environmental factors, including the disruption of circadian sleep/wake cycles (Gale et al., 2011). Although the disruption of circadian sleep/wake cycles has traditionally been associated with mRNA oscillations, a recent study implicates the RBP Thyroid Hormone Receptor-Associated Protein3 (THRAP3) as a regulator of alternative-splicing. This study demonstrated that THRAP3 regulated circadian clock-dependent alternative splicing by binding to and regulating alternative splicing of key exocytosis factors (Marcheva et al., 2020).
In addition to transcriptome wide changes in alternative splicing, several groups have explored the alternative splicing of specific genes involved in β cell development, function, and survival. Of note, several Maturity Diabetes of the Youth (MODY) and T2D associated genes such as HNF-1α, GCK, and TCF7L2 have splicing variants (Prokunina-Olsson et al., 2009). While TCF7L2 isoforms are not significantly altered in T2D (Prokunina-Olsson et al., 2009), both HNF-1α and GCK diabetes associated alleles result in alternative splicing variation (Cappelli et al., 2009; Lorini and D’Annunzio, 2009), and isoforms of HNF-1α are associated with differential efficiency in insulin gene regulation (Harries et al., 2006; Cappelli et al., 2009). T2D associated alterations in alternative splicing of HNF-1α and several other β cell genes are also reviewed by Dlamini et al. (2017). Based on recent findings in large transcriptomic analyses, this field is likely to grow dramatically.
N6-methyladenosine (m6A) methylation is one of the most prevalent post-transcriptional RNA modifications and is regulated by a set of RNA-binding proteins – writers, readers, and erasers (Figure 1; Zhang et al., 2019). These modifications are introduced by a group of specialized methyltransferases (“writers”) including METTL3 and METTL14. The m6A modifications can confer differential stability, changes in alternative splicing, subcellular localization, and translation efficiency (Zhang et al., 2019). The m6A modifications are often bound by a group of RBPs called readers to perform these differential functions. Finally, a third group of RBPs, including FTO (Fat Mass and Obesity-associated gene), are referred to as erasers and can remove m6A marks. A recent review by Zhang et al. (2019) details the current knowledge on mechanism and function of m6A methylation.
With respect to the β cell, a recent publication by De Jesus et al. (2019) describes the differential m6A methylation observed in T2D. RNA-Seq analysis and fluorescent labeling of human T2D islets compared to healthy controls revealed differential expression of key m6A modulators (METTL3, METTL14, ALKBH5, and YTHDF1). This finding was supported by an independent study showing decreased Mettl3/14 expression in diabetic db/db mice and type 2 diabetes patients (Wang et al., 2020). The decreased expression of m6A writers (METTL3 and METTL14) resulted in differential methylation of 6,078 sites in 4,155 genes (FDR < 0.05). The hypomethylation of genes in the T2D islets are associated with cell-cycle progression, insulin secretion, and the insulin/IGF-AKT-PDX1 pathway. This study also replicated T2D phenotypes and the m6A methylome in METTL3 or METTL14 deficient EndoC-βH1 cells and Mettl14 β-cell-specific knockout mice. Another group generated a similar β cell specific knockout of Mettl14 in mice and observed that these mice display glucose intolerance, decreased GSIS and decreased β cell mass due to β cell death under normal conditions (Liu et al., 2019). These phenotypes are exaggerated in mice fed a high fat diet (Liu et al., 2019). Together these studies indicate a role for METTL14 and m6A modifications in the function and survival of β cells.
In the context of pancreas development, m6A writers (METTL3/14) are critical for β cell expansion and maturation but appear to be dispensable for the differentiation and maturation of other endocrine cell types (Wang et al., 2020). Wang et al. (2020) also showed that in the developing pancreas loss of Mettl3 or Mettl14 from endocrine progenitors independently results in hyperglycemia around weaning, but that loss of both methyltransferases results in significant hyperglycemia and hypo-insulinemia by 2-weeks of age. This functional defect is in part due to decreased proliferation and increased cell death, similar to what had been observed in the previously described β cell specific knockout mice. Additionally, using RNA-Seq and m6A Me-RIP-Seq, the authors concluded that Mettl3/14 directly regulates the β cell maturation factor, MAFA, to promote stability. Other groups have also studied the effects of m6A modifications, particularly in adipogenesis, that could contribute to the pathogenesis of T2D (Gerken et al., 2007; Chu et al., 2008; Ben-Haim et al., 2015; Shen et al., 2015; Wood et al., 2016).
