 Ranran Zhao1
Ranran Zhao1 Yuan Wang
Yuan Wang Jianning He
Jianning He- 1College of Animal Science and Technology, Qingdao Agricultural University, Qingdao, China
- 2Qufu Animal Husbandry and Veterinary Technical Service Center, Qufu, China
- 3China Animal Health and Epidemiology Center, Qingdao, China
- 4College of Animal Science, Inner Mongolia Agricultural University, Hohhot, China
Long non-coding RNAs (lncRNA) and microRNAs (miRNA) are new found classes of non-coding RNAs (ncRNAs) that are not translated into proteins but regulate various cellular and biological processes. In this study, we conducted a transcriptomic analysis of ncRNA and mRNA expression in Aohan fine wool sheep (AFWS) at different growth stages (embryonic day 90, embryonic day 120, and the day of birth), and explored their relationship with wool follicle growth. In total, 461 lncRNAs, 106 miRNAs, and 1,009 mRNAs were found to be differentially expressed during the three stages of wool follicle development. Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were performed to clarify the roles of the differentially expressed lncRNA, miRNA, and mRNA in the different stages of wool follicle development. Quantitative real-time PCR (qRT-PCR) was used to validate the results of RNA-seq analysis. lncRNA (MSTRG.223165) was found to act as a competing endogenous RNA (ceRNA) and may participate in wool follicle development by acting as an miR-21 sponge. Network prediction implicated the MSTRG.223165-miR-21-SOX6 axis in the wool follicle development. The targeting relationships of miR-21 with SOX6 and MSTRG.223165 were validated in dual-luciferase assays. This is the first report indicating the association of the lncRNA–miRNA–mRNA network with wool follicle development in AFWS. This study provides new insights into the regulation of the wool follicle growth and represents a solid foundation for wool sheep breeding programs.
Introduction
The quality and yield of mammalian hair is determined by the characteristics and structure of hair follicles (HF) (Mann, 1968; Nixon et al., 1996). The Aohan fine wool sheep (AFWS) is an ancient representative of this breed-type that originated in Northeast of China. This breed is characterized by stable genetic performance, strong adaptability, and is suitable for breeding in arid areas. The wool has good textile technology performance, with a wool fineness of approximately 22 μm, and a hair length exceeding 9 cm (Wang et al., 1996). It is an excellent raw material for manufacturing fine textiles and has high economic value. The morphological structure and development of HF have important effects on wool properties. HFs are complex structures that are classified as primary hair follicles (PF) and secondary hair follicles (SF) depending on the structural characteristics and developmental stage. Compared with the SF, the diameter of the PF and hair bulbs are larger, and both have complete sebaceous glands. The SF develops later than the PF, and the diameter of the hair follicles and hair bulbs are smaller, with only a few SF having a single sebaceous gland (Carter and Hardy, 1943; Schinckel, 1953). In fine wool sheep, SFs determine fine wool production and have an important effect on fiber diameter.
In sheep breeding, wool length and fineness combined with other indicators are important standards for measuring wool quality in the textile industry. Studies of the genes that regulate HF development have become a focus of research aimed at improving wool quality and increasing wool production (Cui et al., 2010; Hamanaka et al., 2013). Studies on the growth and characteristics of sheep HF have been reported since the 1950s, and HF development is well-understood at the cellular level (Hardy and Lyne, 1956; Moore et al., 1998). Studying the molecular mechanisms that regulate the morphogenesis of secondary wool follicles in AFWS, and finding regulatory factors and functional genes that specifically regulate their growth and development will provide a reliable scientific basis for improving wool quality using molecular breeding techniques.
RNA-sequencing (RNA-seq) has been widely used to evaluate in gene expression patterns in different species or different stages of growth in the same species. This approach has been used in combination with disciplines such as differential gene expression analysis, new transcript prediction, untranslated region (UTR) analysis, alternative splicing analysis, Simple Short Repeat (SSR)/Single Nucleotide Polymorphism (SNP) analysis and non-coding RNA (ncRNA) analysis (Schliebner et al., 2014; Thakur and Randhawa, 2018). NcRNAs include transfer RNA (tRNA), microRNA (miRNA), circular RNA (circRNA), and long non-coding RNA (lncRNA). These ncRNAs cannot be translated into protein, but regulate gene expression, cell function and biological processes at the level of transcription and post transcription (Huttenhofer et al., 2005). lncRNAs represent a class of RNA molecules longer than 200 nucleotides (nt) that was first discovered by high-throughput sequencing of mouse full-length cDNA (Okazaki et al., 2002; Mercer and Mattick, 2013). miRNAs are short (19–25 nt), single-stranded ncRNAs that regulate the expression of target genes at the post-transcriptional level by partially binding to the 3′-UTR region of target mRNA, thereby playing an important role in many biological activities (Ambros, 2004; Bartel, 2004). Since the initial discovery in C. elegans (Lee et al., 1993), 38,589 miRNAs have been identified to date1. CircRNAs exist as a covalently closed loop structure, without the 5′-end cap structure or 3′-poly(A) tail (Li et al., 2018). Some circRNAs such as circZNF609 (Pamudurti et al., 2017) and circ-FBXW7 (Legnini et al., 2017) can be translated into proteins.
In recent years, scientists have discovered a new regulatory mechanism that involves competing endogenous RNA (ceRNA). Two types of RNA transcripts regulate their expression by competitively combining with miRNAs, forming a large-scale miRNA-based transcriptional regulatory network. In this study, lncRNA was found to competitively bind miRNA with mRNA via this regulatory mechanism, thereby regulating the expression of miRNA target genes after transcription.
RNA-sequencing (RNA-seq) has been used in to study a wide variety of processes including sheep reproduction (Legnini et al., 2017), sheep muscle growth (Cao et al., 2018), and liver function (Li et al., 2019). Using this approach, it was shown thatmiR-27b promotes the proliferation of sheep skeletal muscle by targeting the myostatin gene (Zhang et al., 2018). Lnc-SEMT promotes sheep myoblast differentiation in vitro and increases the number of muscle fibers (Wei et al., 2018). Many circRNAs have been identified in the sheep pituitary, and screening of the differentially expressed circRNAs in the prenatal and postnatal pituitary has provided an improved understanding of the function of circRNAs in the pituitary (Li et al., 2017). Studies have also identified mRNAs and lncRNAs in the uterus of multi-tailed and single-tailed small-tailed sheep (Ovis aries) and RNAs such as MSTRG.134747 and UGT1A1 may play a role in the molecular mechanism of sheep fertility (La et al., 2019). However, there are few reports about the mechanism underlying regulation of the lncRNA–miRNA–mRNA network in wool sheep.
In the present study, we evaluated the histological changes in skin tissues at embryonic day 90 (E90d), at embryonic day 120 (E120d), and birth (Birth) to clarify the morphological changes of wool follicles in different stages of growth. To investigate the function of ceRNA regulation in skin wool follicle development, we explored the expression profiles of lncRNA, mRNA, and miRNA in sheep by RNA-seq. The interactions between miRNAs, mRNAs, and lncRNAs that were differentially expressed in three growth stages were determined. We constructed ceRNA regulatory networks and discovered some interesting ceRNA interactions during three stages of hair follicle development, revealing potential regulatory mechanisms, and providing the foundation for further studies on the development of AFWS wool follicles.
Materials and Methods
Sheep Selection and Skin Tissue Preparation
Aohan fine wool sheep is a breed of sheep farmed in China for its excellent wool and meat and strong adaptability. The experimental sheep were raised in the AFWS stud farm of Inner Mongolia Autonomous Region, and fed according to the farm’s feeding plan. Nine healthy Aohan fine wool ewes (aged 3–5 years) were fed under the same conditions, subjected to estrus treatment during September, and artificially fertilized from the same ram. On embryonic day 90 (E90d) and embryonic day 120 (E120d) after fertilization, and the day of birth (Birth), samples of shoulder skin tissue with (diameter 2 cm) were collected from nine sheep (three samples at each stage).
The ewes and lambs were anesthetized with sodium pentobarbital (25 mg/kg) by intravenous injection. After sample collection, the ewes and newborn lambs were released, whereas the fetuses from E90d and E120d were placed, still under anesthesia, inside a closed chamber, and sacrificed by carbon dioxide inhalation. The anesthesia procedure was performed following published protocols (Bennett, 2005). Samples were immediately placed in liquid nitrogen for RNA-Seq and qRT-PCR analysis.
To study the morphological changes of primary and secondary hair follicles in the three stages of development, we collected shoulder fur samples into 4% paraformaldehyde, and prepared paraffin-embedded sections that were stained with hematoxylin–eosin (HE) for histological evaluation.
RNA Isolation and Library Construction
Total RNA was isolated from skin samples used TRIzol (Life Technologies, CA, United States). The RNA samples were evaluated for degradation and impurities by 1% agarose electrophoresis. An Agilent 2100 Bioanalyzer and Agilent RNA 6000 Nano Kit were used to assess RNA concentration and integrity (Agilent Technologies, CA, United States), and purity was determined using a NanoDrop 2000 (NanoDrop Technologies, Wilmington, DE, United States). The samples were sequenced and samples with RNA Integrity Number (RIN) scores >7 were used in subsequent analyses. The sheep genome oar_v4.0 was selected as the reference genome in this study. High -throughput sequencing was performed by Annoroad technologies (Beijing, China).
lncRNA Library Construction and Sequencing
The lncRNA library was constructed from total RNA (3 μg) using different index tags with the super directional RNA library prep kit (NEB, Ipswich, United States) according to the manufacturer’s instructions. First, the ribosomal rRNA was removed using a kit, and the RNA fragment was transformed into a short fragment. Short sequence RNA was used as template and six base random primers (random hexamers) were to synthesize the first-strand of cDNA. The second strand of cDNA was then synthesized. Following purification with a QIA Quick PCR kit, eluted with EB buffer, followed by end-repair and the add base (A) and a sequencing connector, the target fragments were recovered by agarose gel electrophoresis. The double-stranded cDNA was digested by uracil-DNA glycosylase and amplified by PCR. Finally, the target fragments of the complete library were recovered by agarose gel electrophoresis.
miRNA Library Construction and Sequencing
Total RNA samples were digested to generate fragments of 18–30 nt or 15–35 nt RNA and collected by agarose gel electrophoresis; the ends of the isolated RNA fragments were ligated and then reverse-transcribed into cDNA, then PCR amplification was performed to establish a sequencing library. The qualified sequencing libraries were sequenced using the Illumina platform (Illumina X Ten) with the SE50sequencing strategy.
