 Yun Tuo
Yun Tuo Wuying Chu3
Wuying Chu3 Jianshe Zhang
Jianshe Zhang Lingsheng Bao
Lingsheng Bao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 24 July 2020
Sec. Evolutionary and Population Genetics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00714
Spinibarbus caldwelli is an omnivorous cyprinid fish that is distributed widely in China. To investigate the adaptive evolution of S. caldwelli, the muscle transcriptome was sequenced by Illumina HiSeq 4000 platform. A total of 80,447,367 reads were generated by next-generation sequencing. Also, 211,386 unigenes were obtained by de novo assembly. Additionally, we calculated that the divergence time between S. caldwelli and Sinocyclocheilus grahami is 23.14 million years ago (Mya). And both of them diverged from Ctenopharyngodon idellus 46.95 Mya. Furthermore, 38 positive genes were identified by calculating Ka/Ks ratios from 9225 orthologs. Among them, several immune-related genes were identified as positively selected, such as POLR3B, PIK3C3, TOPORS, FASTKD3, CYPLP1A1, and UACA. Our results throw light on the nature of the natural selection of S. caldwelli and contribute to future immunological and transcriptome studies.
Since the discovery of nucleotide sequence variations (Aquadro and Greenberg, 1983), the question of how adaptive natural selection affects the genome, directly or indirectly, has received wide attention. One of the most significant effects of adaptive natural selection is the impact at the polymorphic level and recombination rate (Corbettdetig et al., 2015). As reported previously, levels of polymorphism and recombination decrease proportionally with respect to increase of natural selection forces (Kaplan et al., 1989; Barton, 1998). Furthermore, there might be different kinds of mutations, as single nucleotide substitution was not the only kind found associated with adaptive selection, such as the number of gene copies (Schrider et al., 2013), the infix of transposable elements (Gonzalez et al., 2008), and large scale inversions (Stefansson et al., 2005; Kirkpatrick and Kern, 2012).
After decades of research, natural selection is an adaptive response to long-term environmental changes (Messer and Petrov, 2013). Fortunately, taking advantage of high-throughput sequencing technology, positively selected genes can be quickly identified from abundant homologous genes. Next-generation sequencing (RNA-seq) has been developed from high-throughput technology, which can supply genetic data without background information of a species. Even if there is no reference genome, the high-quality sequences can still be obtained by de novo assembly. For example, by comparing transcriptome data, Ma et al. (2016) have analyzed the adaptive evolution of six catfish species from an altitude gradient. Schartl et al. (2013) have studied the adaptability of the platyfish through genome comparison. Both studies obtained large numbers of gene sequences by high-throughput sequencing.
The current perspective is that positive gene selection is usually associated with immunity and reproduction (Jansa et al., 2003). For example, by transcriptome sequencing, several immune genes show evidence for natural selection in Parachromis managuensis (Zhong et al., 2018). Global analysis of transcriptome sequences in Aldrovandia affinis, a deep-sea fish, has identified a set of genes related to cytoskeletal structures, which means that DNA repair and treatment of genetic information can contribute to adaptation in the deep-sea environment (Lan et al., 2018). Transcriptome analysis in Danio albolineatus and Danio choprae has highlighted the accelerated evolution of immune genes, such as MPG, GTF2E1, REL, and STAT6 in D. choprae and MYCN, ADORA2A, and CYP17A1 in D. albolineatus (Bao and Xia, 2017). These studies stressed that these naturally selected genes were usually related to immunity and resistance in fish. Searching for typical genome changes among species will facilitate the comprehension of biological mechanisms and the genetic basis of adaptation.
As a cultured fish, S. caldwelli has brilliant economic prospects, which are attributed to its fast growth and high environmental adaptability. To understand the biogeographical process of S. caldwelli, the CYTB gene was first employed to identify the biogeographic affinity of S. caldwelli from different river basins in China (Tang et al., 2003). Then, the molecular and morphological evidence was used to show that S. caldwelli is a valid species (Tang et al., 2005). Genetic polymorphisms have been studied in Spinibarbus hollandi, Spinibarbus sinensis, and Spinibarbus denticulatus by using Random Amplification of Polymorphic DNA (Zhu et al., 2006). Furthermore, Wang et al. (2013) have analyzed the molecular variance and phylogenetic relationships in 61 specimens of S. caldwelli based on mitochondrial CYTB (Huang et al., 2008). However, studies on the characteristics of adaptive changes at genome level in S. caldwelli are rare.
In this study, to find the genes associated with resistance and adaptation, a transcriptome sequencing approach was applied to S. caldwelli. The Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Swiss-Prot databases were employed for function annotation and phylogenetic analysis provided evidence for natural selection. Orthologs were obtained by comparing the genomic data of S. caldwelli and six other species. Then, genes under positive selection were identified from the numerous orthologous genes using a branch-site model. The results offered new evidence for positive selection and environmental adaptation in S. caldwelli at the genomic level.
The S. caldwelli used for experiments were captured from the Jinjiang River and collected from a local market in Yichun, Jiangxi Province, China. Dorsal muscles of fish were sampled from three individual fishes. The tissue samples were quickly frozen by liquid nitrogen and stored at −80°C. All experiments were carried out following the Animal Ethics Committee of Hunan Agricultural University.
Total RNA was extracted following the instruction of RNAiso Plus (Takara Bio Inc., Shiga, Japan). RNA purity was measured using OD260/280 values using Nanodrop 2000 (Thermo Fisher Scientific Inc., Waltham, MA, United States). The integrity of RNA was assessed using an Agilent 2100 (Agilent Technologies, Inc., Santa Clara, CA, United States). A cDNA library was generated using NEBnext Ultra RNA library prep kit (New England Biolabs, Inc., Ipswich, MA, United States). mRNA transcriptions were enriched by Magnetic Beads Oligo (dT) and sectioned using fragmentation buffer (Life Technologies, Inc., Carlsbad, CA, United States). The first cDNA strand was synthesized using random hexamer primers, and second chain synthesis was performed using DNA polymerase I and RNase H (Life Technologies, Inc.). Subsequently, the outcomes were purified by AMPure XP system (Beckham Coulter, Inc., Brea, CA, United States) and amplified by PCR to build a sequence library. At last, a Bioanalyzer 2100 system (Agilent Technologies, Inc.) was used to assess the quality of the samples and dilute them to a 1.5 μg/mL concentration. The cDNA library was sequenced on a Hiseq 4000 platform (Illumina Inc., San Diego, CA, United States) by the paired-end reads method.
The raw reads were assessed using CASAVA (v1.8, Illumina Inc.) and filtered to produce clean reads. First, the adaptors were removed. Second, the reads with an N ratio of more than 10% were removed. Finally, low-quality reads (Qphred value of <20 and >50% bases) were trimmed, with a Qphred cut-off value of −10 log10(e). Short reads obtained by sequencing were assembled using Trinity software (Grabherr et al., 2011) and the unigenes used for subsequent analysis. TransDecoder was used to identify candidate coding regions based on the following criteria: (1) An ORF that meets the minimum limit length (200 bp) can be found in the transcript sequence. (2) The logarithm likelihood score was greater than 0. (3) The logarithm likelihood score of the first open reading frame (ORF) is the maximum compared with the other 5. (4) If the candidate ORF was completely involved in other ORFs, we only report the longest ORF.
The unigenes and contigs were annotated in several public databases, including Swiss-Prot, KEGG, and GO. Swiss-Prot and KEGG annotation was performed using BLAST 2.2.28 + software (Camacho et al., 2009) with E-values of le-5 and le-10, respectively. GO annotation was performed using the BlAST2GO v2.5 software and the E-value threshold at le-6 (Conesa and Gotz, 2008).
The coding DNA sequence (CDS) of the unigenes was picked out using TransDecoder software. The unigenes noted by Swiss-Prot were translated after CDS extraction. Then, the obtained ORFs were used to search for orthologous sequences.
