 Beenish Rahat
Beenish Rahat Taqveema Ali
Taqveema Ali Divika Sapehia
Divika Sapehia Aatish Mahajan
Aatish Mahajan Jyotdeep Kaur
Jyotdeep Kaur
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 11 August 2020
Sec. Epigenomics and Epigenetics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00844
The circulating cell-free nucleic acids (ccfNAs) are a mixture of single- or double-stranded nucleic acids, released into the blood plasma/serum by different tissues via apoptosis, necrosis, and secretions. Under healthy conditions, ccfNAs originate from the hematopoietic system, whereas under various clinical scenarios, the concomitant tissues release ccfNAs into the bloodstream. These ccfNAs include DNA, RNA, microRNA (miRNA), long non-coding RNA (lncRNA), fetal DNA/RNA, and mitochondrial DNA/RNA, and act as potential biomarkers in various clinical conditions. These are associated with different epigenetic modifications, which show disease-related variations and so finding their role as epigenetic biomarkers in clinical settings. This field has recently emerged as the latest advance in precision medicine because of its clinical relevance in diagnostic, prognostic, and predictive values. DNA methylation detected in ccfDNA has been widely used in personalized clinical diagnosis; furthermore, there is also the emerging role of ccfRNAs like miRNA and lncRNA as epigenetic biomarkers. This review focuses on the novel approaches for exploring ccfNAs as epigenetic biomarkers in personalized clinical diagnosis and prognosis, their potential as therapeutic targets and disease progression monitors, and reveals the tremendous potential that epigenetic biomarkers present to improve precision medicine. We explore the latest techniques for both quantitative and qualitative detection of epigenetic modifications in ccfNAs. The data on epigenetic modifications on ccfNAs are complex and often milieu-specific posing challenges for its understanding. Artificial intelligence and deep networks are the novel approaches for decoding complex data and providing insight into the decision-making in precision medicine.
The diagnostic platform utilizing the detection of biomarkers in various body fluids called “liquid biopsy” can revolutionize precision medicine. Precision medicine is aimed at attaining better-personalized care by the development of the latest diagnostic and prognostic methods that consider individual variability (Kaur et al., 2017). Liquid biopsy is being utilized for non-invasive prognostic and predictive purposes. Efficient and reliable markers within the body fluids can help in personalized treatment decisions for monitoring disease and survival. ccfNAs have emerged as such markers for screening, diagnosis, prognosis, management, and treatment of various cancers; autoimmune, neurological, and mitochondrial diseases; prenatal diagnosis; diagnosis of pregnancy-related complications (Pos et al., 2018); diabetes; inflammation; rheumatoid arthritis; stroke; and trauma (Swarup and Rajeswari, 2007). An increased amount of ccfNAs is observed in these disorders, making liquid biopsies more sensitive, rapid, accurate, and preferable alternatives for various invasive diagnostic methods (Pos et al., 2018).
ccfNAs present in blood circulation include cell-free genomic DNAs (ccfDNAs) and cell-free mtDNA (Kohler et al., 2009; Thierry et al., 2016) and cell-free RNAs (ccfRNAs) including protein-coding messenger RNA (mRNA), regulatory non-coding RNAs like microRNAs (miRNAs), long non-coding RNAs (lncRNAs), circular RNAs, and RNAs involved in translation like transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs) (Pos et al., 2018).
The ccfNAs (DNAs and RNAs) are generally released into the blood circulation either by apoptosis, necrosis, or active secretion. In healthy persons, the origin of ccfNAs is mainly attributed to lymphoid and myeloid tissues (Snyder et al., 2016), while in the case of various clinical conditions, the associated or the affected tissues would release the extra amount of ccfNAs into blood (Swarup and Rajeswari, 2007; Devonshire et al., 2014) in a pattern specific to the pathophysiological condition (Hunter et al., 2008; Noferesti et al., 2015).
Various genetic as well as epigenetic biomarkers have been explored for ccfNA-based liquid biopsy. As genetic biomarkers are less consistent and provide more variability across studies, epigenetic markers, which are more generalized between samples, present as a promising alternative for early diagnosis and monitoring of the diseases. These epigenetic marks are tissue specific and reflect the pattern of disease progression (Zeng et al., 2019). Furthermore, epigenetic biomarkers are dynamic with most techniques required for analysis of these biomarkers that are already available in clinical laboratories.
The use of epigenetic marks has revolutionized the field of non-invasive molecular diagnosis replacing traditional screening and treatment methods. These assays have great potential in future precise patient care. The epigenetic marks for ccfNAs reflect the pattern specific for the tissue contributing to these ccfNAs. Therefore, the use of epigenetic markers can help in the diagnosis of various diseases even before the onset of actual symptoms and hence help in better management of the disease. Precision medicine has improved health care by the identification of different stages/subsets of diseases, precise diagnosis, and treatment. Furthermore, the development of advanced analytical software techniques like machine learning and artificial intelligence can enhance precision medicine (Ahlquist, 2018; Beltran-Garcia et al., 2019). These are used in combination with next-generation sequencing to identify novel ccfNA-based epigenetic markers.
Reliable markers are required to guide personalized treatment decisions for monitoring disease progression and survival. The presence of epigenetic marks on ccfNAs specific to a particular clinical condition is widely being explored to advance personalized medicine. A perfect epigenetic marker for precision medicine should be able to detect the disease with high sensitivity, predict the risk of disease development and its progression, and monitor the therapeutic response of the patient (Beltran-Garcia et al., 2019). ccfDNAs are associated with various epigenetic marks (Schwarzenbach et al., 2011) like DNA methylation, hydroxymethylcytosine (5hmC), and posttranslational modifications of histones. In addition, nucleosome positioning and occupancy on ccfDNAs have exhibited high sensitivity and specificity in liquid biopsy-based methods for disease detection and classification.
The 5-methylcytosine (5mC) modification at CpG dinucleotides is the most abundant form of DNA methylation. It plays an important role in the regulation of gene expression and is widely used as an epigenetic biomarker for ccfDNA-based assays. DNA methylation has replaced many genetic mutation- or protein-based markers. These 5mC biomarkers are also valuable in identifying tissue-specific methylation to estimate tumor burden and tissue of origin in ccfDNAs. In addition to 5mC, 5-hydroxymethylcytosine (5hmC) is also used as an epigenetic mark on ccfDNAs (Zeng et al., 2019). 5hmC is created by the oxidation of 5mC by 10–11 translocation (Tet) proteins. Although 5hmC is far less abundant compared to 5mC, it is more distinctly distributed among different transcriptionally active regions, which emphasizes its potential as a diagnostic marker. Genome-wide analysis of 5hmC pattern can provide more information about the potential of this epigenetic marker for ccfDNAs (Zeng et al., 2019).
Nucleosome positioning has emerged as a recent biomarker to distinguish the tissue of origin of ccfDNA based on derived nucleosome maps. Snyder et al., 2016 performed deep sequencing on ccfDNAs and observed a distinct pattern of nucleosome positioning between healthy persons and cancer patients correlating with the tissues of origin (Snyder et al., 2016). This emphasizes the use of nucleosome maps, which consist of occupancy of transcription factor and nucleosome as the epigenetic marks to distinguish normal versus cancer ccfDNAs. Hence, nucleosome positioning can also be used to identify various cancers that generally require invasive biopsies for definitive diagnosis. Moreover, genome-wide nucleosome positioning of ccfDNAs is utilized to infer pathological states of multiple disease types. A comprehensive public database called cell-free epigenome atlas (CFEA) provides the epigenome profile of ccfDNAs from various human diseases and can help in a better understanding of collected data (Yu et al., 2020). ccfDNA are generally associated with nucleosomes and histone proteins. Histone proteins are posttranslationally modified at amino acid residues located on their N- and C-terminal tails. These modifications act as epigenetic marks that can specifically distinguish disease-related ccfDNAs in blood samples. Various types of histone modifications are associated with the development and pathogenesis of human diseases (Zhao and Shilatifard, 2019).
In addition to DNA markers, RNA markers like mRNAs, miRNAs, lncRNAs, and circRNAs are also getting attention in the focus of clinical research (Pos et al., 2018).
Most of the currently available diagnostic tests based on ccfNAs use either DNA methylation markers or the differential expression of miRNAs. These biomarkers are relatively easily detected and estimated using accessible techniques like methyLight, methyl-specific PCR, methylation-sensitive high-resolution melting, and pyrosequencing (García-Giménez et al., 2017). DNA methylation specific to fetal and tumor DNA has been reported in pregnant women and cancer patients, respectively (Wong et al., 1999; Poon et al., 2002). The pattern of the methylation in these ccfDNAs has been traced back to their tissue of origin (Lun et al., 2013; Sun et al., 2017). Differentially methylated markers have been reported in ccfDNAs like INS promoter 1 in diabetes and REG1A and CUX2 genes in pancreatic cancer (Lehmann-Werman et al., 2016). Promoter methylation of SERPINB5, RASSF1A, and STAT5A act as epigenetic fetal markers in maternal blood (Chim et al., 2005; Chan et al., 2006; Rahat et al., 2016a).
The various diagnostic approaches to study the epigenetic modifications in the nucleic acids include methylated CpG island recovery assay (MIRA) and MethylCap that rely on methyl-CpG-binding domains (MBD) to capture methylated DNA after DNA fractionation either by restriction digestion or sonication (Mitchell et al., 2011). These methods can also be combined with microarray or NGS technologies (MethylCap-seq) to identify biomarkers for cancer diagnosis and DNA methylation maps of cancer genomes (Simmer et al., 2012). Reduced representation bisulfite sequencing (RRBS) (Meissner et al., 2005) is an efficient method for absolute quantification of the methylation status of more than one million CpG sites at single base-pair resolution, covering regions of moderate to high CpG density (Lee et al., 2014). New techniques such as whole-genome bisulfite sequencing (WGBS) allows for an unbiased assessment of DNA methylation at single-base resolution with full coverage of more than 28 million CpG sites in the human genome, and by using this technique in the clinical settings, relevant biomarkers were identified in colorectal and breast cancers and certain types of leukemia (Berman et al., 2011).
Some of the techniques are used in clinical settings, like parallel shotgun sequencing and targeted sequencing (Norwitz and Levy, 2013) for non-invasive prenatal testing, WGS for fetal gene detection (Lo et al., 2010), and cancer personalized profiling by deep sequencing (CAPP-seq) to quantify circulating tumor DNA (Newman et al., 2014).
Despite the advancement of the techniques to study epigenetic modifications, the use of epigenetic biomarkers present on ccfNAs is limited due to their lower levels in the blood circulation. In the case of cancer, WGS is applied to only 5–10% of cell-free tumor DNA depending on the copy number. Mostly targeted methylation sequencing is carried out in such cases, which has a greater potential for the detection of lower levels of ccfNA in patients with early-stage disease.
Chromatin-based ChIP-seq experiments are revolutionizing our understanding of the complexes associated with chromatin dynamics. Ongoing advances such as nano-ChIP-seq allow ChIP-seq to be analyzed from far fewer cells necessary for embryology and development studies (Nakato and Shirahige, 2017). The emergence of ChIP-exo that digests the ends of DNA fragments not bound to protein is quite promising (Furey, 2012). However, the application of these techniques to identify biomarkers is limited due to the expertise and cost associated.
ChIP-seq also provides critical information on other chromatin modifiers, such as histone marks and the enzymes that modify these marks in diseases such as cancer. ChIP-seq has identified the role of aberrant H3K79 methylation by the methyltransferase DOT1L in mixed lineage leukemia (MLL)-rearranged leukemias (Bernt et al., 2011). In addition to ChiP-seq, different techniques like ChIP-PCR, ELISA-based assays, or mass spectrometry are used to detect and quantify histone modifications on ccfNAs in serum or plasma (Adli and Bernstein, 2011).
The detection and quantification of ccfNAs, viz. RNA, DNA, fetal DNA, fetal RNA, mtDNA, and mitochondrial RNA and miRNA levels in body fluids are of clinical significance. These ccfNAs have the potential to act as biomarkers for diagnosis as well as prognosis of various diseases (Fleischhacker and Schmidt, 2007; Breitbach et al., 2012), such as different cancers, obstetric, autoimmune, neurological, and mitochondrial diseases, as well as prenatal diagnosis, etc. (Kandel, 2012; Shaw et al., 2012). Although the most studied area of epigenetics is DNA methylation, yet in the clinical setting, there are only a few methylation markers. Various blood- or tissue-based cohort well-powered studies have recently shown that changes in the DNA methylation are not only observed frequently in cancers but also in other broad range of complex diseases including neurodegenerative, metabolic, autoimmune, and inflammatory diseases although at a lower frequency (Tost, 2016).
DNA methylation analysis of ccfDNA might provide a valuable option in some cases when the blood–brain barrier is temporarily disrupted. It was recently demonstrated by the detection of unmethylated fragments of MBP3 and WM1, specific for oligodendrocytes in about 75% of patients with relapsing multiple sclerosis (Zachariah et al., 2009). cfRNAs are also present in the patient’s serum/plasma in addition to ccfDNAs. Higher levels of circulatory RNases were observed in cancers and various diseases like cerebral attack, preeclampsia, etc., and surprisingly, RNA found in the circulation was found to be stable (Umu et al., 2018). Changes in the expression of intracellular miRNA have been causally linked with many diseases that include cancer (Esquela-Kerscher and Slack, 2006), cardiovascular diseases (Navickas et al., 2016), neurodegenerative diseases (Gupta et al., 2017), etc. Such changes in expression of miRNA are either similar or distinct in the serum of a particular set of patients, thus enabling miRNA detection in serum as biomarkers of human diseases (Backes et al., 2016). Therefore, ccfNAs play a prominent role in the pathogenesis and diagnosis of various diseases. Further research is required in this field to ensure the widespread application of these markers in clinical settings.
Every year, about 14 million new cases of cancer are reported (excluding skin cancer other than melanoma) that cause about 8.8 million deaths, accounting for 15.7% of deaths in a year (Ferlay et al., 2019). An estimated number of more than 1.8 million new cancer cases are likely to be diagnosed, and 606,520 cancer deaths are expected in the United States in 2020, which deciphers almost 1,660 deaths per day (Siegel et al., 2020). The six major hallmarks of cancer (Hanahan and Weinberg, 2000) are uncontrolled cell growth and division, programmed cell death avoidance, invasion, metastasis, and angiogenesis. The diagnosis of cancer usually occurs following the appearance of signs or symptoms or through screening and investigations like X-rays, blood tests, endoscopy, CT scans, etc. Biopsy tissue examination indicates the type of proliferating cells, genetic abnormalities, and histological grade, and other characteristics. Therefore, advanced measures such as estimating prognosis, risk assessment for early diagnosis, biomarkers, and observing the response to therapy can lead to successful treatment, positive outcomes, and improvement of the quality of life for patients. The tissue biopsy-matched ccfDNA is considered as surrogate marker due to its release from the tumor sites (De Mattos-Arruda et al., 2013). It is proven to be a non-invasive, rapid, and sensitive marker for diagnosis, prognosis, and therapy response monitoring in different cancers (Volik et al., 2016). In addition, the integrity of ccfDNA (extent of ccfDNA fragmentation) may be utilized as a promising biomarker for diagnosis and prognosis of cancer (Madhavan et al., 2014).
Serum or plasma ccfNA serves as a “liquid biopsy,” which is useful for various applications in diagnostics and avoids the necessity for biopsy of tumor tissue. The levels of ccfNA in blood and lymphatic circulation are affected by degradation, clearance, and various other physiological events. Liver and kidney clear nucleic acids from the blood, and they have a half-life of different time intervals in the circulation that varies from 15 min to several hours (Fleischhacker and Schmidt, 2007). miRNAs appear to be extremely stable, but their rate of clearance from the blood is not well studied in cancer patients thus owing to the uniqueness of this research area.
ccfDNAs consists of both genomic DNA (gDNA) as well as mtDNA. There is a production of longer uneven fragments of DNA by necrosis in cancer patients and shorter DNA fragments from apoptosis. Hence, increased levels of longer DNA fragments in the bloodstream have been targeted as a potential marker for the presence of malignant tumor DNA (Arko-Boham et al., 2019). Tumor cells are the origin of ccfDNA in the blood of cancer patients (Stroun et al., 1989). Aberrations specific to tumors like oncogene and tumor suppressor gene mutations (Wang et al., 2004), methylation of DNA (Fujiwara et al., 2005), and instability of microsatellite DNA (Shaw et al., 2000) were recognized in ccfDNA. Tumorigenesis and its progression are monitored by the change in various epigenetic modifications. Patients with different types of malignancies have methylated DNA in their serum or plasma. One of the most important methods for analyzing malignancy is by detecting the presence of methylated ccfDNA in cancer patients.
