 Junsheng Yang
Junsheng Yang Xiaotong Yang
Xiaotong Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet., 23 July 2020
Sec. Genetics of Aging
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00754
This article is part of the Research TopicThe Functional Roles of Protein Assemblies in AgingView all 4 articles
Formation of intracellular mutant Huntingtin (mHtt) aggregates is a hallmark of Huntington’s disease (HD). The mechanisms underlying mHtt aggregation, however, are still not fully understood. A few recent studies indicated mHtt undergoes phase transition, bringing new clues to understand how mHtt aggregates assemble. Here in this mini review, we will summarize these findings with a focus on the factors that affect mHtt phase transition. We will also discuss the possible pathological roles of mHtt phase separation in HD.
Huntington’s disease (HD) is a genetic neurodegenerative disease caused by expanded CAG triplet repeats in the first exon of HTT gene (MacDonald et al., 1993). A biological hallmark of HD is the formation of intracellular insoluble protein aggregates (inclusions) found in the brains of affected patients (DiFiglia et al., 1997; Becher et al., 1998; Gutekunst et al., 1999). In vivo and in vitro studies in the past two decades confirmed that these aggregates are composed of mutant Huntingtin (mHtt) alone or together with other proteins [reviewed in Arrasate and Finkbeiner (2012) and Jimenez-Sanchez et al. (2017)]. This is partially because the expanded CAG repeats in mutated HTT translate to an expanded polyglutamine (polyQ) sequence in mHtt and makes it prone to misfolding and aggregating (Scherzinger et al., 1997, 1999). Whether these aggregates found in HD patients are toxic, benign or even protective, however, has been long debated. On the one hand, the correlation between polyQ length, aggregation appearance and HD age-of-onset indicates potential toxicity of mHtt aggregates (Davies et al., 1997; Ordway et al., 1997; Becher et al., 1998; Perutz and Windle, 2001). On the other hand, the formation of mHtt aggregates has also been reported to be separable from cell death and might even be beneficial for cell survival (Saudou et al., 1998; Kim et al., 1999; Arrasate et al., 2004).
While the debate goes on, the field’s understanding of mHtt aggregation and toxicity, nonetheless, has been evolving as well. It is now known that mHtt proteins exist in the forms of monomers, soluble oligomers, and insoluble polymers both in vivo and in vitro. The formation of mHtt oligomers and polymers from monomers is polyQ length- and time-dependent, likely in a monomer→oligomer→polymer order (Poirier et al., 2002; Iuchi et al., 2003; Takahashi et al., 2008; Lajoie and Snapp, 2010; Legleiter et al., 2010; Olshina et al., 2010; Marcellin et al., 2012; Kim et al., 2016). mHtt species of similar sizes can manifest conformational polymorphism. For monomers and oligomers, mHtt species recognized by the monoclonal antibody 3B5H10 shows higher neurotoxicity than the others (Miller et al., 2011; Peters-Libeu et al., 2012). Structural biology indicates these 3B5H10-recognized mHtt species adopt a more compact conformation. A later work found that they are resistant to selective autophagy and thus have a lower degradation rate (Fu et al., 2017). Whether this is directly linked to their compact structure is not yet clear. Interestingly, the insoluble mHtt polymers/aggregates also have diverse conformations. A study using synchrotron-based infrared microspectroscopy revealed that mHtt aggregates from adult and juvenile HD patients’ brains display three conformations (non-amyloid, amyloid with β-sheet/unordered, and amyloid exclusively with β-sheet), among which the amyloid with β-sheet/unordered structures is distributed positively correlated with brain regions affected by HD (Andre et al., 2013). Another study in immortalized mouse striatal STHdhQ7/Q7 cells found that overexpressed mutant huntingtin fragments can form two types of inclusions, the tightly packed fibrillar inclusion and the loosely packed globular inclusion, based on their morphological appearance under super-resolution microscopy and fluorescence lifetime imaging microscopy (FLIM)-Förster resonance energy transfer (FRET) (Caron et al., 2014). The authors also found that these two types of inclusions have different dynamic properties, in that the fibrillar inclusion does not exchange protein with the soluble phase while the globular inclusion does. Similarly, a later study using an Htt exon1 biosensor that can distinguish protein structures based on their accessibility to biarsenical dyes revealed that mHtt exon1 product (mHttex1) forms disordered inclusions early and then convert to β-sheet-rich amyloid over time (Ramdzan et al., 2017). Taken together, accumulating evidence indicates that mHtt forms conformationally distinct monomers, polymers and inclusions (MacDonald et al., 1993); mHtt species with certain conformation(s) are more toxic than the others (DiFiglia et al., 1997); different mHtt conformations may convert from one to another (Becher et al., 1998). The mechanism(s) underlying their formation and possible conversion, however, is still largely unknown.