After filtering out alternatively spliced transcripts of each gene, only the longest transcripts were retained. The orthologous gene families were identified using the OrthoMCL (v2.0.9) pipeline (Conesa and Gotz, 2008) between S. caldwelli and six other species (Astyanax mexicanus, Danio rerio, Ctenopharyngodon idellus, Latimeria chalumnae, Xiphophorus maculatus, and Sinocyclocheilus grahami). Firstly, we screened the longest transcripts of each gene and then performed BLASTp alignment of all protein sequences of the selected species (E-value ≤ 1e−5). Lastly, the homologous gene families were obtained using the Markov clustering (MCL) algorithm.
After gene family clustering, single-copy genes were used for phylogenetic analysis. Firstly, sequence matrices were initially aligned with MAFFT 7.0 (Katoh and Standley, 2013), then the amino acid sequence was converted to CDS and refined using Gblocks (Gutiérrez-Larruscain et al., 2018). Finally, the GTRGAMMA model was employed to analyze the data with raxml method. Bootstrap was set to 100 and L. chalumnae was used as the outgroup specie.
We estimated divergence time with the Bayesian method MCMCTree in PAML v4.9e (Yang, 2007). Fourfold degenerate positions were extracted by CDS sequence alignment of 4126 single-copy orthologous genes that are shared by S. caldwelli and other fishes. The parameter of MCMCTREE was set as follows: clock = 2, RootAge ≤ 434.42, model = 7, BDparas = 110, kappa_gamma = 62, alpha_gamma = 11, rgene_gamma = 23.18, sigma2_gamma = 11.3. And the divergence times of C. idellus [36.15–64.69 million years ago (Mya)] (Li et al., 2013; Wang et al., 2013), X. maculatus (210.16–256.54 Mya) (Betancur-R et al., 2013; Chen et al., 2013), and L. chalumnae (424.83–445.83 Mya) (Nakatani et al., 2011; Betancur-R et al., 2015) were added as fossil calibrations.
According to the high similarity of homologous genes, a BLASTp search was employed to align the protein sequences of these seven species (E-value ≤ 1e−5) to identify orthologous sequences.
The detection of natural selection was performed using CODEML software in the PAML package using the branch-site model (Zhang et al., 2005). A Chi-square test was employed to verify the significance of obtained positively selected genes. Genes under positive selection were assigned to Swiss-Prot and KEGG categories to identify their related functions.
In this study, after removing the low-quality reads, 80,447,367 reads with 22.41 Gb of clean bases were ultimately obtained. By de novo assembly of these clean reads, 387,437 contigs, with N50 of 1077 bp and the average length of 620 bp, were generated. Furthermore, 211,386 unigenes with N50 of 706 bp and the average length of 548 bp have been assembled. contig and unigene data are outlined in Supplementary Table S1 and their length distribution is shown in Figure 1. All raw data can be downloaded in the NCBI Sequence Read Archive database (Accession No. PRJNA563944).
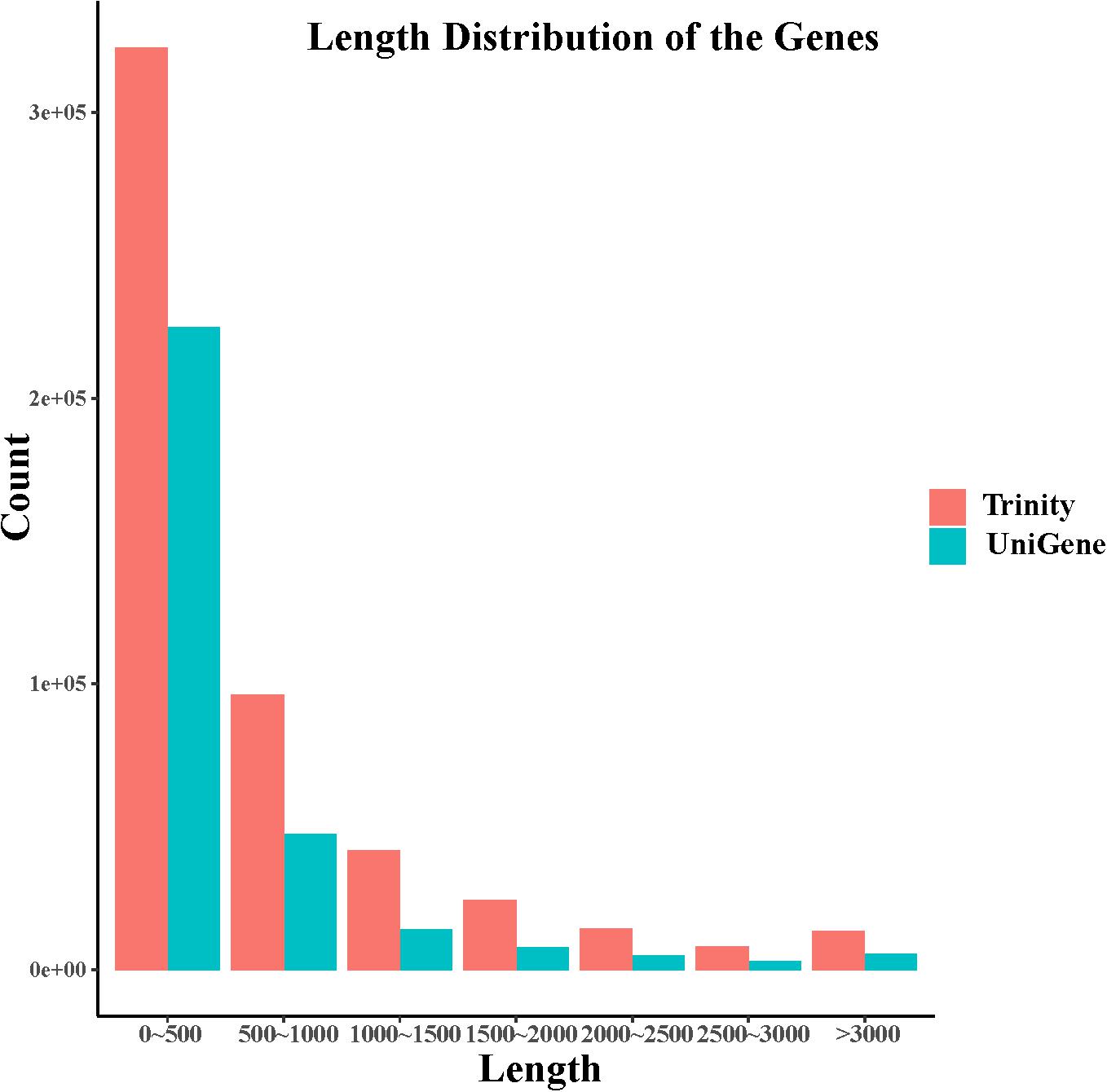
Figure 1. Length distribution of contigs and unigenes derived from S. caldwelli.
A total of 63,755 ORFs were identified by TransDecoder (Supplementary Table S2). Also, 61,337 unigenes (30.16% of total unigenes) from S. caldwelli were annotated in three public databases (Swiss-Prot, GO, and KEGG; Table 1).
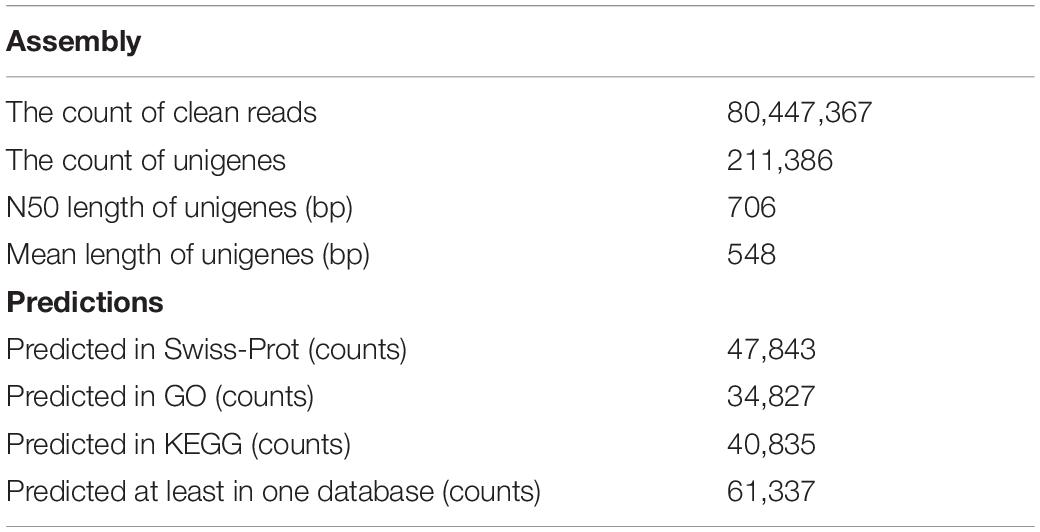
Table 1. Summary of transcriptome assembly from S. Caldwelli.
