 Faiza Hina
Faiza Hina Gulbar Yisilam1
Gulbar Yisilam1 Chengxin Fu
Chengxin Fu- 1Laboratory of Systematic and Evolutionary Botany and Biodiversity, College of Life Sciences, Zhejiang University, Hangzhou, China
- 2Department of Botany, University of Wisconsin–Madison, Madison, WI, United States
The moonseed genus Menispermum L. (Menispermaceae) is disjunctly distributed in East Asia and eastern North America. Although Menispermum has important medicinal value, genetic and genomic information is scarce, with very few available molecular markers. In the current study, we used Illumina transcriptome sequencing and de novo assembly of the two Menispermum species to obtain in-depth genetic knowledge. From de novo assembly, 53,712 and 78,921 unigenes were generated for M. canadense and M. dauricum, with 37,527 (69.87%) and 55,211 (69.96%) showing significant similarities against the six functional databases, respectively. Moreover, 521 polymorphic EST-SSRs were identified. Of them, 23 polymorphic EST-SSR markers were selected to investigate the population genetic diversity within the genus. The newly developed EST-SSR markers also revealed high transferability among the three examined Menispermaceae species. Overall, we provide the very first transcriptomic analyses of this important medicinal genus. In addition, the novel microsatellite markers developed here will aid future studies on the population genetics and phylogeographic patterns of Menispermum at the intercontinental geographical scale.
Introduction
Menispermum L. (moonseed) is a small genus (∼2 spp.) of deciduous climbing woody lianas in the family Menispermaceae. The genus exhibits a classic East Asian-Eastern North American disjunct pattern, with M. dauricum DC. native to East Asia and M. canadense L. occurring in eastern North America (Xiang et al., 2000; Ortiz et al., 2016). Both species are a typical element of temperate forests in East Asia/eastern North America, where they are widespread and abundant (Londre and Schnitzer, 2006).
Menispermum canadense (Canadian moonseed) is widely distributed in the lowland forests of eastern North America, west to Manitoba and Oklahoma (Purrington and Horn, 1993), while M. dauricum (Siberian moonseed) is widespread from eastern China to Siberia, Korea, and Japan. Rhizomes of M. dauricum are used as a crude drug in Traditional Chinese Medicine (TCM) known as “Bei-Dou-Gen” or “Bian-Fu-Ge-Gen” in China, which is useful for treating tonsillitis, rheumatic arthralgia, diarrhea, dysentery, gastroenteritis, cardiovascular, and thrombosis disorders. More than 25 phenolic alkaloids have been isolated from Menispermum, of which several are common in both species. Phenolic alkaloids from M. dauricum had broad pharmacological effects, for instance, anti-inflammation, anti-thrombosis, antioxidant, analgesia, treating high blood pressure and cardiovascular disorder, anti-arrhythmic, shielding neurons of cerebral ischemia, anti-tumor, and improving learning disabilities (Su et al., 2004, 2007; Guo et al., 2015). However, we still lack overall genetic knowledge on Menispermum, despite its medicinal importance.
Microsatellites (or simple sequence repeats, SSRs) are characterized by ample distribution in the genome, high polymorphism, co-dominance, reproducibility, and testable neutrality, and have thus been commonly used for assessing the genetic similarity among individuals or closely related taxa (Estoup et al., 1999; Guichoux et al., 2011). Microsatellites can be classified into genomic SSRs (gSSR) and expressed sequence tag SSRs (EST-SSR), depending on whether they are recovered from random genomic sequences or transcribed RNA sequences (Song et al., 2012). EST-SSRs are derived from the most conserved regions of the genome and thus have a higher transferability rate across the related species than that of gSSRs. Microsatellites have been extensively used in genomics, genetic diversity evolution, phylogenetics, comparative genetic mapping, and molecular breeding (Naghavi et al., 2007). So far, no microsatellite markers have been developed for either M. canadense or M. dauricum.
Transcriptome, or RNA sequencing (RNA-seq), technology is an efficient molecular technique for studying the evolution of species, determining differentially expressed genes, exploring the population dynamics, and comparative genetic mapping (Guo et al., 2015; Rai et al., 2016; Arora and Narula, 2017). De novo transcriptome assembly has become a revolutionary mode for high-throughput sequencing in life sciences research (Mardis, 2008; Davey et al., 2011). Transcriptome sequencing not only allows quick and comprehensive analyses of the plant genome but also presents an easy and effective way of developing a large number of EST-SSR markers (Lister et al., 2009; Tan et al., 2013).
To further understand the genetic and genomic background of Menispermum, we sequenced the transcriptomes of both M. canadense and M. dauricum, and we aim to (i) characterize their transcriptomes, (ii) develop polymorphic EST-SSRs in Menispermum, and (iii) test their transferability in the family Menispermaceae. The transcriptomic analyses presented here will offer foundational genetic information for future research, utilization, and protection of Menispermum.
Results
Transcriptome Sequencing and de novo Assembly
In this research, about 53.79 and 57.15 Mb raw reads were generated for M. canadense and M. dauricum, respectively. The raw reads were deposited in the NCBI (National Centre for Biotechnology Information) SRA (Sequence Read Archive) database (M. canadense SRP166813 and M. dauricum: SRP166814). Through rigorous quality checking and data filtering, 44.60 and 45.22 Mb high-quality clean reads were left, respectively. Through de novo assembly, 86,883 and 104,064 transcripts were obtained with a mean length of 799 and 945 bp in M. canadense and M. dauricum, respectively (Table 1). The transcripts (Supplementary Figure S1) were then clustered into unigenes with total lengths ranging from 300 to >3,000 bp (unigenes <300 bp length were discarded) in both M. canadense and M. dauricum. In M. canadense, of all 53,712 unigenes, 14,579 were of 300 bp, while 1,569 were longer than 3,000 bp (Figure 1). From 78,921 unigenes of M. dauricum, 15,550 had 300 bp length, and only 3,015 were longer than 3,000 bp. Unigenes present an ample source of information for the identification of genes and molecular markers.
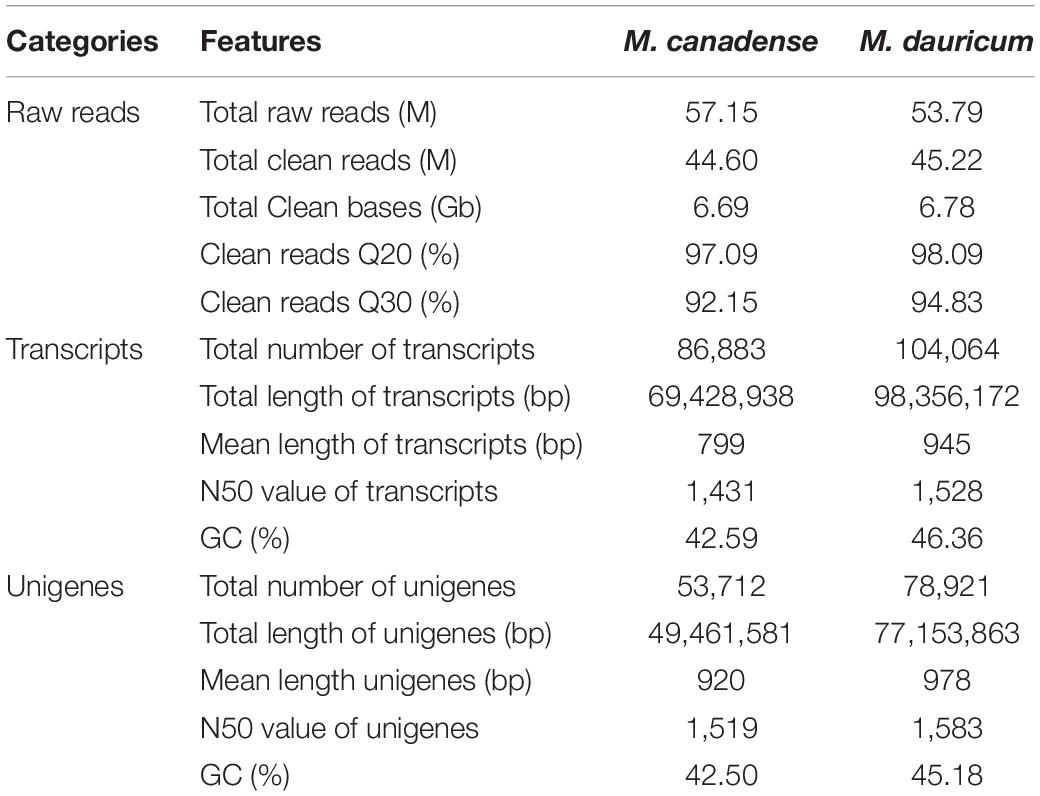
Table 1. Summary of de novo transcriptome assembly of Menispermum canadense and M. dauricum.
