
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Genet. , 06 March 2020
Sec. Livestock Genomics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00201
 Mun Hua Tan1,2*
Mun Hua Tan1,2* Han Ming Gan1,2
Han Ming Gan1,2 Yin Peng Lee1,2
Yin Peng Lee1,2 Frederic Grandjean3Laurence J. Croft1,2
Frederic Grandjean3Laurence J. Croft1,2 Christopher M. Austin1,2,4,5
Christopher M. Austin1,2,4,5The Decapoda represent a highly speciose and diverse group of crustaceans that provide important ecosystem services across mainly marine and freshwater environments, with many of the larger species being of significant commercial importance for fisheries and aquaculture industries. The value of generating and using genomic resources for key crustacean species and groups is widely appreciated for a variety of purposes including phylogenetic and population studies, selective breeding programs and broodstock management (Tan et al., 2016; Grandjean et al., 2017; Wolfe et al., 2019; Zenger et al., 2019; Zhang et al., 2019). However, comprehensive genomic studies are particularly limited for decapod crustaceans (Zenger et al., 2019; Zhang et al., 2019), compared to other aquatic invertebrate groups. Most genomic-related studies on decapod crustaceans have focused on mitogenome recovery and transcriptomics studies, with few genome assemblies attempted and most of these on commercially-important species (Song et al., 2016; Yuan et al., 2018; Zhang et al., 2019). Of the decapod species for which genome assemblies have been published and are publicly available, the quality is highly variable and BUSCO completeness poor with the exception of Procambarus virginalis (87.0%) and Litopeneaus vannamei (84.6%) (Table 1). It is now apparent that the assembly and annotation of decapod genomes are highly challenging due to the need to deal with large repetitive genomes, often with high heterozygosity, and should not to be undertaken by the faint-hearted or on a shoe-string budget (Zhang et al., 2019).
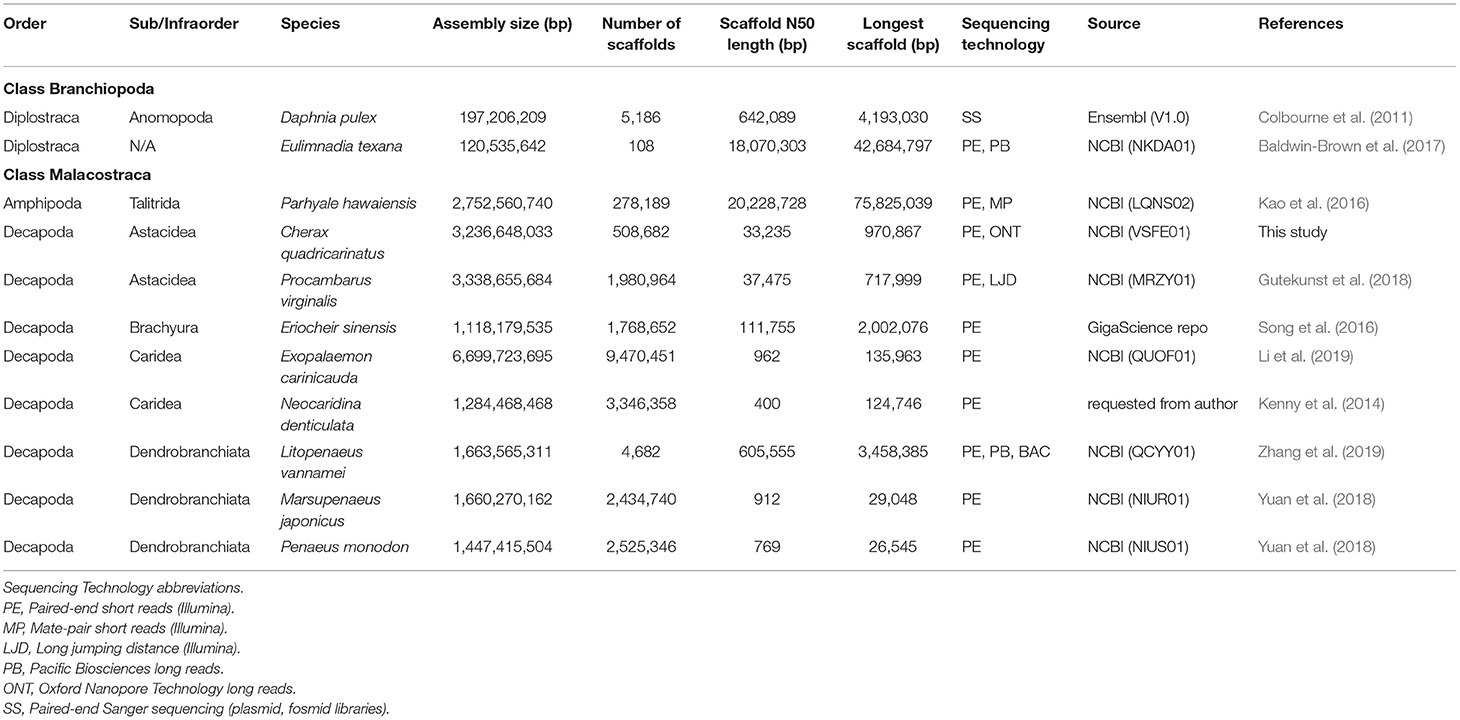
Table 1. Genomes used in this comparative study.
The lack of genomic resources coupled with limited understanding of the molecular basis of gene expression and phenotypic variation will continue to limit advances in aquaculture-based productivity of decapods. Understanding the molecular basis of phenotypic variation and gene function is therefore important for selective breeding programs for traits such as increased growth and disease resistance (Zenger et al., 2019). Similarly, whole genome assemblies support genome-wide association studies (GWAS) to identify trait-specific loci and for genomic-based selective breeding.
