
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet., 31 January 2020
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.01399
 Donghu Zhou1,2,3†
Donghu Zhou1,2,3† Saiping Qi1,2,3†
Saiping Qi1,2,3† Wei Zhang2,3,4,5Lina Wu2,3,4,5Anjian Xu1,2,3Xiaojin Li1,2,3Bei Zhang1,2,3Yanmeng Li1,2,3Siyu Jia1,2,3Hejing Wang1,2,3
Wei Zhang2,3,4,5Lina Wu2,3,4,5Anjian Xu1,2,3Xiaojin Li1,2,3Bei Zhang1,2,3Yanmeng Li1,2,3Siyu Jia1,2,3Hejing Wang1,2,3 Jidong Jia2,3,4,5Xiaojuan Ou2,3,4,5Jian Huang1,2,3,4,5*Hong You1,2,3,4,5* on behalf of China Registry of Genetic/Metabolic Liver Diseases (CR-GMLD) Group
Jidong Jia2,3,4,5Xiaojuan Ou2,3,4,5Jian Huang1,2,3,4,5*Hong You1,2,3,4,5* on behalf of China Registry of Genetic/Metabolic Liver Diseases (CR-GMLD) GroupRotor syndrome, a rare autosomal-recessive genetic disorder characterized by conjugated hyperbilirubinemia, is caused by biallelic pathogenic variants in both SLCO1B1 and SLCO1B3 genes. Long interspersed nuclear elements (LINEs) make up about 17% of the human genome and insertion of LINE-1 in genes can result in genetic diseases. In the current study, we examined SLCO1B1 and SLCO1B3 genes in two Chinese patients diagnosed with Rotor syndrome based on laboratory tests. In one patient, a novel exon 4 inversion variant was identified. This variant may have been induced by LINE-1 retrotransposon insertion into SLCO1B3 intron 3, and was identified using genome walking. Splicing assay results indicated that the exon inversion, resulting in SLCO1B3 exon 4 (122 bp) exclusion in the mature mRNA, might generate a premature termination codon. Here, we describe an exon inversion contributing to the molecular etiology of Rotor syndrome. Our results may inform future diagnoses and guide drug prescriptions and genetic counseling.
Rotor syndrome is a benign condition that presents with conjugated hyperbilirubinemia (Jirsa et al., 1993) and is inherited in an autosomal-recessive manner. The prevalence of Rotor syndrome is unknown but is estimated to be quite low (<1:1,000,000). It is unknown if the prevalence of Rotor syndrome varies among different populations. Rotor syndrome diagnosis is established when the patient presents with isolated, predominantly conjugated hyperbilirubinemia without other liver injury and with typical cholescintigraphy findings. The clinical features of Rotor syndrome usually begin shortly after birth or in childhood. Although no specific treatment is required for Rotor syndrome, it needs to be clearly differentiated from other inherited bilirubin clearance disorders that can present with either conjugated or unconjugated hyperbilirubinemia, including Gilbert syndrome, Dubin-Johnson syndrome, and Crigler-Najjar syndrome (types I and II). Thus, the genetic detection of pathogenic variants is indispensable for distinguishing Rotor syndrome from other hereditary metabolic diseases, especially Dubin-Johnson syndrome, if no typical histopathological evidence is available.
Rotor syndrome is caused by homozygous or compound heterozygous pathogenic variants in both SLCO1B1 and SLCO1B3 genes. Such pathogenic variants result in complete functional deficiency of OATP1B1 and OATP1B3 proteins (van de Steeg et al., 2012) and inhibition of conjugated bilirubin uptake into the liver. At least one wild-type SLCO1B1 or SLCO1B3 allele prevents Rotor syndrome. Only a few studies have reported co-occurring pathogenic variants in SLCO1B1 and SLCO1B3 in patients from Japan, the Philippines, and Saudi Arabia and in patients of mixed central European descent (van de Steeg et al., 2012; Kagawa et al., 2015).
In human genome, LINE-1 is the only autonomous active transposable element, and comprise ~17% of the draft sequence (Lander et al., 2001). Insertion of the LINE-1 into gene loci can cause various forms of genetic instability, including extra nucleotide insertions, exon deletions, or chromosomal inversion (Symer et al., 2002; Wheelan et al., 2005; Zemojtel et al., 2007; Cordaux and Batzer, 2009; Adney et al., 2019). Here, we report two patients with Rotor syndrome in whom several pathogenic SLCO1B1 and SLCO1B3 variants were identified. Moreover, we identified a LINE-1-induced inversion variant through genome walking analysis.
