
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 25 May 2020
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00492
 Jie-Yuan Jin1,2,3Pan-Feng Wu1Ji-Qiang He1
Jie-Yuan Jin1,2,3Pan-Feng Wu1Ji-Qiang He1 Liang-Liang Fan3,4Zhuang-Zhuang Yuan3Xiao-Yang Pang1
Liang-Liang Fan3,4Zhuang-Zhuang Yuan3Xiao-Yang Pang1 Ju-Yu Tang1*
Ju-Yu Tang1* Li-Yang Zhang2,3*
Li-Yang Zhang2,3*Background: Hereditary sensory and autonomic neuropathies (HSANs) are a rare and severe group of sensory axonal neuropathies. HSANs have been classified into eight groups based on mode of inheritance, clinical features, and the involved genes. HSAN-VI, perhaps the most notable type, is an autosomal recessive disease, which manifests as the severely impaired pain sensitivity, autonomic disturbances, distal myopathy, spontaneous or surgical amputations, and sometimes early death. Mutations in DST have been identified as the cause of HSAN-VI. DST encodes dystonin, a member of the plakin protein family that is involved in cytoskeletal filament networks. Dystonin has seven major isoforms in nerve, muscle, and epithelium.
Material and Methods: The present study investigated a Chinese family with HSAN and explored potential pathogenic variants using whole-exome sequencing (WES). Variants were screened and filtered through bioinformatics analysis and prediction of variant pathogenicity. Co-segregation analysis was subsequently conducted.
Results: We identified compound heterozygous variants of DST (c.3304G>A, p.V1102I and c.13796G>A, p.R4599H) in two patients.
Conclusion: We reported on a Chinese family with HSAN-VI family and detected the disease-causing variants. Our description expands the spectrum of known DST variants and contributes to the clinical diagnosis of HSAN-VI.
Hereditary sensory and autonomic neuropathies (HSANs), also known as hereditary sensory neuropathies (HSNs), are a rare and severe group of sensory axonal neuropathies (Davidson et al., 2012). The cardinal clinical features of HSAN are various sensory and autonomic dysfunctions characterized by the loss of pain and temperature sensations and, in some subtypes, positive sensory symptoms (Leipold et al., 2013). Patients with HSANs might also exhibit altered sweating (hyperhidrosis or anhidrosis), cardiovascular dysregulation, dyskinesia, and gastrointestinal dysmotility (Hepburn et al., 2014; Woods et al., 2015). Other complications include ulcers, infections, osteomyelitis, self-mutilation, and spontaneous or surgical amputations (Phatarakijnirund et al., 2016). HSANs are classified into eight groups based on inheritance, clinical features, and the involved genes (Supplementary Table 1) (Davidson et al., 2012; Heimer et al., 2016; Yozu et al., 2016).
Variants in 18 genes and one locus have been identified as causing HSAN. DST was reported to be associated with HSAN-VI (OMIM 614653) in three unrelated families (Edvardson et al., 2012; Manganelli et al., 2017; Fortugno et al., 2019). All HSAN-VI patients presented severely impaired pain sensitivity, autonomic disturbances, and distal myopathy, and some died in early childhood (Edvardson et al., 2012; Manganelli et al., 2017). DST is located on 6p12.1 and encodes dystonin, a member of the plakin protein family that is involved in cytoskeletal filament networks (Pool et al., 2005). DST has four different promoters and many alternative splicing sites that produce different transcripts, including three major neuronal isoforms (dystonin-a1, a2, and a3), three muscular isoforms (dystonin-b1, b2, and b3), and one epithelial isoform (dystonin-e) (Jefferson et al., 2006; Kunzli et al., 2016). Dst variants in mice cause dystonia musculorum (dt), a neurodegenerative disease characterized by progressive degeneration of sensory neurons (Brown et al., 1995).
Here, we report on a family with HSAN-VI in China, in which the HSAN-VI patients harbor compound heterozygosity of DST consisting of two missense variants (NM_001144769: c.3304G>A, p.V1102I and c.13796G>A, p.R4599H). These two variants were inherited from the parents. To the best of our knowledge, this combination of variants has not been previously reported.
The Review Board of Xiangya Hospital of the Central South University approved this research (approval number: 20190038). Written informed consent was obtained from the patients and their guardians, and all subjects consented to participate in this study and to the publication of the images. Blood was collected from the proband and related family members. Segregation analysis was performed for all family members based on whole-exome sequencing (WES) results.
Thermal thresholds (cold, warm) were evaluated using a thermal sensory analyzer (TSA 2001, Israel). The base temperature was 32°C, the range was from 0 to 50°C, and the rate of change was 1°C/s. The bilateral hypothenar muscles and dorsum pedis were tested 5 times each at 15 s intervals.
