
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 27 August 2020
Sec. Cancer Genetics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00829
 Zhe-Yu Hu1,2*†
Zhe-Yu Hu1,2*† Liping Liu1,2†Ning Xie1,2Jun Lu1,2Zhentian Liu3Yu Tang1,2
Liping Liu1,2†Ning Xie1,2Jun Lu1,2Zhentian Liu3Yu Tang1,2 Yikai Wang4Jianbo Yang1,5,6*Quchang Ouyang1,2*
Yikai Wang4Jianbo Yang1,5,6*Quchang Ouyang1,2*PALB2 is an important BRCAx candidate for familial breast cancers (FBC). PALB2 pathogenic variants (PVs) may not to conform to “two hit” paradigm. However, a recent study demonstrates that in the majority PALB2 germline mutant breast cancers, the loss of heterozygosity (LOH) and somatic point mutations are the “second hit.” This study aimed to investigate the second hits in germline PALB2 mutations in breast cancers. We screened out 28 germline PALB2-mutation carriers among 480 familial cancer patients (including 143 FBC patients) in Geneplus database pool. Of the 143 patients with FBC, 10 had mono-allelic PALB2 germline mutations. All these germline PALB2 mutations were high-risk stop-gain, frameshift, or splicing mutations that concentrated in EX5–EX9 and might led to truncated proteins, severe functional defects and malignant phenotype. The hotspots were c.1057A[3 > 2] and c.3114-1G > A. Other mutations included c.389delA, c.2068C > T, c.2167_2168delAT, c.2629delT and c.2968G > T. Only one FBC patient has PALB2 somatic mutation and two patients had LOH of PALB2. All germline PALB2 mutations were high-risk mutations, whereas the somatic PALB2 mutations were moderate-risk missense mutations. We also distinguished PALB2 “novel mutations” from “reported mutations.” In conclusion, germline PALB2 mutation should be put into the context of future screening.
Nearly the one-eighth of females develop breast cancer over the course of their lifetime (Owens et al., 2019). Approximately 5–27% of breast cancers are hereditary (Lichtenstein et al., 2000). BRCA1 was the first gene identified as a susceptibility gene for hereditary breast cancer (HBC) (Torchard et al., 1994). Soon after, BRCA2 was also identified as a susceptibility gene for HBC (Collins et al., 1995). Not all HBCs involve BRCA1/2 mutations. 70–80% of HBCs involve non-BRCA1/2 (BRCAx) mutations (Hedenfalk et al., 2003; Keeney et al., 2017). Instead, ATM, CHD8, CDH1, RAD50, CHEK2, and PALB2 are found to harbor germline mutations conferring high to moderate risk for BRCAx HBC (Aloraifi et al., 2015; Li et al., 2017; Tavera-Tapia et al., 2017). Most of those genes are related to DNA damage repair and their mutations in embryo are thought to increase the risk of HBC by 20–80%. BRCAx HBCs may develop the secondary somatic BRCA1/2 aberrations (including point mutations and hypermethylation) (Alvarez et al., 2005). In a study of 656 families, no convincing evidence has been found to verify the risk effect of the epigenetic modifier and known germline breast cancer driver gene mutations (Li et al., 2017). But in a most recent study of 524 families with germline PALB2 variants (PVs), PALB2 was confirmed as a major breast cancer susceptibility gene, and its germline PVs also associates with ovarian, pancreatic and male breast cancers (Yang et al., 2020).
In 2006, PALB2 has been identified as the partner and localizer of BRCA2 (Xia et al., 2006) and a susceptibility gene for HBC (Rahman et al., 2007). Overall, 35% of females carrying PALB2 mutations are expected to develop breast cancer before 70 years of age, and 58% of carriers with a family history of breast cancer are expected to develop the disease (Antoniou et al., 2014). By binding to the BRCA1 and BRCA2 proteins, PALB2 protein facilitates homologous recombination repair (HRR) for DNA double strand breaks (DSBs) (Ripperger et al., 2009; Wiltshire et al., 2020).
The “second hit” theory of tumor-suppressor genes (TSGs) suggests that the loss of function (LOF) mutations on both alleles of a given TSG are necessary for tumorigenesis (Knudson, 1971; Smith et al., 2016). For example, some HBC patients have mutations in both PALB2 alleles (PALB2-NULL), of which one is inherited and the other is a somatic point mutation and epigenetic modification (Potapova et al., 2008; Bouwman et al., 2011; Scott et al., 2016; Lee et al., 2018). However, in some cases, PALB2 seems not to conform to that theory. For example, some HBC patients have one heterozygous germline PALB2 mutation (PALB2-HET), with one normal wild-type PALB2 allele. Interestingly, there was no significant difference in the HRD scores between PALB2 heterozygotes and null tumors (Lee et al., 2018). These PALB2-HET HBC patients have more defects in homologous recombination repair (HRR) than patients with sporadic breast cancer. In addition, HRD mutational signatures are predominant in some PALB2 heterozygous carriers. The fact that most the PALB2-HET BC tumors also exhibit known cancer drivers suggests either tumor evolution or this demonstrates the well known phenomenon of differential positive selection (Martincorena et al., 2017).
In the haploinsufficient paradigm, malignancy can be induced by the mutation on one allele of a dose-dependent TSG (Gilad et al., 2010). In a mouse model, homozygous PALB2 knockout is lethal; malignancies are only developed in heterozygous PALB2 knockout mutants (Bouwman et al., 2011). In an Australian study (Lee et al., 2018), PALB2-HET patients have much more high-risk germline PALB2 frameshift variants than PALB2-NULL patients (80 vs. 30%), suggesting that serious PALB2 defects on one allele might be enough to induce malignant phenotype. In a Chinese study, high-risk loss-of-function (LOF) mutations (frameshift and splicing mutations) were detected rarely in patients with sporadic breast cancer (0.56%), but more frequently in HBC patients (1.31%) (Zhang et al., 2017). Thus, we hypothesize that heterozygous germline PALB2 LOF mutations cause PALB2 functional haploinsufficiency, leading to HRR impairment and HBC.