Very recently, several studies carried out in vitro in yeast and mammalian cells indicated that the formation of mHtt assemblies is mediated by phase separation and phase transition (Peskett et al., 2018; Posey et al., 2018; Aktar et al., 2019). These findings suggested the involvement of a new mechanism in the formation of mHtt aggregates and thus provided new clues to understand their polymorphism in conformation and toxicity. We will summarize these works and discuss their possible pathological relevance in the following sections.
The term “phase” in physics is used to describe a thermodynamic system composed of materials with uniform physical properties. The change from one phase to another is coined phase transition (e.g., a transition from liquid phase to solid phase). Liquid–liquid phase separation (LLPS) is a special kind of phase transition that refers to the separation of a solution into two distinct co-existing phases, whereas one is solute-enriched and the other is solute-absent (Alberti, 2017) (also see Figure 1). Although the idea that cytoplasm to be a mixture of liquid with suspending droplets of different chemical properties can be tracked back to as early as 1899 (Wilson, 1899), the intracellular phase separation was not experimentally demonstrated until Brangwynne et al. (2009) found that the P granule is liquid-like and formed by LLPS. The same group also revealed that nucleoli also have liquid-like behavior (Brangwynne et al., 2011). During the past decade, the intracellular phase separation of more biological macromolecules has been discovered, suggesting that phase separation might be a common mechanism for cells to regulate essential biological processes [reviewed in Alberti (2017), Gerlich (2017), Shin and Brangwynne (2017), Boeynaems et al. (2018)]. In line with this notion, aberrant phase separation is proposed to contribute to the pathology of diseases (Hyman et al., 2014; Cable et al., 2019; Verdile et al., 2019).
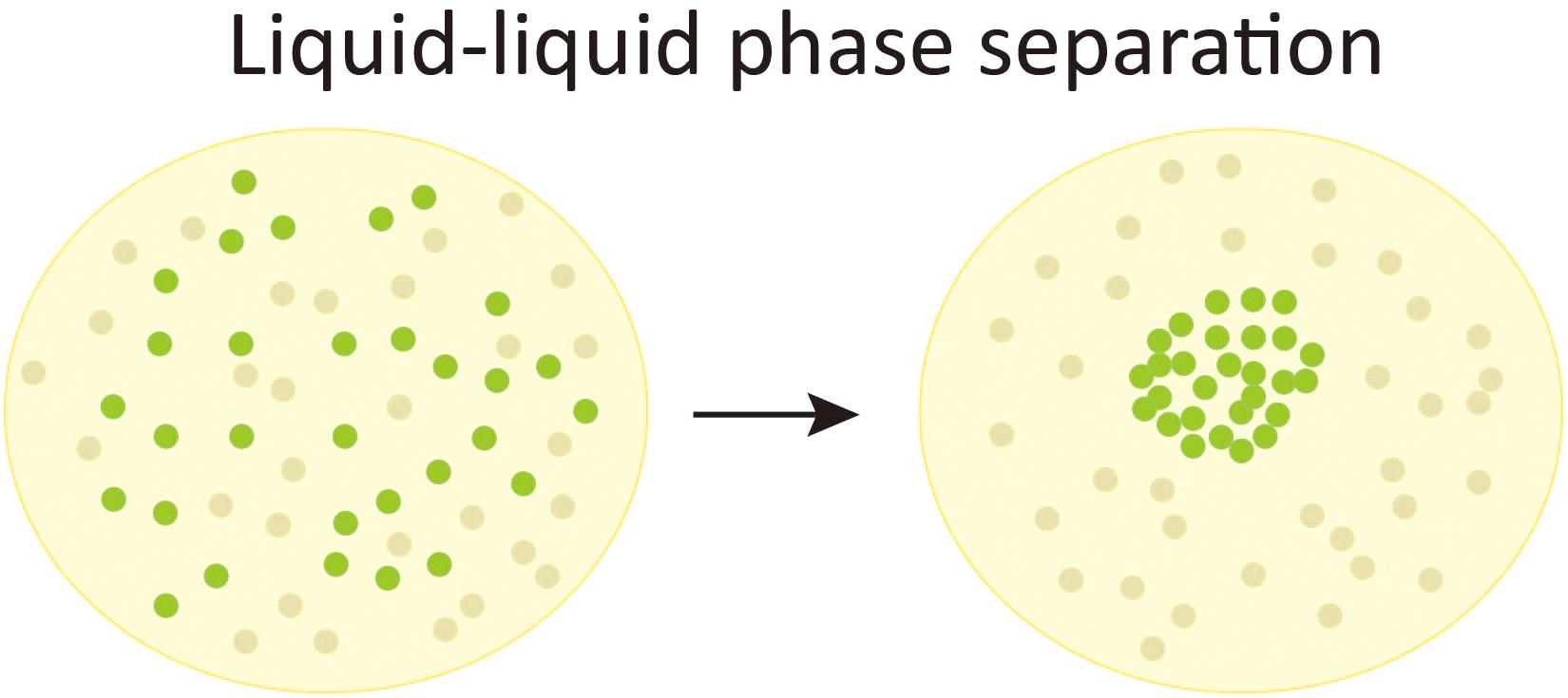
Figure 1. Liquid-liquid phase separation. A schematic chart of liquid-liquid phase separation (LLPS). In a solution composed of evenly distributed solute molecules (the green bubbles) and solvent molecules (gray bubbles), when LLPS occurs, the solute molecules condense to form a membraneless liquid-like phase and leaves a surrounding region absent of the solute molecules.