Identification of lncRNAs
Quality Control and Transcriptome Assembly
Illumina raw reads results were stored in FASTQ (fq). The original sequence contained the sequencing linker sequences and low quality sequences. To ensure the quality of the information analysis data, we filtered the original offline sequence data to obtain high quality clean reads for subsequent analysis. Data quality control involved removal of contaminated reads, low quality reads, reads containing N ratios >5%, and reads that matched the ribosomal RNA. The RNA-seq data filtered for each sample was compared with the genome by HiSAT2 (Kim et al., 2015). Fragments per kilobase million (FPKM) was used to estimate gene expression levels quantitatively according to the following formula: FPKM=, where F is the number of fragments covered by each gene, N is the sequence of the total alignment, and L is the length of the gene. The FPKM method eliminates the effects of gene length and sequencing differences on gene expression.
lncRNA Coding Potential Prediction and Target Gene Prediction
lncRNA is a long-chain ncRNA (>200 bp) classified as lincRNA (intergenic RNA), intronic lncRNA, anti-sense lncRNA, sense lncRNA, and bidirectional lncRNA according to the positional relationship with the coding sequence. When conducting novel lncRNA screening, the three basic criteria were applied:(1) Transcript length ≥200 bp and number of exons is ≥2; (2) Coverage for each transcript was calculated and transcripts with <5 were screened out in all samples; and (3) Known mRNAs and other ncRNAs were screened. Following this initial screening of lncRNA information, four analytical methods were used to predict its coding potential: Coding-Non-Coding Index (CNCI) analysis (Sun et al., 2013), Coding Potential Calculator (CPC) analysis (Lei et al., 2007), PFAM protein domain analysis (Bateman et al., 2002; Mistry et al., 2007), and Coding Potential Assessment Tool (CPAT) analysis (Wang et al., 2013). The results predicted by the four software were used as the subsequent lncRNA analysis data set.
Identification of miRNAs
The raw data for miRNA were stored in FASTQ (fq) format. To ensure the accuracy of our subsequent analysis, the clean reads were first mapped to the reference genome (oar_v4.0) using Bowtie software (V1.1.2) (Langmead et al., 2009), and the number of total clean reads and their alignment ratios were compared with the sequence of the specified species in the miRBase database (Release 21). The sequences matching different regions in each sample were obtained, and the expression level, sequence, length and features such as secondary structure of the known miRNAs were identified. The clean reads, which were not annotated as known miRNAs, were compared to the ncRNA sequences in Rfam (13.0) (Burge et al., 2013) to enable annotation of rRNA, tRNA, snRNA, snoRNA, and other ncRNAs.
Identification of Differentially Expressed RNAs
Three AFWS were selected as biological replicates, to identify differentially expressed lncRNAs, mRNAs, and miRNAs. The Fragments per Kilobase per Million Mapped Fragments (FPKM) values (Trapnell et al., 2010) were used to estimate the expression of lncRNA and mRNA, and Transcripts Per Million (TPM) values (Zhou et al., 2010) were used to assess miRNA expression. Differential expression analysis of lncRNA, mRNA and miRNA was performed using DEseq (Anders and Huber, 2010). The treatment group was compared with the reference group. For lncRNA and mRNA, log2Ratio| ≥ 1 and q < 0.05 were used as screening conditions for significant DE genes. For miRNA, pval ≤ 0.05, padj ≤ 0.05 and | log2(Fold_change)| ≥ 1 are screening conditions for DE miRNAs.
Quantitative Real-Time PCR
We used the CFX96 Real-Time System (BioRad, CA, United States) to validate the expression levels of DE lncRNAs, DE mRNAs, and DE miRNAs. The primer sequences of the selected lncRNAs, mRNAs and miRNAs are listed in Supplementary Table S1. For lncRNA and mRNA, total RNA was synthesized used as a template to synthesize cDNA using Transcriptor First-Strand cDNA Synthesis Kit (Roche, Australia). For miRNA, the reverse primer of the miRNAs was provided in the Mir-XTM miRNA first-Strand Synthesis Kit (TaKaRa, Dalian, China) and cDNA was synthesized. The housekeeping gene GAPDH was used to normalize the expression levels of the lncRNAs and mRNAs, and U6 was used to normalize the expression levels of miRNAs. Quantitative real-time PCR was carried out in a 20-μL reaction mixture containing 10 μL 2 × iTaqTM Universal SYBR@ Green Supermix (BioRad, CA, United States), 1 μL cDNA, 8 μL ddH2O, and 0.5 μL forward and reverse primers. The following thermocycling program was used: 95°C for 10 min; 45 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 10 s; 72°C for 6 min. At least three samples were analyzed for each developmental stage (E90d, E120d, Birth) and each sample was analyzed in three independent reactions. The relative expression levels were calculated using the 2–ΔΔCt method.
Target Gene Prediction, GO Annotation, and KEGG Analysis
The DE lncRNA was subjected to cis and trans target analysis, and its function was indirectly predicted by the target gene. The principle of cis target gene prediction is that the function of lncRNA is related to the protein coding gene adjacent to the genomic locus, and the protein coding gene of the adjacent position (50 Kb upstream and downstream) of the lncRNA is screened as a cis target gene. The principle of trans target gene prediction is that the function of lncRNA is related to a co-expressed gene, and the trans target gene is screened according to the correlation coefficient of lncRNA and mRNA expression level (correlation coefficient corr ≥ 0.9).
miRNA Family Analysis and Target Gene Prediction
miRNA conservation is manifested as the existence of miRNAs of the same family in many closely related species. To identify the phenomenon of conservation in each species, miRNA family analysis is performed using the miRBase reference for known miRNAs. Three target prediction software packages, PITA, miRanda (Enright et al., 2003), and Target Scan, were used to predict miRNA target genes, retaining at least the target genes present in two prediction software.
Gene Ontology enrichment and KEGG pathway analysis of DE mRNA and DE lncRNA and DE miRNA target genes were performed to investigate their biological functions. GO functional analysis were used Blast2GO (Conesa et al., 2005) and KOBAS were used for GO enrichment and KEGG pathway analysis. In GO enrichment, genes are annotated according to three ontologies: molecular function (MF), the cellular component (CC), and biological process (BP). KEGG pathway analysis was then conducted to explore the significantly enriched pathways of the genes. In GO terms and KEGG pathways, q < 0.05 was considered to indicate a term and pathway for significant enrichment of mRNA.
Construction of ncRNAs Regulatory Networks
MiRanda, PITA and Target Scan software were used to predict the mRNA and lncRNA targets of miRNAs. The miRNA–mRNA, lncRNA–miRNA, and lncRNA–miRNA–mRNA networks were constructed according to the predicted miRNA–mRNA and miRNA–lncRNA pairs using Cytoscape software (Shannon et al., 2003). Predicted miRNA–mRNA and miRNA–lncRNA regulatory pairs were based on differential expression during the three developmental periods. CeRNA networks were constructed based on the DE mRNAs and DE lncRNAs with the same miRNA binding sites.
Dual-Luciferase Assay
Dual-luciferase reported assays were used to verify the predicted SOX6 and MSTRG.223165 as targets of miR-21. We cloned the wild-type (WT) and mutant 3′-UTRs of the SOX6 and MSTRG.223165 mRNAs containing the miR-21 target site into the psiCHECK-2 reporter plasmid to generate psiCHECK-2-SOX6-3′UTR WT, psiCHECK-2-SOX6-3′UTR-mutant, psiCHECK-2-MSTRG.223165-3′UTR WT and psiCHECK-2-MSTRG.223165-3′UTR-mutant. These constructs were then cotransfected into 293T cells with miR-21 mimics or negative control respectively, and luciferase activity was measured 48 h later. The transfected cells were disrupted by the addition of 100 μl passive lysis buffer. A sample (20 μl) of the lysis mix was then added to 100 μl Luciferase Assay Reagent II (LAR II). The Firefly luciferase activity was determined as the internal control and 100 μl Stop & Glo® Reagent (Luciferase Assay Reagent, Promega) was added to determine the Renilla luciferase activity of the reporter gene.
Results
Hair Follicle Growth Process
The HE staining showed that at E90d, most of the PF had already developed, and some of the earlier-developed PF had visible columnar structures. SF were observed around some of the PF. PF occur earlier, with larger and longer hair follicles, and accessory structures such as sweat glands, sebaceous glands, and the arrector pili muscles. SF are smaller and grow near the epidermis of the PF (Figures 1A,B). At E120d, the SF has increased in number, and were separated from the PF but arranged in parallel (Figures 1C,D). At birth, some SF mature and penetrate the body surface (Figures 1E,F). The results of this study were consistent with those of Roger (Rogers, 2006) and Liu (Liu et al., 2015), indicating that these three stages are typical periods of SF development; these results support the accuracy and representativeness of the sample collection.