As a counterpoint to the m6A writers METTL3/14, FTO is an m6A eraser. FTO is expressed in a variety of cell and tissue types including endocrine cells (Taneera et al., 2015; DiGruccio et al., 2016; The Tabula Muris Consortium et al., 2018). Whole body knockouts and nervous system specific knockouts of Fto in mice result in postnatal growth deficiencies (Gao et al., 2010), however, its specific function in β cells is debated. In T2D human islets, FTO expression is reduced (Kirkpatrick et al., 2010; Taneera et al., 2018) and the FTO gene has decreased DNA methylation (Dayeh et al., 2014). Several groups have investigated the functional role of FTO in pancreatic islets using different experimental models. Overexpression of FTO in rat INS-1 cells appeared to affect first wave insulin secretion (Russell and Morgan, 2011), whereas overexpression in mouse MIN6 cells resulted in the inhibition of GSIS without changes in insulin gene expression (Fan et al., 2015). Given the discrepant results of these studies, in addition to the caveats associated with overexpression studies, perhaps the more relevant functional assessment was the use of siRNA knockdown to deplete FTO in an engineered human insulin secretion reporter rat β cell line (GRINCH) (Taneera et al., 2018). In these experiments, depletion of FTO resulted in decreased insulin mRNA expression and insulin secretion (Taneera et al., 2018).
While these emerging studies are beginning to highlight the relevance of m6A RNA methylation in β cell function, and the potential contribution of alterations in this RNA modification to the onset of diabetes, the underlying mechanism and direct targets of m6A methylation and demethylation that contribute to the observed functional changes have yet to be determined. Future studies of the islet cell-specific changes in RNA modifications that result from defects in the m6A pathway will provide critical new information about the regulation of islet function in normal and disease conditions.
Traditionally, endocrine cells have been molecularly defined by their cell-specific transcriptomes. Furthermore, validation of human diabetes GWAS studies have relied primarily on gene expression changes associated with disease. More recently, however, large scale high-throughput sequencing efforts have revealed the previously unappreciated importance of co- and post-transcriptional RNA regulation in the specification and function of differentiated cells. These high-resolution sequencing technologies have not only identified changes in expression levels, they have unveiled numerous RNA modifications and alternative splicing events within each of the islet endocrine cell populations and in individual β cells. This discovery has prompted investigation into the many RBPs that are expressed in the pancreatic islet and those that become dysregulated in β cells undergoing stress conditions that mimic diabetes (Table 1). Each of these RBPs can have hundreds of targets and affect multiple pathways within a cell in both physiological and pathophysiological conditions (Keene, 2007; Hogan et al., 2008; Lukong et al., 2008; Blanchette et al., 2009; Li et al., 2014), making their potential impact on cellular identity and function pervasive. This also implies that there are many more layers of regulation in the β cell, particularly mediated by RBPs, that have yet to be explored. The knowledge gained from understanding regulation of β cell development and function at the level of mRNA modifications could be immensely useful to optimize protocols to generate insulin-secreting β-like cells from human stem cells (Hrvatin et al., 2014), which is currently a promising method of replacing β cell loss in T1D. Furthermore, characterization of dysregulated splicing events could open therapeutic opportunities to correct specific mRNAs using antisense oligonucleotide (ASO) technologies. ASOs are small synthetic nucleotide sequences that can target specific mRNA transcripts to target and eliminate anomalous splice variants (Schoch and Miller, 2017). Treatment with ASOs has become increasingly promising for treating neurodegenerative diseases (Schoch and Miller, 2017) and could be an innovative mechanism to correct aberrant splicing defects occurring in T2D β cells. Overall, the study of RBPs and RNA modifications are primed to be the next frontier of mechanisms that regulate β cell formation, function, and in the development of novel therapeutics.
NM researched the literature and wrote the manuscript. LS researched the literature and edited the manuscript. Both authors contributed to the article and approved the submitted version.
Funding to LS was provided from the NIH R01 DK111405, R01 DK082590, and P30DK116073 (DRC). Support was also provided from the University of Colorado RNA Biosciences Initiative (RBI).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank members of the Sussel lab for critical reading of the manuscript and useful discussions.
Alvelos, M. I., Juan-Mateu, J., Colli, M. L., Turatsinze, J. V., and Eizirik, D. L. (2018). When one becomes many-alternative splicing in beta-cell function and failure. Diabetes Obes. Metab. 20(Suppl. 2), 77–87. doi: 10.1111/dom.13388
Auweter, S. D., Oberstrass, F. C., and Allain, F. H. (2007). Solving the structure of PTB in complex with pyrimidine tracts: an NMR study of protein-RNA complexes of weak affinities. J. Mol. Biol. 367, 174–186. doi: 10.1016/j.jmb.2006.12.053
Baltrusch, S. (2016). Mitochondrial network regulation and its potential interference with inflammatory signals in pancreatic beta cells. Diabetologia 59, 683–687. doi: 10.1007/s00125-016-3891-x
Baralle, F. E., and Giudice, J. (2017). Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 18, 437–451. doi: 10.1038/nrm.2017.27
Barthson, J., Germano, C. M., Moore, F., Maida, A., Drucker, D. J., Marchetti, P., et al. (2011). Cytokines tumor necrosis factor-alpha and interferon-gamma induce pancreatic beta-cell apoptosis through STAT1-mediated Bim protein activation. J. Biol. Chem. 286, 39632–39643. doi: 10.1074/jbc.m111.253591
Ben-Haim, M. S., Moshitch-Moshkovitz, S., and Rechavi, G. (2015). FTO: linking m6A demethylation to adipogenesis. Cell Res. 25, 3–4. doi: 10.1038/cr.2014.162
Blanchette, M., Green, R. E., MacArthur, S., Brooks, A. N., Brenner, S. E., Eisen, M. B., et al. (2009). Genome-wide analysis of alternative pre-mRNA splicing and RNA-binding specificities of the Drosophila hnRNP A/B family members. Mol. Cell. 33, 438–449. doi: 10.1016/j.molcel.2009.01.022
Brereton, M. F., Iberl, M., Shimomura, K., Zhang, Q., Adriaenssens, A. E., Proks, P., et al. (2014). Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat Commun. 5:4639.
