 Yixi Sun1,2,3
Yixi Sun1,2,3 Yuqin Luo1,2,3Yeqing Qian1,2,3
Yuqin Luo1,2,3Yeqing Qian1,2,3 Min Chen1,2,3Liya Wang1,2,3Hongge Li1,2,3Yu Zou4
Min Chen1,2,3Liya Wang1,2,3Hongge Li1,2,3Yu Zou4 Minyue Dong1,2,3*
Minyue Dong1,2,3*- 1Department of Reproductive Genetics, Women’s Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 2Key Laboratory of Reproductive Genetics, Ministry of Education, Zhejiang University, Hangzhou, China
- 3Key Laboratory of Women’s Reproductive Health of Zhejiang Province, Zhejiang University, Hangzhou, China
- 4Department of Diagnostic Radiology, Women’s Hospital, School of Medicine, Zhejiang University, Hangzhou, China
Skewed X-chromosome inactivation (XCI) plays an important role in the phenotypic heterogeneity of X-linked disorders. However, the role of skewed XCI in XCI-escaping gene SHOX regulation is unclear. Here, we focused on a heterozygous deletion of SHOX gene enhancer with clinical heterogeneity. Using SNP array, we detected that the female proband with Leri-Weill dyschondrosteosis (LWD) carried an 857 kb deletion on Xp22.3 (encompassing SHOX enhancer) and a 5,707 kb large-fragment deletion on Xq25q26. XCI analysis revealed that the X-chromosome with the Xq25q26 large-fragment deletion was completely inactivated, which forced the complete activation of the other X-chromosome carrying SHOX enhancer deletion. While the Xp22.3 deletion locates on the escaping XCI region, under the combined action of skewed XCI and escaping XCI, transcription of SHOX gene was mainly from the activated X-chromosome with SHOX enhancer defect, involving in the formation of LWD phenotype. Interestingly, this SHOX enhancer deletion was inherited from her healthy mother, who also demonstrated completely skewed XCI. However, the X-chromosome with SHOX enhancer deletion was inactivated, and the normal X-chromosome was activated. Combing with escaping XCI, her phenotype was almost normal. In summary, this study was a rare report of SHOX gene enhancer deletion in a family with clinical heterogeneity due to skewed inactivation of different X-chromosomes, which can help in the genetic counseling and prenatal diagnosis of disorders in females with SHOX defect.
Introduction
The short stature homeobox gene (SHOX), locating in the pseudoautosomal region (PAR1) of the short arm of the X and Y chromosomes, is one of the major growth genes in humans. In 1997, the SHOX gene was linked with the occurrence of short stature in Turner syndrome for the first time (Rao et al., 1997). Subsequently, SHOX haploinsufficiency has been demonstrated in individuals exhibiting different phenotypes, ranging from idiopathic short stature (ISS) to Lėri-Weill dyschondrosteosis (LWD) (Fukami et al., 2016). In LWD, the classic clinical features observed are short stature and Madelung deformity, which are also characterized by abnormal alignment of the radius, ulna, and carpal bones of the wrist (Seki et al., 2014). Additionally, a loss of both copies of SHOX results in the occurrence of Langer mesomelic dysplasia (LMD), which is a more severe disorder (Shears et al., 2002; Zinn et al., 2002).
The genetic defects underlying SHOX haploinsufficiency include copy-number variations (CNVs) and mutations, which not only occur in the coding region, but also in the regulatory elements of the SHOX gene (Binder, 2011). Among the regulatory elements, highly conserved non-coding DNA elements (CNEs) located several hundred kilobases downstream of SHOX, they have previously been identified as enhancers (Sabherwal et al., 2007). Numerous studies have shown that the SHOX gene enhancer plays an important role in the regulation of this gene to achieve optimal transcriptional efficiency. Furthermore, certain defects in the coding region along with deletions in the SHOX gene enhancer can also cause ISS or LWD, but the combined proportion in which both need to occur has not been well defined, since highly variable phenotypes are observed even if the same mutations run with in a family (Binder et al., 2004). Thus, a detailed study to determine the cause of these phenotypic differences was required.
X-chromosome inactivation (XCI) plays an important role in the phenotypic heterogeneity of X-linked disorders in females (Plenge et al., 2002; Renault et al., 2011; Sankaran et al., 2015; Torres and Puig, 2017; Juchniewicz et al., 2018). Due to skewed XCI, the same mutation in X-linked genes may result in the different phenotypes (Orstavik, 2009). While it was reported that skewed XCI might affect the phenotypes of patients with Xp22.3 complex rearrangement (Suzuki et al., 2016). However, the role of skewed XCI in the XCI-escaping gene SHOX was unclear. In this study, we have evaluated a case in which deletion of the SHOX enhancer was observed among members of a family with clinical heterogeneity due to differently skewed XCI. In this family, the proband with heterozygous deletion of the SHOX enhancer was a patient with LWD, although her mother harboring the same deletion was found to be almost healthy.
Case Presentation
The proband II2 is a 35-year-old Chinese woman with a short stature (150 cm, −1.1SD), bilateral Madelung deformity, bowing of the radius, and mesomelia affecting the arms, in particular, which was a typical LWD phenotype (Figure 1A). However, her parents and other family members were found to be healthy. It was observed that the mother was healthy with a height of 163cm (+1.1SD) and completely normal arms (Figure 1B).