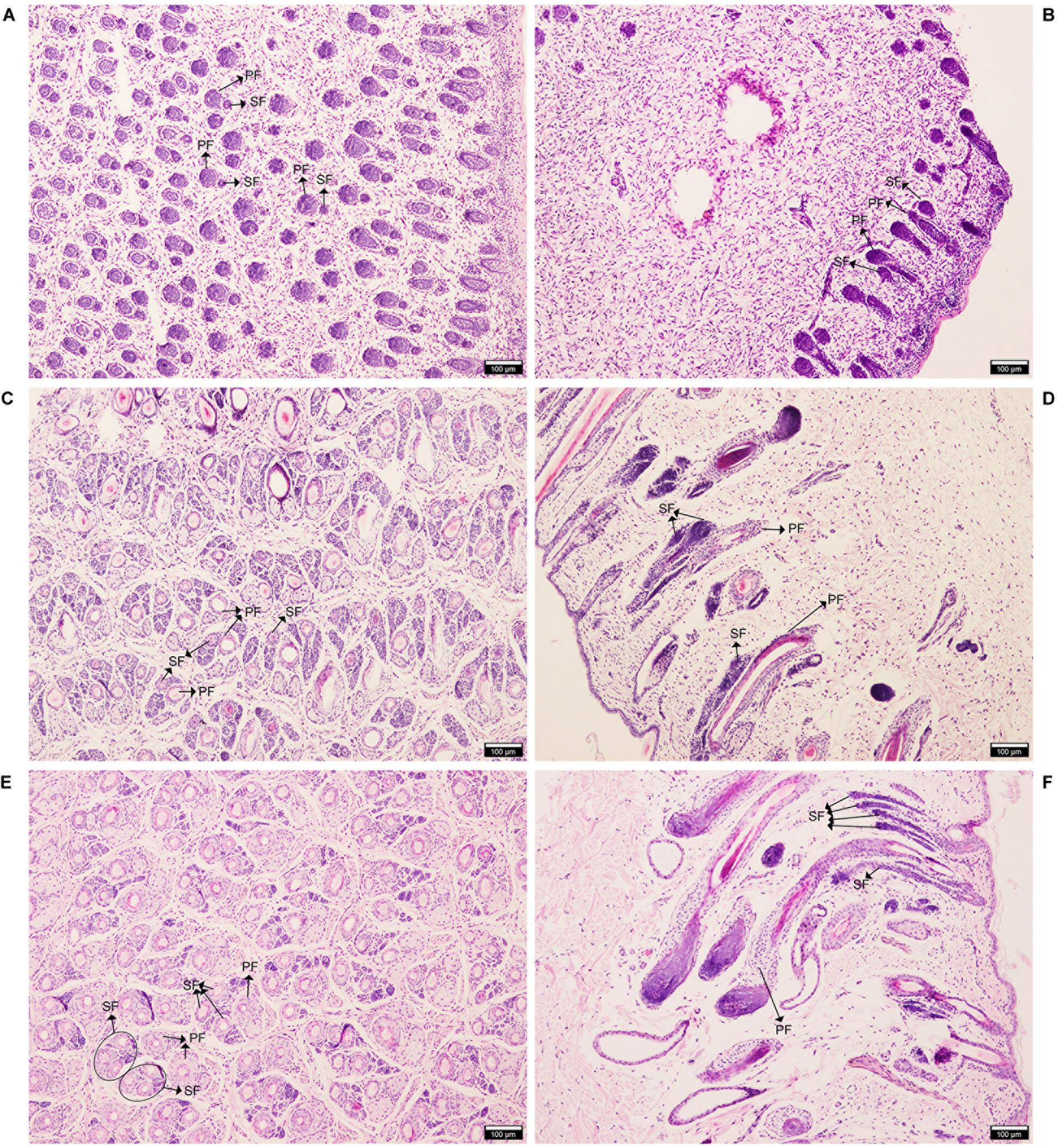
Figure 1. Hematoxylin and eosin (HE) staining of AFWS wool follicles at three developmental stages. Horizontal and longitudinal sections of primary and secondary wool follicles. (A,B) E90d, (C,D) E120d, (E,F) At birth. PF, primary wool follicle; SF, secondary wool follicle.
Identification of lncRNAs in AFWS Skin
In the analysis results of lncRNA, a total of 895,800,158 raw reads were produced from the Illumina PE 150 platform. After the raw data was filtered, 832,283,504 clean reads were obtained for subsequent analysis. We screened novel lncRNA for three types: lincRNA, intronic lncRNA, and anti-sense lncRNA. Subsequently, CNCI analysis (Sun et al., 2013), CPC analysis (Lei et al., 2007), PFAM protein domain analysis (Bateman et al., 2002; Mistry et al., 2007) and CPAT analysis (Wang et al., 2013) were conducted to determine the coding potential of the selected novel lncRNAs; the non-coding transcripts identified using these methods are shown in Figure 2. In total, 13,148 lncRNAs predicted using these four prediction methods were selected for subsequent analysis.
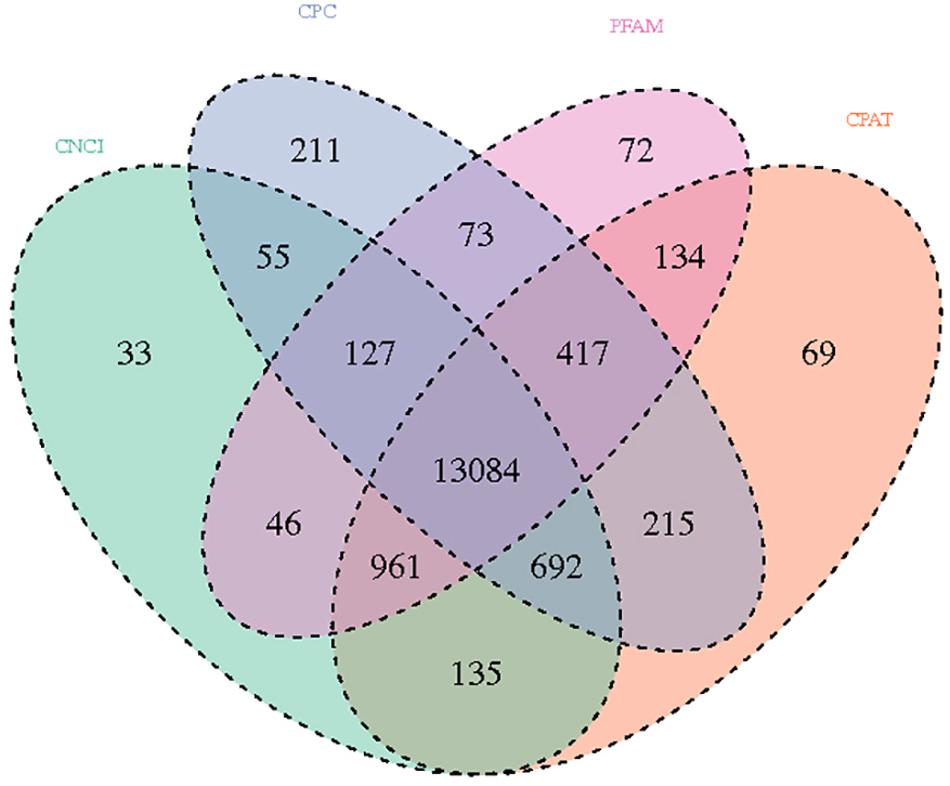
Figure 2. Venn diagram showing the total and unique lncRNA numbers predicted by CNCI, CPC, FPAM, and CPAT.
Comparative Analysis of lncRNA and mRNA
Transcription length, exon number, expression level and tissue specificity were used to compare the differences and characteristics of lncRNA and mRNA. Length distribution analysis that lncRNA was longer than mRNA (Supplementary Figure S1B), while the number and expression level of mRNAs were higher than those of lncRNAs (Supplementary Figures S1A,C). The Jensen–Shannon divergence (JS) score is an indicator of tissue expression bias, with higher scores indicating the more uneven gene expression in tissues. The JS score showed that the tissue specificity of mRNA was stronger than that of lncRNA (Supplementary Figure S1D).
The prediction of cis and trans target genes for differentially expressed lncRNAs was used to understand the function of the lncRNAs in the different growth stages. In the Birth_E90d comparison, 50 potential cis targets genes were identified for 26 lncRNAs, while 578 potential trans target genes were identified for 32 lncRNAs. In the Birth_E120d comparison, 155 potential cis targets genes were identified for 78 lncRNAs, while 1269 potential trans target genes were identified for 101 lncRNAs. In theE120d_E90d comparison, 424 potential targets cis genes were identified for 224 lncRNAs, while 2,256 potential trans target genes were identified for 235 lncRNAs. We also found that some lncRNAs have more than 50 target genes; such as MSTRG.421324, which predominantly target genes in the KRT family, according to previous reports, the KRT family is closely related to goat hair follicle growth (Gao et al., 2016), and dysregulation of KRT can cause hair disorders (Donet et al., 2008). LOC106990846 has 207 trans target genes, including TCF12, VEGFA, FOX13, and SMAD family genes (Potter et al., 2006; Kandyba et al., 2014; Du et al., 2018; Zhang et al., 2019b), all of which are involved in regulating the growth and differentiation of hair follicles. In addition, MSTRG.447490 and gene24735 target PRR9, which plays a regulatory role in HF keratinization and hair shaft differentiation (Gao et al., 2016). Detailed interactions between lncRNAs and mRNAs are listed in Supplementary Table S2.
Identification of miRNAs in AFWS Skin
From the RNA-seq results, 152 known miRNAs from 77 precursors were identified (Supplementary Table S3). By comparing the sequence of the species specified in the miRBase database (Release 21), we obtained the sequences of 432 novel miRNAs and their precursor sequences (Supplementary Table S4).
Identification of DE lncRNAs, mRNAs, and miRNAs
We used DEseq (Anders and Huber, 2010) to analyze the DE lncRNA, DE mRNA and DE miRNA according to the following screening criteria: DE lncRNA and DE mRNA, | log2Ratio| ≥ 1, and q < 0.05 and DE miRNA, P < 0.05 and | log2(Fold_change)| ≥ 1. Volcano plots, clustering maps, and Venn diagrams were used to describe the distribution of the DE lncRNAs, DE mRNAs, and DE miRNA among the three groups (Supplementary Figures S2–S4). The numbers of DE lncRNAs, DE mRNAs, and DE miRNA are listed in Table 1. A total of 41 DE lncRNAs were identified in the Birth_E90d comparison (upregulated: 35, downregulated: 6), 144 in the Birth_E120d comparison (upregulated: 125, downregulated: 19), and 384 in the E120d_E90d (upregulated: 144, downregulated: 240). A total 54 DE mRNAs were identified in the Birth_E90d (upregulated: 18, downregulated: 36), 340 in the Birth_E120d comparison (upregulated: 259, downregulated: 81), and 950 in the E120d_E90d comparison (upregulated: 306, downregulated: 644). A total 18 DE lncRNAs were identified in the Birth_E90d comparison (upregulated: 8, downregulated: 10), 31 in the Birth_E120d comparison (upregulated: 28, downregulated: 3), and 95 in the E120d_E90d comparison (upregulated: 14, downregulated: 81).
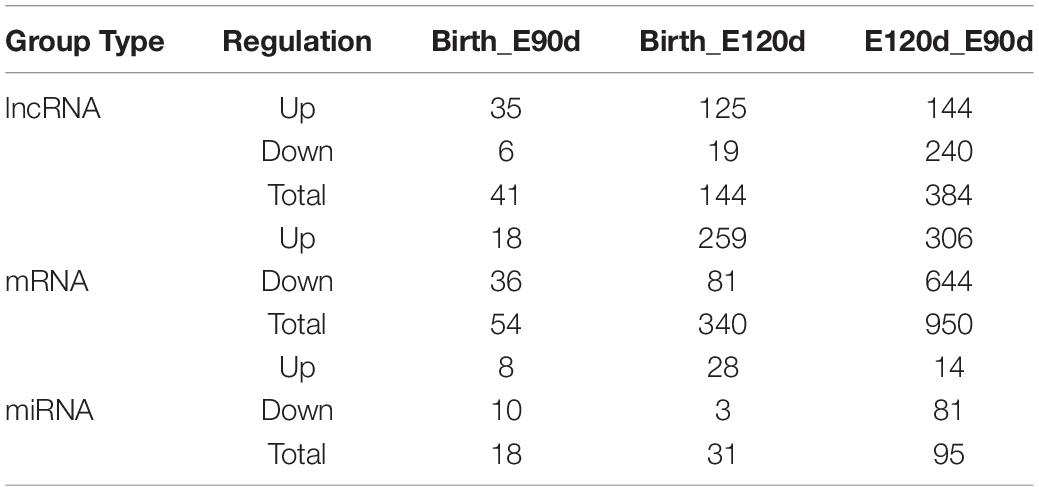
Table 1. Differentially expressed lncRNA, mRNA, and miRNA.
