
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 10 January 2020
Sec. Evolutionary and Population Genetics
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.01293
 Michel David Bohórquez1,2†
Michel David Bohórquez1,2† Diego Ordoñez3,4†
Diego Ordoñez3,4† Carlos Fernando Suárez2,5
Carlos Fernando Suárez2,5 Belén Vicente6
Belén Vicente6 Carmen Vieira6
Carmen Vieira6 Julio López-Abán6
Julio López-Abán6 Antonio Muro6Iván Ordóñez2
Antonio Muro6Iván Ordóñez2 Manuel Alfonso Patarroyo2,5*
Manuel Alfonso Patarroyo2,5*Bovine leukocyte antigens (BoLA) have been used as disease markers and immunological traits in cattle due to their primary role in pathogen recognition by the immune system. A higher MHC allele diversity in a population will allow presenting a broader peptide repertoire. However, loss of overall diversity due to domestication process can decrease a population’s peptide repertoire. Within the context of zebu and taurine cattle populations, BoLA-DRB3 genetic diversity in Spanish Morucha and Colombian Normande cattle was analyzed and an approach to estimate functional diversity was performed. Sequence-based typing was used for identifying 29, 23, 27, and 28 alleles in Spanish Morucha, Nariño-, Boyacá-, and Cundinamarca-Normande cattle, respectively. These breeds had remarkably low heterozygosity levels and the Hardy-Weinberg principle revealed significant heterozygote deficiency. FST and DA genetic distance showed that Colombian Normande populations had greater variability than other phenotypically homogeneous breeds, such as Holstein. It was also found that Spanish Morucha cattle were strongly differentiated from other cattle breeds. Spanish Morucha had greater divergence in the peptide-binding region regarding other cattle breeds. However, peptide-binding region covariation indicated that the potential peptide repertoire seemed equivalent among cattle breeds. Despite the genetic divergence observed, the extent of the potential peptide repertoire in the cattle populations studied appears to be similar and thus their pathogen recognition potential should be equivalent, suggesting that functional diversity might persist in the face of bottlenecks imposed by domestication and breeding.
The major histocompatibility complex (MHC) is a primary component of the adaptive immune system that offers a unique alternative for addressing both immunological and evolutionary biological issues. The MHC consists of a group of loci in jawed vertebrates encoding molecules which are fundamental for regulating the immune response (Klein et al., 2007). Class I and class II genes comprise one subset of these loci encoding the cell surface glycoproteins necessary for T-lymphocytes to recognize antigenic peptides on cell surfaces (Rock et al., 2016). While class I molecules are expressed by all nucleated cells and present peptides from intracellular proteins to CD8+ T-cells, class II molecules are expressed by professional antigen-presenting cells and present peptides derived from extracellular proteins to CD4+ T-cells, thereby playing a key role in defence against pathogens (Klein et al., 2007; Neefjes et al., 2011).
The MHC in cattle (also known as bovine leukocyte antigen-BoLA) is located on chromosome 23 and has the overall structure characterizing other mammals’ MHC (Takeshima and Aida, 2006). DR and DQ genes encode the molecules within the BoLA class II region that bind the peptides which will be presented to T-lymphocytes, representing the main class II restriction elements for CD4+ T-cells. The BoLA-DQ region comprises DQA and DQB loci, which may vary in number depending on the haplotype; this enables additional diversity by means of intrahaplotype and interhaplotype pairing of DQA and DQB molecules (Andersson and Rask, 1988; Glass et al., 2000; Norimine and Brown, 2005). BoLA-DR consists of the monomorphic BoLA-DRA locus and three DRB loci, of which the BoLA-DRB3 gene is the only one known to be fully functional (Burke et al., 1991).
BoLA-DRB3 is the most polymorphic bovine MHC gene; 136 different alleles have been reported to date (Maccari et al., 2017). Such polymorphisms are mainly located in the β1 domain’s peptide-binding region (PBR), which is encoded by exon 2 and has been used for defining BoLA-DRB3 alleles (Sigurdardottir et al., 1991). Because the amino acids (aa) forming the PBR determine MHC-presented peptides’ binding affinity, different alleles will bind a different repertoire of peptides, thus influencing immune response variability (Baxter et al., 2009; van Deutekom and Kesmir, 2015). Similarities between BoLA-DRB3 PBR are used as criteria for developing in silico pan-specific methods for predicting MHC–peptide-binding affinity (Nielsen et al., 2008; Hoof et al., 2009). BoLA-DRB3 divergence can be used for estimating the size of peptide-binding repertoire. Thus, individuals or populations having highly divergent BoLA-DRB3 alleles will have a broader peptide-binding repertoire than those having very similar alleles (Klein et al., 2007; Lenz et al., 2013). Furthermore, peptide-binding differences among populations could be estimated by correlating variation patterns among their BoLA-DRB3 allele repertoires.
Ascertaining cattle’s BoLA-DRB3 allele frequency distribution in different regions worldwide has been used for executing infectious disease control programmes (Maillard et al., 2003) and can be applied to developing vaccines having a wider range of protection (Patarroyo et al., 2011). Different BoLA-DRB3 alleles have been associated with variations in susceptibility to infectious diseases (Dietz et al., 1997; Acosta-Rodriguez et al., 2005; Martinez et al., 2006; Nascimento et al., 2006; Kulberg et al., 2007; Juliarena et al., 2008; Nikbakht et al., 2016; Carignano et al., 2017), vaccine responses (Garcia-Briones et al., 2000; Rupp et al., 2007; Baxter et al., 2009; Gowane et al., 2013) and production traits (Sharif et al., 1999). BoLA-DRB3 genetic diversity has been characterized in both widespread and autochthonous creole cattle breeds (Takeshima et al., 2001; Takeshima et al., 2002; Takeshima et al., 2003; Baxter et al., 2008; Wang et al., 2008; Giovambattista et al., 2013; Takeshima et al., 2014; Takeshima et al., 2015a).
MHC diversity is of great interest for breeders, population geneticists and evolutionary biologists, considering that different degrees and MHC variability patterns reflect evolutionary processes such as adaptation, selection (natural, sexual or artificial) and drift within and between populations (Goszczynski et al., 2014; Takeshima et al., 2014). Several studies have shown that decreased MHC variability might be caused by population bottlenecks (Bollmer et al., 2007; Bollmer et al., 2011; Mason et al., 2011; Zhang et al., 2016). On the contrary, a high level of diversity could be maintained by balancing selection driven by pathogens or other mechanisms due to MHC function regarding pathogen recognition, despite extreme population bottlenecks (Edwards and Hedrick, 1998; Garrigan and Hedrick, 2001; Aguilar et al., 2004; Borg et al., 2011; Moutou et al., 2013; Newhouse and Balakrishnan, 2015). Although functional polymorphism may be narrower than genetic polymorphism in pigs (Moutou et al., 2013), most studies on domestic animals’ MHC have focused on genetic polymorphism; however, it remains unknown whether bottlenecks associated with domestication and breeding could have reduced MHC functional diversity.
Despite advances in characterizing BoLA-DRB3 genetic diversity in cattle, information about allele frequency is not available for some breeds (only for a few out of over 800 breeds of cattle recognized worldwide via sequence-based typing) (Takeshima et al., 2018). The few MHC alleles in domestic animals contrasts with those of other species (Nino-Vasquez et al., 2000; Suarez et al., 2006; Lopez et al., 2014; Maccari et al., 2017; Shiina et al., 2017). However, a smaller amount of alleles does not necessarily mean reduced functionality, in fact two species can have very similar functional repertoire size despite varying regarding the amount of alleles (Suarez et al., 2017).
This study has thus been aimed at determining whether domestic cattle breeding has decreased actual MHC variability. MHC diversity of two new breeds has been characterized. The Normande breed was chosen out of convenience. Spanish Morucha, a breed having relatively low human intervention, has been used here as an approach to what could be considered a natural cattle population. BoLA-DRB3 genetic diversity, structure, differentiation, and selective pressure in Colombian Normande and Spanish Morucha breeds was characterized and compared to worldwide taurine and zebu breeds. Each cattle breed’s BoLA-DRB3 PBR variability and covariation was then analyzed as an approach to compare potential peptide-binding repertoire size and indication of functional diversity. Genetic divergence patterns based on population genetics and PBR variability analysis were similar to that observed for functional divergence; however, the potential peptide-binding repertoire was very similar for all breeds. These results provide an insight into MHC evolution and contribute toward efforts at describing bovine MHC diversity according to breed and location.
Whole blood was collected aseptically from the coccygeal vein of 165 Normande and Morucha cattle. Considering that Colombian and Spanish cattle farming is characterized by extensive livestock production systems having a relatively few animals per herd, phenotypically purebred animals were sampled from the regions having the greatest amount of purebred cattle in Colombia (Normande) and Spain (Morucha), sampling the most representative amount of farms from each region (Table 1). The herds and purebred animals analyzed were sampled randomly, avoiding related individuals/farms for each region. Normande cattle samples came from Colombia’s Nariño (n = 30), Cundinamarca (n = 41), and Boyacá departments (n = 40). Morucha cattle samples (n = 54) came from Spain’s Salamanca province. A PureLink Genomic DNA Mini Kit (Invitrogen, Carlsbad, CA, USA) was used for extracting genomic DNA (gDNA), following the manufacturer’s instructions. Previously reported data regarding further 17 taurine and zebu populations from Asia and South America was also included for comparison (Takeshima et al., 2003; Giovambattista et al., 2013; Takeshima et al., 2015a; Takeshima et al., 2015b; Takeshima et al., 2018). Table 1 gives general information about the populations analyzed. The Universidad de Ciencias Aplicadas y Ambientales (UDCA, Bogotá) Research Ethics Committee (Minute No. 201901) approved this study.