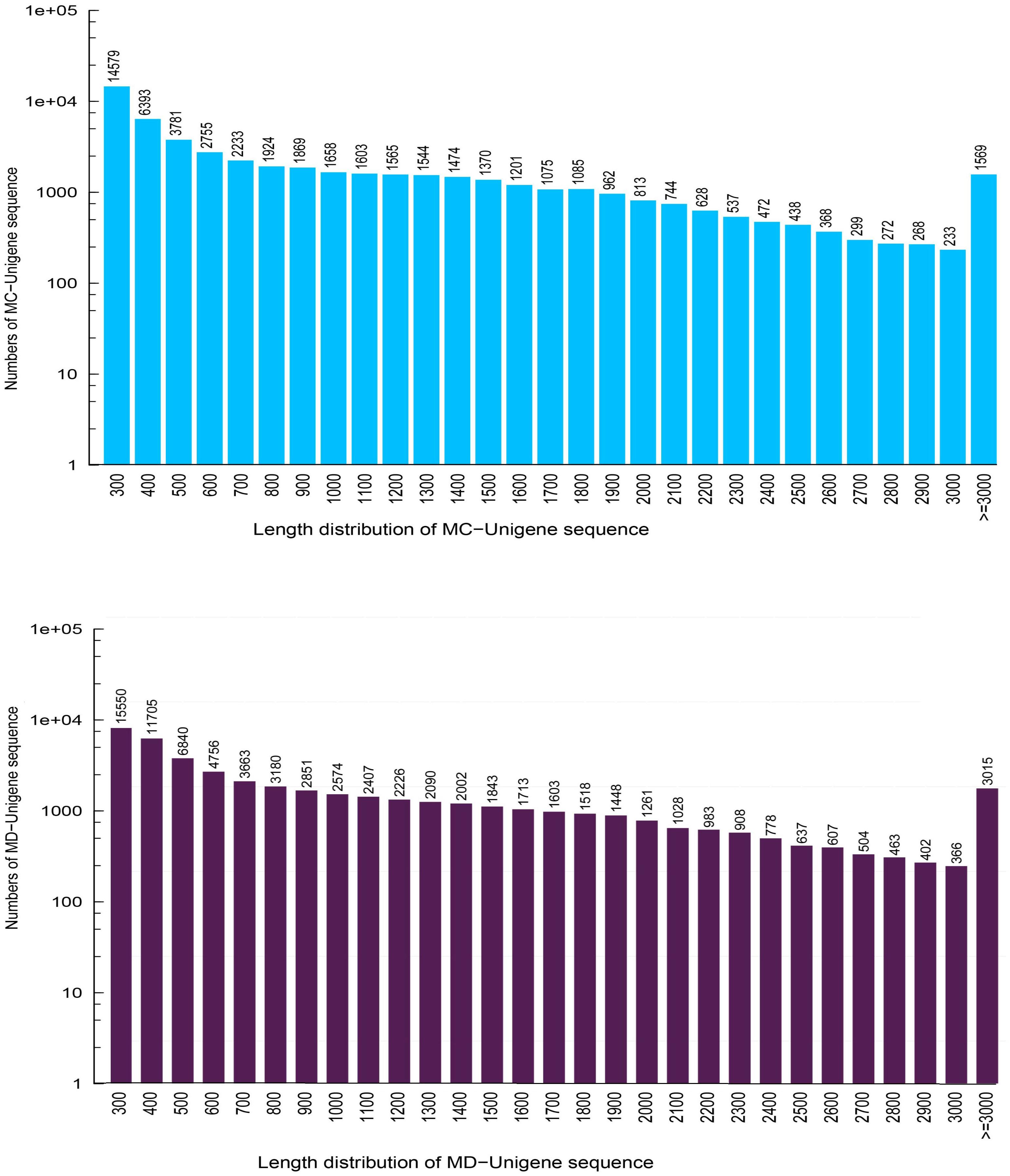
Figure 1. Length distribution of all unigenes in Menispermum canadense (MC) and M. dauricum (MD). The x-axis represents the lengths of all the unigenes, and the y-axis represents the numbers of unigenes with certain length.
Functional Annotation of Unigenes
De novo assembled unigenes of M. canadense and M. dauricum were functionally annotated against the six functional public databases, Nt, Nr, COG, GO, KEGG, and Swiss–Prot. All nucleotide sequences were obtained by splicing, and the BLAST algorithm (E < 1E-5) was used for comparison and to get a similar sequence and corresponding annotation.
Of M. canadense and M. dauricum unigenes, 35,842 (66.73%) and 52,180 (66.11%) show significant homology with the proteins in the Nr database, while 24,302 (45.25%) and 40,554 (51.38%) of the control sequences match with the entries in the Nt database (Table 2), respectively. As for the species distribution of Nr annotations (Figure 2A) of M. canadense and M. dauricum (Figure 2B), Nelumbo nucifera (Nelumbonaceae) has the highest similarity score (53.49 and 42.3%, respectively), followed by Vitis vinifera (Vitaceae; 12.01 and 9.4%) and Theobroma cacao (Malvaceae; 2.7 and 1.9%).
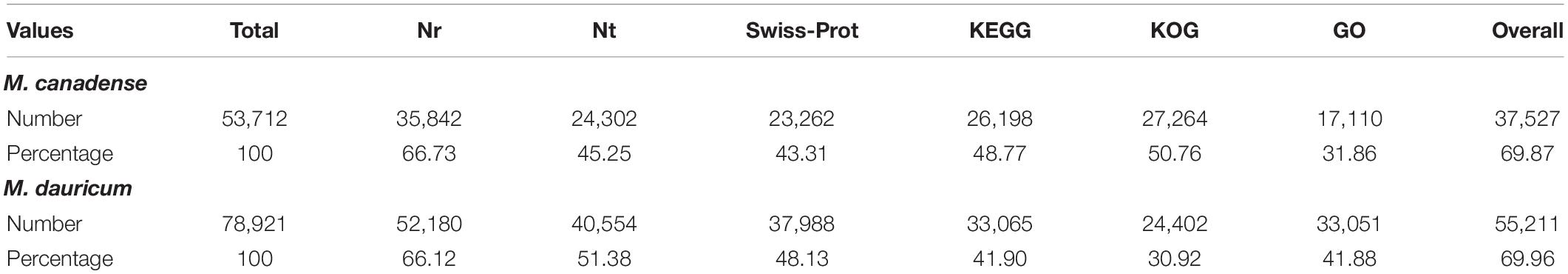
Table 2. Unigenes functional annotation summary of Menispermum canadense and M. dauricum, against 6 functional databases.
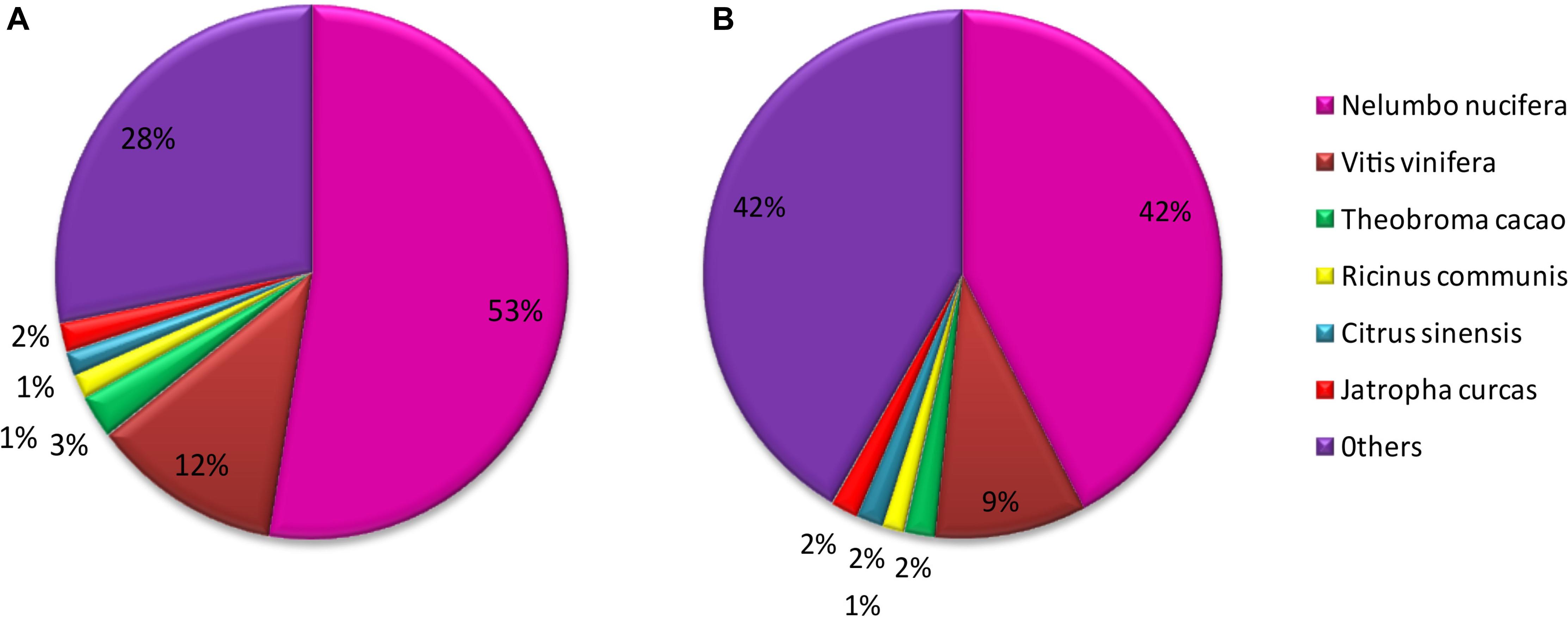
Figure 2. Characteristics of homology analysis in Menispermum unigenes against the non-redundant protein database with an E-value of 10–5. (A) Species based distribution of the top BLASTx hits for 548 each assembled unigenes in M. canadense. (B) Species based distribution of the top BLASTx hits for each assembled unigenes in M. dauricum.
Further, we annotated the unigenes of both Menispermum species with the COG database and calculated the unigene distribution based on the 25 functional groups, including cellular structure, metabolic functions, signal transduction, etc. In M. canadense, general function prediction (6,861, 19%) represents the largest group, followed by signal transduction mechanism with 3,649 genes (10%, Figure 3A), while in M. dauricum, general function prediction, with 7,432 genes (16%), is again the largest group, followed by transcription (4,333 genes, 9%), and translation, ribosomal structure, and biogenesis (4,011 genes, 8%). Cell motility is the smallest group in M. canadense, while in contrast in M. dauricum, nuclear structures and extracellular structures are classified as the smallest groups.