Two patients with Rotor syndrome (patient R1, male, 15 years old and patient R2, male, 26 years old) were recruited from the China Registry for Genetics/Metabolic Liver Disease. Both patients underwent a standard biochemical and liver histopathology examination (Jirsa et al., 1993), and displayed conjugated hyperbilirubinemia (patient R1: total bilirubin (TBil) 103 μmol/L, direct bilirubin (DBil) 59.7 μmol/L; patient R2: TBil 101 μmol/L, DBil 96 μmol/L). Their livers were histologically normal. The patients both provided written informed consent, and this study was approved by the Ethics Committee of Beijing Friendship Hospital, Capital Medical University.
Genomic DNA was extracted using the Blood Genomic DNA Kit (CoWin Biosciences, Beijing, China) following the manufacturer’s instructions. All SLCO1B1 and SLCO1B3 exons, and at least 50 bp of flanking sequence, were amplified using primers listed in Table S1. PCR amplification was performed in a PCR cycler (ProFlex; Applied Biosystems, CA, USA). The products were sequenced in the forward and reverse orientations by Tianyi Huiyuan Bioscience and Technology Inc. (Beijing, China).
The obtained sequences were aligned to the relevant reference sequences (SLCO1B1, NG_011745.1 or SLCO1B3, NG_032071.1) and variants were detected using Mutation Surveyor 5.01 software (Dong and Yu, 2011; Minton et al., 2011). The clinical and predicted pathogenicity of the variants were determined using VarCards (http://varcards.biols.ac.cn/) (Li et al., 2018).
A genome walking strategy was used to identify the unknown sequence between SLCO1B3 exons 3 and 5 in patient R2. The Genome Walking Kit (TaKaRa Bio, Shiga, Japan) was used according to the manufacturer's instructions. The random primers used (AP primers) were purchased from TaKaRa, and the sequences of the 12 specific primers (SP primers) used are listed in Table S1. The amplification products obtained were separated by agarose gel electrophoresis and extracted from the gel. The products were inserted into a simple T-vector for sequencing. Splice site analysis was performed using Human Splicing Finder online software (http://www.umd.be/HSF/).
The pET-01 vector was used for both exon-trapping and as a splicing reporter. The wild-type or inverted exon 4 and the flanking intronic sequences (250 bp) were cloned into the pET-01 vector using BamHI and NotI restriction enzyme sites. The plasmids were transfected into cultured 293T and Huh-7 cells (Lipofectamine LTX Reagent, Thermo Fisher Scientific, CA, USA). After 24 h, the cells were harvested and total RNA was isolated using TRIzol RNA isolation reagents (Invitrogen, CA, USA). RNA was reverse-transcribed to cDNA using a cDNA synthesis kit (Transcriptor First Strand cDNA Synthesis Kit, Roche Diagnostic Systems, NJ, USA) following the manufacturer's instructions. The primers were designed to amplify the splicing products of the pET-01 vector (Table S1). After PCR amplification, agarose gel electrophoresis was used to determine amplicon length and to deduce whether exon 4 was present in the mature mRNA.
All SLCO1B1 and SLCO1B3 exons, and at least 50 bp of flanking intronic sequences, were amplified from genomic DNA obtained from patients R1 and R2 using the primers listed in Table S1. All expected PCR products were obtained with the exception of SLCO1B3 exon 4 in patient R2. Three pathogenic point mutations were identified in SLCO1B1 and SLCO1B3 following Sanger sequencing, including two nonsense mutations and one splicing mutation. Both patients were homozygous for all three genetic variants identified (Figures 1A–C and Table 1). In addition, three SNP sites were identified in SLCO1B1 (Table S2). The clinical and predicted pathogenicity of the variants were integrated and obtained from VarCards.