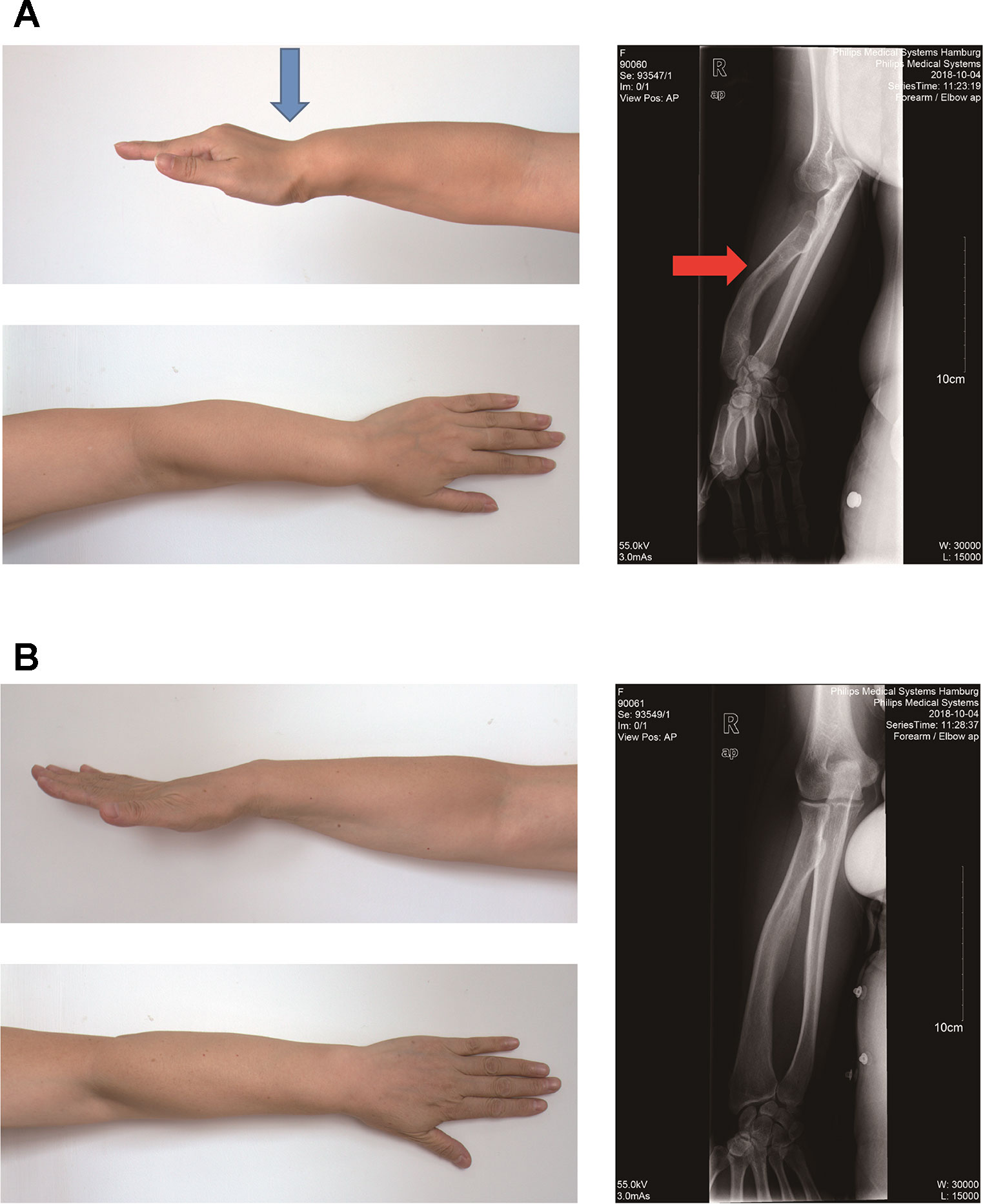
Figure 1 Photograph of the forearm of the proband II2 and mother I2. (A). Photograph of the forearm of the proband II2 showing a Madelung deformity and bowing of the radius (blue and red arrows). This photograph shows the curve and shortening of the forearm, hand and wrist, the structure of which appears like that of a dinner fork. (B). Photograph of the forearm of the mother I2 showing a normal phenotype.
The proband II2 arriving at our hospital was a pregnant female who was in her 14th week of gestation requesting for a non-invasive prenatal testing (NIPT). Triple tests on chromosomes 13, 18, and 21 showed a low risk in NIPT; however, a 6 Mb deletion in the long arm of the X-chromosome (Z-score was −19.43) was detected, which was anticipated to have a maternal origin (Figure 2A). In order to confirm the risk of this genetic disorder to the fetus, amniocentesis was subsequently conducted. Karyotyping and SNP array were performed and showed the presence of an 857 kb deletion in Xp22.33 (chrX: 784,064–1,640,746) in the male fetus III1. To verify the results, SNP array was also performed on samples of the pregnant woman II2 and her husband II1. Surprisingly, not only a deletion of 857 kb in Xp22.33, but also a 5,707 kb deletion (chrX: 127,915,006–133,621,667) in Xq25q26.3 was observed in the woman, the location of which was consistent with the result of the fetal NIPT (Figures 2B, C and Supplementary Table 2). SNP array results have been deposited in Gene Expression Omnibus (GEO), the accession number is GSE138489, as appended below: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138489. To explore the reason underlying the phenotypic differences between the proband II2 with LWD and her healthy mother I2, we also performed a PCR-based HUMARA assay to assess XCI patterns.