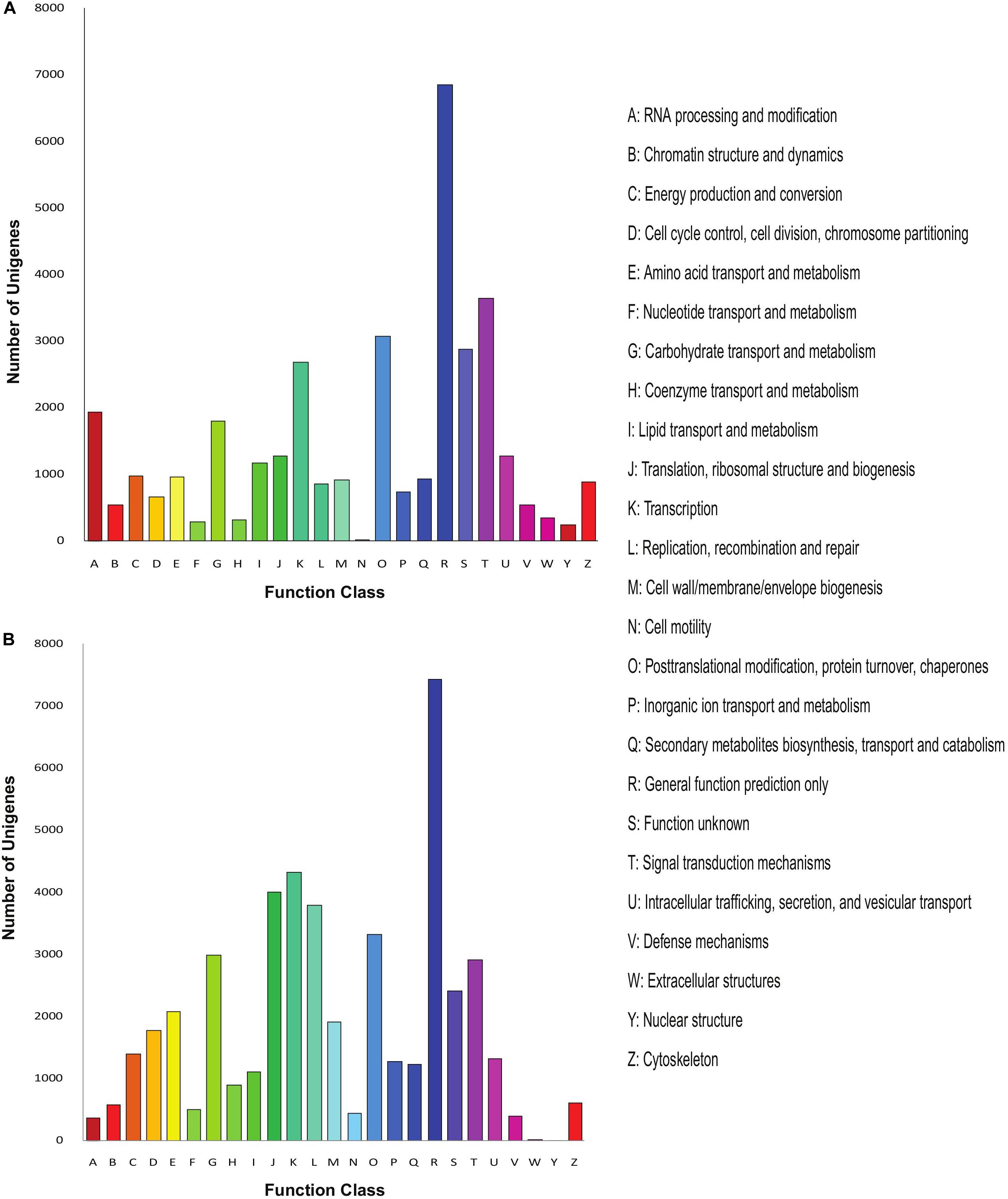
Figure 3. COG analysis of the unigene sequences of Menispermum. (A) COG analysis of the unigene sequences of M. canadense. (B) COG analysis of the unigene sequences of M. dauricum. The x-axis indicates the function class while y-axis indicates the number of unigenes in specific functional group.
The unigenes aligned to the Nr database were further annotated to the GO database. We annotated 17,110 (31.86%) unigenes into three major categories: biological process (7,167, 28%), cellular component (8,436, 33%), and molecular functions (10,056, 39%), with 54 subcategories (Figure 4A). Most of the unigenes in the biological processes are specified for the metabolic process (8,748) and cellular processes (8,303), while catalytic activity (8,456) and binding (7,550) are the major subcategories in the metabolic functions. In M. dauricum, 33,051 unigenes were assigned to GO functional groups (Figure 4B). The distribution of ontology categories is consistent with most of the sequenced plant transcriptomes, such as Vaccinium cyanococcus (Rowland et al., 2012) and Salix integra (Shi et al., 2016). Mostly sequenced unigenes are accountable for the fundamental biological metabolism processes, cell, and cell parts demonstrated from the GO annotation for Menispermum.
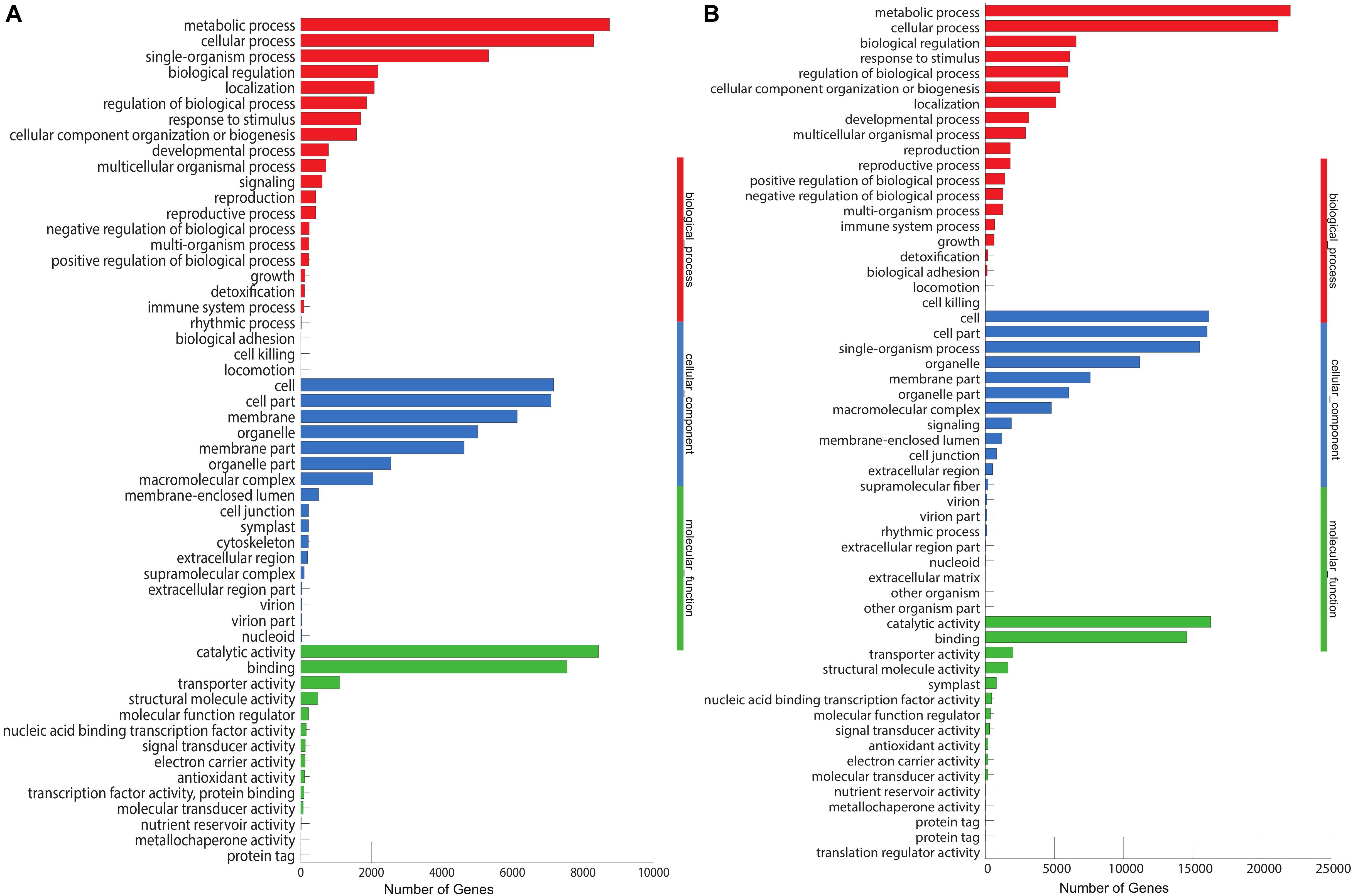
Figure 4. Gene Ontology (GO classification of assembled unigenes of Menispermum. (A) A summary of GO analysis of the M. canadense. (B) Summary of GO analysis of the M. dauricum. The y-axis indicates genes in the specific categories of the three main categories while x-axis indicates the number of genes in the category.
Menispermum canadense and M. dauricum transcriptomes were also analyzed against the KEGG database, with 26,198 (48.77%) and 33,065 (41.09%) unigenes identified, respectively. Unigenes are significantly assigned to 135 metabolic pathways. In M. canadense, metabolic pathways are categorized into six main divisions (Figure 5A), of which metabolisms is the largest division (with 14,503 genes), followed by genetic information processing (5,450) and environmental information processing (1,477), cellular processing (1,000), organismal systems (721), and human diseases (92) pathways. M. dauricum also shows the same divisions, with 25,643 genes in the largest category, metabolisms, followed by genetic information process with 11,382 genes (Figure 5B). In addition, 23,262 (43.31%) and 37,988 (48.13%) unigenes match with the Swiss-Prot database in M. canadense and M. dauricum, respectively. These functional annotations present valuable information for understanding gene structures and functions and developmental and biochemical pathways in Menispermum.