This study was approved by the Ethics Committee at Beijing Geneplus Institute and Hunan Cancer Hospital. All patients provided informed consent for genetic analysis of their genomic DNA (gDNA) and circulating tumor DNA (ctDNA). 480 familial cancer patients (including 143 familial breast cancer patients) were assessed by specialist Familial Cancer Clinic and determined to be sufficiently strong to be eligible for clinical genetic testing by local criteria. In addition, 196 sporadic advanced breast cancer (ABC) patients without family history were also assessed. The somatic and germline pathogenic variants using a 1021-gene panel (Hu et al., 2018).
The somatic PALB2 mutations were also investigated in 986 invasive breast cancer samples from TCGA-BRCA project and in 3,090 breast cancer samples from cBioPortal database.
To detect the germline PALB2 variants, gDNA was extracted from peripheral blood cells using a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Peripheral blood samples were collected in Streck tubes (Streck, Omaha, NE, United States) and centrifuged within 72 h to separate the plasma from peripheral blood cells. For somatic variants detection, ctDNA was extracted from the plasma using a QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany). Both DNA extractions were performed according to the manufacturer’s instructions, as described previously (Hu et al., 2018).
Sequencing libraries of gDNA and ctDNA were prepared using the DNA Library Preparation Kit for Illumina (New England Biolabs, Ipswich, MA, United States). Custom biotinylated oligonucleotide probes (IDT, Coralville, IA, United States) covering the exons of 1,021 genes that are highly mutated in 12 common solid tumors were used for hybrid capture, as described previously (Yang et al., 2017). The Illumina HiSeq 3000 Sequencing System (Illumina, San Diego, CA, United States) was used for DNA sequencing with a 2 × 101-bp paired-end strategy, as described previously (Hu et al., 2018).
Terminal adaptor sequences were removed from the raw sequencing data. Subsequently, reads with more than 50% low-quality bases, or more than 50% undefined bases, were discarded. The remaining reads were mapped to the reference human genome (hg19) using the Burrows-Wheel Aligner (BWA)1 with default parameters. Picard’s Mark Duplicates tool2 was used to identify duplicate reads. Local realignment and quality recalibration were performed using The Gene Analysis Toolkit3 (GATK, version 3.4-46-gbc02625). Single-nucleotide variants and small insertions and deletions were called using the MuTect2 algorithm4 (version 1.1.4), and further filtration and validation were performed according to established criteria (Yang et al., 2017). The Contra algorithm5 (v2.0.8) was used to identify somatic copy-number alterations defined using the ratio between the adjusted depths of ctDNA and control gDNA. After automatic calling, candidate variants were manually validated using an online visualization tool6 (Integrative Genomics Viewer, IGV).
PALB2 LOH was also assessed in patients cohorts. Heterozygous germline single-nucleotide polymorphisms (SNP) across the PALB2 locus were identified similarly to a recent study (Lee et al., 2018). Alternate PALB2 allele frequencies were determined by comparing the gDNA and ctDNA sequencing results and used to infer the PALB2 LOH status.
Different mutational processes generate unique combinations of mutation types, termed “mutational signatures.” The mutational signatures were compared between 10 PALB2-associated breast tumors and 30 sporadic breast tumors (without any germline mutations or familial history of breast cancer). The somatic single-nucleotide variations were divided into six groups (T > A, T > C, T > G, C > A, C > G, C > T) and 96 subgroups according to the trinucleotide context. The mutational signatures were quantified using the deconstructSigs package (Rosenthal et al., 2016) with deconvolution methods based on the 30 mutational signatures created by COSMIC (Alexandrov et al., 2013).
All statistical analyses were conducted by using SAS 9.4. All tests of hypotheses were two-tailed and conducted at a significance level of 0.05 and at a marginal significant level of 0.15.
As shown in Table 1 and Table 2, among the 143 patients with familial breast cancers (FBCs), heterozygous germline PALB2 mutations were detected in 10 patients. c.3114-1G > A [IVS10 splicing variant] was the most frequent mutation (Table 2). c.3114-1G > A [IVS10 splicing variant] and c.1057A [3 > 2][p.K353Nfs∗3] mutations were common in familial breast cancer. All of the mono-allelic PALB2 germline mutations detected were high-risk LOF mutations. In these tables, we distinguished PALB2 “novel mutations” from the “reported mutations.”
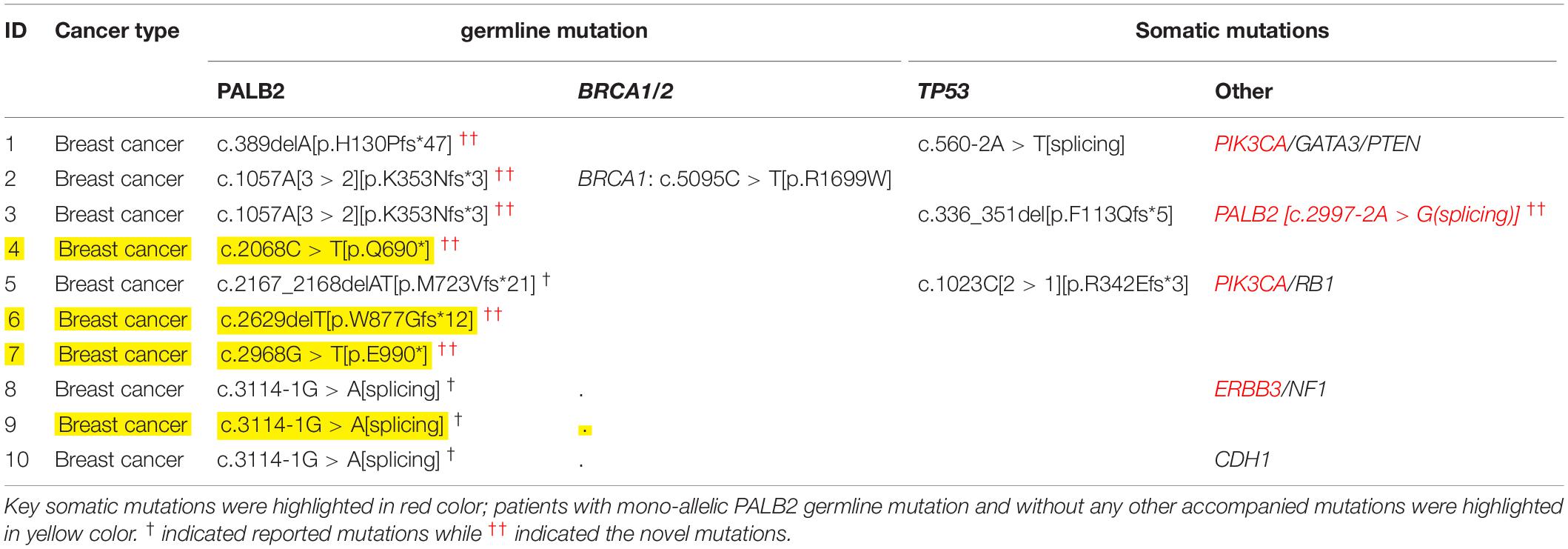
Table 1. Germline and somatic mutations in 10 patients with FBCs and heterozygous germline PALB2 mutations.