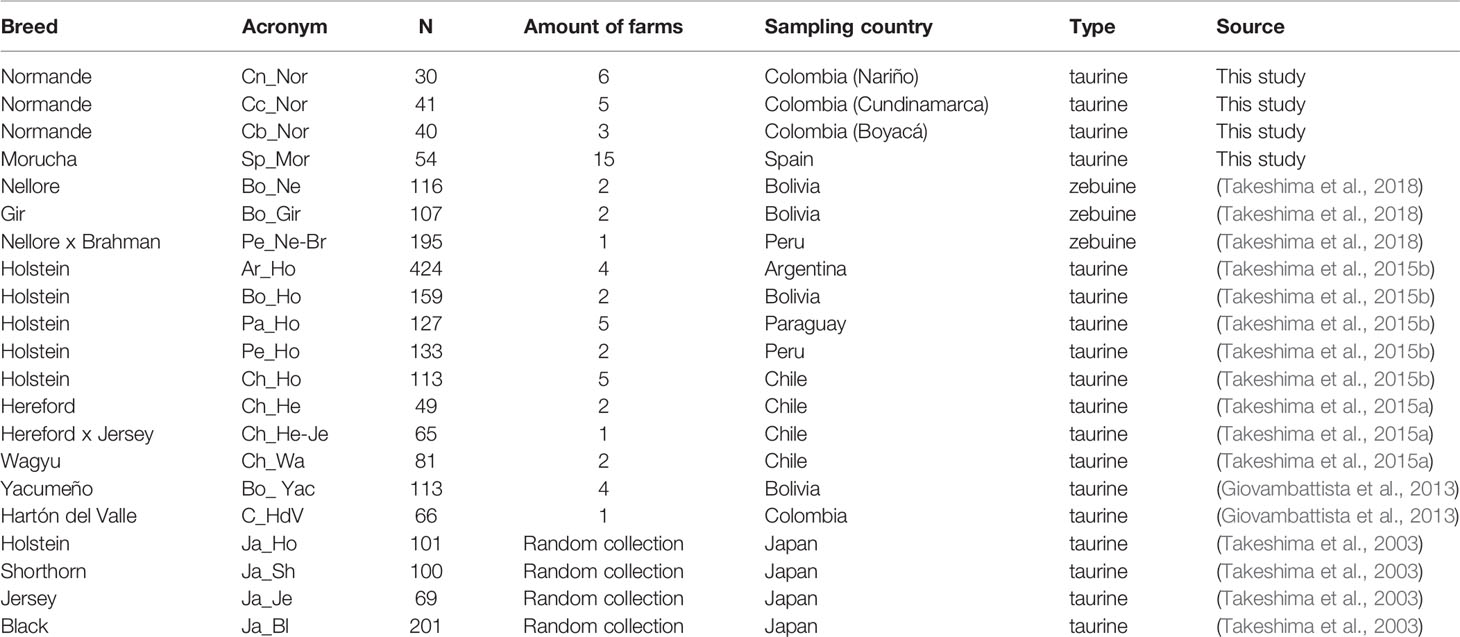
Table 1 General information regarding the 21 cattle populations analyzed in this study.
Sequence-based typing was used for identifying BoLA-DRB3 alleles. The BoLA-DRB3 gene’s exon 2 was amplified by polymerase chain reaction (PCR), using a high-fidelity polymerase along with DRB3F and DRB3R primers (Ledwidge et al., 2001). The PCR reaction mixture contained 1X Pfx amplification buffer, 300 μM of each dNTP, 0.45 μM of each primer, 1 mM MgSO4, 1 U Platinum Pfx DNA Polymerase (Invitrogen), and 50 ng gDNA in a 50 μl final volume. Two independent reactions were performed for each sample, following Lenz and Becker’s recommendations (Lenz and Becker, 2008) to avoid the formation of chimeric products; the thermal profile was as follows: a denaturation step at 94°C for 5 min followed by 30 cycles of 94°C for 30 s, 64°C for 30 s, and 68°C for 1 min. The Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA) was used for purifying PCR products which were then sequenced using a BigDye Terminator kit (Macrogen, Seoul, South Korea and Sequencing Service of the University of Salamanca, Spain) in both directions (using the DRB3F and DRB3R primers).
Another two PCR products were purified and ligated independently into pELMO vector for those animals to which a genotype could not be assigned from the PCR products sequences (Ramos et al., 2017). The ligated PCR products were used for transforming Escherichia coli TOP10 chemically competent cells (Invitrogen); colony PCR, with the DRB3F and DRB3R primers, was used for screening the resulting colonies for positive recombinant clones. The colony PCR reaction mixture contained 1X NH4 reaction buffer, 250 μM of each dNTP, 0.25 μM of each primer, 1.5 mM MgCl2, 1 U BIOLASE DNA polymerase (Bioline, London, UK) in a final 10 μl volume. A Zyppy Plasmid Miniprep Kit (Zymo Research Corporation, Irvine, CA, USA) was used for plasmid extraction, following the manufacturer’s instructions. A BigDye Terminator kit (Macrogen) was used for sequencing clones in both directions, using ccdBsec-F and ccdBsec-R external primers (Ramos et al., 2017). At least eight clones were sequenced for each animal.
CLC Main Workbench v.3.6.5 software (CLC bio, Aarhus, Denmark) was used for assembling each independent PCR and clone sequences (manually edited if necessary) and heterozygous positions were identified for producing a consensus containing IUPAC ambiguity codes. The genotype for each animal was assigned by comparing these sequences with BoLA-DRB3 allele sequences reported in the IPD-MHC database (Maccari et al., 2017) using HAPLOFINDER (Miltiadou et al., 2003), according to Baxter et al., 2008.
Allele frequencies and the amount of alleles (Na) were obtained by direct counting; 95% confidence intervals (95%CI) were calculated for allele frequencies, according to Fung and Keenan (Fung and Keenan, 2014), allowing for a small sample and population size. Observed heterozygosity (ho) and unbiased expected heterozygosity (he) according to Nei (Nei and Chesser, 1983) were estimated using Arlequin v.3.5 software for population genetic analysis (Excoffier and Lischer, 2010). Five randomized datasets of 30 animals were used to calculate ho to confirm that no sampling size effect would alter diversity estimates. FIS statistic (the correlation of alleles within an individual relative to the subpopulation) (Weir and Cockerham, 1984) included in Genepop v.4.7.0 was used for estimating potential departures from Hardy-Weinberg equilibrium, using the exact test of significance (Rousset, 2008).
Arlequin v.3.5 was used for calculating the amount of polymorphic sites (S), nucleotide diversity (π), and the average amount of pairwise nucleotide differences between populations (Nei and Li, 1979). MEGAX software (Kumar et al., 2018) was used for calculating the average amount of synonymous (dS) and nonsynonymous (dN) substitutions per site by Nei-Gojobori’s method with Jukes-Cantor correction and Z-test was used for assessing dN/dS ratio significance (Nei and Gojobori, 1986).
Normande population structure was evaluated first because samples from this breed were obtained from three geographically defined regions having local breeding practices which could have differentiated cattle populations. Population structure and genetic differentiation between populations were evaluated by estimating FST statistics (the correlation of randomly chosen alleles within the same subpopulation relative to the entire population), as described by Weir and Cockerham (Weir and Cockerham, 1984) using Arlequin v.3.5 (Pairwise FST) and Genepop v.4.7.0. (overall FST). POPTREE2 (Takezaki et al., 2010) was used for estimating genetic distances DA (Nei, 1978) from allele frequencies and constructing dendrograms using the NJ algorithm (Takezaki et al., 2010). PAST software v.3.2 (Hammer et al., 2001) was used for assigning confidence levels to branch nodes by bootstrapping DA genetic distances, with 10,000 replicates. PAST software v.3.2 was used for metric multidimensional scaling (MDS) analysis, based on DA genetic distances, and principal component analysis (PCA), based on allele frequencies.
GeneDoc (Nicholas et al., 1997) was used for calculating identity and similarity percentages (assessed by the BLOSUM62 substitution matrix) for the 31 putative positions constituting the MHC-DRB PBR (Suarez et al., 2006). Percentage similarity was calculated for all observed genotypes within populations; five randomized datasets of 30 individuals from each population were used for one-way ANOVA and Bonferroni tests, after using STATA for proving normal distribution and homoscedasticity (StataCorp, 2017).
WebLogo software (Crooks et al., 2004) was used for creating a logo of PBR positions for each population, using the BLOSUM62 substitution matrix, including a color similarity scheme. Each position’s frequency depended on the allele frequency observed for each population, considering only those having greater than 5% frequency. Covariation was estimated by constructing a vector having 620 coordinates (31 positions × 20 possible aa) for each population. Another Multidimensional Analysis Package (amap R 3.4.3) (R_Core_Team, 2019) was used for calculating Pearson correlation coefficients (PCC) between vectors. The R 3.4.3 stats package (R_Core_Team, 2019) was used to perform a metric MDS on the correlation distance matrix so obtained.
Sequence-based typing identified 29 alleles in Spanish Morucha, 23 in Nariño, 27 in Boyacá, and 28 in Cundinamarca Normande cattle (Table 2; Supplementary Data Sheet 1). No new alleles were observed. Eight (BoLA-DRB3*001:01, 002:01, 005:01, 007:01, 010:01, 012:01, 014:01:01 and 048:02) high-frequency alleles (> 5%) were identified in Normande cattle, accounting for 55.6% of all alleles. Six of these alleles were found to be high-frequency alleles in at least two out of three regions. BoLA-DRB3*001:01 and BoLA-DRB3*002:01 (16.1% cumulative frequency) occurred with high frequency in all three Colombian regions.
