 Manvi Sharma
Manvi Sharma Yuanyuan Li
Yuanyuan Li Matthew L. Stoll
Matthew L. Stoll Trygve O. Tollefsbol
Trygve O. Tollefsbol
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 15 January 2020
Sec. Nutrigenomics
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.01329
This article is part of the Research TopicRole of Epigenetic Modifications on Diet-Induced Metabolic DiseasesView all 16 articles
Metabolic diseases are becoming an alarming health issue due to elevated incidences of these diseases over the past few decades. Various environmental factors are associated with a number of metabolic diseases and often play a crucial role in this process. Amongst the factors, diet is the most important factor that can regulate these diseases via modulation of the gut microbiome. The gut microbiome participates in multiple metabolic processes in the human body and is mainly responsible for regulation of host metabolism. The alterations in function and composition of the gut microbiota have been known to be involved in the pathogenesis of metabolic diseases via induction of epigenetic changes such as DNA methylation, histone modifications and regulation by noncoding RNAs. These induced epigenetic modifications can also be regulated by metabolites produced by the gut microbiota including short-chain fatty acids, folates, biotin and trimethylamine-N-oxide. In addition, studies have elucidated the potential role of these microbial-produced metabolites in the pathophysiology of obesity and diabetes. Hence, this review focuses on the interactions between the gut microbiome and epigenetic processes in the regulation and development of obesity and diabetes, which may have potential as a novel preventive or therapeutic approach for several metabolic and other human diseases.
The gut microbiota consists of trillions of microorganisms including bacteria, archaea, viruses, and eukaryotes present in the intestine of humans (Allin et al., 2015). Generally, the gut microbiota is populated by Bacteroidetes, Firmicutes, Actinobacteria, Proteobacteria, Verrucomicrobia, Tenericutes, and Lentisphaerae as the predominant phyla. The major genera are Pseudomonas, Streptococcus, Prevotella, Fusobacteria, Veillonella, Haemophilus, Neisseria, Porphyromonas, Bacteroides, Clostridium, Faecalibacterium, Eubacterium, Ruminococcus, Peptococcus, Peptostreptococcus, Lactobacillus, Streptomyces, and Bifidobacterium (Marlicz et al., 2018; Neu and Pammi, 2018; Rea et al., 2018). Another similar term, gut microbiome, refers to the total genomes of gut microbiota and is often used to describe the entity of microbial functions encoded by gut microbiota (Schlaeppi and Bulgarelli, 2015). The advents in gene sequencing have revealed that the genome of human gut microbial communities (~3 million genes) is more than 100 times as large as the human genome (Gill et al., 2006), while the human:bacterial cells ratio is thought to be approximately 1:1 (Sender et al., 2016).
A neonate may procure its microbiota from the environment during delivery and also from its mother via breastfeeding (Dominguez-Bello et al., 2010). Studies have shown that neonates born through normal deliveries have an early and abundant composition of Lactobacillus, Bacteroides, and Prevotella; however, neonates born with cesarean deliveries have a delay in onset or lower levels of Bacteroides, Bifidobacteria, and Lactobacillus and predominately have colonization of Clostridium difficile, Clostridium perfringens, and Escherichia coli (Gronlund et al., 1999; Tsuji et al., 2012; Bokulich et al., 2016; Nagpal et al., 2016; Nagpal et al., 2017). These Lactobacillus species are known as probiotics due to their health-promoting properties and preventive properties in various metabolic diseases such as obesity and diabetes (Azad et al., 2018). However, C. difficile and C. perfringens bacteria are known for production of toxins, which can cause lethal diseases such as food poisoning, infection, diarrhea and colitis in humans (Noren, 2010).
During the early developmental phase, dietary factors also play a pivotal role in shaping the microbiota (Cotillard et al., 2013). For example, the gut microbiota of breast-fed neonates is mainly dominated by Bifidobacteria, Lactobacillus, Staphylococcus, and Streptococcus as compared to neonates that were on infant formulas, who have higher numbers of Bacteroides, Clostridia, and Proteobacteria (Favier et al., 2002; Bokulich et al., 2016). Various other studies have also elucidated that diet has a strong impact on maturation as well as maintenance of the gut microbiome (De Filippo et al., 2010; David et al., 2014) and potentially on the health of an individual (Camilleri et al., 2019). In addition to dietary factors, other potential variables such as antibiotic intake and infections can also influence gut microbiota (Qin and Wade, 2018) and can result in the disproportion and reduction of microbial biodiversity which is known as gut dysbiosis. These factors influencing the development of the gut microbiota are depicted in Figure 1.
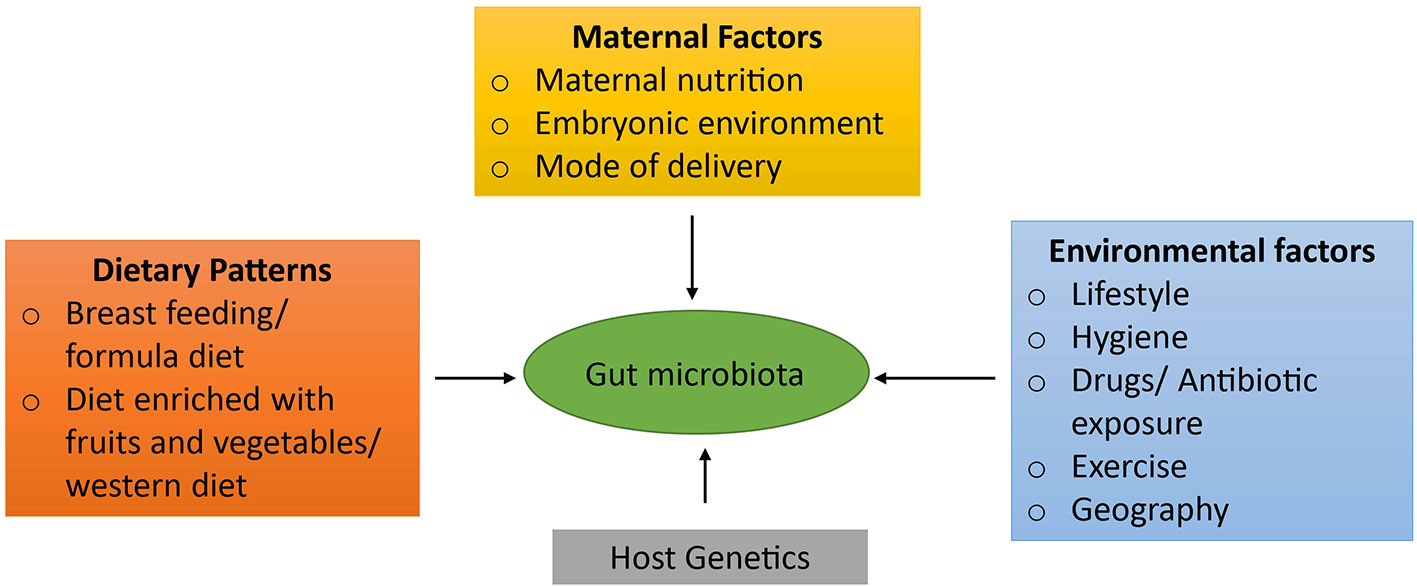
Figure 1 Factors influencing the development of gut microbiota. Numerous factors play a paramount role in the development of gut microbiota. Initially, the gut microbiota is acquired from a mother during the pregnancy via maternal nutrition, embryonic environment, and the mode of delivery. In addition, dietary patterns plays the most crucial role in maturation as well as maintenance of the gut flora such as breast feeding, formula diet, the composition of diet: diet rich in fruits and vegetables or western diet. Other factors that also contribute to its shaping are individual's genetic factors and environmental factors such as drug or antibiotic intake, infections, life style patterns, migration to a different location, etc.
A newborn infant's microbiota starts to develop according to dietary and environmental factors, but lacks an abundant diversity of commensal microorganisms. Over the first few years of life, the young child's microbiota undergoes substantial alterations, ultimately transitioning to a more mature pattern by age 3 or 4, by which time the pace of change slows down (Koenig et al., 2011; Yatsunenko et al., 2012; Stewart et al., 2018). During adulthood, in the absence of major environmental perturbations, the microbiota appears to be stable for many years, perhaps the whole lifespan (Mehta et al., 2018). These microbiota and their functions represent a “core gut microbiome,” which consists of particular types of gut microbial species, which resides in every individual and is mainly responsible for the correct functioning of a gut microbial ecosystem (Turnbaugh and Gordon, 2009; Qin et al., 2010). Gut commensal communities impact several metabolic functions within the human body such as digestion, nutrients absorption, regulation of intestinal hormones secretion, modulation of intestinal immunity and inflammatory processes, synthesis of vitamins, amino acids, and various metabolites such as short-chain fatty acids (SCFAs), choline, and lipids (Nicholson et al., 2012; Allin et al., 2015; Devaux and Raoult, 2018). In addition to the metabolic functions, the gut microbiome also shapes gene expression in the host. Through various microbial-derived metabolites, gut bacteria can influence the host metabolism by inducing epigenetic alterations of key genes which modulate the initiation and progression of diseases (Yuille et al., 2018). Therefore, gut dysbiosis has been associated with an increasing incidence of conditions such as metabolic diseases such as type 2 diabetes (Allin et al., 2015) and obesity (Ley et al., 2006); inflammatory diseases such as inflammatory bowel disease (Pascal et al., 2017) and rheumatoid arthritis (Maeda and Takeda, 2017); liver diseases (Janssen et al., 2017); and cancers (Lofgren et al., 2011; Yu et al., 2017).
With the increasing prevalence of metabolic disorders such as obesity and diabetes in Western countries, it is essential to focus on the interplay between epigenetic mechanisms and gut microbial composition in the induction of these diseases that might provide a novel therapeutic approach for prevention and treatment. Hence, we will discuss the potential roles of the gut microbiome and epigenetics in the pathophysiology and pathogenesis of obesity and diabetes (type 1 and type 2).
Epigenetics is the study of phenotypic changes secondary to alterations in gene expression that do not directly arise from changes in the underlying DNA sequence. In eukaryotes, epigenetic mechanisms primarily involve DNA methylation, posttranscriptional histone modifications, chromatin restructuring, and regulation of gene expression by noncoding RNAs (Paul et al., 2015). These epigenetic mechanisms can be regulated by cross-talk of microbial metabolites, external factors such as diet, antibiotics and also by other environmental factors (pH, oxygen, and temperature), resulting in the modulation of a large number of human metabolic diseases (Romano and Rey, 2018).
The noncoding RNA (ncRNA) are RNA transcripts that are not translated into the proteins (Dempsey et al., 2018). The alterations in ncRNA have been known to contribute to various diseases such as obesity, diabetes, neurodegenerative diseases, liver diseases, and lung diseases (Devaux and Raoult, 2018). The major types of ncRNAs are microRNA (miRNA), short-interfering RNAs (siRNAs), piwi-interacting RNAs (piRNAs), and long noncoding RNAs (lncRNAs). lncRNAs modify gene expression via the formation of complexes with chromatin-modifying protein and by functioning as signaling and guide molecules (Devaux and Raoult, 2018). A few studies have focused on the role of the gut microbiome in regulation of lncRNA gene expression in the host. In order to obtain in-depth knowledge about their correlation, Liang et al. performed a bioinformatics study on characterization of lncRNA regulated by gut microbiome in intestinal epithelial tissues of mice (Liang et al., 2015). The comparison between germ-free mice with the conventional (re-colonized with mice microbiota) and gnotobiotic mice (recolonized with either E. coli or E. coli-expressing bile salt hydrolase) showed that only six lncRNAs were commonly upregulated. These lncRNAs were also found to be overexpressed in immune organs such as thymus and spleen, which may reflect their crucial role in immune processes. Furthermore, this study also elucidated that lncRNA expression profiles successfully differentiated gontobiotic mice from conventional mice based on their gut microbial composition. Despite the less available information on the association of lncRNA and gut microbiota, this study provided novel insight of microbial-regulated lncRNA expression. This study may also enhance understanding of the potential roles played by lncRNA expression that are regulated by gut microbiota and in-turn influence metabolic disorders such as obesity and diabetes. However, this study had a limitation with respect to determining whether the expression of lncRNAs was regulated by the gut microbial communities of the host (Liang et al., 2015). Another study by Dempsey et al. evaluated lncRNA expression in various tissues- liver, duodenum, jejunum, ileum, white adipose tissue, brown adipose tissue, colon, and skeletal muscle from germ-free and conventional mice (Dempsey et al., 2018). They found, in the absence of microbial communities (i.e., germ-free mice), the lncRNAs were differentially regulated in distal (liver, muscle, and fat) tissues as well as in proximal (intestinal) tissues. Most of the lncRNAs which were found to be regulated by gut microbiome were present in an essential metabolic organ, namely, the jejunum. Overall, this is the first study that demonstrated that the gut microbiome is important for the lncRNA expression in the various metabolic and other organs (Dempsey et al., 2018). These studies implicate a potential role played by the gut microbiome in regulation of lncRNA expression. However, more investigations in this context should be conducted to further clarify the role of this association of host-microbe interactions with the pathogenesis of obesity and diabetes.