Freshwater crayfishes make up an important group of Decapod crustaceans (Crandall and De Grave, 2017) naturally occurring on all continents, with the exception of Antarctica and Africa (Toon et al., 2010; Bracken-Grissom et al., 2014). They reach their greatest species richness in Australia and North America. A number of species are subject to aquaculture and recreational activities, indigenous fishers for food security and bait fisheries for anglers, and others are of conservation concern due to a range of activities threatening vulnerable freshwater environments (Piper, 2000; Richman et al., 2015; CABI, 2019). The genomic architecture and karyotype evolution of freshwater crayfish is also of interest as they have some of the largest chromosome numbers (2n = 200) recorded for invertebrate species (Tan et al., 2004). In Australia, freshwater crayfish species from the genus Cherax Erichson, known as smooth yabbies, include the largest commercial crayfish species in the world (Austin, 1987, 1996; Austin and Knott, 1996). Of these, the economically-important and best known is the red claw crayfish, Cherax quadricarinatus von Martens, distributed widely across northern Australia (Austin, 1996; FAO, 2019). This species is capable of growing to greater than 200 grams making it the second largest commercial species of Cherax (Austin, 1987) behind the marron (C. cainii Austin) (Austin and Ryan, 2002). The popularity of the red claw as an aquaculture and ornamental species and its adaptability has resulted in widespread translocation, resulting in an extensive global distribution and it is acknowledged as a major invasive species of inland aquatic ecosystems in the tropics (Ahyong and Yeo, 2007; Larson and Olden, 2013; Saoud et al., 2013). Due to its large size and ease with which it can be maintained in captivity, C. quadricarinatus is also increasingly being used as a model organism to address fundamental questions relevant to molecular biology, physiology, functional genomics and cell biology (Fernández et al., 2012; Pamuru et al., 2012; Ventura et al., 2019).
The genetics of C. quadricarinatus has been well studied using PCR-based approaches and recently, next generation sequencing has been used for mitogenomic and transcriptomic studies (Baker et al., 2008; Gan et al., 2016; Tan et al., 2016). In this paper, we present the first genome assembly for a southern hemisphere crayfish using a hybrid assembly approach utilizing long Nanopore reads and short Illumina reads, which has proven to be an efficient and effective approach for the assembly and annotation of species with large and repetitive genomes (Austin et al., 2017; Tan et al., 2018; Gan et al., 2019; Sánchez-Herrero et al., 2019), but has only been minimally explored to support decapod assemblies (Van Quyen et al., 2020).
We then benchmark our assembly against seven other published decapod genome assemblies and present a preliminary phylogenomic analysis for decapod crustaceans. Lastly, we use these data to investigate the occurrence and diversification of nuclear mitochondrial DNA (NUMTs)1 in decapod genomes, which is directly relevant to debates on the veracity of the most common approach to the DNA barcoding of life for molecular-based taxonomic identification and biodiversity assessment based on the mitochondrial cytochrome oxidase 1 (cox1) gene region (Hebert et al., 2003; Cristescu, 2014; deWaard et al., 2018).
Tissue samples from two male C. quadricarinatus individuals were collected from Northern Territory in Australia, DWN1 from Rapid Creek and M2R2 from Charles Darwin University Aquaculture Centre, both located in Darwin following institutionally endorsed ethical, biosecurity and safety guidelines. Genomic DNA was extracted using E.Z.N.A.® Tissue DNA Kit (Omega Bio-tek) from tail muscle tissue. One microgram of DWN1 was sent to Macrogen, Korea for PCR-free library preparation and sequencing on the Illumina HiSeq platform. Additional Illumina sequencing on the Illumina NovaSeq 6000 system was also performed at the Deakin Genomics Centre using a PCR-based library constructed with NEBNext Ultra Illumina library preparation kit. Using tissue samples from DWN1 and M2R2 individuals, for each Nanopore library, ~1 μg of gDNA as measured by Qubit Fluorometer (Invitrogen) was processed using the LSK108 library preparation kit followed by sequencing on the R9.4 MinION flowcell. Nanopore data were base called with Albacore versions compatible with kits and flow cells used (discontinued, was available on https://community.nanoporetech.com) and adapter-trimmed with the Porechop tool (Wick, 2017).
Raw reads were pre-processed prior to assembly. The fastp tool (–poly_g_min_len 1) (Chen et al., 2018) was used to trim polyG sequences from the 3' end of reads generated on the Illumina NovaSeq platform. All short reads were trimmed with Trimmomatic (Bolger et al., 2014) to eliminate adapters (ILLUMINACLIP:2:30:10) and low quality sequences (AVGQUAL:20), and subsequently assembled with the Platanus assembler (Kajitani et al., 2014) using default parameters. The repeat content and large size of this genome necessitated scaffolding of the assembly with several data types; these included the use of short and long reads in order to span gaps of various sizes, in addition to using data from a previous C. quadricarinatus transcriptome project (Tan et al., 2016) to stitch together scaffolds containing coding genes. A first level of scaffolding was performed with Platanus, which scaffolds the assembled contigs and further closes gaps with insert size and sequence information from short paired-end libraries. Subsequently, as second level of scaffolding was accomplished with long Nanopore reads using LINKS (Warren et al., 2015), which was run for 20 iterations with multiple distance values (d) and step of sliding window (t), in addition to set values for k-mer (-k 19), minimum number of links (-l 5) and maximum link ratio between two best pairs (-a 0.3). Detailed parameters used in each iteration is available as Data Sheet 1. Resulting scaffolds were polished with pilon v1.22 (–fix all) (Walker et al., 2014) for five iterations. A third scaffolding step was performed using C. quadricarinatus RNA-seq reads previously generated (ERP004477) (Tan et al., 2016). HISAT2 was used to align RNA-seq reads to the assembly (Kim et al., 2019), information from which was used by BESST (Sahlin et al., 2014) for further scaffolding. All assemblies were evaluated for “completeness” based on the detection of single copy conserved genes (arthropoda_odb9) with BUSCO (Simão et al., 2015).
Jellyfish (Marçais and Kingsford, 2011) was used to count the occurrence of 19-, 21- and 25-mers in pre-processed short reads, generating k-mer frequency histograms that were uploaded to the GenomeScope webserver (max kmer coverage disabled) (Vurture et al., 2017) for estimations of haploid genome size, heterozygosity, and repetitive content.
A first iteration of the MAKER v2.31.10 annotation pipeline (Cantarel et al., 2008) was run to produce initial gene models based on transcript and protein hints (est2genome = 1; protein2genome = 1) from C. quadricarinatus transcriptome sequences (Tan et al., 2016), protein sequences from UniProt-SwissProt (Consortium, 2018) and an additional set of proteins from four other published decapod genomes: Eriocheir sinensis (Song et al., 2016), Litopenaeus vannamei (Zhang et al., 2019), Marsupenaeus japonicas, and Penaeus monodon (Yuan et al., 2018). These gene models were used to train two ab initio gene predictors, SNAP (Korf, 2004) and AUGUSTUS (Stanke et al., 2006), which were provided in a second MAKER iteration. Gene models resulting from this iteration were again used to retrain SNAP and AUGUSTUS before a third and final iteration of MAKER. Genes with Annotation Edit Distance values (AED) ≤ 0.5 were retained (Eilbeck et al., 2009). A small AED value points to a lesser degree of variance between the predicted gene and evidences/hints used in prediction. For functional annotation, DIAMOND (Buchfink et al., 2014) was used to find homology between the predicted genes and known proteins in the UniProtKB (Swiss-Prot and TrEMBL) database (Consortium, 2018). The predicted protein sequences were also scanned for motifs, signatures, and protein domains using InterProScan (Jones et al., 2014).