Mechanical pain perception was evaluated using a calibrated monofilament with a bending force of 95 mN that was connected to a sharp non-penetrating probe (50-mm tip). This was applied 10 times for 1–2 s each time. The percentage of stimuli perceived as painful and the pain magnitude using a visual analog scale were recorded. Three null stimuli were randomly applied during testing to evaluate subject reliability (Nolano et al., 2008).
Genomic DNA was extracted with a DNeasy blood and tissue kit (Qiagen, Valencia, CA, USA). The Novogene Bioinformatics Institute (Beijing, China) performed exome capture, high-throughput sequencing, and common filtering. All the exomes were captured by means of Agilent SureSelect Human All Exon V5 kits and sequenced with the Illumina HiSeq 2000 platform. After filtering of common variants (allele frequency > 0.05) from the YanHuang genome (http://yh.genomics.org.cn/), Exome Aggregation Consortium (http://exac.broadinstitute.org/), 1000 Genomes Project (https://www.genome.gov/27528684/1000-genomes-project/), Genome Aggregation (http://gnomad.broadinstitule.org), dbSNP (https://www.ncbi.nlm.nih.gov/SNP/), and ESP (http://evs.gs.washington.edu/EVS/) databases; unique single-nucleotide polymorphisms (SNPs); and insertions/deletions (indels) were detected in the subjects. These variants were screened against the list of genes previously identified as being associated with HSAN (Supplementary Table 1) to identify the variants in known causative genes. Then, MutationTaster (http://www.mutationtaster.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and SIFT (http://provean.jcvi.org/index.php) were used to predict whether the variants affected protein structure and function. Gene function, inheritance pattern, clinical phenotype, and pathogenicity were annotated according to Online Mendelian Inheritance in Man (OMIM) (https://www.omim.org) and American College of Medical Genetics classification (Richards et al., 2015).
Primer pairs were designed via DNASTAR (DST c.3304G>A f: TCAAGATTTCCCGCCTCATAAA, DST c.3304G>A r: GCACACAGATGGGACCATAG; DST c.13796G>A f: CCTGCTTGAAACTGACAGTGTT, DST c.13796G>A r: CCCATATAACATGGTCAGTTTG); the sequences of the primers will be provided upon request. The target fragments were amplified via polymerase chain reaction (PCR) and characterized with an ABI 3100 Genetic Analyzer (ABI, Foster City, CA).
Total RNA was extracted using an RNA extraction kit provided by Qiagen (Germany) and then stored at −80°C.
The total RNA was sent to Novogene (Beijing, China) for transcriptome sequencing using the Illumina HiSeq 4000 sequencing platform. Data were compared and analyzed using a high-efficiency combination of node calculations by Dell and stored by Isilon. Genes with a p < 0.05, an adjusted p-value (padj) < 0.05 and a −1 < log2 fold change > 1 were considered differentially expressed genes.
The total RNA was reverse transcribed using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, USA) and then subjected to qPCR by 2× SYBR Green qPCR Mix (Thermo Fisher Scientific, USA).
Data were processed and graphed using GraphPad Prism 5 software and tested to determine significant differences (p < 0.05).
We identified a Chinese family with HSAN (Figure 1A). The proband (II:1) was admitted to the Department of Orthopedics, Xiangya Hospital of Central South University, for an ulcer near the left ankle and diagnosed with HSAN. The proband was a 14-year-old boy with an identical twin brother (II:2). They were born at term via cesarean section. The height and weight of the proband were 145 cm (<3%) and 26.5 kg (<3%), respectively, without any growth hormone imbalances, and his brother was 150 cm (<3%) tall, and weighed 29.0 kg (<3%), suggesting that they had growth retardation (without hormonal abnormalities).