Another type of ncRNA, miRNAs, also play crucial roles in maintaining metabolic homeostasis and in development of obesity and insulin resistance (Davalos et al., 2011; Trajkovski et al., 2011), This has also been shown in a recent study by Virtue et al. that focused on the interplay between miRNAs and development of obesity through gut microbial population in germ-free and conventionally housed specific pathogen-free mice (Virtue et al., 2019). The investigators found that the miR-181a and miR-181b expression were increased in epididymal white adipose tissues obtained from conventional mice as compared to germ-free mice, which implicates the importance of gut microbiota in the regulation of miRNA. In order to confirm this regulation, they colonized germ-free mice with conventional mice microbiota and found similar results, that is, upregulation of miR-181a and miR-181b in epididymal white adipocytes. The investigators hypothesized that the potential mechanism behind the regulation of miR-181 might be via gut microbial-produced metabolites. This leads to the finding that tryptophan-derived metabolites negatively regulate miR-181 expression in white adipocytes, which further influence the pathways of adiposity, energy balance and insulin sensitivity. In addition, this study also revealed the imbalance of the microbiota—miR-181 axis is vital for obesity and insulin resistance development (Virtue et al., 2019). Therefore, this novel study elaborates that the gut microbiota might serve as a key regulator for functioning of miRNAs especially in white adipocyte tissue, which have a major impact on the development of obesity and insulin resistance. However, this study has not provided the role of specific gut microbial species that may be critically involved in the regulation of miR-181 expression. Further well-designed studies are required for elucidating the role of the gut microbiome on miRNA in the etiology of obesity as well as in diabetes.
DNA methylation refers to the inclusion of a methyl group (-CH3) on the carbon-5 of the cytosine ring in cytosine-guanine dinucleotide-rich (CpG) regions (Romano and Rey, 2018). The enzymes, DNA methyltransferases (DNMTs) catalyze this process and also regulate the gene expression. These DNMTs are highly sensitive to the availability of nutrients that can also be affected by the metabolic activities of the microbial species present in the gut (Romano and Rey, 2018). The major metabolic activity involves synthesis of metabolites that can modulate the epigenome by participating in one-carbon metabolism (Mischke and Plosch, 2013). Metabolites such as folate, vitamin B12, betaine, and choline are potentially involved in the synthesis of 6-methyltetrahydrofolate, which is a methyl group donor, for the generation of S-adenosylmethionine (SAM) that participates in DNA methylation processes (Kovacheva et al., 2007; Crider et al., 2012; Kok et al., 2015; Zeisel, 2017; Mahmoud and Ali, 2019). These methyl donor nutrients are found to be regulated by specific gut microbial communities such as Lactobacillus and Bifidobacteria that are known for folate production (Strozzi and Mogna, 2008; Rossi et al., 2011). In order to understand the role played by Lactobacillus and Bifidobacterium in regulation of diabetes, a study by Murri et al. found their levels were reduced in type 1 diabetic healthy Caucasian children (Murri et al., 2013). These studies show an important association between gut microbial communities and DNA methylation mechanisms that may regulate diabetes.
Other crucial metabolites, such as SCFAs are synthesized in the gut by the fermentation of nondigestible carbohydrates by certain microbes. Some of the major SCFAs including butyrate can also influence DNA methylation processes by inducing phosphorylation of ERK (MAP kinase1), which results in down-regulation of DNMT1 and consequently demethylation of tumor suppressor genes including RARB2, p21, and p16 (Sarkar et al., 2011). The amount of butyrate synthesis depends on both dietary intake and synthesis by gut microbial communities. Various bacteria are known for butyrate production including Megasphaera, Odoribacter, Eubacterium, Peptoniphilus, Fusobacterium, Coprococcus, Porphyromonas, Faecalibacterium, Anaerotruncus, Clostridium, Subdoligranulum, and Roseburia (Demehri et al., 2016). The butyrate-producing bacteria, Clostridium, was found to be present in abundant levels in p21-p-luc male mice, which were treated with a high-fat diet (HFD) diet for thirty weeks when compared with mice on a normal diet (Yoshimoto et al., 2013), indicating the potential role of diet in impacting the gut microbiota and downstream metabolic processes. Similarly, the number of Clostridium was increased in type 1 diabetic children (Murri et al., 2013). These studies suggest an important role by gut microbiota in regulation of obesity and diabetes via production of gut metabolites.
Several other crucial nutrients also play a vital role in the regulation of the DNA methylation process (Liu et al., 2019). For instance, choline is a water-soluble vitamin-like nutrient found in various food sources such as meat, grain, milk, egg, and their derived products. Gut communities can metabolize choline into several metabolites that impact human health, such as trimethylamine (TMA) (Romano et al., 2015). TMA can be further metabolized by flavin monooxygenase (FMO) enzymes into trimethylamine-N-oxide (TMAO) (Baker and Chaykin, 1960), which has been linked to obesity and diabetes (Fennema et al., 2016; Velasquez et al., 2016; Tang and Hazen, 2017). This choline derived metabolite, TMAO was also found to be involved in vast production of ROS (Sun et al., 2016), that in-turn can influence epigenetic programming as it can lead to deamination or depurination of nucleic acids, which may trigger DNA repair mechanisms and replacement with a nonmethylated cytosine (Avila et al., 2015). To investigate the relationship between choline and metabolic disorders via DNA methylation, a study was conducted on germ-free C57BL/6 female mice (Romano et al., 2017). These mice were divided into two different groups based on the colonization in gut; a) with choline-utilizing bacteria, or b) with bacteria unable to utilize choline and unable to produce TMA. Both mouse groups were kept on HFD supplemented with choline for several weeks, which resulted in lower methylation levels in heart, colon, brain, and liver tissues in the first group of mice as compared to the other group. In addition, the mice colonized with choline-utilizing bacteria displayed adiposity features. It was concluded that choline-utilizing bacteria compete for choline uptake with the host, resulting in lower levels of choline and methyl donor in the host, ultimately making the host more susceptible toward metabolic disorders (Romano et al., 2017). These studies implicate the association of choline in modulating epigenetic machinery that might act as a key player in the pathophysiology of metabolic diseases. However, more studies are required to confirm the exact mechanisms induced by choline on modulating the DNA methylation process.
Histones are proteins that are wound by DNA in the nucleus to form the condensed chromatin and mainly comprise of four families: H1, H2A, H2B, H3, and H4. Histones are prone to various modifications including acetylation, methylation, phosphorylation, SUMOylation, poly-ADP ribosylation, biotinylation, ubiquitination, citrullination, and proline isomerization (Bernstein et al., 2007; Paul et al., 2015). Out of all modifications, histone methylation, acetylation, and deacetylation are known to play key roles in the induction and progression of various disorders. The histone methylation process involves the addition of methyl groups to the histone proteins by histone methyltransferases (HMTs) enzymes. Histone methylation can either lead to transcription activation (e.g., H3K4) or transcription inactivation (e.g., H3K9, H3K27), depending on the specific residue and modifications (Orouji and Utikal, 2018). A recent study by Tateishi et al. showed a correlation of histone methylation with obesity and hyperlipidemia in mice. They found that impairment in Jhdm2a function (a H3K9-specific histone demethylase) in Jhdm2a knockout mice resulted in altered β-adrenergic-stimulated glycerol release and oxygen absorption in brown fat. This study also revealed that β-adrenergic activation induces Jhdm2a binding to the PPAR responsive element of the Ucp1 gene, an important gene involved in energy balance and that leads to a decrease in H3K9me2, which contributed to obese phenotypes such as fat deposition and rise in lipid content (Tateishi et al., 2009).
Histone acetyltransferases (HATs) enzymes catalyze the transfer of acetyl groups from acetyl-CoA to the amino-terminal lysine residues on histone proteins (Roth et al., 2001). The histone acetylation process can be regulated by various gut-microbial derived metabolites such as SCFAs (Krautkramer et al., 2016; Qin and Wade, 2018). It has been found that supplementation with acetate raised the acetylation levels of brain histones H3 at lysine 9 and H4 at lysine 8 and 16 that resulted in neuroglial activation and decline in the cholinergic cell (a nerve cell) in a rat model of LPS-induced neuroinflammation (Soliman and Rosenberger, 2011). A recent study by Wang et al. investigated the impact of lentinan, a polysaccharide derived from mushroom, on the intestinal microbiota of piglets that were challenged with E. coli lipopolysaccharide-induced intestine injury. They found that lentinan supplementation increased SCFAs levels including butyrate, propionate, iso-butyrate, and isovalerate in the cecum, which further led to a rise in H3 histone acetylation and a decline in intestinal inflammation (Wang et al., 2019b). It has been evident that the chromatin state of several tissue constituents such as colonic cells, can be modulated by SCFAs produced in the gut (Krautkramer et al., 2016).
Histone deacetylases (HDACs) constitute a class of enzymes that remove an acetyl group from the amino-terminal lysine residues of histones resulting in compacted chromatin (Yuille et al., 2018). Histone deacetylation is primarily associated with transcriptional inactivation and overexpression of HDACs and has been linked to a number of neurological and inflammatory diseases. Overall, 13 HDACs have been found in humans, which are classified into four classes- Class I contains HDACs 1, 2, 3, and 8; Class IIa consists of HDACs 4, 5, 7, and 9; Class IIb contains HDACs 6 and 10; Class III is comprised of Sirt1-Sirt7 and Class IV consists of HDAC 11. Each of these plays an important role in cell survival, proliferation and differentiation which can also influence tumorigenesis (Yuille et al., 2018). HDAC inhibitors have well-known potential to act as therapeutic agents in various diseases. The gut microbiome can modulate the activity of HDACs via production of epigenetic metabolites such as the SCFAs. Butyrate and propionate have been identified as potential contributors to HDAC inhibition (Marlicz et al., 2018). Butyrate is essential for maintaining homeostasis in the gut and is also important in the regulation of many processes such as epigenetic mechanisms, lipogenesis, gluconeogenesis, and inflammatory conditions. Among various epigenetic modifications, butyrate is specifically known as a class I and class II HDAC inhibitor (Marlicz et al., 2018). An interesting study focused on 79 distinct commensal human gut bacteria to investigate the connection between SCFA profiles and HDAC inhibitory properties. These findings revealed that three butyrate-producing bacterial strains: Megasphaera massiliensis MRx0029, Roseburia intestinalis MRx0071, and Bariatricus massiliensis MRx1342, manifested the highest inhibition of HDAC activity. In addition, M. massiliensis produced significant levels of valeric acid and hexanoic acid, which are medium-chain fatty acids. It was also reported that valeric acid and butyrate cumulatively showed inhibition against Class I HDACs- HDACs1, 2, 3 8, particularly HDAC2 (Yuille et al., 2018).
Obesity is a leading disorder that involves accumulation of excessive fat in the body. Obesity has been found to be linked with multiple conditions including cardiovascular diseases, diabetes, metabolic disorders, and cancers (Kopelman, 2000). There are various factors that have been shown to play a key role in the pathophysiology and pathogenesis of obesity such as genetic susceptibility, dietary patterns, ethnic differences, antibiotic intake, and environmental factors.