Publicly-available genome assemblies for seven published decapod species (Eriocheir sinensis, Exopalaemon carinicauda, Litopenaeus vannamei, Marsupenaeus japonicus, Neocaridina denticulata, Penaeus monodon, Procambarus virginalis) (Kenny et al., 2014; Song et al., 2016; Gutekunst et al., 2018; Yuan et al., 2018; Li et al., 2019; Zhang et al., 2019) and three other non-decapod crustaceans (Daphnia pulex, Eulimnadia texana, Parhyale hawaiensis) (Colbourne et al., 2011; Kao et al., 2016; Baldwin-Brown et al., 2017) were downloaded (sources in Table 1) and compared in terms of genome and assembly sizes, repeat content and completeness. “Completeness” of a genome was assessed with BUSCO (Simão et al., 2015) based on the arthropoda_odb9 databases. To obtain repeat profiles, repeat families were identified de novo using RepeatModeler (Smit and Hubley, 2019), which uses RepeatScout (Price et al., 2005) and RECON (Bao and Eddy, 2002) to scan for repeats. The output from this step contains a set of consensus sequences from each identified repeat family that was used by RepeatMasker (Smit et al., 2019) to mask repeats within the assembly.
The eight assembled genomes for C. quadricarinatus, Er. sinensis, Ex. carinicauda, L. vannamei, M. japonicus, N. denticulata, Pe. monodon, and Pr. virginalis were also scanned for the presence of putative NUMTs. The blastn tool (Altschul et al., 1990) was used to align the mitochondrial cox1 gene of each species to its corresponding genome (exception: Procambarus clarkii cox1 was used since the full cox1 sequence for Pr. virginalis is unavailable on NCBI). Each output was filtered to eliminate alignments based on the following criteria:
1. Alignment length <100 nucleotides.
2. Alignment length that spans 95% of a genomic scaffold.
3. Alignment at the edge of a genomic scaffold.
An illustration of how these filters were applied is provided in Data Sheet 2. Sequences of these putative NUMTs were extracted based on alignment start and stop coordinates and were aligned with MAFFT (Katoh and Standley, 2013). IQ-TREE (Nguyen et al., 2014) was used to perform model testing and construct a maximum-likelihood (ML) phylogenetic tree (-m TESTMERGE).
A ML tree was also constructed based on orthologous protein sequences predicted by BUSCO. This tree consists of six decapods (C. quadricarinatus, Er. sinensis, L. vannamei, M. japonicus, Pe. monodon, Pr. virginalis) with D. pulex, Eu. texana and Pa. hawaiensis as non-decapod outgroup species, rooted with Drosophila melanogaster. Since this analysis used sequences predicted by BUSCO, N. denticulata and Ex. carinicauda with low BUSCO completeness were excluded from this analysis. Briefly, protein sequences within each orthologous group (based on metazoa_odb9 and arthropoda_odb9) were aligned with MAFFT (Katoh and Standley, 2013) and trimmed with Gblocks (Castresana, 2000). Only multiple sequence alignments containing sequences from all 10 species were retained and concatenated into a supermatrix with FASconCat-G (Kück and Longo, 2014). IQ-TREE (Nguyen et al., 2014) was used for initial model testing and construction of the ML tree (-m TESTMERGE), with support values indicated by the SH-like approximate likelihood ratio test (SH-aLRT) (Guindon et al., 2010) and ultrafast bootstrap (UFBoot) (Minh et al., 2013) (-alrt 1000 -bb 1000).
This study produced 191 Gbp of HiSeq data (2 × 100 bp) and 774 Gbp of NovaSeq data (2 × 150 bp) in addition to 36 Gbp of Nanopore reads (average: 3,419 bp) to assist with scaffolding. Kmer-based methods estimated a 5 Gbp size for the Cherax quadricarinatus genome (Data Sheet 3), a value that is within the 3.82 to 6.06 Gbp size range that has been reported for species in the Infraorder Astacidea (Gregory, 2020) (Figure 1A). Based on this estimate, data generated in this study yields sequencing depths of 193× and 7× of short and long reads, respectively. Sequence data is available in the Sequence Read Archive (SRA) on NCBI (BioProject: PRJNA559771).
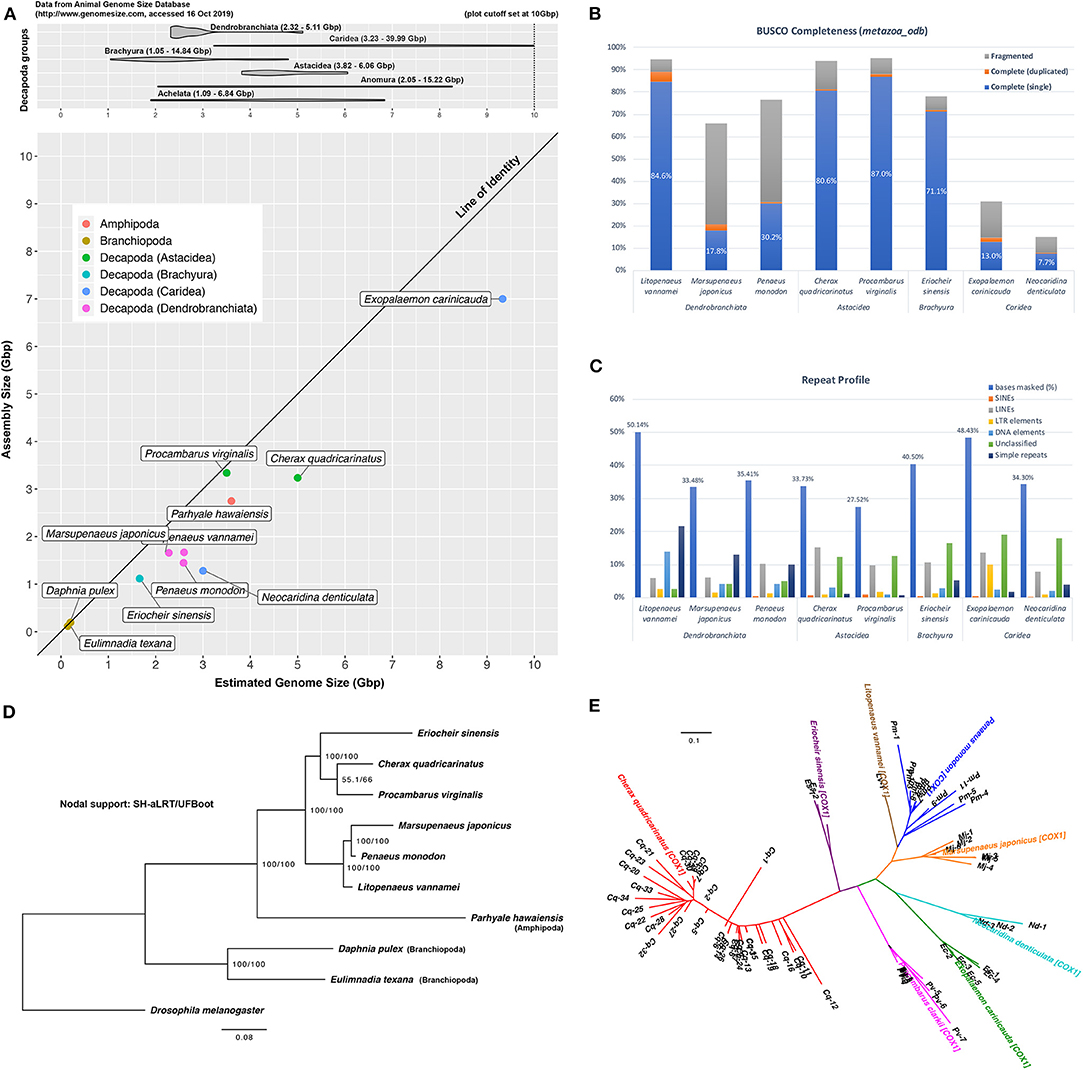
Figure 1. Cherax quadricarinatus and other published and publicly-available decapod genomes. (A) Top: range of genome sizes of decapod species in various sub- and infra-orders, based on information from the Animal Genome Size Database. Bottom: discrepancy between assembly and expected genome sizes of current available decapod genomes. (B) Genome “completeness” based on the arthropoda_odb9 BUSCO dataset. (C) Masked repetitive regions of each genome and profiles of interspersed repeats. (D) Maximum-likelihood (ML) tree based on BUSCO predictions, rooted with D. melanogaster, with SH-aLRT/UFBoot values as indication of nodal support. (E) ML tree based on putative NUMTs identified from decapod genome assemblies.