For early diagnosis of colorectal cancer (CRC), analysis of promoter hypermethylation in blood and fecal DNA has the potential to be used as a non-invasive test, and efforts are made for clinical application of these molecular markers. Various studies have observed MGMT, RASSF2A, Wif-1, NGFR, and SEPT9 as aberrantly methylated genes used as diagnostic biomarkers in patients with CRC (Lee et al., 2009; Powrozek et al., 2014). Several potential methylation biomarkers have been found that differentiate plasma from breast cancer patients and that from control subjects (Hoque et al., 2006). Remarkably, two independent studies recognized CST6 as being methylated differentially between breast cancer and control plasma samples (Radpour et al., 2011; Chimonidou et al., 2013). For lung cancer, an early focus was to search methylated CDKN2A as a plasma diagnostic biomarker. Studies observed hypermethylation of CDKN2A in the plasma of patients with lung cancer as compared to cancer-free controls (Zhang et al., 2011). SHOX2 was identified as a potential biomarker in a retrospective study done by researchers from the Theracode, a diagnostic firm (Kneip et al., 2011). A recent study, by a group, as part of the Australian Pancreatic Cancer Genome Initiative (APGI), has observed elevated levels of aberrant methylation in the important cell signaling pathways, thus suggesting its possibility as a disease biomarker. They worked on a group of six candidate genes, NPTX2, SARP2, UCHL1, ppENK, CDKN2A, and RASSF1A, and observed differential methylation in the promoters of all the genes in pancreatic cancer and healthy controls except in CDKN2A promoter, which was methylated differentially between pancreatic cancer patients and those having chronic pancreatitis (Park et al., 2012). Epigenetic events in the progression of cancer include the promoter region hypermethylation of the genes, pi-class GSTP1, and APC, which are the most common somatic genome abnormalities in colorectal and prostate cancer (Ellinger et al., 2008). RASSF1A, RARB, SEPT9, ESR1, and CDKN2A are the important methylated genes that have shown utility in prognosis using ccfDNA assays in many patients. Methylation of histones is an active process with vital roles in differentiation and development. Tumorigenesis also occurs due to aberrant levels of histone methylation. The promoters associated with H3K4 are primarily trimethylated by SET1A and SET1B. SET1A plays a vital role in oncogenic function in breast cancer metastasis, lung cancer, and colorectal cancer (Zhao and Shilatifard, 2019). Table 1 presents the frequently hypermethylated genes in various cancer types.
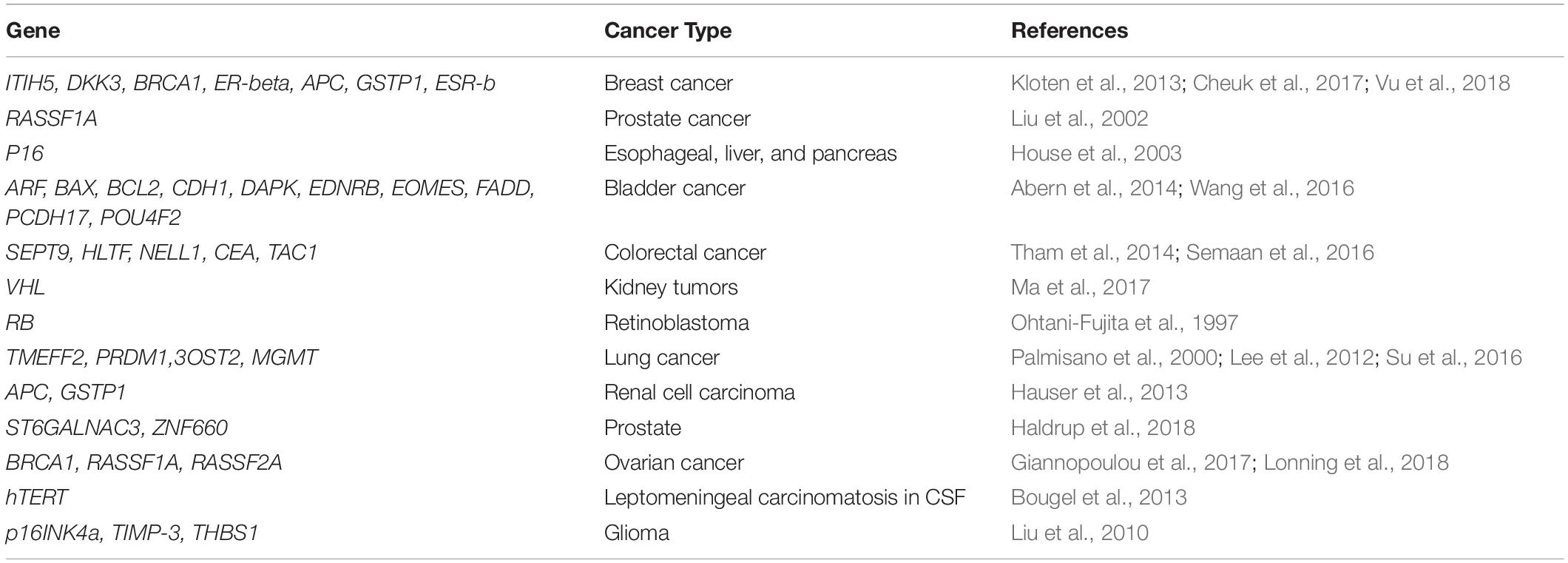
Table 1. Frequently hypermethylated genes in various cancer types.
In various cancers, miRNA expression dysregulation has been observed that suggests its role in many processes necessary for the progression of cancer like proliferation, cell death, metastasis, and resistance to treatment (Iorio and Croce, 2012). During the development of the liver, miRNA expression changes dynamically. miR-500 is one such oncofetal miRNA that is important for the diagnosis of hepatocellular carcinoma (Yamamoto et al., 2009). Lately, in non-small cell lung cancer (NSCLC), miR-1246 and miR-1290 were recognized as tumor-initiating and cell-specific miRNAs (Zhang et al., 2016). miR-1290 was found to be a significant prognostic factor for OSCC patients based on Cox regression analysis. In addition, miR-1290 could serve as a valuable biomarker in OSCC patients to predict the clinical response to chemoradiotherapy (Lin et al., 2018). A study by Alhasan et al., 2016 showed a serum signature of 5-miRNAs (miR-135a, miR-106a, miR-200c, miR-605, and miR-433) predicted a very high-risk prostate cancer (Alhasan et al., 2016). Expression levels of miR-21, miR-23b, miR-200c, and miR-200b were upregulated in metastatic breast cancer when compared to early breast cancer patients, therefore supporting the notion that ccfmiRNAs presents a tool with the crucial diagnostic and prognostic implication in breast cancer (Papadaki et al., 2019). Furthermore, a study discovered that increased miR-122 expression was significantly associated with a reduction in the overall survival as well as progression-free survival in breast cancer patients (Saleh et al., 2019). Elevation in the levels of serum miR-29, miR-122, miR-155, and miR-192 was observed in cholangiocarcinoma. Although miRNAs levels before surgery were inappropriate as survival prognostic marker; however, postsurgery decrease in the serum miR-122 levels was significantly linked with better patient prognosis (Loosen et al., 2019).
ccfDNA analysis is a non-invasive process that allows day to day patient follow-up and monitoring of response toward treatment (Gorges et al., 2012). Both genetic and epigenetic changes are exhibited by ccfDNA (Stroun et al., 2001). The study of these changes might provide valuable information to mold the choice of treatment by clinicians given the limitations of the novel targeted therapies.
Abnormal hypermethylation at CpG islands occurs rarely in non-malignant and normally differentiated cells, so the release of DNA from tumor cells can be found with a prominent extent of sensitivity, even when the excess of DNA is released from normal cells, and this characterizes its potential clinical application (Wong et al., 2001). In this context, promoter region hypermethylation of INK4A occurs very early in the progression of hepatocellular carcinoma (HCC), and hence, it serves as a valuable biomarker for non-invasive diagnosis as well as prediction of response to therapy (Huang et al., 2014).
In the MYCN-amplified neuroblastoma progression, MYCN is detected in circulating DNA. This phenomenon was found to be associated strongly with the quick progression of tumors and poor outcomes (Combaret et al., 2002). Loss of heterozygosity (LOH) and abnormal methylation at the promoter region of MYCN were detected using ccfDNA, which showed elevated levels in patients of high-grade glioma. Detection of promoter region hypermethylation of MYOD1 in serum may serve as a potential prognostic marker for discriminating patients of cervical cancer at high risk for lymph node metastasis or relapse (Widschwendter et al., 2004).
Moreover, the investigation of circulating miRNAs presents great potential in revealing new insights into their role in therapy and diagnosis. miRNA serum signatures (miR-345 -5p, miR-330 -3p, and miR-9 -3p) were found to be significantly upregulated in patients of prostate cancer (PCa) when compared to healthy individuals. The role of miR-345-5p to act as an oncomir through CDKN1A targeting makes it a potential target for PCa therapeutically (Tinay et al., 2018).
Immunotherapy is a rapidly developing therapy in many cancers because of various advantages over standard chemotherapy. Identification of significant miRNAs that provides a foresight of response in cancer immunotherapy would enable better patient selection and enhancement of therapeutic efficacy and provide a novel target (Antonia et al., 2004; Chen et al., 2008). miRNA-21 is a cell-free oncogenic miRNA, which has been known as a potential regulator of STAT3, and thus, it could be detected in various tumors (Ji et al., 2009). Thus, circulating miRNA-21 can act as a biomarker for response in cancer immunotherapy (Wu et al., 2012).
ccfDNAs in glioma were associated with differential methylation levels of MGMT, cyclin-dependent kinase inhibitor 2A, multiple tumor suppressor 1 p16/(INK4a), p73, and retinoic acid receptor beta (RARb) (Balana et al., 2003; Weaver et al., 2006; Wakabayashi et al., 2009). All these studies propose a crucial role of epigenetic marks in ccfNAs in cancer-targeted therapy as well as pathogenesis.
Precision oncology is an approach that includes the molecular profiling of tumors to identify effective therapeutic strategies. A clinical research program initiated by The Englander Institute for Precision Medicine (EIPM) in 2013 uses whole-exome sequencing of metastatic and primary tumors to identify individualized therapeutic options and to help guide clinical decision making, by prospective follow-up of patients (Rennert et al., 2016). Oncology is the obvious choice for heightening the impact of precision medicine. Several targeted therapies have been developed that have shown profound benefits. Recently, novel immunological approaches produced insightful responses (Snyder et al., 2014).
In addition, the identification of epigenetic biomarkers leads to more precise disease prognosis, especially in therapeutic areas that are linked with a high degree of variability concerning survival (Van Neste et al., 2017). Research carried out in several cancers like glioblastoma reveals that levels of 5hmC are critical in the regulation of genes having a crucial role in disease and show that global reduction in 5hmC over the genome leads to poor clinical outcomes in these patients (Johnson et al., 2016).
Epigenetic changes introduced common genetic mutations in an in vitro model of lung cancer (Vaz et al., 2017). Epigenetic-based diagnostics can detect early disease signals and thus can provide possibilities for clinical intervention before the progression of symptoms.
The detection of ccfNAs could be exploited by targeted therapies approved lately and eventually benefit the patients. Scrutinizing cancers by analyzing ccfNA dynamics in blood or serum is an innovative and emerging research area. As far as the existing research advancement and the growth of the medical industry are concerned, we consider that ccfNA assays may be employed for real-time personalized treatments in the future for cancer patients, based on their ccfNAs or ccfDNA methylation levels, for diagnosis and prognosis. Nevertheless, there is much scope for improvement before the application of this technology in clinical settings.
During pregnancy, the apoptosis/necrosis of trophoblasts arising from syncytiotrophoblast is the prime source of the release of ccf-fetal-NAs into the maternal blood (Litton et al., 2009). The presence of ccf-fetal-NAs has paved the way for non-invasive prenatal diagnosis and early prediction of pregnancy-related complications (Lo et al., 1997, 1998). The use of ccf-fetal-NAs has gradually replaced invasive techniques like amniocentesis or chorionic villus sampling (Serr et al., 2017). ccf-fetal-DNA comprises 10–15% of the maternal ccfDNA (Wang et al., 2013) and can be efficiently detected at the fifth week of gestation (Guibert et al., 2003). The amount of ccf-fetal-DNA in maternal blood increases progressively throughout pregnancy (Birch et al., 2005).
Prenatal diagnosis is an established practice for the management of pregnancy as well as avoidance of prenatal/neonatal deaths. The leading causes for such deaths are genetic disorder, birth defects, congenital malformations, and chromosomal abnormalities like trisomy 21 (Down’s syndrome), 18 (Edward’s syndrome), and 13 (Patau syndrome), and sex chromosome aneuploidies like monosomy X (Turner syndrome) (Carlson and Vora, 2017). Therefore, successful management of pregnancy demands efficient and timely prenatal diagnosis to determine the outcome of pregnancy. Timely detection of neural tube defects is already providing early prenatal treatment resulting in better neonatal outcomes (Adzick et al., 2011).
ccf-fetal-DNA is clinically used for the detection of fetal sex and multiple anomalies based on paternally inherited mutations (Bianchi, 1998). Recent studies have discovered many fetal epigenetic biomarkers for ccf-fetal-NA-based liquid biopsies in clinical samples that have demonstrated high clinical potential in disease diagnosis, prognosis, and pregnancy management. These epigenetic modifications are specific to the fetus and help to distinguish fetal nucleic acids from maternal nucleic acids (Jones and Takai, 2001). Clinical testing of recently developed fetal epigenetic markers can help in the proper management of personalized care. The first reported use of fetal-derived epigenetic marker in maternal body fluids had come from Poon et al. (2002) who utilized an imprinted H19/Igf2 locus based on parent-of-origin-specific methylation, and the maternal and the paternal copies of the gene were distinguished in maternal blood (Poon et al., 2002). Based on the placental origin of ccf-fetal-DNA having placenta-specific DNA methylation pattern, the genomic regions that show differential methylation between the placenta and the maternal blood cells can act as a marker for fetal DNA in maternal blood. The promoter region of maspin (SERPINB5) is the first such reported universal fetal DNA marker, with detectable hypomethylation, in the background of hypermethylated maternal sequences. The fetal origin of these hypomethylated maspin has been confirmed by the clearance of these sequences within 24 h of delivery (Chim et al., 2005). The clinical use of hypomethylated maspin is limited by the required bisulfite treatment of ccf-fetal-DNA, as this treatment can degrade around 95% of the DNA (Grunau et al., 2001), thus decreasing the amount of already low levels of fetal DNA in maternal blood. Such limitation was overcome by the detection of fetal-derived hypermethylated RASSF1A in maternal blood for prenatal diagnosis (Chan et al., 2006; Hyland et al., 2009; Tounta et al., 2011b). The maternal hypomethylated RASSF1A ccfDNA can be removed by treatment with methylation-sensitive restriction enzyme digestion, leaving behind fetal hypermethylated RASSF1A ccf-fetal-DNA (Chan et al., 2006). Various other fetal-derived differentially methylated sequences have also shown a similar potential to act as fetal DNA epigenetic markers in maternal blood (Table 2).
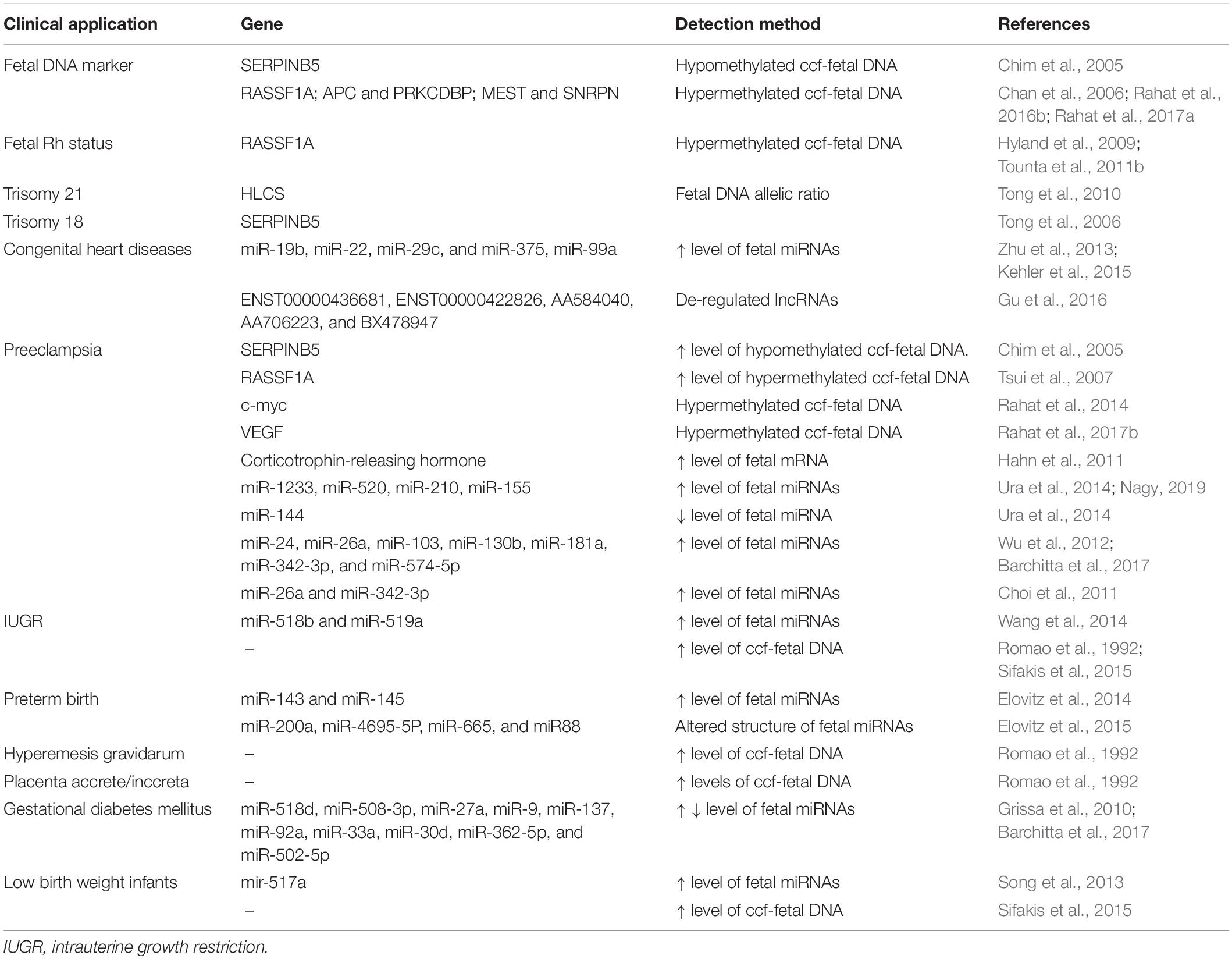
Table 2. Clinical application of ccf-fetal nucleic acids in prenatal diagnosis and pregnancy-related diseases.
ccf-fetal-DNA methylation markers have the potential of being used as both quantitative as well as qualitative markers in prenatal diagnosis. As qualitative markers, these are used to estimate the false positives during the determination of fetal gender, Rh status, and paternally inherited polymorphisms (Chan et al., 2006), while as quantitative markers, these can estimate the levels of ccf-fetal-DNA in maternal plasma. Such an application of ccf-fetal-DNA finds its use in the detection of chromosomal aneuploidies (Lun et al., 2008). Based on the location of the maspin gene on chromosome 18, hypomethylated fetal maspin has been used to calculate the allelic ratio to diagnose trisomy 18 with 100% sensitivity (Tong et al., 2006). Fetal trisomy 21 was detected by analyzing chromosomal dosage via targeting of fetal hypermethylated HLCS sequences in the combination of microfluidics digital PCR. RASSF1A on chromosome 3 and ZFY on the Y chromosome were used as references (Tong et al., 2010). Fragmentation pattern of ccf-fetal-DNA in maternal plasma has been successfully used for enrichment method in size separation manner on agarose gel electrophoresis (Ramezanzadeh et al., 2017).