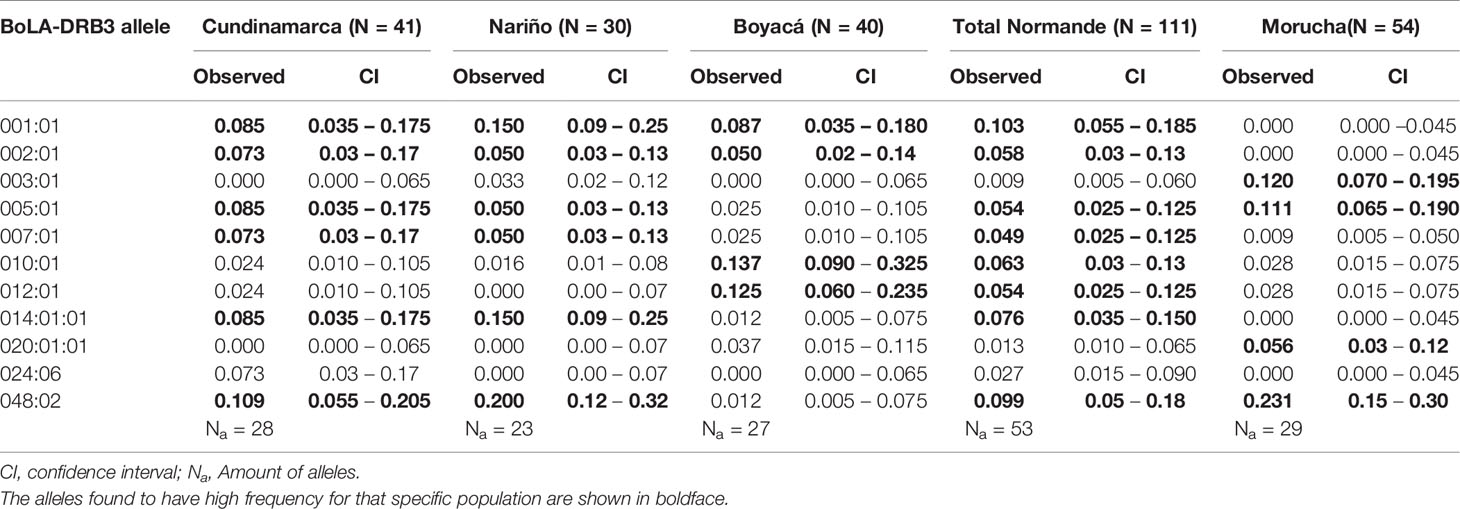
Table 2 High frequency alleles (>5%) ound fin Cundinamarca Normande, Nariño Normande, Boyacá Normande, overall Normande, and Spanish Morucha.
Regarding Morucha cattle, only four alleles (BoLA-DRB3*003:01, 005:01, 020:01:01 and 048:02) occurred with high frequency, accounting for 51.9% of all alleles. Only BoLA-DRB3*005:01 and BoLA-DRB3*048:02 were shared by both breeds as high-frequency alleles. Supplementary Table 1 gives complete information about allele frequencies.
Overall FST (FST = 0.022) and pairwise FST calculated within the Normande breed indicated that the three populations were differentiated at a similar degree to that seen between breeds, and therefore these three Colombian Normande populations were considered separately for diversity analysis.
We determined the observed (ho) and expected (he) heterozygosity and the fixation index (FIS) in Morucha and Normande cattle populations and compared them to those reported for other cattle breeds to examine BoLA-DRB3 locus variability and potential departures from Hardy-Weinberg equilibrium. No relation with the original sample size was detected when ho was calculated from randomized datasets of 30 animals, indicating that this estimate (and its derivatives) can be used to compare genetic diversity in populations having unequal sized samples. For the three Normande populations as well as the Morucha population, he was higher than ho with a significant heterozygote deficiency (Table 3). All three Normande populations as well as the Morucha had the lowest ho compared to the other cattle populations, excluding the Chilean Hereford-Jersey population, although having similar he levels. These populations also had the greatest deviations from Hardy-Weinberg equilibrium (FIS).
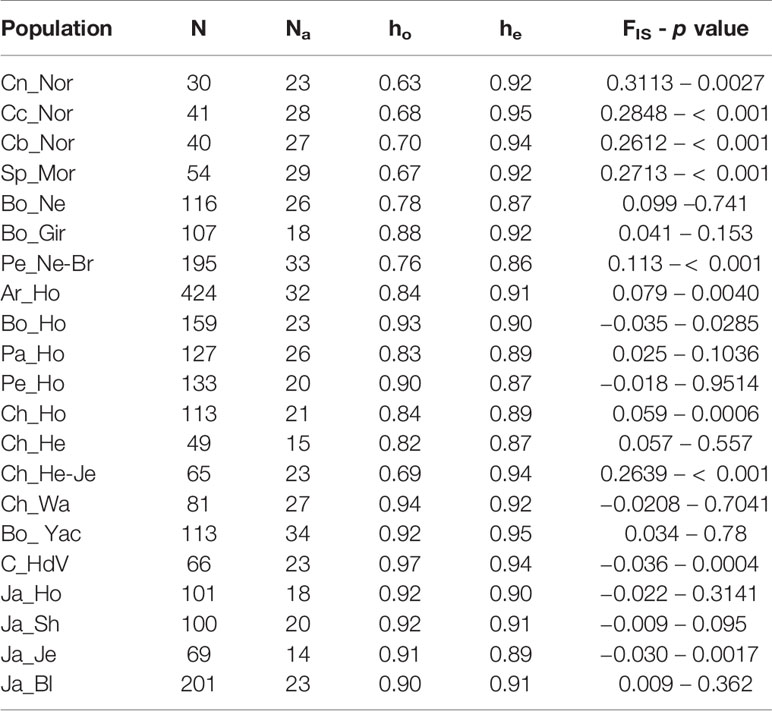
Table 3 Amount of individuals (N), amount of alleles (Na), observed (ho), and expected (he) heterozygosity and Hardy-Weinberg equilibrium, as evaluated by FIS coefficient, for 21 cattle populations.
The amount of polymorphic sites (S) and nucleotide diversity (π) were estimated to evaluate nucleotide genetic variability within populations (Table 4). The four populations had intermediate S values (61 for Spanish Morucha and Cundinamarca Normande to 65 for Nariño Normande and Boyacá Normande), while the π value was the lowest in Spanish Morucha (0.0711) and intermediate in the three Normande populations (0.0790 to 0.0817).
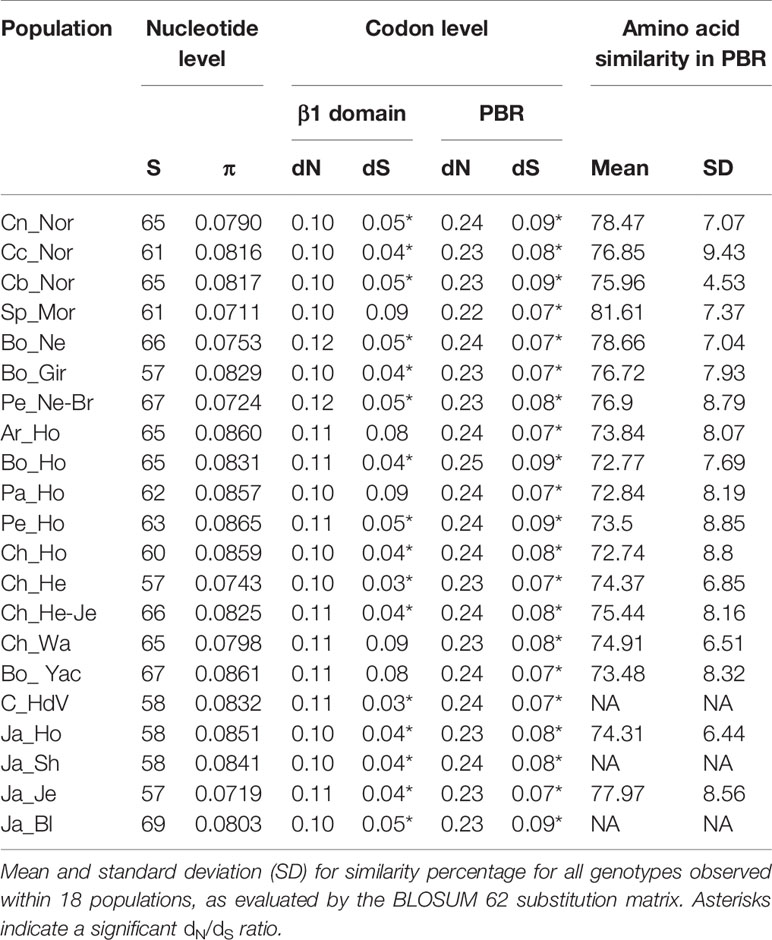
Table 4 Values for the amount of polymorphic sites (S), nucleotide diversity (π), mean amount of nonsynonymous (dN) and synonymous (dS) nucleotide substitutions per site, in 21 cattle populations.
Nucleotide genetic variability between populations was also assessed calculating the average amount of pairwise nucleotide differences (Figure 1A; Supplementary Data Sheet 2), which ranged from -0.0367 (between Chilean Holstein and Peruvian Holstein) to 4.73 (between Chilean Hereford-Jersey and Peruvian Nellore-Brahman). Nucleotide differences between populations can be negative in rare cases where nucleotide diversity is high because it implies subtracting variability within populations from total variability (Nei and Li, 1979).