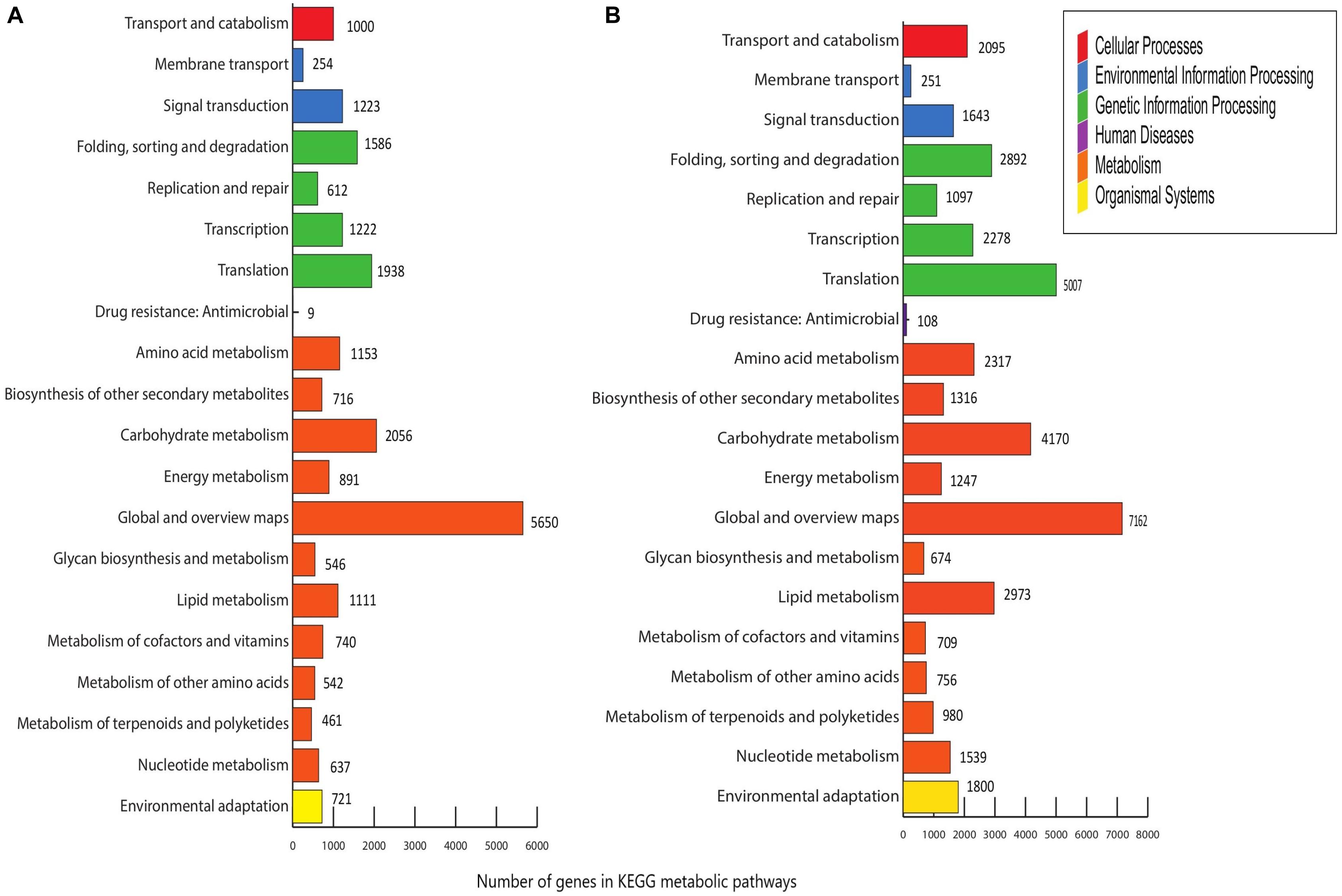
Figure 5. KEGG metabolic pathways of Menispermum. (A) Metabolic pathways of assembled unigenes of M. canadense. (B) Metabolic pathways of assembled unigenes of M. dauricum. The y-axis is the name of metabolic pathway, and the x-axis is the ratio of the number of the genes.
Identification of Candidate Polymorphic EST-SSR
Five hundred and twenty-one candidate polymorphic EST-SSRs were successfully mined out from the transcriptomes of M. canadense and M. dauricum, with a mean length of 20 bp (Supplementary Table S1). Of these EST-SSRs, tri-nucleotide repeats (TNR) are the most abundant repeat type (307; 59.38%), followed by di- (DNR; 197; 38.10%), tetra- (TTR; 10; 1.93%), penta- (PNRs; 1; 0.19%), and hexa- (HNR; 2; 0.39%) nucleotide repeats (Figure 6). Among the DNR, AG/CT (74; 19.3%) is quite dominant, followed by CT/GA (50) and AT/TA (32). AAG/CTT is the most abundant unit for TNR (23.6%), followed by CCG/GGC and AAC/GTT. Interestingly, there is only one CG motif, four CCG, and 11 CGG units in Menispermum.
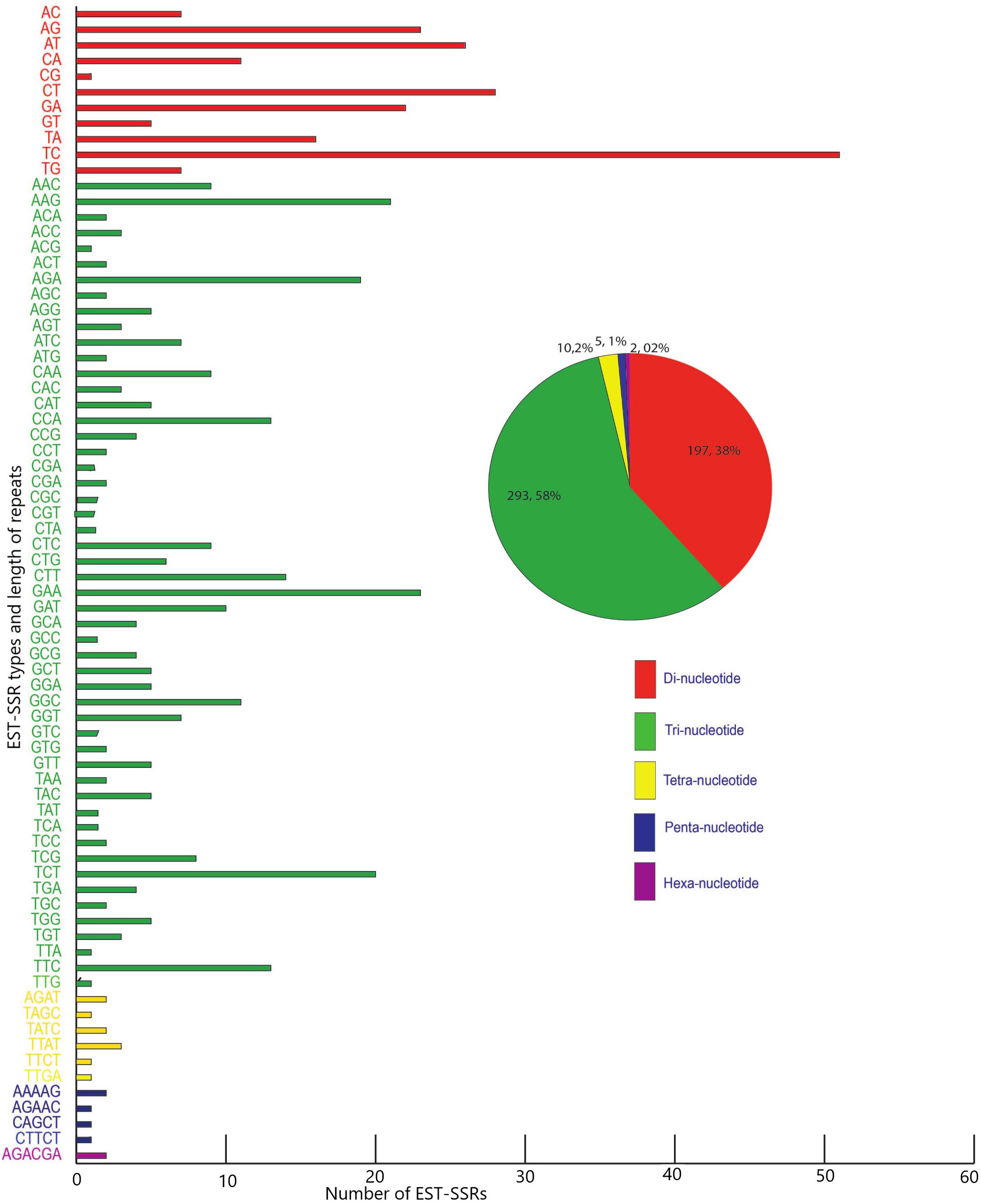
Figure 6. Frequency and distribution of candidate polymorphic EST-SSR from the transcriptome of Menispermum canadense and M. dauricum.
EST-SSR Primer Design, Validation, and Cross-Species Transferability
From the RNA-seq data of M. canadense and M. dauricum, we identified 521 candidate polymorphic EST-SSRs (Supplementary Table S1). The SSR loci were then evaluated by the following criteria: zero missing rate value, 1–2 transferability value, repeat motif ≥3 bp, no primer dimmers, no hairpin structures, and no mismatches. After screening, 23 primer pairs were selected and successfully amplified, which yielded PCR products of the expected size with clear bands. Moreover, these primers were then amplified across 60 individuals from each Menispermum species. The number of alleles ranged from 2 to 8, with a mean of 3.70 alleles per locus. Functional annotations were characterized in the 23 developed polymorphic SSRs of Menispermum, and they mostly belong to general function prediction, followed by carbohydrate transport and metabolism (Supplementary Table S2).
The observed heterozygosity (Ho) varies from 0.059 to 0.957, and expected heterozygosity (He) ranges from 0.195 to 0.729 (Table 3). The polymorphism information content (PIC) varies from 0.181 to 0.69, with a mean of 0.51. Allelic richness (RS) across the six populations from East Asia to North America (Table 4) ranges from 1.80 (AH) to 3.41 (BJ), with the total number of detected alleles (NA) varied from 40 (AH) to 75 (BJ). At the regional level, the mean estimates of within-population diversity (e.g., RS, He, Ho) are different in United States (RS = 2.58, He = 0.506, Ho = 0.403) and China (RS = 2.40, He = 0.247, Ho = 0.281). The population BJ is found to depart from Hardy–Weinberg equilibrium significantly (P < 0.05; Table 4).