Various next-generation sequencing and high-throughput techniques have catalyzed the identification of newer and novel fetal epigenetic markers further advancing non-invasive prenatal diagnosis. The microarray-based approach has identified many fetal epigenetic markers with differential methylation between chorionic villus samples and maternal blood, on chromosomes 21, 13, and 18 for aneuploidy detection (Chu et al., 2009). Combining high-resolution tiling oligonucleotide array with methylated DNA immunoprecipitation (MeDiP) has helped in a genome-wide screen for detecting the differential methylated sites between placental tissue and maternal blood cells. It has detected various new fetal epigenetic markers on chromosomes 21, 13, and 18 (Papageorgiou et al., 2009). Whole-genome bisulfite sequencing has further identified many clinically useful novel fetal-specific methylated CpG sites (Lun et al., 2013). Latest techniques like high-resolution methylation-specific bead chip microarray (Hatt et al., 2015) and GeneChip Human Promoter 1.0R Array (Wang et al., 2017) identified many differentially methylated CpG sites between maternal blood cells and chorionic villi, which can help in better and efficient prenatal diagnosis and the expansion of its application in other disorders.
More recently, non-coding RNAs like miRNAs, lncRNAs, and circRNAs are in the focus of the clinical research for prenatal diagnosis. Several placenta-specific miRNAs are differentially expressed within the placenta and are also secreted during pregnancy from the trophoblast layer of the placenta (Table 2). These are located in clusters on chromosomes 14 and 19 (C14MC, C19MC, and miR-371-3) (Nagy, 2019). Highly stable placental miRNAs were detected in maternal plasma. These can help in tracking gene regulation in the placenta (Chim et al., 2008). ccf-fetal miRNAs in the maternal blood act as expression-based novel epigenetic markers in prenatal diagnosis. miRNA microarray-based screen has detected many differentially expressed ccf-fetal miRNAs in maternal serum to diagnose congenital heart defects, which can be especially helpful in personalized care (Gu et al., 2019). Circular RNA and lncRNA are used in prenatal diagnosis for congenital heart diseases (Nagy, 2019). Early diagnosis of congenital heart diseases is beneficial to reduce morbidity and mortality. Sequencing by Oligonucleotide Ligation and Detection (SOLiD) has been used to identify congenital heart disease-related miRNAs in maternal blood (Zhu et al., 2013; Kehler et al., 2015). Gu et al. (2016) has reported many lncRNAs related to congenital heart diseases. The clinical use of these lncRNAs is extensively studied (Gu et al., 2016).
The use of artificial intelligence platforms and machine learning in combination to analyze genome-wide DNA methylation data has helped to identify epigenomic predictors for cerebral palsy in newborns with better sensitivity and specificity. These epigenetic predictors provided more mechanistic information about the pathogenesis of cerebral palsy (Bahado-Singh et al., 2019).
Certain pregnancy-related complications are associated with poor placental growth and development, which is usually also accompanied by aggravated trophoblastic apoptosis, resulting in the release of increased amounts of ccf-fetal-NAs into maternal blood. ccf-fetal-NAs are especially important for precision medicine being useful for clinical diagnosis and management of these complications, as these precede the actual clinical symptoms of the disease (Sifakis et al., 2015). The quantification of ccf-fetal-DNA levels in maternal blood might serve as an indicator of some developing abnormality; however, the absolute concentration of ccf-fetal-DNA varies with maternal weight and ethnicity and fluctuates throughout pregnancy (Tounta et al., 2011a), which warrants the need for a disease-specific qualitative marker. Aberrations in the levels of epigenetic marks present in these fetal DNA fragments serve as a valuable alternative for the diagnosis and management of pregnancy-related complications. The ccf-fetal-DNA present in maternal blood has been explored to predict such placental abnormality-linked pregnancies, like intrauterine growth restriction (IUGR); preeclampsia; hemolysis, elevated liver enzyme levels, and low platelet levels (HELLP) syndrome; preterm labor; hyperemesis gravidarum (severe morning sickness); placenta accrete; and placenta inccreta (Romao et al., 1992).
IUGR, defined by less than fifth percentile fetal weight, may or may not be associated with preeclampsia. Early detection of preeclampsia is highly beneficial for the proper management of preeclampsia, which is highly important for both the developing fetus and the mother (Bauer et al., 2006). The development of preeclampsia is also depicted by the increased level of unmethylated fetal SERPINB5 (Chim et al., 2005) in maternal blood. Similarly, hypermethylated fetal RASSF1A (Tsui et al., 2007) sequences are also elevated in preeclamptic blood. Hypermethylated c-myc and VEGF observed specifically in ccf-fetal-DNA in preeclampsia patients are the epigenetic markers, which can diagnose preeclampsia without the requirement of quantitative estimation of ccf-fetal-DNA levels. These sequences can be used both for the diagnosis as well as prognosis of preeclampsia (Rahat et al., 2014, 2017b). Such ccf-fetal-DNA-based epigenetic markers can be beneficial for early prediction and the personalized management of the disease. Fetal DNA epigenetic markers are likely to show potential as diagnostic markers in other complicated pregnancies accompanied by quantitative aberrations of ccf-fetal-DNA (Tsui et al., 2010).
Several fetal-derived mRNAs and miRNAs also as serve as diagnostic and prognostic markers for preeclampsia, preterm births, IUGR, spontaneous abortions, and low birth weight infants. A list of clinical applications of ccf-fetal nucleic acids in prenatal diagnosis and pregnancy-related diseases are given in Table 2. In addition, miRNAs involved in impaired trophoblast migration and invasion (miR-195, miR-276C, miR-278a-5p, and miR-210), impaired angiogenesis (miR-210, miR-21, and miR-22), and dysregulation of the maternal immune system are associated with preeclampsia (Skalis et al., 2019). Aberrant expression of circular miRNAs reported in gestational diabetes mellitus can serve as potential biomarkers for early diagnosis (Barchitta et al., 2017). Microarray analysis has also identified many miRNAs related to gestational diabetes mellitus (Grissa et al., 2010).
The use of microarray and next-generation sequencing can help to identify more ccf-fetal-RNA markers (Ferrari et al., 2008). Extensive research is required on different non-coding RNAs to be utilized in clinical settings for early diagnosis of pregnancy-related disorders.
The major obstacles in the field of ccf-fetal-NAs for diagnosis of prenatal and pregnancy-related complications are the requirements for proper standardized protocols for sample processing, detection methods, data analysis, and appropriate quality controls. Low concentration and fragmented pattern of ccf-fetal-NAs further demand the development of novel technologies for the proper utilization of ccf-fetal-NAs for diagnostics. The combined use of next-generation sequencing and bio-informative analysis could facilitate large-scale comprehensible screening and identification of promising next-generation non-invasive epigenetic biomarker in ccf-fetal-NAs. The screening for novel and disease-specific epigenetic markers on ccf-fetal-NAs in maternal blood will not only help in early diagnosis but also in providing proper personalized care. The ccf-fetal-NA-based diagnostic techniques provide new highly sensitive and specific avenues in clinical settings. These have already replaced invasive diagnostic sampling reducing pregnancy risks. ccf-fetal DNA is already being used in clinical settings, while the use of lncRNAs, circular RNAs, and miRNAs are in the research phase and could soon be used for clinical diagnosis of many fetal- and pregnancy-related disorders.
Intensive research is required in this area based on large populations to develop new clinical applications of fetal epigenetic marks in maternal blood. Additional mechanistic studies are required to identify the epigenetic changes behind the fetal–maternal complications, which can provide more insight into the possible epigenetic marks for non-invasive diagnosis.
In autoimmune diseases, there are abnormal immune responses to healthy body tissues. In the United States, about 8% of the population (24 million) are affected by autoimmune diseases, where women are more commonly affected as compared to men (Fairweather and Rose, 2004). Nearly 80 different types of autoimmune diseases are known (Hayter and Cook, 2012). The appearance of disease symptoms in adulthood makes the diagnosis of autoimmune immune diseases difficult. Celiac disease, Graves’ disease rheumatoid arthritis, systemic lupus erythematosus, diabetes mellitus type 1, inflammatory bowel disease, and multiple sclerosis are some well-known autoimmune diseases (Hohlfeld et al., 2016).
Witebsky’s et al. (1957) postulates were formulated for the first time for the diagnosis of autoimmune diseases. Accumulating evidence has shown that there is a significant role of epigenetic modifications in the development and progression of autoimmune diseases. With the benefits of ease of detection and the ability to analyze disease activity, specific epigenetic modifications can be proposed as novel biomarkers in autoimmune diseases.
In autoimmune diseases like other several pathological conditions, the presence of ccfDNA, has been observed, thus developing the interest of using them as a potential biomarker. Many pieces of evidence have shown that there is the presence of abnormal DNA demethylation in peripheral blood mononuclear cells (PBMCs) and CD4+ T cells of lupus patients (Javierre et al., 2010; Jeffries et al., 2011; Hewagama et al., 2013). Hypomethylation status of two sites, CpG site1 (Chr1: 79,085,222) and CpG site 2 (Chr1: 79,085,250; cg06872964), within the promoter region of IFI44L (IFN-induced protein 44-like) were identified as biomarkers for the diagnosis of SLE and further validated in the Chinese population consisting of 1,144 lupus patients, 1,350 healthy subjects, 429 RA, patients and 199 patients of primary Sjögren’s syndrome (pSS), as well as in a European cohort (Zhao et al., 2016). DNA methylation levels thus can not only distinguish active patients from inactive ones but importantly also indicate the activity of autoimmune diseases.
DNA methylation might be a good parameter, different from genetic and protein biomarkers, to serve as a predictive biomarker. Reduced DNA methylation at the IL-6 and ERa promoters in PBMCs in RA patients is associated with overexpressed IL-6 and hyperactive ERa signaling (Nile et al., 2008; Liao et al., 2012; Liu et al., 2014). Aberrant epigenetic modifications also have been evidenced to be associated with systemic sclerosis (SSc) disease. In this context, abnormal global and gene-specific DNA demethylation (e.g., at CD11a, CD70, and CD40L) and several hypermethylated genes (PRF1, CD11a, FoxP3, CD70, and CDKN2A) in whole blood have been observed from South Africans with SSc (Matatiele et al., 2015). Besides, the Th17-related genes, like RORC1 and RORC2, are hypomethylated from SSc patients in PBMCs and further correlated with inflammatory parameters (Almanzar et al., 2016). However, these aforementioned alterations are not validated as predicted biomarkers as yet, so extensive work is required in this direction in the future. Overall, these studies suggest that changes in circulating DNA methylation levels, observed in autoimmune diseases (Rainer et al., 2003), can act as an important tool to monitor the response of the treatment and predict progression of the disease and patient’s stratification according to different stages in the disease.
Like cell-free DNA, a few studies have also reported the role of cell-free RNAs in autoimmune diseases. Recent studies have revealed that there are ∼600 circulatory RNAs differentially expressed in the PBMCs in patients suffering from rheumatoid arthritis (Zheng et al., 2017). Similarly, over 200 circulatory RNAs in the plasma of SLE patients and 400 in the PBMCs of relapsing–remitting multiple sclerosis patients (RR-MS) were found in comparison to healthy controls (Cardamone et al., 2017). Such an abundance of different circulatory RNAs can help improve the efficiency of clinical diagnosis by their combined detection with other transitional markers.
Apart from these two types of ccfNAs, other forms like miRNA, long non-coding RNA, and mtDNA also have tremendous potential in future screening, diagnosis, and prognosis of autoimmune diseases. Many studies have identified miRNAs that are abnormally expressed in lupus. However, target genes have been identified only for a few of them. In a recent study, circulating miRNA profile was identified in patients with autoimmune disease as well as in Treg-depleted mice model. Results of this study from quantitative reverse transcription PCR (qRT-PCR) quantification and analysis of receiver operating characteristic (ROC) curve were able to determine a total of six miRNAs (miR-551b, miR-34cmiR-448, miR-9, miR-148, and miR-124) in the mouse models with T-reg depletion and three miRNA (miR-448, miR-124, and miR-551b) in patients with RA, SLE, Sjogren’s syndrome (SS), and ulcerative colitis (UC), leading to a conclusion that they could serve as valuable specific biomarkers in these diseases.
As the optimal source of biomarkers, many other circulating miRNAs also have been identified to be correlated with lupus. Among them, miR-146a and miR-155 are the first miRNAs that have been described as decreased in lupus serum (Wang et al., 2010). In subsequent studies, the serum levels of miR-200a, miR-200b, miR-200c, miR-429, miR-205, miR-192, miR-126, miR-16, miR-451, miR-223, miR-21, and miR-125a-3p (Wang et al., 2011, 2012) were found to be abnormally expressed in SLE and correlated with disease activity.
Another inspiring observation is that miR-126 has been reported to regulate DNA methylation in lupus T cells by targeting DNMT1 (Zhao et al., 2011), supporting the idea that lupus T cells are switched on by DNA hypomethylation via miRNAs (Ceribelli et al., 2011). Future studies in this direction can establish that not only circulatory DNA or RNA but also circulating miRNAs can also represent potential universal epigenetic biomarkers for autoimmune diseases (Jin et al., 2013).
At present, the treatment for autoimmune diseases is primarily based on immunosuppressive as well as anti-inflammatory agents, which mostly include humanized monoclonal antibodies, engineered biologics, and fusion proteins. Such treatment options are particularly generated against some signaling pathways or are selective for a certain subset of cells in the immune systems. The effectiveness of such types of treatments is for short duration only and not even antigen-specific in some cases. Moreover, chronic administration of these agents leads to the common side effects of increased incidence of infections and general immunosuppression (Tavakolpour, 2017). By utilizing the disease-specific epigenetic marks on the ccfNAs, for non-invasive detection, monitoring, and screening of autoimmune disorders, it will become feasible to offer personalized medicine to manage the autoimmune diseases in the future. A list of ccfNAs in autoimmune diseases is presented in Table 3.
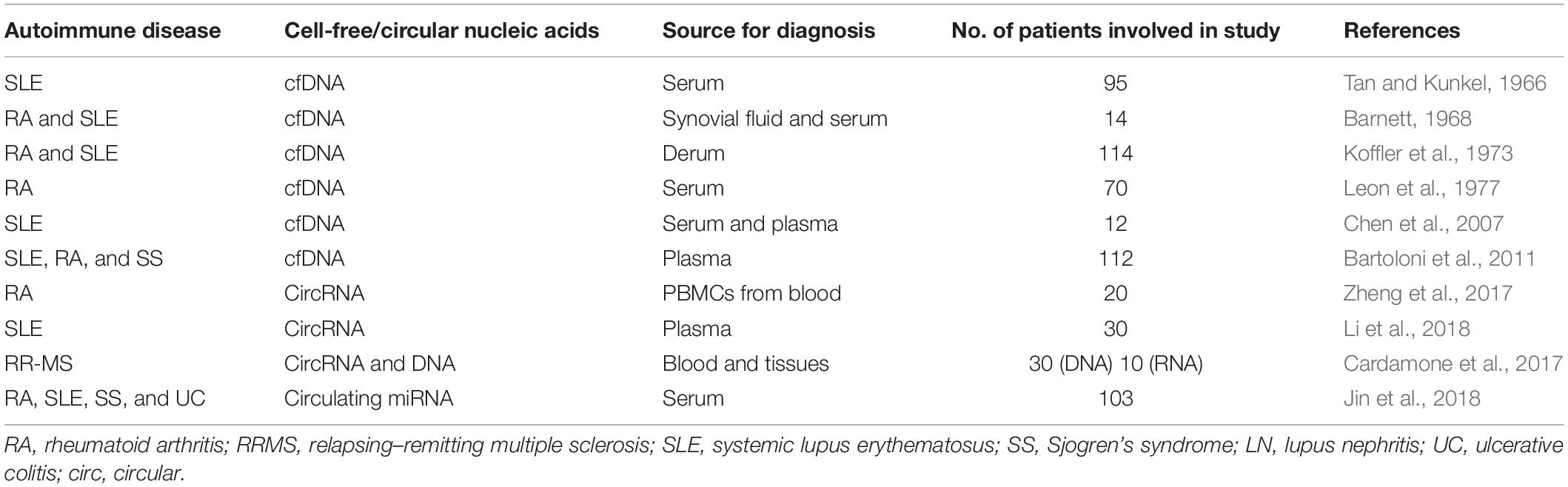
Table 3. Cell-free nucleic acids in autoimmune diseases.