The GO database illustrated the experimental results regarding molecular functions, biological processes, and cellular components (Figure 2 and Supplementary Table S3). The top two GO terms in the molecular function hierarchy were catalytic activity and binding, which included 8338 and 6403 unigenes, respectively. Among the biological process hierarchy, cellular, single-organism, and metabolic processes represented the majority of the unigenes (4169, 2788, and 2602, respectively). Besides, in the cellular component hierarchy, the GO terms “cell,” “cell part,” and “organelle” contained the most abundant unigenes (2638, 2638, and 1998, respectively). Additionally, more genes can be found per orthogroup in S. caldwelli than in others, and this might be an overestimation issue by Trinity, which is often reluctant to combine multiple isoforms (Cabau et al., 2017).
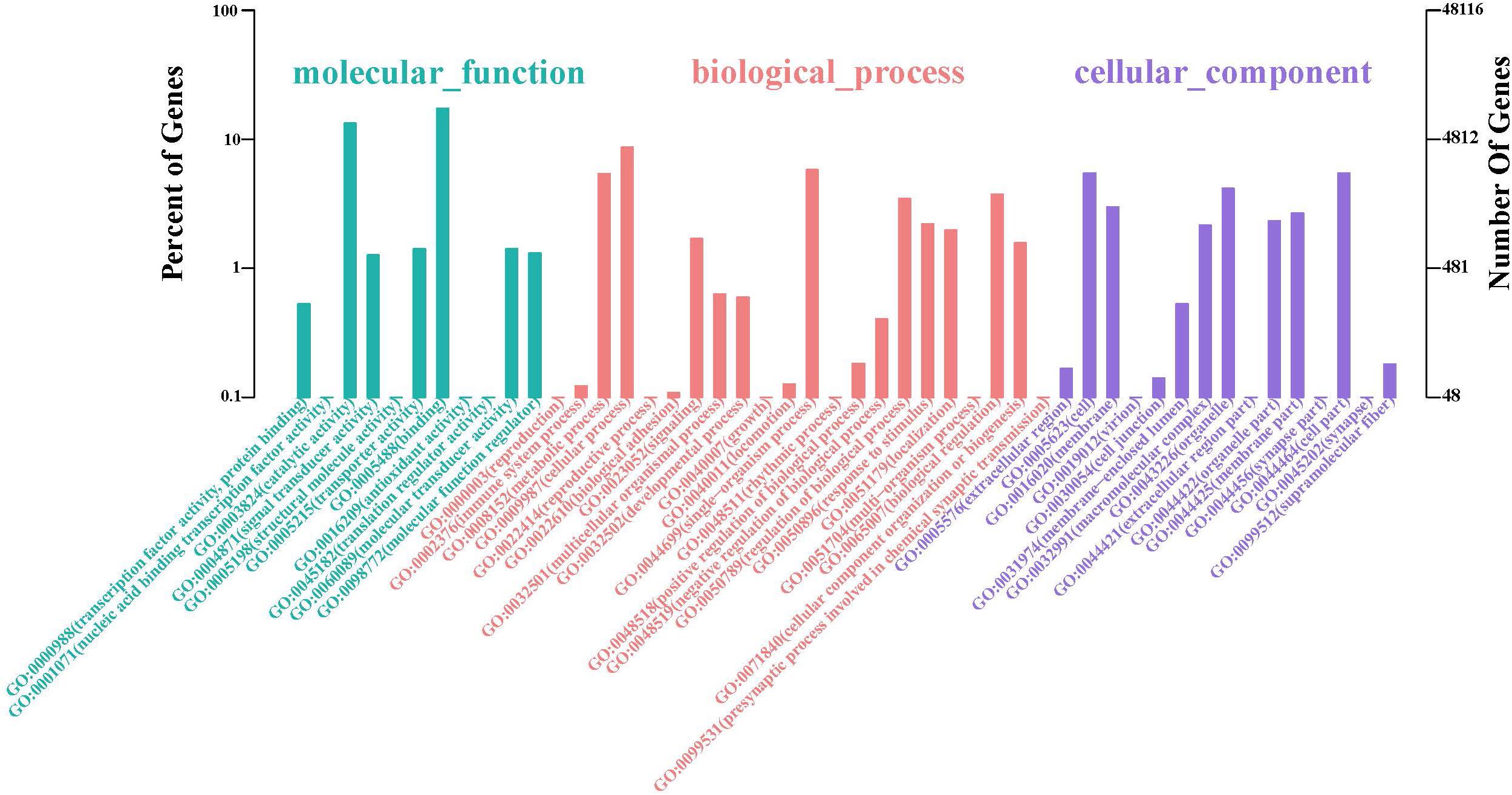
Figure 2. Annotation of unigenes of S. caldwelli from the GO database.
In the obtained S. caldwelli transcriptome, the KEGG pathway database annotated and sorted the unigene functions into five categories, including cellular process pathways, environmental information processing, genetic information processing, metabolism, and organismal systems (9193, 8785, 4544, 7098, and 3162 unigenes, respectively; Figure 3). In the KEGG sub-classification, the categories of signal transduction; transport and catabolism; cellular community-eukaryotes; cell growth and death; and folding, sorting, and degradation (6866, 3002, 2823, 2405, and 1816 unigenes, respectively) were the five major pathways.