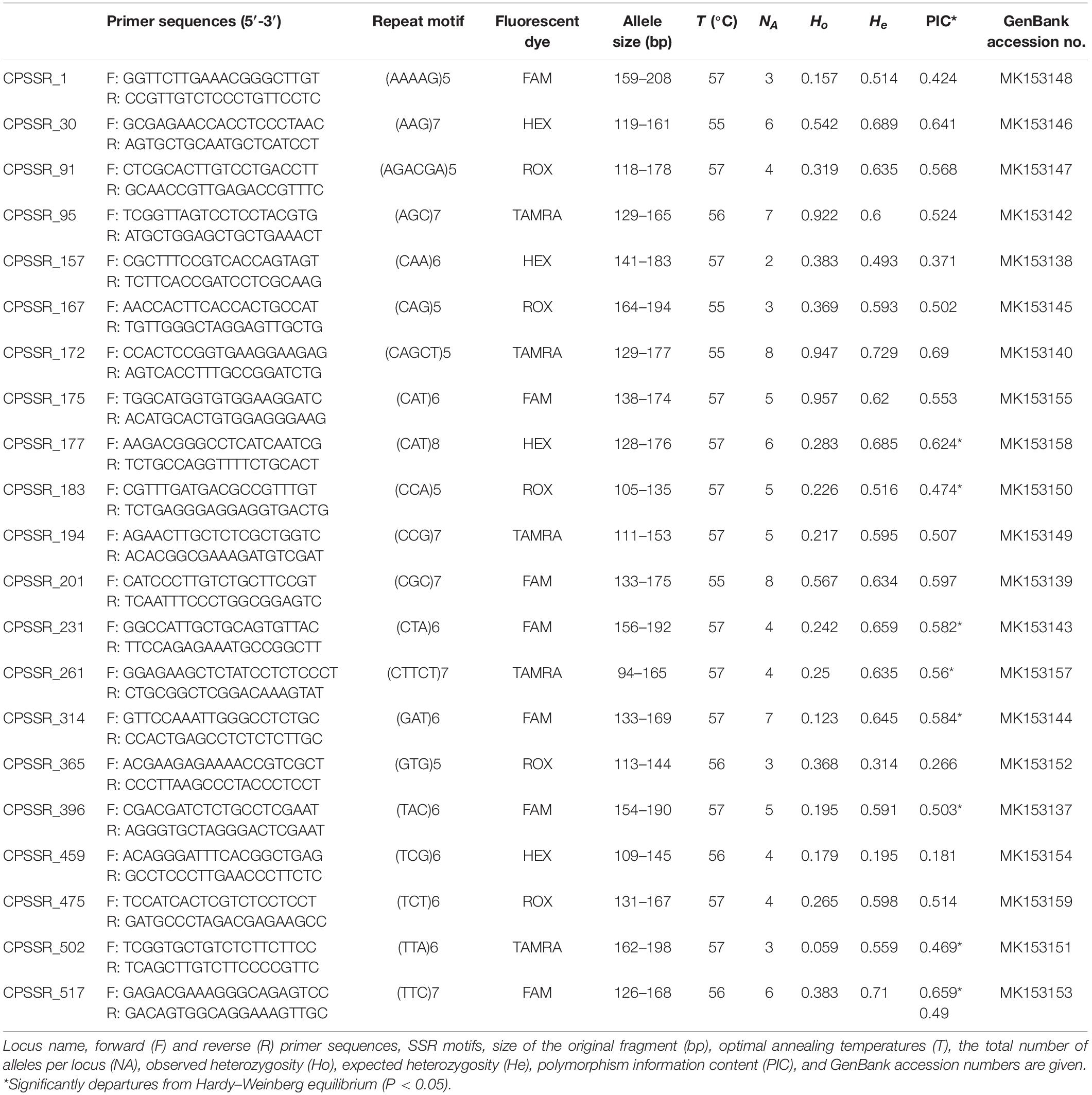
Table 3. Characteristics of 23 newly developed polymorphic microsatellite loci in Menispermum.
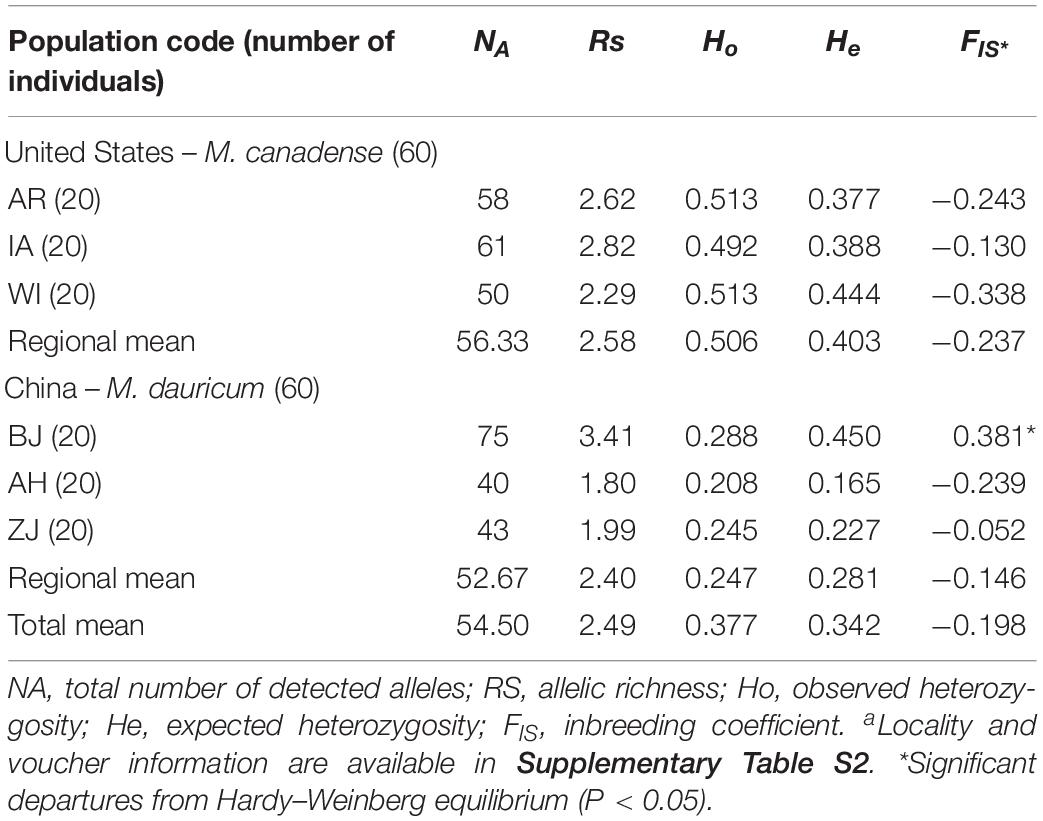
Table 4. Genetic parameters of the 23 microsatellite loci within six populations of Menispermum.a
In the current study, 23 candidate polymorphic EST-SSRs were developed to assess the cross-species transferability in three species Cocculus orbiculatus, Sinomenium acutum, Stephania tetrandra from the Menispermaceae. The transferability ratios ranged from 85 to 90%.
Discussion
In the current study, using RNA-seq technology on the Illumina HiSeq 2500 platform, we have characterized the transcriptomes of both Menispermum species, M. canadense, and M. dauricum. In spite of their medicinal value (Liu et al., 2009), there is a deficiency in genetic knowledge of the genus Menispermum, and there are no available SSR markers.
The mean length and N50 of all unigenes of M. canadense were 920 bp and 1,519 bp, and those of M. dauricum were 945 bp and 1,528 bp, respectively. In comparison to previous studies, the mean length and N50 of all unigenes in Menispermum are similar to those in Curcuma longa (910 bp, 1,515 bp; Annadurai et al., 2013) but much higher than those in Ipomoea batatas (581 bp, 765 bp; Wang et al., 2010), Cicer arietinum (523 bp, 900 bp; Garg et al., 2011), and Sesamum indicum (629 bp, 947 bp; Wei et al., 2011). Additionally, the mean length is slightly lower than in Zantedeschia rehmannii (1,038 bp, 1,476 bp; Wei et al., 2016) and Euphorbia fischeriana (1,066 bp, 1,500 bp; Barrero et al., 2011). Previous studies suggest that longer mean lengths of the unigenes and larger N50 values signify accurate and effective transcriptome assembly (Chen et al., 2015; Li et al., 2018). The longer unigenes with high sequencing depth in Menispermum will be valuable for exploring the gene functions and the molecular mechanisms.
The unigene annotation rate was much higher in M. dauricum (producing 55,211 out of 78,921 significant hits) than in M. canadense (37,527 out of 53,712), with percentages of 69.96 and 69.87, respectively (Table 2). Based on Nr function annotation, the ratio of unigene similarity for M. canadense, 53.49% of annotated unigenes, exhibited similarity to Nelumbo nucifera (Indian lotus), which is higher than M. dauricum, with 42.3% (Figure 2). The limited whole chloroplast genome sequences and transcriptome sequences within public databases for Menispermum could influence the annotation efficiency. The gene function database (GO) predicts the physiological role of the unigenes (Kumar et al., 2014). In M. canadense, GO classified into 54 subcategories of three main categories at ratios of 23, 17, and 14, while in M. dauricum, it divided into 57 subcategories, with 23, 19, and 15, respectively, in the three main categories (Figure 3). This result is also comparable with prior studies on C. alismatifolia (Taheri et al., 2019), in which unigenes were grouped into 51 subcategories within three categories at 23, 17, and 11, and Rhododendron rex (Zhang et al., 2017), with 62 subcategories at 25, 20, and 17. In addition, several unigenes identified in the annotated GO database are responsible for cold shock (K09250), heat tolerance (K09419), and osmotic stress (GO:0009266) in Menispermum (Mutlu et al., 2013; Zhang et al., 2017). These results illustrate the involvement of diverse molecular functional unigenes in varied metabolic pathways.