Hybrid assembly of this genome resulted in a 3.24 Gbp final assembly contained in 508,682 scaffolds, with a N50 length of 33,235 bp (Table 1). Details of each assembly at each scaffolding level are available in Data Sheet 4. The BUSCO tool reports the presence of 81.3% and 12.8% of complete and fragmented arthropod BUSCOs, respectively. These values are comparable to the completeness evaluation of other recently sequenced decapod genomes that are relatively much smaller (L. vannamei, Pr. virginalis, Er. sinensis) (Table 1, Figure 1B). GenomeScope profiles display a small shoulder on k-mer distributions, indicating some level of heterozygosity within the genome and estimated an average heterozygosity level of 0.44%, which translates into approximately 1 mutation in 230 bp (Data Sheet 3). This is on par with the heterozygosity level of 0.53% reported for the marbled crayfish, Pr. virginalis (Gutekunst et al., 2018), and is higher than in prawns with both M. japonicus and Pe. monodon, both with 0.19% and 0.21% heterozygosity, respectively (Yuan et al., 2018). In addition, 33.73% of this assembly was masked for repeats, with a large proportion of interspersed repeats being long interspersed nuclear elements (LINEs), representing a repeat profile that is similar to that of Pr. virginalis, another crayfish species (Figure 1C). While the report of only 5.9% missing BUSCO genes in this C. quadricarinatus assembly suggests that majority of the gene space is present, the assembly reported in this study makes up only 64.8% of the expected 5 Gbp genome with 85.2% of short reads successfully mapped back to the assembly, indicating that there remains regions of this large genome that are missing from the assembly. This is not unusual for decapod genomes as they are rarely assembled to their full size, as seen from data points in Figure 1A, which mostly fall well below the “line of identity” between assembly vs. estimated genome size. The exception being Pr. virginalis with an assembled size close to its 3.5 Gbp genome size. This study tackles the ambitious assembly of a gigantic genome riddled with large repetitive structures and heterozygous regions that pose serious challenges to computational and bioinformatic resources, but there are clear benefits to hybrid assembly as an efficient method to add to the limited pool of genomic resources available for the order Decapoda. This assembly is available on NCBI (BioProject: PRJNA559771, WGS: VSFE00000000). It is noteworthy that this study has incorporated the largest dataset of long Nanopore reads to date for a decapod genome assembly (36 Gbp, ~7×), the other being the recent updated assembly of the black tiger prawn genome that incorporated the use of 2.5 Gbp of long Nanopore reads (~1.25×) (Van Quyen et al., 2020). The only other study to use long reads in decapod whole genome research is by Zhang et al. (2019) who utilized PacBio reads to support the assembly of the genome for the Pacific white shrimp L. vannamei.
An average of 95.4% of RNA-seq reads from five C. quadricarinatus tissue types in Tan et al. (2016) were successfully aligned to the assembly. The annotation process predicted a total of 19,494 protein-coding genes, with an average gene length of 9,768 bp containing an average of 4.4 exons per gene. This number of predicted genes is within the range of that typically reported for other decapod crustaceans (Er. sinensis: 14436, M. japonicus: 16716, Pe. monodon: 18100, Pr. virginalis: 21772, L. vannamei: 25596). Of the annotated protein-coding genes, 88% are functionally annotated based on homology to existing UniProt protein sequences or through identification of protein domains and signatures. Protein and transcript sequences, BLAST homology alignments and InterProScan results are available as Data Sheet 5.
Alignment and trimming of 97 orthologous genes resulted in a supermatrix of 16,679 amino acid characters. Maximum-likelihood analysis based on these protein sequences generated the tree shown in Figure 1D. Rooted with the fruit fly, the phylogeny recovers branchiopods and amphipods as outgroup species to the decapod species. Shrimp and prawn species (L. vannamei, M. japonicus, Pe. monodon) are clustered in a clade as the Dendrobranchiata, which in turn forms a sister relationship with a second clade consisting of the other three decapod species. In this latter clade, crayfish species are placed as sister taxa (C. quadricarinatus and Pr. virginalis), consistent with their taxonomic status as species within Astacidea, but with surprisingly weak nodal support (SH-aLRT 55.1%, UFBoot 66.0%). These relationships are consistent with findings reported in mitogenome- and nuclear-based studies (Bracken et al., 2009; Shen et al., 2013; Tan et al., 2019; Wolfe et al., 2019). Alignment and phylogenetic tree produced in this analysis are available as Data Sheet 6.