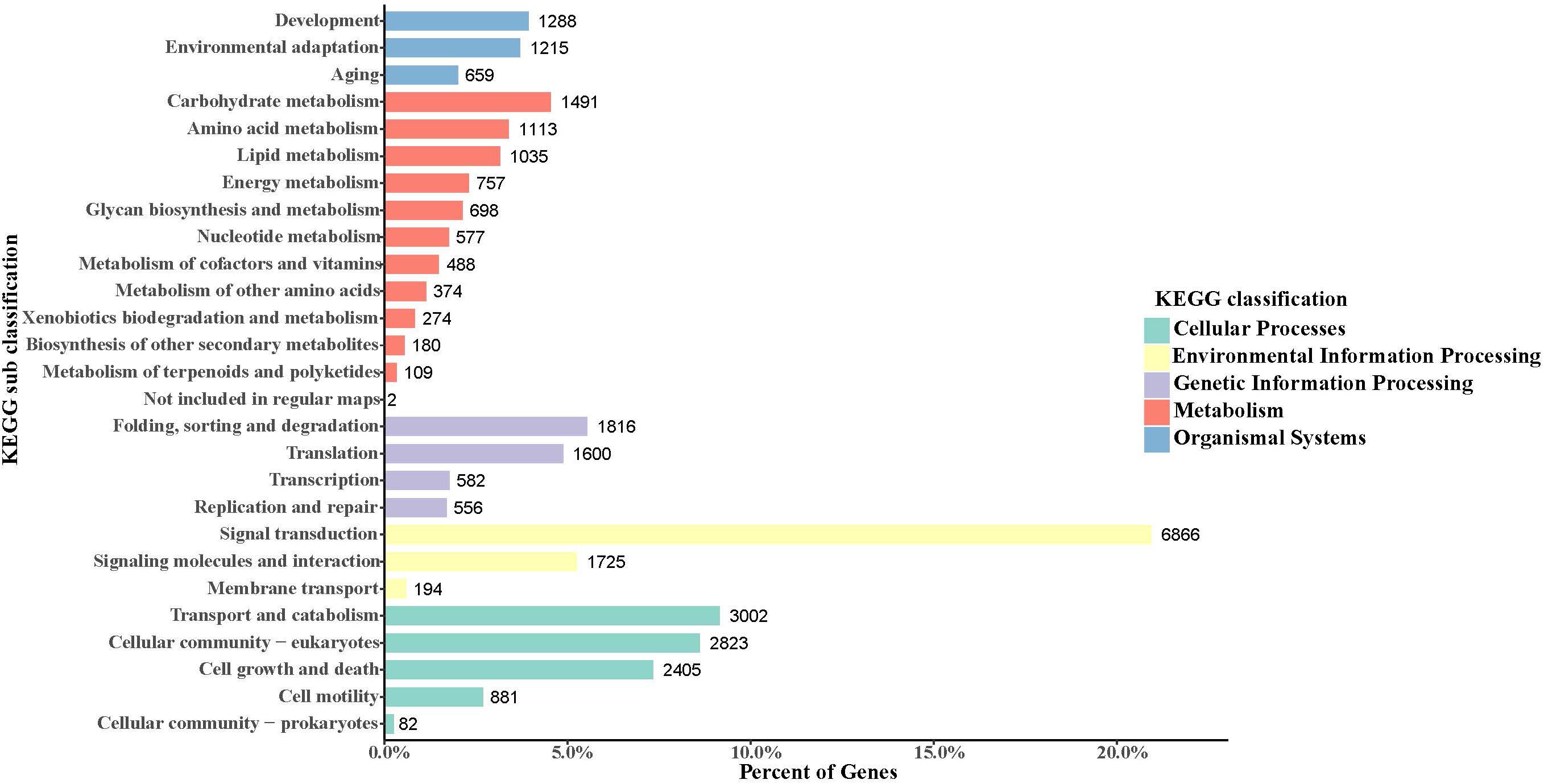
Figure 3. Annotation of unigenes of S. caldwelli in the KEGG pathway database. From top to bottom by color: organismal systems, blue; metabolism, red; genetic information processing, purple; environmental information processing, yellow; cellular processes, green.
Through analyses of gene families between S. caldwelli and six species, 16,132 gene families containing 37,187 genes were clustered. Among them, 2436 gene families were unique in S. caldwelli (Figure 4A). Furthermore, S. caldwelli and S. grahami had the largest number of shared gene families (12,427 gene families) among these species of fish (Figure 4B).
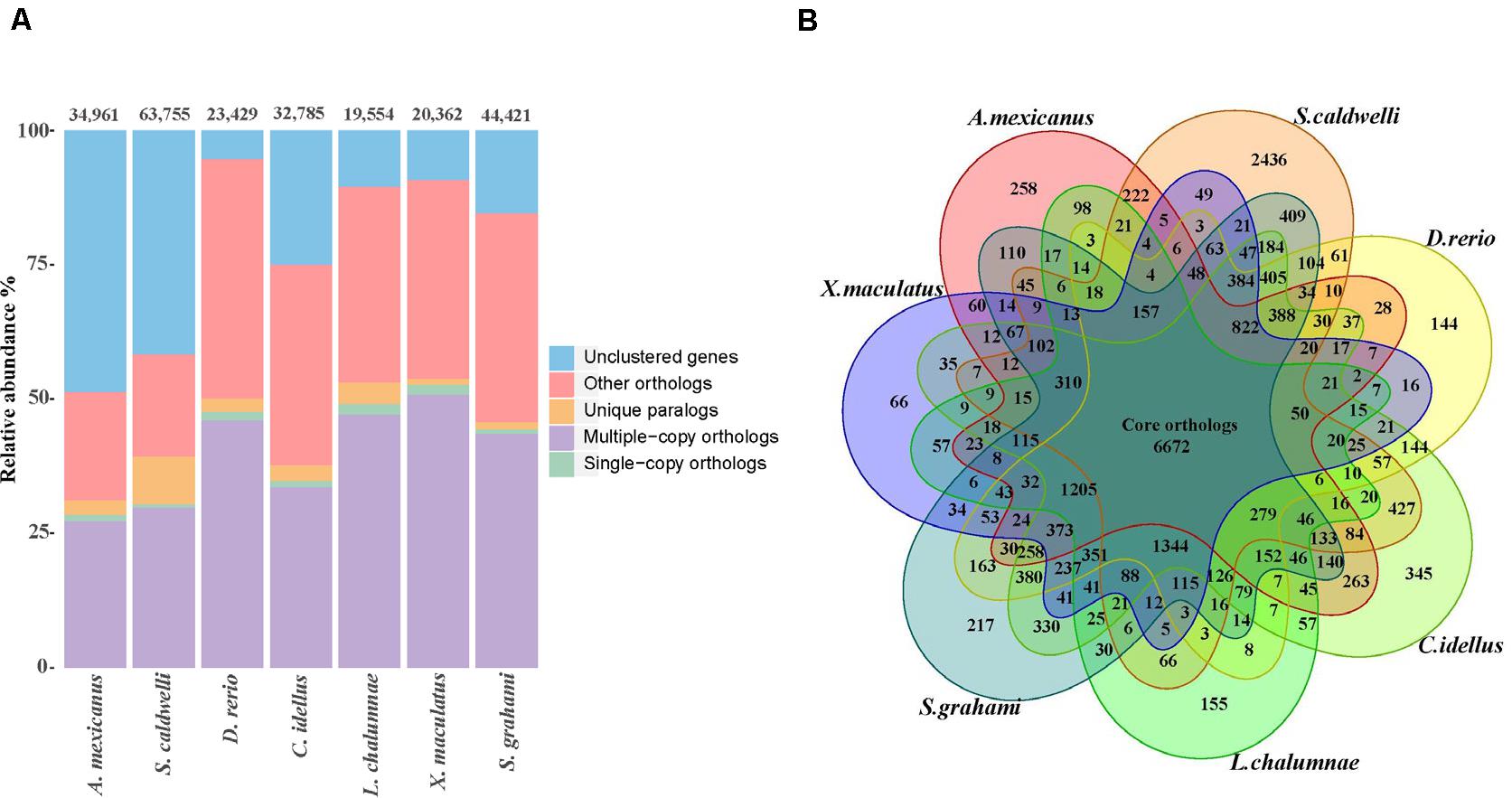
Figure 4. (A) The number of genes in the seven fish species, displaying the great quantity of S. caldwelli genes in comparison withS. grahami and others. The count of multiple copy orthologs from S. caldwelli was high. (B) Comparison of orthologous genes of S. caldwelli and six fish species by Venn diagram.
According to the molecular clock theory, the codons are replaced with each other in an almost constant ratio over time. But the encoded amino acid does not change with the substitution of the third base of the fourfold degenerate synonymous sites (neutral evolution) (Kimura, 1983). Therefore, the fourfold degenerate synonymous sites in single-copy gene families were usually used to estimate the divergence time between species. In this study, we calculated that the divergence time between S. caldwelli and S. grahami is 23.14 Mya. And both of them diverged from C. idellus 46.95 Mya (Figure 5).