Of all KEGG pathways, global and overview maps were dominant, followed by carbohydrate metabolism, translation, folding, sorting and degradation, in M. canadense, while in M. dauricum, global and overview maps were dominant, followed by translation, carbohydrate metabolism, and lipid metabolism. Moreover, 135 pathways mapped against the KEGG database in M. canadense, which is higher than for M. dauricum, which significantly matched with only 128 pathways. Our findings generated rich genetic and genomic information that will facilitate further research in biochemistry, gene function discovery, physiological genetics, molecular genetics, and the biological pathways in Menispermum or related species.
Microsatellites have been extensively used in forensics, phylogeography, genomic mapping, population genetics, conservation genetics, molecular breeding, and determining parentage over the past several decades (Ellegren, 2000; Esselink et al., 2004). The frequency and distribution of microsatellites are dependent on an array of factors such as the size of the dataset, tools, and criteria used for mining. EST-SSRs are from the transcribed DNA regions and exhibit major advantages for their presence in the functional genes, consistent amplification efficiency, and high cross-species transferability rates (Scott et al., 2000; Gupta et al., 2003). To our knowledge, no SSR markers are available in Menispermum. The current study presents the first mining and development of EST-SSRs in Menispermum. To date, several tools have been used for the assessment of SSR and polymorphism, such as MISA (Thiel, 2003), SSRLocator (Da Maia et al., 2008), and GMATA (Wang and Wang, 2016). However, they all have some deficiencies, such as long running time, poor capacity for a large dataset, a need to manually screen for polySSR, or inability to dealing with large genomes or multiple individuals. CandiSSR (Xia et al., 2016) enables users to find the putative polymorphic SSR from several species with great ease and efficiency from the genomes and also from the transcriptomes. In the current study, we used CandiSSR for the development of putative polymorphic EST-SSR markers. From the transcriptome of the two species of Menispermum, 521 polymorphic EST-SSR markers were proficiently mined. The most dominant nucleotide repeat motifs are AG/CT (19.3%) and AAG/CTT (23.6%) for di- and trinucleotide repeat motifs, which are similar to Heveabra siliensis (rubber tree) (Li et al., 2012) and Sesamum indicum L. (sesame) (Zhang et al., 2012). Trinucleotides are the most abundant repeat motifs in Menispermum because, in open reading frames (ORFs), there may be no hindrance from the insertions, deletions, or any mutations within the translated regions, whereas frameshift mutation may restrict the development of other types of SSR motifs (Metzgar et al., 2000; Chen et al., 2015). Our results robustly support and expand the belief that there is a low GC content in eudicots, because only one CG, four CCG, and eleven CGG motifs were found. The rarity of GC/CG, CCG/CGG repeat units (Kumpatla and Mukhopadhyay, 2005; Chen et al., 2015) has been reported in plenty of dicotyledonous plants, for example, Medicago truncatula (Eujayl et al., 2004), Raphanus sativus (Wang et al., 2013), and Ipomoea batatas (Wang et al., 2011). Previous studies on dicotyledonous plants (Morgante et al., 2002; Kumpatla and Mukhopadhyay, 2005) also illustrated that the tri-nucleotide AAG motif may be significantly prominent, such as in Cucumis sativus (Hu et al., 2010), Ricinus communis (Tan et al., 2014), and Sesamum indicum (Wei et al., 2011).
In the present study, 23 polymorphic EST-SSR markers were selected to evaluate the genetic diversity among Menispermum populations. In the developed polymorphic EST-SSRs, the number of alleles ranges from 2 to 8 with a mean of 4.86, Ho = 0.38, He = 0.58, and PIC = 0.49 (Table 3), which depict a high level of polymorphism in Menispermum. Besides, M. canadense demonstrated higher genetic diversity, with NA = 56.33 and RS = 2.58, than M. dauricum, with NA = 4.50 and RS = 2.49 (Table 4). The genetic diversity patterns found here may help in our future study on the East Asia-North America disjunctive distribution of Menispermum.
The 23 polymorphic EST-SSR markers were also tested for transferability in three other Menispermaceae species. The primers were successfully amplified with multiple bands in all the species except for SR1. The transferability ratio was 85–90%, which is comparable or higher than that obtained in Festuca arundinacea (92%; Saha et al., 2004), Medicago truncatula (89%; Eujayl et al., 2004), Epimedium sagittatum (85.7%; Zeng et al., 2010), and Cumumis melo (12.7%; Fernandez-Silva et al., 2008). EST-SSRs depict a higher transferability ratio owing to the high conservation of genetic markers in comparison to gSSRs, which are derived through genomic libraries. Hence, the high transferability across the family Menispermaceae presents valuable information resources for the development of molecular markers and evolutionary studies. Furthermore, the novel polymorphic EST-SSR markers and the characterization of Menispermum transcriptomes will offer a comprehensive source of specific genes along with in-depth knowledge of their pathways.
Materials and Methods
RNA Isolation, Construction of cDNA Library, Illumina Sequencing and Reads Filtering
Fresh leaves from M. canadense and M. dauricum were sampled, cleaned, and placed in liquid nitrogen until RNA extraction. RNA was isolated by using an RNeasy Plant kit (Qiagen Bioinformatics, Germany), and quality and quantity were evaluated with an Agilent 2100 Bioanalyzer (Agilent Technologies, United States) and NanoDropTM (Agilent RNA 6000 Nano Kit) to determine the purity of the RNA samples. Preparation of two cDNA libraries was then performed using a NEBNext Ultra TM RNA-seq Library Preparation Kit (New England Bio Labs, United States). Oligo-(dT) magnetic beads were used to isolate poly-A from the total RNA. cDNA fragments were purified by MinElute Kit (Qiagen Bioinformatics, Germany) and resolved with the EB buffer for the reparation of the end with the addition of single nucleotide A (Adenine). The first-strand cDNA was synthesized by random hexamer primers and superscript III reverse transcriptase. After the second-strand cDNA preparation, end-repaired and dA-tailed fragments connected with the sequencing adapters. Adapter ligated cDNA libraries (about 500 bp) were amplified and sequenced through the Illumina HiSeq 2500 platform (Illumina Inc., BGI, Shenzhen, China). Raw data (2 × 150 bp paired-end reads) filtration was done by FASTX-TOOLKIT (Gordon and Hannon, 2010) version 0.0.14, which removed the reads with adapters with more than 5% unknown bases (N) and low-quality reads with quality less than 15% and greater than 20%. After filtering, the remaining reads were clean reads.
De novo Assembly and Annotation
TRINITY (Grabherr et al., 2011) version 2.0.6 was employed to perform the de novo assembly with clean reads. Paired-end reads were mapped to contigs from the same transcripts and distances. The resulting sequences with the lowest number of Ns were generated as unigenes. To avoid redundancy, TGICL (Pertea et al., 2003) version 2.0.6 (-l 40 -c 10 -v 25 -O ’-repeat_stringency 0.95 - minmatch 35 -minscore 35’) was then used to cluster 86,883 and 104,064 transcripts into 53,712 and 78,921 non-redundant unigenes in M. canadense and M. dauricum, respectively. The raw sequencing data from both of the species were deposited in the Sequence Read Archive (SRA) of NCBI (National Center for Biotechnology Information and their accession numbers were acquired. BLASTN version 2.2.23 with default parameters1 and BLASTX (e-value <10–5) were used to align Unigenes to the Nt, Nr2, KOG3, KEGG4, and Swiss-Prot5 databases to perform the annotations, while Blast2GO version2.5.06 was used for Nr annotation to perform the GO annotation.
EST-SSR Marker Development, Mining, and Primer Design
Through CandiSSR (Xia et al., 2016), we successfully mined 521 candidate polymorphic EST-SSRs with a 20-bp mean length (Supplementary Table S1) by comparing the unigenes of two Menispermum species. In CandiSSR, the parameters were set to sequence length 100, blast e-value 1e–10 cutoff, 95 cutoff blast identity, and 95 cutoff blast coverage. Based on the Primer3 package (Koressaar and Remm, 2007), primers were generated automatically for each SSR locus. Optimal polySSR primers were selected under three criteria: (1) missing rate value = zero, (2) transferability value = 1; (3) repeat motif ≥3 bp. Primer dimers, hairpin structures, or mismatches were checked in OLIGO (Rychlik, 2007) version 7 (Molecular Biology Insights Inc., Colorado Springs, CO, United States). With this strict screening strategy, we sorted out 23 primer pairs for amplification. Electrophoresis peaks were assessed through GeneMarker (Holland and Parson, 2011) version 2.4.0. In total, 23 primer pairs that had high variations and stable repetition were selected for further analysis. The primer sequences analyzed in this study were submitted to GenBank (Table 3).
EST-SSR Amplification and Transferability in Cross-Species
To perform PCR, a forward primer with an M13 sequence (5′−TGTAAAACGACGGCCAGT−3′) at the 5′ end was synthesized for all loci, and four fluorescents (FAM, ROX, HEX, and TAMRA) were labeled with a universal M13 primer (5′−TGTAAAACGACGGCCAGT−3′). The two currently recognized Menispermum species were selected for population genetic studies. Within the genus, three populations from M. canadense ranging from North America to Canada and three from M. dauricum endemic to China were selected. Three populations with 20 individuals from each species (Supplementary Table S3) were tested for population genetics analyses. DNA was extracted from silica gel-dried leaves through Plant DNAzol (Invitrogen Life Technologies). The DNA quality was assessed through electrophoresis on the gel with 0.8% agarose colored with 1% GelRed (Biotium) with the reference of a 1,000-bp marker (TaKaRa, Dalian, Liaoning, China), on the basis of the integrity and intensity of the band on the gel.