A few recent studies indicate that mHtt undergoes phase transition to form higher-ordered assemblies, both in vitro and in cells.
Using a combination of solubility analysis, right-angle static light scattering and transmission electron microscopy, Posey et al. (2018) reported that purified huntingtin N-terminal fragments (Htt-NTF) formed three phases distinguished by their saturation concentration, size, and shape. More specifically, the authors designated them as the M phase (consists of soluble monomers and oligomers), the S phase (mainly consists of bigger soluble aggregate spheres sized about 25 nm in diameter), and the F phase (mainly consists of insoluble fibrillar aggregates). Htt-NTF can separate into different phases when its concentration goes above a construct-specific saturation concentration.
In another study published in 2018, Peskett et al. (2018) used correlative light and electron microscopy (CLEM) and time-lapse fluorescence microscopy to study the aggregation of Httex1-GFP proteins. They observed that Httex1-GFP forms dim liquid-like and bright solid-like assemblies, in vitro, in yeast cells as well as in mammalian cells. More interestingly, the authors also found that the liquid-like Httex1-GFP assemblies are formed by LLPS and can convert into solid-like assemblies by phase transition.
Aktar et al. (2019) reported that in yeast cells, mHttex1-GFP form bright membrane-less spherical phase-separated inclusion bodies (IBs) with uniform GFP intensity. However, based on the timescale in FRAP experiments, the authors concluded that the nature of IBs they observed are not “fully liquid” nor solid but rather gel-like. Besides the IBs, the authors also observed clusters of smaller inclusions (which they named cluster like inclusions or CLIs) that are asymmetrical in shape and GFP intensity, in a small proportion of cells. Moreover, the authors also tracked the random movements of small mHttex1-GFP particles and propose that their collision and coalescence lead to the development of mHtt inclusions.
Taken together, the above-mentioned studies all indicated that Httex1 manifest phase transition, supported by evidence from thermo-kinetics, light scattering, quantitative fluorescent microscopy, and electron microscopy experiments. However, besides the common findings pointing to mHtt phase transition, these studies used quite different methodology and focused on different aspects of mHtt phase behavior (see Table 1 for summary). For instance, Posey et al. (2018) studied sub-micron-sized Htt species and factors that affect their thermo-kinetic properties with biochemical and biophysical approaches, while Peskett et al. (2018) and Aktar et al. (2019) observed micron-sized Htt inclusions and their liquid- or solid-like properties mainly with fluorescent microscopy assisted by quantitative analysis. Considering the diverse sizes and conformations of mHtt species, thorough works with multiple mutually supportive approaches are needed to further explore the phase properties of mHtt assemblies. Nonetheless, these latest studies have shed new light to the understanding of the polymorphism of mHtt species at different scales.

Table 1. Summary of mHtt phase behavior.
Diverse factors are known to affect mHtt aggregation; whether they can also modulate mHtt phase transition was also explored by in the studies discussed in the previous section.
Posey et al. (2018) characterized the saturation concentrations of soluble mHtt-NTF for its S and F phases and found that the sequence flanking the mHtt polyQ region affects the phase boundaries. Specifically, the 17N-terminal amino acids of mHtt (N17) lower the fibrillar-phase saturation concentration cF, indicating N17 facilitates mHtt-NTF phase separation in vitro, while the proline-rich region downstream of polyQ sequence (i.e., P-rich region) increases cF, suggesting it lowers the driving force for mHtt-NTF phase separation. The authors also studied in depth how profilin, a small actin-binding protein, may contribute to the modulation of mHtt-NTF phase separation. They observed that profilin preferably binds to the soluble monomers and oligomers of mHtt-NTF, thereby destabilize mHtt-NTF aggregates. Moreover, computer simulation suggests profilin interacts with mHtt-NTF through a combined effect of specific binding with the P-rich region and auxiliary binding with the polyQ sequence. Interestingly, mHtt-NTF with longer polyQ (40Q vs. 30Q) showed higher affinity with profilin in protein binding assays.