The concept of DNA barcoding, by which a short universal DNA sequence is used to discriminate among species, is being very widely used to support taxonomic identification, detection of cryptic species and for establishing a reference database to support ecological, biodiversity and conservation-related studies (Ratnasingham and Hebert, 2007). A relatively recent and potentially powerful application of barcoding is the use of the rapidly increasing COI database as a reference for supporting environmental DNA metabarcoding studies (Deiner et al., 2017; Günther et al., 2018). However, DNA barcoding has its critics (Moritz and Cicero, 2004; Collins and Cruickshank, 2013), with a persistent concern being the impact of mitochondrial DNA copies in the nuclear genome (Bensasson et al., 2001; Hazkani-Covo et al., 2010) on the veracity of the DNA barcoding methodology, potentially making DNA barcoding unreliable for certain taxonomic groups (Sorenson and Quinn, 1998), including crustaceans (Song et al., 2008).
NUMTs have now been reported in a diversity of animal groups including crustaceans, with cytochrome oxidase 1 (cox1) NUMTs found in planktonic copepods (Bucklin et al., 1999) and snapping shrimp (Williams and Knowlton, 2001). Nguyen et al. (2002) and Munasinghe et al. (2003) reported the first NUMTs for crayfish, from the genus Cherax, and Song et al. (2008) for species of Orconectes. Our study extends the findings of Song et al. (2008) in that we identify cox1 pseudogenes in each of the decapod genome assemblies we have examined, but find the number to be widely variable among taxa. This analysis identified the most cox1 pseudogenes in C. quadricarinatus (34 insertion sites), followed by Pe. monodon (11), Pr. virginalis (8), M. japonicus (6), Ex. carinicauda (5), N. denticulata (3), Er. sinensis (2), and L. vannamei (1) (Figure 1E). NUMTs identified from these decapod species form monophyletic groups by species in the phylogeny, suggesting that the cox1 mitochondrial gene was integrated into each nuclear genome subsequent to the divergence of species in this analysis, with either multiple independent transfer events or further evolution of NUMTs within each species through duplication events. The NUMT phylogenetic tree is available in Data Sheet 6.
In a broad-based study of NUMTs in 85 eukaryotic genomes, Hazkani-Covo et al. (2010) found a correlation with genome size. While finding the highest number of NUMTs in the red claw crayfish, with the second largest decapod genome assembled, is consistent with this observation, the evidence for a similar overall trend is not so clear across all the decapod species. In this context, it should be noted that we only have a relatively small sampling of decapod genomes and the varying assembly quality and volume of sequence data among studies makes testing this hypothesis fraught at this stage. There are also several caveats associated with this analysis. The identification process is dependent on the search methods and implemented filters, which if too conservative, can limit detection and exclude true NUMTs with highly divergent and variable sequences, especially if the time of insertion from the mitogenome occurred millions of years ago (Tsuji et al., 2012). It is also unknown whether other published decapod studies have been post-processed to exclude scaffolds containing mitochondrial genes during the submission process to databases. Nevertheless, results from this analysis provide an insight into the prevalence of NUMTs in decapod genomes, encouraging further exploration into the evolution of NUMTs in future studies.
Following on from mitogenomic and transcriptomic studies, we present the first draft genome for the red claw crayfish (Cherax quadricarinatus) based on relatively large volumes of short and long genomic reads from Illumina and Oxford Nanopore (ONT) platforms. While the assembly is relatively fragmented, it is much better than many of those currently available for decapod species, and the quality of the annotation is equivalent to other more recently-sequenced decapod genomes. Due to the very large size and repetitive structures of the red claw genome, the assembly was highly challenging. However, we demonstrated the value of long Nanopore reads and a hybrid assembly approach for improving an assembly based on short reads alone (Data Sheet 4), which gives encouragement for tackling other crayfish and crustacean taxa with large and complex genomes. This draft genome will be an important and valuable resource to support ongoing comparative genomic, phylogenomics and molecular-based breeding studies for aquaculture, conservation and biodiversity-related studies and can be approved upon over time, with the generation of additional long read data. However, a major challenge still remains in relation to the computational resources needed to assemble large repetitive genomes from predominately short reads, even when aided with long reads (Lewin et al., 2018). Computationally, assembly, scaffolding and polishing processes to achieve the final draft genome took almost 85 processor-weeks, and annotation required another 100 processor-weeks. A worthwhile next step would be to investigate the efficacy of a long-read led crayfish genome assembly, now feasible as result of declining costs and improved accuracy of long read sequencing, and which should be less expensive and lead to greatly reduced processor time for assembly tasks.
The datasets generated for this study can be found in the NCBI BioProject PRJNA559771, BioSamples SAMN12558911 and SAMN12885547 (WGS accession: VSFE00000000; SRA accessions: SRR10214688, SRR10416208, SRR10416210, SRR10445782, SRR10445783, SRR10445784, SRR10484712).
CA, HG, and LC conceived the project. CA collected the samples. HG and YL performed the sequencing. MT analyzed the data. MT and CA wrote the manuscript. FG and LC modified the manuscript. All authors read and approved the final version of the manuscript.
Funding for this study was provided by the Deakin Genomics Centre, Deakin University (Australia), by the Centre National de la Recherche Scientifique and the University of Poitiers (France) and the Monash University Malaysia Tropical Medicine and Biology Platform, Monash University (Malaysia).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research utilized computational resources and services provided by the National Computational Infrastructure (NCI), which was supported by the Australian Government. We thank the Charles Darwin University Aquaculture Centre and the Museum and Art Gallery of the Northern Territory for assistance in obtaining samples. We would also like to thank Robert Ruge of Deakin University for assistance and use of the SIT HPC Cluster.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00201/full#supplementary-material
1. ^The natural integration of DNA from mitochondria into the nuclear genome resulting in nuclear copies of mitochondrial pseudogenes (NUMTs).
Ahyong, S. T., and Yeo, D. C. J. (2007). Feral populations of the Australian Red-Claw crayfish (Cherax quadricarinatus von Martens) in water supply catchments of Singapore. Biol. Invasions 9, 943–946. doi: 10.1007/s1s0530-007-90944-0
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0S022-2836(05)803600-2
Austin, C. (1987). Diversity of Australian freshwater crayfish increases potential for aquaculture. Aust. Fish. 46, 30–31.