Neurological disorders include diseases associated with central as well as the peripheral nervous system. These disorders include Alzheimer’s disease (AD), epilepsy, other dementias, Parkinson’s disease (PD), and traumatic disorders of the nervous system (Amor et al., 2014).
Symptoms associated with chronic neurodegenerative diseases occur late after the beginning of the pathology due to the compensatory potential of the brain that has been demonstrated in various studies. Any treatment is difficult in the later stages of neurodegenerative diseases due to the massive death of neuronal cells (Sperling et al., 2011; Pillai and Cummings, 2013). The recent ability to detect neurological biomarkers in the blood is due to technological advances in detection. The advances related to the use of ccfNA in neurological disorders are as follows.
Cell-free DNA acts as a marker for traumatic brain injury (TBI) and neurodegenerative diseases. The blood–brain barrier gets disrupted and leaky after TBI and neurodegenerative diseases (Carvey et al., 2009), which makes ccfNAs as potential markers for disease as well as injury (Lehmann-Werman et al., 2016). This includes changes in levels of ccfNA overall and also ccfNA markers specifically associated with the brain (Zlokovic, 2011).
Specific cell-free miRNA levels (mir-34c) involved in apoptosis and survival caspase cascade in plasma of Alzheimer’s patients are known for the prediction of disease (Bhatnagar et al., 2014). An analysis presented by Tan et al. (2014) showed that miRNAs are involved in the processes in pathogenesis associated with AD: amyloid-β accumulation, toxicity associated with tau proteins, inflammation, as well as neuronal cell death (Tan et al., 2014). In AD patients, activation, as well as inhibition of expression of miR-9, was found, which is enriched in the brain (Jin et al., 2013). miR-133b was downregulated in the midbrain of PD patients (Kim et al., 2007) as well as in mouse models of PD (Harraz et al., 2011; Filatova et al., 2012). However, studies are required to ascertain the presence of these miRNAs in circulation in these patients and their evaluation for potential biomarkers.
DNA methylation has a significant involvement in several neurodegenerative diseases (Al-Mahdawi et al., 2016). Increases and decrease in both 5mC and 5hmC at global levels have been identified in different diseases including AD (Al-Mahdawi et al., 2014). 5mC and 5hmC are identified as potential epigenetic markers in various neurodegenerative diseases both at global and locus-specific levels (Xiao et al., 2012; Sandi et al., 2013). However, further investigations are required for using 5mC and 5hmC as epigenetic biomarkers in cell-free circulating nucleic acids.
Previous studies have identified increased brain-derived ccfDNA in the serum of patients after traumatic brain injury (Lehmann-Werman et al., 2016). Rhomboid 5 Homolog 2 (RHBDF2) was found to be differentially methylated in the central nervous system (CNS) in Alzheimer’s disease (De Jager et al., 2014). In addition, a differentially methylated region located in the promoter–enhancer region of the RHBDF2 gene was identified in amyotrophic lateral sclerosis (ALS) patients in ccfDNA in the plasma (Mendioroz et al., 2018). Thus, liquid biopsy may be applied to living patients as a source of potential epigenetic biomarkers for neurodegenerative disorders.
Levels of miR-146a and miR-155, the proinflammatory miRNAs, were found to be high in cerebrospinal fluid (CSF) of AD patients along with miR-9 and miR-125b that are enriched in neurons (Alexandrov et al., 2012). Differential expression of miR-15b and miR-21 was found in CSF from patients with primary CNS lymphoma, gliomas, and brain metastasis (Baraniskin et al., 2012). miR-451 was detected in CSF microparticles after brain injury (Pigati et al., 2010). A list of ccfNAs studied in neurological diseases is provided in Table 4.
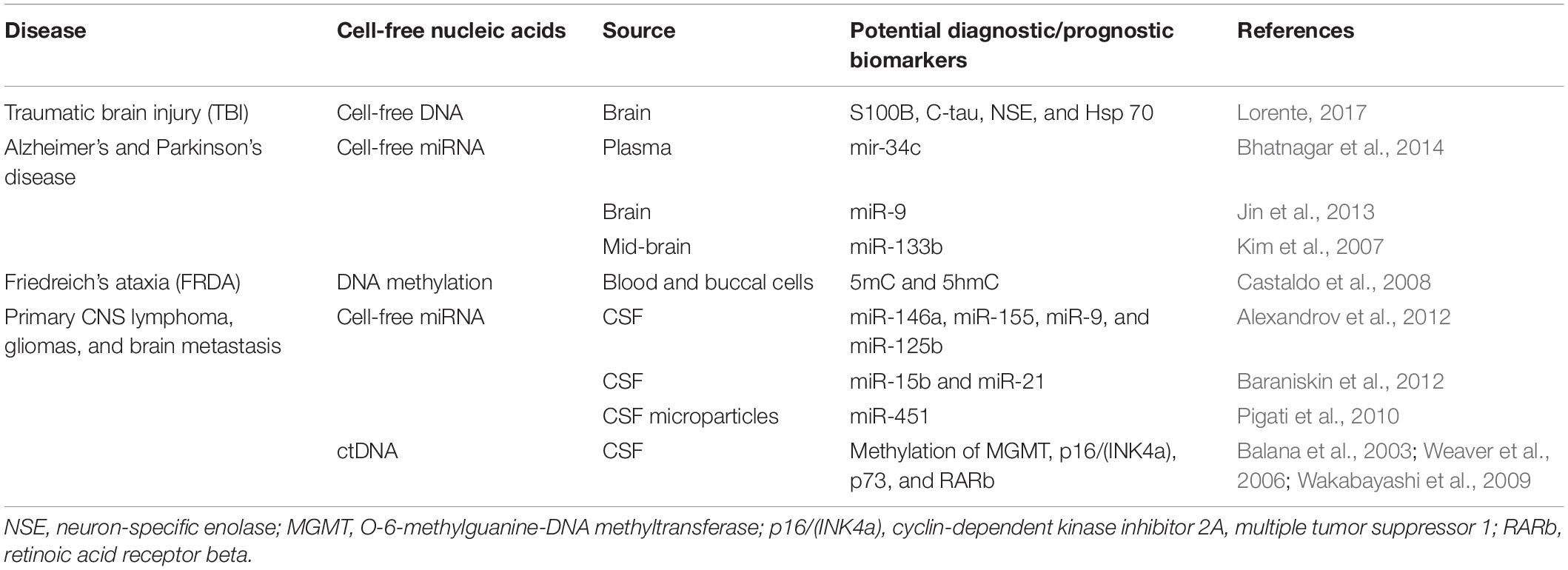
Table 4. Circulating cell-free nucleic acids (ccfNAs) in neurological diseases.
In mitochondrial disorders, mitochondria fail to function properly and are not able to generate enough energy required for the body. These diseases are the chronic ones due to genetic causes, often inherited from the previous generation (Khan et al., 2015). Mitochondrial diseases can affect multiple organs of the body (Al-Enezi et al., 2008) and, in many conditions, can lead to secondary mitochondrial dysfunction like Lou Gehrig’s disease, diabetes, muscular dystrophy, Alzheimer’s disease, and cancer (Niyazov et al., 2016).
Commonly known mitochondrial diseases are mitochondrial myopathy, Leigh syndrome, Leber’s hereditary optic neuropathy (LHON), myoclonic epilepsy with ragged red fibers (MERRFs), myoneurogenic gastrointestinal encephalopathy (MNGIE), mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), mitochondrial myopathy encephalomyopathy lactic acidosis stroke-like symptoms (MELAS), neuropathy ataxia retinitis pigmentosa and ptosis (NARP), and Friedreich’s ataxia (Khan et al., 2015).
The diagnosis of mitochondrial diseases is difficult because it affects multiple organs, and hence, patients exhibit a variety of symptoms. Moreover, there is no single diagnostic or laboratory test that can accurately confirm a mitochondrial disease (Parikh et al., 2015). Therefore, investigation of ccfDNA or mtDNA in the plasma of patients independently can be a better biomarker.
Unlike nuclear DNA, mtDNA molecules are arranged in clusters, called nucleoids, which are tethered to the mitochondrial membrane and are devoid of histones (Holt, 2010). Earlier observations had suggested that mitochondria lacked the machinery required for DNA methylation. However, several strands of evidence later, including lower frequency of CG dinucleotides by bioinformatic analysis and modulation of mtDNA methylation in response to oxidative stress (Rebelo et al., 2009), suggested the other way. Additionally, DNA methylation of nuclear mitochondrial genes may play an important role in the understanding of mitochondrial disorders.
There has been some evidence that some types of histones do localize to the mitochondrial membrane, where mtDNA is tethered (Choi et al., 2011). In the nuclear DNA, histone modifications are important in transcriptional control that can be altered in diseases affecting nuclear-encoded mitochondrial proteins, of which Friedreich ataxia is an example. This disorder is caused by transcriptional silencing due to certain histone modifications of the FXN gene, which encodes a mitochondrial protein involved in the biosynthesis of iron–sulfur clusters (Rai et al., 2010). In one of the studies, total mtDNA in plasma was quantified and found to be high in Friedreich’s ataxia patients, which opened up its possible role as a blood-based biomarker (Swarup et al., 2011).
Serum microRNAs like miR-1275, miR-149, miR-1, miR-133a, miR-133b, miR-145, miR-206, miR-208a, miR-208b, miR499, and miR-206 are reported in some of the studies to diagnose the muscle mitochondrial dysfunction (Cacchiarelli et al., 2011; Endo et al., 2013; Hu et al., 2014). In addition, in some metabolic diseases, where the mitochondria are not functioning properly like brown adipogenesis (Zhang et al., 2015), non-alcoholic fatty liver disease (Leti et al., 2015), and diabetes (Raffort et al., 2015), distinctive patterns of microRNA have been observed. In another study, mutations in cybrid cells identified the role of microRNA-9/9∗ pattern in different mitochondrial disorders, e.g., mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) and myoclonic epilepsy with ragged-red fiber (MERRF) (Meseguer et al., 2015).
Hence, apart from the quantification of mtDNA in FRDA and serum microRNA profiles to assess mitochondrial myopathies, there is no available data on ccfNA epigenetic marker in mitochondrial diseases. Working on circulating mtDNA poses many challenges. mtDNA is highly polymorphic, which makes introducing targeted mutations into the mitochondria and generation of cellular or animal models of mitochondrial disorders quite challenging.
In addition, the gene panels required for the molecular diagnosis of mitochondrial disorders constitute an expensive approach. Therefore, the determination of epigenetic marks on ccfNA in plasma can have a diagnostic as well as prognostic potential in mitochondrial diseases. Thus, once the technical hurdles to study circulating mtDNA are taken care of by technology advancement, there is a strong motivation to explore the role of epigenetic mechanisms in mtDNA disease, as epigenetic factors may serve to explain the observed phenotypic heterogeneity, variable penetrance, and pronounced environmental triggers in this group of disorders.
There is a high impact of technologies such as high-performance computing as well as biological databases, like artificial intelligence (AI), machine learning (ML), and neural network, in the field of health care. Epigenetic data have traits like chemical and biological stability over time that make it open to ML (Xiong et al., 2015). Large-scale data-rich repositories such as The Cancer Genome Atlas (TCGA), BLUEPRINT, and the ENCODE association provide large amounts of samples to employ comprehensive, high-throughput statistical analysis of differentially methylated regions with biological relevance (Yuan et al., 2011; Munsell et al., 2015). Artificial intelligence and machine learning tools have especially found scope in cancer precision medicine by offering cancer patients personalized care. These methods help to decipher weak signals in the blood circulation at the early stages of cancer and provide a real-time assessment of cancer treatment. Nearly all datasets consist of DNA methylation profiles derived from peripheral blood, meaning that patients will only be required to provide a small blood sample. These can detect minute quantities of tumor DNA in blood and analyze their epigenetic marks for cancer monitoring.
Various programs have been generated to provide useful information for proper diagnosis. The Graphite is a bioconductor package to convert pathway topology to gene network (Sales et al., 2012). The micrographite package, for instance, provides a process to amalgamate mRNA and microRNA data via their association to canonical pathways (Calura et al., 2014). This approach has been beneficial in recognizing key microRNAs in primary myelofibrosis (Calura et al., 2016c), myeloma (Calura et al., 2016a), and ovarian cancer (Calura et al., 2016b). Another program, Mergeomics (multidimensional data integration to identify pathogenic perturbations to biological systems), combines data from epigenetic, genomic, and transcriptional association studies through a process of functional enrichment, which is used as the base for network construction; nevertheless, this tool has not been used in the context of cancer (Shu et al., 2016). Based on the multiomics data, Netboost is a network reconstruction method having statistical dependency and employs a commutable approach to lessen dimensionality. This system has been utilized for the categorization and survival study of acute myeloid leukemia data (Schlosser et al., 2020). Pair-wise relationships among various omics layers are identified by AMARETTO, which decides on cancer driver genes by taking into consideration frequently altered genes at the genome or epigenome level with functional consequences (Champion et al., 2018). Another tool MAGIA is used for the rebuilding of microRNA and transcription factor regulatory routes and has been employed for the scrutiny of expression and regulatory mechanisms in the NCI60 cell panel. As personalized genomic medicine pierces the age of “Big Data,” these would lead to uncovering of novel biomarkers on cancer indicators in the blood linked to particular disease states offered by machine learning algorithms. ML algorithms will assist with assessing the effects of various biomarkers concurrently and reveal top order interactions between biomarkers that would not be feasible to devise manually. A cancer genomics company, GRAIL, has launched a large-scale study, the Circulating Cell-Free Genome Atlas (CCGA), which uses machine learning to create a huge, representative library of cancer mutations and healthy mutations using data from ccfDNA and white blood cell genomes, to train their cancer screening algorithms (Cohn et al., 2019). With the availability of more data from clinical trials, this system can fine tune its algorithm to improve its diagnostic acumen.
Furthermore, artificial neural networks, which mimic the neurons of the brain, are functioning to interpret the data and provide the basis of machine learning. Disorders of neurodevelopment start early in childhood and have an impact on a diversity of functional domains as well as executive and cognitive function, social and language function, also behavior control, and motor function (Uddin et al., 2016; Thapar et al., 2017). Various other diagnoses include autism spectrum disorder (ASD) (Jiang et al., 2013), intellectual disability (ID) (Maulik et al., 2011), attention-deficit hyperactivity disorder (ADHD) (Lionel et al., 2011), and movement disorders (Huisman-Van Dijk et al., 2016). Consequently, the initiation of technologies dependent on transcriptome sequencing led to the foundation of the Allen developmental human brain atlas, profiling of the non-coding elements in the human genome by ENCODE database and the Human Cell Atlas (Kang et al., 2011; Regev et al., 2017). Recently, AI approaches have revealed by far reasonable success in neurodegenerative diseases (NDDs) by improving genetic diagnostics. The implementation of the Human Splicing Code is one of the first ML algorithms that demonstrate persuasive evidence of correctly categorizing disease-causing variants as well as those that are intronic. This process applies a Bayesian ML algorithm and has been illustrated in spinal muscular atrophy and pathogenic missense variants in ASD (Zhou and Troyanskaya, 2015). AI approaches are vital to explain the hidden arrangement in phenotype and genetic heterogeneity. NDDs are characterized by both phenotypic and genetic heterogeneity. For instance, cognitive function is impacted by 15q13.3 microdeletion syndrome and is found to be linked with heterogenous phenotypes that include speech delay (16%), epilepsy/seizure (57%), and ASD (11%) (Lowther et al., 2015). Although, biology enlightens this nosological evolution; however, more stress needs to be given on using AI approaches on major-scale datasets to authenticate or confront existing categorization paradigms.
The utility of epigenetic alterations in ccfNA in various diseases as diagnostic and prognostic markers as well as therapeutic targets has been summarized in Figure 1. Liquid biopsy is fast replacing the invasive methods for the diagnosis and prognosis of various diseases. This method offers the possibility to separate and identify ccfNAs for their utility for screening, diagnostic, prognosis, or for selecting therapeutic options. The different types of ccfNAs being evaluated are ccfDNA, ccfRNA, ccfmtDNA, ccfmiRNA, ccflncRNA, etc. With the development of newer technologies for isolation of small amounts of ccfNAs and detection of the specific signatures on these, ccfDNA are already in clinical practice for a few diseases. Furthermore, recent advances in the field have shifted the focus from determining the quantity, SNPs, mutations of the ccfNAs to analyzing the epigenetic signatures like methylated sequences and nucleosome positioning, which are specific to the particular clinical condition. The epigenetic markers on the ccfNAs are widely being explored to further advance the field of personalized medicine. However, the genetic or epigenetic markers related to ccfNAs have paved the way in clinical practice mostly in cancer and prenatal screening only. For various other diseases like neurological, autoimmune, and mitochondrial diseases, there are limited data, most of which are limited to research findings only. There is a need to have a comprehensive data analysis of the epigenetic markers in ccfNAs in different physiological and pathological conditions and further testing of the selected markers in large population-based studies and disease cohorts. The field of epigenetic markers in ccfNAs holds tremendous potential in the field of precision medicine.