Peskett et al. (2018) also looked at polyQ length and the P-rich region for their roles in mHtt phase transition. In both yeast and mammalian cells, mHttex1 with different length of polyQ (25, 43, and 97) can all form liquid-like assemblies, but a polyQ expansion (43 and 97) is required for solid-like assemblies’ formation. In vitro, however, even mHttex1 with 25Q can also undergo liquid to solid transition. P-rich region assists the mHttex1 to form assemblies in yeast, whether this is also the case in mammalian cells is unclear. The authors also tested whether electrostatic interactions play a role in mHtt phase separation and found that even very high salt concentration (up to 1M) had little effect on mHtt-25QP droplet formation in vitro. In their yeast experiments, the authors also looked at the yeast prion [RNQ+] and found it not required for mHttex1 phase separation.
Aktar et al. (2019) checked phase transition of mHttex1 with different polyQ length (25Q and 72Q) as well. Consistent with previous reports (Krobitsch and Lindquist, 2000; Meriin et al., 2002) and Peskett et al.’s work, mHttex1-25Q did not form inclusions. Besides, Hsp104, a yeast disaggregase, is required for inclusion formation as reported before (Krobitsch and Lindquist, 2000; Meriin et al., 2002). Interestingly, Aktar et al. (2019) did not see mHttex1-72Q forming any liquid-like or solid-like assemblies in rnq1Δ strains.
In summary, several factors can affect mHtt phase transition. Expanded polyQ is required for mHtt phase transition in both studies with mHttex1-GFP under fluorescent microscopy. The effect of the flanking sequences, however, looks somewhat controversial. The P-rich region lowers the driving force for phase separation in vitro and facilitates profilin in suppressing mHttex1 aggregation (Posey et al., 2018), but promotes intracellular mHttex1 phase separation in yeast (Peskett et al., 2018). One possible explanation for this discrepancy is, compared with the in vitro aggregation system, the intracellular milieu contains many more mHtt-interacting proteins potentially affecting mHtt phase separation. Profilin, Hsp104, and Rnq1 are some examples, although the exact mechanisms by which they modulate mHtt phase separation still requires further studies. It’s worth mentioning that although Peskett et al. (2018) found the [RNQ+] prion is not required for mHttex1 phase separation, Aktar et al. (2019) reported yeast cells lacking the RNQ1 gene were unable to form mHtt assemblies. Because these two experiments were carried out in different yeast strains with different [RNQ+] prion-forming properties, it is thus still unclear whether the non-prion form of Rnq1 protein assists mHtt phase separation.
Besides the intrinsic and intracellular factors explored in the above-mentioned studies, there are also factors that may affect mHtt phase transition in intercellular manners. mHtt aggregates were first reported to have prion-like properties such as templated misfolding in vitro and intercellular propagation in co-cultured cells [reviewed in Pearce and Kopito (2018)]. Later studies reported that mHtt aggregates also underwent intercellular transfer in mouse brains and contribute to HD pathology phenotypes (Pecho-Vrieseling et al., 2014; Jeon et al., 2016). A more recent work using a FRET-based mHtt aggregate seeding assay indicates a strong correlation between mHtt species’ seeding activity and HD pathology progression (Ast et al., 2018). It is thus crucial to explore whether and how an mHtt aggregate “seed” obtained via intercellular transfer may affect the local mHtt phase transition. In addition, recent studies in Alzheimer’s disease (AD) cells suggest that microglia are capable of uptake tau “seeds” but may promote their intercellular propagation via incomplete degradation (Spanic et al., 2019). Whether similar mechanisms also contribute to mHtt aggregation intercellularly remain to be explored.
Although the discovery of phase transition of mHtt may bring exciting new directions in HD research, a few key questions need to be answered to understand its pathological relevance.
First, it is crucial to know whether phase transition is a universal mechanism underlying mHtt aggregation in all cells, especially in neurons. Studies so far were carried out in vitro, in yeast cells and in HEK cells. It is critical to confirm the current findings in more relevant HD cell models such as neuronal cell lines or iPSC-derived neurons (Geater et al., 2018). Cell-type-specific difference of mHtt aggregation has been reported in mice HD models (Jansen et al., 2017). Microglia from frontal cortex and striatum have much lower frequency of nuclear mHtt aggregation than neurons and other glia cells from the same brain areas, despite of similar mHtt expression levels. The still unclear mechanisms underlying the cell-type-specific mHtt aggregation might also affect mHtt phase transition.
Second, mHtt is known to form aggregates in both the cytoplasm and nucleus, which are of very different biomolecule environment. Moreover, it is proposed that cytosolic and nuclear mHtt aggregates contribute differently to toxicity (Jimenez-Sanchez et al., 2017). It is thus necessary to compare cytosolic and nuclear mHtt phase transition, if applicable, to understand their physiological consequences. For example, HD is known to manifest nucleolar dysfunction and impaired rDNA transcription (Lee et al., 2014). Very recently, Frottin et al. found that the nucleolar granular component (GC) also undergoes phase separation and can serve as a membrane-less protein quality control compartment to temporarily store denatured proteins and prevent them from irreversible aggregation (Frottin et al., 2019). But the capacity of the nucleolus to buffer proteinaceous folding stress is limited. When overloaded, the GC liquid phase will go through a transition to solid phase and subsequently lead to nucleolar dysfunction. Whether intranuclear mHtt affect GC phase separation, for instance, by disrupting the process directly or by overwhelming its buffering capacity, will be a very interesting question worth testing.