Diet is a vital factor in the establishment of the composition of gut microbiota, which cross-talks with the intestine, and participates in the generation of signals to communicate with distal organs such as the liver. It therefore plays an integral role in shaping the host's metabolism (Schroeder and Backhed, 2016). Differences in dietary patterns can lead to alterations in the composition and function of microbial communities, resulting in change in fermentation processes, energy consumption, sensation, and permeability in a manner that results in weight gain (Kumar et al., 2014; Devaux and Raoult, 2018). Beside these, recent evidence suggests that diets strongly influence epigenetic processes that are linked to obesity development (Qian et al., 2017). Numerous studies have indicated that the shifts in microbial communities mainly due to consumption of a “Western” diet, which is high in fat and carbohydrate, result in predisposition toward obesity. Specifically, a decrease of Bacteroidetes and an increase of Firmicutes levels have been linked with the consumption of a western diet (Ley et al., 2006). These bacterial communities have been known to directly influence the epigenetic reprogramming via DNA methylation. As an illustration, Kumar et al. reported that the infants born from mothers who had a higher Firmicutes gut composition showed altered DNA methylation as compared to infants born from mothers with higher composition of Bacteroidetes in the gut. This study also reported that the differentially methylated genes of infants whose mothers had high levels of Firmicutes were positively correlated to cardiovascular diseases, inflammation, obesity, and abnormal lipid metabolism (Kumar et al., 2014).
In contrast, studies have shown that certain plant-based diets have been linked to a particular spectrum of bacteria that ameliorates development of obesity. For example, a recent study focused on the impact of ginger on gut microbiota and prevention of obesity in C57BL/6J mice (Wang et al., 2019a). The mice were divided into four groups based on the feeding of normal diet or HFD diet with or without ginger. It was found the mice on the HFD and ginger diets showed a decrease in weight, as well as diminished low-grade inflammation and insulin resistance. The microbiota profile associated with these changes included abundance of Bifidobacterium genus and major SCFA-producer bacteria such as Alloprevotella and Allobaculum. To address whether these gut microbial differences were responsible for the metabolic improvements, the investigators performed fecal microbiota transplantation into mice whose microbiota was depleted by antibiotics. The transplants were technically successful, insofar as they recapitulated the microbiota profiles of the donor mice. Importantly, the mice that received microbiota from the HFD and ginger diet group showed a decrease in body weight, body mass and improvement in glucose tolerance as compared to recipients of microbiota from the HFD group. This study confirms the association of particular microbial species with the ginger-supplemented diet and provides an underlying mechanism of prevention of obesity via these microbial species (Wang et al., 2019a). Multiple other species of bacteria may beneficially impact metabolic processes. For example, Bifidobacterium has been known to impact metabolic events such as insulin resistance, low-grade inflammation, and obesity in mice (Zhang et al., 2017). Similarly, the SCFA-producers Alloprevotella and Allobaculum have been found to be associated with improvement in obesity and insulin resistance (Zhang et al., 2015). Also, the SCFAs produced from these bacteria such as butyrate and propionate are known to strongly influence molecular pathways that impact obesity development (Lin et al., 2012). The major SCFAs such as butyrate, propionate and acetate bind to G-protein coupled receptors (GPRs) - GPR41 (Free Fatty Acid Receptor 3) and GPR43 (Free Fatty Acid Receptor 2) expressed on the intestinal mucus layer of immune cells, liver cells, and adipose tissue (Brahe et al., 2013), resulting in decrease in weight gain, less intake of food and liposysis inhibition in adipose tissues (Lu et al., 2016). In addition to these, various other studies have focused on changes in intestinal composition with obesity and its role in metabolic mechanisms and epigenetics as highlighted in Table 1. Therefore, diets may act as a bridge that links the gut microbiome and host metabolism and that contributes to altered health outcomes through, at least in part, regulation of epigenetic mechanisms.
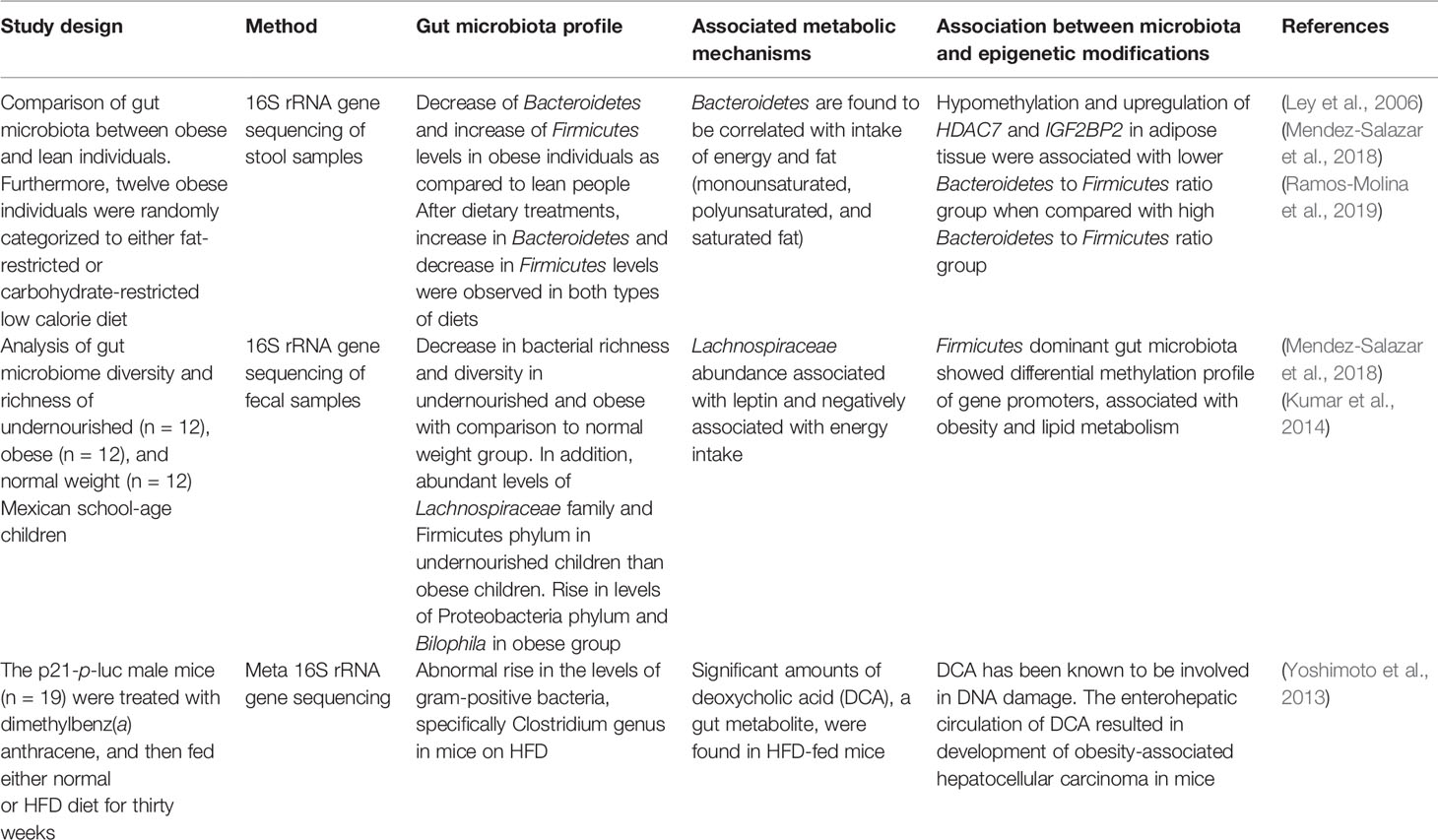
Table 1 The alterations in gut microbiota in obesity and its role in metabolic mechanisms and epigenetics.
The direct association between diet, gut bacteria, and epigenetic factors that leads to obesity development have been highlighted in Figure 2. Epigenetic mechanisms such as histone modifications and DNA methylations play a key role in the development of obesity (Qian et al., 2017; Lieber et al., 2018). As a study found that loss of HDAC6 has been associated with a rise in acetylation of a protein called “cell death–inducing DNA fragmentation factor subunit α-like effector C,” which further led to an increase in lipid droplet storage and ultimately body weight gain (Qian et al., 2017). DNA methylation also plays a promising role in regulation of obesity; this was demonstrated in a study involving administration of Lactobacillus rhamnosus and Bifidobacterium lactis to pregnant women, which resulted in a decrease in the methylation of the FTO and MC4R promoters in the women and their children (Vahamiko et al., 2019). The significance of these findings is that these genes such as FTO has been associated with weight gain and BMI; and is well-known risk factor of obesity (Claussnitzer et al., 2015; Qi et al., 2015). Also, MC4R gene involves in the key metabolic processes such as regulation of food consumption and energy balance; and the abnormalities in this gene can lead to decrease in satiety and onset of obesity (Fani et al., 2014; Rovite et al., 2014). Overall, this study has provided an important insight that supplementation of probiotics during pregnancy can modulate DNA methylation of promoter region of genes that are associated with weight gain and obesity in mothers and their children (Vahamiko et al., 2019). In the future, more research will be required to elaborate on the interaction between the gut microbiome and DNA methylation modifications that may play key roles in regulation of obesity.
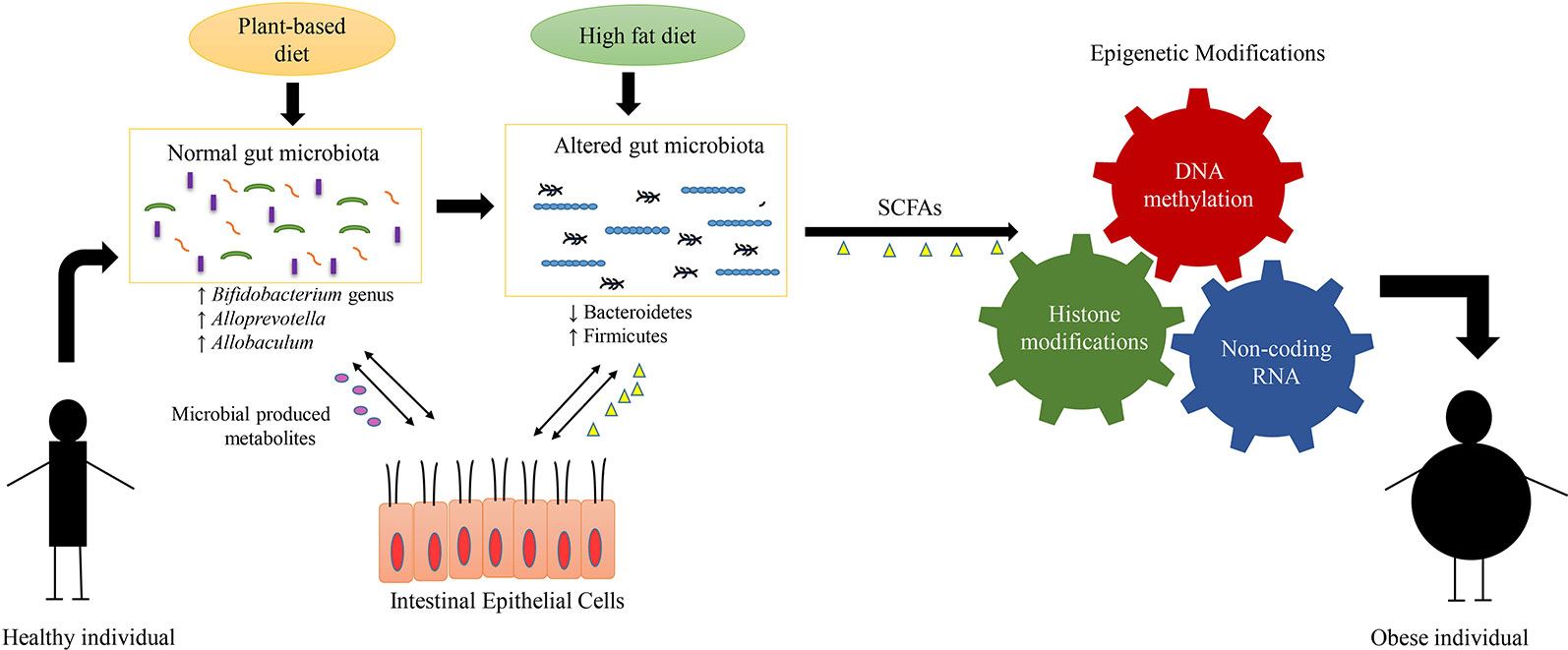
Figure 2 Interplay between diet, gut microbiome, epigenetic mechanisms, and obesity. The alterations in composition of microbial communities mainly arise due to difference in dietary patterns. The plant-based diets have been known to be associated with diverse and particular gut flora such as Bifidobacterium genus, Alloprevotella, and Allobaculum. The diverse composition of gut microbiota results in production of various metabolites such as short-chain fatty acids (SCFA). These gut microbial-produced metabolites interact with the epithelial cells of the host and help to maintain the host metabolism. On the other hand, the diets enriched in fat and carbohydrate, result in lowered gut diversity and alterations in the composition of gut microbiota such as decrease in levels of Bacteroidetes and increase in levels of Firmicutes. This gut dysbiosis (alterations in composition and function of gut bacteria) produce metabolites that induce specific epigenetic alterations such as DNA methylation, histone modification and noncoding RNA, which in-turn regulate the development of obesity.