Austin, C. (1996). Systematics of the freshwater crayfish genus Cherax erichson (Decapoda: Parastacidae) in Northern and Eastern Australia: electrophoretic and morphological variation. Aust. J. Zool. 44, 259–296. doi: 10.1071/ZO9O960259
Austin, C., and Knott, B. (1996). Systematics of the freshwater crayfish genus Cherax erichson (Decapoda: Parastacidae) in South-Western Australia: electrophoretic, morphological and habitat variation. Aust. J. Zool. 44, 223–258. doi: 10.1071/ZO9O960223
Austin, C. M., and Ryan, S. G. (2002). Allozyme evidence for a new species of freshwater crayfish of the genus Cherax Erichson (Decapoda : Parastacidae) from the south-west of Western Australia. Invertebr. Syst. 16, 357–367. doi: 10.1071/IT0T1010
Austin, C. M., Tan, M. H., Harrisson, K. A., Lee, Y. P., Croft, L. J., Sunnucks, P., et al. (2017). De novo genome assembly and annotation of Australia's largest freshwater fish, the Murray cod (Maccullochella peelii), from illumina and nanopore sequencing read. Gigascience 6, 1–6. doi: 10.1093/gigascience/gix0x63
Baker, N., de Bruyn, M., and Mather, P. B. (2008). Patterns of molecular diversity in wild stocks of the redclaw crayfish (Cherax quadricarinatus) from northern Australia and Papua New Guinea: impacts of Plio-Pleistocene landscape evolution. Freshw. Biol. 53, 1592–1605. doi: 10.1111/j.13655-2427.2008.01996.x
Baldwin-Brown, J. G., WeekWeeks, S. C., and Long, A. D. (2017). A new standard for crustacean genomes: the highly contiguous, annotated genome assembly of the clam shrimp Eulimnadia texana reveals HOX gene order and identifies the sex chromosome. Genome Biol. Evol. 10, 143–156. doi: 10.1093/gbe/evx2x80
Bao, Z., and Eddy, S. R. (2002). Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269, 1269–1276. doi: 10.1101/gr.88502
Bensasson, D., Zhang, D.-X., Hartl, D. L., and Hewitt, G. M. (2001). Mitochondrial pseudogenes: evolution's misplaced witnesses. Trends Ecol. Evol. 16, 314–321. doi: 10.1016/S0S169-5347(01)021511-6
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu1u70
Bracken, H. D., Toon, A., Felder, D. L., Martin, J. W., Finley, M., Rasmussen, J., et al. (2009). The decapod tree of life: compiling the data and moving toward a consensus of decapod evolution. Arthropod Syst. Phylogeny 67, 99–116.
Bracken-Grissom, H. D., Ahyong, S. T., Wilkinson, R. D., Feldmann, R. M., Schweitzer, C. E., Breinholt, J. W., et al. (2014). The emergence of lobsters: phylogenetic relationships, morphological evolution and divergence time comparisons of an ancient group (Decapoda: Achelata, Astacidea, Glypheidea, Polychelida). Syst. Biol. 63, 457–479. doi: 10.1093/sysbio/syu0u08
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Bucklin, A., Guarnieri, M., Hill, R., Bentley, A., and Kaartvedt, S. (1999). “Taxonomic and systematic assessment of planktonic copepods using mitochondrial COI sequence variation and competitive, species-specific PCR,” in Molecular Ecology of Aquatic Communities, eds J. P. Zehr and M. A. Voytek (Dordrecht: Springer), 239–254.
Cantarel, B. L., Korf, I., Robb, S. M. C., Parra, G., Ross, E., Moore, B., et al. (2008). MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196. doi: 10.1101/gr.6743907
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a0a26334
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i8i84–i8i90. doi: 10.1093/bioinformatics/bty5y60
Colbourne, J. K., Pfrender, M. E., Gilbert, D., Thomas, W. K., Tucker, A., Oakley, T. H., et al. (2011). The ecoresponsive genome of Daphnia pulex. Science 331, 555–561. doi: 10.1126/science.1197761
Collins, R. A., and Cruickshank, R. H. (2013). The seven deadly sins of DNA barcoding. Mol. Ecol. Resour. 13, 969–975. doi: 10.1111/17555-0998.12046
Consortium, T. U. (2018). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D5D06–D5D15. doi: 10.1093/nar/gky1y049
Crandall, K. A., and De Grave, S. (2017). An updated classification of the freshwater crayfishes (Decapoda: Astacidea) of the world, with a complete species list. J. Crustacean Biol. 37, 615–653. doi: 10.1093/jcbiol/rux0x70
Cristescu, M. E. (2014). From barcoding single individuals to metabarcoding biological communities: towards an integrative approach to the study of global biodiversity. Trends Ecol. Evol. 29, 566–571. doi: 10.1016/j.tree.2014.08.001
Deiner, K., Bik, H. M., Mächler, E., Seymour, M., Lacoursière-Roussel, A., Altermatt, F., et al. (2017). Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Mol. Ecol. 26, 5872–5895. doi: 10.1111/mec.14350
deWaard, J. R., Levesque-Beaudin, V., deWaard, S. L., Ivanova, N. V., McKeown, J. T. A., Miskie, R., et al. (2018). Expedited assessment of terrestrial arthropod diversity by coupling Malaise traps with DNA barcoding. Genome 62, 85–95. doi: 10.1139/gen-20188-0093
Eilbeck, K., Moore, B., Holt, C., and Yandell, M. (2009). Quantitative measures for the management and comparison of annotated genomes. BMC Bioinformatics 10:67. doi: 10.1186/14711-21055-10-67
Fernández, M. S., Bustos, C., Luquet, G., Saez, D., Neira-Carrillo, A., Corneillat, M., et al. (2012). Proteoglycan occurrence in gastrolith of the crayfish Cherax quadricarinatus (Malacostraca: Decapoda). J. Crustacean Biol. 32, 802–815. doi: 10.1163/193724012X22X6X49804
Gan, H. M., Tan, M. H., and Austin, C. M. (2016). The complete mitogenome of the red claw crayfish Cherax quadricarinatus (Von Martens, 1868) (Crustacea: Decapoda: Parastacidae). Mitochondrial DNA Part A 27, 385–386. doi: 10.3109/19401736.2014.895997
Gan, H. M., Tan, M. H., Austin, C. M., Sherman, C. D. H., Wong, Y. T., Strugnell, J., et al. (2019). Best foot forward: Nanopore long reads, hybrid meta-Assembly, and haplotig purging optimizes the first genome assembly for the Southern Hemisphere blacklip abalone (Haliotis rubra). Front. Genet. 10:889. doi: 10.3389/fgene.2019.00889