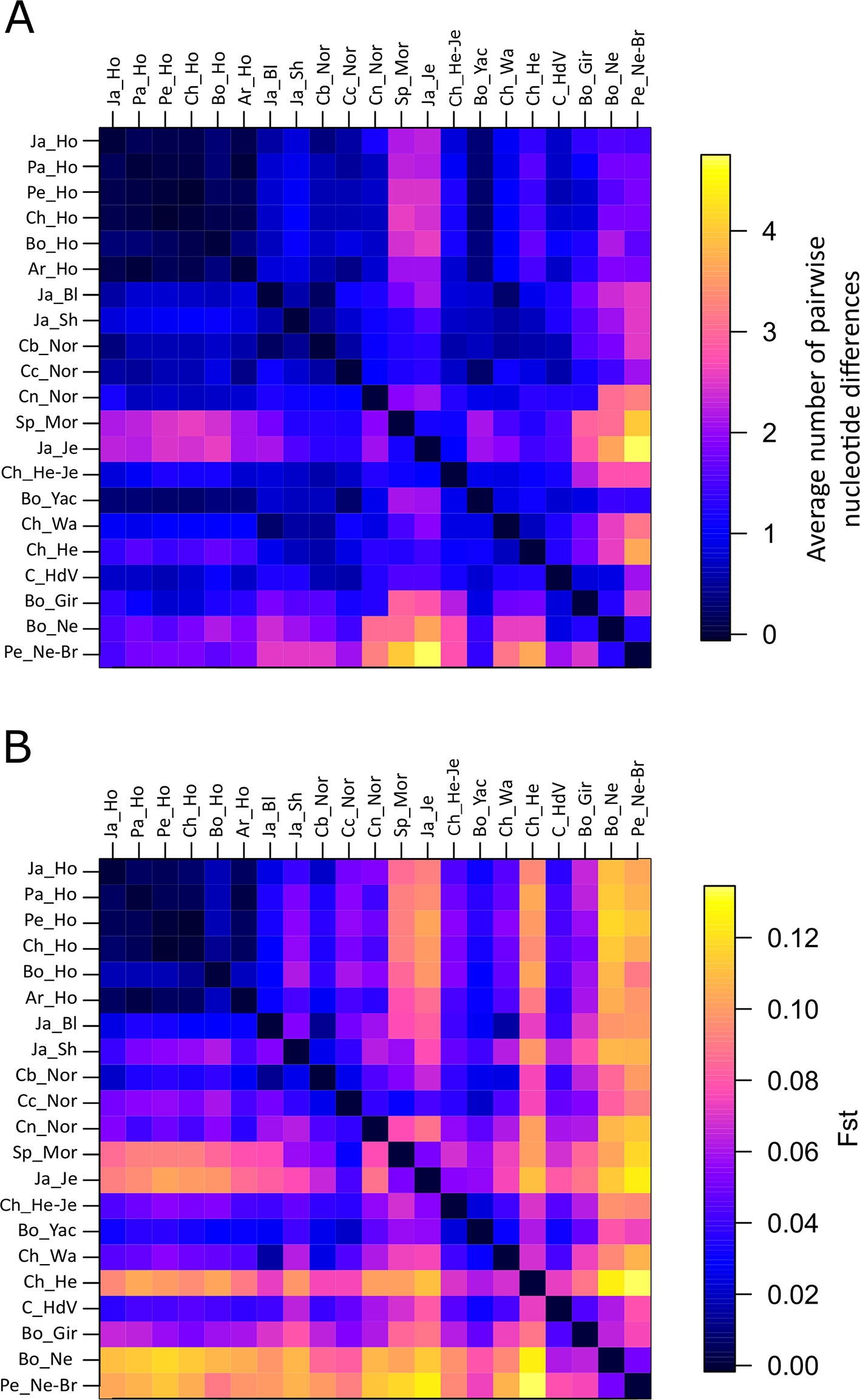
Figure 1 Heat map of the calculated average amount of pairwise nucleotide differences (A) and (B) pairwise FST among 21 cattle populations. Identical values are shown above and below the diagonal.
Holstein populations had rather low differentiation from each other in the average amount of pairwise nucleotide differences. Bo_Yac was observed to have little differentiation from Holstein populations. Four comparisons had the highest nucleotide variability values (Peruvian Nellore-Brahman with Japanese Jersey, Peruvian Nellore-Brahman with Spanish Morucha, Bolivian Nellore with Japanese Jersey, and Peruvian Nellore-Brahman with Chilean Hereford). The remaining comparisons had intermediate values. By contrast with Normande populations, Spanish Morucha had strong differentiation from Holstein and zebu populations, similar to that observed in Japanese Jersey. Boyacá Normande was more differentiated from Cundinamarca Normande than from Nariño Normande. Nariño Normande had the highest differentiation from zebu populations.
Codon selection was evaluated using the dN/dS ratio. Selection pattern was very similar for Normande and Spanish Morucha populations, as well as between all populations (Table 4). Considering the whole β1 domain, the estimated nonsynonymous substitution rate (dN) was 0.10, in both Spanish Morucha and Normande cattle and the synonymous substitutions rate (dS) ranged from 0.04 in Cundinamarca Normande to 0.09 in Spanish Morucha, with a significant dN/dS ratio being found in most populations. The difference between dN and dS was more prominent when only considering PBR codons and all populations had a significant dN/dS ratio. Spanish Morucha had the lowest dN value of all populations (0.22) while sharing the lowest dS value (0.07) with other populations.
Pairwise FST values were used for assessing genetic differentiation between populations (Figure 1B; Supplementary Data Sheet 3). Pairwise comparisons ranged from −0.0010 (Chilean Holstein with Peruvian Holstein) to 0.1338 (Chilean Hereford with Peruvian Nellore-Brahman). Similar to that seen regarding the amount of pairwise nucleotide differences, the lowest FST values were observed when comparing Holstein populations to one another. Nevertheless, except for Bolivian Gir, zebu populations had marked differentiation compared to taurine populations. Similar to that seen in Chilean Hereford and Japanese Jersey, Spanish Morucha was differentiated more from other taurine populations than Normande (0.0688 average pairwise FST in Spanish Morucha and 0.0470 in Normande). Nariño Normande was also observed to be most divergent from zebu populations and Boyacá Normande was less differentiated from taurine populations than Boyacá Normande and Nariño Normande. The highest differentiation was observed between Bolivian Nellore with Holstein populations and when comparing a group consisting of Japanese Jersey, Spanish Morucha, and Chilean Hereford to Bolivian Nellore and Peruvian Nellore-Brahman. The remaining comparisons had an intermediate level of differentiation.
The DA genetic distance matrix was used to construct a dendrogram, using the NJ algorithm (Figure 2A and Supplementary Data Sheet 4). DA genetic distances were also used for depicting the relationship between populations in two dimensions by MDS analysis (Figure 2B). Dendrograms and MDS gave a similar distribution pattern, only differing regarding the clustering of groups not well supported by bootstrap (Japanese Black, Japanese Shorthorn, Boyacá Normande, Chilean Hereford-Jersey, Bo_Yac, Chilean Wagyu, Colombian Hartón del Valle and Chilean Hereford). MDS analysis had a 0.799 R-square (RSQ), indicating that representation in two coordinates gave a good description of real BoLA-DRB3 variability. Zebu populations were clearly separated from taurine populations. Despite the two types of cattle having a heterogeneous distribution, lacking any clear clustering except for the Holstein group, we were able to identify four clusters which were relatively well supported by bootstrap (>90). The zebu group (Bolivian Nellore, Peruvian Nellore-Brahman and Bolivian Gir), the Holstein group (Japanese Holstein, Peruvian Holstein, Peruvian Holstein, Chilean Holstein, Bolivian Holstein and Argentinian Holstein), a group formed by Cundinamarca Normande and Nariño Normande and a group formed by Spanish Morucha and Japanese Jersey. Boyacá Normande was noted to be closer to the other taurine populations than Cundinamarca Normande and Nariño Normande.
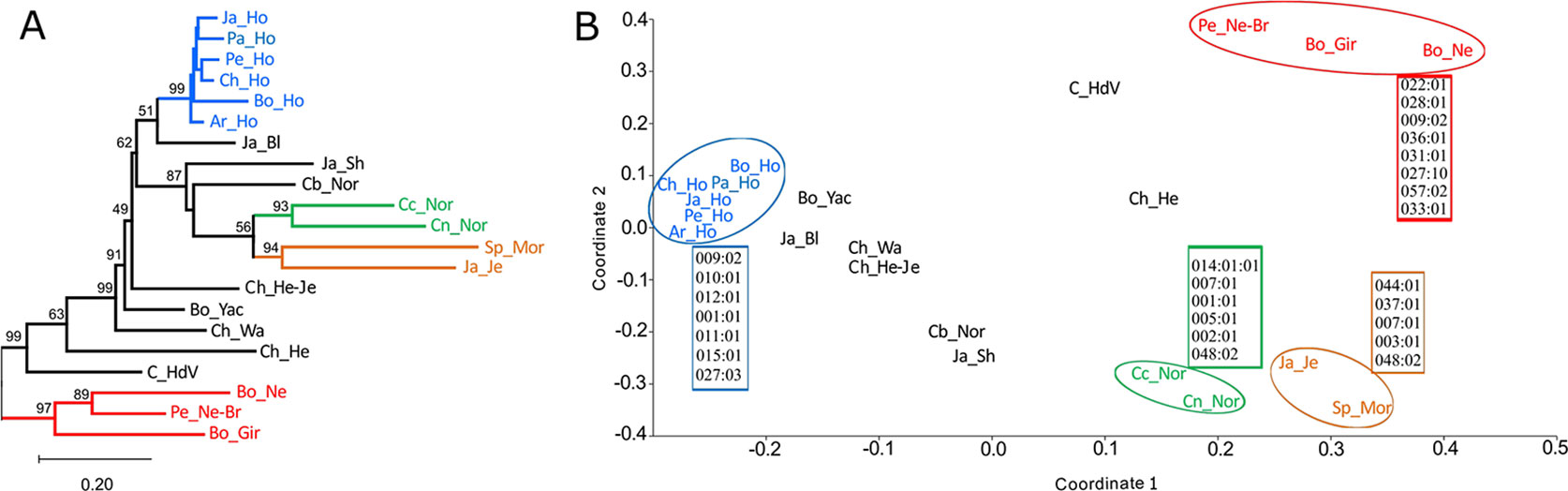
Figure 2 Dendrogram (A) and multidimensional scaling (MDS) analysis (B) based on DA genetic distances. Alleles are indicated which contributed to population grouping, as identified by principal component analysis (PCA). Groups well-supported by bootstrap in MDS are circled in the dendrogram.