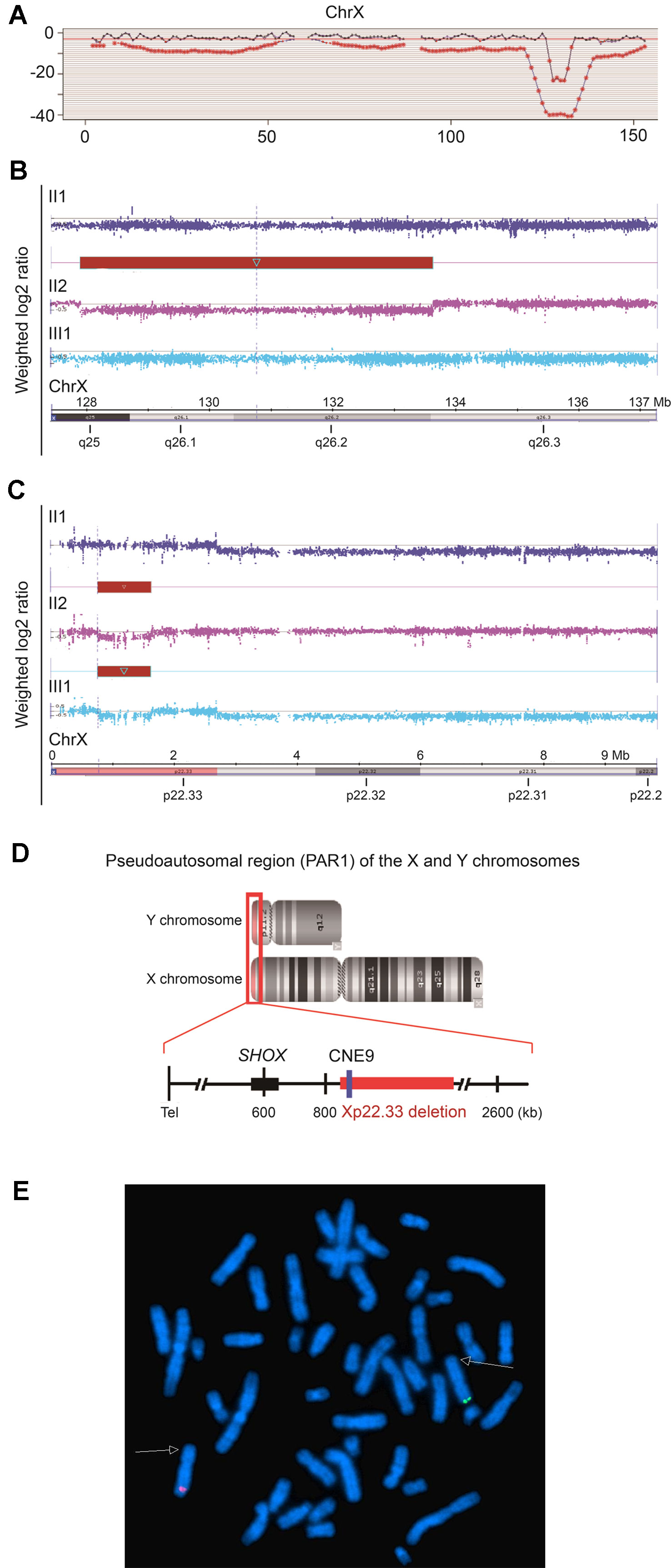
Figure 2 Characteristics of the 5,707 kb and 857 kb deletions of Xq25q26.3 and Xp22.33, respectively. (A). Results of cfDNA screening of the pregnant proband. cfDNA screening study of the maternal plasma, illustrating an uncertain 6 Mb deletion in the long arm of the X-chromosome (128M–133M), Z score = −19.43. (B and C). SNP array analysis of the fetus (III1) and the couples (II1, II2). (B). The red bar indicates a heterozygous 5,707 kb deletion in Xq25q26.3 (chrX: 127,915,006–133,621,667) in the fetus (III1). (C). The red bar indicates a heterozygous 857 kb deletion in Xp22.33 (chrX: 784,064–1,640,746) in the fetus (III1) and the pregnant proband (II2). (D). Pseudoautosomal region (PAR1) of the X and Y chromosomes. The 857 kb deletion in Xp22.33 (red bar) was located 164 kb downstream of the SHOX gene (chrX: 585,079–620,146), including the evolutionarily conserved CNE9 (blue bar), which was the SHOX gene enhancer. (E). For the proband II2, FISH experiment showed that the Xp22.3 deletion (detected by the probe RP11-1119O18, Spectrum Green) and the Xq25q26 deletion (detected by the probe RP11-313D19, Spectrum Red) located on the different X chromosomes, respectively.
Finally, the fetus III1 with Xp22.33 deletion was born, his length was 50cm (−0.2 SD), weight 3.3 kg (+0 SD). On the latest examination at 8 months, his developmental milestones were almost normal, and his length was 72 cm (+0.3 SD), weight 11.7 kg (+2.2 SD).
This study was carried out in accordance with the recommendations of Ethics Committee of Women’s Hospital, School of Medicine Zhejiang University, and an informed consent was acquired from all the participants of this study in accordance with the Declaration of Helsinki. The study protocol was approved by the Review Board of the Women’s Hospital, School of Medicine, Zhejiang University in China.
Materials and Methods
Materials and methods were in Supplementary Materials and Supplementary Table 1.
Results
In order to explore the possible causes of the disease, we analyzed the two deletions (857 kb and 5,707 kb).The 5,707 kb deletion in Xq25q26.3 included 29 Online Mendelian Inheritance in Man (OMIM) genes. Among these, 9 genes (bold) were found to be morbid and might be associated with many X-linked diseases (Supplementary Table 2) but not with skeletal development. Additionally, the 857 kb deletion in Xp22.33 included 11 OMIM genes in the pseudoautosomal region (PAR1) of the X and Y chromosomes (Figures 2B–D and Supplementary Table 2). Interestingly, the SHOX gene enhancer, which is closely associated with skeletal development, was also located in the deletion region of Xp22.33. Further analysis revealed that it was located 164 kb downstream of the SHOX gene (chrX: 585,079–620,146) and included the evolutionarily conserved non-coding DNA element 9 (CNE9), which was the SHOX gene enhancer. We speculated that the 857 kb deletion in Xp22.33 had contributed to the SHOX gene mutation resulting in the LWD phenotype in case of the proband.