Buckanovich, R. J., and Darnell, R. B. (1997). The neuronal RNA binding protein Nova-1 recognizes specific RNA targets in vitro and in vivo. Mol. Cell. Biol. 17, 3194–3201. doi: 10.1128/mcb.17.6.3194
Cappelli, A., Tumini, S., Consoli, A., Carinci, S., Piersanti, C., Ruggiero, G., et al. (2009). Novel mutations in GCK and HNF1A genes in Italian families with MODY phenotype. Diabetes Res. Clin. Pract. 83, e72–e74. doi: 10.1016/j.diabres.2008.12.007
Castelo-Branco, P., Furger, A., Wollerton, M., Smith, C., Moreira, A., and Proudfoot, N. (2004). Polypyrimidine tract binding protein modulates efficiency of polyadenylation. Mol. Cell. Biol. 24, 4174–4183. doi: 10.1128/mcb.24.10.4174-4183.2004
Christiansen, J., Kolte, A. M., Hansen, T., and Nielsen, F. C. (2009). IGF2 mRNA-binding protein 2: biological function and putative role in type 2 diabetes. J. Mol. Endocrinol. 43, 187–195. doi: 10.1677/jme-09-0016
Chu, X., Erdman, R., Susek, M., Gerst, H., Derr, K., Al-Agha, M., et al. (2008). Association of morbid obesity with FTO and INSIG2 allelic variants. Arch. Surg. 143, 235–240;discussion 241.
Cnop, M., Abdulkarim, B., Bottu, G., Cunha, D. A., Igoillo-Esteve, M., Masini, M., et al. (2014). RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes Metab. Res. Rev. 63, 1978–1993. doi: 10.2337/db13-1383
Dayeh, T., Volkov, P., Salo, S., Hall, E., Nilsson, E., Olsson, A. H., et al. (2014). Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 10:e1004160. doi: 10.1371/journal.pgen.1004160
De Jesus, D. F., Zhang, Z., Kahraman, S., Brown, N. K., Chen, M., Hu, J., et al. (2019). m(6)A mRNA methylation regulates human beta-cell biology in physiological states and in type 2 diabetes. Nat. Metab. 1, 765–774. doi: 10.1038/s42255-019-0089-9
DiGruccio, M. R., Mawla, A. M., Donaldson, C. J., Noguchi, G. M., Vaughan, J., Cowing-Zitron, C., et al. (2016). Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol. Metab. 5, 449–458. doi: 10.1016/j.molmet.2016.04.007
Dlamini, Z., Mokoena, F., and Hull, R. (2017). Abnormalities in alternative splicing in diabetes: therapeutic targets. J. Mol. Endocrinol. 59, R93–R107.
Eizirik, D. L., Sammeth, M., Bouckenooghe, T., Bottu, G., Sisino, G., Igoillo-Esteve, M., et al. (2012). The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 8:e1002552. doi: 10.1371/journal.pgen.1002552
Eliasson, L., and Esguerra, J. L. S. (2020). MicroRNA networks in pancreatic islet cells: normal function and type 2 diabetes. Diabetes Metab. Res. Rev. 69, 804–812. doi: 10.2337/dbi19-0016
Fairbrother, W. G., Yeh, R., Sharp, P. A., and Burge, C. B. (2002). Predictive Identification of Exonic Splicing Enhancers in Human Genes. Science 297, 1007–1013. doi: 10.1126/science.1073774
Fan, H. Q., He, W., Xu, K. F., Wang, Z. X., Xu, X. Y., and Chen, H. (2015). FTO inhibits insulin secretion and promotes NF-kappaB activation through positively regulating ROS production in pancreatic beta cells. PLoS One 10:e0127705. doi: 10.1371/journal.pone.0127705
Fred, R. G., Bang-Berthelsen, C. H., Mandrup-Poulsen, T., Grunnet, L. G., and Welsh, N. (2010). High glucose suppresses human islet insulin biosynthesis by inducing miR-133a leading to decreased polypyrimidine tract binding protein-expression. PLoS One 5:e10843. doi: 10.1371/journal.pone.0010843
Fred, R. G., Mehrabi, S., Adams, C. M., and Welsh, N. (2016). PTB and TIAR binding to insulin mRNA 3′- and 5′UTRs; implications for insulin biosynthesis and messenger stability. Heliyon 2:e00159. doi: 10.1016/j.heliyon.2016.e00159
Fred, R. G., Sandberg, M., Pelletier, J., and Welsh, N. (2011). The human insulin mRNA is partly translated via a cap- and eIF4A-independent mechanism. Biochem. Biophys. Res. Commun. 412, 693–698. doi: 10.1016/j.bbrc.2011.08.030
Fujimoto, K., Hanson, P. T., Tran, H., Ford, E. L., Han, Z., Johnson, J. D., et al. (2009). Autophagy regulates pancreatic beta cell death in response to Pdx1 deficiency and nutrient deprivation. J. Biol. Chem. 284, 27664–27673. doi: 10.1074/jbc.m109.041616