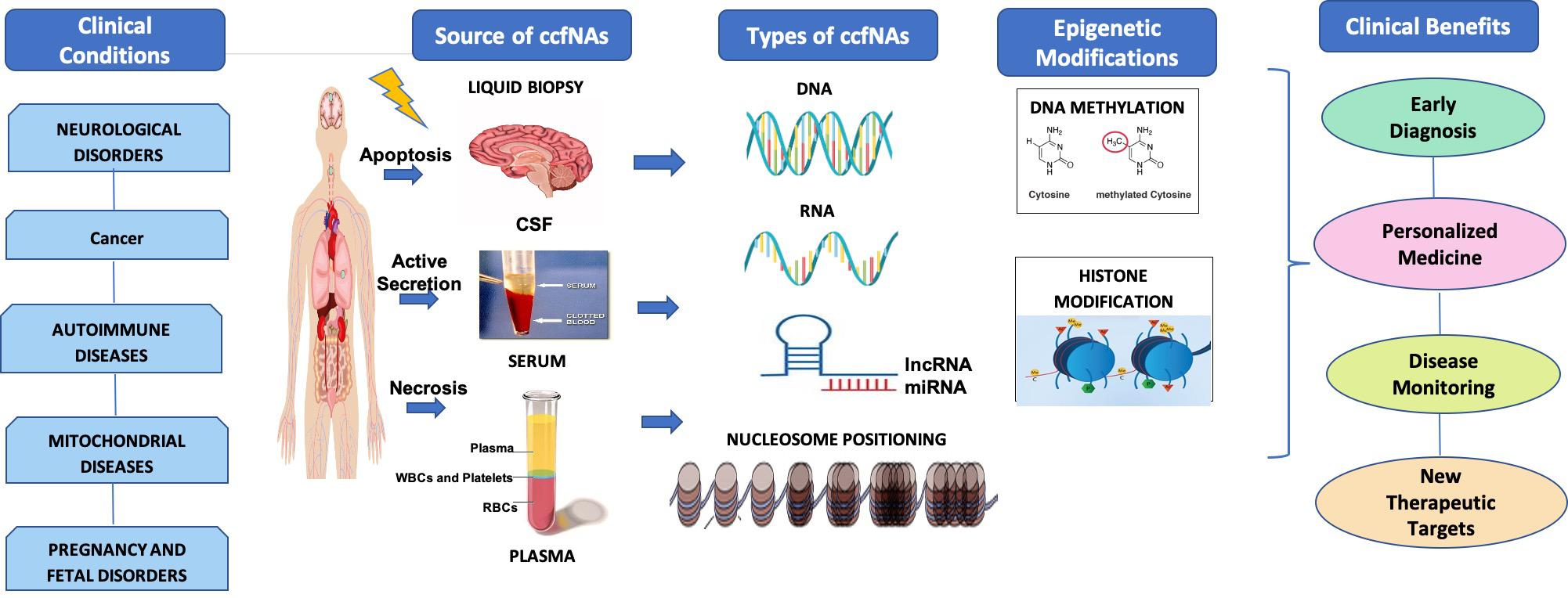
Figure 1. Under various clinical conditions such as neurological disorder, cancer, autoimmune diseases, mitochondrial diseases, and pregnancy and fetal disorders, circulating cell-free nucleic acids (ccfNAs) are released into body fluids like serum, plasma, and cerebrospinal fluids by apoptosis and necrosis. ccfNAs are of various types. The important ones are DNA, RNA, long non-coding RNAs (lncRNAs), and microRNA (miRNA) and have been observed to have disease-specific epigenetic modifications. These act as diagnostic and prognostic markers as well as therapeutic targets providing significant clinical benefits.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
5hmC, 5-hydroxy methyl cytosine; ccfDNAs, circulating cell-free deoxyribonucleic acids; ccf-fetal-NAs, circulating cell-free fetal nucleic acids; ccfmiRNAs, circulating cell-free miRNAs; ccfNAs, circulating cell-free nucleic acids; ccfRNAs, circulating cell-free ribonucleic acids; mtDNA, mitochondrial DNA.
Abern, M. R., Owusu, R., and Inman, B. A. (2014). Clinical performance and utility of a DNA methylation urine test for bladder cancer. Urol. Oncol. 32, 51.e21–51.e26. doi: 10.1016/j.urolonc.2013.08.003
Adli, M., and Bernstein, B. E. (2011). Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat. Protoc. 6, 1656–1668. doi: 10.1038/nprot.2011.402
Adzick, N. S., Thom, E. A., Spong, C. Y., Brock, J. W. III, Burrows, P. K., Johnson, M. P., et al. (2011). A randomized trial of prenatal versus postnatal repair of myelomeningocele. N. Engl. J. Med. 364, 993–1004.
Ahlquist, D. A. (2018). Universal cancer screening: revolutionary, rational, and realizable. NPJ Precis. Oncol. 2:23.
Alexandrov, P. N., Dua, P., Hill, J. M., Bhattacharjee, S., Zhao, Y., and Lukiw, W. J. (2012). microRNA (miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int. J. Biochem. Mol. Biol. 3, 365–373.
Alhasan, A. H., Scott, A. W., Wu, J. J., Feng, G., Meeks, J. J., Thaxton, C. S., et al. (2016). Circulating microRNA signature for the diagnosis of very high-risk prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 113, 10655–10660. doi: 10.1073/pnas.1611596113
Al-Enezi, M., Al-Saleh, H., and Nasser, M. (2008). Mitochondrial disorders with significant ophthalmic manifestations. Middle East Afr. J. Ophthalmol. 15, 81–86. doi: 10.4103/0974-9233.51998
Al-Mahdawi, S., Virmouni, S. A., and Mark, A. P. (2016). “DNA Methylation in Neurodegenerative Diseases,” in Epigenetic Biomarkers and Diagnostics, ed. J. L. García-Giménez (Cambridge, MA: Academic Press).
Al-Mahdawi, S., Virmouni, S. A., and Pook, M. A. (2014). The emerging role of 5-hydroxymethylcytosine in neurodegenerative diseases. Front. Neurosci. 8:397. doi: 10.3389/fnins.2014.00397
Almanzar, G., Klein, M., Schmalzing, M., Hilligardt, D., El Hajj, N., Kneitz, H., et al. (2016). Disease Manifestation and Inflammatory Activity as Modulators of Th17/Treg Balance and RORC/FoxP3 Methylation in Systemic Sclerosis. Int. Arch. Allergy Immunol. 171, 141–154. doi: 10.1159/000450949
Amor, S., Peferoen, L. A., Vogel, D. Y., Breur, M., Van Der Valk, P., Baker, D., et al. (2014). Inflammation in neurodegenerative diseases–an update. Immunology 142, 151–166. doi: 10.1111/imm.12233
Antonia, S., Mule, J. J., and Weber, J. S. (2004). Current developments of immunotherapy in the clinic. Curr. Opin. Immunol. 16, 130–136. doi: 10.1016/j.coi.2004.01.012
Arko-Boham, B., Aryee, N. A., Blay, R. M., Owusu, E. D. A., Tagoe, E. A., Doris Shackie, E.-S., et al. (2019). Circulating cell-free DNA integrity as a diagnostic and prognostic marker for breast and prostate cancers. Cancer Genet. 235–236, 65–71. doi: 10.1016/j.cancergen.2019.04.062
Backes, C., Meese, E., and Keller, A. (2016). Specific miRNA disease biomarkers in blood, serum and plasma: challenges and prospects. Mol. Diagn. Ther. 20, 509–518. doi: 10.1007/s40291-016-0221-4
Bahado-Singh, R. O., Vishweswaraiah, S., Aydas, B., Mishra, N. K., Guda, C., and Radhakrishna, U. (2019). Deep Learning/Artificial Intelligence and Blood-Based DNA Epigenomic Prediction of Cerebral Palsy. Int. J. Mol. Sci. 20:2075. doi: 10.3390/ijms20092075
Balana, C., Ramirez, J. L., Taron, M., Roussos, Y., Ariza, A., Ballester, R., et al. (2003). O6-methyl-guanine-DNA methyltransferase methylation in serum and tumor DNA predicts response to 1,3-bis(2-chloroethyl)-1-nitrosourea but not to temozolamide plus cisplatin in glioblastoma multiforme. Clin. Cancer Res. 9, 1461–1468.
Baraniskin, A., Kuhnhenn, J., Schlegel, U., Maghnouj, A., Zollner, H., Schmiegel, W., et al. (2012). Identification of microRNAs in the cerebrospinal fluid as biomarker for the diagnosis of glioma. Neuro Oncol. 14, 29–33. doi: 10.1093/neuonc/nor169
Barchitta, M., Maugeri, A., Quattrocchi, A., Agrifoglio, O., and Agodi, A. (2017). The role of miRNAs as biomarkers for pregnancy outcomes: a comprehensive review. Int. J. Genomics 2017:8067972.
Barnett, E. V. (1968). Detection of nuclear antigens (DNA) in normal and pathologic human fluids by quantitative complement fixation. Arthritis Rheum. 11, 407–417. doi: 10.1002/art.1780110306
Bartoloni, E., Ludovini, V., Alunno, A., Pistola, L., Bistoni, O., Crino, L., et al. (2011). Increased levels of circulating DNA in patients with systemic autoimmune diseases: a possible marker of disease activity in Sjogren’s syndrome. Lupus 20, 928–935. doi: 10.1177/0961203311399606
Bauer, M., Hutterer, G., Eder, M., Majer, S., Leshane, E., Johnson, K. L., et al. (2006). A prospective analysis of cell-free fetal DNA concentration in maternal plasma as an indicator for adverse pregnancy outcome. Prenat. Diagn. 26, 831–836. doi: 10.1002/pd.1513
Beltran-Garcia, J., Osca-Verdegal, R., Mena-Molla, S., and Garcia-Gimenez, J. L. (2019). Epigenetic IVD tests for personalized precision medicine in cancer. Front. Genet. 10:621. doi: 10.3389/fgene.2019.00621
Berman, B. P., Weisenberger, D. J., Aman, J. F., Hinoue, T., Ramjan, Z., Liu, Y., et al. (2011). Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 44, 40–46. doi: 10.1038/ng.969
Bernt, K. M., Zhu, N., Sinha, A. U., Vempati, S., Faber, J., Krivtsov, A. V., et al. (2011). MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20, 66–78. doi: 10.1016/j.ccr.2011.06.010
Bhatnagar, S., Chertkow, H., Schipper, H. M., Yuan, Z., Shetty, V., Jenkins, S., et al. (2014). Increased microRNA-34c abundance in Alzheimer’s disease circulating blood plasma. Front. Mol. Neurosci. 7:2. doi: 10.3389/fnmol.2014.00002
Bianchi, D. W. (1998). Fetal DNA in maternal plasma: the plot thickens and the placental barrier thins. Am. J. Hum. Genet. 62, 763–764. doi: 10.1086/301809
Birch, L., English, C. A., O’donoghue, K., Barigye, O., Fisk, N. M., and Keer, J. T. (2005). Accurate and robust quantification of circulating fetal and total DNA in maternal plasma from 5 to 41 weeks of gestation. Clin. Chem. 51, 312–320. doi: 10.1373/clinchem.2004.042713
Bougel, S., Lhermitte, B., Gallagher, G., De Flaugergues, J. C., Janzer, R. C., and Benhattar, J. (2013). Methylation of the hTERT promoter: a novel cancer biomarker for leptomeningeal metastasis detection in cerebrospinal fluids. Clin. Cancer Res. 19, 2216–2223. doi: 10.1158/1078-0432.ccr-12-1246
Breitbach, S., Tug, S., and Simon, P. (2012). Circulating cell-free DNA: an up-coming molecular marker in exercise physiology. Sports Med. 42, 565–586. doi: 10.2165/11631380-000000000-00000
Cacchiarelli, D., Legnini, I., Martone, J., Cazzella, V., D’Amico, A., Bertini, E., et al. (2011). miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol. Med. 3, 258–265. doi: 10.1002/emmm.201100133
Calura, E., Bisognin, A., Manzoni, M., Todoerti, K., Taiana, E., Sales, G., et al. (2016a). Disentangling the microRNA regulatory milieu in multiple myeloma: integrative genomics analysis outlines mixed miRNA-TF circuits and pathway-derived networks modulated in t(4;14) patients. Oncotarget 7, 2367–2378. doi: 10.18632/oncotarget.6151
Calura, E., Martini, P., Sales, G., Beltrame, L., Chiorino, G., D’incalci, M., et al. (2014). Wiring miRNAs to pathways: a topological approach to integrate miRNA and mRNA expression profiles. Nucleic Acids Res. 42:e96. doi: 10.1093/nar/gku354
Calura, E., Paracchini, L., Fruscio, R., Difeo, A., Ravaggi, A., Peronne, J., et al. (2016b). A prognostic regulatory pathway in stage I epithelial ovarian cancer: new hints for the poor prognosis assessment. Ann. Oncol. 27, 1511–1519. doi: 10.1093/annonc/mdw210
Calura, E., Pizzini, S., Bisognin, A., Coppe, A., Sales, G., Gaffo, E., et al. (2016c). A data-driven network model of primary myelofibrosis: transcriptional and post-transcriptional alterations in CD34+ cells. Blood Cancer J. 6:e439. doi: 10.1038/bcj.2016.47
Cardamone, G., Paraboschi, E. M., Rimoldi, V., Duga, S., Solda, G., and Asselta, R. (2017). The characterization of GSDMB splicing and backsplicing profiles identifies novel isoforms and a circular RNA that are dysregulated in multiple sclerosis. Int. J. Mol. Sci. 18:576. doi: 10.3390/ijms18030576
Carlson, L. M., and Vora, N. L. (2017). Prenatal Diagnosis: Screening and Diagnostic Tools. Obstet. Gynecol. Clin. North Am. 44, 245–256.
Carvey, P. M., Hendey, B., and Monahan, A. J. (2009). The blood-brain barrier in neurodegenerative disease: a rhetorical perspective. J. Neurochem. 111, 291–314. doi: 10.1111/j.1471-4159.2009.06319.x
Castaldo, I., Pinelli, M., Monticelli, A., Acquaviva, F., Giacchetti, M., Filla, A., et al. (2008). DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J. Med. Genet. 45, 808–812. doi: 10.1136/jmg.2008.058594
Ceribelli, A., Yao, B., Dominguez-Gutierrez, P. R., and Chan, E. K. (2011). Lupus T cells switched on by DNA hypomethylation via microRNA? Arthritis Rheum. 63, 1177–1181. doi: 10.1002/art.30192
Champion, M., Brennan, K., Croonenborghs, T., Gentles, A. J., Pochet, N., and Gevaert, O. (2018). Module analysis captures pancancer genetically and epigenetically deregulated cancer driver genes for smoking and antiviral response. EBioMedicine 27, 156–166. doi: 10.1016/j.ebiom.2017.11.028
Chan, K. C., Ding, C., Gerovassili, A., Yeung, S. W., Chiu, R. W., Leung, T. N., et al. (2006). Hypermethylated RASSF1A in maternal plasma: a universal fetal DNA marker that improves the reliability of noninvasive prenatal diagnosis. Clin. Chem. 52, 2211–2218. doi: 10.1373/clinchem.2006.074997
Chen, J. A., Meister, S., Urbonaviciute, V., Rodel, F., Wilhelm, S., Kalden, J. R., et al. (2007). Sensitive detection of plasma/serum DNA in patients with systemic lupus erythematosus. Autoimmunity 40, 307–310. doi: 10.1080/08916930701356317
Chen, X., Ba, Y., Ma, L., Cai, X., Yin, Y., Wang, K., et al. (2008). Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 18, 997–1006.
Cheuk, I. W., Shin, V. Y., and Kwong, A. (2017). Detection of methylated circulating DNA as noninvasive biomarkers for breast cancer diagnosis. J. Breast Cancer 20, 12–19.
Chim, S. S., Shing, T. K., Hung, E. C., Leung, T. Y., Lau, T. K., Chiu, R. W., et al. (2008). Detection and characterization of placental microRNAs in maternal plasma. Clin. Chem. 54, 482–490. doi: 10.1373/clinchem.2007.097972
Chim, S. S., Tong, Y. K., Chiu, R. W., Lau, T. K., Leung, T. N., Chan, L. Y., et al. (2005). Detection of the placental epigenetic signature of the maspin gene in maternal plasma. Proc. Natl. Acad. Sci. U.S.A. 102, 14753–14758. doi: 10.1073/pnas.0503335102
Chimonidou, M., Tzitzira, A., Strati, A., Sotiropoulou, G., Sfikas, C., Malamos, N., et al. (2013). CST6 promoter methylation in circulating cell-free DNA of breast cancer patients. Clin. Biochem. 46, 235–240. doi: 10.1016/j.clinbiochem.2012.09.015
Choi, Y. S., Hoon Jeong, J., Min, H. K., Jung, H. J., Hwang, D., Lee, S. W., et al. (2011). Shot-gun proteomic analysis of mitochondrial D-loop DNA binding proteins: identification of mitochondrial histones. Mol. Biosyst. 7, 1523–1536.
Chu, T., Burke, B., Bunce, K., Surti, U., Allen Hogge, W., and Peters, D. G. (2009). A microarray-based approach for the identification of epigenetic biomarkers for the noninvasive diagnosis of fetal disease. Prenat. Diagn. 29, 1020–1030. doi: 10.1002/pd.2335
Cohn, A. L., Seiden, M., Kurtzman, K. N., Hubbell, E., Gross, S., Venn, O., et al. (2019). The circulating cell-free genome atlas (CCGA) study: follow-up (F/U) on non-cancer participants with cancer-like cell-free DNA signals. J. Clin. Oncol. 37, 5574–5574. doi: 10.1200/JCO.2019.37.15_suppl.5574
Combaret, V., Audoynaud, C., Iacono, I., Favrot, M. C., Schell, M., Bergeron, C., et al. (2002). Circulating MYCN DNA as a tumor-specific marker in neuroblastoma patients. Cancer Res. 62, 3646–3648.