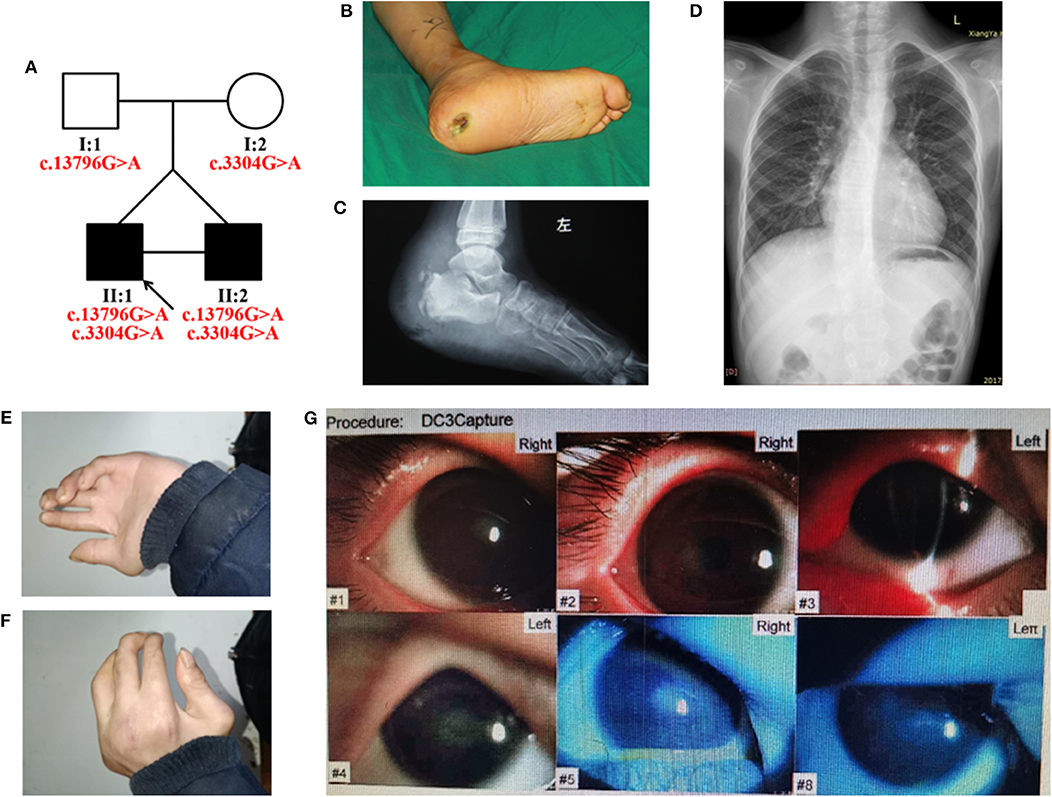
Figure 1. (A) Pedigree of the HSAN-VI family with segregation analysis. The black symbols represent the affected members, and the arrow indicates the proband. Red text represents the variants. (B–G) Aspects of the phenotype of the proband. The proband has an ankle ulcer (B), necrosis of the astragalus (C), moderate scoliosis (D), phalangeal joint contracture (E,F), and cataract (G).
The proband had severely impaired pain sensitivity (Table 1). He consistently could not perceive noxious thermal/gelid stimuli or innocuous warm stimuli, which had resulted in the ankle ulcer and necrosis of the astragalus (Figures 1B,C). A nerve conduction study revealed peripheral nerve damage in the extremities, especially of the sensory nerves (Supplementary Table 2). The proband had moderate scoliosis (with a Cobb angle of 33°) without centrum malformation (Figure 1D). The younger brother also exhibited HSAN, but his condition was clearly milder. The brothers presented with arthrogryposes of the fingers, foot extroversion, knee recurvation deformities, and myasthenia; hence, they achieved independent ambulation at 3 years of age (Figures 1E,F). The proband had thin blue scleras and corneal ulcers and was subsequently diagnosed with a cataract (Figure 1G). He also displays urinary incontinence. Furthermore, the proband was diagnosed with vascular structure abnormalities via centesis at 18 months of age. Their parents (I:1 and I:2) were unaffected.
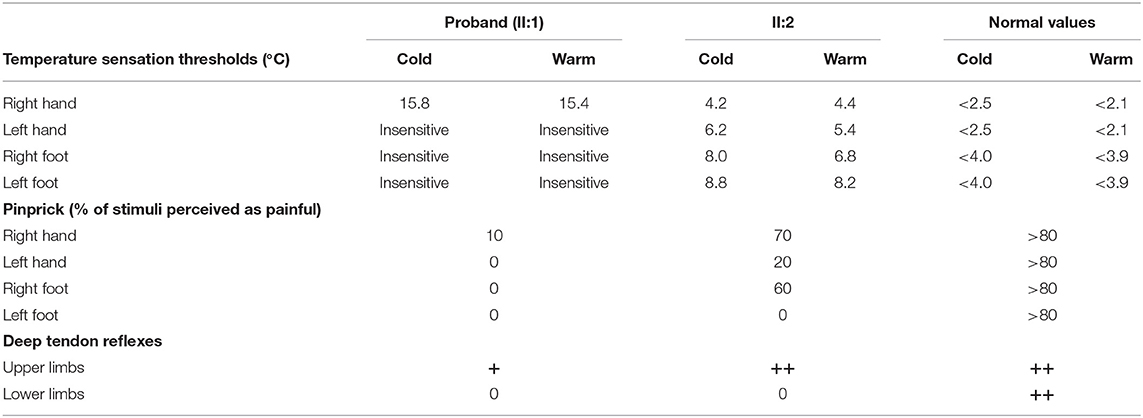
Table 1. Quantitative sensory testing and deep tendon reflexes.