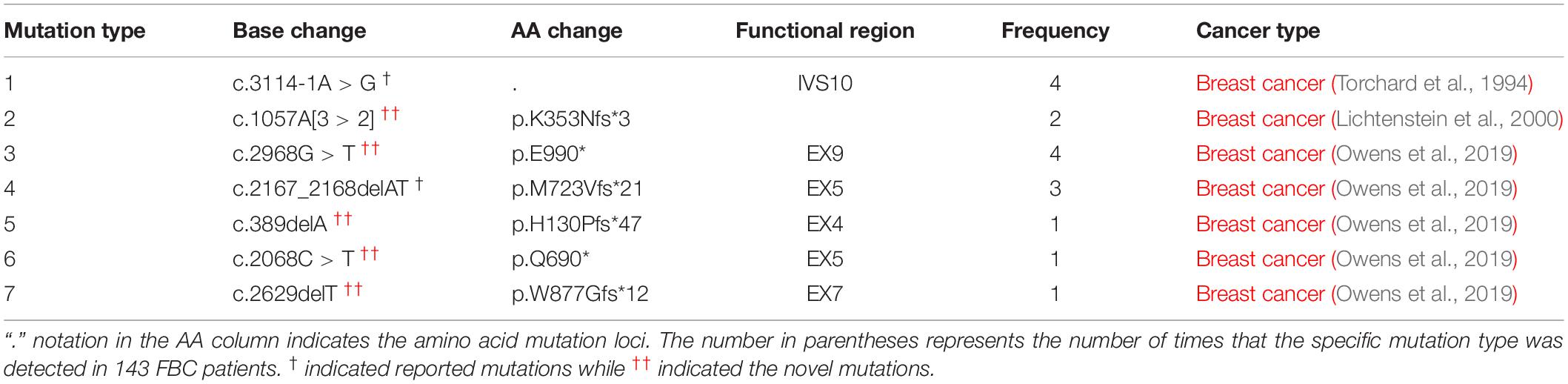
Table 2. Germline PALB2 mutations detected in gDNA of 10 patients with FBCs.
While all 18 PALB2-associated patients with familial cancers other than FBCs had additional somatic mutations, four of the 10 PALB2-associated patients with familial breast cancer had no additional somatic or germline mutations (Tables 1, 3). Compared with the other cancer types, familial breast cancer was less likely to have an additional somatic or germline mutations accompanying a germline PALB2 mutation (6/10 vs. 18/18, Fisher’s exact test, p = 0.02; Table 3).
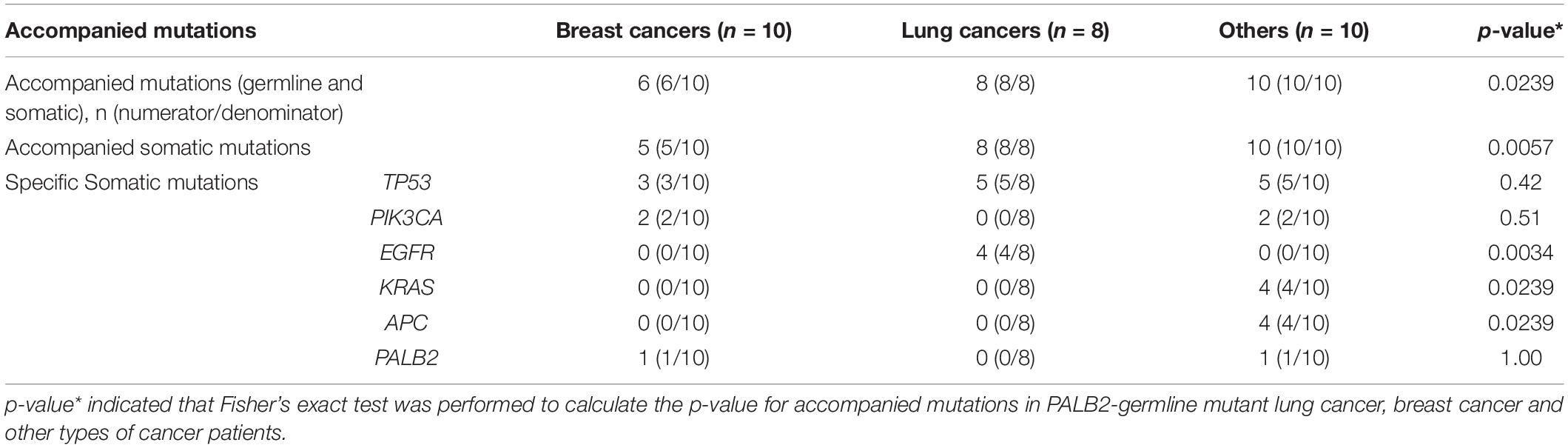
Table 3. Somatic and other germline mutations accompanied with PALB2 germline pathogenic variants.