Several studies have implicated the interplay between inflammation and obesity. For instance, the inflammatory cytokines such as interleukin 1, interleukin 6, tumor necrosis factor, and C-reactive protein were found to be associated with the obesity markers- BMI, waist circumference, or percentage body fat (Marques-Vidal et al., 2012). Obesity is also associated with inflammatory conditions, such as juvenile psoriatic arthritis. These individuals were found to have a greater risk of obesity as compared to healthy children as well as children with other forms of juvenile arthritis. (Samad et al., 2018). Low-grade inflammation may contribute significantly to obesity development as high levels of activated CD8+T cells and aggravated immune response were observed in adipose tissue of HFD-fed mice (Nishimura et al., 2009). Regulatory T (Treg) cells, a subpopulation of T-helper cells, maintains balance between proinflammatory and antiinflammatory immune responses. Studies have shown dependency of Treg cell on the intestinal communities as they acquire signals from the gut microbiota (Romano-Keeler et al., 2012). Forkhead box P3 (Foxp3) is a transcription factor expressed by Treg cells, that plays a crucial role in the development and function of Treg cells (Hori et al., 2003). This Foxp3 protein is regulated posttranslationally by lysine acetylation by HDACs and HDAC inhibition (Wang et al., 2015). Therefore, the products of gut microbiota such as SCFA, which act as HDAC inhibitors, may be involved in Treg cell differentiation and ameliorate inflammatory states such as obesity. For example, butyrate plays an essential role in the differentiation of Treg cells via increasing the acetylation of noncoding regions of the Foxp3 locus (Furusawa et al., 2013). It specifically acts as a HDAC inhibitor in intestinal epithelial cells and thereby alters metabolic functions. For example, HDAC3 knockout mice did not show obesity features (improvement in glucose tolerance and insulin levels) despite being on the same HFD as wild-type C57BL6 mice. Further analysis of intestinal epithelial cells from HDAC3 knockout mice showed altered expression of Chka, Mttp, Apoa1, and Pck1 (an enzyme involved in gluconeogenesis and glyceroneogenesis processes in adipose (Franckhauser et al., 2002)), which are associated with multiple metabolic processes in the intestinal epithelial cells. Thereafter, butyrate supplementation in control mice resulted in weight loss and refinement of metabolic functions (Whitt et al., 2018).
From the past decade, there has been a significant increase in the incidence of Diabetes (Zhou et al., 2016). Type 1 diabetes is an autoimmune disease that arises when T-cell mediated destruction of insulin-producing β cells occurs (Gianani and Eisenbarth, 2005), whereas type 2 diabetes is a chronic disease which commences when insulin resistance develops in the body (Chen et al., 2011). Due to acquired insulin resistance, more insulin, which is produced by the pancreas, is required. However, the pancreas fails to generate enough insulin, which in turn increases the blood glucose level (Larsen et al., 2010). A large number of factors such as environmental, genetic and lifestyle factors influence both type 1 and type 2 diabetes (Qin et al., 2012). Various studies have provided evidence of a direct relationship between epigenetic mechanisms and the gut microbiome in the etiology of both types of diabetes (Allin et al., 2015).
Recent experimental data from human studies have shown that the gut microbial composition has a crucial role in the development of type 1 diabetes (Brown et al., 2011; Giongo et al., 2011). As also highlighted in Table 2, the study by Giongo et al. focused on the children at high risk for developing type 1 diabetes based upon at-risk HLA types (Giongo et al., 2011). They were followed prospectively for the development of antibodies associated with diabetes and the microbiota of children who ultimately developed antibodies was compared to that of the children who did not develop antibodies. They found that prior to disease onset, there were alterations in gut microbiota such as reduction in Firmicutes and increase in Bacteroidetes and decreased diversity in the infants who produced antibodies (Giongo et al., 2011). However, whether the observed microbial difference arose due to differences in dietary patterns of children is not yet known. Similarly, another study by Murri et al. focused on the gut microbial profile of children diagnosed with type 1 diabetes and healthy children (Murri et al., 2013). The results of this study revealed increased levels of Bacteroidetes, Clostridium spp. and Veillonella as well as decline in Lactobacillus, Bifidobacterium, Blautia, and Prevotella in diabetic children when compared with healthy children. These alterations in gut microbiota were independent of dietary patterns, as no significant difference between the dietary habits was observed between both groups of children (Murri et al., 2013). The resulting limited gut microbial diversity creates an imbalance of the microbial ecosystem and essential processes in the gut which may reduce the diverse diet digestion capacity that eventually leads to a decrease in energy levels in type 1 diabetes patients and cause them to be more prone toward diseases (Giongo et al., 2011). The knowledge acquired from this study may allow interventions that can cure or delay autoimmunity in patients by changing the gut microbiota through epigenetic regulations.
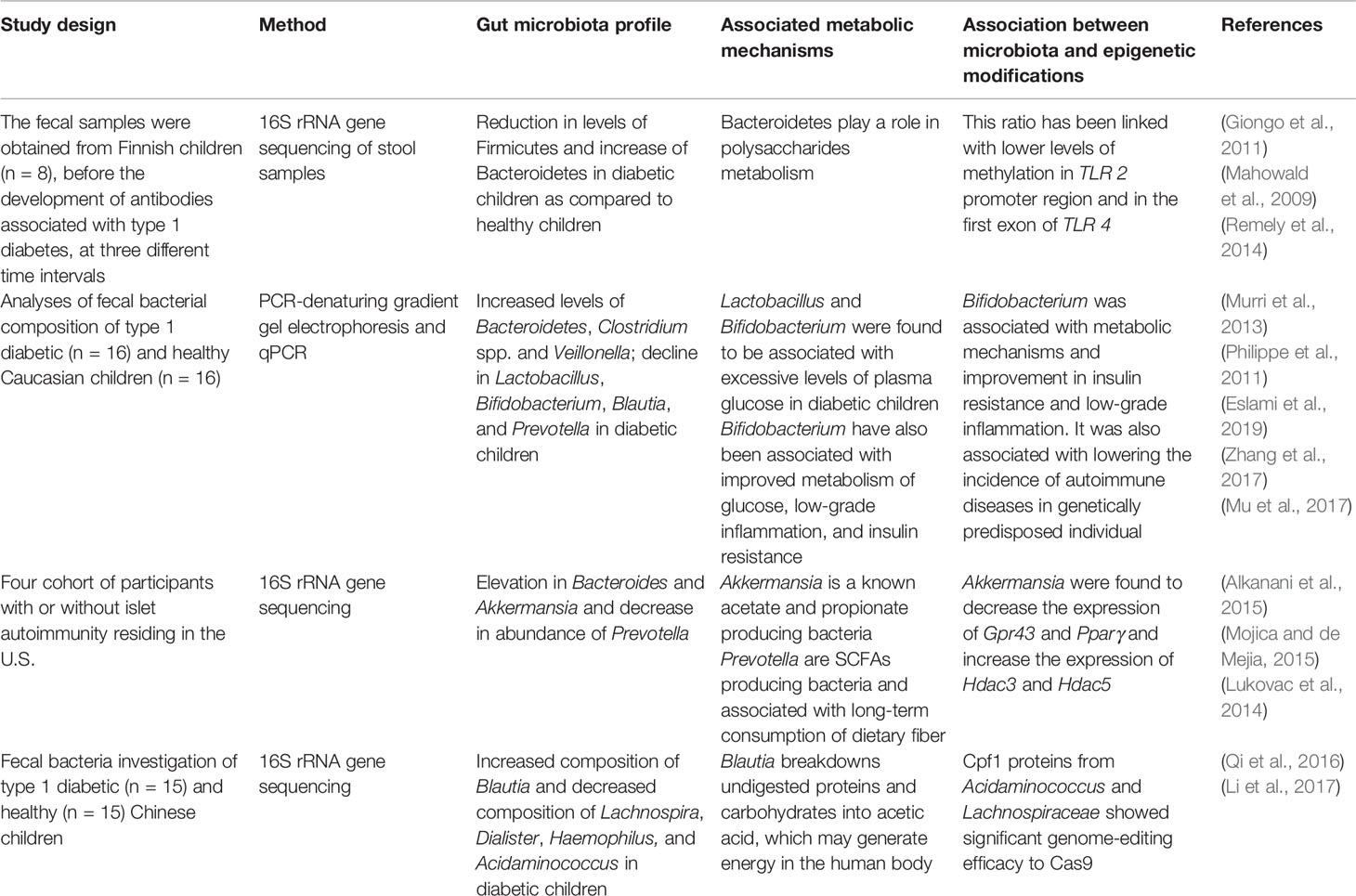
Table 2 Role of the gut microbiome in induction of type 1 diabetes and the association between gut microbiota, metabolic mechanisms, and epigenetic modifications.
Additional evidence has shown that the intestinal microbiota community is also associated with type 2 diabetes. As highlighted in Table 3, a study in the Chinese population revealed a decrease in the Roseburia and Faecalibacterium species and increase in Escherichia coli in type 2 diabetes patients as compared to nondiabetic control subjects (Qin et al., 2012). Both Roseburia and Faecalibacterium species are SCFAs-producing bacteria and also possess antiinflammatory properties (Zhang et al., 2013; Remely et al., 2014). These microbial-produced SCFA metabolites such as butyrate are important in epigenetic modulations, such as HDAC inhibition (Donohoe et al., 2014). As aforementioned, butyrate also plays an essential role in the differentiation of Treg cells through acetylation of noncoding regions of the Foxp3 locus (Furusawa et al., 2013). Various other studies also investigated the relationship between alterations in gut microbiota in type 2 diabetes and their associated metabolic and epigenetic mechanisms which are detailed in Table 3. Over the past years, it has become increasingly apparent that epigenetic modifications such as DNA methylation are associated with type 2 diabetes. Genome-wide association studies have revealed the correlation of single nucleotide polymorphisms of type 2 diabetes with defect in secretion of insulin, which corresponds to abnormality in pancreatic islet cells (Ruchat et al., 2009). Therefore, DNA methylation levels of CpG sites and the transcriptome in pancreatic islets were analyzed and compared between type 2 diabetes patients and normal individuals (Dayeh et al., 2014). This study detected 1,649 CpG sites and 853 genes, including KCNQ1, TCF7L2, and FTO, with differential DNA methylation in islets from type 2 diabetes patients (Dayeh et al., 2014). The findings also include 102 genes exhibiting differential DNA methylation as well as differential gene expression in islets from type 2 diabetes. Those genes include CDKN1A, PDE7B, SEPT9, and EXOC3L2, which were found to be key genes in regulating insulin secretion in β-cells and glucagon secretion in pancreatic α-cells (Dayeh et al., 2014). This study suggests that epigenetic mechanisms may contribute significantly to the regulation and developmental process of insulin resistance and type 2 diabetes via pancreatic cells.