After filtering the common variants from the 1000 Genomes Project, YH, ExAC, dbSNP132, and ESP databases, 571 unique SNPs were detected in the proband. The variants were filtered against 18 genes known to be involved in HSAN (Supplementary Table 1), and a set of three variants in two genes were identified in the proband (Table 2). By analyzing the bioinformatic predictions, inheritance patterns, OMIM clinical phenotypes, and American College of Medical Genetics classification (Richards et al., 2015) of these three genes, we strongly suspected that variants in DST (c.3304G>A, p.V1102I and c.13796G>A, p.R4599H) were the causative genetic variants in these patients.
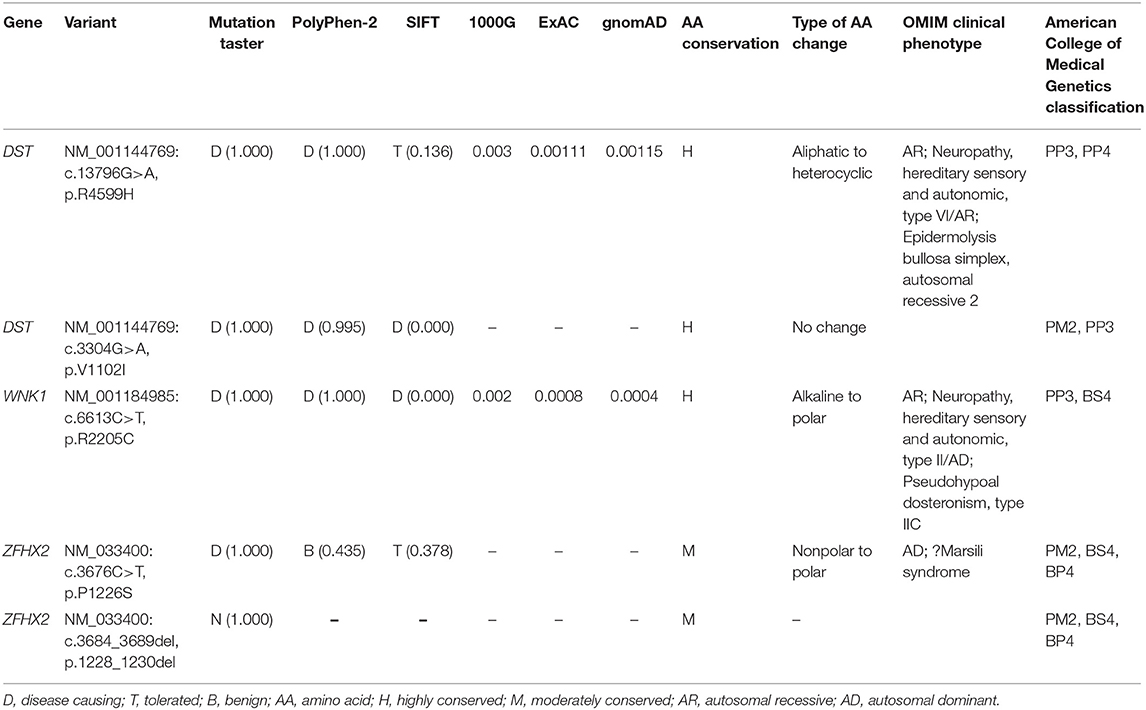
Table 2. Variants identified by WES in combination with HSAN-related gene filtering in the present family.
Sanger sequencing showed that the DST variant (c.3304G>A, p.V1102I) in the identical twins was inherited from their mother and the other (c.13796G>A, p.R4599H) was inherited from their father (Figures 2A,B). This finding indicates that the compound heterozygosity co-segregated within the affected family members. Neither variant was identified in the 200 control cohorts previously studied by our group. The amino acid sequence alignment analysis suggested that these two variants were both located in a highly evolutionarily conserved site (Figures 2C,D).