De Jager, P. L., Srivastava, G., Lunnon, K., Burgess, J., Schalkwyk, L. C., Yu, L., et al. (2014). Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 17, 1156–1163. doi: 10.1038/nn.3786
De Mattos-Arruda, L., Cortes, J., Santarpia, L., Vivancos, A., Tabernero, J., Reis-Filho, J. S., et al. (2013). Circulating tumour cells and cell-free DNA as tools for managing breast cancer. Nat. Rev. Clin. Oncol. 10, 377–389. doi: 10.1038/nrclinonc.2013.80
Devonshire, A. S., Whale, A. S., Gutteridge, A., Jones, G., Cowen, S., Foy, C. A., et al. (2014). Towards standardisation of cell-free DNA measurement in plasma: controls for extraction efficiency, fragment size bias and quantification. Anal. Bioanal. Chem. 406, 6499–6512. doi: 10.1007/s00216-014-7835-3
Ellinger, J., Bastian, P. J., Jurgan, T., Biermann, K., Kahl, P., Heukamp, L. C., et al. (2008). CpG island hypermethylation at multiple gene sites in diagnosis and prognosis of prostate cancer. Urology 71, 161–167. doi: 10.1016/j.urology.2007.09.056
Elovitz, M. A., Anton, L., Bastek, J., and Brown, A. G. (2015). Can microRNA profiling in maternal blood identify women at risk for preterm birth? Am. J. Obstet. Gynecol. 212, 782.e1–782.e5. doi: 10.1016/j.ajog.2015.01.023
Elovitz, M. A., Brown, A. G., Anton, L., Gilstrop, M., Heiser, L., and Bastek, J. (2014). Distinct cervical microRNA profiles are present in women destined to have a preterm birth. Am. J. Obstet. Gynecol. 210, 221.e1–221.e11. doi: 10.1016/j.ajog.2013.12.043
Endo, K., Weng, H., Naito, Y., Sasaoka, T., Takahashi, A., Fukushima, Y., et al. (2013). Classification of various muscular tissues using miRNA profiling. Biomed. Res. 34, 289–299. doi: 10.2220/biomedres.34.289
Esquela-Kerscher, A., and Slack, F. J. (2006). Oncomirs - microRNAs with a role in cancer. Nat. Rev. Cancer 6, 259–269. doi: 10.1038/nrc1840
Fairweather, D., and Rose, N. R. (2004). Women and autoimmune diseases. Emerg. Infect. Dis. 10, 2005–2011.
Ferlay, J., Colombet, M., Soerjomataram, I., Mathers, C., Parkin, D. M., Pineros, M., et al. (2019). Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 144, 1941–1953. doi: 10.1002/ijc.31937
Ferrari, M., Cremonesi, L., and Galbiati, S. (2008). 10. Circulating nucleic acids as diagnostic tool. EJIFCC 19, 68–74.
Filatova, E. V., Alieva, A., Shadrina, M. I., and Slominsky, P. A. (2012). MicroRNAs: possible role in pathogenesis of Parkinson’s disease. Biochemistry 77, 813–819. doi: 10.1134/s0006297912080020
Fleischhacker, M., and Schmidt, B. (2007). Circulating nucleic acids (CNAs) and cancer–a survey. Biochim. Biophys. Acta 1775, 181–232. doi: 10.1016/j.bbcan.2006.10.001
Fujiwara, K., Fujimoto, N., Tabata, M., Nishii, K., Matsuo, K., Hotta, K., et al. (2005). Identification of epigenetic aberrant promoter methylation in serum DNA is useful for early detection of lung cancer. Clin. Cancer Res. 11, 1219–1225.
Furey, T. S. (2012). ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 13, 840–852. doi: 10.1038/nrg3306
García-Giménez, J. L., Mena-Molla, S., Beltran-Garcia, J., and Sanchis-Gomar, F. (2017). Challenges in the analysis of epigenetic biomarkers in clinical samples. Clin. Chem. Lab. Med. 55, 1474–1477. doi: 10.1515/cclm-2016-1162
Giannopoulou, L., Chebouti, I., Pavlakis, K., Kasimir-Bauer, S., and Lianidou, E. S. (2017). RASSF1A promoter methylation in high-grade serous ovarian cancer: a direct comparison study in primary tumors, adjacent morphologically tumor cell-free tissues and paired circulating tumor DNA. Oncotarget 8, 21429–21443. doi: 10.18632/oncotarget.15249
Gorges, T. M., Schiller, J., Schmitz, A., Schuetzmann, D., Schatz, C., Zollner, T. M., et al. (2012). Cancer therapy monitoring in xenografts by quantitative analysis of circulating tumor DNA. Biomarkers 17, 498–506. doi: 10.3109/1354750x.2012.689133
Grissa, O., Yessoufou, A., Mrisak, I., Hichami, A., Amoussou-Guenou, D., and Grissa, A. (2010). Growth factor concentrations and their placental mRNA expression are modulated in gestational diabetes mellitus: possible interactions with macrosomia. BMC Pregnancy Childbirth 10:7. doi: 10.1186/1471-2393-10-7
Grunau, C., Clark, S. J., and Rosenthal, A. (2001). Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 29, E65–E65.
Gu, H., Chen, L., Xue, J., Huang, T., Wei, X., Liu, D., et al. (2019). Expression profile of maternal circulating microRNAs as non-invasive biomarkers for prenatal diagnosis of congenital heart defects. Biomed. Pharmacother. 109, 823–830. doi: 10.1016/j.biopha.2018.10.110
Gu, M., Zheng, A., Tu, W., Zhao, J., Li, L., Li, M., et al. (2016). Circulating LncRNAs as novel, non-invasive biomarkers for prenatal detection of fetal congenital heart defects. Cell. Physiol. Biochem. 38, 1459–1471. doi: 10.1159/000443088
Guibert, J., Benachi, A., Grebille, A. G., Ernault, P., Zorn, J. R., and Costa, J. M. (2003). Kinetics of SRY gene appearance in maternal serum: detection by real time PCR in early pregnancy after assisted reproductive technique. Hum. Reprod. 18, 1733–1736. doi: 10.1093/humrep/deg320
Gupta, P., Bhattacharjee, S., Sharma, A. R., Sharma, G., Lee, S. S., and Chakraborty, C. (2017). miRNAs in Alzheimer Disease - A Therapeutic Perspective. Curr. Alzheimer Res. 14, 1198–1206.
Hahn, S., Rusterholz, C., Hosli, I., and Lapaire, O. (2011). Cell-free nucleic acids as potential markers for preeclampsia. Placenta 32(Suppl.), S17–S20.
Haldrup, C., Pedersen, A. L., Ogaard, N., Strand, S. H., Hoyer, S., Borre, M., et al. (2018). Biomarker potential of ST6GALNAC3 and ZNF660 promoter hypermethylation in prostate cancer tissue and liquid biopsies. Mol. Oncol. 12, 545–560. doi: 10.1002/1878-0261.12183
Harraz, M. M., Dawson, T. M., and Dawson, V. L. (2011). MicroRNAs in Parkinson’s disease. J. Chem. Neuroanat. 42, 127–130.
Hatt, L., Aagaard, M. M., Graakjaer, J., Bach, C., Sommer, S., Agerholm, I. E., et al. (2015). Microarray-based analysis of methylation status of CpGs in placental DNA and maternal blood DNA–potential new epigenetic biomarkers for cell free fetal DNA-Based diagnosis. PLoS One 10:e0128918. doi: 10.1371/journal.pone.0128918
Hauser, S., Zahalka, T., Fechner, G., Muller, S. C., and Ellinger, J. (2013). Serum DNA hypermethylation in patients with kidney cancer: results of a prospective study. Anticancer Res. 33, 4651–4656.
Hayter, S. M., and Cook, M. C. (2012). Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun. Rev. 11, 754–765. doi: 10.1016/j.autrev.2012.02.001
Hewagama, A., Gorelik, G., Patel, D., Liyanarachchi, P., Mccune, W. J., Somers, E., et al. (2013). Overexpression of X-linked genes in T cells from women with lupus. J. Autoimmun. 41, 60–71. doi: 10.1016/j.jaut.2012.12.006
Hohlfeld, R., Dornmair, K., Meinl, E., and Wekerle, H. (2016). The search for the target antigens of multiple sclerosis, part 1: autoreactive CD4+ T lymphocytes as pathogenic effectors and therapeutic targets. Lancet Neurol. 15, 198–209. doi: 10.1016/S1474-4422(15)00334-8
Holt, I. J. (2010). Zen and the art of mitochondrial DNA maintenance. Trends Genet. 26, 103–109. doi: 10.1016/j.tig.2009.12.011
Hoque, M. O., Feng, Q., Toure, P., Dem, A., Critchlow, C. W., Hawes, S. E., et al. (2006). Detection of aberrant methylation of four genes in plasma DNA for the detection of breast cancer. J. Clin. Oncol. 24, 4262–4269. doi: 10.1200/jco.2005.01.3516
House, M. G., Herman, J. G., Guo, M. Z., Hooker, C. M., Schulick, R. D., Lillemoe, K. D., et al. (2003). Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann. Surg. 238, 423–431.
Hu, J., Kong, M., Ye, Y., Hong, S., Cheng, L., and Jiang, L. (2014). Serum miR-206 and other muscle-specific microRNAs as non-invasive biomarkers for Duchenne muscular dystrophy. J. Neurochem. 129, 877–883. doi: 10.1111/jnc.12662
Huang, G., Krocker, J. D., Kirk, J. L., Merwat, S. N., Ju, H., Soloway, R. D., et al. (2014). Evaluation of INK4A promoter methylation using pyrosequencing and circulating cell-free DNA from patients with hepatocellular carcinoma. Clin. Chem. Lab. Med. 52, 899–909.
Huisman-Van Dijk, H. M., Schoot, R., Rijkeboer, M. M., Mathews, C. A., and Cath, D. C. (2016). The relationship between tics, OC, ADHD and autism symptoms: a cross- disorder symptom analysis in Gilles de la Tourette syndrome patients and family-members. Psychiatry Res. 237, 138–146. doi: 10.1016/j.psychres.2016.01.051
Hunter, M. P., Ismail, N., Zhang, X., Aguda, B. D., Lee, E. J., Yu, L., et al. (2008). Detection of microRNA expression in human peripheral blood microvesicles. PLoS One 3:e3694. doi: 10.1371/journal.pone.0003694
Hyland, C. A., Gardener, G. J., Davies, H., Ahvenainen, M., Flower, R. L., Irwin, D., et al. (2009). Evaluation of non-invasive prenatal RHD genotyping of the fetus. Med. J. Aust. 191, 21–25. doi: 10.5694/j.1326-5377.2009.tb02668.x
Iorio, M. V., and Croce, C. M. (2012). MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 4, 143–159. doi: 10.1002/emmm.201100209
Javierre, B. M., Fernandez, A. F., Richter, J., Al-Shahrour, F., Martin-Subero, J. I., and Rodriguez-Ubreva, J. (2010). Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 20, 170–179. doi: 10.1101/gr.100289.109
Jeffries, M. A., Dozmorov, M., Tang, Y., Merrill, J. T., Wren, J. D., and Sawalha, A. H. (2011). Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics 6, 593–601. doi: 10.4161/epi.6.5.15374
Ji, J., Shi, J., Budhu, A., Yu, Z., Forgues, M., Roessler, S., et al. (2009). MicroRNA expression, survival, and response to interferon in liver cancer. N. Engl. J. Med. 361, 1437–1447. doi: 10.1056/nejmoa0901282
Jiang, Y. H., Yuen, R. K., Jin, X., Wang, M., Chen, N., Wu, X., et al. (2013). Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 93, 249–263.
Jin, F., Hu, H., Xu, M., Zhan, S., Wang, Y., Zhang, H., et al. (2018). Serum microRNA profiles serve as novel biomarkers for autoimmune diseases. Front. Immunol. 9:2381. doi: 10.3389/fimmu.2018.02381
Jin, X. F., Wu, N., Wang, L., and Li, J. (2013). Circulating microRNAs: a novel class of potential biomarkers for diagnosing and prognosing central nervous system diseases. Cell. Mol. Neurobiol. 33, 601–613. doi: 10.1007/s10571-013-9940-9
Johnson, K. C., Houseman, E. A., King, J. E., Von Herrmann, K. M., Fadul, C. E., and Christensen, B. C. (2016). 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat. Commun. 7:13177.
Jones, P. A., and Takai, D. (2001). The role of DNA methylation in mammalian epigenetics. Science 293, 1068–1070. doi: 10.1126/science.1063852
Kandel, E. S. (2012). Mutations in circulating mitochondrial DNA: Cassandra of oral cancer? Oncotarget 3, 664–665. doi: 10.18632/oncotarget.567
Kang, H. J., Kawasawa, Y. I., Cheng, F., Zhu, Y., Xu, X., Li, M., et al. (2011). Spatio-temporal transcriptome of the human brain. Nature 478, 483–489.
Kaur, J., Rahat, B., Thakur, S., and Kaur, J. (2017). “Trends in precision medicine,” in Progress and Challenges in Precision Medicine, eds M. Verma and D. Barh (London: Academic Press), 269–299. doi: 10.1016/b978-0-12-809411-2.00015-5
Kehler, L., Biro, O., Lazar, L., Rigo, J. Jr., and Nagy, B. (2015). Elevated hsa-miR-99a levels in maternal plasma may indicate congenital heart defects. Biomed. Rep. 3, 869–873. doi: 10.3892/br.2015.510
Khan, N. A., Govindaraj, P., Meena, A. K., and Thangaraj, K. (2015). Mitochondrial disorders: challenges in diagnosis & treatment. Indian J. Med. Res. 141, 13–26. doi: 10.4103/0971-5916.154489
Kim, J., Inoue, K., Ishii, J., Vanti, W. B., Voronov, S. V., Murchison, E., et al. (2007). A MicroRNA feedback circuit in midbrain dopamine neurons. Science 317, 1220–1224. doi: 10.1126/science.1140481
Kloten, V., Becker, B., Winner, K., Schrauder, M. G., Fasching, P. A., Anzeneder, T., et al. (2013). Promoter hypermethylation of the tumor-suppressor genes ITIH5, DKK3, and RASSF1A as novel biomarkers for blood-based breast cancer screening. Breast Cancer Res. 15:R4.
Kneip, C., Schmidt, B., Seegebarth, A., Weickmann, S., Fleischhacker, M., Liebenberg, V., et al. (2011). SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. J. Thorac. Oncol. 6, 1632–1638. doi: 10.1097/jto.0b013e318220ef9a
Koffler, D., Agnello, V., Winchester, R., and Kunkel, H. G. (1973). The occurrence of single-stranded DNA in the serum of patients with systemic lupus erythematosus and other diseases. J. Clin. Invest. 52, 198–204. doi: 10.1172/jci107165
Kohler, C., Radpour, R., Barekati, Z., Asadollahi, R., Bitzer, J., Wight, E., et al. (2009). Levels of plasma circulating cell free nuclear and mitochondrial DNA as potential biomarkers for breast tumors. Mol. Cancer 8:105. doi: 10.1186/1476-4598-8-105
Lee, B. B., Lee, E. J., Jung, E. H., Chun, H. K., Chang, D. K., Song, S. Y., et al. (2009). Aberrant methylation of APC, MGMT, RASSF2A, and Wif-1 genes in plasma as a biomarker for early detection of colorectal cancer. Clin. Cancer Res. 15, 6185–6191. doi: 10.1158/1078-0432.ccr-09-0111
Lee, S. M., Park, J. Y., and Kim, D. S. (2012). Methylation of TMEFF2 gene in tissue and serum DNA from patients with non-small cell lung cancer. Mol. Cells 34, 171–176. doi: 10.1007/s10059-012-0083-5
Lee, Y. K., Jin, S., Duan, S., Lim, Y. C., Ng, D. P., Lin, X. M., et al. (2014). Improved reduced representation bisulfite sequencing for epigenomic profiling of clinical samples. Biol. Proced. Online 16:1. doi: 10.1186/1480-9222-16-1
Lehmann-Werman, R., Neiman, D., Zemmour, H., Moss, J., Magenheim, J., Vaknin-Dembinsky, A., et al. (2016). Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. U.S.A. 113, E1826–E1834.
Leon, S. A., Ehrlich, G. E., Shapiro, B., and Labbate, V. A. (1977). Free DNA in the serum of rheumatoid arthritis patients. J. Rheumatol. 4, 139–143.
Leti, F., Malenica, I., Doshi, M., Courtright, A., Van Keuren-Jensen, K., Legendre, C., et al. (2015). High-throughput sequencing reveals altered expression of hepatic microRNAs in nonalcoholic fatty liver disease-related fibrosis. Transl. Res. 166, 304–314. doi: 10.1016/j.trsl.2015.04.014
Li, H., Li, K., Lai, W., Li, X., Wang, H., Yang, J., et al. (2018). Comprehensive circular RNA profiles in plasma reveals that circular RNAs can be used as novel biomarkers for systemic lupus erythematosus. Clin. Chim. Acta 480, 17–25. doi: 10.1016/j.cca.2018.01.026
Liao, J., Liang, G., Xie, S., Zhao, H., Zuo, X., Li, F., et al. (2012). CD40L demethylation in CD4(+) T cells from women with rheumatoid arthritis. Clin. Immunol. 145, 13–18. doi: 10.1016/j.clim.2012.07.006
Lin, L. H., Chang, K. W., Kao, S. Y., Cheng, H. W., and Liu, C. J. (2018). Increased plasma circulating cell-free DNA could be a potential marker for oral cancer. Int. J. Mol. Sci. 19:E3303.