Gale, J. E., Cox, H. I., Qian, J., Block, G. D., Colwell, C. S., and Matveyenko, A. V. (2011). Disruption of circadian rhythms accelerates development of diabetes through pancreatic beta-cell loss and dysfunction. J. Biol. Rhythms 26, 423–433. doi: 10.1177/0748730411416341
Gao, X., Shin, Y. H., Li, M., Wang, F., Tong, Q., and Zhang, P. (2010). The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS One 5:e14005. doi: 10.1371/journal.pone.0014005
Garcia-Blanco, M. A., Jamison, S. F., and Sharp, P. A. (1989). Identification and purification of a 62,000-dalton protein that binds specifically to the polypyrimidine tract of introns. Genes Dev. 3, 1874–1886. doi: 10.1101/gad.3.12a.1874
Gehman, L. T., Meera, P., Stoilov, P., Shiue, L., O’Brien, J. E., Meisler, M. H., et al. (2012). The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev. 26, 445–460. doi: 10.1101/gad.182477.111
Gerken, T., Girard, C. A., Tung, Y. C., Webby, C. J., Saudek, V., Hewitson, K. S., et al. (2007). The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 318, 1469–1472. doi: 10.1126/science.1151710
Gerstberger, S., Hafner, M., and Tuschl, T. (2014). A census of human RNA-binding proteins. Nat. Rev. Genet. 15, 829–845. doi: 10.1038/nrg3813
Gold, G., Gishizky, M. L., and Grodsky, G. M. (1982). Evidence that glucose “marks” beta cells resulting in preferential release of newly synthesized insulin. Science 218, 56–58. doi: 10.1126/science.6181562
Good, A. L., Haemmerle, M. W., Oguh, A. U., Doliba, N. M., and Stoffers, D. A. (2019). Metabolic stress activates an ERK/hnRNPK/DDX3X pathway in pancreatic beta cells. Mol. Metab. 26, 45–56. doi: 10.1016/j.molmet.2019.05.009
Good, A. L., and Stoffers, D. A. (2020). Stress-induced translational regulation mediated by RNA binding proteins: key links to beta-cell failure in diabetes. Diabetes Metab. Res. Rev. 69, 499–507. doi: 10.2337/dbi18-0068
Halban, P. A. (1982). Differential rates of release of newly synthesized and of stored insulin from pancreatic islets. Endocrinology 110, 1183–1188. doi: 10.1210/endo-110-4-1183
Harries, L. W., Ellard, S., Stride, A., Morgan, N. G., and Hattersley, A. T. (2006). Isomers of the TCF1 gene encoding hepatocyte nuclear factor-1 alpha show differential expression in the pancreas and define the relationship between mutation position and clinical phenotype in monogenic diabetes. Hum. Mol. Genet. 15, 2216–2224. doi: 10.1093/hmg/ddl147
Hentze, M. W., Castello, A., Schwarzl, T., and Preiss, T. (2018). A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 19, 327–341. doi: 10.1038/nrm.2017.130
Hinman, M. N., and Lou, H. (2008). Diverse molecular functions of Hu proteins. Cell Mol. Life. Sci. 65, 3168–3181. doi: 10.1007/s00018-008-8252-6
Hogan, D. J., Riordan, D. P., Gerber, A. P., Herschlag, D., and Brown, P. O. (2008). Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 6:e255. doi: 10.1371/journal.pbio.0060255
Hong, Y., Tak, H., Kim, C., Kang, H., Ji, E., Ahn, S., et al. (2020). RNA binding protein HuD contributes to beta-cell dysfunction by impairing mitochondria dynamics. Cell Death Differ. 27, 1633–1643. doi: 10.1038/s41418-019-0447-x
Hrvatin, S., O’Donnell, C. W., Deng, F., Millman, J. R., Pagliuca, F. W., DiIorio, P., et al. (2014). Differentiated human stem cells resemble fetal, not adult, beta cells. Proc. Natl. Acad. Sci. U.S.A. 111, 3038–3043. doi: 10.1073/pnas.1400709111
Jacko, M., Weyn-Vanhentenryck, S. M., Smerdon, J. W., Yan, R., Feng, H., Williams, D. J., et al. (2018). Rbfox splicing factors promote neuronal maturation and axon initial segment assembly. Neuron 97, 853–868.e6. doi: 10.1016/j.neuron.2018.01.020