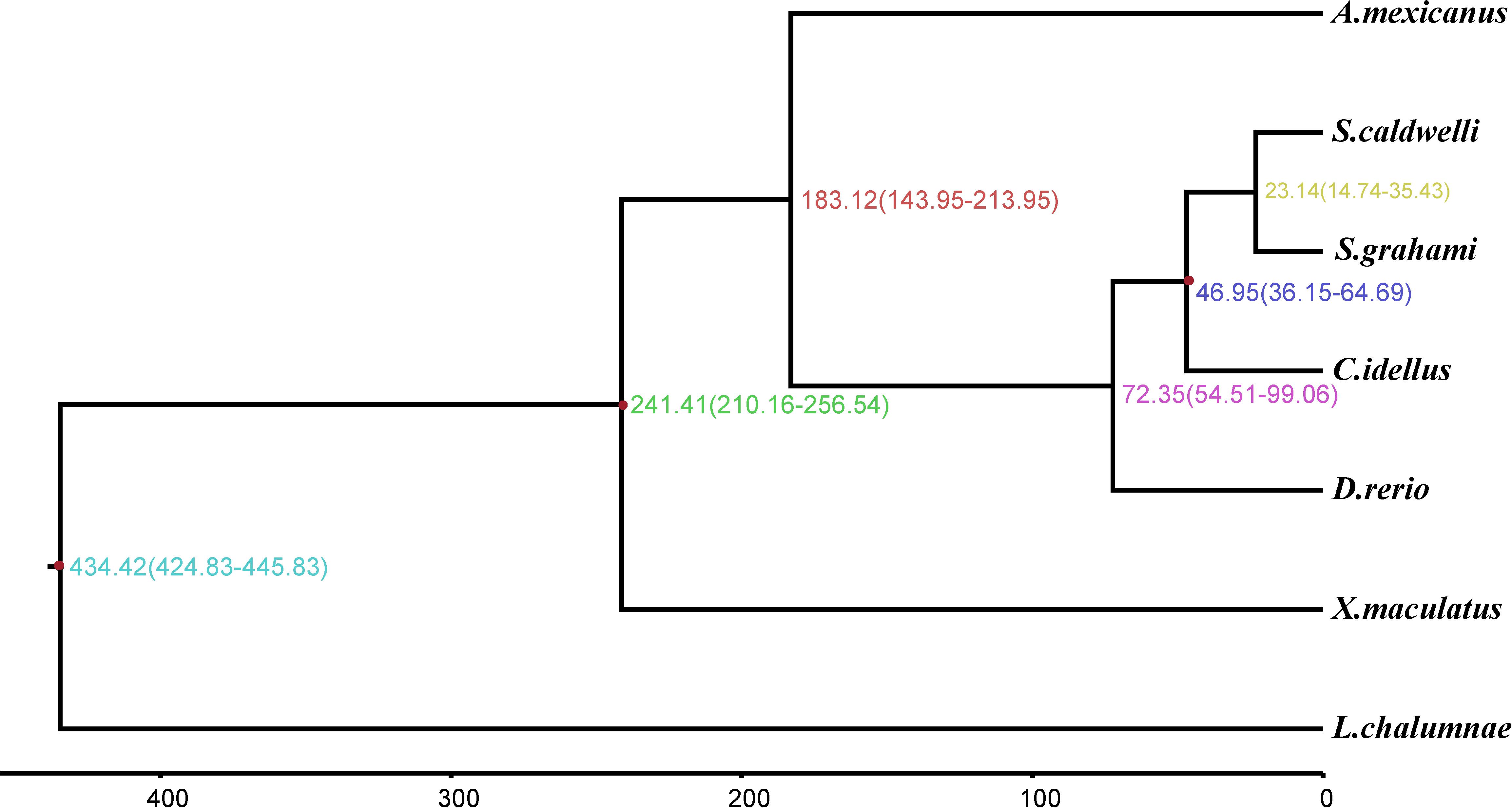
Figure 5. The phylogenetic tree and molecular timing of S. caldwelli and other fish species, showing S. caldwelli had diverged 23.4 million years ago. A total of 13,568 fourfold degenerate positions are used to compute the tree and infer the divergence dates.
To further understand the evolutionary dynamics of S. caldwelli, BLASTp (E-value ≤ 1e−5) searching among S. caldwelli and other six species identified a total of 9225 putative orthologs.
Moreover, by the branch-site model, 38 positively selected genes were found and all of them annotated in the Swiss-Prot database (Supplementary Table S4).
GO categories were employed to annotate these genes. It was found that the most abundant GO terms of positively selected genes in S. caldwelli were “cellular process” and “binding.” Also, there were positively selected genes identified by the term “response to stimulus” (Figure 6A).
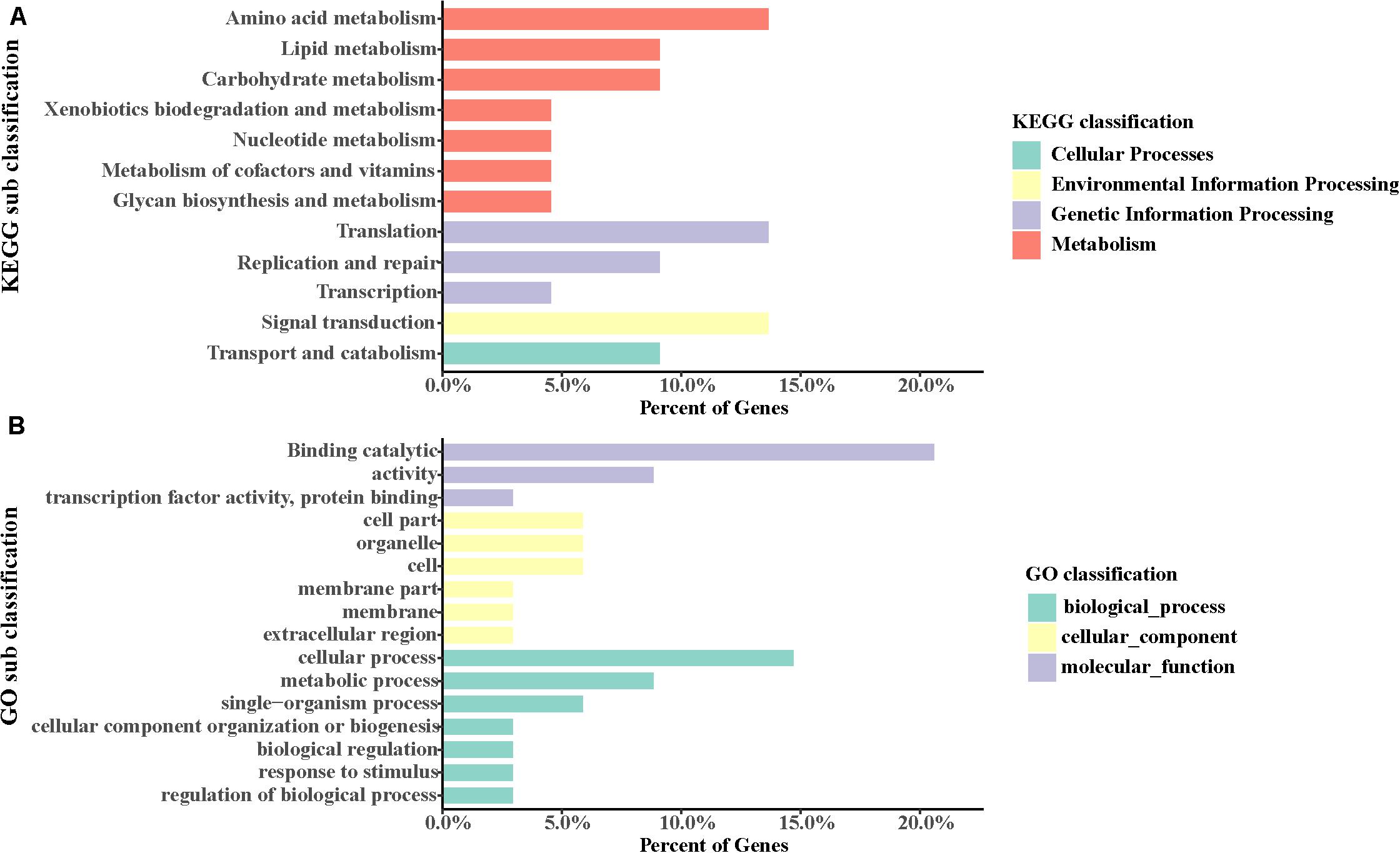
Figure 6. (A,B) Positive gene assignments in GO and KEGG pathway, respectively.
Also, using the KEGG pathway database, some genes were identified that might have accelerated evolution. The most two abundant pathways with accelerated evolution genes were “amino acid metabolism” and “translation” (Figure 6B).