Details of the differentially expressed lncRNAs, mRNAs and miRNA among the three developmental stages are listed in Supplementary Tables S5–S7. It was found that novel lncRNAs, such as MSTRG.617489, MSTRG.136252, MSTRG.571405, and MSTRG.223165, and known lncRNAs, such as, LOC105613955, LOC105616569, and LOC105606646, were significantly differentially expressed and had high FPKM, indicating that these lncRNAs play a role during the growth of hair follicles. Moreover, several mRNAs, such as VCAN, KRT, HOXC13, and PRR9 as well as miRNAs such as the miR-200 family, let-7 family, miR-148a, and miR-143, detected in this study, which may have important roles in HF growth and development and were therefore implicated in these processes in AFWS.
Validation of RNA-Seq Results by qRT-PCR
To validate the RNA-seq results, we randomly selected four highly expressed lncRNAs, four mRNAs, and four miRNAs and detected their expression levels in the E90d, E120d, and Birth samples by qRT-PCR, the results were in accordance with the RNA-seq data, the correlation results for all RNAs were r > 0.8, which demonstrated the high reliability of the RNA-seq data (Figure 3).
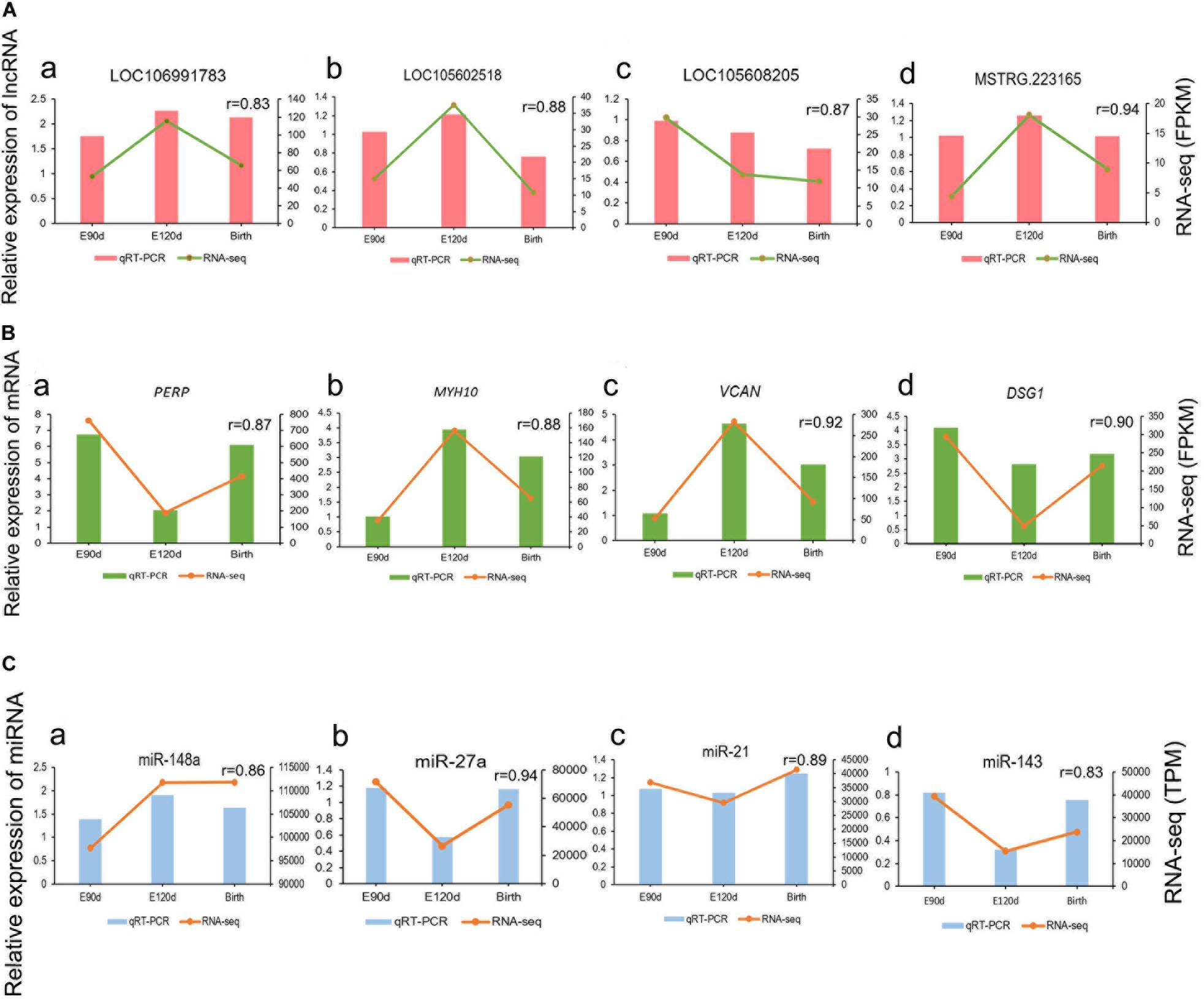
Figure 3. Validation of differentially expressed lncRNAs (A), mRNAs (B), and miRNAs (C) at the E90d, E120d, and Birth stages by qRT-PCR.
GO Enrichment and KEGG Pathway Analysis
lncRNA function is related to the adjacent protein coding gene at the genomic locus; therefore, the 50-kb protein coding genes upstream and downstream of the lncRNA were screened for GO and KEGG enrichment. Several GO items were found to be significantly enriched in the experimental groups (Supplementary Table S8). Some GO terms were not significantly enriched but related to skin and hair follicles; for example, hair follicle morphogenesis (GO:0031069), hair cycle process (GO:0022405), and positive regulation of hair follicle cell proliferation (GO:0071338). The KEGG pathways associated with target genes of DE lncRNAs between E90d, E120d and Birth of the HF development included the PI3K-Akt, MAPK, FoxO, and cGMP-PKG signaling pathways.
Gene Ontology annotation of DE mRNAs is shown in Supplementary Table S9. The GO terms identified include hair follicle growth terms, such as hair follicle morphogenesis (GO:0031069), hair cycle phase (GO:0044851), hair cycle (GO:0042633), negative regulation of hair cycle (GO:0042636), skin morphogenesis (GO:0043589), and hair cell differentiation (GO:0035315). According to the KEGG pathway analysis of the DE mRNAs, cell adhesion molecules (CAMs) and tight junctions were associated with the growth of skin/hair.
Finally, in the GO enrichment analysis, target genes of DE miRNAs (Supplementary Table S10) were found to be related to aspects of HF development, such as hair cell differentiation (GO:0035315), skin development (GO:0043588), regulation of hair follicle development (GO:0051797), hair cycle process (GO:0022405), hair follicle development and (GO:0001942). According to the KEGG pathway analysis of the DE miRNAs, the cAMP, PI3K-Akt, and AMPK signaling pathways were related to HF development. The detailed information of DE lncRNAs, DE mRNAs, DE miRNAs were listed in Supplementary Table S11.
Construction of the DE lncRNA–DE miRNA–DE mRNA Network
To increase our understanding of the molecular regulation mechanism of hair follicle growth, we investigated the relationship between lncRNA, miRNA, and mRNA by constructing the DE lncRNA-DE miRNA-DE mRNA network (Supplementary Figures S5, S6). Several interesting lncRNA–miRNA pairs were revealed, such as miR-200a -MSTRG.313133, miR-21/miR-143 and their target lncRNA, MSTRG.172759. The miRNA–mRNA network indicated that several regulatory interactions, such as miR-150-KRT1, miR-370-3p and its target gene KRT32 (gene15074), KRT35 (gene15075), VCAN (gene10153), and FOXN1 (gene14402), may be important in hair follicle development. Subsequently, according to the principle of ceRNA regulation, we identified target mRNAs and target lncRNAs with the same miRNA binding sites to construct the lncRNA–miRNA–mRNA regulatory network (Supplementary Figure S7). In this model, miRNAs form the center of the network with lncRNA as the bait, and mRNA as the target, suggesting that lncRNA acts as a sponge of miRNA to regulate gene expression. In short, lncRNA and mRNA have the same expression trends that are opposite expression to that of miRNA. In this study, we identified 14 ceRNA pairs that shared 10 miRNAs, such as MSTRG.222885-miR-152-SPON1, MSTRG.223165-miR-21-SOX6, and MSTRG.308950-miR-103-IGF2R, all of which warrant further investigation (Supplementary Table S12).
After further analysis, we found significant differences in the expression levels of miR-21 during the three stages of HF development (Birth-E120d and E120d-E90d), with significantly higher expression at Birth compared with that at E120d, and also at E90d compared with that at E120d. At the same time, we also found significant difference is SOX6 expression in the E120d-E90d and Birth-E120d groups, with the lowest expression at E90d, and the highest at E120d. Previous studies have shown that miR-21 participates in the growth of animal hair follicles (Zhai et al., 2019), and the SOX family was reported to play a role in cutaneous melanoma (Villani et al., 2017; Noto et al., 2019).
From the results of our study, MSTRG.223165 was identified as a ceRNA for miR-21, which targets SOX6. Therefore, we speculated that MSTRG.223165-miR-21-SOX6 plays a role in the growth of sheep wool follicles. Dual-luciferase assays showed that miR-21 reduced luciferase activity by binding to target sites on MSTRG.223165 and the SOX6 3′UTR (Figure 4). These interactions between lncRNA, miRNA, and mRNA indicate a potential regulatory mechanism in skin development and AFWS hair follicle growth.