Although no other patients were exhibiting the LWD phenotype in her family, we detected the gene dosage of CRLF2 and CNE9 (the SHOX gene enhancer in the deletion region of Xp22.33), AIFM1 and FRMD7 (deletion region of Xq25q26.3) of the female proband and her parents by qPCR (Figure 3). The Xp22.33 regions were both located in PAR1 of the X and Y chromosomes. Data obtained from a normal female was used as the control, and the dosages of CNE9 and CRLF2 were normal in I1 and II1, and half in I2, II2, and III1 (Figures 3A, B). On the other hand, the Xq25q26.3 region existed only in the X chromosome. The gene dosages of AIFM1 and FRMD7 were normal in female I2, and half in I1, II1, II2, and III1 (Figures 3C, D). Above all, the Xp22.33 deletion in the proband II2 was inherited from her healthy mother I2, and the Xq25q26.3 deletion occurred de novo.
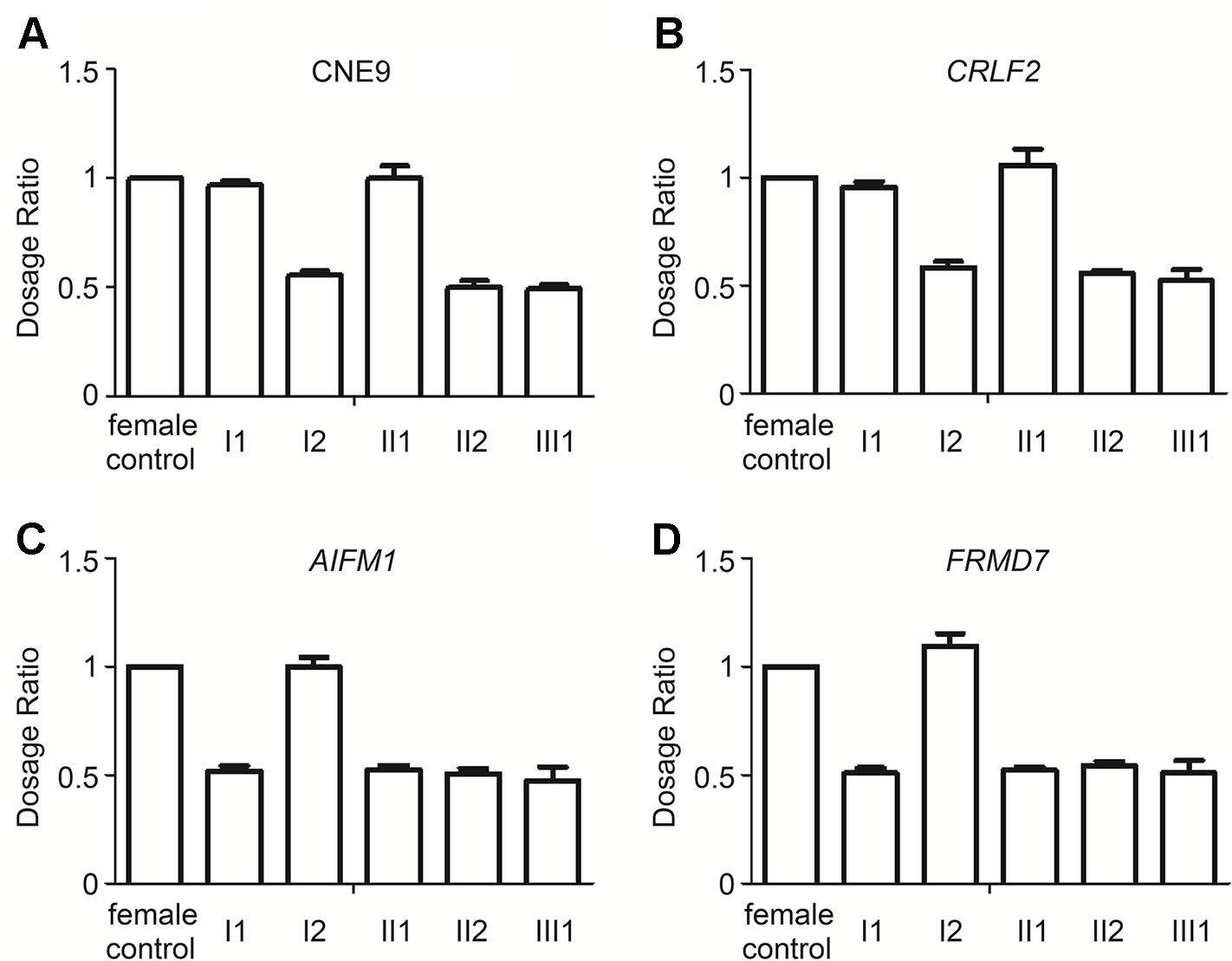
Figure 3 Relative ratio of the Xp22.33 (CNE9 and CRLF2) and Xq25q26.3 (AIFM1 and FRMD7) regions in the family by qPCR. (A and B). Xp22.33 region (CNE9 and CRLF2) was located in PAR1 of the X and Y chromosomes, including the SHOX enhancer. Dosages in normal female were equal, Xp22.33 region dosages in I1 and II1 were normal, and those in I2, II2, III1 with Xp22.33 deletion were half. (C and D) The AIFM1 and FRMD7 were present only in the X-chromosome. The AIFM1 and FRMD7 dosage in I2 were normal, while that in I1, II1, II2, III1 with Xq25q26.3 deletion was half. From the above data, it was elucidated that the Xp22.33 deletion in the proband II2 was inherited from her healthy mother I2, and the Xq25q26.3 deletion occurred de novo.
We further explored the reason underlying the phenotypic differences between the proband II2 with LWD and her healthy mother I2. As presented in Figure 4, a PCR-based HUMARA assay was performed to assess XCI patterns in II2 and I2. After digestion with the methylation-sensitive restriction enzyme HpaII, it was found that only the inactive X-chromosome could synthesize PCR products. The origin of the inactivated X-chromosome was determined by segregation analysis. The undigested PCR product of II2 gave two peaks of 274 bp and 280 bp, respectively. A single peak representing 274 bp was observed for the HpaII-digested product (Figure 4B), which indicated the inactivated allele inherited from her father I1, in whose samples one peak of 274 bp was assayed by PCR. The assay of the undigested PCR product of the mother I2 gave two peaks of 280 bp and 283 bp. A single peak of 280 bp was obtained by assaying the HpaII-digested product, which was different from that of the inactivated allele of II2. The above results showed that complete (100%) skewing of XCI was found in the proband II2 and her mother I2, which was different from that of the inactivated X-chromosome (Figures 4C, D). Segregation analysis revealed that the normal X-chromosome from her father I1 was completely inactivated (Figure 4A).