Somatic TP53 mutations were the most common type of mutation accompanying germline PALB2 mutations. Somatic EGFR mutations were frequent in lung cancers, whereas somatic KRAS and APC mutations were frequent in colorectal cancers (Supplementary Table S1 and Table 3). In PALB2-associated breast cancers, the accompanying mutations included germline BRCA1 mutation and somatic TP53, PIK3CA, PALB2, ERBB3, and RB1 mutations (Tables 1, 3).
Among 143 FBCs, ten patients with familial breast cancer had heterozygous germline PALB2 mutations, all of which were high-risk LOF mutations (Table 1). One of the 10 PALB2-associated patients had a somatic PALB2 mutation; four others had somatic mutations in other genes, including TP53, PIK3CA, and ERBB3; one had two germline mutations (BRCA1 and PALB2); and in the remaining four patients, heterozygous germline PALB2 mutation was the only genetic event. Among 18 PALB2 tumors other than FBCs, only one had somatic PALB2 variant.
PALB2 LOH was evaluated in 28 PALB2-associated tumors. Only four out of 28 tumors had true LOH at the PALB2 allele, including 2/10 breast tumors (Supplementary Tables S2, S3). By screening ctDNA from 10 PALB2-associated HBCs and 30 sporadic ABCs, we found that the PALB2-associated tumors had a different mutational signature from the sporadic tumors (Figure 1). Mutational signature 3, which is related to defective HRR (Nik-Zainal et al., 2016), was present in 4 of PALB2-associated breast tumors, including all three PALB2-NULL BC cases, but absent among sporadic breast tumors (Supplementary Figure S1–S2). Besides the mutational signature 3, the mutational signature R1 was also common in PALB2-associated breast tumors.
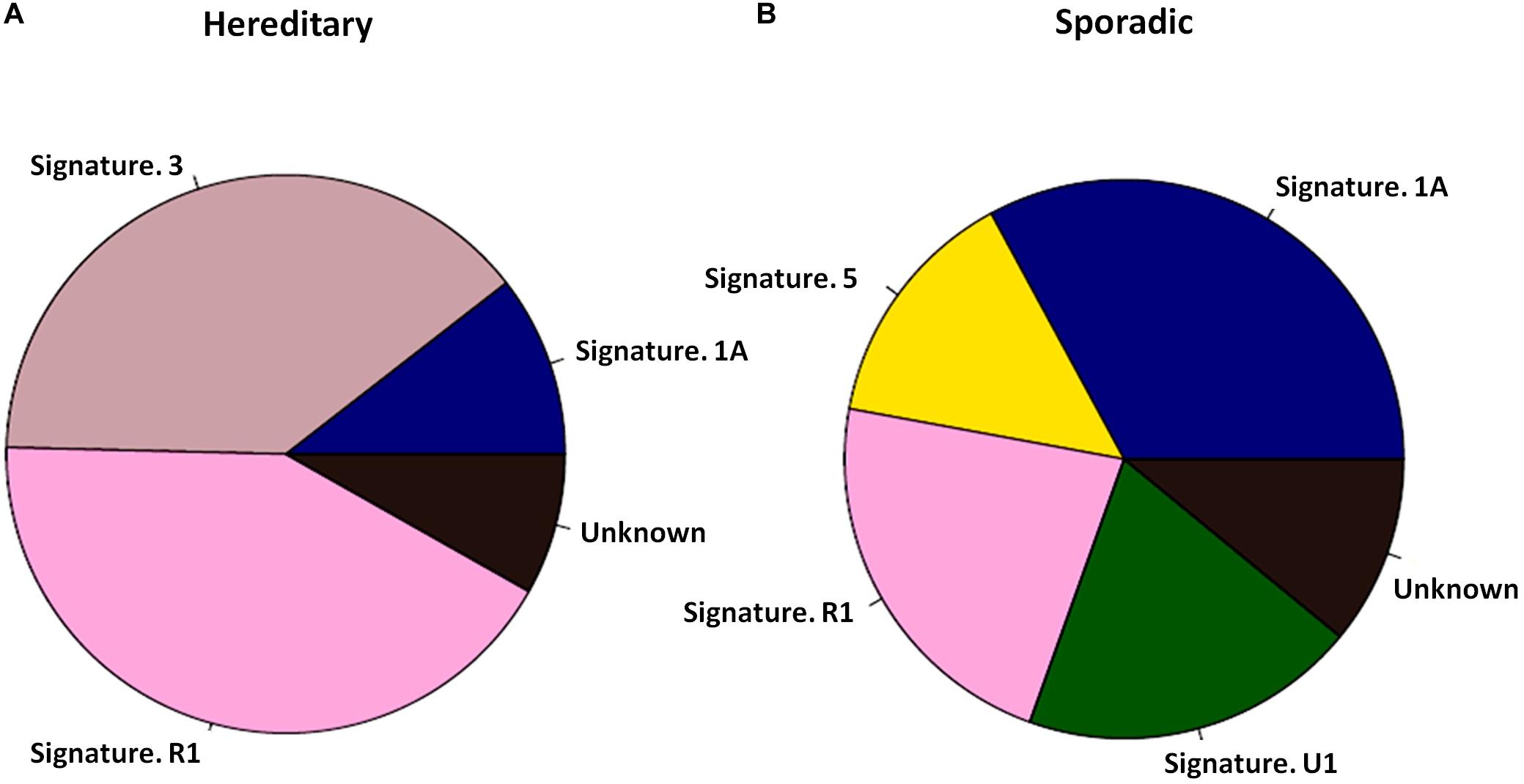
Figure 1. Mutational signatures of 10 PALB2 BC tumors and 30 sporadic BC tumors. (A) Pie diagram exhibiting the weighted contribution of each reference signature according to COSMIC exhibiting signs of mutational signature R1 and mutational signature 3 as the largest contributors in PALB2 BC tumors. (B) Pie diagram exhibiting the weighted contribution of each reference signature according to COSMIC exhibiting signs of mutational signature 1A, R1, and 5 as the largest contributors in sporadic BC tumors.