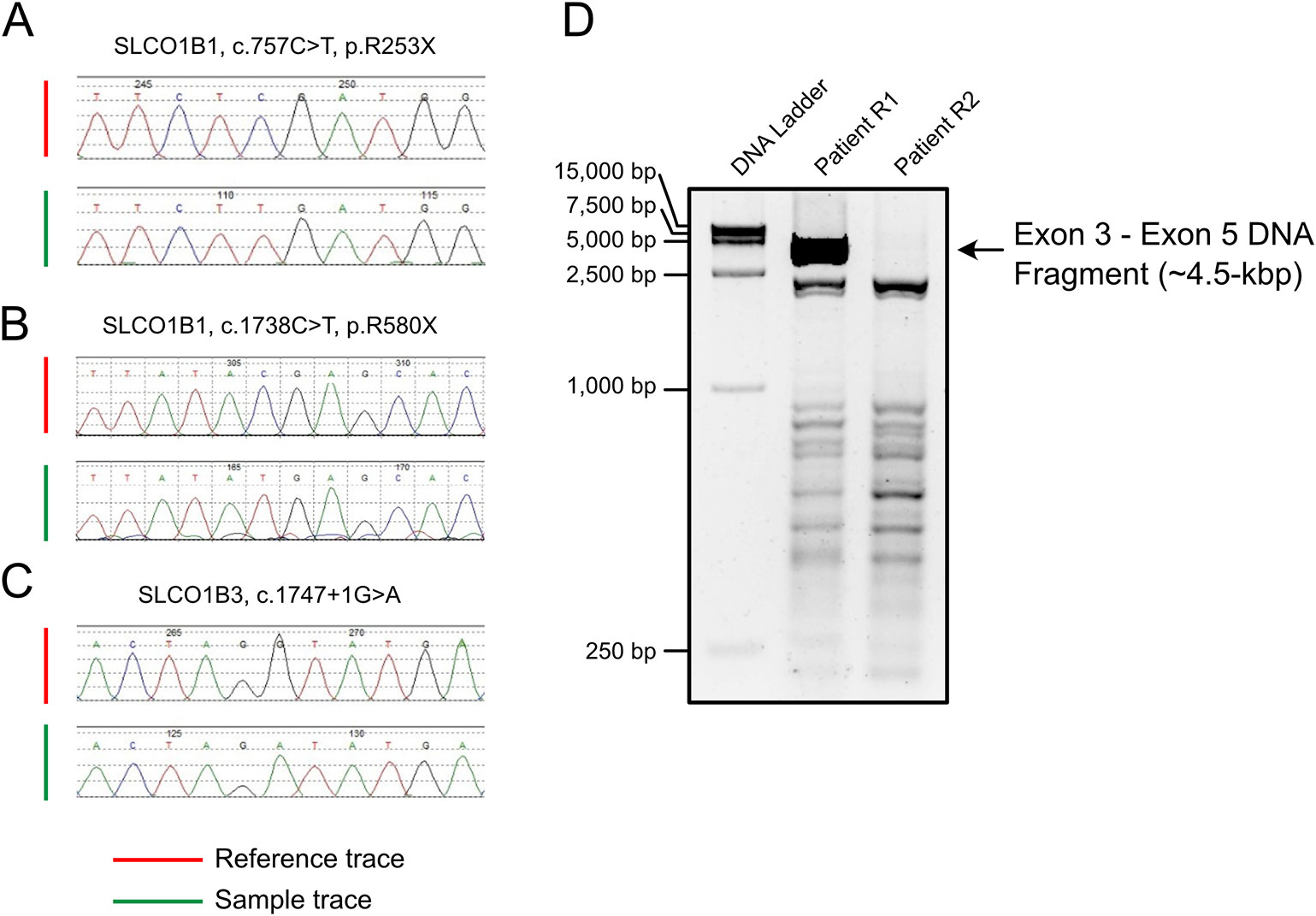
Figure 1 PCR amplification and Sanger sequencing of the target region in both patients with Rotor syndrome. (A–C) The sequences of three pathogenic point mutations. (D) Long-range PCR using the exon 3 forward primer and exon 5 reverse primer failed to produce the predicted ~4.5-kbp exon 3 to exon 5 amplification product.
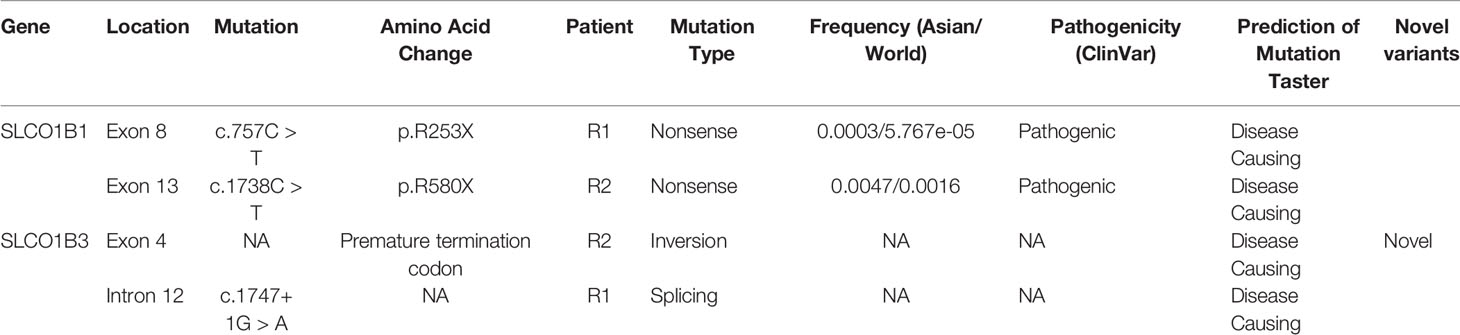
Table 1 Pathogenic SLCO1B1 and SLCO1B3 variants identified in patients with Rotor syndrome.