Third, according to the reports so far, the expanded polyQ is required for mHtt to form solid-like assemblies but not for some liquid like assemblies. In other words, wild-type Htt (wtHtt) can also form liquid-like assemblies. Considering most HD patients are heterozygous that expresses a mixture of wtHtt and mHtt, it is closely relevant to study possible interactions between wtHtt and mHtt in their phase transition. The interaction between wtHtt and mHtt has been reported to affect disease progression and mHtt toxicity (Ho et al., 2001; Leavitt et al., 2001; Aziz et al., 2009; Saleh et al., 2014). Htt with wide-type polyQ length (25Q) was also found in mHtt (103Q) aggregates (Duennwald et al., 2006). Whether these reported interactions involve phase transition will be another interesting question to explore.
In summary, the discovery of phase transition of mHtt revealed a new mechanism involved in mHtt aggregation. Understanding the pathological relevance of different mHtt phases (e.g., the cytotoxicity of liquid phase and solid phase) and the factors that affect their transition, such as profilin that stabilize soluble mHtt phase and destabilize mHtt aggregates, will be crucial in identifying potential targets to interfere mHtt phase transition. Antagonizing the pathological mHtt phase transitions and correcting the normal phase transition could be a new strategy in treating the disease.
It’s been almost three decades since the discovery of the causing gene of HD and the identity of aggregates found in patients. However, the mechanisms of mHtt aggregation and toxicity are still not fully understood. The discovery of the phase transition of mHtt added a new mechanism of mHtt aggregation and will help us to have a better understanding of the underlying molecular events. Considering that Htt with regular polyQ length can also go through LLPS, it is possible that de-regulated phase transition of mHtt also contribute to the pathology of the disease.
Moreover, accumulating evidence indicate that phase transition is used by the cell to regulate diverse intracellular biological processes such as autophagy (Sun et al., 2018; Fujioka et al., 2020) and protein quality control (Frottin et al., 2019; Yasuda et al., 2020), both are compromised in HD. Can phase transition be a new mechanism that mHtt affect normal cellular “daily life”? In other words, is it possible that mHtt undermine essential cellular functions via disrupting their phase transition-mediated events? Furthermore, can the phase transition of mHtt interfere with phase transition of other biological macromolecules? Some hundreds of proteins were found in mHtt aggregates (Hosp et al., 2017), could some of them be a result of mHtt-disrupted phase transition? Given the case that profilin affects the phase boundaries of mHtt, this possibility looks conceivable and is certainly worth testing, at least for mHtt-interacting proteins.
Although intracellular phase transition was confirmed not for long, it has already been reported to be involved in a number of diseases features protein aggregation, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD) (Murakami et al., 2015; Patel et al., 2015; Monahan et al., 2017), AD (Ambadipudi et al., 2017; Wegmann et al., 2018). and HD (this review). Phase transition thus may serve as a new targetable process for treating these diseases (Verdile et al., 2019). However, like the case of HD discussed in this review, this can only base on a thorough knowledge of phase separation in general and phase transition of the specific target protein.
JY and XY wrote, reviewed, and edited the article. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of Zhejiang Province of China (LY20H090019) and the Young Talent Grants from the Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals at ZJUT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to colleagues whose works are not cited due to space limitations.