PCR amplifications were performed through the two−step PCR protocol (Schuelke, 2000) by following the Tsingke PCR kit protocol (Tsingke Biotech Company, Beijing, China). In the first step, a final volume of 25 μL PCR mixture was obtained containing 1.0 μL DNA template, 12.5 μL Mix AmpliTaqGold 360 (Thermofisher Biotech Company, Applied Biosystems, Foster City, CA, United States), 9.5 μL of distilled water, and 1.0 μL of forward primer (which was produced with an 18-bp M13 tail 5′-TGTAAAACGACGGCCAGT-3′ at the 5′ end) and 1.0 μL reverse primers. The PCR amplification procedure was initial denaturation for 5 min at 95°C, 35 cycles of 30 s at 95°C, 45 s at annealing temperature (optimal primer temperature TM), 30 s syntheses at 72°C, and finally a 10-min extension period at 72°C with a 4°C holding temperature. In the second step, a 30-μL final volume was obtained containing 3 μL of the first PCR products, 1 μL fluorescent-labeled (FAM, ROX, HEX, TAMRA) universal M13 primer and 1 μL of reverse primer, 15 μL PCR Mix, and 10 μL distilled water. SSR loci were amplified under the following conditions: 2 min at 94°C; 35 cycles of 94°C for 60 s, 30 min at 57°C, 60 s at 72°C, and finally a 10-min extension step at 72°C and a 4°C holding temperature. Electrophoresis peaks were assessed through Gene Marker version 2.4.0 (Soft Genetics, State College, PA, United States). A total of 23 primer pairs demonstrating stable repeatability with high variations were picked for further analysis.
Cross-species transferability was tested among three Menispermaceae species, Cocculus orbiculatus, Sinomenium acutum, and Stephania tetrandra. Five individuals from each were assessed using the same DNA extraction and PCR procedure. A gene marker was used to assess the peaks.
Population Genetic Diversity Analysis
Overall genetic parameters, i.e., the number of alleles, observed and expected heterozygosities, and PIC (polymorphism information content) were calculated for the assessment of genetic polymorphism per locus using CERVUS (Kalinowski et al., 2007) version 3.0.3 and GenAlEx (Peakall and Smouse, 2006) version 6.5. The significance of deviations from Hardy–Weinberg equilibrium, given by FIS deviation, were tested by FSTAT (Goudet, 2001) version 2.9.3.
Data Availability Statement
The datasets generated for this study can be found in the M. canadense: SRP166813, M. dauricum: SRP166814, SSR loci: MK153148, MK153146, MK153147, MK153142, MK153138, MK153145, MK153140, MK153155, MK153158, MK153150, MK153149, MK153139, MK153143, MK153157, MK153144, MK153152, MK153137, MK153154, MK153159, MK153151, MK153153.
Author Contributions
CF, PL, and FH made substantial contributions to the conception and design of this research. In particular, PL and SW collected the materials. FH performed the experiments, wrote the manuscript, organized the contents of the article, and prepared figures. FH and GY analyzed the data. PL, SW, and CF revised the manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (Grant No. 31970225), the Zhejiang Provincial Natural Science Foundation (Grant No. LY19C030007), and the NSFC-NSF Dimensions of Biodiversity Program (Grant No. 31461123001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00380/full#supplementary-material
FIGURE S1 | The length distribution of the transcripts in Menispermum canadense (MC) and M. dauricum (MD). The x-axis represents the lengths of all the trinity sequences, and the y-axis represents the numbers of trinity sequences with certain length.
TABLE S1 | Designed EST-SSR from CandiSSR.
TABLE S2 | EST-SSR functional annotation in Menispermum.
TABLE S3 | Locality and voucher information for Menispermum used in this study. Voucher specimens are deposited at the herbarium of Zhejiang University (HZU), Hangzhou, Zhejiang, China.
Footnotes
- ^ http://blast.ncbi.nlm.nih.gov/Blast.cgi
- ^ ftp://ftp.ncbi.nlm.nih.gov/blast/db
- ^ http://www.ncbi.nlm.nih.gov/KOG
- ^ http://www.genome.jp/keg
- ^ http://ftp.ebi.ac.uk/pub/databases/swissprot
- ^ http://www.blast2go.com
References
Annadurai, R. S., Neethiraj, R., Jayakumar, V., Damodaran, A. C., Rao, S. N., Katta, M. A., et al. (2013). De Novo transcriptome assembly (NGS) of Curcuma longa L. rhizome reveals novel transcripts related to anticancer and antimalarial terpenoids. PLoS One 8:e56217. doi: 10.1371/journal.pone.0056217
Arora, L., and Narula, A. (2017). Gene editing and crop improvement using CRISPR-Cas9 system. Front. Plant Sci. 8:1932. doi: 10.3389/fpls.2017.01932
Barrero, R. A., Chapman, B., Yang, Y., Moolhuijzen, P., Keeble-Gagnère, G., Zhang, N., et al. (2011). De novo assembly of Euphorbia fischeriana root transcriptome identifies prostratin pathway related genes. BMC Genomics 12:600. doi: 10.1186/1471-2164-12-600
Chen, H., Liu, L., Wang, L., Wang, S., Somta, P., and Cheng, X. (2015). Development and validation of EST-SSR markers from the transcriptome of adzuki bean (Vigna angularis). PLoS One 10:e0131939. doi: 10.1371/journal.pone.0131939
Da Maia, L. C., Palmieri, D. A., De Souza, V. Q., Kopp, M. M., de Carvalho, F. I. F., and Costa de Oliveira, A. (2008). SSR locator: tool for simple sequence repeat discovery integrated with primer design and PCR simulation. Int. J. Plant Genom. 2008:412696.
Davey, J. W., Hohenlohe, P. A., Etter, P. D., Boone, J. Q., Catchen, J. M., and Blaxter, M. L. (2011). Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 12:499. doi: 10.1038/nrg3012
Ellegren, H. (2000). Microsatellite mutations in the germline: implications for evolutionary inference. Trends Genet. 16, 551–558. doi: 10.1016/s0168-9525(00)02139-9
Esselink, G., Nybom, H., and Vosman, B. (2004). Assignment of allelic configuration in polyploids using the MAC-PR (microsatellite DNA allele counting—peak ratios) method. Theor. Appl. Genet. 109, 402–408. doi: 10.1007/s00122-004-1645-5
Estoup, A., Cornuet, J., Goldstein, D., and Schlötterer, C. (1999). “Microsatellites: evolution and applications,” in Microsatellite Evolution: Inferences from Population Data, eds D. B. Goldstein and C. Schlotterer (New York, NY: Oxford University Press), 49–64.
Eujayl, I., Sledge, M. K., Wang, L., May, G. D., Chekhovskiy, K., Zwonitzer, J. C., et al. (2004). Medicago truncatula EST-SSRs reveal cross-species genetic markers for Medicago spp. Theor. Appl. Genet. 108, 414–422. doi: 10.1007/s00122-003-1450-6
Fernandez-Silva, I., Eduardo, I., Blanca, J., Esteras, C., Pico, B., Nuez, F., et al. (2008). Bin mapping of genomic and EST-derived SSRs in melon (Cucumis melo L.). Theor. Appl. Genet. 118, 139–150. doi: 10.1007/s00122-008-0883-3
Garg, R., Patel, R. K., Tyagi, A. K., and Jain, M. (2011). De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 18, 53–63. doi: 10.1093/dnares/dsq028
Gordon, A., and Hannon, G. (2010). FASTX-TOOLKIT, Version 0.0. 14. Computer Program and Documentation Distributed by the Author. Available online at: http://hannonlab.cshl.edu/fastx_toolkit (accessed February 1, 2017).
Goudet, J. (2001). FSTAT Version 2.9. 3, a Program to Estimate and Test Gene Diversities and Fixation Indices. Available at http://www.unil.ch/izea/softwares/fstat.html (accessed March 1, 2019).
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29:644. doi: 10.1186/1471-2105-12-S14-S2
Guichoux, E., Lagache, L., Wagner, S., Chaumeil, P., Léger, P., Lepais, O., et al. (2011). Current trends in microsatellite genotyping. Mol. Ecol. Res. 11, 591–611. doi: 10.1111/j.1755-0998.2011.03014.x
Guo, Q., Su, H., Jiang, Q., Qi, X., Su, U., and Wang, Z. (2015). Phenolic alkaloids from Menispermum dauricum reduce inflammatory reaction and ischemic brain damage in cerebral ischemia rats. Monatshefte Chem. Chem. Monthly 146, 501–509. doi: 10.1007/s00706-014-1359-6
Gupta, P. K., Rustgi, S., Sharma, S., Singh, R., Kumar, N., and Balyan, H. (2003). Transferable EST- SSR markers for the study of polymorphism and genetic diversity in bread wheat. Mol. Genet. Genomics 270, 315–323. doi: 10.1007/s00438-003-0921-4
Holland, M. M., and Parson, W. (2011). GeneMarker® HID: a reliable software tool for the analysis of forensic STR data. J. Forensic Sci. 56, 29–35. doi: 10.1111/j.1556-4029.2010.01565.x
Hu, J.-B., Zhou, X.-Y., and Li, J.-W. (2010). Development of novel EST-SSR markers for cucumber (Cucumis sativus) and their transferability to related species. Sci. Hortic. 125, 534–538. doi: 10.1016/j.scienta.2010.03.021
Kalinowski, S. T., Taper, M. L., and Marshall, T. C. (2007). Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 16, 1099–1106. doi: 10.1111/j.1365-294x.2007.03089.x
Koressaar, T., and Remm, M. (2007). Enhancements and modifications of primer design program Primer3. Bioinformatics 23, 1289–1291. doi: 10.1093/bioinformatics/btm091
Kumar, S., Shah, N., Garg, V., and Bhatia, S. (2014). Large scale in-silico identification and characterization of simple sequence repeats (SSRs) from de novo assembled transcriptome of Catharanthus roseus (L.) G. Don. Plant Cell Rep. 33, 905–918. doi: 10.1007/s00299-014-1569-8
Kumpatla, S. P., and Mukhopadhyay, S. (2005). Mining and survey of simple sequence repeats in expressed sequence tags of dicotyledonous species. Genome 48, 985–998. doi: 10.1139/g05-060
Li, D., Deng, Z., Qin, B., Liu, X., and Men, Z. (2012). De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics 13:192. doi: 10.1186/1471-2164-13-192
Li, X., Li, M., Hou, L., Zhang, Z., Pang, X., and Li, Y. (2018). De novo transcriptome assembly and population genetic analyses for an endangered chinese Endemic Acer miaotaiense (Aceraceae). Genes 9:378. doi: 10.3390/genes9080378
Lister, R., Gregory, B. D., and Ecker, J. R. (2009). Next is now: new technologies for sequencing of genomes, transcriptomes, and beyond. Curr. Opin. Plant Biol. 12, 107–118. doi: 10.1016/j.pbi.2008.11.004
Liu, Q.-N., Zhang, L., Gong, P.-L., Yang, X.-Y., and Zeng, F.-D. (2009). Inhibitory effects of dauricine on early afterdepolarizations and L-type calcium current. Can. J. Physiol. Pharm. 87, 954–962. doi: 10.1139/y09-090
Londre, R. A., and Schnitzer, S. A. (2006). The distribution of lianas and their change in abundance in temperate forests over the past 45 years. Ecology 87, 2973–2978. doi: 10.1890/0012-9658(2006)87
Mardis, E. R. (2008). Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 9, 387–402. doi: 10.1146/annurev.genom.9.081307.164359
Metzgar, D., Bytof, J., and Wills, C. (2000). Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 10, 72–80.