The InnateDB database Breuer et al. (2013) was employed to identify the positively selected genes associated with the immune system. Consequently, six immune system genes were found, including DNA-directed RNA polymerase III subunit RPC2 (POLR3B), phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3), E3 ubiquitin-protein ligase Topors (TOPORS), fast kinase domain-containing protein 3 (FASTKD3), cytochrome P450 1A1 (CYP1A1), and Uveal autoantigen with coiled-coil domains and ankyrin repeats protein (UACA) (Table 2).

Table 2. Positively selected genes associated with immune from InnateDB.
Spinibarbus caldwelli is a medium to large-sized fish, which belongs to the order Cypriniformes, family Cyprinidae, and genus Spinibarbus and is widely distributed in the rivers of southern China (Tang et al., 2003). Previous studies on the genetic information of S. caldwelli have been mainly based on a few genes. For example, the CYTB gene was first employed to identify the biogeographic affinity of S. caldwelli from different river basins in China. Then, the molecular and morphological evidence was used to show that S. caldwelli is a valid species (Tang et al., 2005). Furthermore, Huang et al. (2008) have analyzed the mitochondrial DNA diversity of S. caldwelli. They tested the molecular variance and genetic diversity among 61 specimens of S. caldwelli by using mitochondrial CYTB. The results showed that the diversity in S. caldwelli was lower than other cyprinids (Huang et al., 2008). In this study, the molecular-clock approach predicted divergence between S. caldwelli and S. grahami is 23.14 Mya with confidence interval 14.74–35.43. Both of them diverged from C. idellus 46.95 Mya with confidence interval 36.15–64.69. These results were consistent with previous studies and further refined the speciation time of S. caldwelli.
Previous studies of various species, such as fish (Xiao et al., 2015), insects (Roux et al., 2014), and bats (Hawkins et al., 2019), have suggested that positively selected genes can be related to immune function (Elmer et al., 2010), energy supplying (Ma et al., 2016), collagen production (Hawkins et al., 2019), DNA repair (Lan et al., 2018), and response to hypoxia (Ma et al., 2016). Ren et al. (2014) found GATA-binding protein 4 (GATA4), Transducin (beta)-like 3 (TBL3), PHD finger protein 13 (PHF13), and Neutral cholesterol ester hydrolase 1 (NCEH1) in Erythroculter ilishaeformis exhibit strong positive selection metabolic processes and development. Zou et al. (2014) compared the transcriptome of Elopichthys bambusa with Hypophthalmichthys molitrix and Megalobrama amblycephala, revealing multiple candidate adaptive genes in each species, such as zona pellucida glycoprotein 2 in E. bambusa and zebrafish vitelline envelope protein in M. amblycephala. In Gymnocypris selincuoensis, zona pellucida sperm binding protein 3 (Zp3) and homeobox transcription factor Nanog (Nanog) associated with reproduction may be involved in adaption to the strong ultraviolet (UV) radiation (Feng et al., 2019). The genome-wide study on Gymnocypris przewalskii showed that positive selection on genes involved in energy metabolism, immune system, and hypoxia response contributed to highland adaption (Tong et al., 2017). Meanwhile, 138 positively selected genes were detected in a deep-sea fish (A. affinis), associating with cytoskeleton structures, DNA repair, and genetic information processing (Lan et al., 2018).
There are plentiful studies focusing on the positive selection of immune-related genes. To illustrate, by transcriptome sequencing, Zhong et al. (2018) screened 105 positively selected genes in P. managuensis. Among them, the genes encoding for phosphoinositide 3-kinase adapter protein 1, Ras-related protein Rap-1b, complement factor I, and probable ATP-dependent RNA helicase DHX58 were related to immune adaptation. Li et al. (2019) found that B cell-mediated immunity, chemokine-mediated signaling pathway, and immunoglobulin mediated immune response were the positively selected biological processes in big head carp, whereas those in silver carp mainly included the antigen processing and presentation, defense response to fungus, and detection of bacteria. Chen et al. (2008) revealed that TLR9 gene was positive selection in teleosts. Xiao et al. (2015) gave evidence that several immune-related genes, such as Notch2 and Nfatc3b, were identified as positively selected genes in tilapia. The study in Leuciscus waleckii also showed that most of the genes were associated with stress adaption and immunity among 61 positive selection genes (Xu et al., 2013). Xu et al. (2016) identified 40 full-length miiuy croaker MHC class IIA (Mimi-DAA) functional alleles from 26 miiuy croaker individuals and found 22 positively selected sites on the Mimi-DAA alleles. Among them, five sites were related to the binding of peptide antigen, indicating that these selected residues may play a crucial role in immune function (Xu et al., 2016).
To identify the potential genes involved in natural selection, the positively selected genes were screened. A total of 38 positively selected genes were identified (Figure 6). The most abundant KEGG pathways with positively selected genes included “amino acid metabolism,” “translation,” and “signal transduction.” These genes were assigned to GO categories subsequently. It has been indicated by the analysis that several positively selected loci or genes were found in multiple biological process in S. caldwelli. Therefore “binding catalytic” and “cellular process” were the most abundant GO terms associated with positively selected genes. Meanwhile, “response to stimulus” was also found with positively selected genes in S. caldwelli (Figure 6B).
By searching in the InnateDB database, several candidate genes associated with the immune system were found in S. caldwelli, including POLR3B, PIK3C3, TOPORS, FASTKD3, CYP1A1, and UACA. POLR3B is an antiviral molecule, which has been reported capable of confusing purine metabolism and has something to do with Alzheimer’s disease (Ansoleaga et al., 2015). A study on the relationship of brain immune genes with social behavior in strains of inbreeding rat shows that POLR3B is connected to behavioral processes that are essential to their social activities (Ma et al., 2015). Yee et al. (2007) reported that the causative mutation of POLR3B can jeopardize digestive organ development in zebrafish. PIK3C3 is an enzyme that generates phosphatidylinositol 3-phosphate (PI3P), a membrane component, in zebrafish. Lack of PIK3C3 causes intestinal inflammation and injury (Zhao et al., 2018). Kariuki et al. (2010) have found that the PIK3C3 locus is related to morbidity from systemic lupus erythematosus (SLE). TOPORS is a topoisomerase-I and p53-binding protein that is dynamically related to PML nuclear bodies (Rajendra et al., 2004). It has been predicted that TOPORS is a tumor suppressor in colon cancer and other malignancies (Rajendra et al., 2004). Besides, Chakarova et al. (2010) noted that TOPORS regulates retinal development in zebrafish. FASTKD3, a necessary component of mitochondrial respiration, belongs to the FAST kinase domain-containing protein (FASTKD) family, and therefore has a relationship with cell energy modulation (Simarro et al., 2010). CYP1A1 activates polycyclic aromatic hydrocarbons, which are turned into reactive intermediates associated with toxicity and carcinogenesis (Shigeyuki et al., 2006). It also plays an important role in detoxification (Shigeyuki et al., 2006). Interestingly, CYP1A1 and CYP17A1 previously reported in D. albolineatus (Bao and Xia, 2017) are both related to Cytochrome P450, arranged by substrate type pathway. UACA has been considered to be a potential target autoantigen in Vogt-Koyanagi-Harada, Behçet’s disease, and sarcoidosis, consequently leading to panuveitis (Yamada et al., 2001). Interestingly, despite that several immune-related genes have been found under selection, our dataset failed to identify other loci that are usually under selection in fish related to the immune system like MhC. This may be attributed to difficulty among detection in the presence of the reduced expression of these (genes) loci when using muscular tissue. It may also point to some specificity of the muscular immune system.
In this article, the muscle transcriptome of S. caldwelli was obtained through de novo assembly of its transcriptome and screening for accelerated evolution genes using the branch-site model. Based on this data, we calculated that the divergence time between S. caldwelli and S. grahami is 23.14 Mya. And both of them diverged from C. idellus 46.95 Mya. Thirty-eight positively selected loci and genes were identified in S. caldwelli. Several genes were found related to immune functions. These results demonstrated the accelerated genetic evolution of these immune genes. The present results provided new insight into evolutionary pressures and significant in the understanding of function and immunology of S. caldwelli.