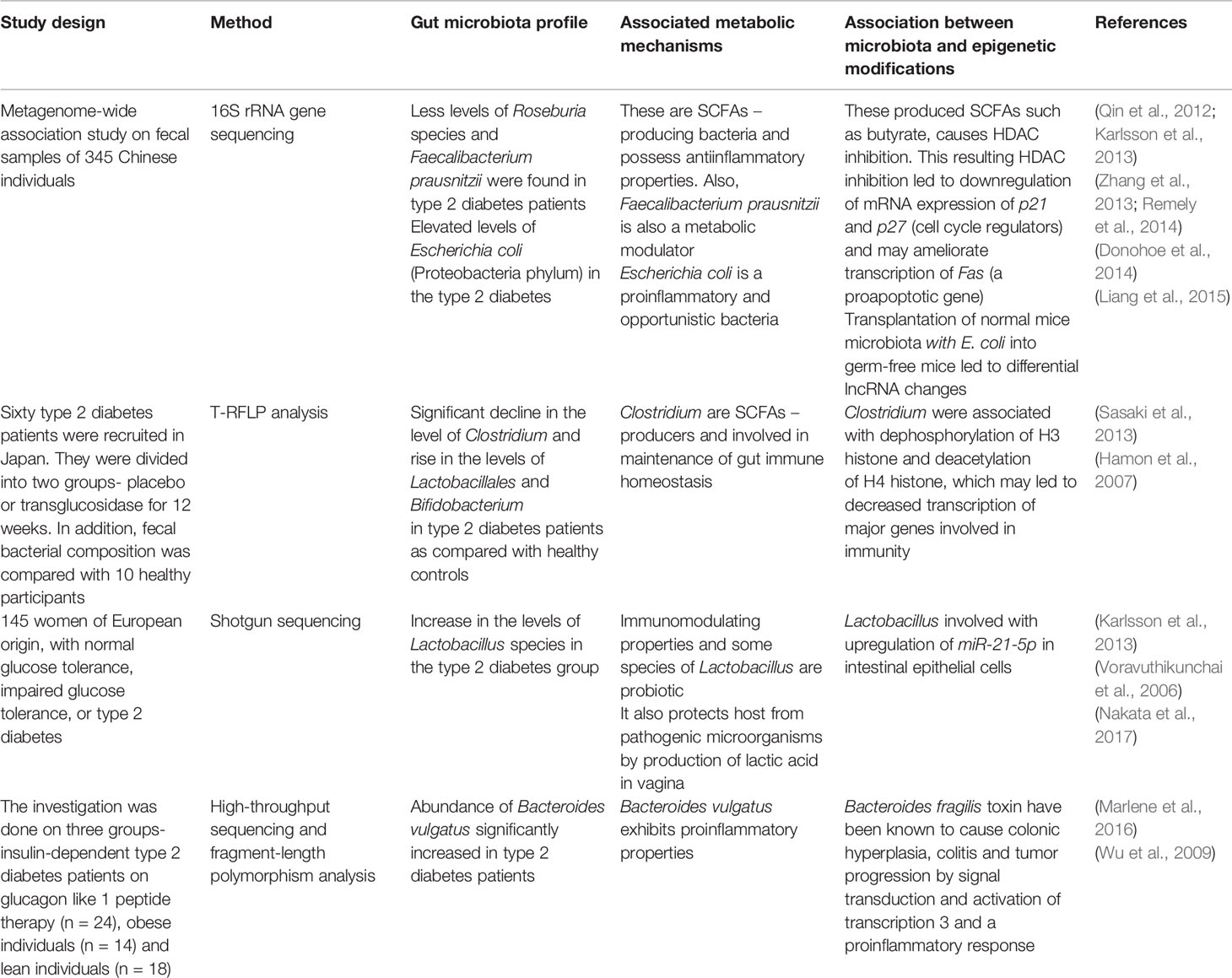
Table 3 Interplay between gut microbiota, metabolic mechanisms, epigenetic modifications, and type 2 diabetes.
The link between the gut microbiome and host metabolism is also shown in Figure 3. The gut microbiome can modulate the host metabolism in multiple ways and may result in insulin resistance and type 2 diabetes. First, with an abundance of gram-negative bacteria such as Escherichia coli, it has been hypothesized that the detachment of lipolysaccharide (LPS), the outer layer of gram-negative bacteria, may induce proinflammation in type 2 diabetes as well as in obesity (Allin et al., 2015). Secreted lipopolysaccharide binds with Toll-like receptor 4 (TLR4) and activates proinflammatory immune pathways, which leads to low-grade inflammation and further decreased insulin sensitivity (Cani et al., 2007). The decline in microbial gene richness was also found to be associated with low-grade inflammation (Cotillard et al., 2013). Second, three main types of SCFAs, butyrate, propionate, and acetate, produced from the fermentation of dietary fibers by gut bacteria, can influence glucose and energy metabolism of the host. Acetate and propionate are known to participate in essential metabolic processes such as glyconeogenesis and lipogenesis in the liver. Butyrate acts as an essential energy substrate for colonic mucosal cells and exhibits a positive effect on insulin sensitivity (Gao et al., 2009). These SCFAs bind to GPR41 and GPR43, leading to various effects depending on the types of cells affected (Brahe et al., 2013). In immune cells, the binding of SCFAs with GPRs results in lesser development of inflammation. However, it contributes to an increase in the secretions of GLP1 and PYY from L-cells (an enteroendocrine cell) in the colon, which improves insulin sensitivity (Lin et al., 2012). It has been demonstrated that enhancing the levels of GLP-1 by altering the composition of the gut microbiota with exposure to antibiotics leads to improved glucose tolerance, insulin resistance and a rise in beneficial metabolites such as succinic acid (Hwang et al., 2015). In addition, SCFAs are found to regulate glucose metabolism by intestinal gluconeogenesis.
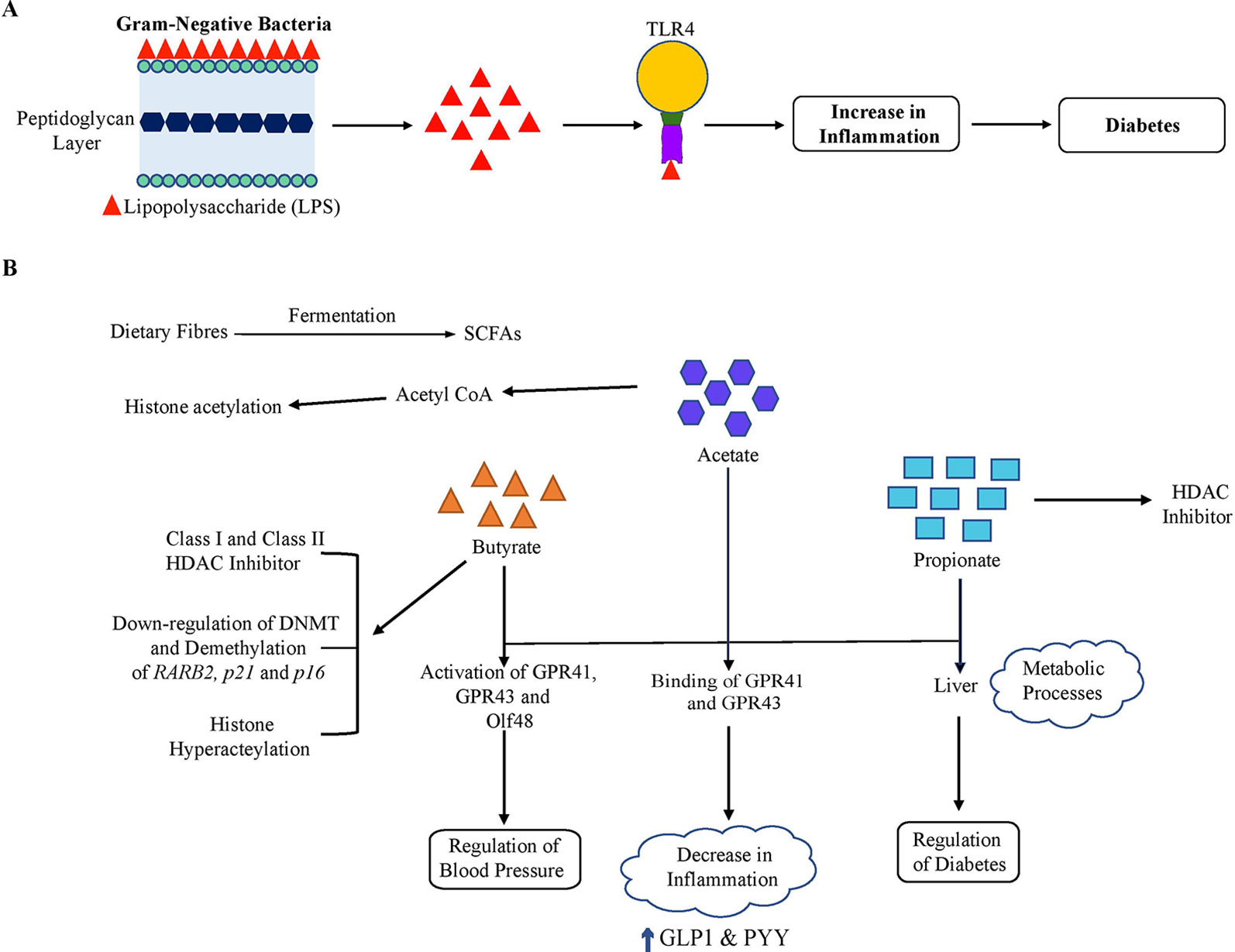
Figure 3 Gut microbiome and host metabolism. The gut microbiome may alter host metabolism through many mechanisms. Of these, two important mechanisms are illustrated. (A) Lipopolysaccharide (LPS). LPS originates from the outer membrane of Gram-negative bacteria and binds to Toll-like receptor 4 (TLR4), which results in low-grade inflammation and thus a decline in insulin sensitivity. (B) Short-chain fatty acids (SCFA). The gut microbiota ferments dietary fibers to SCFAs, including acetate, butyrate, and propionate. Acetate and propionate participate in essential metabolic processes such as gluconeogenesis and lipogenesis in the liver. In addition, SCFAs bind to the G protein-coupled receptors GPR41 and GPR43 resulting in various effects depending on the cellular types affected. In immune cells, this signaling led to a decrease in the inflammation and resulted in an increase in GLP1 and PYY levels in enteroendocrine L-cells, which improve insulin sensitivity overall. Also, these SCFAs activate GPR41, GPR43, and Olfr78 expressed in the kidney. Olfr78 induces SCFA-mediated release of renin which leads to rise in blood pressure. On the other hand, GPR43 resists this change in blood pressure by vasodilatory action. SCFAs are involved in the induction of epigenetic alterations. Butyrate is a known class I and class II HDAC inhibitor. Butyrate can also affect DNA methylation and demethylation of some tumor suppressor genes (RARB2, p21, and p16) and is involved in acetylation of histone H3. Propionate is also a contributor to HDAC inhibition. In addition, acetate has been found to be involved in increasing histone acetylation via transferring an acetyl group from acetyl-CoA.
SCFAs also play a crucial role in the regulation of blood pressure by renin secretion as also shown in Figure 2. The SCFAs induce activation of GPR41, GPR43, and Olfr78, an olfactory receptor expressed in the kidney. Olfr78 also participates in the secretion of renin, induced by SCFA from afferent arterioles that lead to a rise in the blood pressure. High blood pressure is a feature of metabolic syndrome, which includes metabolic alterations such as obesity, hypertension, glucose intolerance, dyslipidemia, diabetes, etc (Schillaci et al., 2004; Lin et al., 2009). However, GPR43 resists this change in blood pressure by vasodilatory action (Pluznick et al., 2013; Pluznick, 2014). In adipocytes, binding of SCFA with GPR43 contributes to enhanced metabolism by restricting fat accumulation in adipose tissue (Kimura et al., 2013). Thus, microbial-produced SCFAs may beneficially regulate glucose metabolism which correlate with a low risk of diabetes and also regulate the blood pressure.