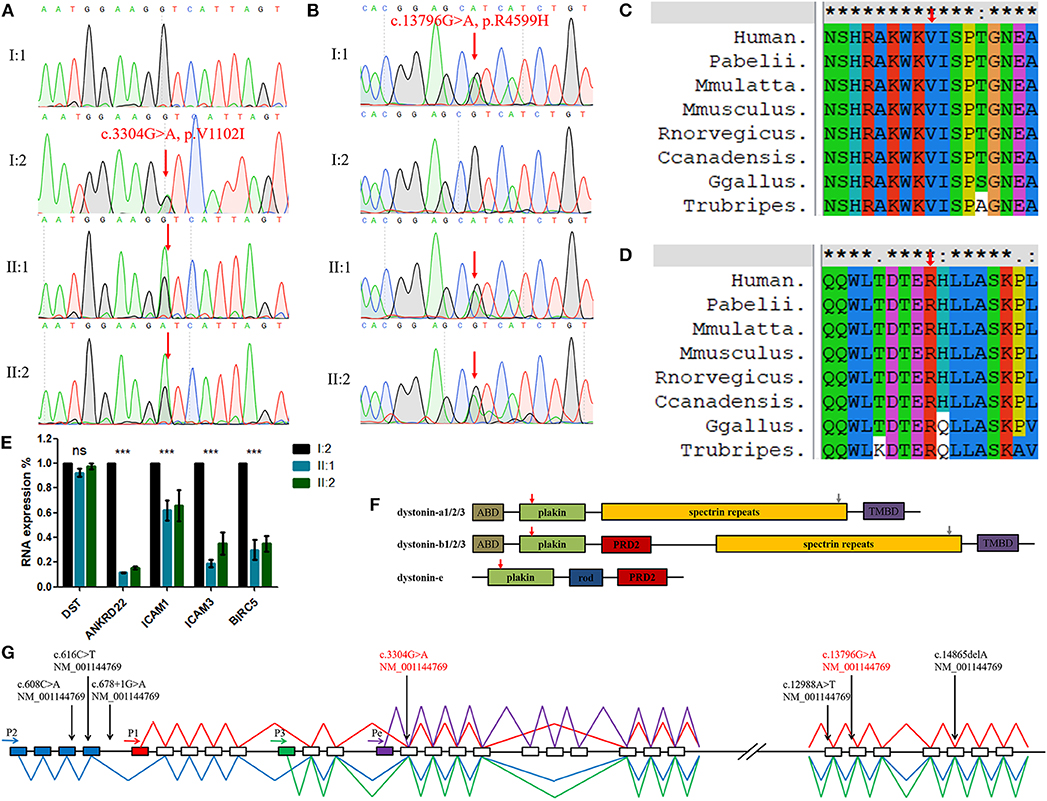
Figure 2. (A,B) Sequencing results of the DST variants. Sequence chromatograms indicate compound heterozygous variants (c.3304G>A, p.V1102I and c.13796G>A, p.R4599H) in the family with HSAN-VI. (C,D) DST peptide sequences surrounding the mutated residues (p.V1102I and p.R4599H) with multiple interspecies alignments generated by ClustalX. The mutation position is indicated with a red arrow. (E) The results of qPCR. “***”, statistical significance; “ns”, no statistical significance. (F) The structures of three major tissue-specific dystonin isoforms. “ABD”, an actin-binding domain; “plakin”, a plakin domain; “rod”, a rod domain; “PRD2”, the plectin repeats; “spectrin repeats”, spectrin repeats; and “TMBD”, a combination of an EF hand domain and a growth arrest-specific 2 protein-related region. The red (V1102I) and gray (R4599H) arrows indicate the mutated AA site. (G) Schematic diagram of DST and the variants identified in HSAN-VI patients. Four putative promoters are indicated by horizontal arrows. Blue boxes indicate the unique exons of isoform-2, the red box indicates the unique exon of isoform-1, the green box indicates the unique exon of isoform-3, and the purple box indicates the unique exon of isoform-e. The blue line represents the transcript of isoform-2, the red line represents the transcript of isoform-1, the green line represents the transcript of isoform-3, and the purple line represents the transcript of isoform-e. Full vertical arrows show the positions of the variants of DST in the gene. The red text represents the present variants.
By sequencing the peripheral blood RNA from I:2, II:1, and II:2 and excluding immunity-related proteins, we detected the down-regulated expression of ANKRD22, ICAM1/3, and BIRC5 in the HSAN-VI patients, suggesting changes in cell adhesion and the cytoskeleton, which is in accordance with the expectation that DST variants can alter cytoskeletal filament networks (Figure 2E and Supplementary Figure 1). However, we did not observe any change in DST expression by RNAseq or qPCR (Figure 2E).
We report on a Chinese family with HSAN-VI for the first time, revealing an autosomal recessive defect in DST. This report further demonstrates a correlation between DST and HSAN-VI. Notably, the severities of the sensory and autonomic dysfunctions in the two patients were significantly different, even though they are identical twins who experienced the same conditions until the older brother started to seek treatment. This may be a result of competition for nutrition and space during embryonic development and indicates obvious phenotypic heterogeneity. It has previously been reported that the same DST variants lead to family members with different levels of neuronal dysfunction (Manganelli et al., 2017). In addition, the limb phenotypes in one HSAN-VI patient are often different, let alone different individuals. We have also broadened the knowledge of HSAN-VI symptoms, and our results suggest that growth retardation, scoliosis, and corneal ulcerations may be parts of the phenotypes.