Allele frequencies were used for PCA and these results were compared to MDS to identify which alleles contributed most to differentiate or cluster cattle populations. Groups clearly identified in both MDS and in the first two PC were depicted as having the major alleles contributing to such grouping (Figure 2B and Supplementary Data Sheet 5).
Population distribution in the first two principal components (PC) (Supplementary Figure 1), accounting for 44.6% of total variance, was very similar to MDS. Boyacá Normande was located toward the Holstein variability region in the first PC and was clearly differentiated from Spanish Morucha, and the other Normande, which were located toward zebu populations’ variability region. Boyacá Normande differentiation in the first PC was explained by the high BoLA-DRB3*010:01, 012:01, 001:01, and 011:01 allele frequency. The different position of Cundinamarca Normande and Nariño Normande regarding Spanish Morucha in the first PC was explained by BoLA-DRB3*007:01, 002:01, 014:01:01, and 001:01 alleles. Holstein populations were clearly differentiated in the first PC by high BoLA-DRB3*009:02, 010:01, 012:01, 001:01, 011:01, and 015:01 allele frequency. Alleles BoLA-DRB3*048:02, 002:01, 003:01, 005:01, 007:01, and 008:01 explained the grouping of Cundinamarca Normande, Nariño Normande, Spanish Morucha, and Japanese Jersey in the second PC. Moreover, BoLA-DRB3*012:01, 016:01, 010:01, and 011:01 alleles differentiated Boyacá Normande, while BoLA-DRB3*048:02, 001:01, 014:01:01, and 024:06 alleles differentiated Cundinamarca Normande from Nariño Normande.
The clearest differentiation between zebu and taurine populations was observed in the second PC and was mainly explained by BoLA-DRB3*036:01, 028:01, 009:02, 031:01, 027:10, 057:02, 033:01, and 022:01 alleles, enriched in zebu populations. Spanish Morucha clustered with Japanese Jersey in the first two PC, enriched with BoLA-DRB3*048:02, 044:01, 037:01, 007:01, and 003:01. Nariño Normande and Cundinamarca Normande were grouped by BoLA-DRB3*014:01:01, 007:01, 001:01, 005:01, 002:01, and 048:02 alleles. Several alleles occurring with low frequency, unique to some populations (low correlation in the PCA), contributed to differentiating these populations (BoLA-DRB3*020:09, 024:02, 024:04, 012:02, 020:10, 024:01, 030:22, and 078:01 in Spanish Morucha, BoLA-DRB3*043:02, 022:04, 022:05, 024:07, 028:03, 029:01, and 051:01 in Nariño Normande, BoLA-DRB3*019:02, 034:03, and BoLA-DRB3*075:03 in Cundinamarca Normande) (Supplementary Data Sheet 5).
PBR position similarity was calculated for the alleles reported in the populations used in this study and from previous reports (Takeshima et al., 2003; Giovambattista et al., 2013; Takeshima et al., 2015a; Takeshima et al., 2015b; Takeshima et al., 2018) to further analyze BoLA-DRB3 diversity (Table 4, Supplementary Data Sheet 6). Normande cattle had 78.47% similarity for Nariño, 76.85% for Cundinamarca, and 75.96% for Boyacá. Spanish Morucha similarity was 81.61%, this being the highest value for all groups evaluated here. Holstein populations had an average 74% similarity, Bolivian Holstein (72.77%), and Paraguayan Holstein (72.84%) having the lowest PBR similarity. Average similarity for zebu populations was 76.81% (76.90% for Peruvian Nellore-Brahman, 76.72% for Bolivian Gir, and 78.66% for Bolivian Nellore).
Five randomized sets of 30 animals were constructed to reduce possible sampling effects, considering the clustering observed in diversity analysis results. The groups compared were Holsteins, zebu populations, Nariño Normande, Cundinamarca Normande, Boyacá Normande, and Spanish Morucha. Significant differences between the evaluated groups were found for all randomized datasets (≤0.05 p-value). The Bonferroni test gave significant differences between Spanish Morucha and Holsteins populations (≤0.05 p-value) for all randomizations. Holstein populations’ percentage similarity was lower than for Spanish Morucha (81.61%), indicating that Spanish Morucha had less diversity in the PBR. When identity in PBR positions was assessed, we found that 129 out of 140 alleles had a different PBR.
The logo representation of PBR had remarkable similarities among populations, having very similar substitution patterns (Figure 3A). PBR logos had highly variable positions (11, 13, 37, 70, 71, and 74), some of which were shared by several pockets and usually had nonconservative substitutions. Covariation analysis of PBR aa sequence logos for all cattle populations (Figure 3B) showed that most variability was due to differences in aa frequencies and not aa variations in the same position.

Figure 3 Peptide-binding region (PBR) logos (A), Pearson correlation coefficient (B) and multidimensional scaling (MDS) matrix (C) based on BoLA-DRB3 pocket position variability among populations. In A, similar colors indicate conservative and different colors nonconservative aa changes according to the BLOSUM 62 substitution matrix. B shows identical values above and below the diagonal. * Positions shared by more than one pocket.
Global covariation between PBR positions was high (PCC = 0.92). Three groups could be discriminated (Figures 3B, C). The first contained Cundinamarca Normande, Nariño Normande, Chilean Hereford, Japanese Jersey, Chilean Hereford-Jersey, and Spanish Morucha (group PCC = 0.94). The second included Japanese Shorthorn, Chilean Wagyu, Japanese Black, Boyacá Normande, Paraguayan Holstein, Argentinian Holstein, Japanese Holstein, Peruvian Holstein, Chilean Holstein, Bolivian Holstein, Bo_Yac, Bolivian Gir, and Colombian Hartón del Valle (group PCC = 0.96) and the third consisted of Peruvian Nellore-Brahman and Bolivian Nellore (group PCC = 0.93). All taurine populations and zebu Bo-Gir had an average 0.94 PCC. Although these groups were different regarding previous analysis, similar groups were found, distinguishing zebu populations (Peruvian Nellore-Brahman – Bolivian Nellore), Holstein populations and the Spanish Morucha - Japanese Jersey and Cundinamarca Normande – Nariño Normande association. Overall covariation gave Spanish Morucha as the most differentiated population between taurine breeds, similar to zebu Bolivian Nellore and Peruvian Nellore-Brahman.
MHC gene analysis can provide information about domestic species’ level of overall genetic diversity. While several studies have shown decreased MHC variability in association with population bottlenecks (Bollmer et al., 2007; Bollmer et al., 2011; Mason et al., 2011; Zhang et al., 2016), others have found that high variability persists despite extreme population bottlenecks (Edwards and Hedrick, 1998; Garrigan and Hedrick, 2001; Aguilar et al., 2004; Borg et al., 2011; Moutou et al., 2013; Newhouse and Balakrishnan, 2015). The BoLA-DRB3 gene is the most polymorphic bovine MHC class II DR gene and is the only one having been shown to be fully functional, although the total amount of alleles is low compared to species such as humans (Shiina et al., 2017). This study was therefore aimed at describing BoLA-DRB3 genetic diversity in two new cattle breeds and analyzed whether domestic cattle breeding has decreased actual MHC variability.
Several measures of genetic diversity, such as hs or FIS, have corrections for sample size (Nei and Chesser, 1983). As expected, ho estimated for random datasets did not depend on original sample size. This was because ho is an unbiased estimator of parametric value (Nei and Chesser, 1983) and is mainly determined by the sampling method used. Therefore, the lower ho and higher FIS value for Spanish Morucha and Colombian Normande compared to the other cattle populations (Takeshima et al., 2003; Giovambattista et al., 2013; Takeshima et al., 2015a; Takeshima et al., 2015b; Takeshima et al., 2018) indicated heterozygote deficiency for these two breeds.
Drift and selection interact to configure the allele distribution observed in populations. MHC (a nonneutral marker) allele distribution would have been expected to be that of balancing selection with excess heterozygotes (Hughes and Nei, 1988; Hughes and Yeager, 1998; Takeshima et al., 2008). Nevertheless, the heterozygote deficiency observed in Spanish Morucha and Colombian Normande suggested that other evolutionary processes, such as inbreeding and/or bottlenecks, would have been acting on these populations. Genetic improvement regarding dairy (Normande) or beef production (Morucha) traits or bottlenecks in these breeds’ origin might have contributed to the low diversity observed and high level of inbreeding inferred (>0.2 in Normande and Morucha) for the BoLA-DRB3 locus. Decreased MHC diversity can result in a narrower spectrum of pathogens which a population can recognize, less viability and even loss of pregnancies due to maternal-foetal interactions (Sommer, 2005). However, functional diversity could be preserved despite few alleles being involved.
BoLA-DRB3 divergence between Colombian Normande populations was even higher in some cases than the divergence observed between breeds on different continents (e.g., Spanish Morucha and Japanese Jersey). BoLA-DRB3 divergence between Normande populations can be highlighted compared to that for Holstein populations which are not so differentiated, despite also being phenotypically homogeneous. Comparing French and Colombian Normande cattle might provide insights into Normande populations’ regional divergence because the French Normande breed is slightly variable and is considered a completely closed breed (Danchin-Burge et al., 2012; Porter et al., 2016).