The gut microbiome is regulated by multiple factors such as diet, environment, genetics and epigenetics. Several studies have implicated the interactions of the gut microbiome with the host epigenome, which shows a potential role of the gut microbiome in the regulation of host metabolism. The modulation by the gut microbiome to the host epigenome may be due to direct and frequent contact with the host as well as due to various microbial-derived metabolites produced in the gut. For instance, SCFAs produced in the gut, predominately acetate, butyrate, and propionate, interact with cell surface receptors and with the epithelial and submucosal layers of the colon thereby influencing obesity and diabetes outcome. Although SCFAs are crucial to alter the epigenetic processes of the host through DNA methylation as well as histone modifications, further studies are needed to elucidate the underlying molecular mechanisms and their biological properties in the hosts. For example, which bacterial species have symbiotic relationships, and in what manner do their metabolites participate in specific metabolic processes in humans? Hence, future studies focusing on specific gut microbial metabolites that affect the host epigenome will give new insights into the health and metabolic disease of humans. In addition, the studies focusing on epigenetic mechanisms of the gut microbiome and its influence in obesity and diabetes are emerging. This could hold a promising future in uncovering novel therapeutic mechanisms that may restore the altered intestinal microbiome to a healthy condition and assist in the prevention and treatment of obesity as well as diabetes.
MS and TT conceived of the review article and participated in all drafts of the manuscript. MS wrote the first draft of the manuscript with guidance from TT. YL and MLS participated in editing of several drafts. TT performed final editing and approval of the manuscript. All authors read and approved the final draft.
This work was supported in part by grants from the National Institute of Health (NCI R01CA178441, NCI R01CA204346, NCCIH K01AT009373, and NIDDK P30DK056336).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge generous support from Kayla Lewis and critical comments from Itika Arora and Huixin Wu at the University of Alabama at Birmingham.
SCFA, Short-chain fatty acid; TMA, Trimethylamine; FMO, Flavin monooxygenase; TMAO, Trimethylamine-N-oxide; ncRNA, noncoding RNA; HFD, High fat diet; miRNA, microRNA; siRNA, short-interfering RNA; piRNA, piwi-intreacting RNA; lncRNA, long noncoding RNA; CpG, cytosine-guanine dinucleotide-rich; DNMT, DNA methyltransferase; SAM, S-adenosylmethionine; HAT, Histone acetyltransferase; HDAC, Histone deacetylase; Treg, regulatory T cells; ROS, Reactive oxygen species; LPS, Lipolysaccharide; TLR4, Toll-like receptor 4; GPR, G-protein coupled receptor.
Alkanani, A. K., Hara, N., Gottlieb, P. A., Ir, D., Robertson, C. E., Wagner, B. D., et al. (2015). Alterations in Intestinal Microbiota Correlate With Susceptibility to Type 1 Diabetes. Diabetes 64 (10), 3510–3520. doi: 10.2337/db14-1847
Allin, K. H., Nielsen, T., Pedersen, O. (2015). Mechanisms in endocrinology: Gut microbiota in patients with type 2 diabetes mellitus. Eur. J. Endocrinol. 172 (4), R167–R177. doi: 10.1530/eje-14-0874
Avila, J. G., Echeverri, I., de Plata, C. A., Castillo, A. (2015). Impact of oxidative stress during pregnancy on fetal epigenetic patterns and early origin of vascular diseases. Nutr. Rev. 73 (1), 12–21. doi: 10.1093/nutrit/nuu001
Azad, M. A. K., Sarker, M., Li, T., Yin, J. (2018). Probiotic Species in the Modulation of Gut Microbiota: An Overview. BioMed. Res. Int. 2018, 9478630–9478630. doi: 10.1155/2018/9478630
Baker, J., Chaykin, S. (1960). The biosynthesis of trimethylamine-N-oxide. Biochimica et Biophysica Acta 41 (3), 548–550. doi: 10.1016/0006-3002(60)90062-7
Bernstein, B. E., Meissner, A., Lander, E. S. (2007). The mammalian epigenome. Cell 128 (4), 669–681. doi: 10.1016/j.cell.2007.01.033
Bokulich, N. A., Chung, J., Battaglia, T., Henderson, N., Jay, M., Li, H., et al. (2016). Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 8 (343), 343ra382. doi: 10.1126/scitranslmed.aad7121
Brahe, L. K., Astrup, A., Larsen, L. H. (2013). Is butyrate the link between diet, intestinal microbiota and obesity-related metabolic diseases? Obes. Rev. 14 (12), 950–959. doi: 10.1111/obr.12068
Brown, C. T., Davis-Richardson, A. G., Giongo, A., Gano, K. A., Crabb, D. B., Mukherjee, N., et al. (2011). Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PloS One 6 (10), e25792. doi: 10.1371/journal.pone.0025792
Camilleri, M., Lyle, B. J., Madsen, K. L., Sonnenburg, J., Verbeke, K., Wu, G. D. (2019). Role for diet in normal gut barrier function: developing guidance within the framework of food-labeling regulations. Am. J. Physiol. Gastrointest. Liver Physiol. 317 (1), G17–g39. doi: 10.1152/ajpgi.00063.2019
Cani, P. D., Amar, J., Iglesias, M. A., Poggi, M., Knauf, C., Bastelica, D., et al. (2007). Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56 (7), 1761–1772. doi: 10.2337/db06-1491
Chen, L., Magliano, D. J., Zimmet, P. Z. (2011). The worldwide epidemiology of type 2 diabetes mellitus–present and future perspectives. Nat. Rev. Endocrinol. 8 (4), 228–236. doi: 10.1038/nrendo.2011.183
Claussnitzer, M., Dankel, S. N., Kim, K. H., Quon, G., Meuleman, W., Haugen, C., et al. (2015). FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med. 373 (10), 895–907. doi: 10.1056/NEJMoa1502214
Cotillard, A., Kennedy, S. P., Kong, L. C., Prifti, E., Pons, N., Le Chatelier, E., et al. (2013). Dietary intervention impact on gut microbial gene richness. Nature 500 (7464), 585–588. doi: 10.1038/nature12480
Crider, K. S., Yang, T. P., Berry, R. J., Bailey, L. B. (2012). Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate's role. Adv. Nutr. 3 (1), 21–38. doi: 10.3945/an.111.000992
Davalos, A., Goedeke, L., Smibert, P., Ramirez, C. M., Warrier, N. P., Andreo, U., et al. (2011). miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. U.S.A. 108 (22), 9232–9237. doi: 10.1073/pnas.1102281108
David, L. A., Maurice, C. F., Carmody, R. N., Gootenberg, D. B., Button, J. E., Wolfe, B. E., et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505 (7484), 559–563. doi: 10.1038/nature12820
Dayeh, T., Volkov, P., Salo, S., Hall, E., Nilsson, E., Olsson, A. H., et al. (2014). Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PloS Genet. 10 (3), e1004160. doi: 10.1371/journal.pgen.1004160
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. United States America 107 (33), 14691–14696. doi: 10.1073/pnas.1005963107
Demehri, F. R., Frykman, P. K., Cheng, Z., Ruan, C., Wester, T., Nordenskjold, A., et al. (2016). Altered fecal short chain fatty acid composition in children with a history of Hirschsprung-associated enterocolitis. J. Pediatr. Surg. 51 (1), 81–86. doi: 10.1016/j.jpedsurg.2015.10.012
Dempsey, J., Zhang, A., Cui, J. Y. (2018). Coordinate regulation of long non-coding RNAs and protein-coding genes in germ-free mice. BMC Genomics 19 (1), 834. doi: 10.1186/s12864-018-5235-3
Devaux, C. A., Raoult, D. (2018). The Microbiological Memory, an Epigenetic Regulator Governing the Balance Between Good Health and Metabolic Disorders. Front. In Microbiol. 9, 1379–1379. doi: 10.3389/fmicb.2018.01379
Dominguez-Bello, M. G., Costello, E. K., Contreras, M., Magris, M., Hidalgo, G., Fierer, N., et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. 107 (26), 11971–11975. doi: 10.1073/pnas.1002601107
Donohoe, D. R., Holley, D., Collins, L. B., Montgomery, S. A., Whitmore, A. C., Hillhouse, A., et al. (2014). A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discovery 4 (12), 1387–1397. doi: 10.1158/2159-8290.Cd-14-0501
Eslami, M., Yousefi, B., Kokhaei, P., Hemati, M., Nejad, Z. R., Arabkari, V., et al. (2019). Importance of probiotics in the prevention and treatment of colorectal cancer. J. Cell Physiol. 234 (10), 17127–17143. doi: 10.1002/jcp.28473
Fani, L., Bak, S., Delhanty, P., van Rossum, E. F., van den Akker, E. L. (2014). The melanocortin-4 receptor as target for obesity treatment: a systematic review of emerging pharmacological therapeutic options. Int. J. Obes. (Lond) 38 (2), 163–169. doi: 10.1038/ijo.2013.80
Favier, C. F., Vaughan, E. E., De Vos, W. M., Akkermans, A. D. (2002). Molecular monitoring of succession of bacterial communities in human neonates. Appl. Environ. Microbiol. 68 (1), 219–226. doi: 10.1128/aem.68.1.219-226.2002
Fennema, D., Phillips, I. R., Shephard, E. A. (2016). Trimethylamine and Trimethylamine N-Oxide, a Flavin-Containing Monooxygenase 3 (FMO3)-Mediated Host-Microbiome Metabolic Axis Implicated in Health and Disease. Drug Metab. Dispos. 44 (11), 1839–1850. doi: 10.1124/dmd.116.070615
Franckhauser, S., Muñoz, S., Pujol, A., Casellas, A., Riu, E., Otaegui, P., et al. (2002). Increased Fatty Acid Re-esterification by PEPCK Overexpression in Adipose Tissue Leads to Obesity Without Insulin Resistance. Diabetes 51 (3), 624–630. doi: 10.2337/diabetes.51.3.624
Furusawa, Y., Obata, Y., Fukuda, S., Endo, T. A., Nakato, G., Takahashi, D., et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504 (7480), 446–450. doi: 10.1038/nature12721
Gao, Z., Yin, J., Zhang, J., Ward, R. E., Martin, R. J., Lefevre, M., et al. (2009). Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58 (7), 1509–1517. doi: 10.2337/db08-1637
Gianani, R., Eisenbarth, G. S. (2005). The stages of type 1A diabetes: 2005. Immunol. Rev. 204, 232–249. doi: 10.1111/j.0105-2896.2005.00248.x
Gill, S. R., Pop, M., Deboy, R. T., Eckburg, P. B., Turnbaugh, P. J., Samuel, B. S., et al. (2006). Metagenomic analysis of the human distal gut microbiome. Science 312 (5778), 1355–1359. doi: 10.1126/science.1124234
Giongo, A., Gano, K. A., Crabb, D. B., Mukherjee, N., Novelo, L. L., Casella, G., et al. (2011). Toward defining the autoimmune microbiome for type 1 diabetes. Isme J. 5 (1), 82–91. doi: 10.1038/ismej.2010.92
Gronlund, M. M., Lehtonen, O. P., Eerola, E., Kero, P. (1999). Fecal microflora in healthy infants born by different methods of delivery: permanent changes in intestinal flora after cesarean delivery. J. Pediatr. Gastroenterol. Nutr. 28 (1), 19–25. doi: 10.1097/00005176-199901000-00007
Hamon, M. A., Batsche, E., Regnault, B., Tham, T. N., Seveau, S., Muchardt, C., et al. (2007). Histone modifications induced by a family of bacterial toxins. Proc. Natl. Acad. Sci. U.S.A. 104 (33), 13467–13472. doi: 10.1073/pnas.0702729104
