 Roger Colobran1,2,3*
Roger Colobran1,2,3*- 1Immunology Division, Department of Cell Biology, Physiology and Immunology, Hospital Universitari Vall d'Hebron, Vall d'Hebron Research Institute, Autonomous University of Barcelona, Barcelona, Spain
- 2Department of Clinical and Molecular Genetics, Hospital Universitari Vall d'Hebron, Barcelona, Spain
- 3Jeffrey Model Foundation Excellence Center, Barcelona, Spain
A Commentary on
Bradykinin-Mediated Angioedema: An Update of the Genetic Causes and the Impact of Genomics
by Marcelino-Rodriguez, I., Callero, A., Mendoza-Alvarez, A., Perez-Rodriguez, E., Barrios-Recio, J., Garcia-Robaina, J. C., et al. (2019). Front. Genet. 10:900. doi: 10.3389/fgene.2019.00900
Hereditary angioedema (HAE) is a rare autosomal-dominant genetic disease characterized by recurrent episodes of edema of the deep dermis and mucosal/submucosal tissues, and less frequently, potentially life-threatening laryngeal edema. The estimated worldwide prevalence of HAE is 1:50,000, but this figure may be even higher, as patients with this condition are often misdiagnosed or undiagnosed (Bernstein, 2018). The genetic bases of HAE were initially associated with mutations in SERPING1, the gene that encodes the C1-inhibitor (C1-INH) protein. In the absence of adequate functional C1-INH, the plasma contact system is insufficiently regulated, with consequent generation of bradykinin, the primary mediator of swelling in HAE. Now, more than 20 years since this genetic cause was identified (Bissler et al., 1997), the latest update includes 748 different SERPING1 variants associated with HAE (Ponard et al., 2019). Nonetheless, in addition SERPING1, which still accounts for the majority of genetic defects underlying HAE, an increasing number of new causal genes have been identified, especially in the last few years. The genes that cause HAE in patients with normal C1-INH levels are coagulation factor 12 gene (F12) (discovered in 2006) (Dewald and Bork, 2006), angiopoietin-1 (ANGPT1) and plasminogen gene (PLG) (discovered in 2018) (Bafunno et al., 2018; Bork et al., 2018b), and kininogen-1 gene (KNG1) (discovered in 2019) (Bork et al., 2019).
I read with great interest the article by Marcelino-Rodriguez et al. on the genetic causes of bradykinin-mediated angioedema (Marcelino-Rodriguez et al., 2019). These authors comprehensively reviewed all HAE causal genes, including those most recently described, and emphasized the crucial role of next generation sequencing (NGS), specifically whole-exome sequencing (WES), in these recent discoveries. When they dealt with PLG, one of the recently described HAE causal genes, they focused on the p.Lys330Glu mutation, a recurrent mutation described in more than 20 independent families from different European countries and Japan (Belbézier et al., 2018; Bork et al., 2018b; Germenis et al., 2018; Yakushiji et al., 2018; Recke et al., 2019), and also mentioned the p.Lys311Glu variant, described in three patients by Dewald (2018). I would like to clarify that Lys330Glu and Lys311Glu are actually two different names for the same genetic variant. Lys330Glu refers to amino acid residue 330 of the primary translated protein, and Lys311Glu to amino acid residue 311 of the mature protein. The main reason for this discrepancy is the 19-amino-acid signal peptide present in the PLG protein, which allows it to be secreted to the extracellular region (Figure 1). The HGVS (Human Genome Variation Society) recommendations for describing sequence variants state the following: “A protein reference sequence should represent the primary translation product, not a processed mature protein, and thus includes the starting methionine, any signal peptide sequences, etc.” (den Dunnen et al., 2016). Therefore, the correct name for this PLG mutation at the protein level is p.Lys330Glu.