Long-range PCR using the forward primer for exon 3 and reverse primer for exon 5 failed to produce the predicted ~4.5-kbp exon 3–5 product in patient R2 (Figure 1D). We hypothesized that the missing target product may be the result of a DNA rearrangement around the fourth exon in SLCO1B3. To investigate this, genome walking analysis was performed (both forward and reverse) to identify the unknown sequences between exons 3 and 5. Specific primers were designed around exon 3 or 5 (Figure 2A). The amplification products were separated by agarose gel electrophoresis, extracted from the gel, and inserted into a simple T-vector for Sanger sequencing. Further genome walking analysis was then performed based on the newly identified DNA sequence. The results showed that SLCO1B3 exon 4, 1,042 bp of 5′ sequence, and the 3′ GTAAGAATTAATAGTGACAGT (chr12: 20,859,975-20,861,159), were reversed end-to-end in the homozygous state (Figure 2B and Supplemental File S1).
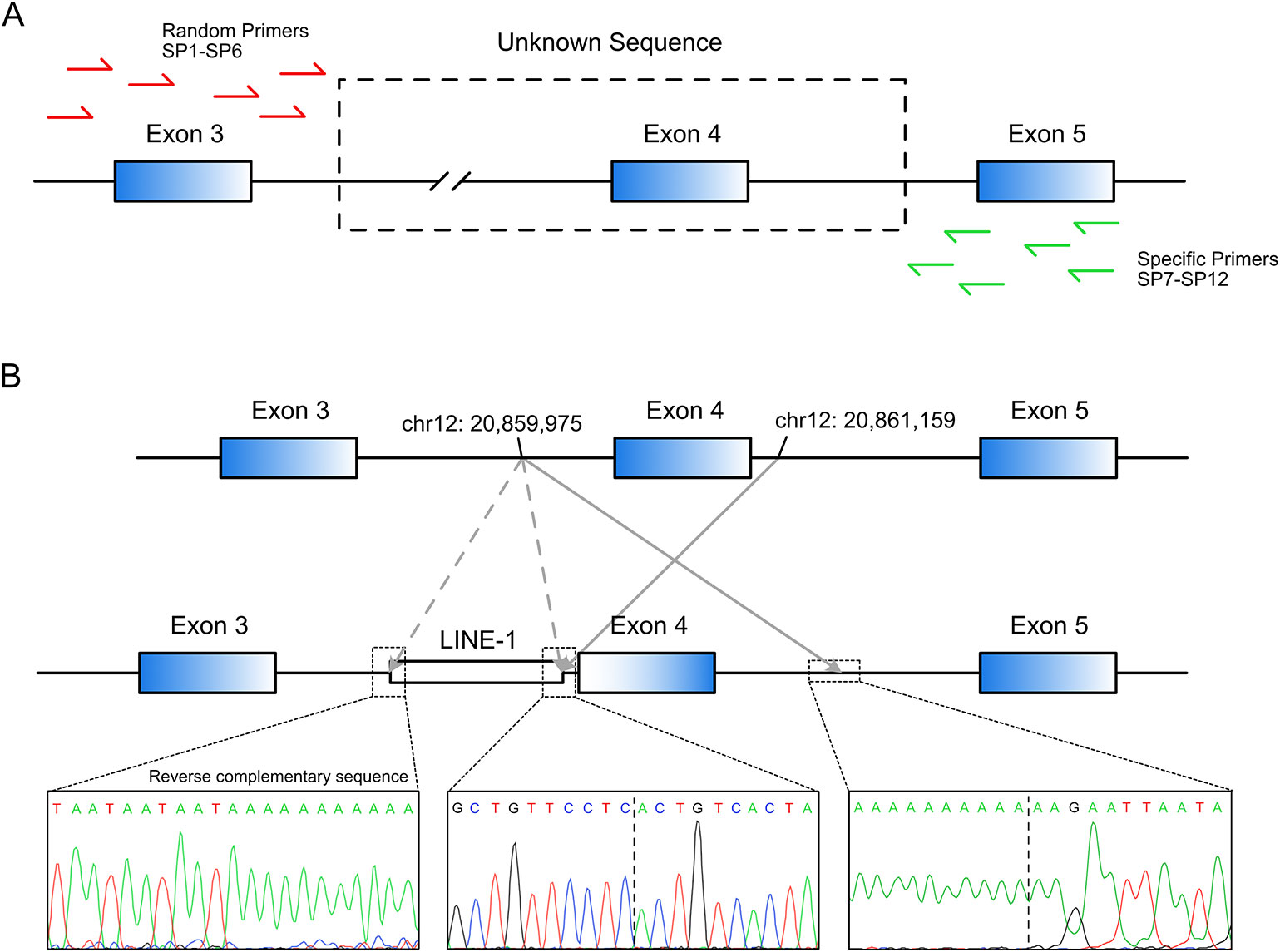
Figure 2 LINE-1-induced DNA inversion identified in SLCO1B3. (A) Genome walking analysis was used to identify the unknown sequence between SLCO1B3 exons 3 and 5 in Patient R2. (B) The LINE-1 insertion reversed the fourth exon of SLCO1B3, 1,040 bp of 5′ sequence, and the 3′ GTAAGAATTAATAGTGACAGT, end-to-end in the homozygous state.