Aktar, F., Burudpakdee, C., Polanco, M., Pei, S., Swayne, T. C., Lipke, P. N., et al. (2019). The huntingtin inclusion is a dynamic phase-separated compartment. Life Sci. Alliance 2:e201900489. doi: 10.26508/lsa.201900489
Alberti, S. (2017). Phase separation in biology. Curr. Biol. 27, R1097–R1102. doi: 10.1016/j.cub.2017.08.069
Ambadipudi, S., Biernat, J., Riedel, D., Mandelkow, E., and Zweckstetter, M. (2017). Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 8:275. doi: 10.1038/s41467-017-00480-0
Andre, W., Sandt, C., Dumas, P., Djian, P., and Hoffner, G. (2013). Structure of inclusions of Huntington’s disease brain revealed by synchrotron infrared microspectroscopy: polymorphism and relevance to cytotoxicity. Anal. Chem. 85, 3765–3773. doi: 10.1021/ac400038b
Arrasate, M., and Finkbeiner, S. (2012). Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11. doi: 10.1016/j.expneurol.2011.12.013
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810. doi: 10.1038/nature02998
Ast, A., Buntru, A., Schindler, F., Hasenkopf, R., Schulz, A., Brusendorf, L., et al. (2018). mHTT seeding activity: a marker of disease progression and neurotoxicity in models of Huntington’s Disease. Mol. Cell 71, 675–688e6. doi: 10.1016/j.molcel.2018.07.032
Aziz, N. A., Jurgens, C. K., Landwehrmeyer, G. B., Group, E. R. S., van Roon-Mom, W. M. C., van Ommen, G. J. B., et al. (2009). Normal and mutant HTT interact to affect clinical severity and progression in Huntington disease. Neurology 73, 1280–1285. doi: 10.1212/WNL.0b013e3181bd1121
Becher, M. W., Kotzuk, J. A., Sharp, A. H., Davies, S. W., Bates, G. P., Price, D. L., et al. (1998). Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol. Dis. 4, 387–397. doi: 10.1006/nbdi.1998.0168
Boeynaems, S., Alberti, S., Fawzi, N. L., Mittag, T., Polymenidou, M., Rousseau, F., et al. (2018). Protein phase separation: a new phase in cell biology. Trends Cell Biol. 28, 420–435. doi: 10.1016/j.tcb.2018.02.004
Brangwynne, C. P., Eckmann, C. R., Courson, D. S., Rybarska, A., Hoege, C., Gharakhani, J., et al. (2009). Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732. doi: 10.1126/science.1172046
Brangwynne, C. P., Mitchison, T. J., and Hyman, A. A. (2011). Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. U.S.A. 108, 4334–4339. doi: 10.1073/pnas.1017150108
Cable, J., Brangwynne, C., Seydoux, G., Cowburn, D., Pappu, R. V., Castaneda, C. A., et al. (2019). Phase separation in biology and disease-a symposium report. Ann. N. Y. Acad. Sci. 1452, 3–11. doi: 10.1111/nyas.14126
Caron, N. S., Hung, C. L., Atwal, R. S., and Truant, R. (2014). Live cell imaging and biophotonic methods reveal two types of mutant huntingtin inclusions. Hum. Mol. Genet. 23, 2324–2338. doi: 10.1093/hmg/ddt625
Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A., et al. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548. doi: 10.1016/s0092-8674(00)80513-9
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Duennwald, M. L., Jagadish, S., Giorgini, F., Muchowski, P. J., and Lindquist, S. (2006). A network of protein interactions determines polyglutamine toxicity. Proc. Natl. Acad. Sci. U.S.A. 103, 11051–11056. doi: 10.1073/pnas.0604548103
Frottin, F., Schueder, F., Tiwary, S., Gupta, R., Korner, R., Schlichthaerle, T., et al. (2019). The nucleolus functions as a phase-separated protein quality control compartment. Science 365, 342–347. doi: 10.1126/science.aaw9157
Fu, Y., Wu, P., Pan, Y., Sun, X., Yang, H., Difiglia, M., et al. (2017). A toxic mutant huntingtin species is resistant to selective autophagy. Nat. Chem. Biol. 13, 1152–1154. doi: 10.1038/nchembio.2461
Fujioka, Y., Alam, J. M., Noshiro, D., Mouri, K., Ando, T., Okada, Y., et al. (2020). Phase separation organizes the site of autophagosome formation. Nature 578, 301–305. doi: 10.1038/s41586-020-1977-6
Geater, C., Hernandez, S., Thompson, L., and Mattis, V. B. (2018). Cellular models: HD patient-derived pluripotent stem cells. Methods Mol. Biol. 1780, 41–73. doi: 10.1007/978-1-4939-7825-0_4
Gerlich, D. W. (2017). Cell organization by liquid phase separation. Nat. Rev. Mol. Cell Biol. 18:593. doi: 10.1038/nrm.2017.93
Gutekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S., Jones, R., et al. (1999). Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J. Neurosci. 19, 2522–2534. doi: 10.1523/jneurosci.19-07-02522.1999
Ho, L. W., Brown, R., Maxwell, M., Wyttenbach, A., and Rubinsztein, D. C. (2001). Wild type Huntingtin reduces the cellular toxicity of mutant Huntingtin in mammalian cell models of Huntington’s disease. J. Med. Genet. 38, 450–452. doi: 10.1136/jmg.38.7.450