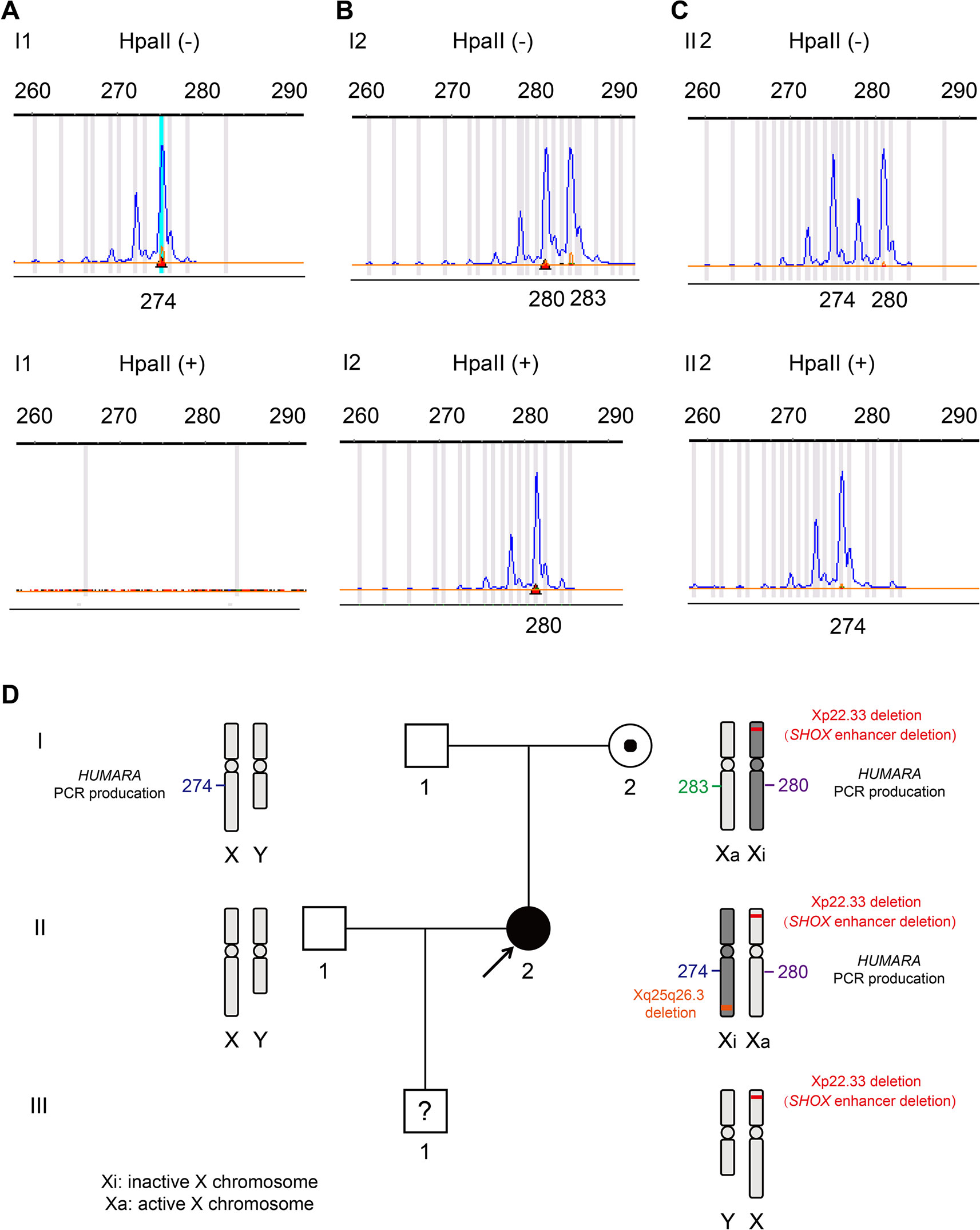
Figure 4 X-chromosome inactivation (XCI) pattern and linkage analyses were based on the polymorphic CAG repeat in exon 1 of the gene for androgen receptor (HUMARA). (A). A peak of 274 bp for HUMARA was observed by assaying the undigested PCR product of I1 and no peaks were observed for the HpaII digested product. (B). The undigested PCR product of I2 gave two peaks of 280 and 283 bp, each, while only one peak of 280 bp was observed with the HpaII digested product. I2 exhibited 100% skewing of XCI, and the inactivated X-chromosome was linked with the 280 bp peak of the HUMARA PCR products. (C). The undigested PCR product of the proband II2 gave two peaks of 274 and 280 bp. One X-chromosome linked with the 280 bp peak of HUMARA was inhibited from the mother I2 and the other from the father I1. The product of HpaII digestion gave only one peak of 274 bp. II2 also demonstrated 100% skewing of XCI, but the inactivated X-chromosome was linked with the 274 bp peak of AR, which is different from that of the mother I2. (D). Schematic diagram of Xp22.33 and Xq25q26.3 deletions, and HUMARA PCR products in the pedigree. It can be seen that Xp22.33 and Xq25q26.3 deletions in II2 are located on different X-chromosomes. Xp22.33 deletion (SHOX enhancer deletion) of I2 occurred in the X-chromosome, whose allele was linked with the 280 bp peak of HUMARA PCR. The X-chromosome was inactivated and delivered to the proband I2, but her X-chromosome was activated.