Splice site analysis indicated that the original splice acceptor and splice donor sites were destroyed after the inversion of exon 4 (Figure 3A), causing complete loss of exon 4 (122 bp) in the mature SLCO1B3 mRNA. We constructed a splicing-reporter vector (pET-01) containing either the wild-type or inverted exon 4, and 250 bp of flanking intronic sequences (Figure 3B). RT-PCR showed that the inverted exon 4 was absent after the splicing of the pre-mRNA, both in 293T and Huh-7 cells. Thus, for SLCO1B3 gene, a premature stop codon might generated (Figure 3C and Supplemental File S2).
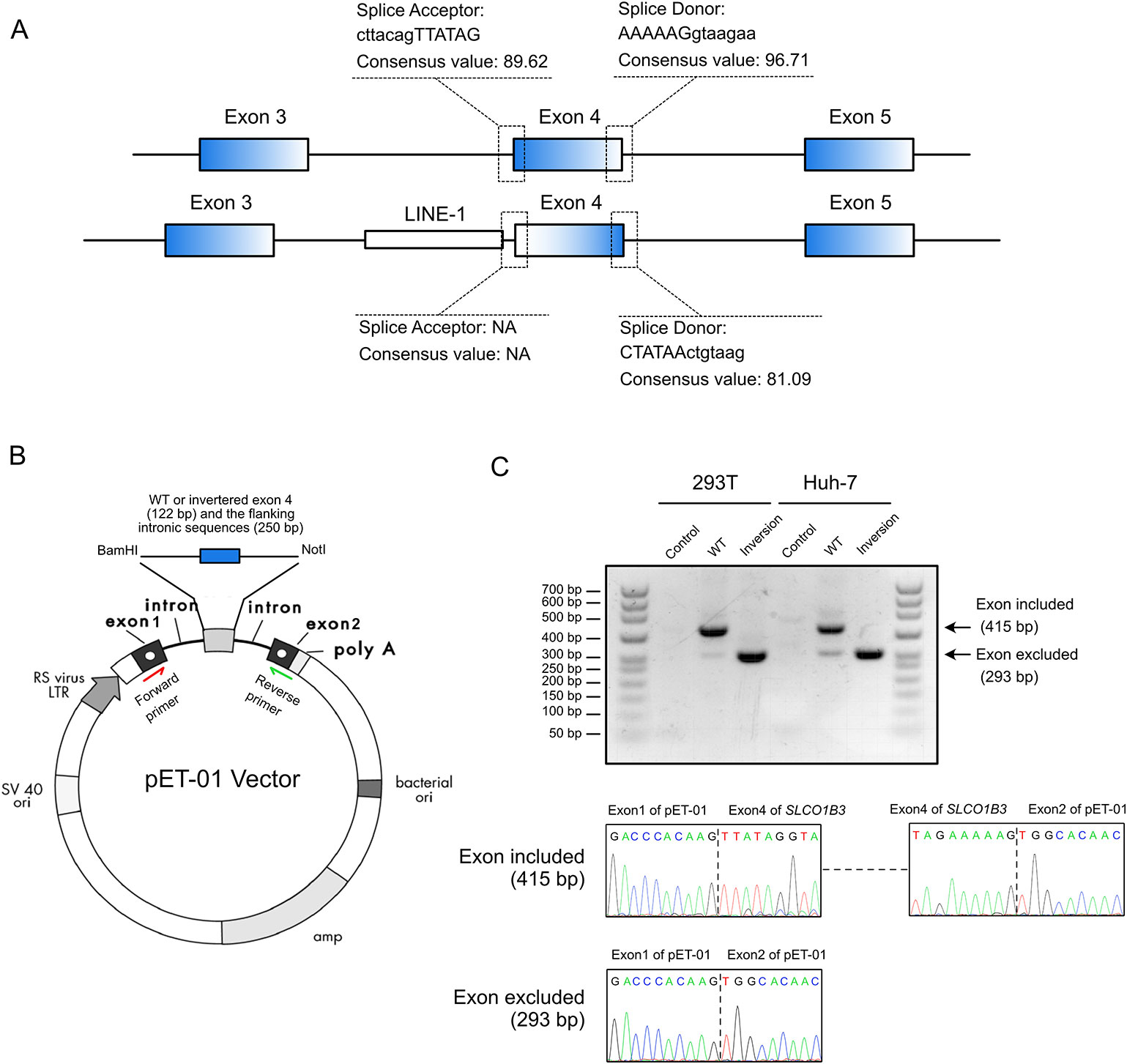
Figure 3 The inverted exon 4 was absent in the mature mRNA. (A) The splice acceptor and donor sites were destroyed after the inversion of exon 4. (B) The construction of pET-01 vector containing either the wild-type or inverted exon 4 of SLCO1B3 (Reproduced from the handbook of pET-01 vector, MoBiTec, www.mobitec.com). (C) The splicing assay indicated the absent of the fourth exon of SLCO1B3 in mature mRNA.
Next, we investigated how the DNA inversion was induced. Further sequence analysis revealed that a reverse-complementary LINE-1 retrotransposon (~6 kbp) was inserted upstream of the inverted DNA fragment (Figure 2B). This was confirmed by long-range PCR (~10 kbp product) and Sanger sequencing (Supplemental File S1). LINE-1 was reported to be able to suppress gene transcription and induce exonization and exon skipping (Ostertag