Hosp, F., Gutierrez-Angel, S., Schaefer, M. H., Cox, J., Meissner, F., Hipp, M. S., et al. (2017). spatiotemporal proteomic profiling of Huntington’s Disease inclusions reveals widespread loss of protein function. Cell Rep. 21, 2291–2303. doi: 10.1016/j.celrep.2017.10.097
Hyman, A. A., Weber, C. A., and Julicher, F. (2014). Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 30, 39–58. doi: 10.1146/annurev-cellbio-100913-013325
Iuchi, S., Hoffner, G., Verbeke, P., Djian, P., and Green, H. (2003). Oligomeric and polymeric aggregates formed by proteins containing expanded polyglutamine. Proc. Natl. Acad. Sci. U.S.A. 100, 2409–2414. doi: 10.1073/pnas.0437660100
Jansen, A. H., van Hal, M., Op den Kelder, I. C., Meier, R. T., de Ruiter, A. A., Schut, M. H., et al. (2017). Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia 65, 50–61. doi: 10.1002/glia.23050
Jeon, I., Cicchetti, F., Cisbani, G., Lee, S., Li, E., Bae, J., et al. (2016). Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 132, 577–592. doi: 10.1007/s00401-016-1582-9
Jimenez-Sanchez, M., Licitra, F., Underwood, B. R., and Rubinsztein, D. C. (2017). Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med. 7:a024240. doi: 10.1101/cshperspect.a024240
Kim, M., Lee, H. S., LaForet, G., McIntyre, C., Martin, E. J., Chang, P., et al. (1999). Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J. Neurosci.. 19, 964–973. doi: 10.1523/jneurosci.19-03-00964.1999
Kim, Y. E., Hosp, F., Frottin, F., Ge, H., Mann, M., Hayer-Hartl, M., et al. (2016). Soluble oligomers of PolyQ-expanded huntingtin target a multiplicity of key cellular factors. Mol. Cell 63, 951–964. doi: 10.1016/j.molcel.2016.07.022
Krobitsch, S., and Lindquist, S. (2000). Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 1589–1594. doi: 10.1073/pnas.97.4.1589
Lajoie, P., and Snapp, E. L. (2010). Formation and toxicity of soluble polyglutamine oligomers in living cells. PLoS One 5:e15245. doi: 10.1371/journal.pone.0015245
Leavitt, B. R., Guttman, J. A., Hodgson, J. G., Kimel, G. H., Singaraja, R., Vogl, A. W., et al. (2001). Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am. J. Hum. Genet. 68, 313–324. doi: 10.1086/318207
Lee, J., Hwang, Y. J., Ryu, H., Kowall, N. W., and Ryu, H. (2014). Nucleolar dysfunction in Huntington’s disease. Biochim. Biophys. Acta 1842, 785–790. doi: 10.1016/j.bbadis.2013.09.017
Legleiter, J., Mitchell, E., Lotz, G. P., Sapp, E., Ng, C., DiFiglia, M., et al. (2010). Mutant huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 285, 14777–14790. doi: 10.1074/jbc.M109.093708
MacDonald, M. E., Ambrose, C. M., Duyao, M. P., Myers, R. H., Lin, C., Srinidhi, L., et al. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585
Marcellin, D., Abramowski, D., Young, D., Richter, J., Weiss, A., Marcel, A., et al. (2012). Fragments of HdhQ150 mutant huntingtin form a soluble oligomer pool that declines with aggregate deposition upon aging. PLoS One 7:e44457. doi: 10.1371/journal.pone.0044457
Meriin, A. B., Zhang, X., He, X., Newnam, G. P., Chernoff, Y. O., and Sherman, M. Y. (2002). Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 157, 997–1004. doi: 10.1083/jcb.200112104
Miller, J., Arrasate, M., Brooks, E., Libeu, C. P., Legleiter, J., Hatters, D., et al. (2011). Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Chem. Biol. 7, 925–934. doi: 10.1038/nchembio.694
Monahan, Z., Ryan, V. H., Janke, A. M., Burke, K. A., Rhoads, S. N., Zerze, G. H., et al. (2017). Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 36, 2951–2967. doi: 10.15252/embj.201696394
Murakami, T., Qamar, S., Lin, J. Q., Schierle, G. S., Rees, E., Miyashita, A., et al. (2015). ALS/FTD mutation-induced Phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88, 678–690. doi: 10.1016/j.neuron.2015.10.030
Olshina, M. A., Angley, L. M., Ramdzan, Y. M., Tang, J., Bailey, M. F., Hill, A. F., et al. (2010). Tracking mutant huntingtin aggregation kinetics in cells reveals three major populations that include an invariant oligomer pool. J. Biol. Chem. 285, 21807–21816. doi: 10.1074/jbc.M109.084434
Ordway, J. M., Tallaksen-Greene, S., Gutekunst, C. A., Bernstein, E. M., Cearley, J. A., Wiener, H. W., et al. (1997). Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell 91, 753–763. doi: 10.1016/s0092-8674(00)80464-x
Patel, A., Lee, H. O., Jawerth, L., Maharana, S., Jahnel, M., Hein, M. Y., et al. (2015). A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077. doi: 10.1016/j.cell.2015.07.047