Six of 196 (6/196) sporadic ABC patients had heterozygous germline PALB2 mutations, and other three patients (3/196) had somatic PALB2 mutations (Supplementary Figure S3). The rate of heterozygous germline PALB2 mutation was much higher than the rate of somatic PALB2 mutation (6/196 vs. 3/196). All six germline PALB2 mutations were high-risk LOF mutations (Supplementary Figure S3A), including two c.3114-1G > A splicing mutations, two frameshift mutations (p.W877Gfs∗12 and p.K353Nfs∗3), and two stop-gain mutations (p.E990∗). None of the six patients with germline PALB2 mutations had any somatic PALB2 mutations. In four of those patients, the heterozygous germline PALB2 mutation was the only genetic mutation identified. In the resting two patients, one was accompanied by a germline BRCA1 mutation; the other one had somatic mutations in other genes, such as NF1 and ERBB2.
The three somatic PALB2 mutations were moderate-risk missense mutations (p.L763F, p.E53K, and p.P5S, Supplementary Figure S3B). As shown in Supplementary Table S4, ABC patients with somatic PALB2 mutations had high tumor mutation burden (TMB), while germline mutant patients exhibited low (even U) TMB. In follow-ups, we found that all three PALB2 somatic mutant patients progressed within 6 months after PALB2 somatic mutations were detected. However, among PALB2 germline mutant patients, only one progressed within 6 months, and the rest did not progress within 6 months (Supplementary Table S4).
The frequency of somatic PALB2 mutations among patients with invasive breast carcinoma in the TCGA database (TCGA-BRCA) was 1.12% (11/986; Supplementary Figure S4A). Across all cancer types, there were 209 patients in the TCGA database with a total of 158 unique somatic PALB2 mutations, most of which (134/158) were missense mutations (Supplementary Figure S4B). High-risk frameshift mutations p.N280Tfs∗8 and p.M296∗ were the most frequent PALB2 mutations in the TCGA database (appearing in five and six patients, respectively; Supplementary Figures S4B,C). Of the high-risk PALB2 mutations, only the p.Q1146∗ stop-gain mutation appeared in TCGA-BRCA patients. Among 3,090 patients with breast cancer in the cBioPortal database, only 25 (25/3,090) had somatic PALB2 mutations; 20 were missense mutations and five were high-risk mutations, including four stop-gain mutations (E12∗, E667∗, Q1146∗, and Q822∗) and one frameshift mutation (I1035Mfs∗6).
Splicing mutation c.3114-1G > A in patient ID182 was inherited from her father (male health carrier, Supplementary Figure S5), indicating that sporadic breast cancer with PALB2 mono-allelic mutation should be recognized as hereditary breast cancer. None of patient ID18’s family members had history of malignancies. But, by testing gDNA and ctDNA mutations in her family members, her father and sister were both germline PALB2 c.3114-1G > A heterozygote mutant healthy carrier Supplementary Figure S5. These results showed a parental heredity of germline PALB2 heterozygote mutation in a non-familial breast cancer patient. The risk of PALB2 c.3114-1G > A mutation carriers to have cancers was listed in Supplementary Table S5. The life-time risk of breast cancer for PALB2 normal population is 12.4% for females. But among PALB2 c.3114-1G > A mutation carriers, this risk increased to 33–58% for females. Germline PALB2 c.3114-1G > A heterozygote mutation was a pathological gene in this family. Thus, even this family has no history of evidenced malignancies, patient ID18 was definitely a hereditary breast cancer patient.
In this study, we summarized the main findings from patient samples and database (Supplementary Figure S4). All of the detected heterozygous germline PALB2 LOF mutations were high-risk LOF mutations, whereas most of the PALB2 somatic mutations were moderate-risk missense mutations. The rate of heterozygous germline PALB2 LOF mutation was much higher than the rate of somatic PALB2 mutation. Most of the heterozygous germline PALB2 LOF mutations were not accompanied by PALB2 somatic mutations or LOH.
A recent study found the risk of female breast cancer in families with PALB2 pathogenic variants to be 7.18 fold higher than controls (Yang et al., 2020). PALB2 pathogenic variants significantly increased the risk of breast cancers, ovarian cancer, pancreatic cancer and male breast cancers. Other associations were excluded (e.g., Colon, prostate) or not yet evaluated (lung). In this study, the germline PALB2 aberrations in familial lung, colon or prostate cancer might be “incidental findings.” In Supplementary Table S6, all the listed PALB2 variations were with uncertain significance. Thus in these cases, it is possible that the PALB2 variant is not “causative” of the cancer. This may also reflect in the differences in the somatic landscape among cancers.
Heterozygous germline PALB2 LOF mutations were previously shown to be associated with familial breast cancer with a prevalence of about 1% (Kurian et al., 2019). In the SEER database, PALB2 mutations are among the prevalent pathogenic variants in breast cancer (Kurian et al., 2019). In our study, germline PALB2 heterozygous mutations were detected in non-familial breast cancer patients (Supplementary Figure S5). Heterozygous germline PALB2 LOF mutation c.3114-1G > A [splicing] was reported previously in sporadic breast cancer in Chinese patients (Zhang et al., 2017). This mutation was common not only in breast cancer but also in stomach carcinoma (Supplementary Table S1), suggesting a context of future screening in families with other cancers.
Hereditary cancer is caused by genetic mutations that pass from parents to children. Sometimes, hereditary cancer might be mis-identified as sporadic cancer because of a failure to recognize a mono-allelic mutation as a hereditary driver of cancer. Actually, sporadic cancers have been reported to be influenced by germline mutations or downstream effectors of susceptible germline mutations or in pathways that involve known susceptibility genes (Jazaeri et al., 2002; Potapova et al., 2008; Lolas Hamameh et al., 2017). Mono-allelic germline PALB2 high-risk LOF mutation led to defects in homologous recombination repair (HRR) and caused breast cancer phenotype in our detected PALB2-associated patients, supporting the haploinsufficiency hypothesis for PALB2. Based this hypothesis, we recommended sporadic breast cancers with mono-allelic susceptibility gene mutations to be diagnosed as hereditary breast cancer (HBC), and also recommended their family members to take genetic screen and counseling.