Spanish Morucha is an autochthonous cattle breed from the black Iberian group which is restricted to the Northwest of the Iberian Peninsula; it was initially used for draft, milk and beef but is now solely used for meat production. It has moderate inbreeding rates and a population structure restricting mating within herds (Canas-Alvarez et al., 2014). Nevertheless, a restricted gene flow regarding other autochthonous Spanish breeds having geographical proximity during their history and similar production systems might have moulded Spanish Morucha genetic diversity (Canas-Alvarez et al., 2015). As such, this breed has had relatively low human intervention and can thus be considered close to a natural cattle population.
Spanish Morucha has been shown to be more differentiated from other taurine and zebu breeds. Likewise, Japanese Jersey and Chilean Hereford pairwise FST values for Spanish Morucha were the highest among taurine populations. A similar trend was found when analyzing the sequence information for each allele through the average amount of pairwise nucleotide differences, Spanish Morucha and Japanese Jersey being more differentiated. BoLA-DRB3*048:02 high frequency was the main reason for Spanish Morucha and Japanese Jersey clustering. As European cattle were introduced to improve Japanese native breeds, a similar BoLA-DRB3 gene pool in Iberian and Jersey founder populations or the direct introduction of Iberian cattle along with similar selective pressures might have led to these two cattle populations’ characteristic divergence, despite these two breeds having different production features and purposes.
The frequency of an MHC allele or group of alleles might become increased in cattle populations having the same selective pressures imposed by similar production systems, even in different geographic locations. From the analysis of previously published data (Takeshima et al., 2015a), we found that Holstein populations were enriched with alleles reported to be associated with resistance (BoLA-DRB3*011:01 and BoLA-DRB3*012:01) and susceptibility (BoLA-DRB3*001:01 and BoLA-DRB3*015:01) to mastitis (Yoshida et al., 2009b; Yoshida et al., 2009a). The Holstein breed has been submitted to intense selective pressure due to milk production traits which can make it susceptible to mastitis but, at the same time, there is also pressure for selecting resistant individuals. These two forces might thus create high frequencies of alleles associated with susceptibility and resistance to be in balance. However, it is worth noting that Japanese Black (a beef breed) was also enriched by these alleles. No particular enrichment with this kind of alleles was observed for Normande or Morucha cattle.
The greatest variability in the β1 domain is located in PBR positions, thereby determining the peptides to which an allele can bind. In agreement with other diversity analysis in which Spanish Morucha had lower nucleotide and codon variability, variability in the PBR was lower and could indicate that the magnitude of this population’s peptide repertoire could be significantly smaller than that of other cattle. Holstein populations shared similar PBR constitution and covariation patterns by contrast with Normande ones, which had a more differentiated PBR. Despite zebu populations being well-differentiated, Bolivian Gir appears to have a BoLA-DRB3 allele distribution similar to that of taurine breeds, thus grouping them with taurine populations. High correlation coefficients in PBR aa sequence logo analysis indicated that all breeds tended to have similar peptide repertoires, despite genetic differentiation. This suggested that domestication and/or breeding have not decreased functional MHC variability. Indeed, the fact that Spanish Morucha has lesser variability suggests that breeding might have increased both genetic and functional variation.
Great diversity was observed when analysing BoLA-DRB3 identity in the PBR, despite having fewer alleles compared to other species, such as primates (Suarez et al., 2006), i.e., even though having fewer alleles, potential peptide repertoire size in bovines is similar to that for other species. This means that cattle have 129 distinct PBR (based on sequence identity) and that infectious disease control programmes should be designed as if there were 129 different alleles instead of 140 (136 in the IPD Database and four reported elsewhere). A similar amount of PBR sequences has been determined in humans and owl monkeys (Suarez et al., 2017). This is especially important when it comes to peptide-based vaccine design which might be more efficacious when the peptide-MHC complex has potentially greater affinity as this can be increased by modifying peptides to bind to MHC molecules [reviewed in (Patarroyo et al., 2011)].
However, allele frequency would be the main guide regarding population control programmes based on MHC diversity. For example, based on allele frequency distribution, 67% to 73% of the peptide repertoire in Holstein populations would be determined only by seven alleles and this information could be used when designing peptide-based vaccines. A potential limitation of such approach is that the MHC-DR PBR positions used have been documented by X-ray crystallography just in humans and mice. Nevertheless, several of these positions have been associated with vaccine responses and susceptibility or resistance to infectious diseases (Garcia-Briones et al., 2000; Baxter et al., 2009; Rastislav and Mangesh, 2012; Carignano et al., 2017) and thus analysis based on these positions is reasonable.
Despite genetic divergence being observed between cattle populations, their peptide repertoires seem to be functionally equivalent. The great similarities and covariation among populations regarding the BoLA-DRB3 PBR might signify that BoLA-DRB3 peptide repertoire size is very similar among the cattle populations analyzed here and thus breeding has not reduced cattle’s MHC functional diversity. Although cattle have fewer alleles than species such as some primates, PBR sequence identity supports the idea that the potential peptide repertoire between these species is equivalent and functional diversity can persist despite population bottlenecks. In other words, less genetic MHC diversity (fewer alleles) does not mean loss of functional diversity. Genetic improvement regarding breeding and domestication appears to have produced three different Colombian Normande populations based on BoLA-DRB3 genetic diversity. When analysing these populations regarding the diversity described for other cattle, it was noted that the forces which have contributed toward moulding BoLA-DRB3 locus diversity in Colombian Normande have produced remarkable differentiation compared to other phenotypically homogeneous breeds, such as Holstein. It is also remarkable that Spanish Morucha was shown to be strongly differentiated in all types of analysis.
All datasets generated for this study are included in the article/Supplementary Material.
The animal study was reviewed and approved by The Universidad de Ciencias Aplicadas y Ambientales (UDCA, Bogotá) Research Ethics Committee (Minute No. 201901) approved this study.
MB, DO, CS, and MP designed the study. MB, DO, and CV carried out experimental procedures. MB, DO, CS, BV, CV, JL-A, AM, IO, and MP analyzed the data and wrote the manuscript. AM and MP supervised the study. All authors approved the final version of the manuscript.
This study was partially supported by the Diputación de Salamanca, Caja Rural de Salamanca and the Instituto de Salud Carlos III, ISCIII, Spain (www.isciii.es) under grants: RICET RD16/0027/0018 and PI16/01784. MB was partially financed trough a scholarship from the Microbiology Postgraduate Program, Universidad Nacional de Colombia. DO was financed through a scholarship from the PhD Program in Biomedical and Biological Sciences, Universidad del Rosario.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Doctor Alejandro Garavito and the Colombian Normande Breeders Association for their support in obtaining Cundinamarca Normande samples and José Manuel Sánchez Recio from the Genealogic National Morucha Breeders Association for obtaining the Spanish Morucha samples. We would also like to thank to Jason Garry for extensively reviewing the manuscript.
ANOVA, analysis of variance; BoLA, bovine leukocyte antigen; MDS, multidimensional scaling analysis; MHC, major histocompatibility complex; PBR, peptide-binding region; aa, amino acid; PCA, principal component analysis; PCC, Pearson correlation coefficient.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01293/full#supplementary-material
Acosta-Rodriguez, R., Alonso-Morales, R., Balladares, S., Flores-Aguilar, H., Garcia-Vazquez, Z., Gorodezky, C. (2005). Analysis of BoLA class II microsatellites in cattle infested with Boophilus microplus ticks: class II is probably associated with susceptibility. Vet. Parasitol. 127, 313–321. doi: 10.1016/j.vetpar.2004.10.007
Aguilar, A., Roemer, G., Debenham, S., Binns, M., Garcelon, D., Wayne, R. K. (2004). High MHC diversity maintained by balancing selection in an otherwise genetically monomorphic mammal. Proc. Natl. Acad. Sci. U. S. A. 101, 3490–3494. doi: 10.1073/pnas.0306582101
Andersson, L., Rask, L. (1988). Characterization of the MHC class II region in cattle. The number of DQ genes varies between haplotypes. Immunogenetics 27, 110–120. doi: 10.1007/bf00351084
Baxter, R., Hastings, N., Law, A., Glass, E. J. (2008). A rapid and robust sequence-based genotyping method for BoLA-DRB3 alleles in large numbers of heterozygous cattle. Anim. Genet. 39, 561–563. doi: 10.1111/j.1365-2052.2008.01757.x