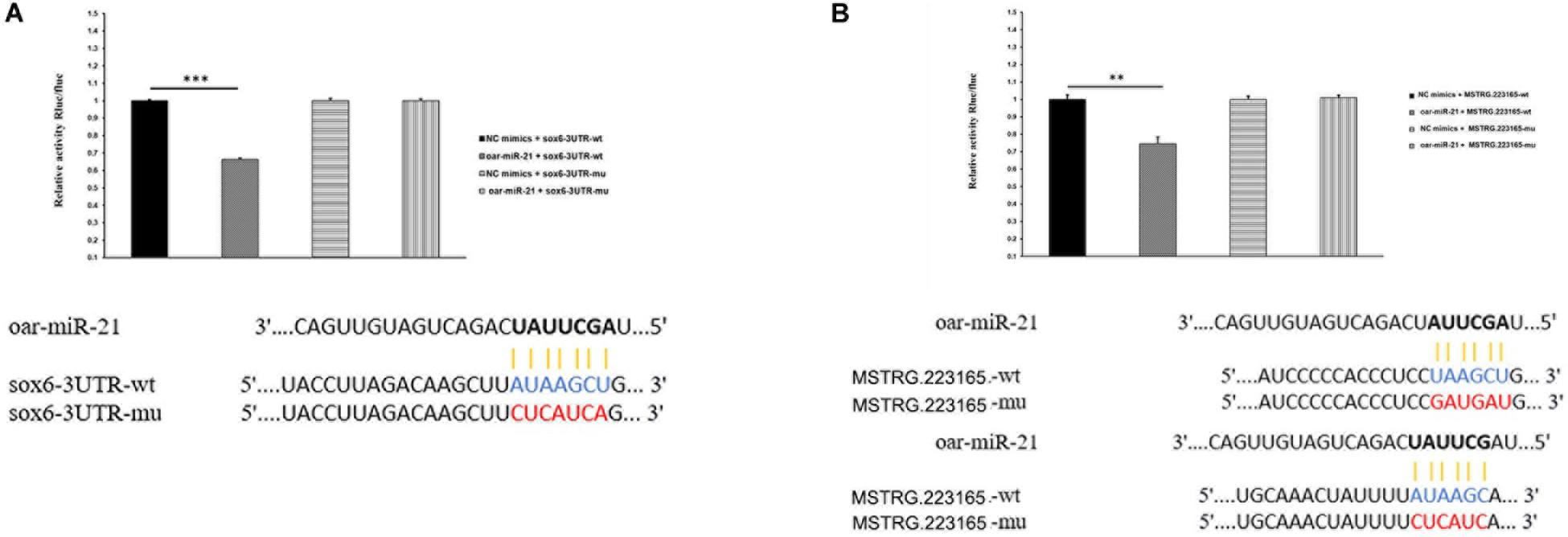
Figure 4. (A) miR-21 putative binding sites in SOX6 3′-UTR. Pairing schematic of SOX6 3′-UTR and miR-21; blue text indicates wild-type sites, and red text indicates mutated sites in the psiCHECK-2 reporter plasmid. The constructs were cotransfected with miR-21 mimics (or negative control) into 293T cells and dual-luciferase assays were performed 48h after transfection. (Data represent mean ± SD, **P < 0.01, ***P < 0.001.) (B) miR-21 putative binding sites in MSTRG.223165 3′-UTR. Pairing schematic of MSTRG.223165 3′-UTR and miR-21; blue text indicates wild-type sites, and red text indicates mutated sites in the psiCHECK-2 reporter plasmid. The constructs were cotransfected with miR-21 mimics (or negative control) into 293T cells and dual-luciferase assays were performed 48 h after transfection. (Data represent mean ± SD, **P < 0.01, ***P < 0.001.)
Discussion
In many mammals, hair follicles undergo a variety of morphological changes at different times, with certain regularities that exhibit periodicity (anagen, catagen, telogen). Hair growth has been studied in many animal models, including mice (Hansen et al., 1984; Slominski et al., 1994; Kwack et al., 2019), American mink (Zhang et al., 2019a), rabbit (Zhao et al., 2019), yak (Song et al., 2019), and goat (Meyer et al., 1997). Following development from stem cells, hair follicles undergo rapid and cyclical growth, and are surrounded by dermal fibroblasts. During the catagen stage, HFs are surrounded by interfollicular dermal fibroblasts, and the hair bulb starts to shrink. Finally, HFs enter the telogen stage, the hair shaft stops growing, and that hair falls out (Stenn and Paus, 2001). Although many animals have similar hair growth patterns, they may be affected by environmental factors, such as light, season, and temperature (Sun et al., 2019; Zhang et al., 2020). In mammals, SF produce fine hair, which determines wool quality. HE staining showed that PF grew at E90d, while SF were smaller. PF and SF were separated at E120d, and SF matured at Birth, this model supports our research.
In recent years, studies have shown that ncRNAs play an important role in biological processes (Suh, 2018; Tosar et al., 2018), including hair follicle growth cycle (Fu et al., 2014). We used RNA-sequencing technology to identify the lncRNA, mRNA and miRNA molecules expressed in sheep skin during different HF development across two fetal (E90d, E120d) and one postnatal stages (Birth). We detected 461 lncRNAs, 1,009 mRNAs and 106 miRNAs, which were differentially expressed in all comparison groups (Birth-E90, Birth-E120d, and E120d-E90d). We also investigated the expression patterns and functions of lncRNAs, mRNAs and miRNAs to clarify their interactions and relationships with sheep HF development. Several lncRNAs related to hair follicle growth have been reported, such as lnc-000133 and lncRNA-HOTAIR, which participate in hair follicle growth in Cashmere goats (Jiao et al., 2019; Zheng et al., 2019), although the specific functions of lncRNA in hair follicle development remain to be fully elucidated. Studies have shown that mRNAs and miRNAs, such as FZD4, FZD9, DKK1, BAMBI, miR-143, and the miR-200 family, play roles in hair follicle development. In accordance with a previous report, we found that FZD4, FZD9, DKK1, and BAMBI were differentially expressed during hair follicle development in fine wool sheep (Chen, 2018). miR-143 was detected in our study and has also been reported in association with the HF growth of HU sheep (Huttenhofer et al., 2005). Reports have indicated that the miR-200 family may be a key miRNA in hair follicle development (Hoefert et al., 2018; Liu et al., 2018); thus, we speculate that miR-200b, which was identified in our study, may play a role in the growth of HF of AFWS. Our results were consistent with these reports, provide guidance for our research.
In this study, we identified 493 cis and 2,415 trans targets in 461 differentially expressed lncRNAs, 1,009 DE mRNAs, and 3,738 target genes for 111 differentially expressed miRNAs. GO annotation and KEGG pathway analysis performed to explore the potential functions and underlying mechanism of these differentially expressed lncRNAs, mRNAs, and miRNAs. GO has a total of three ontologies that describe the molecular function of the gene (MF), the location of the cell (CC), and the biological process involved (BP). KEGG is a database that can be used to decipher the genome by predicting the role of cellular activities in organisms. Our analysis showed that multiple GO terms and signaling pathways form complex mechanisms that regulate the growth of hair follicles. These GO terms included hair follicle development (GO:0001942), hair cycle (GO:0042633), and regulation of hair cycle (GO:0042634).
KEGG pathways include the FoxO, cGMP-PKG, cAMP, PI3K-Akt, and AMPK signaling pathways. Previous studies have shown that these pathways participate in, or regulate the growth process of wool follicles (Lv et al., 2017; Jiankui et al., 2019; Ranran et al., 2020). It was found that BRAF is enriched in CAMP and FoxO signaling pathways, and PIK3R3 is enriched in the CAMP, JAK-STAT, and AMP signaling pathways. In this study, we found that BRAF and PIK3R3 were significantly differentially expressed and downregulated in E120d-E90d. BRAF inhibitors are involved in the development of hair curls and have been used treat hair loss as well as in hair care products (Keating and Dasanu, 2017). In mice with spontaneous fur mutations, researchers found that PIK3R3 may affect cell cycle, immune response and skin development during mouse growth (Ji et al., 2017). We also found that MSTRG.172760, MSTRG.421324, MSTRG.482102, MSTRG.535526, MSTRG.610044, MSTRG.653547, MSTRG.722949, MSTRG.730613 and gene3930 all target gene360 (PIK3R3). MSTRG.173186, MSTRG.225489, and MSTRG.617489 target gene8419 (BRAF), suggesting that these lncRNAs regulate hair follicle growth through gene360 (PIK3R3) and gene8419 (BRAF), which are therefore implicated as candidate genes for hair follicle growth.
miRNA–lncRNA and miRNA–mRNA networks provide clues for analyzing gene regulation. In our study, we found several interesting networks, such as miR-200a-MSTRG.313133, miR-21/miR-143 and their target lncRNA MSTRG.172759. Among them, the miRNAs were associated with hair growth, and their target gene lncRNAs were expressed at high levels with differential expression patterns during the growth. It has been reported that the miR-200 family regulates cell adhesion in the hair germ cells and influence morphogenesis of the hair follicle (Hoefert et al., 2018). miR-21 may affect hair follicle growth by regulating its target genes (Zhai et al., 2019), and miR-143 is a candidate gene in the regulation of hair follicle growth in Hu sheep (Gao et al., 2017). Thus, this network may play a role in regulating the growth of hair follicles. In addition, miRNA–mRNA networks, such as miR-150-KRT1, miR-370-3p and its target gene KRT32 (gene15074), KRT35 (gene15075), VCAN (gene10153), FOXN1 (gene14402) and their target genes have been studied in relation to hair follicle or skin growth. miR-150 participates in the regulation the formation of hair follicle melanoma (Wang et al., 2019). miR-370-3p acts as a target of circRNA_NEK6 and plays a role in proliferation of thyroid carcinoma (Chen et al., 2018). The KRT family is related to the degree of curl and shape of horsehair (Morgenthaler et al., 2017). VCAN is a marker gene for hair papilla cells, and its growth rate is not affected by external light intensity (Jampa-Ngern et al., 2017). FOXN1 may affect the growth of secondary hair follicles in Cashmere goats (Zhang et al., 2019c).