The sequencing data in our article has been uploaded to the NCBI Sequence Read Archive database (SRR10123035).
The animal study was reviewed and approved by the Animal Care and Use Committee (IACUC) of Hunan Agricultural University (File code: ACC2017007).
TX and JZ: methodology. LB: software. YT: validation, writing—original draft preparation, and writing—review and editing. WC: formal analysis and supervision. JC: investigation. TX: resources, project administration, and funding acquisition. LC: data curation. JZ: visualization. All authors contributed to the article and approved the submitted version.
This study was supported by Chinese Special Fund for Agro-scientific Research in the Public Interest (201303056–7), the innovation and demonstration of breeding Mode in Pond healthy Colleges and Universities (2017NK1032), the Scientific Research Fund of Hunan Provincial Education Department, grant number (18C0757), and the Science and Technology Project of Changsha, China, grant number (KC1809009).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00714/full#supplementary-material
Ansoleaga, B., Jove, M., Schluter, A., Garciaesparcia, P., Moreno, J., Pujol, A., et al. (2015). Deregulation of purine metabolism in Alzheimer’s disease. Neurobiol. Aging 36, 68–80.
Aquadro, C. F., and Greenberg, B. D. (1983). Human mitochondrial dna variation and evolution: analysis of nucleotide sequences from seven individuals. Genetics 103, 287–312.
Bao, L. S., and Xia, J. L. (2017). Global analysis of transcriptome sequences highlights accelerated evolution of immune genes in Danio choprae and Danio albolineatus. Fish Shellfish Immunol. 66, 390–397. doi: 10.1016/j.fsi.2017.05.011
Barton, N. H. (1998). The effect of hitch-hiking on neutral genealogies. Genet. Res. 72, 123–133. doi: 10.1017/s0016672398003462
Betancur-R, R., Ortí, G., and Pyron, R. A. (2015). Fossil-based comparative analyses reveal ancient marine ancestry erased by extinction in ray-finned fishes. Ecol. Lett. 18, 441–450. doi: 10.1111/ele.12423
Betancur-R, R., Broughton, R. E., Wiley, E. O., Carpenter, K., López, J. A., Li, C., et al. (2013). The tree of life and a new classification of bony fishes. PLoS Curr. 5:ecurrents.tol.53ba26640df0ccaee75bb165c8c26288. doi: 10.1371/currents.tol.53ba26640df0ccaee75bb165c8c26288
Breuer, K., Foroushani, A. B. K., Laird, M. R., Chen, C., Sribnaia, A., Lo, R., et al. (2013). InnateDB: systems biology of innate immunity and beyond-recent updates and continuing curation. Nucleic Acids Res. 41, 1228–1233. doi: 10.1093/nar/gks1147
Cabau, C., Escudié, F., Djari, A., Guiguen, Y., Bobe, J., and Klopp, C. J. P. (2017). Compacting and correcting Trinity and Oases RNA-Seq de novo assemblies. PeerJ 5:e2988. doi: 10.7717/peerj.2988
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chakarova, C. F., Khanna, H., Shah, A. Z., Patil, S. B., Sedmak, T., Murga-Zamalloa, C. A., et al. (2010). TOPORS, implicated in retinal degeneration, is a cilia-centrosomal protein. Hum. Mol. Genet. 20, 975–987. doi: 10.1093/hmg/ddq543
Chen, J. S., Wang, T., Tzeng, T., Wang, C., and Wang, D. J. F. (2008). Evidence for positive selection in the TLR9 gene of teleosts. Fish Shellfish Immunol. 24, 234–242. doi: 10.1016/j.fsi.2007.11.005
Chen, W. J., Lavoué, S., and Mayden, R. L. J. E. (2013). Evolutionary origin and early biogeography of otophysan fishes (Ostariophysi: Teleostei). Evolution 67, 2218–2239. doi: 10.1111/evo.12104
Conesa, A., and Gotz, S. (2008). Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 619832–619832.
Corbettdetig, R. B., Hartl, D. L., and Sackton, T. B. (2015). Natural selection constrains neutral diversity across a wide range of species. PLoS Biol. 13:e1002112. doi: 10.1371/journal.pbio.1002112
Elmer, K. R., Fan, S., Gunter, H. M., Jones, J. C., Boekhoff, S., Kuraku, S., et al. (2010). Rapid evolution and selection inferred from the transcriptomes of sympatric crater lake cichlid fishes. Mol. Ecol. 19, 197–211. doi: 10.1111/j.1365-294x.2009.04488.x
Feng, X., Jia, Y., Zhu, R., Chen, K., and Chen, Y. (2019). Characterization and analysis of the transcriptome in Gymnocypris selincuoensis on the Qinghai-Tibetan Plateau using single-molecule long-read sequencing and RNA-seq. DNA Res. 26, 353–363. doi: 10.1093/dnares/dsz014
Gonzalez, J., Lenkov, K., Lipatov, M., Macpherson, J. M., and Petrov, D. A. (2008). High rate of recent transposable element–induced adaptation in Drosophila melanogaster. PLoS Biol. 6:e251. doi: 10.1371/journal.pbio.0060251
Grabherr, M., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gutiérrez-Larruscain, D., Santos-Vicente, M., Anderberg, A. A., Rico, E., and Martínez-Ortega, M. M. J. T. (2018). Phylogeny of the Inula group (Asteraceae: Inuleae): evidence from nuclear and plastid genomes and a recircumscription of Pentanema. Taxon 67, 149–164. doi: 10.12705/671.10
Hawkins, J. A., Kaczmarek, M. E., Müller, M. A., Drosten, C., Press, W. H., and Sawyer, S. L. (2019). A metaanalysis of bat phylogenetics and positive selection based on genomes and transcriptomes from 18 species. Proc. Natl. Acad. Sci. U.S.A. 116, 11351–11360. doi: 10.1073/pnas.1814995116
Huang, Z., Huang, L., Lin, X., and Ling, W. (2008). Studies on protease activities of the nemertean Procephalothrix simulus. Period. Ocean Univ. China 38, 259–262.
Jansa, S. A., Lundrigan, B. L., and Tucker, P. K. (2003). Tests for positive selection on immune and reproductive genes in closely related species of the murine genus mus. J. Mol. Evol. 56, 294–307. doi: 10.1007/s00239-002-2401-6
Kaplan, N. L., Hudson, R. R., and Langley, C. H. (1989). The “hitchhiking effect” revisited. Genetics 123, 887–899.
Kariuki, S. N., Franek, B. S., Mikolaitis, R. A., Utset, T. O., Jolly, M., Skol, A. D., et al. (2010). Promoter variant of PIK3C3 is associated with autoimmunity against Ro and Sm epitopes in African-American lupus patients. BioMed Res. Int. 2010:826434.
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kimura, M. (1983). The Neutral Theory of Molecular Evolution. Cambridge: Cambridge University Press.
Kirkpatrick, M., and Kern, A. D. (2012). Where’s the money? Inversions, genes, and the hunt for genomic targets of selection. Genetics 190, 1153–1155. doi: 10.1534/genetics.112.139899
Lan, Y., Sun, J., Xu, T., Chen, C., Tian, R., Qiu, J. W., et al. (2018). De novo transcriptome assembly and positive selection analysis of an individual deep-sea fish. BMC Genomics 19:394.
Li, G., Zhao, Y., Guo, S., Liu, B., Chen, Y., Sun, X., et al. (2019). Comparative analysis of spleen transcriptome detects differences in evolutionary adaptation of immune defense functions in bighead carp and silver carp. Fish Shellfish Immunol. 84, 148–157. doi: 10.1016/j.fsi.2018.09.077
Li, Y., Ren, Z., Shedlock, A. M., Wu, J., Sang, L., Tersing, T., et al. (2013). High altitude adaptation of the schizothoracine fishes (Cyprinidae) revealed by the mitochondrial genome analyses. Gene 517, 169–178. doi: 10.1016/j.gene.2012.12.096
Ma, L., Piirainen, S., Kulesskaya, N., Rauvala, H., and Tian, L. (2015). Association of brain immune genes with social behavior of inbred mouse strains. J. Neuroinflamm. 12:75.