Dystonin is a very large cytoskeletal cross-linking protein that interacts with actin and microtubule networks, protein complexes, membrane-bound organelles, and cellular membranes (Ferrier et al., 2014). In humans, DST is 500 kb in length and encompasses more than 100 exons (Ferrier et al., 2013). DST has four tissue-specific promoters and undergoes many alternative splicing events leading to the expression of numerous isoforms in the central and peripheral nervous systems (dystonin-a), muscle (dystonin-b), and epidermis (dystonin-e) (Kunzli et al., 2016). Promoter-1,−2, and−3 are common promoters active in the nerves and muscle, whereas promoter-e drives tissue-specific dystonin expression in the epidermis (Young et al., 2006). The structure of dystonin-a includes an actin-binding domain (ABD), a plakin domain, spectrin repeats (SRs), and a microtubule-binding domain (MTBD) consisting of an EF-hand pair and a growth arrest-specific 2 (Gas2) domain (Figure 2F) (Suozzi et al., 2012). In contrast to dystonin-a, dystonin-b has two extra plectin repeat domains (PRD2) between the plakin domain and the SRs (Ferrier et al., 2013). Dystonin-e is the smallest, consisting of a plakin domain, a rod domain, and a PRD2 domain (Fine and Mellerio, 2009). The variant (c.3304G>A, p.V1102I) in our patients is located in the plakin domain. This domain plays an important role in cell junctions, and p.V1102I may have effects on its structure and function. The other variant (c.13796G>A, p.R4599H) lies in the spectrin repeats, which give dystonin molecules elasticity to adapt to different membrane structures. The MUpro database (https://www.ics.uci.edu/~baldig/mutation.html) predicted that p.R4599H could decrease protein stability (ΔΔG = −1.074) based on both the value and sign of the energy using a support vector machine (SVM) and sequence information only.
We detected reduced expression of ANKRD22, ICAM1/3, and BIRC5 by RNAseq and qPCR in the proband and his brother. ANKRD22 is a mitochondrial protein and may be associated with schizophrenia (Fromer et al., 2014). DST may affect ANKRD22 to regulate neuronal physiological activities. ICAMs play important roles in cell proliferation, differentiation, motility, and trafficking (Rosette et al., 2005). The change in ICAM1/3 expression suggests an abnormality in cell adhesion. Moreover, DST has been verified can alter cell adhesion and migration (Fortugno et al., 2019). BIRC5 is a member of the inhibitor family of apoptosis and participates in the organization of the central spindle by associating with polymerized microtubules (Kamran et al., 2017). Its reduced expression suggests a reduction in cell proliferation. DST can interact with the cytoskeleton, and its loss of function can inhibit cell proliferation. DST may regulate or interact with ICAMs and BIRC5 to participate in cellular activities and individual development.
The RNAseq and qPCR results did not detect an abnormal expression of DST, which may be because these variants alter the function of the protein, not its expression. Unfortunately, lacking an antibody and patient tissue samples, we cannot further verify our results in regard to alterations of DST expression and function by western blotting.
The c.3304G>A variant has not been reported previously. For the c.13796G>A variant, a frequency of 0.00115 has been reported in the global population. However, in East Asians, the frequency may reach 0.01199. Given this low frequency compared with the frequency in other populations (0.00003 in South Asians, 0.00028 in Latinos, 0.00016 in Europeans, and 0.00132 in Africans) (data from GnomAD), we think that this is the result of a phenomenon similar to a founder effect. It is noted that no carriers have been found to be homozygous for this variation. Given that HSAN-VI is a recessive hereditary disease, the existence of heterozygous carriers does not mean that the c.13796G>A variant is benign. We rechecked the WES results and did not find any variants more credible than the DST variants (Supplementary Figure 2, Supplementary Tables 3,4). In particular, we focused on variants in known HSAN-related genes, including SPTLC1, SPTLC2, ATL1, DNMT1, ATL3, WNK1, FAM134B, KIF1A, SCN9A, IKBKAP, NTRK1, NGFB, DST, SCN11A, PRDM12, CCT5, TECPR2, and ZFHX2. However, no other significant variants were identified (Davidson et al., 2012; Heimer et al., 2016; Yozu et al., 2016).