Figure 4D gives aschematic diagram of Xp22.33 and Xq25q26.3 deletions, and PCR product of HUMARA in the pedigree. Only Xp22.33 deletion (not Xq25q26.3 deletion) derived from II2 was seen in III1. Meanwhile, the FISH detection showed that the deletions of Xp22.33 and Xq25q26.3 in II2 were located on different X-chromosomes (Figure 2E). Both, Xp22.33 deletion (SHOX enhancer deletion) and 280 bp peak of HUMARA PCR products in II2, were derived from I2, which were linked to the same X-chromosome. However, the allele which gave a peak of 280 bp after HUMARA PCR was found to be activated in II2, but inactivated in I2 (Figures 4B, C).
Discussion
Microdeletions in the region downstream of SHOX have been reported as the most common genetic defects in patients with LWD (Benito-Sanz et al., 2006; Marchini et al., 2016). Several evolutionarily conserved non-coding elements located downstream of SHOX (for example CNE4, CNE5, ECR1, and ECS4/CNE9), are known to act as enhancers (Benito-Sanz et al., 2005; Fukami et al., 2006; Huber et al., 2006; Rappold et al., 2007), and interactions between the SHOX gene and CNEs have been verified using in vitro and in vivo assays (Kenyon et al., 2011; Verdin et al., 2015). Moreover, the deletion of our case also included the limb enhancer with 563 basepair (bp) (chrX: 827,128–827,691), which had specific activity in the limb regions where SHOX functions, and it also contributed to the pathogenicity of deletions downstream of SHOX (Skuplik et al., 2018). In addition, microdeletions in the region that is further downstream of the previously known CNEs have recently been identified in patients with LWD features or short stature (Chen et al., 2009). Therefore, we concluded that the Xp22.33 deletion resulted in SHOX gene defect and contributed to the manifestation of abnormal skeletal phenotype in the proband. Interestingly, the proband was a patient with LWD, but her mother harboring the Xp22.33 deletion was almost normal.
Individuals with SHOX defect (for example CNE9 deletion) have a phenotype ranging from normal to LWD (Benito-Sanz et al., 2012; Bunyan et al., 2013; Marchini et al., 2016). Phenotypic differences have been explained by several hypotheses. For example, as a genetic modifier, CYP26C1 variants can effect clinical manifestations of SHOX deficiency (Montalbano et al., 2016). For another example, it has described skewed X-inactivation patterns, which were caused by X-chromosomal rearrangements, can also effect phenotypes of Xp22.3 defect (Suzuki et al., 2016; Ogushi et al., 2019), which gives us an inspiration.
This further indicated the presence of differently skewed XCI in the proband and her mother, which suggested that clinical heterogeneity resulting from the deletion of the SHOX gene enhancer was caused by the skewed XCI. While SHOX is also an XCI-escaping gene (Carrel and Willard, 2005; Blaschke and Rappold, 2006; Sun et al., 2017). Skewed XCI and escaping XCI are both involved in our case. Expressions of the XCI-escaping genes on two X chromosome were not always equal, XCI-escaping genes in active X chromosome (Xa) were frequently in the state of preferential expression. LAURA CARREL et al. has indicated that the XCI-escaping gene REP1 was also expressed from the inactive chromosome (Xi), but the level of expression relative to Xa was reduced, it showed that skewed XCI were also existed in XCI-escaping genes (Carrel and Willard, 1999). In addition, Nathalie Fieremans et al. has also indicated that escaping XCI was often partial and incomplete with a lower expression from the inactive X chromosome. Skewed XCI of the XCI-escaping genes DDX3X and SMC1A were revealed in intellectual disability female patients (Fieremans et al., 2016). So we speculated that the SHOX enhancer was similar, based on the skewed XCI and escaping XCI theory, we can explain the current case reasonably.
According to one-hit of skewed XCI, defect in a single X-chromosome results in its inactivation, in order to remedy the defect. A large proportion of female carriers of severe X-linked disorders are asymptomatic and have severely skewed XCI, probably because of selectively mediated favorable skewing, thus suggesting preferential X-inactivation against the chromosome that harbors the mutation (Orstavik, 2009), for example ATR-X syndrome (Gibbons et al., 1992), dyskeratosis congenital (Ferraris et al., 1997), X-linked agammaglobulinemia (Moschese et al., 2000), and severe combined immunodeficiency (Li et al., 1998). In our study, X-chromosomes with deletion of the SHOX gene enhancer were completely inactivated in the proband’s mother I2, and the activated chromosomes were normal. Meanwhile, due to XCI-escaping, we speculated that transcription of SHOX was mainly from the activated X chromosome, which was normal. So it was observed that I2 was almost healthy and did not exhibit severe LWD phenotype.
According to the two-hit model of skewed XCI, if both the X-chromosomes are defective, the mutated X-chromosome with a more harmful mutation will be completely inactivated to avoid its adverse reaction, forcing the other to become active (Plenge et al., 2002; Orstavik, 2009). The hypothesis of “female X-linked two-hit model” has been used to support studies on multiple disorders, including X-linked intellectual disability (Plenge et al., 2002), MECP2 duplication (Fieremans et al., 2014), and the Wiskott-Aldrich syndrome (Daza-Cajigal et al., 2013).
In our study, a deletion of 857 kb (including deletion of the SHOX gene enhancer) was found on one of the X-chromosomes of the proband II2, and another 5,707 kb deletion on the other. The larger deletion was likely more harmful. Hence, the X-chromosome with the 5,707 kb deletion was totally inactivated, and the other one with the SHOX gene enhancer deletion (857 kb deletion) was completely activated. Meanwhile, due to locating on XCI-escaping region, transcription of SHOX was partially from the inactive chromosome, but it was mainly origin from the active X-chromosome with SHOX gene enhancer deletion. Combing effect of skewed XCI and escaping XCI, the proband II2 presented severe LWD phenotype.