and Kazazian, 2001; Chen et al., 2006; Kalliokoski and Niemi, 2009). Therefore, we believe that the identified LINE-1 insertion caused the observed inversion of the entire exon 4 and the adjacent intronic sequence in the SLCO1B3 gene of patient R2.
To date, only 26 and 10 pathogenic variants have been identified in SLCO1B1 and SLCO1B3, respectively (based on the Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/). Here, for the first time, we report SLCO1B1 and SLCO1B3 gene mutations in Chinese patients with Rotor syndrome. Unlike several other hereditary liver metabolic diseases, such as Wilson disease and hereditary hemochromatosis (Lv et al., 2016; Li et al., 2019), the hotspots for SLCO1B1 and SLCO1B3 mutations are quite similar between different populations. We identified one nonsense variant (SLCO1B1, c.757C > T, p.R253X) and one splicing variant (SLCO1B3, c.1747+1G > A) in patient R1, and one nonsense variant (SLCO1B1, c.1738C > T, p.R580X) and an exon 4 inversion in the SLCO1B3 gene of patient R2. With the exception of the exon 4 inversion, these identified variants have been reported previously (Kagawa et al., 2015). Genome walking analysis showed that a 1,185 bp DNA segment, containing the entire exon 4 and adjacent intron sequence, was reversed end-to-end in SLCO1B3. The inversion was most likely induced by the insertion of a LINE-1 retrotransposon into SLCO1B3 intron 3.