We summarized all of the DST variants previously identified in HSAN-VI patients (Figure 2G) (Edvardson et al., 2012; Manganelli et al., 2017; Fortugno et al., 2019). All of the variants in patients with HSAN-VI involve dystonin-a, which is expressed in nerves, and dystonin-b, which is expressed in muscle. Consistently, all of these patients display neuropathy and myopathy. Our patients also have similar phenotypes. In contrast, variants that affect dystonin-e cause autosomal recessive epidermolysis bullosa simplex (EBS) (Groves et al., 2010). The relationships between DST genotypes and their phenotypes require further study.
In summary, the present study identified a novel compound heterozygous DST genotype consisting of two missense variants (c.3304G>A, p.V1102I and c.13796G>A, p.R4599H) in identical twins with HSAN-VI. The identification of these variants expands the spectrum of known DST variants and further demonstrates that DST is associated with HSAN-VI. Our report describes new phenotypes that can assist in the clinical diagnosis of HSAN-VI.
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA632884.
The Review Board of Xiangya Hospital of the Central South University approved this research. Written informed consent was obtained from the patients and their guardians, and all subjects consented to this study and the publication of the images.
J-QH, P-FW, and X-YP collected and provided clinical information. J-YJ, L-LF, and Z-ZY carried out whole exome sequencing and data analysis. L-YZ and J-YT designed experiments and wrote the manuscript.
This work was supported by the National Science and Technology Major Project of the Ministry of Science and Technology of China (2017ZX10103005-006), the National Natural Science Foundation of China (81370394), the Emergency Project of Prevention and Control for COVID-19 of Central South University (160260003), the Fundamental Research Funds for Central Universities of Hunan Province (CX20190104), and the Fundamental Research Funds for Central Universities of Central South University (CX20190104).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the patients and their families for participating in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00492/full#supplementary-material
Brown, A., Bernier, G., Mathieu, M., Rossant, J., and Kothary, R. (1995). The mouse dystonia musculorum gene is a neural isoform of bullous pemphigoid antigen 1. Nat. Genet. 10, 301–306. doi: 10.1038/ng0795-301
Davidson, G., Murphy, S., Polke, J., Laura, M., Salih, M., Muntoni, F., et al. (2012). Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort. J. Neurol. 259, 1673–1685. doi: 10.1007/s00415-011-6397-y
Edvardson, S., Cinnamon, Y., Jalas, C., Shaag, A., Maayan, C., Axelrod, F. B., et al. (2012). Hereditary sensory autonomic neuropathy caused by a mutation in dystonin. Ann. Neurol. 71, 569–572. doi: 10.1002/ana.23524
Ferrier, A., Boyer, J. G., and Kothary, R. (2013). Cellular and molecular biology of neuronal dystonin. Int. Rev. Cell Mol. Biol. 300, 85–120. doi: 10.1016/B978-0-12-405210-9.00003-5
Ferrier, A., Sato, T., De Repentigny, Y., Gibeault, S., Bhanot, K., O'meara, R. W., et al. (2014). Transgenic expression of neuronal dystonin isoform 2 partially rescues the disease phenotype of the dystonia musculorum mouse model of hereditary sensory autonomic neuropathy VI. Hum. Mol. Genet. 23, 2694–2710. doi: 10.1093/hmg/ddt663
Fine, J. D., and Mellerio, J. E. (2009). Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J. Am. Acad. Dermatol. 61, 387–402; quiz 403–384. doi: 10.1016/j.jaad.2009.03.053
Fortugno, P., Angelucci, F., Cestra, G., Camerota, L., Ferraro, A. S., Cordisco, S., et al. (2019). Recessive mutations in the neuronal isoforms of DST, encoding dystonin, lead to abnormal actin cytoskeleton organization and HSAN type VI. Hum. Mutat. 40, 106–114. doi: 10.1002/humu.23678
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P., et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi: 10.1038/nature12929
Groves, R. W., Liu, L., Dopping-Hepenstal, P. J., Markus, H. S., Lovell, P. A., Ozoemena, L., et al. (2010). A homozygous nonsense mutation within the dystonin gene coding for the coiled-coil domain of the epithelial isoform of BPAG1 underlies a new subtype of autosomal recessive epidermolysis bullosa simplex. J. Invest. Dermatol. 130, 1551–1557. doi: 10.1038/jid.2010.19