Whole-exome or big-panel gene sequencing of gDNA and ctDNA can effectively detect all mutations in known cancer-susceptibility genes in patients with pathologically confirmed tumors. ctDNA testing also allows the evaluation of LOH and mutational signatures. Different combinations of somatic mutations relate to different mutational processes, termed “mutational signature.” The targeted sequencing of mutational signatures could be use to identify genetic risk factors for cancer. For example, mutational signature 3 is associated with germline BRCA1/2 mutations and HRR deficiency (Alexandrov et al., 2015; Nik-Zainal et al., 2016). Mutational signatures 2 and 13 are found to be prominent in breast cancer (Alexandrov et al., 2013); they are related to the local hypermutation cancers, suggesting potentially implicating AID/APOBEC enzymes in cancer process. In our study, mutational signature 3 was more common in PALB2-associated tumors than in other tumors (Figure 1), suggesting a defect in DNA HRR machinery (Davies et al., 2017; Polak et al., 2017) induced by LOF mutations of PALB2 (Nik-Zainal et al., 2016).
All of the heterozygous germline PALB2 mutations that we detected in advanced breast cancers and familial breast cancers in the Geneplus cohort were high-risk LOF mutations, whereas all of the PALB2 somatic mutations were moderate-risk missense mutations. The frequency of PALB2-associated mutations was much higher than that of somatic PALB2 mutations. Most PALB2-HET breast cancers were not accompanied by PALB2 somatic mutations, LOH, or hypermethylation (Lee et al., 2018), which supported the haploinsufficiency hypothesis. However, the lack of hepermethylation data was the limitation of this study.
In 2018, Lee et al. (2018) published a research to demonstrate the molecular basis of PALB2-associated breast cancer. In this article, authors found among 15 PALB2-germline mutant breast cancer patients, 10 were PALB2-NULL patients with both somatic and germline PALB2 mutations and 5 were PALB2-HET patients with only germline PALB2 mutations. Based on their findings, authors suggested that most PALB2-associated breast cancers comformed to “second hit” theory. However, when compared to our findings, we found that all the PALB2-germline mutations in our PALB2-associated breast cancers were high risk frameshift, stopgain or splicing mutations, while most of the PALB2-germline mutations in PALB2-NULL patients (7/10) were moderate missense mutation (Lee et al., 2018). Also, in 5 were PALB2-HET patients in Lee’s paper, most (4/5) PALB2-germline mutations were high-risk frameshift mutations, which consistent with our finding. So, we suggested that PALB2-associated patients with high-risk PALB2-germline mutations might not conform to the “second hit” theory.
In conclusion, while most of the PALB2 somatic mutations were moderate-risk missense mutations, the heterozygous germline PALB2 LOF mutations were high-risk LOF mutations. The heterozygous germline PALB2 LOF mutation was also much more common than the somatic PALB2 mutation in breast cancers. Most of the heterozygous germline PALB2 LOF mutations were not accompanied by PALB2 somatic mutations, LOH, or hypermethylation. Germline PALB2 mutation should be put into the context of future screening, diagnostics and even for poly (ADP-ribose) polymerase (PARP) inhibitors treatment.
The CNGBdb website is https://db.cngb.org/data and the request number is CNP0001128.
The studies involving human participants were reviewed and approved by the Ethics Committee at the Hunan Cancer Hospital and Beijing Geneplus Institute. All patients provided informed consent for genetic analysis of their genomic DNA (gDNA) and circulating tumor DNA (ctDNA). The patients/participants provided their written informed consent to participate in this study.
QO and Z-YH designed this study and interpreted the outcomes regarding surgery and survival. All authors helped perform the data analysis and prepare the manuscript and read and approved the final manuscript. Z-YH wrote the manuscript and performed the data analysis.
This work was support Development and Reform Commission of Hunan Province (CN) Hunan Key Research Project Nos. 2019SK20232 and 2019JJ50356 (Z-YH). The funders supported the study design, data collection and the publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank technique support from Geneplus-Beijing Institute. And we thank the funding support from Hunan Provincial Science and Technology Foundation (2018SK20232 and 2019JJ50356).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00829/full#supplementary-material
FIGURE S1 | Frequency plots of mutational signature of hereditary PALB2 tumors. The proportions of signature 1A (top), signature 3 (medium), and signature R1 (bottom) were 10.5%, 39.1%, and 42.2%, respectively.
FIGURE S2 | Frequency plots of mutational signature of sporadic breast tumors. The proportions of signature 1A (top), signature 5 (medium), and signature R1 (bottom) were 32.9%, 14.2%, and 22.4%, respectively.
FIGURE S3 | Germline (in the red dash rectangles) and somatic mutation profiles of advanced breast cancers with germline or somatic PALB2 mutation. BRCA1 (blue dash line) and PALB2 (red dash line) mutations were highlighted. Dark blue represents the most commonly mutated genes, and light blue represents the least commonly mutated genes. If genes were mutated at the same frequency, they are listed in alphabetic order. (A) Among 196 advanced breast cancers, two triple-negative breast cancer (TNBC) tumors (ID35, ID78), two Luminal B (ID118, ID196), one Luminal A (ID182) and one Her2-positive tumors (ID96) had mono-allelic germline PALB2 mutation. (B) One TNBC (ID163), one Luminal A (ID94) and one Luminal B (ID39) advanced breast cancers had somatic PALB2 mutations.
FIGURE S4 | Somatic PALB2 mutations detected in the TCGA database. (A) Percentages of patients affected by a total of 216 PALB2 mutations across 23 projects in the TCGA database. A total of 209 patients were affected by PALB2 mutations. Somatic PALB2 mutations were detected in 8.09% of patients with uterine corpus endometrial carcinoma and 1.12% of patients with invasive breast carcinoma (TCGS-BRCA). (B) Numbers of patients affected by each of 158 somatic PALB2 variations in the TCGA database. A total of 209 patients were affected by PALB2 mutations. There were 134 missense mutations (red dots). The frameshift mutations (blue dots) p.N280Tfs∗8 and p.M296∗ were the most frequent mutations, affecting six and five patients, respectively). (C) The risk level associated with each mutation in B. The Frameshift and stop-gain mutations were high-risk PALB2 variations (red dots). Missense mutations were moderate-risk variations (blue dots).