Jang, S. K., and Wimmer, E. (1990). Cap-independent translation of encephalomyocarditis virus RNA: structural elements of the internal ribosomal entry site and involvement of a cellular 57-kD RNA-binding protein. Genes Dev. 4, 1560–1572. doi: 10.1101/gad.4.9.1560
Jeffery, N., Richardson, S., Chambers, D., Morgan, N. G., and Harries, L. W. (2019). Cellular stressors may alter islet hormone cell proportions by moderation of alternative splicing patterns. Hum. Mol. Genet. 28, 2763–2774. doi: 10.1093/hmg/ddz094
Jensen, K. B., Dredge, B. K., Stefani, G., Zhong, R., Buckanovich, R. J., Okano, H. J., et al. (2000). Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 25, 359–371. doi: 10.1016/s0896-6273(00)80900-9
Jin, Y., Suzuki, H., Maegawa, S., Endo, H., Sugano, S., Hashimoto, K., et al. (2003). A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 22, 905–912. doi: 10.1093/emboj/cdg089
Johnson, J. M., Castle, J., Garrett-Engele, P., Kan, Z., Loerch, P. M., Armour, C. D., et al. (2003). Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302, 2141–2144. doi: 10.1126/science.1090100
Juan-Mateu, J., Rech, T. H., Villate, O., Lizarraga-Mollinedo, E., Wendt, A., Turatsinze, J. V., et al. (2017). Neuron-enriched RNA-binding proteins regulate pancreatic beta cell function and survival. J. Biol. Chem. 292, 3466–3480. doi: 10.1074/jbc.m116.748335
Kamath, R. V., Leary, D. J., and Huang, S. (2001). Nucleocytoplasmic shuttling of polypyrimidine tract-binding protein is uncoupled from RNA export. Mol. Biol. Cell 12, 3808–3820. doi: 10.1091/mbc.12.12.3808
Keene, J. D. (2007). RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 8, 533–543. doi: 10.1038/nrg2111
Kim, C., Kim, W., Lee, H., Ji, E., Choe, Y. J., Martindale, J. L., et al. (2014). The RNA-binding protein HuD regulates autophagosome formation in pancreatic beta cells by promoting autophagy-related gene 5 expression. J. Biol. Chem. 289, 112–121. doi: 10.1074/jbc.m113.474700
Kirkpatrick, C. L., Marchetti, P., Purrello, F., Piro, S., Bugliani, M., Bosco, D., et al. (2010). Type 2 diabetes susceptibility gene expression in normal or diabetic sorted human alpha and beta cells: correlations with age or BMI of islet donors. PLoS One 5:e11053. doi: 10.1371/journal.pone.0011053
Kiselev, Y., Eriksen, T. E., Forsdahl, S., Nguyen, L. H., and Mikkola, I. (2012). 3T3 cell lines stably expressing Pax6 or Pax6(5a)–a new tool used for identification of common and isoform specific target genes. PLoS One 7:e31915. doi: 10.1371/journal.pone.0031915
Knoch, K. P., Bergert, H., Borgonovo, B., Saeger, H. D., Altkruger, A., Verkade, P., et al. (2004). Polypyrimidine tract-binding protein promotes insulin secretory granule biogenesis. Nat. Cell Biol. 6, 207–214. doi: 10.1038/ncb1099
Knoch, K. P., Meisterfeld, R., Kersting, S., Bergert, H., Altkruger, A., Wegbrod, C., et al. (2006). cAMP-dependent phosphorylation of PTB1 promotes the expression of insulin secretory granule proteins in beta cells. Cell Metab. 3, 123–134. doi: 10.1016/j.cmet.2005.12.008
Krentz, N. A. J., Lee, M. Y. Y., Xu, E. E., Sproul, S. L. J., Maslova, A., Sasaki, S., et al. (2018). Single-cell transcriptome profiling of mouse and hESC-derived pancreatic progenitors. Stem Cell Rep. 11, 1551–1564. doi: 10.1016/j.stemcr.2018.11.008
Kulkarni, S. D., Muralidharan, B., Panda, A. C., Bakthavachalu, B., Vindu, A., and Seshadri, V. (2011). Glucose-stimulated translation regulation of insulin by the 5′ UTR-binding proteins. J. Biol. Chem. 286, 14146–14156. doi: 10.1074/jbc.m110.190553
Lee, E. K., Kim, W., Tominaga, K., Martindale, J. L., Yang, X., Subaran, S. S., et al. (2012). RNA-binding protein HuD controls insulin translation. Mol. Cell 45, 826–835. doi: 10.1016/j.molcel.2012.01.016
Li, B., and Yen, T. S. (2002). Characterization of the nuclear export signal of polypyrimidine tract-binding protein. J. Biol. Chem. 277, 10306–10314. doi: 10.1074/jbc.m109686200