The mechanism of ceRNA regulation has been extensively researched in various diseases (Tang et al., 2019; Tao et al., 2019). Recently, TMPO-AS1 has been identified as a competitive endogenous RNA that promotes tumorigenesis of osteosarcoma by regulating the miR-199a-5p/WNT7B axis, which provides a potential therapeutic target for osteosarcoma patients (Cui and Zhao, 2019). Studies of The Cancer Genome Atlas (TCGA) database showed that lncRNA MATN1-AS1 promotes the progression of gliomas by regulating the miR-200b/c/429-CHD1 axis, suggesting that MATN1-AS1 may be a therapeutic target for glioma (Zhu et al., 2020). In a study of ncRNA and RNA in the cashmere goat hair follicle cycle, Wang et al. (2017) constructed ceRNA networks by RNA-seq and bioinformatics analysis. Based on the results, three ceRNA networks, miR-221-5p-lnc_000679-WNT3, were selected as candidate ceRNA networks that may be involved in the regulation of hair follicle growth. Zhang first studied the regulatory relationship of lncRNA–mRNA in the development of fine wool follicles (Yangfan et al., 2018), which provided a reference for our research. Our experiment is the first study to provide RNA-seq data for hair follicle development from the fetal stage to birth in AFWS, and also the first study on the lncRNA–miRNA–mRNA regulatory network in different development stages of AFWS. The ceRNA results show that some lncRNA–mRNA interact with multiple different miRNAs, and one miRNA can also target multiple mRNAs at the same time. This phenomenon indicates that miRNA occupies a central position in the ceRNA network, linking lncRNA and mRNA. Collation of the target mRNA and target lncRNA data with the same miRNA binding site revealed that interactions such as MSTRG.222885-miR-152-SPON1, MSTRG.223165-miR-21-SOX6, and MSTRG.308950 -miR-103 -IGF2R are involved in the ceRNA network. It was found that SPON1 regulated the growth, differentiation and morphogenesis of hair follicles via Wnt, BMP and other signaling pathways (Muhammad et al., 2019). It has been reported that IGF2R as a target gene of miR-211 activates the MAPK signaling pathway and thereby inhibits the formation of skin melanoma (Dror et al., 2016). MAPK is also a signaling pathway for hair follicle growth and development and IGF2R may also affect hair color and hair follicle growth through this signal pathway (Xiao et al., 2019).
In this study, we mainly studied the network of MSTRG.223165-miR-21-SOX6, in which MSTRG.223165 sponges miR-21, which targets SOX6; thus, this provides important information about the growth of AFWS hair follicles. It has been reported thatmiRNA-21 is an important downstream component of BMP signaling and plays a significant role in skin development (Ahmed et al., 2011). Studies of super merino and small-tailed Han sheep showed that CNKSR2, KLF3 and TNPO1 are the target genes of miR-21, and may therefore be involved in the function of miR-21 in hair follicle development (Zhai et al., 2019). The Sox family is expressed in early hair follicle development, and Sox13, which is essential for epidermal and adnexal development, can be used as a useful marker of early hair follicle development (Noto et al., 2019). SOX2 and SOX18 are critical for determining the type of hair follicle, and Sox18 regulates the normal differentiation of dermal papillae of all hair types (Villani et al., 2017). During the differentiation of neural stem cells, miR-21 expression was increased, and reduced SOX protein expression by binding to SOX2, thereby promoting differentiation (Ni et al., 2014). In our analysis of DE lncRNA-DE miRNA-DE mRNA, we found that MSTRG.223165, miR-21 and SOX6 were correlated, suggesting that MSTRG.223165 regulated the expression of Sox6. Thus, we hypothesize that MSTRG.223165 functions as a regulatory gene in the growth of hair follicles, although the molecular regulation mechanism remains to be clarified.
Conclusion
In this study, we investigated the expression of lncRNAs, mRNAs, and miRNAs in the skin during different developmental stages in AFWS. GO annotation and KEGG pathway analysis were used to identify the candidate lncRNAs, mRNAs, and miRNAs in the developmental stages of the wool follicle growth. We also constructed the ceRNA networks of MSTRG.223165 -miR-21- SOX6 and dual-luciferase assays were used to verify the target relationships in this network to investigate the factors that affect wool follicle growth. These results provide the foundation for further exploration of the molecular mechanism underlying the regulation of wool growth.
Data Availability Statement
Our data is being uploaded to the SRA database, the lncRNA-Seq and mRNA-Seq data was submitted to the SRA database under accession number SRP240733. miRNA-Seq data was submitted to the SRA database under accession number SRP253232.
Ethics Statement
All experimental and surgical procedures involved in this study followed the “Guidelines for Experimental Animals” of the Ministry of Science and Technology (Beijing, China). Operations and Animal Care were sustained by the experimental animal ethics committee of Qingdao Agricultural University.
Author Contributions
RZ, JL, and NL conceived and designed this study. RZ, NL, LrL, and FY participated in sample collection. RZ, JL, and LlL performed the experiments. RZ, YW, FY, and HL analyzed the data and prepared the figures and tables. RZ, NL, and JH wrote the manuscript. All authors reviewed and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 31402047), by the Earmarked Fund for Modern China Wool & Cashmere Technology Research System (CARS-39), by a Project of Shandong Province Higher Educational Science and Technology Program (J18KA136), and by the Project of Shandong Province Agricultural Variety Program (2019LZGC012).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00590/full#supplementary-material
FIGURE S1 | Comparative analysis of lncRNAs and mRNAs. (A) Distribution of exon numbers. (B) Length distribution. (C) Expression levels. (D) JS score density distribution.
FIGURE S2 | Identification of differentially expressed lncRNAs during three developmental stages of AFWS. (A–C) Volcano plots showing the differentially expressed lncRNAs in pairwise comparison groups (upregulated or downregulated). (D) Venn diagram showing the number of overlapping differentially expressed lncRNAs in compared groups. (E) Heatmap of differentially expressed lncRNAs; yellow and blue indicate high and expression levels, respectively.
FIGURE S3 | Identification of differentially expressed mRNA during three developmental stages of AFWS. (A–C) Volcano plots showing the differentially expressed mRNAs in pairwise comparison groups (upregulated or downregulated). (D) Venn diagram showing the number of overlapping differentially expressed mRNAs in compared groups. (E) Heatmap of differentially expressed mRNAs; yellow and blue indicate high and expression levels, respectively.
FIGURE S4 | Identification of differentially expressed miRNA during three developmental stages of AFWS. (A–C) Volcano plots showing the differentially expressed miRNAs in pairwise comparison groups (upregulated or downregulated). (D) Venn diagram showing the number of overlapping differentially expressed miRNAs in compared groups. (E) Heatmap of differentially expressed miRNAs; yellow and blue indicate high and expression levels, respectively.
FIGURE S5 | Interactions between miRNA and mRNAs in AFWS wool follicles. (A) miRNA–mRNA network between birth and E90d. (B) miRNA–mRNA network between birth and E120d. (C) miRNA–mRNA network between E120d and E90d.
FIGURE S6 | Interactions between lncRNAs and miRNAs in AFWS wool follicles. (A) lncRNA–miRNA network between birth and E90d. (B) lncRNA–miRNA network between birth and E120d. (C) lncRNA–miRNA network between E120d and E90d.
FIGURE S7 | Interactions of lncRNA–miRNA–mRNA in AFWS wool follicles. (A) lncRNA–miRNA–mRNA network between birth and E90d. (B) lncRNA–miRNA–mRNA network between birth and E120d. (C) lncRNA–miRNA–mRNA network between E120d and E90d.
TABLE S1 | Detailed information of primers used for qRT-PCR analysis.
TABLE S2 | Target mRNA prediction (cis and trans) of lncRNA.
TABLE S3 | Annotated mature miRNAs and precursors.
TABLE S4 | Novel mature miRNA sequence and precursor sequence.
TABLE S5 | Identification of differentially expressed lncRNAs during three developmental stages of AFWS. (A–C) Volcano plots showing the differentially expressed lncRNAs in pairwise comparison groups (upregulated or downregulated). (D) Venn diagram showing the number of overlapping differentially expressed lncRNAs in compared groups. (E) Heatmap of differentially expressed lncRNAs; yellow and blue indicate high and expression levels, respectively.
TABLE S6 | Identification of differentially expressed mRNA during three developmental stages of AFWS. (A–C) Volcano plots showing the differentially expressed mRNAs in pairwise comparison groups (upregulated or downregulated). (D) Venn diagram showing the number of overlapping differentially expressed mRNAs in compared groups. (E) Heatmap of differentially expressed mRNAs; yellow and blue indicate high and expression levels, respectively.
TABLE S7 | Identification of differentially expressed miRNA during three developmental stages of AFWS. (A–C) Volcano plots showing the differentially expressed miRNAs in pairwise comparison groups (upregulated or downregulated). (D) Venn diagram showing the number of overlapping differentially expressed miRNAs in compared groups. (E) Heatmap of differentially expressed miRNAs; yellow and blue indicate high and expression levels, respectively.
TABLE S8 | Gene ontology annotation of differentially expressed lncRNAs between E90d, E120d, and birth.
TABLE S9 | Gene ontology annotation of differentially expressed mRNAs between E90d, E120d, and birth.
TABLE S10 | Gene ontology annotation of differentially expressed miRNAs between E90d, E120d, and birth.
TABLE S11 | Detailed results of KEGG pathway analysis of lncRNAs, mRNAs, and miRNAs.
TABLE S12 | Fourteen pairs of ceRNAs consistent with the ceRNA regulation mechanism.
Footnotes
References
Ahmed, M. I., Mardaryev, A. N., Lewis, C. J., Sharov, A. A., and Botchkareva, N. V. (2011). MicroRNA-21 is an important downstream component of BMP signalling in epidermal keratinocytes. J. Cell Sci. 124, 3399–3404. doi: 10.1242/jcs.086710
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/s0092-8674(04)00045-5
Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S. R., et al. (2002). The Pfam protein families database. Nucleic Acids Res. 30, 276–280. doi: 10.1093/nar/30.1.276
Burge, S. W., Daub, J., Eberhardt, R., Tate, J., Barquist, L., Nawrocki, E. P., et al. (2013). Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 41, D226–D232. doi: 10.1093/nar/gks1005
Cao, Y., You, S., Yao, Y., Liu, Z. J., Hazi, W., Li, C. Y., et al. (2018). Expression profiles of circular RNAs in sheep skeletal muscle. Asian Australas. J. Anim. Sci. 31, 1550–1557. doi: 10.5713/ajas.17.0563
Carter, H. B., and Hardy, M. H. (1943). Studies in the Biology of the Skin and Fleece of Sheep. Melbourne: H. E. Daw, Government Printer.
Chen (2018). Transcriptome Analysis of Skin in Primary Hair Follicle Substrate Development of Shaggy and Fine Hair Sheep. Master’s thesis, Huazhong Agricultural University, Wuhan.