Ma, X., Dai, W., Kang, J., Yang, L., and He, S. (2016). Comprehensive transcriptome analysis of six catfish species from an altitude gradient reveals adaptive evolution in tibetan fishes. G3 Genes Genomes Genetics 6, 141–148. doi: 10.1534/g3.115.024448
Messer, P. W., and Petrov, D. A. (2013). Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol. Evol. 28, 659–669. doi: 10.1016/j.tree.2013.08.003
Nakatani, M., Miya, M., Mabuchi, K., Saitoh, K., and Nishida, M. J. B. (2011). Evolutionary history of Otophysi (Teleostei), a major clade of the modern freshwater fishes: pangaean origin and Mesozoic radiation. BMC Evol. Biol. 11:177. doi: 10.1186/1471-2148-11-177
Rajendra, R., Malegaonkar, D., Pungaliya, P., Rasheed, Z., Marshall, H., Saleem, A., et al. (2004). Topors, a putative tumor suppressor, functions as a RING-domain-dependent E3 ubiquitin ligase for p53. AACR 64:588.
Ren, L., Tan, X., Xiong, Y., Xu, K., Zhou, Y., Zhong, H., et al. (2014). Transcriptome analysis reveals positive selection on the divergent between topmouth culter and zebrafish. Gene 552, 265–271. doi: 10.1016/j.gene.2014.09.053
Roux, J., Privman, E., Moretti, S., Daub, J. T., Robinsonrechavi, M., and Keller, L. (2014). Patterns of positive selection in seven ant genomes. Mol. Biol. Evol. 31, 1661–1685. doi: 10.1093/molbev/msu141
Schartl, M., Walter, R. B., Shen, Y., Garcia, T., Catchen, J. M., Amores, A., et al. (2013). The genome of the platyfish, Xiphophorus maculatus, provides insights into evolutionary adaptation and several complex traits. Nat. Genet. 45, 567–572. doi: 10.1038/ng.2604
Schrider, D. R., Navarro, F. C. P., Galante, P. A. F., Parmigiani, R. B., Camargo, A. A., Hahn, M. W., et al. (2013). Gene copy-number polymorphism caused by retrotransposition in humans. PLoS Genet. 9:e1003242. doi: 10.1371/journal.pgen.1003242
Shigeyuki, U., Dalton, T. P., Nadine, D., Curran, C. P., Sandrine, D., Miller, M. L., et al. (2006). Oral benzo[a]pyrene in Cyp1 knockout mouse lines: CYP1A1 important in detoxication, CYP1B1 metabolism required for immune damage independent of total-body burden and clearance rate. Mol. Pharmacol. 69, 1103–1114. doi: 10.1124/mol.105.021501
Simarro, M., Gimenezcassina, A., Kedersha, N., Lazaro, J., Adelmant, G., Marto, J. A., et al. (2010). Fast kinase domain-containing protein 3 is a mitochondrial protein essential for cellular respiration. Biochem. Biophys. Res. Commun. 401, 440–446. doi: 10.1016/j.bbrc.2010.09.075
Stefansson, H., Helgason, A., Thorleifsson, G., Steinthorsdottir, V., Masson, G., Barnard, J., et al. (2005). A common inversion under selection in Europeans. Nat. Genet. 37, 129–137. doi: 10.1038/ng1508
Tang, Q., Liu, H., Yang, X., and Nakajima, T. (2005). Molecular and morphological data suggest that Spinibarbus caldwelli (Nichols) (Teleostei: Cyprinidae) is a valid species. Ichthyol. Res. 52, 77–82. doi: 10.1007/s10228-004-0259-x
Tang, Q., Yang, X., and Liu, H. (2003). Biogeographical process of Spinbarbus caldwelli revealed by sequence variations of mitochondrial cytochrome b gene. Acta Hydrobiol. Snica 27, 352–356.
Tong, C., Fei, T., Zhang, C., and Zhao, K. J. B. E. B. (2017). Comprehensive transcriptomic analysis of Tibetan Schizothoracinae fish Gymnocypris przewalskii reveals how it adapts to a high altitude aquatic life. BMC Evol. Biol. 17:74. doi: 10.1186/s12862-017-0925-z
Wang, J., Wu, X., Chen, Z., Yue, Z., Ma, W., Chen, S., et al. (2013). Molecular phylogeny of European and African Barbus and their West Asian relatives in the Cyprininae (Teleostei: Cypriniformes) and orogenesis of the Qinghai-Tibetan Plateau. Chinese Sci. Bull. 58, 3738–3746. doi: 10.1007/s11434-013-5878-z
Xiao, J., Zhong, H., Liu, Z., Yu, F., Luo, Y., Gan, X., et al. (2015). Transcriptome analysis revealed positive selection of immune-related genes in tilapia. Fish Shellfish Immunol. 44, 60–65. doi: 10.1016/j.fsi.2015.01.022
Xu, J., Ji, P., Wang, B., Zhao, L., Wang, J., Zhao, Z., et al. (2013). Transcriptome sequencing and analysis of wild amur ide (Leuciscus waleckii) inhabiting an extreme alkaline-saline lake reveals insights into stress adaptation. PLoS One 8:e59703. doi: 10.1371/journal.pone.0059703
Xu, T., Liu, J., Sun, Y., Zhu, Z., Liu, T. J. D., and Immunology, C. (2016). Characterization of 40 full-length MHC class IIA functional alleles in miiuy croaker: polymorphism and positive selection. Dev. Comp. Immunol. 55, 138–143. doi: 10.1016/j.dci.2015.10.019
Yamada, K., Senju, S., Nakatsura, T., Murata, Y., Ishihara, M., Nakamura, S., et al. (2001). Identification of a novel autoantigen UACA in Patients with Panuveitis. Biochem. Biophys. Res. Commun. 280, 1169–1176. doi: 10.1006/bbrc.2001.4189
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yee, N. S., Gong, W., Huang, Y., Lorent, K., Dolan, A. C., Maraia, R. J., et al. (2007). Mutation of RNA Pol III subunit rpc2/polr3b leads to deficiency of subunit Rpc11 and disrupts zebrafish digestive development. PLoS Biol. 5:e312. doi: 10.1371/journal.pbio.0050312
Zhang, J., Nielsen, R., and Yang, Z. (2005). Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479. doi: 10.1093/molbev/msi237
Zhao, S., Xia, J., Wu, X., Zhang, L., Wang, P., Wang, H., et al. (2018). Deficiency in class III PI3-kinase confers postnatal lethality with IBD-like features in zebrafish. Nat. Commun. 9:2639.
Zhong, H., Zhang, H., Tang, Z., Guo, Z., and Zhou, Y. (2018). Evidence for natural selection of immune genes from Parachromis managuensis by transcriptome sequencing. Biotechnol. Biotechnol. Equ. 32, 1431–1439. doi: 10.1080/13102818.2018.1519377
Zhu, B., Zou, P., Zhong, L., Liu, Z., Peng, L., and Chen, D. (2006). RAPD molecular genetic markers identification in three different species of Spinibarbus fishes. J. Nanchang Univ. 30, 597–600.
Keywords: Spinibarbus caldwelli, transcriptome, natural selection, immune genes, orthologous unigenes
Citation: Tuo Y, Chu W, Zhang J, Cheng J, Chen L, Bao L and Xiao T (2020) Analysis of Natural Selection of Immune Genes in Spinibarbus caldwelli by Transcriptome Sequencing. Front. Genet. 11:714. doi: 10.3389/fgene.2020.00714
Received: 24 September 2019; Accepted: 11 June 2020;
Published: 24 July 2020.
Edited by:
Denis Baurain, University of Liège, BelgiumReviewed by:
Huan Zhong, Guangxi Academy of Fishery Sciences, ChinaCopyright © 2020 Tuo, Chu, Zhang, Cheng, Chen, Bao and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingsheng Bao, MTcxOTA2MjU0QHFxLmNvbQ==; Tiaoyi Xiao, dHl4aWFvMTEyOEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.