Li, X., Kazan, H., Lipshitz, H. D., and Morris, Q. D. (2014). Finding the target sites of RNA-binding proteins. Wiley Interdiscip. Rev. RNA 5, 111–130. doi: 10.1002/wrna.1201
Li, Z., Zhou, M., Cai, Z., Liu, H., Zhong, W., Hao, Q., et al. (2018). RNA-binding protein DDX1 is responsible for fatty acid-mediated repression of insulin translation. Nucleic Acids Res. 46, 12052–12066. doi: 10.1093/nar/gky867
Licatalosi, D. D., and Darnell, R. B. (2010). RNA processing and its regulation: global insights into biological networks. Nat. Rev. Genet. 11, 75–87. doi: 10.1038/nrg2673
Licatalosi, D. D., Mele, A., Fak, J. J., Ule, J., Kayikci, M., Chi, S. W., et al. (2008). HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–469. doi: 10.1038/nature07488
Lin, J. C., Yan, Y. T., Hsieh, W. K., Peng, P. J., Su, C. H., and Tarn, W. Y. (2013). RBM4 promotes pancreas cell differentiation and insulin expression. Mol. Cell. Biol. 33, 319–327. doi: 10.1128/mcb.01266-12
Liu, J., Luo, G., Sun, J., Men, L., Ye, H., He, C., et al. (2019). METTL14 is essential for beta-cell survival and insulin secretion. Biochim Biophys Acta Mol Basis Dis 1865, 2138–2148. doi: 10.1016/j.bbadis.2019.04.011
Lorini, R., and D’Annunzio, G. (2009). The importance of the environmental factors in the outbreak of diabetes mellitus in the pediatric age. Minerva Pediatr. 61, 681–684.
Lou, H., Helfman, D. M., Gagel, R. F., and Berget, S. M. (1999). Polypyrimidine tract-binding protein positively regulates inclusion of an alternative 3′-terminal exon. Mol. Cell. Biol. 19, 78–85. doi: 10.1128/mcb.19.1.78
Lukong, K. E., Chang, K. W., Khandjian, E. W., and Richard, S. (2008). RNA-binding proteins in human genetic disease. Trends Genet. 24, 416–425. doi: 10.1016/j.tig.2008.05.004
Lunde, B. M., Moore, C., and Varani, G. (2007). RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol. 8, 479–490. doi: 10.1038/nrm2178
Magro, M. G., and Solimena, M. (2013). Regulation of beta-cell function by RNA-binding proteins. Mol. Metab. 2, 348–355. doi: 10.1016/j.molmet.2013.09.003
Marcheva, B., Perelis, M., Weidemann, B. J., Taguchi, A., Lin, H., Omura, C., et al. (2020). A role for alternative splicing in circadian control of exocytosis and glucose homeostasis. Genes Dev. 34, 1–17. doi: 10.1101/gad.338178.120
Mitchell, S. A., Spriggs, K. A., Coldwell, M. J., Jackson, R. J., and Willis, A. E. (2003). The Apaf-1 internal ribosome entry segment attains the correct structural conformation for function via interactions with PTB and unr. Mol. Cell. 11, 757–771. doi: 10.1016/s1097-2765(03)00093-5
Nutter, C. A., and Kuyumcu-Martinez, M. N. (2018). Emerging roles of RNA-binding proteins in diabetes and their therapeutic potential in diabetic complications. Wiley Interdiscip. Rev. RNA 9:e1459. doi: 10.1002/wrna.1459
Okano, H. J., and Darnell, R. B. (1997). A hierarchy of Hu RNA binding proteins in developing and adult neurons. J. Neurosci. 17, 3024–3037. doi: 10.1523/jneurosci.17-09-03024.1997
Ortis, F., Naamane, N., Flamez, D., Ladriere, L., Moore, F., Cunha, D. A., et al. (2010). Cytokines interleukin-1beta and tumor necrosis factor-alpha regulate different transcriptional and alternative splicing networks in primary beta-cells. Diabetes Metab. Res. Rev. 59, 358–374. doi: 10.2337/db09-1159
Perez, I., McAfee, J. G., and Patton, J. G. (1997). Multiple RRMs contribute to RNA binding specificity and affinity for polypyrimidine tract binding protein. Biochemistry 36, 11881–11890. doi: 10.1021/bi9711745
Ponthier, J. L., Schluepen, C., Chen, W., Lersch, R. A., Gee, S. L., Hou, V. C., et al. (2006). Fox-2 splicing factor binds to a conserved intron motif to promote inclusion of protein 4.1R alternative exon 16. J. Biol. Chem. 281, 12468–12474. doi: 10.1074/jbc.m511556200
Prokunina-Olsson, L., Welch, C., Hansson, O., Adhikari, N., Scott, L. J., Usher, N., et al. (2009). Tissue-specific alternative splicing of TCF7L2. Hum. Mol. Genet. 18, 3795–3804.