FIGURE S5 | Pedigree for patient ID182. This patient had a heterozygous germline PALB2 mutation but no family history of breast cancer. The patient’s father and sister were healthy carriers of the mutation, indicating parental heredity of a germline PALB2 heterozygote mutation.
TABLE S1 | Germline and somatic mutations in 18 patients with familial cancers other than FBCs and heterozygous germline PALB2 mutations.
TABLE S2, S3 | Analysis of loss of heterozygosity at the PALB2 locus in 28 mono-allelic PALB2 mutant familial cancers patients. Colon cancer (ID5), pulmonary cancer (ID10), breast cancer (ID20 and ID28) had LOH. All this patients were highlighted in red color in Supplementary Table S2.
TABLE S4 | TMB and Prognostic imformation about PALB2 mutant patients. TMB∗ (mut/MB), tumor mutation burden, was divided into three levels (U, Low and High), according to the protocol (Hu et al., 2018). TMB-U represented the variated allele (single nucleotide variants, small insertion and deletion) frequency <3%. TMB-high patients were identified with ≥11 mut/MB (upper quartile of all data). Others were identified as TMB-low patients. Progression∗∗ (Owens et al., 2019) identified patients who recieved standard treatment but progressed within 6 months after the genetic testing; otherwise, progression = 0.
TABLE S5 | Increased cancer risk in carriers of the PALB2 c.3114-1G > A mutation.
TABLE S6 | Charger summary of the detected PALB2 variants.
Alexandrov, L. B., Jones, P. H., Wedge, D. C., Sale, J. E., Campbell, P. J., Nik-Zainal, S., et al. (2015). Clock-like mutational processes in human somatic cells. Nat. Genet. 47, 1402–1407. doi: 10.1038/ng.3441
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A., Behjati, S., Biankin, A. V., et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. doi: 10.1038/nature12477
Aloraifi, F., McDevitt, T., Martiniano, R., McGreevy, J., McLaughlin, R., Egan, C. M., et al. (2015). Detection of novel germline mutations for breast cancer in non-BRCA1/2 families. FEBS J. 282, 3424–3437. doi: 10.1111/febs.13352
Alvarez, S., Diaz-Uriarte, R., Osorio, A., Barroso, A., Melchor, L., Paz, M. F., et al. (2005). A predictor based on the somatic genomic changes of the BRCA1/BRCA2 breast cancer tumors identifies the non-BRCA1/BRCA2 tumors with BRCA1 promoter hypermethylation. Clin. Cancer Res. 11, 1146–1153.
Antoniou, A. C., Casadei, S., Heikkinen, T., Barrowdale, D., Pylkas, K., and Roberts, J. (2014). Breast-cancer risk in families with mutations in PALB2I. N. Engl. J. Med. 371, 497–506. doi: 10.1056/NEJMoa1400382
Bouwman, P., Drost, R., Klijn, C., Pieterse, M., van der Gulden, H., and Song, J. Y. (2011). Loss of p53 partially rescues embryonic development of Palb2 knockout mice but does not foster haploinsufficiency of Palb2 in tumour suppression. J. Pathol. 224, 10–21. doi: 10.1002/path.2861
Collins, N., McManus, R., Wooster, R., Mangion, J., Seal, S., Lakhani, S. R., et al. (1995). Consistent loss of the wild type allele in breast cancers from a family linked to the BRCA2 gene on chromosome 13q12-13. Oncogene 10, 1673–1675.
Davies, H., Glodzik, D., Morganella, S., Yates, L. R., Staaf, J., and Zou, X. (2017). HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 23, 517–525. doi: 10.1038/nm.4292
Gilad, O., Nabet, B. Y., Ragland, R. L., Schoppy, D. W., Smith, K. D., Durham, A. C., et al. (2010). Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 70, 9693–9702. doi: 10.1158/0008-5472.CAN-10-2286
Hedenfalk, I., Ringner, M., Ben-Dor, A., Yakhini, Z., Chen, Y., Chebil, G., et al. (2003). Molecular classification of familial non-BRCA1/BRCA2 breast cancer. Proc. Natl. Acad. Sci. U.S.A. 100, 2532–2537. doi: 10.1073/pnas.0533805100
Hu, Z. Y., Xie, N., Tian, C., Yang, X., Liu, L., and Li, J. (2018). Identifying circulating tumor DNA mutation profiles in metastatic breast cancer patients with multiline resistance. EBioMedicine 32, 111–118. doi: 10.1016/j.ebiom.2018.05.015
Jazaeri, A. A., Yee, C. J., Sotiriou, C., Brantley, K. R., Boyd, J., and Liu, E. T. (2002). Gene expression profiles of BRCA1-linked, BRCA2-linked, and sporadic ovarian cancers. J. Natl. Cancer Inst. 94, 990–1000. doi: 10.1093/jnci/94.13.990
Keeney, M. G., Couch, F. J., Visscher, D. W., and Lindor, N. M. (2017). Non-BRCA familial breast cancer: review of reported pathology and molecular findings. Pathology 49, 363–370. doi: 10.1016/j.pathol.2017.03.002
Knudson, A. G. (1971). Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U.S.A. 68, 820–823. doi: 10.1073/pnas.68.4.820
Kurian, A. W., Ward, K. C., Howlader, N., Deapen, D., Hamilton, A. S., Mariotto, A., et al. (2019). Genetic testing and results in a population-based cohort of breast cancer patients and ovarian cancer patients. J. Clin. Oncol. 37, 1305–1315. doi: 10.1200/JCO.18.01854
Lee, J. E. A., Li, N., Rowley, S. M., Cheasley, D., Zethoven, M., McInerny, S., et al. (2018). Molecular analysis of PALB2-associated breast cancers. J. Pathol. 245, 53–60. doi: 10.1002/path.5055
Li, J., Li, H., Makunin, I., Thompson, B. A., Tao, K., Young, E. L., et al. (2017). Panel sequencing of 264 candidate susceptibility genes and segregation analysis in a cohort of non-BRCA1, non-BRCA2 breast cancer families. Breast Cancer Res. Treat. 166, 937–949. doi: 10.1007/s10549-017-4469-0