Baxter, R., Craigmile, S. C., Haley, C., Douglas, A. J., Williams, J. L., Glass, E. J. (2009). BoLA-DR peptide binding pockets are fundamental for foot-and-mouth disease virus vaccine design in cattle. Vaccine 28, 28–37. doi: 10.1016/j.vaccine.2009.09.131
Bollmer, J. L., Vargas, F. H., Parker, P. G. (2007). Low MHC variation in the endangered Galapagos penguin (Spheniscus mendiculus). Immunogenetics 59, 593–602. doi: 10.1007/s00251-007-0221-y
Bollmer, J. L., Hull, J. M., Ernest, H. B., Sarasola, J. H., Parker, P. G. (2011). Reduced MHC and neutral variation in the Galapagos hawk, an island endemic. BMC Evol. Biol. 11, 1471–2148. doi: 10.1186/1471-2148-11-143
Borg, A. A., Pedersen, S. A., Jensen, H., Westerdahl, H. (2011). Variation in MHC genotypes in two populations of house sparrow (Passer domesticus) with different population histories. Ecol. Evol. 1, 145–159. doi: 10.1002/ece3.13
Burke, M. G., Stone, R. T., Muggli-Cockett, N. E. (1991). Nucleotide sequence and northern analysis of a bovine major histocompatibility class II DR beta-like cDNA. Anim. Genet. 22, 343–352. doi: 10.1111/j.1365-2052.1991.tb00688.x
Canas-Alvarez, J. J., Gonzalez-Rodriguez, A., Martin-Collado, D., Aviles, C., Altarriba, J., Baro, J. A., et al. (2014). Monitoring changes in the demographic and genealogical structure of the main Spanish local beef breeds. J. Anim. Sci. 92, 4364–4374. doi: 10.2527/jas.2013-7420
Canas-Alvarez, J. J., Gonzalez-Rodriguez, A., Munilla, S., Varona, L., Diaz, C., Baro, J. A., et al. (2015). Genetic diversity and divergence among Spanish beef cattle breeds assessed by a bovine high-density SNP chip. J. Anim. Sci. 93, 5164–5174. doi: 10.2527/jas.2015-9271
Carignano, H. A., Beribe, M. J., Caffaro, M. E., Amadio, A., Nani, J. P., Gutierrez, G., et al. (2017). BOLA-DRB3 gene polymorphisms influence bovine leukaemia virus infection levels in Holstein and Holstein x Jersey crossbreed dairy cattle. Anim. Genet. 48, 420–430. doi: 10.1111/age.12566
Crooks, G. E., Hon, G., Chandonia, J. M., Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Danchin-Burge, C., Leroy, G., Brochard, M., Moureaux, S., Verrier, E. (2012). Evolution of the genetic variability of eight French dairy cattle breeds assessed by pedigree analysis. J. Anim. Breed. Genet. 129, 206–217. doi: 10.1111/j.1439-0388.2011.00967.x
Dietz, A. B., Detilleux, J. C., Freeman, A. E., Kelley, D. H., Stabel, J. R., Kehrli, M. E., Jr. (1997). Genetic association of bovine lymphocyte antigen DRB3 alleles with immunological traits of Holstein cattle. J. Dairy Sci. 80, 400–405. doi: 10.3168/jds.S0022-0302(97)75950-2
Edwards, S. V., Hedrick, P. W. (1998). Evolution and ecology of MHC molecules: from genomics to sexual selection. Trends Ecol. Evol. 13, 305–311. doi: 10.1016/s0169-5347(98)01416-5
Excoffier, L., Lischer, H. E. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Fung, T., Keenan, K. (2014). Confidence intervals for population allele frequencies: the general case of sampling from a finite diploid population of any size. PloS One 9, e85925. doi: 10.1371/journal.pone.0085925
Garcia-Briones, M. M., Russell, G. C., Oliver, R. A., Tami, C., Taboga, O., Carrillo, E., et al. (2000). Association of bovine DRB3 alleles with immune response to FMDV peptides and protection against viral challenge. Vaccine 19, 1167–1171. doi: 10.1016/s0264-410x(00)00313-3
Garrigan, D., Hedrick, P. W. (2001). Class I MHC polymorphism and evolution in endangered California Chinook and other Pacific salmon. Immunogenetics 53, 483–489. doi: 10.1007/s002510100352
Giovambattista, G., Takeshima, S. N., Ripoli, M. V., Matsumoto, Y., Franco, L. A., Saito, H., et al. (2013). Characterization of bovine MHC DRB3 diversity in Latin American Creole cattle breeds. Gene 519, 150–158. doi: 10.1016/j.gene.2013.01.002
Glass, E. J., Oliver, R. A., Russell, G. C. (2000). Duplicated DQ haplotypes increase the complexity of restriction element usage in cattle. J. Immunol. 165, 134–138. doi: 10.4049/jimmunol.165.1.134
Goszczynski, D. E., Ripoli, M. V., Takeshima, S. N., Baltian, L., Aida, Y., Giovambattista, G. (2014). Haplotype determination of the upstream regulatory region and the second exon of the BoLA-DRB3 gene in Holstein cattle. Tissue Antigens 83, 180–183. doi: 10.1111/tan.12293
Gowane, G. R., Sharma, A. K., Sankar, M., Narayanan, K., Das, B., Subramaniam, S., et al. (2013). Association of BoLA DRB3 alleles with variability in immune response among the crossbred cattle vaccinated for foot-and-mouth disease (FMD). Res. Vet. Sci. 95, 156–163. doi: 10.1016/j.rvsc.2013.03.001
Hammer, Ø, Harper, D., Ryan, P. D. (2001). PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electronica 4, 1–9.
Hoof, I., Peters, B., Sidney, J., Pedersen, L. E., Sette, A., Lund, O., et al. (2009). NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61, 1–13. doi: 10.1007/s00251-008-0341-z
Hughes, A. L., Nei, M. (1988). Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335, 167–170. doi: 10.1038/335167a0
Hughes, A. L., Yeager, M. (1998). Natural selection and the evolutionary history of major histocompatibility complex loci. Front. Biosci. 26, d509–516. doi: 10.2741/a298
Juliarena, M. A., Poli, M., Sala, L., Ceriani, C., Gutierrez, S., Dolcini, G., et al. (2008). Association of BLV infection profiles with alleles of the BoLA-DRB3.2 gene. Anim. Genet. 39, 432–438. doi: 10.1007/s10528-016-9712-6
Klein, J., Sato, A., Nikolaidis, N. (2007). MHC, TSP, and the origin of species: from immunogenetics to evolutionary genetics. Annu. Rev. Genet. 41, 281–304. doi: 10.1146/annurev.genet.41.110306.130137
Kulberg, S., Heringstad, B., Guttersrud, O. A., Olsaker, I. (2007). Study on the association of BoLA-DRB3.2 alleles with clinical mastitis in Norwegian Red cows. J. Anim. Breed. Genet. 124, 201–207. doi: 10.1111/j.1439-0388.2007.00662.x
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Ledwidge, S. A., Mallard, B. A., Gibson, J. P., Jansen, G. B., Jiang, Z. H. (2001). Multi-primer target PCR for rapid identification of bovine DRB3 alleles. Anim. Genet. 32, 219–221. doi: 10.1046/j.1365-2052.2001.00761.x
Lenz, T. L., Becker, S. (2008). Simple approach to reduce PCR artefact formation leads to reliable genotyping of MHC and other highly polymorphic loci–implications for evolutionary analysis. Gene 427, 117–123. doi: 10.1016/j.gene.2008.09.013
Lenz, T. L., Mueller, B., Trillmich, F., Wolf, J. B. (2013). Divergent allele advantage at MHC-DRB through direct and maternal genotypic effects and its consequences for allele pool composition and mating. Proc. Biol. Sci. 280, 7. doi: 10.1098/rspb.2013.0714
Lopez, C., Suarez, C. F., Cadavid, L. F., Patarroyo, M. E., Patarroyo, M. A. (2014). Characterising a microsatellite for DRB typing in Aotus vociferans and Aotus nancymaae (Platyrrhini). PloS One 9, e96973. doi: 10.1371/journal.pone.009697
Maccari, G., Robinson, J., Ballingall, K., Guethlein, L. A., Grimholt, U., Kaufman, J., et al. (2017). IPD-MHC 2.0: an improved inter-species database for the study of the major histocompatibility complex. Nucleic Acids Res. 45, D860–D864. doi: 10.1093/nar/gkw1050
Maillard, J. C., Berthier, D., Chantal, I., Thevenon, S., Sidibe, I., Stachurski, F., et al. (2003). Selection assisted by a BoLA-DR/DQ haplotype against susceptibility to bovine dermatophilosis. Genet. Sel. Evol. 35, 2003027. doi: 10.1051/gse:2003027
Martinez, M. L., Machado, M. A., Nascimento, C. S., Silva, M. V., Teodoro, R. L., Furlong, J., et al. (2006). Association of BoLA-DRB3.2 alleles with tick (Boophilus microplus) resistance in cattle. Genet. Mol. Res. 5, 513–524.
Mason, R. A. B., Browning, T. L., Eldridge, M. D. B., (Mason2011 co-authors) (2011). Reduced MHC class II diversity in island compared to mainland populations of the black-footed rock-wallaby (Petrogale lateralis lateralis). Conserv. Genet. 12, 91–103. doi: 10.1007/s10592-009-9993-y
Miltiadou, D., Law, A. S., Russell, G. C. (2003). Establishment of a sequence-based typing system for BoLA-DRB3 exon 2. Tissue Antigens 62, 55–65. doi: 10.1034/j.1399-0039.2003.00080.x
Moutou, K. A., Koutsogiannouli, E. A., Stamatis, C., Billinis, C., Kalbe, C., Scandura, M., et al. (2013). Domestication does not narrow MHC diversity in Sus scrofa. Immunogenetics 65, 195–209. doi: 10.1007/s00251-012-0671-8
Nascimento, C., Machado, M., Martinez, M., Barbosa, M., Fonseca, M., Guimarães, M., et al. (2006). Association of the Bovine Major Histocompatibility Complex (BoLA) BoLA-DRB3 gene with fat and protein production and somatic cell score in Brazilian gyr dairy cattle (Bos indicus). Genet. Mol. Biol. 29, 641–647. doi: 10.1590/S1415-47572006000400011
Neefjes, J., Jongsma, M. L., Paul, P., Bakke, O. (2011). Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 11, 823–836. doi: 10.1038/nri3084
Nei, M., Chesser, R. K. (1983). Estimation of fixation indices and gene diversities. Ann. Hum. Genet. 47, 253–259. doi: 10.1111/j.1469-1809.1983.tb00993.x
Nei, M., Gojobori, T. (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426. doi: 10.1093/oxfordjournals.molbev.a040410
Nei, M., Li, W. H. (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U.S.A. 76, 5269–5273. doi: 10.1073/pnas.76.10.5269
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590. doi: 10.0000/PMID17248844
Newhouse, D. J., Balakrishnan, C. N. (2015). High major histocompatibility complex class I polymorphism despite bottlenecks in wild and domesticated populations of the zebra finch (Taeniopygia guttata). BMC Evol. Biol. 15, 015–0546. doi: 10.1186/s12862-015-0546-3
Nicholas, K. B., Nicholas, H. B., Deerfield, D. W. (1997). GeneDoc: Analysis and Visualization of Genetic Variation. EMBNew. News 4, 14.