Lionel, A. C., Crosbie, J., Barbosa, N., Goodale, T., Thiruvahindrapuram, B., Rickaby, J., et al. (2011). Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci. Transl. Med. 3:95ra75. doi: 10.1126/scitranslmed.3002464
Litton, C., Stone, J., Eddleman, K., and Lee, M. J. (2009). Noninvasive prenatal diagnosis: past, present, and future. Mt. Sinai J. Med. 76, 521–528. doi: 10.1002/msj.20153
Liu, B. L., Cheng, J. X., Zhang, W., Zhang, X., Wang, R., Lin, H., et al. (2010). Quantitative detection of multiple gene promoter hypermethylation in tumor tissue, serum, and cerebrospinal fluid predicts prognosis of malignant gliomas. Neuro Oncol. 12, 540–548. doi: 10.1093/neuonc/nop064
Liu, H. W., Lin, H. L., Yen, J. H., Tsai, W. C., Chiou, S. S., Chang, J. G., et al. (2014). Demethylation within the proximal promoter region of human estrogen receptor alpha gene correlates with its enhanced expression: implications for female bias in lupus. Mol. Immunol. 61, 28–37. doi: 10.1016/j.molimm.2014.05.002
Liu, L., Yoon, J. H., Dammann, R., and Pfeifer, G. P. (2002). Frequent hypermethylation of the RASSF1A gene in prostate cancer. Oncogene 21, 6835–6840. doi: 10.1038/sj.onc.1205814
Lo, Y. M. D., Chan, K. C., Sun, H., Chen, E. Z., Jiang, P., Lun, F. M., et al. (2010). Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2:61ra91. doi: 10.1126/scitranslmed.3001720
Lo, Y. M. D., Corbetta, N., Chamberlain, P. F., Rai, V., Sargent, I. L., Redman, C. W., et al. (1997). Presence of fetal DNA in maternal plasma and serum. Lancet 350, 485–487. doi: 10.1016/S0140-6736(97)02174-0
Lo, Y. M. D., Tein, M. S., Lau, T. K., Haines, C. J., Leung, T. N., Poon, P. M., et al. (1998). Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am. J. Hum. Genet. 62, 768–775. doi: 10.1086/301800
Lonning, P. E., Berge, E. O., Bjornslett, M., Minsaas, L., Chrisanthar, R., Hoberg-Vetti, H., et al. (2018). White blood cell BRCA1 promoter methylation status and ovarian cancer risk. Ann. Intern. Med. 168, 326–334.
Loosen, S. H., Lurje, G., Wiltberger, G., Vucur, M., Koch, A., Kather, J. N., et al. (2019). Serum levels of miR-29, miR-122, miR-155 and miR-192 are elevated in patients with cholangiocarcinoma. PLoS One 14:e0210944. doi: 10.1371/journal.pone.0210944
Lorente, L. (2017). Biomarkers associated with the outcome of traumatic brain injury patients. Brain Sci. 7:142. doi: 10.3390/brainsci7110142
Lowther, C., Costain, G., Stavropoulos, D. J., Melvin, R., Silversides, C. K., Andrade, D. M., et al. (2015). Delineating the 15q13.3 microdeletion phenotype: a case series and comprehensive review of the literature. Genet. Med. 17, 149–157. doi: 10.1038/gim.2014.83
Lun, F. M., Chiu, R. W., Sun, K., Leung, T. Y., Jiang, P., Chan, K. C., et al. (2013). Noninvasive prenatal methylomic analysis by genomewide bisulfite sequencing of maternal plasma DNA. Clin. Chem. 59, 1583–1594. doi: 10.1373/clinchem.2013.212274
Lun, F. M., Tsui, N. B., Chan, K. C., Leung, T. Y., Lau, T. K., Charoenkwan, P., et al. (2008). Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc. Natl. Acad. Sci. U.S.A. 105, 19920–19925. doi: 10.1073/pnas.0810373105
Ma, E. G., Bai, Y. F., Cao, W., Cao, Y., Huang, Y. G., Cheng, H. C., et al. (2017). Cytosine 5-hydroxymethylation regulates VHL gene expression in renal clear cell carcinoma. Oncotarget 8, 63780–63787. doi: 10.18632/oncotarget.19070
Madhavan, D., Wallwiener, M., Bents, K., Zucknick, M., Nees, J., Schott, S., et al. (2014). Plasma DNA integrity as a biomarker for primary and metastatic breast cancer and potential marker for early diagnosis. Breast Cancer Res. Treat. 146, 163–174. doi: 10.1007/s10549-014-2946-2
Matatiele, P., Tikly, M., Tarr, G., and Gulumian, M. (2015). DNA methylation similarities in genes of black South Africans with systemic lupus erythematosus and systemic sclerosis. J. Biomed. Sci. 22:34.
Maulik, P. K., Mascarenhas, M. N., Mathers, C. D., Dua, T., and Saxena, S. (2011). Prevalence of intellectual disability: a meta-analysis of population-based studies. Res. Dev. Disabil. 32, 419–436. doi: 10.1016/j.ridd.2010.12.018
Meissner, A., Gnirke, A., Bell, G. W., Ramsahoye, B., Lander, E. S., and Jaenisch, R. (2005). Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877. doi: 10.1093/nar/gki901
Mendioroz, M., Martinez-Merino, L., Blanco-Luquin, I., Urdanoz, A., Roldan, M., and Jerico, I. (2018). Liquid biopsy: a new source of candidate biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 5, 763–768. doi: 10.1002/acn3.565
Meseguer, S., Martinez-Zamora, A., Garcia-Arumi, E., Andreu, A. L., and Armengod, M.-E. (2015). The ROS-sensitive microRNA-9/9* controls the expression of mitochondrial tRNA-modifying enzymes and is involved in the molecular mechanism of MELAS syndrome. Hum. Mol. Genet. 24, 167–184. doi: 10.1093/hmg/ddu427
Mitchell, N., Deangelis, J. T., and Tollefsbol, T. O. (2011). Methylated-CpG island recovery assay. Methods Mol. Biol. 791, 125–133. doi: 10.1007/978-1-61779-316-5_10
Munsell, B. C., Wee, C. Y., Keller, S. S., Weber, B., Elger, C., Da Silva, L. A., et al. (2015). Evaluation of machine learning algorithms for treatment outcome prediction in patients with epilepsy based on structural connectome data. Neuroimage 118, 219–230. doi: 10.1016/j.neuroimage.2015.06.008
Nagy, B. (2019). Cell-free nucleic acids in prenatal diagnosis and pregnancy-associated diseases. EJIFCC 30, 215–223.
Nakato, R., and Shirahige, K. (2017). Recent advances in ChIP-seq analysis: from quality management to whole-genome annotation. Brief. Bioinform. 18, 279–290.
Navickas, R., Gal, D., Laucevicius, A., Taparauskaite, A., Zdanyte, M., and Holvoet, P. (2016). Identifying circulating microRNAs as biomarkers of cardiovascular disease: a systematic review. Cardiovasc. Res. 111, 322–337. doi: 10.1093/cvr/cvw174
Newman, A. M., Bratman, S. V., To, J., Wynne, J. F., Eclov, N. C., Modlin, L. A., et al. (2014). An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 20, 548–554. doi: 10.1038/nm.3519
Nile, C. J., Read, R. C., Akil, M., Duff, G. W., and Wilson, A. G. (2008). Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 58, 2686–2693. doi: 10.1002/art.23758
Niyazov, D. M., Kahler, S. G., and Frye, R. E. (2016). Primary mitochondrial disease and secondary mitochondrial dysfunction: importance of distinction for diagnosis and treatment. Mol. Syndromol. 7, 122–137. doi: 10.1159/000446586
Noferesti, S. S., Sohel, M. M., Hoelker, M., Salilew-Wondim, D., Tholen, E., Looft, C., et al. (2015). Controlled ovarian hyperstimulation induced changes in the expression of circulatory miRNA in bovine follicular fluid and blood plasma. J. Ovarian Res. 8:81.
Norwitz, E. R., and Levy, B. (2013). Noninvasive prenatal testing: the future is now. Rev. Obstet. Gynecol. 6, 48–62.
Ohtani-Fujita, N., Dryja, T. P., Rapaport, J. M., Fujita, T., Matsumura, S., Ozasa, K., et al. (1997). Hypermethylation in the retinoblastoma gene is associated with unilateral, sporadic retinoblastoma. Cancer Genet. Cytogenet. 98, 43–49. doi: 10.1016/s0165-4608(96)00395-0
Palmisano, W. A., Divine, K. K., Saccomanno, G., Gilliland, F. D., Baylin, S. B., Herman, J. G., et al. (2000). Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res. 60, 5954–5958.
Papadaki, C., Stoupis, G., Tsalikis, L., Monastirioti, A., Papadaki, M., Maliotis, N., et al. (2019). Circulating miRNAs as a marker of metastatic disease and prognostic factor in metastatic breast cancer. Oncotarget 10, 966–981. doi: 10.18632/oncotarget.26629
Papageorgiou, E. A., Fiegler, H., Rakyan, V., Beck, S., Hulten, M., Lamnissou, K., et al. (2009). Sites of differential DNA methylation between placenta and peripheral blood: molecular markers for noninvasive prenatal diagnosis of aneuploidies. Am. J. Pathol. 174, 1609–1618. doi: 10.2353/ajpath.2009.081038
Parikh, S., Goldstein, A., Koenig, M. K., Scaglia, F., Enns, G. M., Saneto, R., et al. (2015). Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet. Med. 17, 689–701. doi: 10.1038/gim.2014.177
Park, J. W., Baek, I. H., and Kim, Y. T. (2012). Preliminary study analyzing the methylated genes in the plasma of patients with pancreatic cancer. Scand. J. Surg. 101, 38–44. doi: 10.1177/145749691210100108
Pigati, L., Yaddanapudi, S. C., Iyengar, R., Kim, D. J., Hearn, S. A., Danforth, D., et al. (2010). Selective release of microRNA species from normal and malignant mammary epithelial cells. PLoS One 5:e13515. doi: 10.1371/journal.pone.0013515
Pillai, J. A., and Cummings, J. L. (2013). Clinical trials in predementia stages of Alzheimer disease. Med. Clin. North Am. 97, 439–457. doi: 10.1016/j.mcna.2013.01.002
Poon, L. L., Leung, T. N., Lau, T. K., Chow, K. C., and Lo, Y. M. (2002). Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clin. Chem. 48, 35–41. doi: 10.1093/clinchem/48.1.35
Pos, O., Biro, O., Szemes, T., and Nagy, B. (2018). Circulating cell-free nucleic acids: characteristics and applications. Eur. J. Hum. Genet. 26, 937–945. doi: 10.1038/s41431-018-0132-4
Powrozek, T., Krawczyk, P., Kucharczyk, T., and Milanowski, J. (2014). Septin 9 promoter region methylation in free circulating DNA-potential role in noninvasive diagnosis of lung cancer: preliminary report. Med. Oncol. 31:917. doi: 10.1007/s12032-014-0917-4
Radpour, R., Barekati, Z., Kohler, C., Lv, Q., Burki, N., Diesch, C., et al. (2011). Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS One 6:e16080. doi: 10.1371/journal.pone.0016080
Raffort, J., Hinault, C., Dumortier, O., and Van Obberghen, E. (2015). Circulating microRNAs and diabetes: potential applications in medical practice. Diabetologia 58, 1978–1992. doi: 10.1007/s00125-015-3680-y
Rahat, B., Hamid, A., Ahmad Najar, R., Bagga, R., and Kaur, J. (2014). Epigenetic mechanisms regulate placental c-myc and hTERT in normal and pathological pregnancies; c-myc as a novel fetal DNA epigenetic marker for pre-eclampsia. Mol. Hum. Reprod. 20, 1026–1040. doi: 10.1093/molehr/gau053
Rahat, B., Mahajan, A., Bagga, R., Hamid, A., and Kaur, J. (2017a). Epigenetic modifications at DMRs of placental genes are subjected to variations in normal gestation, pathological conditions and folate supplementation. Sci. Rep. 7:40774.
Rahat, B., Najar, R. A., Hamid, A., Bagga, R., and Kaur, J. (2017b). The role of aberrant methylation of trophoblastic stem cell origin in the pathogenesis and diagnosis of placental disorders. Prenat. Diagn. 37, 133–143. doi: 10.1002/pd.4974
Rahat, B., Thakur, S., Bagga, R., and Kaur, J. (2016a). Epigenetic regulation of STAT5A and its role as fetal DNA epigenetic marker during placental development and dysfunction. Placenta 44, 46–53. doi: 10.1016/j.placenta.2016.06.003
Rahat, B., Thakur, S., Hamid, A., Bagga, R., and Kaur, J. (2016b). Association of aberrant methylation at promoter regions of tumor suppressor genes with placental pathologies. Epigenomics 8, 767–787. doi: 10.2217/epi.16.7
Rai, M., Soragni, E., Chou, C. J., Barnes, G., Jones, S., Rusche, J. R., et al. (2010). Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich’s ataxia patients and in a mouse model. PLoS One 5:e8825. doi: 10.1371/journal.pone.0008825
Rainer, T. H., Wong, L. K., Lam, W., Yuen, E., Lam, N. Y. L., and Metreweli, C. (2003). Prognostic use of circulating plasma nucleic acid concentrations in patients with acute stroke. Clin. Chem. 49, 562–569. doi: 10.1373/49.4.562
Ramezanzadeh, M., Khosravi, S., and Salehi, R. (2017). Cell-free fetal nucleic acid identifier markers in maternal circulation. Adv. Biomed. Res. 6:89. doi: 10.4103/2277-9175.211800
Rebelo, A. P., Williams, S. L., and Moraes, C. T. (2009). In vivo methylation of mtDNA reveals the dynamics of protein-mtDNA interactions. Nucleic Acids Res. 37, 6701–6715. doi: 10.1093/nar/gkp727
Regev, A., Teichmann, S. A., Lander, E. S., Amit, I., Benoist, C., Birney, E., et al. (2017). The human cell atlas. eLife 6:e27041.
Rennert, H., Eng, K., Zhang, T., Tan, A., Xiang, J., Romanel, A., et al. (2016). Development and validation of a whole-exome sequencing test for simultaneous detection of point mutations, indels and copy-number alterations for precision cancer care. NPJ Genom. Med. 1:16019.
Romao, R. M., Levi, J. E., Carvalho, H. B. D., Francisco, R. P., VAmorim Filho, A. G. D., and Amorim Filho, et al. (1992). Use of cell-free fetal nucleic acids in maternal blood for prenatal diagnosis: the reality of this scenario in Brazil. Rev. Assoc. Med. Bras. 58, 615–619.
Saleh, A. A., Soliman, S. E., Habib, M. S. E., Gohar, S. F., and Abo-Zeid, G. S. (2019). Potential value of circulatory microRNA122 gene expression as a prognostic and metastatic prediction marker for breast cancer. Mol. Biol. Rep. 46, 2809–2818. doi: 10.1007/s11033-019-04727-5
Sales, G., Calura, E., Cavalieri, D., and Romualdi, C. (2012). graphite - a Bioconductor package to convert pathway topology to gene network. BMC Bioinformatics 13:20. doi: 10.1186/1471-2105-13-20
Sandi, C., Al-Mahdawi, S., and Pook, M. A. (2013). Epigenetics in Friedreich’s Ataxia: challenges and opportunities for therapy. Genet. Res. Int. 2013: 852080.
Schlosser, P., Knaus, J., Schmutz, M., Dohner, K., Plass, C., Bullinger, L., et al. (2020). Netboost: boosting-supported network analysis improves high-dimensional omics prediction in acute myeloid leukemia and Huntington’s disease. IEEE/ACM Trans. Comput. Biol. Bioinform. doi: 10.1109/TCBB.2020.2983010
Schwarzenbach, H., Hoon, D. S., and Pantel, K. (2011). Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 11, 426–437. doi: 10.1038/nrc3066
Semaan, A., Van Ellen, A., Meller, S., Bergheim, D., Branchi, V., Lingohr, P., et al. (2016). SEPT9 and SHOX2 DNA methylation status and its utility in the diagnosis of colonic adenomas and colorectal adenocarcinomas. Clin. Epigenetics 8:100.
Serr, D. M., Sachs, L., and Danon, M. (2017). The diagnosis of sex before birth using cells from the amniotic fluid (a preliminary report). Bull. Res. Counc. Isr. 5B, 137–138.
Shaw, J. A., Page, K., Blighe, K., Hava, N., Guttery, D., Ward, B., et al. (2012). Genomic analysis of circulating cell-free DNA infers breast cancer dormancy. Genome Res. 22, 220–231. doi: 10.1101/gr.123497.111
Shaw, J. A., Smith, B. M., Walsh, T., Johnson, S., Primrose, L., Slade, M. J., et al. (2000). Microsatellite alterations plasma DNA of primary breast cancer patients. Clin. Cancer Res. 6, 1119–1124.
Shu, L., Zhao, Y., Kurt, Z., Byars, S. G., Tukiainen, T., Kettunen, J., et al. (2016). Mergeomics: multidimensional data integration to identify pathogenic perturbations to biological systems. BMC Genomics 17:874. doi: 10.1186/s12864-016-3198-9
Siegel, R. L., Miller, K. D., and Jemal, A. (2020). Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30.