Lichtenstein, P., Holm, N. V., Verkasalo, P. K., Iliadou, A., Kaprio, J., Koskenvuo, M., et al. (2000). Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 343, 78–85. doi: 10.1056/NEJM200007133430201
Lolas Hamameh, S., Renbaum, P., Kamal, L., Dweik, D., Salahat, M., and Jaraysa, T. (2017). Genomic analysis of inherited breast cancer among Palestinian women: genetic heterogeneity and a founder mutation in TP53. Int. J. Cancer 141, 750–756. doi: 10.1002/ijc.30771
Martincorena, I., Raine, K. M., Gerstung, M., Dawson, K. J., Haase, K., Van Loo, P., et al. (2017). Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041e21. doi: 10.1016/j.cell.2017.09.042
Nik-Zainal, S., Davies, H., Staaf, J., Ramakrishna, M., Glodzik, D., Zou, X., et al. (2016). Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54. doi: 10.1038/nature17676
Owens, D. K., Davidson, K. W., Krist, A. H., Barry, M. J., Cabana, M., and Caughey, A. B. (2019). Medication use to reduce risk of breast cancer: US preventive services task force recommendation statement. JAMA 322, 857–867. doi: 10.1001/jama.2019.11885
Polak, P., Kim, J., Braunstein, L. Z., Karlic, R., Haradhavala, N. J., and Tiao, G. (2017). A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 49, 1476–1486. doi: 10.1038/ng.3934
Potapova, A., Hoffman, A. M., Godwin, A. K., Al-Saleem, T., and Cairns, P. (2008). Promoter hypermethylation of the PALB2 susceptibility gene in inherited and sporadic breast and ovarian cancer. Cancer Res. 68, 998–1002. doi: 10.1158/0008-5472.CAN-07-2418
Rahman, N., Seal, S., Thompson, D., Kelly, P., Renwick, A., and Elliott, A. (2007). PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 39, 165–167. doi: 10.1038/ng1959
Ripperger, T., Gadzicki, D., Meindl, A., and Schlegelberger, B. (2009). Breast cancer susceptibility: current knowledge and implications for genetic counselling. Eur. J. Hum. Genet. 17, 722–731. doi: 10.1038/ejhg.2008.212
Rosenthal, R., McGranahan, N., Herrero, J., Taylor, B. S., and Swanton, C. (2016). DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17:31. doi: 10.1186/s13059-016-0893-4
Scott, C. M., Joo, J. E., O’Callaghan, N., Buchanan, D. D., Clendenning, M., Giles, G. G., et al. (2016). Methylation of breast cancer predisposition genes in early-onset breast cancer: australian breast cancer family registry. PLoS One 11:e0165436. doi: 10.1371/journal.pone.0165436PONE-D-16-34211
Smith, M. J., Urquhart, J. E., Harkness, E. F., Miles, E. K., Bowers, N. L., Byers, H. J., et al. (2016). The contribution of whole gene deletions and large rearrangements to the mutation spectrum in inherited tumor predisposing syndromes. Hum. Mutat. 37, 250–256. doi: 10.1002/humu.22938
Tavera-Tapia, A., Perez-Cabornero, L., Macias, J. A., Ceballos, M. I., Roncador, G., de la Hoya, M., et al. (2017). Almost 2% of Spanish breast cancer families are associated to germline pathogenic mutations in the ATM gene. Breast Cancer Res. Treat. 161, 597–604. doi: 10.1007/s10549-016-4058-7
Torchard, D., Blanchet-Bardon, C., Serova, O., Langbein, L., Narod, S., Janin, N., et al. (1994). Epidermolytic palmoplantar keratoderma cosegregates with a keratin 9 mutation in a pedigree with breast and ovarian cancer. Nat. Genet. 6, 106–110. doi: 10.1038/ng0194-106
Wiltshire, T., Ducy, M., Foo, T. K., Hu, C., Lee, K. Y., Belur Nagaraj, A., et al. (2020). Functional characterization of 84 PALB2 variants of uncertain significance. Genet. Med. 22, 622–632. doi: 10.1038/s41436-019-0682-z
Xia, B., Sheng, Q., Nakanishi, K., Ohashi, A., Wu, J., and Christ, N. (2006). Control of BRCA2 cellular and clinical functions by a nuclear partner. PALB2. Mol. Cell 22, 719–729. doi: 10.1016/j.molcel.2006.05.022
Yang, X., Chu, Y., Zhang, R., Han, Y., Zhang, L., and Fu, Y. (2017). Technical validation of a next-generation sequencing assay for detecting clinically relevant levels of breast cancer-related single-nucleotide variants and copy number variants using simulated cell-Free DNA. J. Mol. Diagn. 19, 525–536. doi: 10.1016/j.jmoldx.2017.04.007
Yang, X., Leslie, G., Doroszuk, A., Schneider, S., Allen, J., and Decker, B. (2020). Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J. Clin. Oncol. 38, 674–685. doi: 10.1200/JCO.19.01907
Keywords: germline PALB2 mutation, hereditary breast cancer, loss of heterozygosity, somatic mutations, mutational signature
Citation: Hu Z-Y, Liu L, Xie N, Lu J, Liu Z, Tang Y, Wang Y, Yang J and Ouyang Q (2020) Germline PALB2 Mutations in Cancers and Its Distinction From Somatic PALB2 Mutations in Breast Cancers. Front. Genet. 11:829. doi: 10.3389/fgene.2020.00829
Received: 30 December 2019; Accepted: 09 July 2020;
Published: 27 August 2020.
Edited by:
Laura Ottini, Sapienza University of Rome, ItalyReviewed by:
Vijai Joseph, Cornell University, United StatesCopyright © 2020 Hu, Liu, Xie, Lu, Liu, Tang, Wang, Yang and Ouyang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhe-Yu Hu, amVubnlkYXZpZEAxNjMuY29t; Jianbo Yang, amlhbmJvX3lhbmdAaG90bWFpbC5jb20=; Quchang Ouyang, b3lxYzE5NjlAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.