Heimer, G., Oz-Levi, D., Eyal, E., Edvardson, S., Nissenkorn, A., Ruzzo, E. K., et al. (2016). TECPR2 mutations cause a new subtype of familial dysautonomia like hereditary sensory autonomic neuropathy with intellectual disability. Eur. J. Paediatr. Neurol. 20, 69–79. doi: 10.1016/j.ejpn.2015.10.003
Hepburn, L., Prajsnar, T. K., Klapholz, C., Moreno, P., Loynes, C. A., Ogryzko, N. V., et al. (2014). Innate immunity. A Spaetzle-like role for nerve growth factor beta in vertebrate immunity to Staphylococcus aureus. Science 346, 641–646. doi: 10.1126/science.1258705
Jefferson, J. J., Leung, C. L., and Liem, R.K. (2006). Dissecting the sequence specific functions of alternative N-terminal isoforms of mouse bullous pemphigoid antigen 1. Exp. Cell Res. 312, 2712–2725. doi: 10.1016/j.yexcr.2006.04.025
Kamran, M., Long, Z. J., Xu, D., Lv, S. S., Liu, B., Wang, C. L., et al. (2017). Aurora kinase A regulates Survivin stability through targeting FBXL7 in gastric cancer drug resistance and prognosis. Oncogenesis 6:e298. doi: 10.1038/oncsis.2016.80
Kunzli, K., Favre, B., Chofflon, M., and Borradori, L. (2016). One gene but different proteins and diseases: the complexity of dystonin and bullous pemphigoid antigen 1. Exp. Dermatol. 25, 10–16. doi: 10.1111/exd.12877
Leipold, E., Liebmann, L., Korenke, G. C., Heinrich, T., Giesselmann, S., Baets, J., et al. (2013). A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 45, 1399–1404. doi: 10.1038/ng.2767
Manganelli, F., Parisi, S., Nolano, M., Tao, F., Paladino, S., Pisciotta, C., et al. (2017). Novel mutations in dystonin provide clues to the pathomechanisms of HSAN-VI. Neurology 88, 2132–2140. doi: 10.1212/WNL.0000000000003992
Nolano, M., Provitera, V., Estraneo, A., Selim, M. M., Caporaso, G., Stancanelli, A., et al. (2008). Sensory deficit in Parkinson's disease: evidence of a cutaneous denervation. Brain 131, 1903–1911. doi: 10.1093/brain/awn102
Phatarakijnirund, V., Mumm, S., Mcalister, W. H., Novack, D. V., Wenkert, D., Clements, K. L., et al. (2016). Congenital insensitivity to pain: fracturing without apparent skeletal pathobiology caused by an autosomal dominant, second mutation in SCN11A encoding voltage-gated sodium channel 1.9. Bone 84, 289–298. doi: 10.1016/j.bone.2015.11.022
Pool, M., Boudreau Lariviere, C., Bernier, G., Young, K. G., and Kothary, R. (2005). Genetic alterations at the Bpag1 locus in dt mice and their impact on transcript expression. Mamm. Genome 16, 909–917. doi: 10.1007/s00335-005-0073-4
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rosette, C., Roth, R. B., Oeth, P., Braun, A., Kammerer, S., Ekblom, J., et al. (2005). Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis 26, 943–950. doi: 10.1093/carcin/bgi070
Suozzi, K. C., Wu, X., and Fuchs, E. (2012). Spectraplakins: master orchestrators of cytoskeletal dynamics. J. Cell Biol. 197, 465–475. doi: 10.1083/jcb.201112034
Woods, C. G., Babiker, M. O., Horrocks, I., Tolmie, J., and Kurth, I. (2015). The phenotype of congenital insensitivity to pain due to the NaV1.9 variant p.L811P. Eur. J. Hum. Genet. 23, 561–563. doi: 10.1038/ejhg.2014.166
Young, K. G., Pinheiro, B., and Kothary, R. (2006). A Bpag1 isoform involved in cytoskeletal organization surrounding the nucleus. Exp. Cell Res. 312, 121–134. doi: 10.1016/j.yexcr.2005.10.002
Keywords: HSAN-VI, DST, scoliosis, dystonin, whole-exome sequencing
Citation: Jin J-Y, Wu P-F, He J-Q, Fan L-L, Yuan Z-Z, Pang X-Y, Tang J-Y and Zhang L-Y (2020) Novel Compound Heterozygous DST Variants Causing Hereditary Sensory and Autonomic Neuropathies VI in Twins of a Chinese Family. Front. Genet. 11:492. doi: 10.3389/fgene.2020.00492
Received: 05 November 2019; Accepted: 20 April 2020;
Published: 25 May 2020.
Edited by:
Musharraf Jelani, Islamia College University, PakistanReviewed by:
Naveed Wasif, University Hospital Schleswig-Holstein, GermanyCopyright © 2020 Jin, Wu, He, Fan, Yuan, Pang, Tang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ju-Yu Tang, dGFuZ2p1eXU3NzQ5QDEyNi5jb20=; Li-Yang Zhang, emhhbmdsaXlhbmdAY3N1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.