Morgante, M., Hanafey, M., and Powell, W. (2002). Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Gen. 30:194. doi: 10.1038/ng822
Mutlu, S., Karadaǧoǧlu, Ö, Atici, Ö, and Nalbantoǧlu, B. (2013). Protective role of salicylic acid applied before cold stress on antioxidative system and protein patterns in barley apoplast. Biol. Plant. 57, 507–513. doi: 10.1007/s10535-013-0322-4
Naghavi, M. R., Mardi, M., Pirseyedi, S. M., Kazemi, M., Potki, P., and Ghaffari, M. R. (2007). Comparison of genetic variation among accessions of Aegilops tauschii using AFLP and SSR markers. Genet. Res. Crop Evol. 54, 237–240. doi: 10.1007/s10722-006-9143-z
Ortiz, R. D. C., Wang, W., Jacques, F., and Chen, Z. J. T. (2016). Phylogeny and a revised tribal classification of Menispermaceae (moonseed family) based on molecular and morphological data. Taxon 65, 1288–1312. doi: 10.12705/656.5
Peakall, R., and Smouse, P. E. (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295. doi: 10.1111/j.1471-8286.2005.01155.x
Pertea, G., Huang, X., Liang, F., Antonescu, V., Sultana, R., Karamycheva, S., et al. (2003). TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19, 651–652. doi: 10.1093/bioinformatics/btg034
Purrington, F., and Horn, D. (1993). Canada moonseed vine (Menispermaceae): Host of four roundheaded wood borers in central Ohio (Coleoptera: Cerambycidae). Pentomol. Soc. Wash. 95, 313–320.
Rai, R., Chauhan, S. K., Singh, V. V., Rai, M., and Rai, G. (2016). RNA-seq analysis reveals unique transcriptome signatures in systemic lupus erythematosus patients with distinct autoantibody specificities. PLoS One 11:e0166312. doi: 10.1371/journal.pone.0166312
Rowland, L. J., Alkharouf, N., Darwish, O., Ogden, E. L., Polashock, J. J., Bassil, N. V., et al. (2012). Generation and analysis of blueberry transcriptome sequences from leaves, developing fruit, and flower buds from cold acclimation through deacclimation. BMC Plant Biol. 12:46. doi: 10.1186/1471-2229-12-46
Rychlik, W. (2007). OLIGO 7 primer analysis software. Methods Mol. Biol. 402, 35–59. doi: 10.1007/978-1-59745-528-2_2
Saha, M. C., Mian, M. A. R., Eujayl, I., Zwonitzer, J. C., Wang, L., and May, G. D. (2004). Tall fescue EST-SSR markers with transferability across several grass species. Theor. Appl. Genet. 109, 783–791. doi: 10.1007/s00122-004-1681-1
Schuelke, M. (2000). An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 18:233. doi: 10.1038/72708
Scott, K. D., Eggler, P., Seaton, G., Rossetto, M., Ablett, E. M., Lee, L. S., et al. (2000). Analysis of SSRs derived from grape ESTs. Theor. Appl. Genet. 100, 723–726. doi: 10.1007/s001220051344
Shi, X., Sun, H., Chen, Y., Pan, H., and Wang, S. (2016). Transcriptome sequencing and expression analysis of cadmium (Cd) transport and detoxification related genes in Cd-accumulating Salix integra. Front. Plant Sci. 7:1577. doi: 10.3389/fpls.2016.01577
Song, Y.-P., Jiang, X.-B., Zhang, M., Wang, Z.-L., Bo, W.-H., An, X.-M., et al. (2012). Differences of EST-SSR and genomic-SSR markers in assessing genetic diversity in poplar. Forest. Stud. China 14, 1–7. doi: 10.1007/s11632-012-0106-5
Su, Y., Su, H., and Sheng, B. (2004). Effect of phenolic alkaloids from Menispermum dauricum on hemodynamics in experimental myocardial ischemia. China Pharm. 7, 83–85.
Su, Y.-M., Zhang, C., Xiao, J.-Y., Gang, H., Wang, Z., and Hua, D. (2007). Effects of PAMD on the proliferation human tumour cells of PC-3 and BT5637. J. Harbin Med. Univ. 2:14.
Taheri, S., Abdullah, T. L., Rafii, M., Harikrishna, J. A., Werbrouck, S. P., Teo, C. H., et al. (2019). De novo assembly of transcriptomes, mining, and development of novel EST-SSR markers in Curcuma alismatifolia (Zingiberaceae family) through Illumina sequencing. Sci. Rep. 9:3047. doi: 10.1038/s41598-019-39944-2
Tan, L.-Q., Wang, L.-Y., Wei, K., Zhang, C.-C., Wu, L.-Y., Qi, G.-N., et al. (2013). Floral transcriptome sequencing for SSR marker development and linkage map construction in the tea plant (Camellia sinensis). PLoS One 8:e81611. doi: 10.1371/journal.pone.0081611
Tan, M., Wu, K., Wang, L., Yan, M., Zhao, Z., Xu, J., et al. (2014). Developing and characterizing Ricinus communis SSR markers by data mining of whole-genome sequences. Mol. Breed. 34, 893–904. doi: 10.1007/s11032-014-0083-6
Thiel, T. (2003). MISA—Microsatellite Identification Tool. Available online at: http://pgrc.ipk-gatersleben.de/misa/ (accessed June 17, 2016).
Wang, X., and Wang, L. (2016). GMATA: an integrated software package for genome-scale SSR mining, marker development and viewing. Front. Plant Sci. 7:1350. doi: 10.3389/fpls.2016.01350
Wang, Y., Pan, Y., Liu, Z., Zhu, X., Zhai, L., Xu, L., et al. (2013). De novo transcriptome sequencing of radish (Raphanus sativus L.) and analysis of major genes involved in glucosinolate metabolism. BMC Genomics 14:836. doi: 10.1186/1471-2164-14-836
Wang, Z., Fang, B., Chen, J., Zhang, X., Luo, Z., Huang, L., et al. (2010). De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics 11:726.
Wang, Z., Li, J., Luo, Z., Huang, L., Chen, X., Fang, B., et al. (2011). Characterization and development of EST-derived SSR markers in cultivated sweetpotato (Ipomoea batatas). BMC Plant Biol. 11:139. doi: 10.1186/1471-2229-11-139
Wei, W., Qi, X., Wang, L., Zhang, Y., Hua, W., Li, D., et al. (2011). Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics 12:451. doi: 10.1186/1471-2164-12-451
Wei, Z., Sun, Z., Cui, B., Zhang, Q., Xiong, M., Wang, X., et al. (2016). Transcriptome analysis of colored calla lily (Zantedeschia rehmannii Engl.) by Illumina sequencing: de novo assembly, annotation and EST-SSR marker development. PeerJ 4:e2378. doi: 10.7717/peerj.2378
Xia, E.-H., Yao, Q.-Y., Zhang, H.-B., Jiang, J.-J., Zhang, L.-P., and Gao, L.-Z. (2016). CandiSSR: an efficient pipeline used for identifying candidate polymorphic SSRs based on multiple assembled sequences. Front. Plant Sci. 6:1171. doi: 10.3389/fpls.2015.01171
Xiang, Q. Y., Soltis, D. E., Soltis, P. S., Manchester, S. R., and Crawford, D. J. (2000). Timing the eastern Asian-eastern North American floristic disjunction: molecular clock corroborates paleontological estimates. Mol. Phylogenet. Evol. 15, 462–472. doi: 10.1006/mpev.2000.0766
Zeng, S., Xiao, G., Guo, J., Fei, Z., Xu, Y., Roe, B. A., et al. (2010). Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC Genomics 11:94. doi: 10.1186/1471-2164-11-94
Zhang, H., Wei, L., Miao, H., Zhang, T., and Wang, C. (2012). Development and validation of genic-SSR markers in sesame by RNA-seq. BMC Genomics 13:316. doi: 10.1186/1471-2164-13-316
Keywords: Menispermum, Illumina high-seq transcriptome, de novo assembly EST-SSR, Menispermum canadense, Menispermum dauricum, population genetic diversity
Citation: Hina F, Yisilam G, Wang S, Li P and Fu C (2020) De novo Transcriptome Assembly, Gene Annotation and SSR Marker Development in the Moon Seed Genus Menispermum (Menispermaceae). Front. Genet. 11:380. doi: 10.3389/fgene.2020.00380
Received: 03 December 2019; Accepted: 27 March 2020;
Published: 08 May 2020.
Edited by:
Lifeng Zhu, Nanjing Normal University, ChinaCopyright © 2020 Hina, Yisilam, Wang, Li and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Faiza Hina, bWZhaXphaGluYUBob3RtYWlsLmNvbQ==; Pan Li, cGFubGlfemp1QDEyNi5jb20=; cGFubGlAemp1LmVkdS5jbg==