Grandjean, F., Tan, M. H., Gan, H. M., Lee, Y. P., Kawai, T., Distefano, R. J., et al. (2017). Rapid recovery of nuclear and mitochondrial genes by genome skimming from Northern Hemisphere freshwater crayfish. Zool. Scripta 46, 718–728. doi: 10.1111/zsc.12247
Gregory, T. R. (2020). “Animal Genome Size Database”. Available online at: http://www.genomesize.com
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq0q10
Günther, B., Knebelsberger, T., Neumann, H., Laakmann, S., and Martínez Arbizu, P. (2018). Metabarcoding of marine environmental DNA based on mitochondrial and nuclear genes. Sci. Rep. 8:1482. doi: 10.1038/s4s1598-018-32917-x
Gutekunst, J., Andriantsoa, R., Falckenhayn, C., Hanna, K., Stein, W., Rasamy, J., et al. (2018). Clonal genome evolution and rapid invasive spread of the marbled crayfish. Nat. Ecol. Evol. 2, 567–573. doi: 10.1038/s4s1559-018-04677-9
Hazkani-Covo, E., Zeller, R. M., and Martin, W. (2010). Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 6:e1e000834. doi: 10.1371/journal.pgen.1000834
Hebert, P. D. N., Cywinska, A., Ball, S. L., and deWaard, J. R. (2003). Biological identifications through DNA barcodes. Proc. Biol. Sci. 270, 313–321. doi: 10.1098/rspb.2002.2218
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu0u31
Kajitani, R., Toshimoto, K., Noguchi, H., Toyoda, A., Ogura, Y., Okuno, M., et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. doi: 10.1101/gr.170720.113
Kao, D., Lai, A. G., Stamataki, E., Rosic, S., Konstantinides, N., Jarvis, E., et al. (2016). The genome of the crustacean Parhyale hawaiensis, a model for animal development, regeneration, immunity and lignocellulose digestion. Elife 5:e2e0062. doi: 10.7554/eLife.20062.057
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst0t10
Kenny, N., Sin, Y., Shen, X., Zhe, Q., Wang, W., Chan, T., et al. (2014). Genomic sequence and experimental tractability of a new decapod shrimp model, Neocaridina denticulata. Mar. Drugs 12, 1419–1437. doi: 10.3390/md1d2031419
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol, 37, 907–915.
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/14711-21055-5-59
Kück, P., and Longo, G. C. (2014). FASconCAT-G: extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 11:81. doi: 10.1186/s1s2983-014-0081-x
Larson, E. R., and Olden, J. D. (2013). Crayfish occupancy and abundance in lakes of the Pacific Northwest, USA. Freshw. Sci. 32, 94–107. doi: 10.1899/12-051.1
Lewin, H. A., Robinson, G. E., Kress, W. J., Baker, W. J., Coddington, J., Crandall, K. A., et al. (2018). Earth BioGenome Project: sequencing life for the future of life. PNAS 115, 4325–4333. doi: 10.1073/pnas.1720115115
Li, J., Lv, J., Liu, P., Chen, P., Wang, J., and Li, J. (2019). Genome survey and high-resolution backcross genetic linkage map construction of the ridgetail white prawn Exopalaemon carinicauda applications to QTL mapping of growth traits. BMC Genomics 20:598. doi: 10.1186/s1s2864-019-5981-x
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr0r11
Minh, B. Q., Nguyen, M. A. T., and von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst0t24
Moritz, C., and Cicero, C. (2004). DNA Barcoding: Promise and Pitfalls. PLoS Biol. 2:e3e54. doi: 10.1371/journal.pbio.0020354
Munasinghe, D. H. N., Murphy, N. P., and Austin, C. M. (2003). Utility of mitochondrial DNA sequences from four gene regions for systematic studies of Australian freshwater crayfish of the genus Cherax (Decapoda: Parastacidae). J. Crustacean Biol. 23, 402–417. doi: 10.1163/200219755-99990350
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2014). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu3u00
Nguyen, T. T. T., Murphy, N. P., and Austin, C. M. (2002). Amplification of multiple copies of mitochondrial cytochrome b gene fragments in the Australian freshwater crayfish, Cherax destructor Clark (Parastacidae: Decapoda). Anim. Genet. 33, 304–308. doi: 10.1046/j.13655-2052.2002.00867.x
Pamuru, R. R., Rosen, O., Manor, R., Chung, J. S., Zmora, N., Glazer, L., et al. (2012). Stimulation of molt by RNA interference of the molt-inhibiting hormone in the crayfish Cherax quadricarinatus. Gen. Comp. Endocrinol. 178, 227–236. doi: 10.1016/j.ygcen.2012.05.007
Piper, L. (2000). Potential for Expansion of the Freshwater Crayfish Industry in Australia. Canberra: RIRDC, 19.
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl. 1), i3i51–i3i58. doi: 10.1093/bioinformatics/bti1i018
Ratnasingham, S., and Hebert, P. D. N. (2007). bold: the barcode of life data system (http://www.barcodinglife.org). Mol. Ecol. Notes 7, 355–364. doi: 10.1111/j.14711-8286.2007.01678.x
Richman, N. I., Böhm, M., Adams, S. B., Alvarez, F., Bergey, E. A., Bunn, J. J. S., et al. (2015). Multiple drivers of decline in the global status of freshwater crayfish (Decapoda: Astacidea). Philos. T. R. Soc. B 370:20140060. doi: 10.1098/rstb.2014.0060
Sahlin, K., Vezzi, F., Nystedt, B., Lundeberg, J., and Arvestad, L. (2014). BESST - efficient scaffolding of large fragmented assemblies. BMC Bioinformatics 15:281. doi: 10.1186/14711-21055-15-281
Sánchez-Herrero, J. F., Frías-López, C., Escuer, P., Hinojosa-Alvarez, S., Arnedo, M. A., Sánchez-Gracia, A., et al. (2019). The draft genome sequence of the spider Dysdera silvatica (Araneae, Dysderidae): a valuable resource for functional and evolutionary genomic studies in chelicerates. Gigascience 8:giz0z99. doi: 10.1093/gigascience/giz0z99
Saoud, I. P., Ghanawi, J., Thompson, K. R., and Webster, C. D. (2013). A review of the culture and diseases of redclaw crayfish Cherax quadricarinatus (Von Martens 1868). J. World Aquac. Soc. 44, 1–29. doi: 10.1111/jwas.12011
Shen, H., Braband, A., and Scholtz, G. (2013). Mitogenomic analysis of decapod crustacean phylogeny corroborates traditional views on their relationships. Mol. Phylogenet. Evol. 66, 776–789. doi: 10.1016/j.ympev.2012.11.002
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv3v51
Smit, A., Hubley, R., and Green, P. (2019). RepeatMasker Open-4.0. 2013–2015. RepeatMasker Home Page.