Nielsen, M., Lundegaard, C., Blicher, T., Peters, B., Sette, A., Justesen, S., et al. (2008). Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PloS Comput. Biol. 4, 1000107. doi: 10.1371/journal.pcbi.1000107
Nikbakht, B. G., Ghorbanpour, R., Esmailnejad, A. (2016). Association of BoLA-DRB3.2 Alleles with BLV Infection Profiles (Persistent Lymphocytosis/Lymphosarcoma) and Lymphocyte Subsets in Iranian Holstein Cattle. Biochem. Genet. 54, 194–207. doi: 10.1007/s10528-016-9712-6
Nino-Vasquez, J. J., Vogel, D., Rodriguez, R., Moreno, A., Patarroyo, M. E., Pluschke, G., et al. (2000). Sequence and diversity of DRB genes of Aotus nancymaae, a primate model for human malaria parasites. Immunogenetics 51, 219–230. doi: 10.1007/s002510050035
Norimine, J., Brown, W. C. (2005). Intrahaplotype and interhaplotype pairing of bovine leukocyte antigen DQA and DQB molecules generate functional DQ molecules important for priming CD4(+) T-lymphocyte responses. Immunogenetics 57, 750–762. doi: 10.1007/s00251-005-0045-6
Patarroyo, M. E., Bermudez, A., Patarroyo, M. A. (2011). Structural and immunological principles leading to chemically synthesized, multiantigenic, multistage, minimal subunit-based vaccine development. Chem. Rev. 111, 3459–3507. doi: 10.1021/cr100223m
Porter, V., Alderson, L., Hall, S. J. G., Sponenberg, D. P. (2016). Mason’s World Encyclopedia of Livestock Breeds and Breeding (Wallingford, UK: CABI Publishing).
R_Core_Team. (2019). R: A Language and Environment for Statistical Computing (Vienna: R Foundation for Statistical Computing).
Ramos, A. E., Munoz, M., Moreno-Perez, D. A., Patarroyo, M. A. (2017). pELMO, an optimised in-house cloning vector. AMB Express 7, 017–0324. doi: 10.1186/s13568-017-0324-2
Rastislav, M., Mangesh, B. (2012). BoLA-DRB3 exon 2 mutations associated with paratuberculosis in cattle. Vet. J. 192, 517–519. doi: 10.1016/j.tvjl.2011.07.005
Rock, K. L., Reits, E., Neefjes, J. (2016). Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 37, 724–737. doi: 10.1016/j.it.2016.08.010
Rousset, F. (2008). genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106. doi: 10.1111/j.1471-8286.2007.01931.x
Rupp, R., Hernandez, A., Mallard, B. A. (2007). Association of bovine leukocyte antigen (BoLA) DRB3.2 with immune response, mastitis, and production and type traits in Canadian Holsteins. J. Dairy Sci. 90, 1029–1038. doi: 10.3168/jds.S0022-0302(07)71589-8
Sharif, S., Mallard, B. A., Wilkie, B. N., Sargeant, J. M., Scott, H. M., Dekkers, J. C., et al. (1999). Associations of the bovine major histocompatibility complex DRB3 (BoLA-DRB3) with production traits in Canadian dairy cattle. Anim. Genet. 30, 157–160. doi: 10.1046/j.1365-2052.1999.00459.x
Shiina, T., Blancher, A., Inoko, H., Kulski, J. K. (2017). Comparative genomics of the human, macaque and mouse major histocompatibility complex. Immunology 150, 127–138. doi: 10.1111/imm.12624
Sigurdardottir, S., Borsch, C., Gustafsson, K., Andersson, L. (1991). Cloning and sequence analysis of 14 DRB alleles of the bovine major histocompatibility complex by using the polymerase chain reaction. Anim. Genet. 22, 199–209. doi: 10.1111/j.1365-2052.1991.tb00670.x
Sommer, S. (2005). The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2, 16. doi: 10.1186/1742-9994-2-16
StataCorp. (2017). Stata Statistical Software. Version Release 15 (College Station, TX: StataCorp LLC).
Suarez, C. F., Patarroyo, M. E., Trujillo, E., Estupinan, M., Baquero, J. E., Parra, C., et al. (2006). Owl monkey MHC-DRB exon 2 reveals high similarity with several HLA-DRB lineages. Immunogenetics 58, 542–558. doi: 10.1007/s00251-006-0127-0
Suarez, C. F., Pabon, L., Barrera, A., Aza-Conde, J., Patarroyo, M. A., Patarroyo, M. E. (2017). Structural analysis of owl monkey MHC-DR shows that fully-protective malaria vaccine components can be readily used in humans. Biochem. Biophys. Res. Commun. 491, 1062–1069. doi: 10.1016/j.bbrc.2017.08.012
Takeshima, S. N., Aida, Y. (2006). Structure, function and disease susceptibility of the bovine major histocompatibility complex. Anim. Sci. J. 77, 138–150. doi: 10.1111/j.1740-0929.2006.00332.x
Takeshima, S., Ikegami, M., Morita, M., Nakai, Y., Aida, Y. (2001). Identification of new cattle BoLA-DRB3 alleles by sequence-based typing. Immunogenetics 53, 74–81. doi: 10.1007/s002510000293
Takeshima, S., Nakai, Y., Ohta, M., Aida, Y. (2002). Short communication: characterization of DRB3 alleles in the MHC of Japanese shorthorn cattle by polymerase chain reaction-sequence-based typing. J. Dairy Sci. 85, 1630–1632. doi: 10.3168/jds.S0022-0302(02)74234-3
Takeshima, S., Saitou, N., Morita, M., Inoko, H., Aida, Y. (2003). The diversity of bovine MHC class II DRB3 genes in Japanese Black, Japanese Shorthorn, Jersey and Holstein cattle in Japan. Gene 316, 111–118. doi: 10.1016/s0378-1119(03)00744-3
Takeshima, S., Matsumoto, Y., Chen, J., Yoshida, T., Mukoyama, H., Aida, Y. (2008). Evidence for cattle major histocompatibility complex (BoLA) class II DQA1 gene heterozygote advantage against clinical mastitis caused by Streptococci and Escherichia species. Tissue Antigens 72, 525–531. doi: 10.1111/j.1399-0039.2008.01140.x
Takeshima, S. N., Miyasaka, T., Polat, M., Kikuya, M., Matsumoto, Y., Mingala, C. N., et al. (2014). The great diversity of major histocompatibility complex class II genes in Philippine native cattle. Meta Gene 2, 176–190. doi: 10.1016/j.mgene.2013.12.005
Takeshima, S. N., Giovambattista, G., Okimoto, N., Matsumoto, Y., Rogberg-Munoz, A., Acosta, T. J., et al. (2015a). Characterization of bovine MHC class II DRB3 diversity in South American Holstein cattle populations. Tissue Antigens 86, 419–430. doi: 10.1111/tan.12692
Takeshima, S. N., Miyasaka, T., Matsumoto, Y., Xue, G., Diaz Vde, L., Rogberg-Munoz, A., et al. (2015b). Assessment of biodiversity in Chilean cattle using the distribution of major histocompatibility complex class II BoLA-DRB3 allele. Tissue Antigens 85, 35–44. doi: 10.1111/tan.12481
Takeshima, S. N., Corbi-Botto, C., Giovambattista, G., Aida, Y. (2018). Genetic diversity of BoLA-DRB3 in South American Zebu cattle populations. BMC Genet. 19, 018–0618. doi: 10.1186/s12863-018-0618-7
Takezaki, N., Nei, M., Tamura, K. (2010). POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Mol. Biol. Evol. 27, 747–745. doi: 10.1093/molbev/msp312
van Deutekom, H. W., Kesmir, C. (2015). Zooming into the binding groove of HLA molecules: which positions and which substitutions change peptide binding most? Immunogenetics 67, 425–436. doi: 10.1007/s00251-015-0849-y
Wang, K., Sun, D., Zhang, Y. (2008). Sequencing of 15 new BoLA-DRB3 alleles. Int. J. Immunogenet. 35, 331–332. doi: 10.1111/j.1744-313X.2008.00772.x
Weir, B. S., Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x
Yoshida, T., Mukoyama, H., Furuta, H., Kondo, Y., Takeshima, S. N., Aida, Y., et al. (2009a). Association of BoLA-DRB3 alleles identified by a sequence-based typing method with mastitis pathogens in Japanese Holstein cows. Anim. Sci. J. 80, 498–509. doi: 10.1111/j.1740-0929.2009.00663.x
Yoshida, T., Mukoyama, H., Furuta, H., Kondo, Y., Takeshima, S. N., Aida, Y., et al. (2009b). Association of the amino acid motifs of BoLA-DRB3 alleles with mastitis pathogens in Japanese Holstein cows. Anim. Sci. J. 80, 510–519. doi: 10.1111/j.1740-0929.2009.00664.x
Keywords: MHC, BoLA-DRB3, genetic diversity, peptide-binding region, genetic resistance, cattle
Citation: Bohórquez MD, Ordoñez D, Suárez CF, Vicente B, Vieira C, López-Abán J, Muro A, Ordóñez I and Patarroyo MA (2020) Major Histocompatibility Complex Class II (DRB3) Genetic Diversity in Spanish Morucha and Colombian Normande Cattle Compared to Taurine and Zebu Populations. Front. Genet. 10:1293. doi: 10.3389/fgene.2019.01293
Received: 29 July 2019; Accepted: 25 November 2019;
Published: 10 January 2020.
Edited by:
Antonio González-Martín, Complutense University of Madrid, SpainReviewed by:
Cedric Gondro, Michigan State University, United StatesCopyright © 2020 Bohórquez, Ordoñez, Suárez, Vicente, Vieira, López-Abán, Muro, Ordóñez and Patarroyo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel Alfonso Patarroyo, bWFwYXRhcnIuZmlkaWNAZ21haWwuY29t
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.