Hori, S., Nomura, T., Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299 (5609), 1057–1061. doi: 10.1126/science.1079490
Hwang, I., Park, Y. J., Kim, Y. R., Kim, Y. N., Ka, S., Lee, H. Y., et al. (2015). Alteration of gut microbiota by vancomycin and bacitracin improves insulin resistance via glucagon-like peptide 1 in diet-induced obesity. FASEB J. 29 (6), 2397–2411. doi: 10.1096/fj.14-265983
Janssen, A. W. F., Houben, T., Katiraei, S., Dijk, W., Boutens, L., van der Bolt, N., et al. (2017). Modulation of the gut microbiota impacts nonalcoholic fatty liver disease: a potential role for bile acids. J. Lipid Res. 58 (7), 1399–1416. doi: 10.1194/jlr.M075713
Karlsson, F. H., Tremaroli, V., Nookaew, I., Bergstrom, G., Behre, C. J., Fagerberg, B., et al. (2013). Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498 (7452), 99–103. doi: 10.1038/nature12198
Kimura, I., Ozawa, K., Inoue, D., Imamura, T., Kimura, K., Maeda, T., et al. (2013). The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 4, 1829. doi: 10.1038/ncomms2852
Koenig, J. E., Spor, A., Scalfone, N., Fricker, A. D., Stombaugh, J., Knight, R., et al. (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. 108 (Supplement 1), 4578–4585. doi: 10.1073/pnas.1000081107
Kok, D. E., Dhonukshe-Rutten, R. A., Lute, C., Heil, S. G., Uitterlinden, A. G., van der Velde, N., et al. (2015). The effects of long-term daily folic acid and vitamin B12 supplementation on genome-wide DNA methylation in elderly subjects. Clin. Epigenet. 7, 121. doi: 10.1186/s13148-015-0154-5
Kopelman, P. G. (2000). Obesity as a medical problem. Nature 404 (6778), 635–643. doi: 10.1038/35007508
Kovacheva, V. P., Mellott, T. J., Davison, J. M., Wagner, N., Lopez-Coviella, I., Schnitzler, A. C., et al. (2007). Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J. Biol. Chem. 282 (43), 31777–31788. doi: 10.1074/jbc.M705539200
Krautkramer, K. A., Kreznar, J. H., Romano, K. A., Vivas, E. I., Barrett-Wilt, G. A., Rabaglia, M. E., et al. (2016). Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 64 (5), 982–992. doi: 10.1016/j.molcel.2016.10.025
Kumar, H., Lund, R., Laiho, A., Lundelin, K., Ley, R. E., Isolauri, E., et al. (2014). Gut microbiota as an epigenetic regulator: pilot study based on whole-genome methylation analysis. MBio 5 (6), e02113-14. doi: 10.1128/mBio.02113-14
Larsen, N., Vogensen, F. K., van den Berg, F. W., Nielsen, D. S., Andreasen, A. S., Pedersen, B. K., et al. (2010). Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PloS One 5 (2), e9085. doi: 10.1371/journal.pone.0009085
Ley, R. E., Turnbaugh, P. J., Klein, S., Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444 (7122), 1022–1023. doi: 10.1038/4441022a
Li, B., Zhao, W., Luo, X., Zhang, X., Li, C., Zeng, C., et al. (2017). Engineering CRISPR-Cpf1 crRNAs and mRNAs to maximize genome editing efficiency. Nat. BioMed. Eng. 1 (5), 0066. doi: 10.1038/s41551-017-0066
Liang, L., Ai, L., Qian, J., Fang, J. Y., Xu, J. (2015). Long noncoding RNA expression profiles in gut tissues constitute molecular signatures that reflect the types of microbes. Sci. Rep. 5, 11763. doi: 10.1038/srep11763
Lieber, A. D., Beier, U. H., Xiao, H., Wilkins, B. J., Jiao, J., Li, X. S., et al. (2018). Loss of HDAC6 alters gut microbiota and worsens obesity. FASEB J. 33, fj201701586R. doi: 10.1096/fj.201701586R
Lin, C.-C., Liu, C.-S., Li, C.-I., Lin, W.-Y., Lai, M.-M., Lin, T., et al. (2009). The relation of metabolic syndrome according to five definitions to cardiovascular risk factors–a population-based study. BMC Public Health 9, 484–484. doi: 10.1186/1471-2458-9-484
Lin, H. V., Frassetto, A., Kowalik, E. J., Jr., Nawrocki, A. R., Lu, M. M., Kosinski, J. R., et al. (2012). Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PloS One 7 (4), e35240. doi: 10.1371/journal.pone.0035240
Liu, L., He, X., Zhao, M., Yang, S., Wang, S., Yu, X., et al. (2019). Regulation of DNA methylation and 2-OG/TET signaling by choline alleviated cardiac hypertrophy in spontaneously hypertensive rats. J. Mol. Cell Cardiol. 128, 26–37. doi: 10.1016/j.yjmcc.2019.01.011
Lofgren, J. L., Whary, M. T., Ge, Z., Muthupalani, S., Taylor, N. S., Mobley, M., et al. (2011). Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 140 (1), 210–220. doi: 10.1053/j.gastro.2010.09.048
Lu, Y., Fan, C., Li, P., Lu, Y., Chang, X., Qi, K. (2016). Short Chain Fatty Acids Prevent High-fat-diet-induced Obesity in Mice by Regulating G Protein-coupled Receptors and Gut Microbiota. Sci. Rep. 6, 37589–37589. doi: 10.1038/srep37589
Lukovac, S., Belzer, C., Pellis, L., Keijser, B. J., de Vos, W. M., Montijn, R. C., et al. (2014). Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. mBio 5 (4), e01438–e01414. doi: 10.1128/mBio.01438-14
Maeda, Y., Takeda, K. (2017). Role of Gut Microbiota in Rheumatoid Arthritis. J. Clin. Med. 6 (6), 60. doi: 10.3390/jcm6060060
Mahmoud, A. M., Ali, M. M. (2019). Methyl Donor Micronutrients that Modify DNA Methylation and Cancer Outcome. Nutrients 11 (3), 608. doi: 10.3390/nu11030608
Mahowald, M. A., Rey, F. E., Seedorf, H., Turnbaugh, P. J., Fulton, R. S., Wollam, A., et al. (2009). Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc. Natl. Acad. Sci. U.S.A. 106 (14), 5859–5864. doi: 10.1073/pnas.0901529106
Marlene, R., Berit, H., Julia, Z., Eva, A., Helmuth, B., Alexander, G. H. (2016). Gut Microbiota of Obese, Type 2 Diabetic Individuals is Enriched in Faecalibacterium prausnitzii, Akkermansia muciniphila and Peptostreptococcus anaerobius after Weight Loss. Endocr. Metab. Immune Disord. - Drug Targets 16 (2), 99–106. doi: 10.2174/1871530316666160831093813
Marlicz, W., Skonieczna-Zydecka, K., Dabos, K. J., Loniewski, I., Koulaouzidis, A. (2018). Emerging concepts in non-invasive monitoring of Crohn's disease. Ther. Adv. Gastroenterol. 11, 1756284818769076. doi: 10.1177/1756284818769076
Marques-Vidal, P., Bochud, M., Bastardot, F., Lüscher, T., Ferrero, F., Gaspoz, J. M., et al. (2012). Association between Inflammatory and Obesity Markers in a Swiss Population-Based Sample (CoLaus Study). Obes. Facts 5 (5), 734–744. doi: 10.1159/000345045
Mehta, R. S., Abu-Ali, G. S., Drew, D. A., Lloyd-Price, J., Subramanian, A., Lochhead, P., et al. (2018). Stability of the human faecal microbiome in a cohort of adult men. Nat. Microbiol. 3 (3), 347–355. doi: 10.1038/s41564-017-0096-0
Mendez-Salazar, E. O., Ortiz-Lopez, M. G., Granados-Silvestre, M. L. A., Palacios-Gonzalez, B., Menjivar, M. (2018). Altered Gut Microbiota and Compositional Changes in Firmicutes and Proteobacteria in Mexican Undernourished and Obese Children. Front. Microbiol. 9, 2494. doi: 10.3389/fmicb.2018.02494
Mischke, M., Plosch, T. (2013). More than just a gut instinct-the potential interplay between a baby's nutrition, its gut microbiome, and the epigenome. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304 (12), R1065–R1069. doi: 10.1152/ajpregu.00551.2012
Mojica, L., de Mejia, E. G. (2015). Characterization and Comparison of Protein and Peptide Profiles and their Biological Activities of Improved Common Bean Cultivars (Phaseolus vulgaris L.) from Mexico and Brazil. Plant Foods Hum. Nutr. 70 (2), 105–112. doi: 10.1007/s11130-015-0477-6
Mu, Q., Kirby, J., Reilly, C. M., Luo, X. M. (2017). Leaky Gut As a Danger Signal for Autoimmune Diseases. Front. In Immunol. 8, 598–598. doi: 10.3389/fimmu.2017.00598
Murri, M., Leiva, I., Gomez-Zumaquero, J. M., Tinahones, F. J., Cardona, F., Soriguer, F., et al. (2013). Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 11, 46. doi: 10.1186/1741-7015-11-46
Nagpal, R., Tsuji, H., Takahashi, T., Kawashima, K., Nagata, S., Nomoto, K., et al. (2016). Sensitive Quantitative Analysis of the Meconium Bacterial Microbiota in Healthy Term Infants Born Vaginally or by Cesarean Section. Front. In Microbiol. 7, 1997–1997. doi: 10.3389/fmicb.2016.01997
Nagpal, R., Tsuji, H., Takahashi, T., Nomoto, K., Kawashima, K., Nagata, S., et al. (2017). Gut dysbiosis following C-section instigates higher colonisation of toxigenic Clostridium perfringens in infants. Benef. Microbes 8 (3), 353–365. doi: 10.3920/bm2016.0216
Nakata, K., Sugi, Y., Narabayashi, H., Kobayakawa, T., Nakanishi, Y., Tsuda, M., et al. (2017). Commensal microbiota-induced microRNA modulates intestinal epithelial permeability through the small GTPase ARF4. J. Biol. Chem. 292 (37), 15426–15433. doi: 10.1074/jbc.M117.788596
Neu, J., Pammi, M. (2018). Necrotizing enterocolitis: The intestinal microbiome, metabolome and inflammatory mediators. Semin. Fetal Neonatal Med. 23 (6), 400–405. doi: 10.1016/j.siny.2018.08.001
Nicholson, J. K., Holmes, E., Kinross, J., Burcelin, R., Gibson, G., Jia, W., et al. (2012). Host-gut microbiota metabolic interactions. Science 336 (6086), 1262–1267. doi: 10.1126/science.1223813
Nishimura, S., Manabe, I., Nagasaki, M., Eto, K., Yamashita, H., Ohsugi, M., et al. (2009). CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 15 (8), 914–920. doi: 10.1038/nm.1964
Noren, T. (2010). Clostridium difficile and the disease it causes. Methods Mol. Biol. 646, 9–35. doi: 10.1007/978-1-60327-365-7_2
Orouji, E., Utikal, J. (2018). Tackling malignant melanoma epigenetically: histone lysine methylation. Clin. Epigenet. 10 (1), 145. doi: 10.1186/s13148-018-0583-z
Pascal, V., Pozuelo, M., Borruel, N., Casellas, F., Campos, D., Santiago, A., et al. (2017). A microbial signature for Crohn's disease. Gut 66 (5), 813–822. doi: 10.1136/gutjnl-2016-313235
Paul, B., Barnes, S., Demark-Wahnefried, W., Morrow, C., Salvador, C., Skibola, C., et al. (2015). Influences of diet and the gut microbiome on epigenetic modulation in cancer and other diseases. Clin. Epigenet. 7, 112. doi: 10.1186/s13148-015-0144-7
Philippe, D., Favre, L., Foata, F., Adolfsson, O., Perruisseau-Carrier, G., Vidal, K., et al. (2011). Bifidobacterium lactis attenuates onset of inflammation in a murine model of colitis. World J. Gastroenterol. 17 (4), 459–469. doi: 10.3748/wjg.v17.i4.459
Pluznick, J. L., Protzko, R. J., Gevorgyan, H., Peterlin, Z., Sipos, A., Han, J., et al. (2013). Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. U.S.A. 110 (11), 4410–4415. doi: 10.1073/pnas.1215927110
Pluznick, J. (2014). A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut. Microbes 5 (2), 202–207. doi: 10.4161/gmic.27492
Qi, Q., Downer, M. K., Kilpelainen, T. O., Taal, H. R., Barton, S. J., Ntalla, I., et al. (2015). Dietary Intake, FTO Genetic Variants, and Adiposity: A Combined Analysis of Over 16,000 Children and Adolescents. Diabetes 64 (7), 2467–2476. doi: 10.2337/db14-1629