Song, H., Buhay, J. E., Whiting, M. F., and Crandall, K. A. (2008). Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. PNAS 105, 13486–13491. doi: 10.1073/pnas.0803076105
Song, L., Bian, C., Luo, Y., Wang, L., You, X., Li, J., et al. (2016). Draft genome of the Chinese mitten crab, Eriocheir sinensis. Gigascience 5:5. doi: 10.1186/s1s3742-016-0112-y
Sorenson, M. D., and Quinn, T. W. (1998). Numts: a challenge for avian systematics and population biology. Auk 115, 214–221. doi: 10.2307/4089130
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34(Suppl. 2), W4W35–W4W39. doi: 10.1093/nar/gkl2l00
Tan, M. H., Austin, C. M., Hammer, M. P., Lee, Y. P., Croft, L. J., and Gan, H. M. (2018). Finding Nemo: hybrid assembly with Oxford Nanopore and Illumina reads greatly improves the clownfish (Amphiprion ocellaris) genome assembly. Gigascience 7, 1–6. doi: 10.1093/gigascience/gix1x37
Tan, M. H., Gan, H. M., Gan, H. Y., Lee, Y. P., Croft, L. J., Schultz, M. B., et al. (2016). First comprehensive multi-tissue transcriptome of Cherax quadricarinatus (Decapoda: Parastacidae) reveals unexpected diversity of endogenous cellulase. Org. Divers. Evol. 16, 185–200. doi: 10.1007/s1s3127-015-02377-3
Tan, M. H., Gan, H. M., Lee, Y. P., Bracken-Grissom, H., Chan, T.-Y., Miller, A. D., et al. (2019). Comparative mitogenomics of the Decapoda reveals evolutionary heterogeneity in architecture and composition. Sci. Rep. 9:1075. doi: 10.1038/s4s1598-019-471455-0
Tan, X., Qin, J. G., Chen, B., Chen, L., and Li, X. (2004). Karyological analyses on redclaw crayfish Cherax quadricarinatus (Decapoda: Parastacidae). Aquaculture 234, 65–76. doi: 10.1016/j.aquaculture.2003.12.020
Toon, A., Pérez-Losada, M., Schweitzer, C. E., Feldmann, R. M., Carlson, M., and Crandall, K. A. (2010). Gondwanan radiation of the Southern Hemisphere crayfishes (Decapoda: Parastacidae): evidence from fossils and molecules. J. Biogeogr. 37, 2275–2290. doi: 10.1111/j.13655-2699.2010.02374.x
Tsuji, J., Frith, M. C., Tomii, K., and Horton, P. (2012). Mammalian NUMT insertion is non-random. Nucleic Acids Res. 40, 9073–9088. doi: 10.1093/nar/gks4s24
Van Quyen, D., Gan, H. M., Lee, Y. P., Nguyen, D. D., Nguyen, T. H., Tran, X. T., et al. (2020). Improved genomic resources for the black tiger prawn (Penaeus monodon). Mar. Genomics 100751. doi: 10.1016/j.margen.2020.100751
Ventura, T., Stewart, M. J., Chandler, J. C., Rotgans, B., Elizur, A., and Hewitt, A. W. (2019). Molecular aspects of eye development and regeneration in the Australian redclaw crayfish, Cherax quadricarinatus. Aquac. Res. 4, 27–36. doi: 10.1016/j.aaf.2018.04.001
Vurture, G. W., Sedlazeck, F. J., Nattestad, M., Underwood, C. J., Fang, H., Gurtowski, J., et al. (2017). GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204. doi: 10.1093/bioinformatics/btx1x53
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e1e12963. doi: 10.1371/journal.pone.0112963
Warren, R. L., Yang, C., Vandervalk, B. P., Behsaz, B., Lagman, A., Jones, S. J. M., et al. (2015). LINKS: scalable, alignment-free scaffolding of draft genomes with long reads. Gigascience 4:35. doi: 10.1186/s1s3742-015-00766-3
Wick, R. (2017). “Porechop”. Github. Available online at: https://github.com/rrwick/Porechop
Williams, S. T., and Knowlton, N. (2001). Mitochondrial pseudogenes are pervasive and often insidious in the snapping shrimp genus Alpheus. Mol. Biol. Evol. 18, 1484–1493. doi: 10.1093/oxfordjournals.molbev.a0a03934
Wolfe, J. M., Breinholt, J. W., Crandall, K. A., Lemmon, A. R., Lemmon, E. M., Timm, L. E., et al. (2019). A phylogenomic framework, evolutionary timeline and genomic resources for comparative studies of decapod crustaceans. Proc. Biol. Sci. 286:2019.0079. doi: 10.1098/rspb.2019.0079
Yuan, J., Zhang, X., Liu, C., Yu, Y., Wei, J., Li, F., et al. (2018). Genomic resources and comparative analyses of two economical penaeid shrimp species, Marsupenaeus japonicus and Penaeus monodon. Mar. Genomics 39, 22–25. doi: 10.1016/j.margen.2017.12.006
Zenger, K. R., Khatkar, M. S., Jones, D. B., Khalilisamani, N., Jerry, D. R., and Raadsma, H. W. (2019). Genomic selection in aquaculture: application, limitations and opportunities with special reference to marine shrimp and pearl oysters. Front. Genet. 9:693. doi: 10.3389/fgene.2018.00693
Keywords: freshwater crayfish, Parastacidae, genome, hybrid assembly, aquaculture, Illumina, Oxford Nanopore
Citation: Tan MH, Gan HM, Lee YP, Grandjean F, Croft LJ and Austin CM (2020) A Giant Genome for a Giant Crayfish (Cherax quadricarinatus) With Insights Into cox1 Pseudogenes in Decapod Genomes. Front. Genet. 11:201. doi: 10.3389/fgene.2020.00201
Received: 03 December 2019; Accepted: 20 February 2020;
Published: 06 March 2020.
Edited by:
Peng Xu, Xiamen University, ChinaReviewed by:
Xiaozhu Wang, Auburn University, United StatesCopyright © 2020 Tan, Gan, Lee, Grandjean, Croft and Austin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mun Hua Tan, bXVuLnRhbkBkZWFraW4uZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.