Sifakis, S., Koukou, Z., and Spandidos, D. A. (2015). Cell-free fetal DNA and pregnancy-related complications (review). Mol. Med. Rep. 11, 2367–2372. doi: 10.3892/mmr.2014.3118
Simmer, F., Brinkman, A. B., Assenov, Y., Matarese, F., Kaan, A., Sabatino, L., et al. (2012). Comparative genome-wide DNA methylation analysis of colorectal tumor and matched normal tissues. Epigenetics 7, 1355–1367. doi: 10.4161/epi.22562
Skalis, G., Katsi, V., Miliou, A., Georgiopoulos, G., Papazachou, O., Vamvakou, G., et al. (2019). MicroRNAs in preeclampsia. Microrna 8, 28–35.
Snyder, A., Makarov, V., Merghoub, T., Yuan, J., Zaretsky, J. M., Desrichard, A., et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199. doi: 10.1056/nejmoa1406498
Snyder, M. W., Kircher, M., Hill, A. J., Daza, R. M., and Shendure, J. (2016). Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 164, 57–68. doi: 10.1016/j.cell.2015.11.050
Song, G. Y., Song, W. W., Han, Y., Wang, D., and Na, Q. (2013). Characterization of the role of microRNA-517a expression in low birth weight infants. J. Dev. Orig. Health Dis. 4, 522–526. doi: 10.1017/s204017441300024x
Sperling, R. A., Jack, C. R. Jr., and Aisen, P. S. (2011). Testing the right target and right drug at the right stage. Sci. Transl. Med. 3:111cm133.
Stroun, M., Anker, P., Maurice, P., Lyautey, J., Lederrey, C., and Beljanski, M. (1989). Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 46, 318–322. doi: 10.1159/000226740
Stroun, M., Lyautey, J., Lederrey, C., Olson-Sand, A., and Anker, P. (2001). About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin. Chim. Acta 313, 139–142. doi: 10.1016/s0009-8981(01)00665-9
Su, Y., Fang, H., and Jiang, F. (2016). Integrating DNA methylation and microRNA biomarkers in sputum for lung cancer detection. Clin. Epigenetics 8:109.
Sun, K., Lun, F. F. M., Jiang, P., and Sun, H. (2017). BSviewer: a genotype-preserving, nucleotide-level visualizer for bisulfite sequencing data. Bioinformatics 33, 3495–3496. doi: 10.1093/bioinformatics/btx505
Swarup, V., and Rajeswari, M. R. (2007). Circulating (cell-free) nucleic acids–A promising, non-invasive tool for early detection of several human diseases. FEBS Lett. 581, 795–799. doi: 10.1016/j.febslet.2007.01.051
Swarup, V., Srivastava, A. K., Padma, M. V., and Rajeswari, M. R. (2011). Quantification of circulating plasma DNA in Friedreich’s ataxia and spinocerebellar ataxia types 2 and 12. DNA Cell Biol. 30, 389–394. doi: 10.1089/dna.2010.1165
Tan, E. M., and Kunkel, H. G. (1966). Characteristics of a soluble nuclear antigen precipitating with sera of patients with systemic lupus erythematosus. J. Immunol. 96, 464–471.
Tan, L., Yu, J. T., Liu, Q. Y., Tan, M. S., Zhang, W., Hu, N., et al. (2014). Circulating miR-125b as a biomarker of Alzheimer’s disease. J. Neurol. Sci. 336, 52–56. doi: 10.1016/j.jns.2013.10.002
Tavakolpour, S. (2017). Towards personalized medicine for patients with autoimmune diseases: opportunities and challenges. Immunol. Lett. 190, 130–138. doi: 10.1016/j.imlet.2017.08.002
Tham, C., Chew, M., Soong, R., Lim, J., Ang, M., Tang, C., et al. (2014). Postoperative serum methylation levels of TAC1 and SEPT9 are independent predictors of recurrence and survival of patients with colorectal cancer. Cancer 120, 3131–3141. doi: 10.1002/cncr.28802
Thapar, A., Cooper, M., and Rutter, M. (2017). Neurodevelopmental disorders. Lancet Psychiatry 4, 339–346.
Thierry, A. R., El Messaoudi, S., Gahan, P. B., Anker, P., and Stroun, M. (2016). Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 35, 347–376. doi: 10.1007/s10555-016-9629-x
Tinay, I., Tan, M., Gui, B., Werner, L., Kibel, A. S., and Jia, L. (2018). Functional roles and potential clinical application of miRNA-345-5p in prostate cancer. Prostate 78, 927–937. doi: 10.1002/pros.23650
Tong, Y. K., Ding, C., Chiu, R. W., Gerovassili, A., Chim, S. S., Leung, T. Y., et al. (2006). Noninvasive prenatal detection of fetal trisomy 18 by epigenetic allelic ratio analysis in maternal plasma: theoretical and empirical considerations. Clin. Chem. 52, 2194–2202. doi: 10.1373/clinchem.2006.076851
Tong, Y. K., Jin, S., Chiu, R. W., Ding, C., Chan, K. C., Leung, T. Y., et al. (2010). Noninvasive prenatal detection of trisomy 21 by an epigenetic-genetic chromosome-dosage approach. Clin. Chem. 56, 90–98. doi: 10.1373/clinchem.2009.134114
Tost, J. (2016). Follow the trace of death: methylation analysis of cell-free DNA for clinical applications in non-cancerous diseases. Epigenomics 8, 1169–1172. doi: 10.2217/epi-2016-0080
Tounta, G., Kolialexi, A., Papantoniou, N., Tsangaris, G. T., Kanavakis, E., and Mavrou, A. (2011a). Non-invasive prenatal diagnosis using cell-free fetal nucleic acids in maternal plasma: progress overview beyond predictive and personalized diagnosis. EPMA J. 2, 163–171. doi: 10.1007/s13167-011-0085-y
Tounta, G., Vrettou, C., Kolialexi, A., Papantoniou, N., Destouni, A., Tsangaris, G. T., et al. (2011b). A multiplex PCR for non-invasive fetal RHD genotyping using cell-free fetal DNA. In Vivo 25, 411–417.
Tsui, D. W., Chan, K. C., Chim, S. S., Chan, L. W., Leung, T. Y., Lau, T. K., et al. (2007). Quantitative aberrations of hypermethylated RASSF1A gene sequences in maternal plasma in pre-eclampsia. Prenat. Diagn. 27, 1212–1218. doi: 10.1002/pd.1897
Tsui, D. W., Chiu, R. W., and Lo, Y. D. (2010). Epigenetic approaches for the detection of fetal DNA in maternal plasma. Chimerism 1, 30–35. doi: 10.4161/chim.1.1.12439
Uddin, M., Pellecchia, G., Thiruvahindrapuram, B., D’abate, L., Merico, D., Chan, A., et al. (2016). Indexing effects of copy number variation on genes involved in developmental delay. Sci. Rep. 6:28663.
Umu, S. U., Langseth, H., Bucher-Johannessen, C., Fromm, B., Keller, A., Meese, E., et al. (2018). A comprehensive profile of circulating RNAs in human serum. RNA Biol. 15, 242–250. doi: 10.1080/15476286.2017.1403003
Ura, B., Feriotto, G., Monasta, L., Bilel, S., Zweyer, M., and Celeghini, C. (2014). Potential role of circulating microRNAs as early markers of preeclampsia. Taiwan J. Obstet. Gynecol. 53, 232–234. doi: 10.1016/j.tjog.2014.03.001
Van Neste, L., Groskopf, J., Grizzle, W. E., Adams, G. W., Deguenther, M. S., Kolettis, P. N., et al. (2017). Epigenetic risk score improves prostate cancer risk assessment. Prostate 77, 1259–1264. doi: 10.1002/pros.23385
Vaz, M., Hwang, S. Y., Kagiampakis, I., Phallen, J., Patil, A., O’hagan, H. M., et al. (2017). Chronic cigarette smoke-induced epigenomic changes precede sensitization of bronchial epithelial cells to single-step transformation by KRAS mutations. Cancer Cell 32, 360–376.e6. doi: 10.1016/j.ccell.2017.08.006
Volik, S., Alcaide, M., Morin, R. D., and Collins, C. (2016). Cell-free DNA (cfDNA): clinical significance and utility in cancer shaped by emerging technologies. Mol. Cancer Res. 14, 898–908. doi: 10.1158/1541-7786.mcr-16-0044
Vu, T. L., Nguyen, T. T., Doan, V. T. H., and Vo, L. T. T. (2018). Methylation profiles of BRCA1, RASSF1A and GSTP1 in vietnamese women with breast cancer. Asian Pac. J. Cancer Prev. 19, 1887–1893.
Wakabayashi, T., Natsume, A., Hatano, H., Fujii, M., Shimato, S., Ito, M., et al. (2009). p16 promoter methylation in the serum as a basis for the molecular diagnosis of gliomas. Neurosurgery 64, 455–461.
Wang, D., Na, Q., Song, W. W., and Song, G. Y. (2014). Altered Expression of miR-518b and miR-519a in the placenta is associated with low fetal birth weight. Am. J. Perinatol. 31, 729–734. doi: 10.1055/s-0033-1361832
Wang, E., Batey, A., Struble, C., Musci, T., Song, K., and Oliphant, A. (2013). Gestational age and maternal weight effects on fetal cell-free DNA in maternal plasma. Prenat. Diagn. 33, 662–666. doi: 10.1002/pd.4119
Wang, G., Tam, L. S., Li, E. K., Kwan, B. C., Chow, K. M., Luk, C. C., et al. (2010). Serum and urinary cell-free MiR-146a and MiR-155 in patients with systemic lupus erythematosus. J. Rheumatol. 37, 2516–2522. doi: 10.3899/jrheum.100308
Wang, G., Tam, L. S., Li, E. K., Kwan, B. C., Chow, K. M., Luk, C. C., et al. (2011). Serum and urinary free microRNA level in patients with systemic lupus erythematosus. Lupus 20, 493–500. doi: 10.1177/0961203310389841
Wang, H., Peng, W., Ouyang, X., Li, W., and Dai, Y. (2012). Circulating microRNAs as candidate biomarkers in patients with systemic lupus erythematosus. Transl. Res. 160, 198–206. doi: 10.1016/j.trsl.2012.04.002
Wang, H. D., Liu, L., Zhao, H. R., Hou, Q. F., Yan, J. B., Shi, W. L., et al. (2017). Detection of fetal epigenetic biomarkers through genome-wide DNA methylation study for non-invasive prenatal diagnosis. Mol. Med. Rep. 15, 3989–3998. doi: 10.3892/mmr.2017.6506
Wang, J. Y., Hsieh, J. S., Chang, M. Y., Huang, T. J., Chen, F. M., Cheng, T. L., et al. (2004). Molecular detection of APC, K- ras, and p53 mutations in the serum of colorectal cancer patients as circulating biomarkers. World J. Surg. 28, 721–726.
Wang, Y., Yu, Y., Ye, R., Zhang, D., Li, Q., An, D., et al. (2016). An epigenetic biomarker combination of PCDH17 and POU4F2 detects bladder cancer accurately by methylation analyses of urine sediment DNA in Han Chinese. Oncotarget 7, 2754–2764. doi: 10.18632/oncotarget.6666
Weaver, K. D., Grossman, S. A., and Herman, J. G. (2006). Methylated tumor-specific DNA as a plasma biomarker in patients with glioma. Cancer Invest. 24, 35–40. doi: 10.1080/07357900500449546
Widschwendter, A., Muller, H. M., Fiegl, H., Ivarsson, L., Wiedemair, A., Muller-Holzner, E., et al. (2004). DNA methylation in serum and tumors of cervical cancer patients. Clin. Cancer Res. 10, 565–571. doi: 10.1158/1078-0432.ccr-0825-03
Witebsky’s, E., Rose, N. R., Terplan, K., Paine, J. R., and Egan, R. W. (1957). Chronic thyroiditis and autoimmunization. J. Am. Med. Assoc. 164, 1439–1447.
Wong, I. H., Lo, Y. M., and Johnson, P. J. (2001). Epigenetic tumor markers in plasma and serum: biology and applications to molecular diagnosis and disease monitoring. Ann. N. Y. Acad. Sci. 945, 36–50. doi: 10.1111/j.1749-6632.2001.tb03862.x
Wong, I. H., Lo, Y. M., Zhang, J., Liew, C. T., Ng, M. H., Wong, N., et al. (1999). Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res. 59, 71–73.
Wu, Z., Lu, H., Sheng, J., and Li, L. (2012). Inductive microRNA-21 impairs anti-mycobacterial responses by targeting IL-12 and Bcl-2. FEBS Lett. 586, 2459–2467. doi: 10.1016/j.febslet.2012.06.004
Xiao, M., Yang, H., Xu, W., Ma, S., Lin, H., Zhu, H., et al. (2012). Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 26, 1326–1338. doi: 10.1101/gad.191056.112
Xiong, H. Y., Alipanahi, B., Lee, L. J., Bretschneider, H., Merico, D., Yuen, R. K., et al. (2015). RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347:1254806.
Yamamoto, Y., Kosaka, N., Tanaka, M., Koizumi, F., Kanai, Y., Mizutani, T., et al. (2009). MicroRNA-500 as a potential diagnostic marker for hepatocellular carcinoma. Biomarkers 14, 529–538. doi: 10.3109/13547500903150771
Yu, F., Li, K., Li, S., Liu, J., Zhang, Y., Zhou, M., et al. (2020). CFEA: a cell-free epigenome atlas in human diseases. Nucleic Acids Res. 48, D40–D44.
Yuan, Q., Zhou, W., Li, S., and Cai, D. (2011). Epileptic EEG classification based on extreme learning machine and nonlinear features. Epilepsy Res. 96, 29–38. doi: 10.1016/j.eplepsyres.2011.04.013
Zachariah, R., Schmid, S., Radpour, R., Buerki, N., Fan, A. X., Hahn, S., et al. (2009). Circulating cell-free DNA as a potential biomarker for minimal and mild endometriosis. Reprod. Biomed. Online 18, 407–411. doi: 10.1016/s1472-6483(10)60100-9
Zeng, C., Stroup, E. K., Zhang, Z., Chiu, B. C., and Zhang, W. (2019). Towards precision medicine: advances in 5-hydroxymethylcytosine cancer biomarker discovery in liquid biopsy. Cancer Commun. 39:12. doi: 10.1186/s40880-019-0356-x
Zhang, H., Guan, M., Townsend, K. L., Huang, T. L., An, D., Yan, X., et al. (2015). MicroRNA-455 regulates brown adipogenesis via a novel HIF1an-AMPK-PGC1α signaling network. EMBO Rep. 16, 1378–1393. doi: 10.15252/embr.201540837
Zhang, W. C., Chin, T. M., Yang, H., Nga, M. E., Lunny, D. P., Lim, E. K., et al. (2016). Tumour-initiating cell-specific miR-1246 and miR-1290 expression converge to promote non-small cell lung cancer progression. Nat. Commun. 7:11702. doi: 10.1038/ncomms11702
Zhang, Y., Wang, R., Song, H., Huang, G., Yi, J., Zheng, Y., et al. (2011). Methylation of multiple genes as a candidate biomarker in non-small cell lung cancer. Cancer Lett. 303, 21–28. doi: 10.1016/j.canlet.2010.12.011
Zhao, M., Zhou, Y., Zhu, B., Wan, M., Jiang, T., Tan, Q., et al. (2016). IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann. Rheum. Dis. 75, 1998–2006.
Zhao, S., Wang, Y., Liang, Y., Zhao, M., Long, H., Ding, S., et al. (2011). MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. 63, 1376–1386. doi: 10.1002/art.30196
Zhao, Z., and Shilatifard, A. (2019). Epigenetic modifications of histones in cancer. Genome Biol. 20:245.
Zheng, F., Yu, X., Huang, J., and Dai, Y. (2017). Circular RNA expression profiles of peripheral blood mononuclear cells in rheumatoid arthritis patients, based on microarray chip technology. Mol. Med. Rep. 16, 8029–8036. doi: 10.3892/mmr.2017.7638
Zhou, J., and Troyanskaya, O. G. (2015). Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 12, 931–934. doi: 10.1038/nmeth.3547
Zhu, S., Cao, L., Zhu, J., Kong, L., Jin, J., Qian, L., et al. (2013). Identification of maternal serum microRNAs as novel non-invasive biomarkers for prenatal detection of fetal congenital heart defects. Clin. Chim. Acta 424, 66–72. doi: 10.1016/j.cca.2013.05.010
Keywords: autoimmune diseases, cancer diagnosis, precision medicine, epigenetic biomarkers, circulating nucleic acids in plasma/serum, prenatal and genetic diagnostics, circulating cell free nucleic acids
Citation: Rahat B, Ali T, Sapehia D, Mahajan A and Kaur J (2020) Circulating Cell-Free Nucleic Acids as Epigenetic Biomarkers in Precision Medicine. Front. Genet. 11:844. doi: 10.3389/fgene.2020.00844
Received: 27 February 2020; Accepted: 13 July 2020;
Published: 11 August 2020.
Edited by:
Rui Henrique, Portuguese Oncology Institute, PortugalReviewed by:
Naoko Hattori, National Cancer Center Research Institute, JapanCopyright © 2020 Rahat, Ali, Sapehia, Mahajan and Kaur. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jyotdeep Kaur, anlvdGRlZXAyMDAxQHlhaG9vLmNvLmlu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.