Chen, F., Feng, Z., Zhu, J., Liu, P., Yang, C., Huang, R., et al. (2018). Emerging roles of circRNA_NEK6 targeting miR-370-3p in the proliferation and invasion of thyroid cancer via Wnt signaling pathway. Cancer Biol. Ther. 19, 1139–1152. doi: 10.1080/15384047.2018.1480888
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Cui, C. Y., Kunisada, M., Piao, Y., Childress, V., Ko, M. S., and Schlessinger, D. (2010). Dkk4 and Eda regulate distinctive developmental mechanisms for subtypes of mouse hair. PLoS One 5:e10009. doi: 10.1371/journal.pone.0010009
Cui, H., and Zhao, J. (2019). LncRNA TMPO-AS1 serves as a ceRNA to promote osteosarcoma tumorigenesis by regulating miR-199a-5p/WNT7B axis. J. Cell. Biochem. 121, 2284–2293. doi: 10.1002/jcb.29451
Donet, E., Bayo, P., Calvo, E., Labrie, F., and Perez, P. (2008). Identification of novel glucocorticoid receptor-regulated genes involved in epidermal homeostasis and hair follicle differentiation. J. Steroid Biochem. Mol. Biol. 108, 8–16. doi: 10.1016/j.jsbmb.2007.05.033
Dror, S., Sander, L., Schwartz, H., Sheinboim, D., Barzilai, A., Dishon, Y., et al. (2016). Melanoma miRNA trafficking controls tumour primary niche formation. Nat. Cell Biol. 18, 1006–1017. doi: 10.1038/ncb3399
Du, K. T., Deng, J. Q., He, X. G., Liu, Z. P., Peng, C., and Zhang, M. S. (2018). MiR-214 regulates the human hair follicle stem cell proliferation and differentiation by targeting EZH2 and Wnt/beta-catenin signaling way in vitro. Tissue Eng. Regen. Med. 15, 341–350. doi: 10.1007/s13770-018-0118-x
Enright, A. J., John, B., Gaul, U., Tuschl, T., Sander, C., and Marks, D. S. (2003). MicroRNA targets in Drosophila. Genome Biol. 5:R1. doi: 10.1186/gb-2003-5-1-r1
Fu, S., Zhao, H., Zheng, Z., Li, J., and Zhang, W. (2014). Melatonin regulating the expression of miRNAs involved in hair follicle cycle of cashmere goats skin. Yi Chuan 36, 1235–1242. doi: 10.3724/SP.J.1005.2014.1235
Gao, W., Sun, W., Yin, J., Lv, X., Bao, J., Yu, J., et al. (2017). Screening candidate microRNAs (miRNAs) in different lambskin hair follicles in Hu sheep. PLoS One 12:e0176532. doi: 10.1371/journal.pone.0176532
Gao, Y., Wang, X., Yan, H., Zeng, J., Ma, S., Niu, Y., et al. (2016). Comparative transcriptome analysis of fetal skin reveals key genes related to hair follicle morphogenesis in cashmere goats. PLoS One 11:e0151118. doi: 10.1371/journal.pone.0151118
Hamanaka, R. B., Glasauer, A., Hoover, P., Yang, S., Blatt, H., Mullen, A. R., et al. (2013). Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci. Signal. 6:ra8. doi: 10.1126/scisignal.2003638
Hansen, L. S., Coggle, J. E., Wells, J., and Charles, M. W. (1984). The influence of the hair cycle on the thickness of mouse skin. Anat. Rec. 210, 569–573. doi: 10.1002/ar.1092100404
Hardy, M. H., and Lyne, A. G. (1956). The pre-natal development of wool follicles in merino sheep. Australian J. Biol. Sci. 9, 423–441.
Hoefert, J. E., Bjerke, G. A., Wang, D., and Yi, R. (2018). The microRNA-200 family coordinately regulates cell adhesion and proliferation in hair morphogenesis. J. Cell Biol. 217, 2185–2204. doi: 10.1083/jcb.201708173
Huttenhofer, A., Schattner, P., and Polacek, N. (2005). Non-coding RNAs: Hope or hype? Trends Genet. 21, 289–297. doi: 10.1016/j.tig.2005.03.007
Jampa-Ngern, S., Viravaidya-Pasuwat, K., Suvanasuthi, S., and Khantachawana, A. (2017). Effect of laser diode light irradiation on growth capability of human hair follicle dermal papilla cells. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2017, 3592–3595. doi: 10.1109/EMBC.2017.8037634
Ji, Z. H., Chen, J., Gao, W., Zhang, J. Y., Quan, F. S., Hu, J. P., et al. (2017). Cutaneous transcriptome analysis in NIH hairless mice. PLoS One 12:e0182463. doi: 10.1371/journal.pone.0182463
Jiankui, W., Kai, C., Zu, Y., Tun, L., Guoying, H., Deping, H., et al. (2019). Transcriptome analysis of improved wool production in skin-specific transgenic sheep overexpressing ovine β-catenin. Int. J. Mol. Sci. 20:620. doi: 10.3390/ijms20030620
Jiao, Q., Yin, R. H., Zhao, S. J., Wang, Z. Y., Zhu, Y. B., Wang, W., et al. (2019). Identification and molecular analysis of a lncRNA-HOTAIR transcript from secondary hair follicle of cashmere goat reveal integrated regulatory network with the expression regulated potentially by its promoter methylation. Gene 688, 182–192. doi: 10.1016/j.gene.2018.11.084
Kandyba, E., Hazen, V. M., Kobielak, A., Butler, S. J., and Kobielak, K. (2014). Smad1 and 5 but not Smad8 establish stem cell quiescence which is critical to transform the premature hair follicle during morphogenesis toward the postnatal state. Stem Cells 32, 534–547. doi: 10.1002/stem.1548
Keating, M., and Dasanu, C. A. (2017). Late-onset robust curly hair growth in a patient with BRAF-mutated metastatic melanoma responding to dabrafenib(Article). J. Oncol. Pharm. Pract. 23, 309–312. doi: 10.1177/1078155216635854
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kwack, M. H., Kim, J. C., and Kim, M. K. (2019). Ectodysplasin-A2 induces apoptosis in cultured human hair follicle cells and promotes regression of hair follicles in mice. Biochem. Biophys. Res. Commun. 520, 428–433. doi: 10.1016/j.bbrc.2019.10.031
La, Y., Tang, J., He, X., Di, R., Wang, X., Liu, Q., et al. (2019). Identification and characterization of mRNAs and lncRNAs in the uterus of polytocous and monotocous Small Tail Han sheep (Ovis aries). PeerJ 7:e6938. doi: 10.7717/peerj.6938
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25.
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854. doi: 10.1016/0092-8674(93)90529-y
Legnini, I., Di Timoteo, G., Rossi, F., Morlando, M., Briganti, F., Sthandier, O., et al. (2017). Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol. Cell 66, 22–37.e9. doi: 10.1016/j.molcel.2017.02.017
Lei, K., Yong, Z., Ye, Z. Q., Liu, X. Q., Zhao, S. Q., Wei, L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349.
Li, C., Li, X., Ma, Q., Zhang, X., Cao, Y., Yao, Y., et al. (2017). Genome-wide analysis of circular RNAs in prenatal and postnatal pituitary glands of sheep. Sci. Rep. 7:16143. doi: 10.1038/s41598-017-16344-y
Li, X., Yang, L., and Chen, L. L. (2018). The biogenesis, functions, and challenges of circular RNAs. Mol. Cell 71, 428–442. doi: 10.1016/j.molcel.2018.06.034
Li, Y., Kong, L., Deng, M., Lian, Z., Han, Y., Sun, B., et al. (2019). Heat stress-responsive transcriptome analysis in the liver tissue of Hu sheep. Genes 10:395. doi: 10.3390/genes10050395
Liu, N., Wang, C., He, J., Cheng, M., Liu, K., Li, L., et al. (2015). Study on hair follicle development and morphological structure in different parts of Aohan fine wool sheep. Chin. J. Anim. Sci. 2015, 1–5.
Liu, Y., Wang, L., Li, X., Han, W., Yang, K., Wang, H., et al. (2018). High-throughput sequencing of hair follicle development-related micrornas in cashmere goat at various fetal periods. Saudi J. Biol. Sci. 25, 1494–1508. doi: 10.1016/j.sjbs.2017.12.009
Lv, X., Sun, W., Yin, J., Ni, R., Su, R., Wang, Q., et al. (2017). An integrated analysis of microRNA and mRNA expression profiles to identify RNA expression signatures in lambskin hair follicles in Hu Sheep. PLoS One 11:e0157463. doi: 10.1371/journal.pone.0157463
Mann, S. J. (1968). The tylotrich (hair) follicle of the American opossum. Anat. Rec. 160, 171–179. doi: 10.1002/ar.1091600203
Mercer, T. R., and Mattick, J. S. (2013). Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 20, 300–307. doi: 10.1038/nsmb.2480
Meyer, W., Saglam, M., Tanyolac, A., Schwarz, R., and Hetzel, U. (1997). [Histochemical and biochemical studies of the circannual aspects of carbohydrate-dependent energy metabolism of the hair follicles of Turkish Angora goats]. Schweiz. Arch. Tierheilkd. 139, 78–83.
Mistry, J., Bateman, A., and Finn, R. D. (2007). Predicting active site residue annotations in the Pfam database. BMC Bioinformatics 8:298. doi: 10.1186/1471-2105-8-298
Moore, G. P. M., Jackson, N., Isaacs, K., and Brown, G. (1998). Pattern and morphogenesis in skin. J. Theor. Biol. 191, 87–94. doi: 10.1006/jtbi.1997.0567
Morgenthaler, C., Diribarne, M., Capitan, A., Legendre, R., Saintilan, R., Gilles, M., et al. (2017). A missense variant in the coil1A domain of the keratin 25 gene is associated with the dominant curly hair coat trait (Crd) in horse. Genet. Sel. Evol. 49:85. doi: 10.1186/s12711-017-0359-5
Muhammad, S. A., Fatima, N., Paracha, R. Z., Ali, A., and Chen, J. Y. (2019). A systematic simulation-based meta-analytical framework for prediction of physiological biomarkers in alopecia. J. Biol. Res. 26:2. doi: 10.1186/s40709-019-0094-x
Ni, Y., Zhang, K., Liu, X., Yang, T., Wang, B., Fu, L., et al. (2014). miR-21 promotes the differentiation of hair follicle-derived neural crest stem cells into Schwann cells. Neural Regen. Res. 9, 828–836.