LINE-1 is the most abundant active autonomous transposon in mammalian genomes (Ostertag and Kazazian, 2001). Most of the recent research focused on the activity and regulation of LINE-1 in cancer and in nervous system, as well as being regulators of gene expression (Thomas et al., 2012; Elbarbary et al., 2016; Burns, 2017; Rodic, 2018; Saleh et al., 2019). Moreover, in earlier investigations, LINE-1 was reported to be able to affect the human genome, generating insertion mutations, altering gene expression, causing genomic instability, and finally, contributing to genetic innovation (Symer et al., 2002; Cordaux and Batzer, 2009). The intronic LINE-1 insertion can induce diseases through transcription suppression and promotion of exon inversion or skipping (Ostertag and Kazazian, 2001; Chen et al., 2006; Lee et al., 2008; Kalliokoski and Niemi, 2009). Similar cases have been previously reported for Rotor syndrome, a LINE-1 insertion was shown to result in the skipping of exon 5 or exons 5–7 and the introduction of stop codons in SLCO1B3 transcripts from six Japanese patients (Kagawa et al., 2015).
Although no specific clinical treatment is required for Rotor syndrome, the absence or dysfunction of the OATP1B1 and OATP1B3 hepatic proteins may have serious consequences for the hepatic uptake and clearance of numerous commonly used drugs, including anticancer drugs and some angiotensin-converting enzyme inhibitors (Kalliokoski and Niemi, 2009; Niemi et al., 2011; Shitara, 2011). Thus, precise identification of SLCO1B1 and SLCO1B3 pathogenic variants by genetic analysis should be used to confirm the diagnosis of Rotor syndrome and to provide guidance for therapeutic drug prescriptions and genetic counseling.
The data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
The two patients provided written informed consent, and this study was approved by the Ethics Committee of Beijing Friendship Hospital, Capital Medical University.
JH and HY designed the study. DZ and SQ analyzed and interpreted the data and wrote the manuscript. SQ, AX, XL, BZ, YL, SJ, and HW performed the experiments. WZ and LW provided patients' samples and clinical data. JJ, XO, JH, and HY advised on the conception and supervised the study. JH revised the paper. All authors vouch for the data and analysis, approve the final version, and agree to publish the manuscript.
This study was supported by grants from the Digestive Medical Coordinated Development Center of Beijing Municipal Administration of Hospitals (No. XXX0101, XXZ0501, XXT03), the National Key Technologies R&D Program (No. 2015BAI13B09), the National Natural Science Foundation of China (No. 81602032, 81800451), and the Beijing Postdoctoral Research Foundation (No. 2018-22-116).
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Natasha Beeton-Kempen, Ph.D., and Rebecca Porter, Ph.D., from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01399/full#supplementary-material
Adney, E. M., Ochmann, M. T., Sil, S., Truong, D. M., Mita, P., Wang, X., et al. (2019). Comprehensive scanning mutagenesis of human retrotransposon LINE-1 identifies motifs essential for function. Genetics. 213 (4), 1401–1414. doi: 10.1534/genetics.119.302601
Burns, K. H. (2017). Transposable elements in cancer. Nat. Rev. Cancer 17 (7), 415–424. doi: 10.1038/nrc.2017.35
Chen, J. M., Ferec, C., Cooper, D. N. (2006). LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease: mutation detection bias and multiple mechanisms of target gene disruption. J. Biomed. Biotechnol. 2006 (1), 56182. doi: 10.1155/JBB/2006/56182
Cordaux, R., Batzer, M. A. (2009). The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 10 (10), 691–703. doi: 10.1038/nrg2640
Dong, C., Yu, B. (2011). Mutation surveyor: an in silico tool for sequencing analysis. Methods Mol. Biol. 760, 223–237. doi: 10.1007/978-1-61779-176-5_14
Elbarbary, R. A., Lucas, B. A., Maquat, L. E. (2016). Retrotransposons as regulators of gene expression. Science 351 (6274), aac7247. doi: 10.1126/science.aac7247
Jirsa, M., Knisely, A. S., Schinkel, A., Kmoch, S. (1993). “Rotor Syndrome,” in GeneReviews®. Eds. Adam, M. P., Ardinger, H. H., Pagon, R. A., Wallace, S. E., Bean, L. J. H., Stephens, K., Amemiya, A. (Seattle WA: University of Washington, Seattle).