Pearce, M. P. M., and Kopito, R. R. (2018). Prion-like characteristics of polyglutamine-containing proteins. Cold Spring Harb. Perspect. Med. 8:a024257. doi: 10.1101/cshperspect.a024257
Pecho-Vrieseling, E., Rieker, C., Fuchs, S., Bleckmann, D., Esposito, M. S., Botta, P., et al. (2014). Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat. Neurosci. 17, 1064–1072. doi: 10.1038/nn.3761
Perutz, M. F., and Windle, A. H. (2001). Cause of neural death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature 412, 143–144. doi: 10.1038/35084141
Peskett, T. R., Rau, F., O’Driscoll, J., Patani, R., Lowe, A. R., and Saibil, H. R. (2018). A liquid to solid phase transition underlying pathological huntingtin Exon1 aggregation. Mol. Cell 70, 588–601e6. doi: 10.1016/j.molcel.2018.04.007
Peters-Libeu, C., Miller, J., Rutenber, E., Newhouse, Y., Krishnan, P., Cheung, K., et al. (2012). Disease-associated polyglutamine stretches in monomeric huntingtin adopt a compact structure. J. Mol. Biol. 421, 587–600. doi: 10.1016/j.jmb.2012.01.034
Poirier, M. A., Li, H., Macosko, J., Cai, S., Amzel, M., and Ross, C. A. (2002). Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 277, 41032–41037. doi: 10.1074/jbc.M205809200
Posey, A. E., Ruff, K. M., Harmon, T. S., Crick, S. L., Li, A., Diamond, M. I., et al. (2018). Profilin reduces aggregation and phase separation of huntingtin N-terminal fragments by preferentially binding to soluble monomers and oligomers. J. Biol. Chem. 293, 3734–3746. doi: 10.1074/jbc.RA117.000357
Ramdzan, Y. M., Trubetskov, M. M., Ormsby, A. R., Newcombe, E. A., Sui, X., Tobin, M. J., et al. (2017). Huntingtin inclusions trigger cellular quiescence, deactivate apoptosis, and lead to delayed necrosis. Cell Rep. 19, 919–927. doi: 10.1016/j.celrep.2017.04.029
Saleh, A. A., Bhadra, A. K., and Roy, I. (2014). Cytotoxicity of mutant huntingtin fragment in yeast can be modulated by the expression level of wild type huntingtin fragment. ACS Chem. Neurosci. 5, 205–215. doi: 10.1021/cn400171d
Saudou, F., Finkbeiner, S., Devys, D., and Greenberg, M. E. (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66. doi: 10.1016/s0092-8674(00)81782-1
Scherzinger, E., Lurz, R., Turmaine, M., Mangiarini, L., Hollenbach, B., Hasenbank, R., et al. (1997). Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558. doi: 10.1016/s0092-8674(00)80514-0
Scherzinger, E., Sittler, A., Schweiger, K., Heiser, V., Lurz, R., Hasenbank, R., et al. (1999). Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc. Natl. Acad. Sci. U.S.A. 96, 4604–4609. doi: 10.1073/pnas.96.8.4604
Shin, Y., and Brangwynne, C. P. (2017). Liquid phase condensation in cell physiology and disease. Science 357:eaaf4382. doi: 10.1126/science.aaf4382
Spanic, E., Langer Horvat, L., Hof, P. R., and Simic, G. (2019). Role of microglial cells in alzheimer’s disease tau propagation. Front. Aging Neurosci. 11:271. doi: 10.3389/fnagi.2019.00271
Sun, D., Wu, R., Zheng, J., Li, P., and Yu, L. (2018). Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 28, 405–415. doi: 10.1038/s41422-018-0017-7
Takahashi, T., Kikuchi, S., Katada, S., Nagai, Y., Nishizawa, M., and Onodera, O. (2008). Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 17, 345–356. doi: 10.1093/hmg/ddm311
Verdile, V., De Paola, E., and Paronetto, M. P. (2019). Aberrant phase transitions: side effects and novel therapeutic strategies in human Disease. Front. Genet. 10:173. doi: 10.3389/fgene.2019.00173
Wegmann, S., Eftekharzadeh, B., Tepper, K., Zoltowska, K. M., Bennett, R. E., Dujardin, S., et al. (2018). Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J 37:e98049. doi: 10.15252/embj.201798049
Wilson, E. B. (1899). The structure of protoplasm. Science 10, 33–45. doi: 10.1126/science.10.237.33
Keywords: huntingtin, phase separation, phase transition, aggregates, Huntington’s disease
Citation: Yang J and Yang X (2020) Phase Transition of Huntingtin: Factors and Pathological Relevance. Front. Genet. 11:754. doi: 10.3389/fgene.2020.00754
Received: 21 March 2020; Accepted: 24 June 2020;
Published: 23 July 2020.
Edited by:
John Tower, University of Southern California, United StatesReviewed by:
Srinivas Ayyadevara, Central Arkansas Veterans Healthcare System Eugene J. Towbin Healthcare Center, United StatesCopyright © 2020 Yang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junsheng Yang, anVuc2hlbmd5QHpqdXQuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.