Ramos-Rodriguez, M., Raurell-Vila, H., Colli, M. L., Alvelos, M. I., Subirana-Granes, M., Juan-Mateu, J., et al. (2019). The impact of proinflammatory cytokines on the beta-cell regulatory landscape provides insights into the genetics of type 1 diabetes. Nat. Genet. 51, 1588–1595. doi: 10.1038/s41588-019-0524-6
Roggli, E., Gattesco, S., Pautz, A., and Regazzi, R. (2012). Involvement of the RNA-binding protein ARE/poly(U)-binding factor 1 (AUF1) in the cytotoxic effects of proinflammatory cytokines on pancreatic beta cells. Diabetologia 55, 1699–1708. doi: 10.1007/s00125-011-2399-7
Russell, M. A., and Morgan, N. G. (2011). Conditional expression of the FTO gene product in rat INS-1 cells reveals its rapid turnover and a role in the profile of glucose-induced insulin secretion. Clin. Sci. 120, 403–413. doi: 10.1042/cs20100416
Sarwade, R. D., Khalique, A., Kulkarni, S. D., Pandey, P. R., Gaikwad, N., and Seshadri, V. (2020). Translation of insulin granule proteins are regulated by PDI and PABP. Biochem. Biophys. Res. Commun. 526, 618–625. doi: 10.1016/j.bbrc.2020.03.106
Sawicka, K., Bushell, M., Spriggs, K. A., and Willis, A. E. (2008). Polypyrimidine-tract-binding protein: a multifunctional RNA-binding protein. Biochem. Soc. Trans. 36, 641–647. doi: 10.1042/bst0360641
Schoch, K. M., and Miller, T. M. (2017). Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron 94, 1056–1070. doi: 10.1016/j.neuron.2017.04.010
Shen, F., Huang, W., Huang, J. T., Xiong, J., Yang, Y., Wu, K., et al. (2015). Decreased N(6)-methyladenosine in peripheral blood RNA from diabetic patients is associated with FTO expression rather than ALKBH5. J. Clin. Endocrinol. Metab. 100, E148–E154.
Shepard, P. J., and Hertel, K. J. (2009). The SR protein family. Genome Biol. 10:242. doi: 10.1186/gb-2009-10-10-242
Singer, R. A., Arnes, L., Cui, Y., Wang, J., Gao, Y., Guney, M. A., et al. (2019). The long noncoding RNA paupar modulates PAX6 regulatory activities to promote alpha cell development and function. Cell Metab. 30:e1098.
Sunters, A., Fernandez de Mattos, S., Stahl, M., Brosens, J. J., Zoumpoulidou, G., Saunders, C. A., et al. (2003). FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J. Biol. Chem. 278, 49795–49805. doi: 10.1074/jbc.m309523200
Szabat, M., Kalynyak, T. B., Lim, G. E., Chu, K. Y., Yang, Y. H., Asadi, A., et al. (2011). Musashi expression in beta-cells coordinates insulin expression, apoptosis and proliferation in response to endoplasmic reticulum stress in diabetes. Cell Death Dis. 2, e232. doi: 10.1038/cddis.2011.119
Taneera, J., Fadista, J., Ahlqvist, E., Atac, D., Ottosson-Laakso, E., Wollheim, C. B., et al. (2015). Identification of novel genes for glucose metabolism based upon expression pattern in human islets and effect on insulin secretion and glycemia. Hum. Mol. Genet. 24, 1945–1955. doi: 10.1093/hmg/ddu610
Taneera, J., Prasad, R. B., Dhaiban, S., Mohammed, A. K., Haataja, L., Arvan, P., et al. (2018). Silencing of the FTO gene inhibits insulin secretion: an in vitro study using GRINCH cells. Mol. Cell. Endocrinol. 472, 10–17. doi: 10.1016/j.mce.2018.06.003
The Tabula Muris Consortium, Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis et al. (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. doi: 10.1038/s41586-018-0590-4
Tillmar, L., Carlsson, C., and Welsh, N. (2002). Control of insulin mRNA stability in rat pancreatic islets. Regulatory role of a 3′-untranslated region pyrimidine-rich sequence. J. Biol. Chem. 277, 1099–1106. doi: 10.1074/jbc.m108340200
Tillmar, L., and Welsh, N. (2002). Hypoxia may increase rat insulin mRNA levels by promoting binding of the polypyrimidine tract-binding protein (PTB) to the pyrimidine-rich insulin mRNA 3′-untranslated region. Mol. Med. 8, 263–272. doi: 10.1007/bf03402152
Ule, J., Stefani, G., Mele, A., Ruggiu, M., Wang, X., Taneri, B., et al. (2006). An RNA map predicting Nova-dependent splicing regulation. Nature 444, 580–586. doi: 10.1038/nature05304
Ule, J., Ule, A., Spencer, J., Williams, A., Hu, J. S., Cline, M., et al. (2005). Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 37, 844–852. doi: 10.1038/ng1610