Nixon, A. J., Broad, L., Saywell, D. P., and Pearson, A. J. (1996). Transforming growth factor-alpha immunoreactivity during induced hair follicle growth cycles in sheep and ferrets. J. Histochem. Cytochem. 44, 377–387. doi: 10.1177/44.4.8601697
Noto, M., Noguchi, N., Ishimura, A., Kiyonari, H., Abe, T., Suzuki, T., et al. (2019). Sox13 is a novel early marker for hair follicle development(Article). Biochem. Biophys. Res. Commun. 862–868. doi: 10.1016/j.bbrc.2018.12.163
Okazaki, Y., Furuno, M., Kasukawa, T., Adachi, J., Bono, H., Kondo, S., et al. (2002). Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420, 563–573. doi: 10.1038/nature01266
Pamudurti, N. R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). Translation of CircRNAs. Mol. Cell 66, 9–21.e7. doi: 10.1016/j.molcel.2017.02.021
Potter, C. S., Peterson, R. L., Barth, J. L., Pruett, N. D., Jacobs, D. F., and Kern, M. J. (2006). Evidence that the satin hair mutant gene Foxq1 is among multiple and functionally diverse regulatory targets for Hoxc13 during hair follicle differentiation. J. Biol. Chem. 281, 29245–29255. doi: 10.1074/jbc.m603646200
Ranran, Z., Nan, L., Fuhui, H., Hegang, L., Jifeng, L., Lanlan, L., et al. (2020). Identification and characterization of circRNAs in the skin during wool follicle development in Aohan fine wool sheep. BMC Genomics 21:187. doi: 10.1186/s12864-020-6599-8
Rogers, G. E. (2006). Biology of the wool follicle: an excursion into a unique tissue interaction system waiting to be re-discovered. Exp. Dermatol. 15, 931–949. doi: 10.1111/j.1600-0625.2006.00512.x
Schinckel, P. G. (1953). Follicle development in the Australian merino. Nature 171, 310–311. doi: 10.1038/171310b0
Schliebner, I., Becher, R., Hempel, M., Deising, H. B., and Horbach, R. (2014). New gene models and alternative splicing in the maize pathogen Colletotrichum graminicola revealed by RNA-Seq analysis. BMC Genomics 15:842. doi: 10.1186/1471-2164-15-842
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Slominski, A., Paus, R., Plonka, P., Chakraborty, A., Maurer, M., Pruski, D., et al. (1994). Melanogenesis during the anagen-catagen-telogen transformation of the murine hair cycle. J. Invest. Dermatol. 102, 862–869. doi: 10.1111/1523-1747.ep12382606
Song, L. L., Cui, Y., Yu, S. J., Liu, P. G., and He, J. F. (2019). TGF-beta and HSP70 profiles during transformation of yak hair follicles from the anagen to catagen stage. J. Cell. Physiol. doi: 10.1002/jcp.28212 [Epub ahead of print].
Stenn, K. S., and Paus, R. (2001). Controls of hair follicle cycling. Physiol. Rev. 81, 449–494. doi: 10.1152/physrev.2001.81.1.449
Suh, N. (2018). MicroRNA controls of cellular senescence. BMB Rep. 51, 493–499. doi: 10.5483/bmbrep.2018.51.10.209
Sun, H., Zhang, Y., Bai, L., Wang, Y., Yang, L., Su, W., et al. (2019). Heat stress decreased hair follicle population in rex rabbits. J. Anim. Physiol. Anim. Nutr. 103, 501–508. doi: 10.1111/jpn.13044
Sun, L., Luo, H., Bu, D., Zhao, G., Yu, K., Zhang, C., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41:e166. doi: 10.1093/nar/gkt646
Tang, X., Feng, D., Li, M., Zhou, J., Li, X., Zhao, D., et al. (2019). Transcriptomic analysis of mRNA-lncRNA-miRNA interactions in hepatocellular carcinoma. Sci. Rep. 9.16096. doi: 10.1038/s41598-019-52559-x
Tao, L., Yang, L., Huang, X., Hua, F., and Yang, X. (2019). Reconstruction and analysis of the lncRNA-miRNA-mRNA network based on competitive endogenous RNA reveal functional lncRNAs in dilated cardiomyopathy. Front. Genet. 10:1149. doi: 10.3389/fgene.2019.01149
Thakur, O., and Randhawa, G. S. (2018). Identification and characterization of SSR, SNP and InDel molecular markers from RNA-Seq data of guar (Cyamopsis tetragonoloba, L. Taub.) roots. BMC Genomics 19:951. doi: 10.1186/s12864-018-5205-9
Tosar, J. P., Rovira, C., and Cayota, A. (2018). Non-coding RNA fragments account for the majority of annotated piRNAs expressed in somatic non-gonadal tissues. Commun. Biol. 1:2. doi: 10.1038/s42003-017-0001-7
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Villani, R., Hodgson, S., Legrand, J., Greaney, J., Wong, H. Y., Pichol-Thievend, C., et al. (2017). Dominant-negative Sox18 function inhibits dermal papilla maturation and differentiation in all murine hair types. Development 144, 1887–1895. doi: 10.1242/dev.143917
Wang, L., Park, H. J., Dasari, S., Wang, S. Q., Kocher, J. P., and Li, W. (2013). CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 41:e74. doi: 10.1093/nar/gkt006
Wang, L. X., Wan, C., Dong, Z. B., Wang, B. H., Liu, H. Y., and Li, Y. (2019). Integrative analysis of long noncoding RNA (lncRNA), microRNA (miRNA) and mRNA expression and construction of a competing endogenous RNA (ceRNA) network in metastatic melanoma. Med. Sci. Monit. 25, 2896–2907. doi: 10.12659/MSM.913881
Wang, S., Ge, W., Luo, Z., Guo, Y., Jiao, B., Qu, L., et al. (2017). Integrated analysis of coding genes and non-coding RNAs during hair follicle cycle of cashmere goat (Capra hircus). BMC Genomics 18:767. doi: 10.1186/s12864-017-4145-0
Wang, W. X., Wang, Q. J., and Wang, S. M. (1996). Correlation analysis of growth and development indexes of Aohan Fine Wool Sheep. Journal of Inner Mongolia University for Nationalities 17–21. (in chinese).
Wei, C., Wu, M., Wang, C., Liu, R., Zhao, H., Yang, L., et al. (2018). Long noncoding RNA Lnc-SEMT modulates IGF2 expression by sponging miR-125b to promote sheep muscle development and growth. Cell. Physiol. Biochem. 49, 447–462. doi: 10.1159/000492979
Xiao, S., Wang, J., Chen, Q., Miao, Y., and Hu, Z. (2019). The mechanism of activated platelet-rich plasma supernatant promotion of hair growth by cultured dermal papilla cells. J. Cosmet. Dermatol. 18, 1711–1716. doi: 10.1111/jocd.12919
Yangfan, N., Shaomei, L., XinTing, Z., Wenshuo, C., Xueer, L., Zhiwei, L., et al. (2018). Transcriptome reveals long non-coding RNAs and mRNAs involved in primary wool follicle induction in carpet sheep fetal skin. Front. Physiol. 9:446. doi: 10.3389/fphys.2018.00446
Zhai, B., Zhang, L., Wang, C., Zhao, Z., Zhang, M., and Li, X. (2019). Identification of microRNA-21 target genes associated with hair follicle development in sheep. PeerJ 7:e7167. doi: 10.7717/peerj.7167
Zhang, H., Shi, Q., Nan, W., Wang, Y., Wang, S., Yang, F., et al. (2019a). Ginkgolide B and bilobalide promote the growth and increase beta-catenin expression in hair follicle dermal papilla cells of American minks. Biofactors 45, 950–958. doi: 10.1002/biof.1562
Zhang, H., Su, Y., Wang, J., Gao, Y., Yang, F., Li, G., et al. (2019b). Ginsenoside Rb1 promotes the growth of mink hair follicle via PI3K/AKT/GSK-3beta signaling pathway. Life Sci. 229, 210–218. doi: 10.1016/j.lfs.2019.05.033
Zhang, Y., Wu, K., Wang, L., Wang, Z., Han, W., Chen, D., et al. (2020). Comparative study on seasonal hair follicle cycling by analysis of the transcriptomes from cashmere and milk goats. Genomics 112, 332–345. doi: 10.1016/j.ygeno.2019.02.013
Zhang, W., Wang, S. Y., Deng, S. Y., Gao, L., Yang, L. W., Liu, X. N., et al. (2018). MiR-27b promotes sheep skeletal muscle satellite cell proliferation by targeting myostatin gene. J. Genet. 97, 1107–1117. doi: 10.1007/s12041-018-0998-5
Zhang, Y., Wang, L., Li, Z., Chen, D., Han, W., Wu, Z., et al. (2019c). Transcriptome profiling reveals transcriptional and alternative splicing regulation in the early embryonic development of hair follicles in the cashmere goat. Sci. Rep. 9:17735. doi: 10.1038/s41598-019-54315-7
Zhao, B., Chen, Y., Hu, S., Yang, N., Wang, M., Liu, M., et al. (2019). Systematic analysis of non-coding RNAs involved in the Angora Rabbit (Oryctolagus cuniculus) hair follicle cycle by RNA sequencing. Front. Genet. 10:407. doi: 10.3389/fgene.2019.00407
Zheng, Y., Wang, Z., Zhu, Y., Wang, W., Bai, M., Jiao, Q., et al. (2019). LncRNA-000133 from secondary hair follicle of Cashmere goat: identification, regulatory network and its effects on inductive property of dermal papilla cells. Anim. Biotechnol. 31, 122–134. doi: 10.1080/10495398.2018.1553788
Zhou, L., Chen, J., Li, Z., Li, X., Hu, X., Huang, Y., et al. (2010). Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS One 5:e15224. doi: 10.1371/journal.pone.0015224
Keywords: Aohan fine wool sheep, long non-coding RNA, microRNA, mRNA, wool follicle, skin development
Citation: Zhao R, Li J, Liu N, Li H, Liu L, Yang F, Li L, Wang Y and He J (2020) Transcriptomic Analysis Reveals the Involvement of lncRNA–miRNA–mRNA Networks in Hair Follicle Induction in Aohan Fine Wool Sheep Skin. Front. Genet. 11:590. doi: 10.3389/fgene.2020.00590
Received: 07 February 2020; Accepted: 15 May 2020;
Published: 09 June 2020.
Edited by:
Jiuzhou Song, University of Maryland, College Park, United StatesReviewed by:
Xiaolong Wang, Northwest A&F University, ChinaLingyang Xu, Institute of Animal Sciences, Chinese Academy of Agricultural Sciences, China
Copyright © 2020 Zhao, Li, Liu, Li, Liu, Yang, Li, Wang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianning He, aGV4aW5neGluZzEwNEAxNjMuY29t