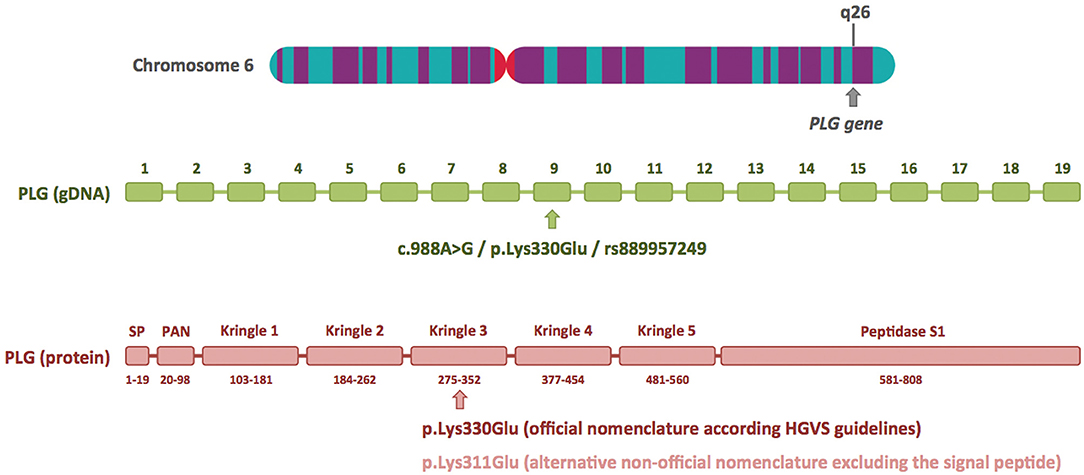
Figure 1. Plasminogen gene structure and location of the c.988A>G/p.Lys330Glu mutation at the DNA and protein level. The plasminogen gene (PLG) has 19 exons and is located on the long arm of chromosome 6 (6q26). The HAE-associated c.988A>G/p.Lys330Glu mutation is located in exon 9 and is included in the dbSNP database (ID rs889957249). The diagram of the plasminogen protein shows its different domains and numbering of the amino acids (based on UniProtKB: P00747). The p.Lys330Glu mutation is located in the kringle 3 domain. The alternative nomenclature, p.Lys311Glu, is based on the mature protein, and it excludes the first 19 amino acids of the signal peptide (SP). The chromosome 6 image was obtained from the Human Genome Project Archive.
At the cDNA level, different nomenclature was used by Bork et al. in the article that first identified this mutation (c.988A>G) and by Georg Dewald (c.1100A>G) (Bork et al., 2018b; Dewald, 2018). Nucleotide positions 988 and 1100 refer to the same position in the sequence of the canonical plasminogen transcript (NBCI reference sequence NM_000301.3): 988 is the position relative to the start codon, whereas 1100 is the position relative to the transcription start site, containing 112 untranslated nucleotides before the first methionine. Again, the HGVS recommendations state: “cDNA numbering starts with ‘c.1' at the A of the ATG translation initiation (start) codon”. Therefore, the correct name for the PLG mutation at the cDNA level is c.988A>G. Although Georg Dewald repeatedly uses the alternative nomenclature to describe the PLG mutation in his report (c.1100A>G/p.Lys311Glu), it must be said that at one point he indicates that this variant can be also named c.988A>G/p.Lys330Glu (Dewald, 2018). However, this brief comment may go unnoticed by readers, and this may have occurred in the case of Marcelino-Rodriguez et al. (2019).
Another confusing factor in reporting the PLG defect in HAE is that in the original article by Bork at al. describing the p.Lys330Glu mutation, the name of the mutation at the cDNA level was incorrect in the abstract and results section. Instead of c.988A>G, it appeared as c.9886A>G several times in the text (likely an involuntary typographic error). The authors noted the error and published a corrigendum several months later (Bork et al., 2018a).
The aim of this commentary is simply to clarify that, to date, only one PLG mutation causing HAE-PLG has been discovered (c.988A>G/p.Lys330Glu), although the aforementioned nomenclature-related factors may give a different impression. Haplotype analysis demonstrated a founder effect that likely explains recurrence of this mutation in more than 20 independent families identified so far (Bork et al., 2018b). The existence of symptom-free carriers indicates that clinical penetrance is incomplete. Although the pathomechanism of HAE-PLG has not been elucidated as yet, there is evidence pointing to bradykinin as the main mediator of edema in this condition; that is, bradykinin-2 receptor antagonist has been effective in shortening acute attacks in several patients (Belbézier et al., 2018; Bork et al., 2018b; Recke et al., 2019). Whether or not other variants in PLG are a cause of HAE remains to be determined. In any case, in the current era of NGS capability, systematic use of the HGVS nomenclature is essential when reporting genetic variants to avoid misunderstandings on dissemination of the information.