No skewed XCI were observed in the male. Perhaps the fetus III1 was too young to present abnormal phenotypes. It was reported that, during childhood, there was probably no relevant additional loss of height in patients with SHOX defect (Binder and Rappold, 1993), while mesomelic disproportion of the skeleton with shortening of the extremities can be evident first until in school-aged children and increase with age in frequency and severity (Ross et al., 2001). Although the phenotype of III1 is now normal, it doesn’t mean that it would be always normal, and final phenotype should be observed for long time.
XCI values depend on the tissues, but a general concordance of XCI patterns was observed among tissues from the same person (Bittel et al., 2008). Recently, Wen-Bin He et al. indicated that XCI pattern of amniocytes can predict the risk of dystrophinopathy in fetal carriers of DMD mutations (He et al., 2019). Therefore, we speculated XCI analysis in amniocytes cells may also contribute to predicting the phenotype of fetus with SHOX defect.
In summary, here we described a rare case of XCI–escaping gene SHOX enhancer deletion in a family with obvious clinical heterogeneity due to skewing inactivation of different X-chromosomes. Furthermore, we underlined the key role of skewed XCI and escaping XCI in the phenotype of X-linked disorders in females. It can help in the genetic counseling and prenatal diagnosis of disorders in females with SHOX defect.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: GSE138489 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138489).
Ethics Statement
The studies involving human participants were reviewed and approved by Review Board of the Women’s Hospital, School of Medicine, Zhejiang University in China. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
MD designed the study. NIPT was carried out by HL; YS, and LW contributed the qPCR detection. YL and YQ performed the karyotyping, FISH and SNP array. XCI analysis was performed by YS and MC. YS and MD wrote the draft manuscript. YZ provided the imagological examination. All co-authors provided feedback on the estimates and contributed to the subsequent versions of the manuscript. All authors read and approved the final version of the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (Grant Nos.81801441 and 81300532), the Key Research and Development Program of the Zhejiang province (Grant No.2019C03025), the National Key Research and Development Program of China (Grant Nos.2016YFC1000703 and 2018YFC1002702).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We also thank Dr. Jiong Gao (BGI Genomics, BGI-Shenzhen, Shenzhen 518083, China) for his assistance in the preparation of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01086/full#supplementary-material
References
Benito-Sanz, S., del Blanco, D. G., Aza-Carmona, M., Magano, L. F., Lapunzina, P., Argente, J., et al. (2006). PAR1 deletions downstream of SHOX are the most frequent defect in a Spanish cohort of Leri-Weill dyschondrosteosis (LWD) probands. Hum. Mutat. 27 (10), 1062. doi: 10.1002/humu.9456
Benito-Sanz, S., Royo, J. L., Barroso, E., Paumard-Hernandez, B., Barreda-Bonis, A. C., Liu, P., et al. (2012). Identification of the first recurrent PAR1 deletion in Leri-Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. J. Med. Genet. 49 (7), 442–450. doi: 10.1136/jmedgenet-2011-100678
Benito-Sanz, S., Thomas, N. S., Huber, C., Gorbenko del Blanco, D., Aza-Carmona, M., Crolla, J. A., et al. (2005). A novel class of Pseudoautosomal region 1 deletions downstream of SHOX is associated with Leri-Weill dyschondrosteosis. Am. J. Hum. Genet. 77 (4), 533–544. doi: 10.1086/449313
Binder, G. (2011). Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm. Res. Paediatr. 75 (2), 81–89. doi: 10.1159/000324105
Binder, G., Rappold, G. A. (1993). “SHOX Deficiency Disorders,” in GeneReviews((R)). Eds. Adam, M. P., Ardinger, H. H., Pagon, R. A., Wallace, S. E., Bean, L. J. H., Stephens, K., Amemiya, A.(Seattle (WA : University of Washington)).
Binder, G., Renz, A., Martinez, A., Keselman, A., Hesse, V., Riedl, S. W., et al. (2004). SHOX haploinsufficiency and Leri-Weill dyschondrosteosis: prevalence and growth failure in relation to mutation, sex, and degree of wrist deformity. J. Clin. Endocrinol. Metab. 89 (9), 4403–4408. doi: 10.1210/jc.2004-0591
Bittel, D.C., Theodoro, M.F., Kibiryeva, N., Fischer, W., Talebizadeh, Z., Butler, M.G. (2008). Comparison of X-chromosome inactivation patterns in multiple tissues from human females. J. Med. Genet. 45 (5), 309–313. doi: 10.1136/jmg.2007.055244.