Qi, C.-J., Zhang, Q., Yu, M., Xu, J.-P., Zheng, J., Wang, T., et al. (2016). Imbalance of Fecal Microbiota at Newly Diagnosed Type 1 Diabetes in Chinese Children. Chin. Med. J. 129 (11), 1298–1304. doi: 10.4103/0366-6999.182841
Qian, H., Chen, Y., Nian, Z., Su, L., Yu, H., Chen, F. J., et al. (2017). HDAC6-mediated acetylation of lipid droplet-binding protein CIDEC regulates fat-induced lipid storage. J. Clin. Invest. 127 (4), 1353–1369. doi: 10.1172/jci85963
Qin, Y., Wade, P. A. (2018). Crosstalk between the microbiome and epigenome: messages from bugs. J. Biochem. 163 (2), 105–112. doi: 10.1093/jb/mvx080
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464 (7285), 59–65. doi: 10.1038/nature08821
Qin, J., Li, Y., Cai, Z., Li, S., Zhu, J., Zhang, F., et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490 (7418), 55–60. doi: 10.1038/nature11450
Ramos-Molina, B., Sanchez-Alcoholado, L., Cabrera-Mulero, A., Lopez-Dominguez, R., Carmona-Saez, P., Garcia-Fuentes, E., et al. (2019). Gut Microbiota Composition Is Associated With the Global DNA Methylation Pattern in Obesity. Front. Genet. 10, 613. doi: 10.3389/fgene.2019.00613
Rea, D., Coppola, G., Palma, G., Barbieri, A., Luciano, A., Del Prete, P., et al. (2018). Microbiota effects on cancer: from risks to therapies. Oncotarget 9 (25), 17915–17927. doi: 10.18632/oncotarget.24681
Remely, M., Aumueller, E., Jahn, D., Hippe, B., Brath, H., Haslberger, A. G. (2014). Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benef. Microbes 5 (1), 33–43. doi: 10.3920/bm2013.006
Romano, K. A., Rey, F. E. (2018). Is maternal microbial metabolism an early-life determinant of health? Lab. Anim. (NY) 47 (9), 239–243. doi: 10.1038/s41684-018-0129-1
Romano, K. A., Vivas, E. I., Amador-Noguez, D., Rey, F. E. (2015). Intestinal Microbiota Composition Modulates Choline Bioavailability from Diet and Accumulation of the Proatherogenic Metabolite Trimethylamine-N-Oxide. mBio 6 (2), e02481–e02414. doi: 10.1128/mBio.02481-14
Romano, K. A., Martinez-Del Campo, A., Kasahara, K., Chittim, C. L., Vivas, E. I., Amador-Noguez, D., et al. (2017). Metabolic, Epigenetic, and Transgenerational Effects of Gut Bacterial Choline Consumption. Cell Host Microbe 22 (3), 279–290.e277. doi: 10.1016/j.chom.2017.07.021
Romano-Keeler, J., Weitkamp, J. H., Moore, D. J. (2012). Regulatory properties of the intestinal microbiome effecting the development and treatment of diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 19 (2), 73–80. doi: 10.1097/MED.0b013e3283514d43
Rossi, M., Amaretti, A., Raimondi, S. (2011). Folate production by probiotic bacteria. Nutrients 3 (1), 118–134. doi: 10.3390/nu3010118
Roth, S. Y., Denu, J. M., Allis, C. D. (2001). Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120. doi: 10.1146/annurev.biochem.70.1.81
Rovite, V., Petrovska, R., Vaivade, I., Kalnina, I., Fridmanis, D., Zaharenko, L., et al. (2014). The role of common and rare MC4R variants and FTO polymorphisms in extreme form of obesity. Mol. Biol. Rep. 41 (3), 1491–1500. doi: 10.1007/s11033-013-2994-4
Ruchat, S. M., Elks, C. E., Loos, R. J., Vohl, M. C., Weisnagel, S. J., Rankinen, T., et al. (2009). Association between insulin secretion, insulin sensitivity and type 2 diabetes susceptibility variants identified in genome-wide association studies. Acta Diabetol. 46 (3), 217–226. doi: 10.1007/s00592-008-0080-5
Samad, A., Stoll, M. L., Lavi, I., Hsu, J. J., Strand, V., Robinson, T. N., et al. (2018). Adiposity in Juvenile Psoriatic Arthritis. J. Rheumatol. 45 (3), 411. doi: 10.3899/jrheum.170598
Sarkar, S., Abujamra, A. L., Loew, J. E., Forman, L. W., Perrine, S. P., Faller, D. V. (2011). Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 31 (9), 2723–2732.
Sasaki, M., Ogasawara, N., Funaki, Y., Mizuno, M., Iida, A., Goto, C., et al. (2013). Transglucosidase improves the gut microbiota profile of type 2 diabetes mellitus patients: a randomized double-blind, placebo-controlled study. BMC Gastroenterol. 13, 81. doi: 10.1186/1471-230x-13-81
Schillaci, G., Pirro, M., Vaudo, G., Gemelli, F., Marchesi, S., Porcellati, C., et al. (2004). Prognostic value of the metabolic syndrome in essential hypertension. J. Am. Coll. Cardiol. 43 (10), 1817–1822. doi: 10.1016/j.jacc.2003.12.049
Schlaeppi, K., Bulgarelli, D. (2015). The plant microbiome at work. Mol. Plant Microbe Interact. 28 (3), 212–217. doi: 10.1094/mpmi-10-14-0334-fi
Schroeder, B. O., Backhed, F. (2016). Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 22 (10), 1079–1089. doi: 10.1038/nm.4185
Sender, R., Fuchs, S., Milo, R. (2016). Revised Estimates for the Number of Human and Bacteria Cells in the Body. PloS Biol. 14 (8), e1002533. doi: 10.1371/journal.pbio.1002533
Soliman, M. L., Rosenberger, T. A. (2011). Acetate supplementation increases brain histone acetylation and inhibits histone deacetylase activity and expression. Mol. Cell Biochem. 352 (1-2), 173–180. doi: 10.1007/s11010-011-0751-3
Stewart, C. J., Ajami, N. J., O'Brien, J. L., Hutchinson, D. S., Smith, D. P., Wong, M. C., et al. (2018). Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562 (7728), 583–588. doi: 10.1038/s41586-018-0617-x
Strozzi, G. P., Mogna, L. (2008). Quantification of folic acid in human feces after administration of Bifidobacterium probiotic strains. J. Clin. Gastroenterol. 42 (Suppl 3 Pt 2), S179–S184. doi: 10.1097/MCG.0b013e31818087d8
Sun, X., Jiao, X., Ma, Y., Liu, Y., Zhang, L., He, Y., et al. (2016). Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 481 (1-2), 63–70. doi: 10.1016/j.bbrc.2016.11.017
Tang, W. H., Hazen, S. L. (2017). Microbiome, trimethylamine N-oxide, and cardiometabolic disease. Transl. Res. 179, 108–115. doi: 10.1016/j.trsl.2016.07.007
Tateishi, K., Okada, Y., Kallin, E. M., Zhang, Y. (2009). Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 458 (7239), 757–761. doi: 10.1038/nature07777
Trajkovski, M., Hausser, J., Soutschek, J., Bhat, B., Akin, A., Zavolan, M., et al. (2011). MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 474 (7353), 649–653. doi: 10.1038/nature10112
Tsuji, H., Oozeer, R., Matsuda, K., Matsuki, T., Ohta, T., Nomoto, K., et al. (2012). Molecular monitoring of the development of intestinal microbiota in Japanese infants. Benef. Microbes 3 (2), 113–125. doi: 10.3920/bm2011.0038
Turnbaugh, P. J., Gordon, J. I. (2009). The core gut microbiome, energy balance and obesity. J. Physiol. 587 (Pt 17), 4153–4158. doi: 10.1113/jphysiol.2009.174136
Vahamiko, S., Laiho, A., Lund, R., Isolauri, E., Salminen, S., Laitinen, K. (2019). The impact of probiotic supplementation during pregnancy on DNA methylation of obesity-related genes in mothers and their children. Eur. J. Nutr. 58 (1), 367–377. doi: 10.1007/s00394-017-1601-1
Velasquez, M. T., Ramezani, A., Manal, A., Raj, D. S. (2016). Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins (Basel) 8 (11), 326. doi: 10.3390/toxins8110326
Virtue, A. T., McCright, S. J., Wright, J. M., Jimenez, M. T., Mowel, W. K., Kotzin, J. J., et al. (2019). The gut microbiota regulates white adipose tissue inflammation and obesity via a family of microRNAs. Sci. Transl. Med. 11 (496), eaav1892. doi: 10.1126/scitranslmed.aav1892
Voravuthikunchai, S. P., Bilasoi, S., Supamala, O. (2006). Antagonistic activity against pathogenic bacteria by human vaginal lactobacilli. Anaerobe 12 (5), 221–226. doi: 10.1016/j.anaerobe.2006.06.003
Wang, L., Liu, Y., Han, R., Beier, U. H., Bhatti, T. R., Akimova, T., et al. (2015). FOXP3+ regulatory T cell development and function require histone/protein deacetylase 3. J. Clin. Invest. 125 (3), 1111–1123. doi: 10.1172/jci77088
Wang, J., Wang, P., Li, D., Hu, X., Chen, F. (2019a). Beneficial effects of ginger on prevention of obesity through modulation of gut microbiota in mice. Eur. J. Nutr. doi: 10.1007/s00394-019-01938-1
Wang, X., Wang, W., Wang, L., Yu, C., Zhang, G., Zhu, H., et al. (2019b). Lentinan modulates intestinal microbiota and enhances barrier integrity in a piglet model challenged with lipopolysaccharide. Food Funct. 10 (1), 479–489. doi: 10.1039/c8fo02438c
Whitt, J., Woo, V., Lee, P., Moncivaiz, J., Haberman, Y., Denson, L., et al. (2018). Disruption of Epithelial HDAC3 in Intestine Prevents Diet-Induced Obesity in Mice. Gastroenterology 155 (2), 501–513. doi: 10.1053/j.gastro.2018.04.017
Wu, S., Rhee, K. J., Albesiano, E., Rabizadeh, S., Wu, X., Yen, H. R., et al. (2009). A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15 (9), 1016–1022. doi: 10.1038/nm.2015
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486 (7402), 222–227. doi: 10.1038/nature11053
Yoshimoto, S., Loo, T. M., Atarashi, K., Kanda, H., Sato, S., Oyadomari, S., et al. (2013). Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499 (7456), 97–101. doi: 10.1038/nature12347
Yu, T., Guo, F., Yu, Y., Sun, T., Ma, D., Han, J., et al. (2017). Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 170 (3), 548–563.e516. doi: 10.1016/j.cell.2017.07.008
Yuille, S., Reichardt, N., Panda, S., Dunbar, H., Mulder, I. E. (2018). Human gut bacteria as potent class I histone deacetylase inhibitors in vitro through production of butyric acid and valeric acid. PloS One 13 (7), e0201073. doi: 10.1371/journal.pone.0201073
Zeisel, S. (2017). Choline, Other Methyl-Donors and Epigenetics. Nutrients 9 (5), 445. doi: 10.3390/nu9050445
Zhang, X., Shen, D., Fang, Z., Jie, Z., Qiu, X., Zhang, C., et al. (2013). Human Gut Microbiota Changes Reveal the Progression of Glucose Intolerance. PloS One 8 (8), e71108. doi: 10.1371/journal.pone.0071108
Zhang, X., Zhao, Y., Xu, J., Xue, Z., Zhang, M., Pang, X., et al. (2015). Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Sci. Rep. 5, 14405. doi: 10.1038/srep14405
Zhang, S., Yang, J., Henning, S. M., Lee, R., Hsu, M., Grojean, E., et al. (2017). Dietary pomegranate extract and inulin affect gut microbiome differentially in mice fed an obesogenic diet. Anaerobe 48, 184–193. doi: 10.1016/j.anaerobe.2017.08.017
Keywords: epigenetic, gut microbiome, diet, metabolic, obesity, diabetes
Citation: Sharma M, Li Y, Stoll ML and Tollefsbol TO (2020) The Epigenetic Connection Between the Gut Microbiome in Obesity and Diabetes. Front. Genet. 10:1329. doi: 10.3389/fgene.2019.01329
Received: 11 February 2019; Accepted: 05 December 2019;
Published: 15 January 2020.
Edited by:
Andrea Masotti, Bambino Gesù Children Hospital (IRCCS), ItalyReviewed by:
Clarissa Gerhauser, German Cancer Research Center (DKFZ), GermanyCopyright © 2020 Sharma, Li, Stoll and Tollefsbol. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Trygve O. Tollefsbol, dHJ5Z3ZlQHVhYi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.