Finally, another inaccuracy regarding the article by Marcelino-Rodriguez et al. can be pointed out. In Table 1, they selected 12 articles described as “genetic studies of acquired bradykinin-mediated angioedema.” However, the first manuscript from this list is, as its title claims (Hereditary Angioedema with Normal C1 Inhibitor and F12 Mutations in 42 Brazilian Families), a study of HAE patients with normal C1-INH and mutations in F12 gene (Veronez et al., 2018), and not a study of acquired angioedema.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
The author's research was funded by Instituto de Salud Carlos III, grant PI17/00660, cofinanced by the European Regional Development Fund (ERDF).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bafunno, V., Firinu, D., D'Apolito, M., Cordisco, G., Loffredo, S., Leccese, A., et al. (2018). Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J. Allergy Clin. Immunol. 141, 1009–1017. doi: 10.1016/j.jaci.2017.05.020
Belbézier, A., Hardy, G., Marlu, R., Defendi, F., Dumestre Perard, C., Boccon-Gibod, I., et al. (2018). Plasminogen gene mutation with normal C1 inhibitor hereditary angioedema: three additional French families. Allergy 73, 2237–2239. doi: 10.1111/all.13543
Bernstein, J. A. (2018). Severity of hereditary angioedema, prevalence, and diagnostic considerations. Am. J. Manage. Care 24, S292–S298. Available online at: https://www.ajmc.com/journals/supplement/2018/new-horizons-diagnosis-treatment-hae/severity-of-hae-prevalence-and-diagnostic-considerations?p=3
Bissler, J. J., Aulak, K. S., Donaldson, V. H., Rosen, F. S., Cicardi, M., Harrison, R. A., et al. (1997). Molecular defects in hereditary angioneurotic edema. Proc. Assoc. Am. Physicians 109, 164–173.
Bork, K., Wulff, K., Rossmann, H., Steinmüller-Magin, L., Braenne, I., Witzke, G., et al. (2019). Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy 74, 2479–2481. doi: 10.1111/all.13869
Bork, K., Wulff, K., Steinmüller-Magin, L., Braenne, I., Staubach-Renz, P., Witzke, G., et al. (2018b). Hereditary angioedema with a mutation in the plasminogen gene. Allergy 73, 442–450. doi: 10.1111/all.13270
Bork, K., Wulff, K., Steinmüller-Magin, L., Braenne, I., Staubach-Renz, P., Witzke, G., et al. (2018a). Corrigendum. Allergy 73:2412. doi: 10.1111/all.13623
den Dunnen, J. T., Dalgleish, R., Maglott, D. R., Hart, R. K., Greenblatt, M. S., McGowan-Jordan, J., et al. (2016). HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 37, 564–569. doi: 10.1002/humu.22981
Dewald, G. (2018). A missense mutation in the plasminogen gene, within the plasminogen kringle 3 domain, in hereditary angioedema with normal C1 inhibitor. Biochem. Biophys. Res. Commun. 498, 193–198. doi: 10.1016/j.bbrc.2017.12.060
Dewald, G., and Bork, K. (2006). Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem. Biophys. Res. Commun. 343, 1286–1289. doi: 10.1016/j.bbrc.2006.03.092
Germenis, A. E., Loules, G., Zamanakou, M., Psarros, F., González-Quevedo, T., Speletas, M., et al. (2018). On the pathogenicity of the plasminogen K330E mutation for hereditary angioedema. Allergy 73, 1751–1753. doi: 10.1111/all.13324
Marcelino-Rodriguez, I., Callero, A., Mendoza-Alvarez, A., Perez-Rodriguez, E., Barrios-Recio, J., Garcia-Robaina, J. C., et al. (2019). Bradykinin-mediated angioedema: an update of the genetic causes and the impact of genomics. Front. Genet. 10:900. doi: 10.3389/fgene.2019.00900
Ponard, D., Gaboriaud, C., Charignon, D., Ghannam, A., Wagenaar-Bos, I. G. A., Roem, D., et al. (2019). SERPING1 mutation update: Mutation spectrum and C1 Inhibitor phenotypes. Hum. Mutat. 41, 38–57. doi: 10.1002/humu.23917
Recke, A., Massalme, E. G., Jappe, U., Steinmüller-Magin, L., Schmidt, J., Hellenbroich, Y., et al. (2019). Identification of the recently described plasminogen gene mutation p.Lys330Glu in a family from Northern Germany with hereditary angioedema. Clin. Transl. Allergy 9:9. doi: 10.1186/s13601-019-0247-x
Veronez, C. L., Moreno, A. S., Constantino-Silva, R. N., Maia, L. S. M., Ferriani, M. P. L., Castro, F. F. M., et al. (2018). Hereditary Angioedema with Normal C1 Inhibitor and F12 Mutations in 42 Brazilian Families. J. Allergy Clin. Immunol. Pract. 6, 1209–1216.e8. doi: 10.1016/j.jaip.2017.09.025
Keywords: hereditary angioedema, plasminogen, mutation, HGVS nomenclature, genetic variant
Citation: Colobran R (2020) Commentary: Bradykinin-Mediated Angioedema: An Update of the Genetic Causes and the Impact of Genomics. Front. Genet. 11:304. doi: 10.3389/fgene.2020.00304
Received: 16 January 2020; Accepted: 13 March 2020;
Published: 03 April 2020.
Edited by:
Anastasios E. Germenis, University of Thessaly, GreeceReviewed by:
Camila Lopes Veronez, University of California, San Diego, United StatesCopyright © 2020 Colobran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger Colobran, rcolobran@vhebron.net