van Arensbergen, J., Garcia-Hurtado, J., Moran, I., Maestro, M. A., Xu, X., Van de Casteele, M., et al. (2010). Derepression of polycomb targets during pancreatic organogenesis allows insulin-producing beta-cells to adopt a neural gene activity program. Genome Res. 20, 722–732. doi: 10.1101/gr.101709.109
Vanzela, E. C., and Cardozo, A. K. (2012). Is ARE/poly(U)-binding factor 1 (AUF1) a new player in cytokine-mediated beta cell apoptosis? Diabetologia 55, 1572–1576. doi: 10.1007/s00125-012-2552-y
Villate, O., Turatsinze, J. V., Mascali, L. G., Grieco, F. A., Nogueira, T. C., Cunha, D. A., et al. (2014). Nova1 is a master regulator of alternative splicing in pancreatic beta cells. Nucleic Acids Res. 42, 11818–11830. doi: 10.1093/nar/gku861
Wang, Y., Sun, J., Lin, Z., Zhang, W., Wang, S., Wang, W., et al. (2020). m(6)A mRNA methylation controls functional maturation in neonatal murine beta cells. Diabetes 69, 1708–1722. doi: 10.2337/db19-0906
Wei, C., Qiu, J., Zhou, Y., Xue, Y., Hu, J., Ouyang, K., et al. (2015). Repression of the central splicing regulator RBFox2 is functionally linked to pressure overload-induced heart failure. Cell Rep. 10, 1521–1533. doi: 10.1016/j.celrep.2015.02.013
Wollerton, M. C., Gooding, C., Robinson, F., Brown, E. C., Jackson, R. J., and Smith, C. W. (2001). Differential alternative splicing activity of isoforms of polypyrimidine tract binding protein (PTB). RNA 7, 819–832. doi: 10.1017/s1355838201010214
Wollerton, M. C., Gooding, C., Wagner, E. J., Garcia-Blanco, M. A., and Smith, C. W. (2004). Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol. Cell. 13, 91–100. doi: 10.1016/s1097-2765(03)00502-1
Wood, A. R., Tyrrell, J., Beaumont, R., Jones, S. E., Tuke, M. A., Ruth, K. S., et al. (2016). Variants in the FTO and CDKAL1 loci have recessive effects on risk of obesity and type 2 diabetes, respectively. Diabetologia 59, 1214–1221. doi: 10.1007/s00125-016-3908-5
Yang, Y. Y., Yin, G. L., and Darnell, R. B. (1998). The neuronal RNA-binding protein Nova-2 is implicated as the autoantigen targeted in POMA patients with dementia. Proc. Natl. Acad. Sci. U.S.A. 95, 13254–13259. doi: 10.1073/pnas.95.22.13254
Yoo, Y. M. (2013). Melatonin-mediated insulin synthesis during endoplasmic reticulum stress involves HuD expression in rat insulinoma INS-1E cells. J. Pineal Res. 55, 207–220. doi: 10.1111/jpi.12064
Zhai, K., Gu, L., Yang, Z., Mao, Y., Jin, M., Chang, Y., et al. (2016). RNA-binding protein CUGBP1 regulates insulin secretion via activation of phosphodiesterase 3B in mice. Diabetologia 59, 1959–1967. doi: 10.1007/s00125-016-4005-5
Zhang, C., Frias, M. A., Mele, A., Ruggiu, M., Eom, T., Marney, C. B., et al. (2010). Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science 329, 439–443. doi: 10.1126/science.1191150
Zhang, C., Fu, J., and Zhou, Y. (2019). A review in research progress concerning m6A methylation and immunoregulation. Front. Immunol. 10:922. doi: 10.3389/fimmu.2019.00922
Zhang, X., Tang, N., Hadden, T. J., and Rishi, A. K. (2011). Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 1813, 1978–1986. doi: 10.1016/j.bbamcr.2011.03.010
Zhong, W., Li, Z., Zhou, M., Xu, T., and Wang, Y. (2018). DDX1 regulates alternative splicing and insulin secretion in pancreatic beta cells. Biochem. Biophys. Res. Commun. 500, 751–757. doi: 10.1016/j.bbrc.2018.04.147
Zhong, X. Y., Wang, P., Han, J., Rosenfeld, M. G., and Fu, X. D. (2009). SR proteins in vertical integration of gene expression from transcription to RNA processing to translation. Mol. Cell. 35, 1–10. doi: 10.1016/j.molcel.2009.06.016
Keywords: pancreatic islet, beta cells, diabetes, RNA processing, RNA binding proteins
Citation: Moss ND and Sussel L (2020) mRNA Processing: An Emerging Frontier in the Regulation of Pancreatic β Cell Function. Front. Genet. 11:983. doi: 10.3389/fgene.2020.00983
Received: 03 July 2020; Accepted: 03 August 2020;
Published: 01 September 2020.
Edited by:
Simona Chera, University of Bergen, NorwayReviewed by:
Ildem Akerman, University of Birmingham, United KingdomCopyright © 2020 Moss and Sussel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lori Sussel, bG9yaS5zdXNzZWxAY3VhbnNjaHV0ei5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.