Kagawa, T., Oka, A., Kobayashi, Y., Hiasa, Y., Kitamura, T., Sakugawa, H., et al. (2015). Recessive inheritance of population-specific intronic LINE-1 insertion causes a rotor syndrome phenotype. Hum. Mutat. 36 (3), 327–332. doi: 10.1002/humu.22745
Kalliokoski, A., Niemi, M. (2009). Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 158 (3), 693–705. doi: 10.1111/j.1476-5381.2009.00430.x
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature 409 (6822), 860–921. doi: 10.1038/35057062
Lee, J., Han, K., Meyer, T. J., Kim, H. S., Batzer, M. A. (2008). Chromosomal inversions between human and chimpanzee lineages caused by retrotransposons. PLoS One 3 (12), e4047. doi: 10.1371/journal.pone.0004047
Li, J., Shi, L., Zhang, K., Zhang, Y., Hu, S., Zhao, T., et al. (2018). VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 46 (D1), D1039–D1048. doi: 10.1093/nar/gkx1039
Li, X., Zhang, W., Zhou, D., Lv, T., Xu, A., Wang, H., et al. (2019). Complex ATP7B mutation patterns in Wilson disease and evaluation of a yeast model for functional analysis of variants. Hum. Mutat. 40 (5), 552–565. doi: 10.1002/humu.23714
Lv, T., Li, X., Zhang, W., Zhao, X., Ou, X., Huang, J. (2016). Recent advance in the molecular genetics of Wilson disease and hereditary hemochromatosis. Eur. J. Med. Genet. 59 (10), 532–539. doi: 10.1016/j.ejmg.2016.08.011
Minton, J. A., Flanagan, S. E., Ellard, S. (2011). Mutation surveyor: software for DNA sequence analysis. Methods Mol. Biol. 688, 143–153. doi: 10.1007/978-1-60761-947-5_10
Niemi, M., Pasanen, M. K., Neuvonen, P. J. (2011). Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 63 (1), 157–181. doi: 10.1124/pr.110.002857
Ostertag, E. M., Kazazian, H. H., Jr. (2001). Biology of mammalian L1 retrotransposons. Annu. Rev. Genet. 35, 501–538. doi: 10.1146/annurev.genet.35.102401.091032
Rodic, N. (2018). LINE-1 activity and regulation in cancer. Front. Biosci. (Landmark Ed) 23, 1680–1686. doi: 10.2741/4666
Saleh, A., Macia, A., Muotri, A. R. (2019). Transposable elements, inflammation, and neurological disease. Front. Neurol. 10, 894. doi: 10.3389/fneur.2019.00894
Shitara, Y. (2011). Clinical importance of OATP1B1 and OATP1B3 in drug-drug interactions. Drug Metab. Pharmacokinet. 26 (3), 220–227. doi: 10.2133/dmpk.DMPK-10-RV-094
Symer, D. E., Connelly, C., Szak, S. T., Caputo, E. M., Cost, G. J., Parmigiani, G., et al. (2002). Human l1 retrotransposition is associated with genetic instability in vivo. Cell 110 (3), 327–338. doi: 10.1016/s0092-8674(02)00839-5
Thomas, C. A., Paquola, A. C., Muotri, A. R. (2012). LINE-1 retrotransposition in the nervous system. Annu. Rev. Cell Dev. Biol. 28, 555–573. doi: 10.1146/annurev-cellbio-101011-155822
van de Steeg, E., Stranecky, V., Hartmannova, H., Noskova, L., Hrebicek, M., Wagenaar, E., et al. (2012). Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J. Clin. Invest. 122 (2), 519–528. doi: 10.1172/JCI59526
Wheelan, S. J., Aizawa, Y., Han, J. S., Boeke, J. D. (2005). Gene-breaking: a new paradigm for human retrotransposon-mediated gene evolution. Genome Res. 15 (8), 1073–1078. doi: 10.1101/gr.3688905
Keywords: Rotor syndrome, SLCO1B1, SLCO1B3, mutation, LINE-1 retrotransposon
Citation: Zhou D, Qi S, Zhang W, Wu L, Xu A, Li X, Zhang B, Li Y, Jia S, Wang H, Jia J, Ou X, Huang J and You H (2020) Insertion of LINE-1 Retrotransposon Inducing Exon Inversion Causes a Rotor Syndrome Phenotype. Front. Genet. 10:1399. doi: 10.3389/fgene.2019.01399
Received: 20 September 2019; Accepted: 20 December 2019;
Published: 31 January 2020.
Edited by:
Lawrence Todd Reiter, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Tatehiro Kagawa, Tokai University Isehara Hospital, JapanCopyright © 2020 Zhou, Qi, Zhang, Wu, Xu, Li, Zhang, Li, Jia, Wang, Jia, Ou, Huang and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Huang, aHVhbmdqMTk2NkBob3RtYWlsLmNvbQ==; Hong You, eW91aG9uZzMwQHNpbmEuY29t
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.