Blaschke, R. J., Rappold, G. (2006). The pseudoautosomal regions, SHOX and disease. Curr. Opin. Genet. Dev. 16 (3), 233–239. doi: 10.1016/j.gde.2006.04.004
Bunyan, D. J., Baker, K. R., Harvey, J. F., Thomas, N. S. (2013). Diagnostic screening identifies a wide range of mutations involving the SHOX gene, including a common 47.5 kb deletion 160 kb downstream with a variable phenotypic effect. Am. J. Med. Genet. A 161A (6), 1329–1338. doi: 10.1002/ajmg.a.35919
Carrel, L., Willard, H. F. (1999). Heterogeneous gene expression from the inactive X chromosome: an X-linked gene that escapes X inactivation in some human cell lines but is inactivated in others. Proc. Natl. Acad. Sci. U.S.A. 96 (13), 7364–7369. doi: 10.1073/pnas.96.13.7364
Carrel, L., Willard, H. F. (2005). X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434 (7031), 400–404. doi: 10.1038/nature03479
Chen, J., Wildhardt, G., Zhong, Z., Roth, R., Weiss, B., Steinberger, D., et al. (2009). Enhancer deletions of the SHOX gene as a frequent cause of short stature: the essential role of a 250 kb downstream regulatory domain. J. Med. Genet. 46 (12), 834–839. doi: 10.1136/jmg.2009.067785
Daza-Cajigal, V., Martinez-Pomar, N., Garcia-Alonso, A., Heine-Suner, D., Torres, S., Vega, A. K., et al. (2013). X-linked thrombocytopenia in a female with a complex familial pattern of X-chromosome inactivation. Blood Cells Mol. Dis. 51 (2), 125–129. doi: 10.1016/j.bcmd.2013.04.004
Ferraris, A. M., Forni, G. L., Mangerini, R., Gaetani, G. F. (1997). Nonrandom X-chromosome inactivation in hemopoietic cells from carriers of dyskeratosis congenita. Am. J. Hum. Genet. 61 (2), 458–461. doi: 10.1016/S0002-9297(07)64075-0
Fieremans, N., Bauters, M., Belet, S., Verbeeck, J., Jansen, A. C., Seneca, S., et al. (2014). De novo MECP2 duplications in two females with intellectual disability and unfavorable complete skewed X-inactivation. Hum. Genet. 133 (11), 1359–1367. doi: 10.1007/s00439-014-1469-6
Fieremans, N., Van Esch, H., Holvoet, M., Van Goethem, G., Devriendt, K., Rosello, M., et al. (2016). Identification of Intellectual Disability Genes in Female Patients with a Skewed X-Inactivation Pattern. Hum. Mutat. 37 (8), 804–811. doi: 10.1002/humu.23012
Fukami, M., Kato, F., Tajima, T., Yokoya, S., Ogata, T. (2006). Transactivation function of an approximately 800-bp evolutionarily conserved sequence at the SHOX 3’ region: implication for the downstream enhancer. Am. J. Hum. Genet. 78 (1), 167–170. doi: 10.1086/499254
Fukami, M., Seki, A., Ogata, T. (2016). SHOX Haploinsufficiency as a Cause of Syndromic and Nonsyndromic Short Stature. Mol. Syndromol. 7 (1), 3–11. doi: 10.1159/000444596
Gibbons, R. J., Suthers, G. K., Wilkie, A. O., Buckle, V. J., Higgs, D. R. (1992). X-linked alpha-thalassemia/mental retardation (ATR-X) syndrome: localization to Xq12-q21.31 by X inactivation and linkage analysis. Am. J. Hum. Genet. 51 (5), 1136–1149.
He, W. B., Du, J., Xie, P. Y., Zhou, S., Zhang, Y. X., Lu, G. X., et al. (2019). X-chromosome inactivation pattern of amniocytes predicts the risk of dystrophinopathy in fetal carriers of DMD mutations. Prenat. Diagn. 39 (8), 603–608. doi: 10.1002/pd.5473
Huber, C., Rosilio, M., Munnich, A., Cormier-Daire, V., French, S. G. M. (2006). High incidence of SHOX anomalies in individuals with short stature. J. Med. Genet. 43 (9), 735–739. doi: 10.1136/jmg.2006.040998
Juchniewicz, P., Kloska, A., Tylki-Szymanska, A., Jakobkiewicz-Banecka, J., Wegrzyn, G., Moskot, M., et al. (2018). Female fabry disease patients and X-chromosome inactivation. Gene 641, 259–264. doi: 10.1016/j.gene.2017.10.064
Kenyon, E. J., McEwen, G. K., Callaway, H., Elgar, G. (2011). Functional analysis of conserved non-coding regions around the short stature hox gene (shox) in whole zebrafish embryos. PloS One 6 (6), e21498. doi: 10.1371/journal.pone.0021498
Li, S. L., Ting, S. S., Lindeman, R., Ffrench, R., Ziegler, J. B. (1998). Carrier identification in X-linked immunodeficiency diseases. J. Paediatr. Child Health 34 (3), 273–279. doi: 10.1046/j.1440-1754.1998.00216.x
Marchini, A., Ogata, T., Rappold, G. A. (2016). A track record on SHOX: from basic research to complex models and therapy. Endocr. Rev. 37 (4), 417–448. doi: 10.1210/er.2016-1036
Montalbano, A., Juergensen, L., Roeth, R., Weiss, B., Fukami, M., Fricke-Otto, S., et al. (2016). Retinoic acid catabolizing enzyme CYP26C1 is a genetic modifier in SHOX deficiency. EMBO Mol. Med. 8 (12), 1455–1469. doi: 10.15252/emmm.201606623
Moschese, V., Orlandi, P., Plebani, A., Arvanitidis, K., Fiorini, M., Speletas, M., et al. (2000). X-chromosome inactivation and mutation pattern in the Bruton’s tyrosine kinase gene in patients with X-linked agammaglobulinemia. Italian XLA Collaborative Group. Mol. Med. 6 (2), 104–113.
Ogushi, K., Hattori, A., Suzuki, E., Shima, H., Izawa, M., Yagasaki, H., et al. (2019). DNA Methylation Status of SHOX-Flanking CpG Islands in Healthy Individuals and Short Stature Patients with Pseudoautosomal Copy Number Variations. Cytogenet. Genome Res. 158, 56–62. doi: 10.1159/000500468
Orstavik, K. H. (2009). X chromosome inactivation in clinical practice. Hum. Genet. 126 (3), 363–373. doi: 10.1007/s00439-009-0670-5
Plenge, R. M., Stevenson, R. A., Lubs, H. A., Schwartz, C. E., Willard, H. F. (2002). Skewed X-chromosome inactivation is a common feature of X-linked mental retardation disorders. Am. J. Hum. Genet. 71 (1), 168–173. doi: 10.1086/341123
Rao, E., Weiss, B., Fukami, M., Rump, A., Niesler, B., Mertz, A., et al. (1997). Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet. 16 (1), 54–63. doi: 10.1038/ng0597-54
Rappold, G., Blum, W. F., Shavrikova, E. P., Crowe, B. J., Roeth, R., Quigley, C. A., et al. (2007). Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J. Med. Genet. 44 (5), 306–313. doi: 10.1136/jmg.2006.046581
Renault, N. K., Renault, M. P., Copeland, E., Howell, R. E., Greer, W. L. (2011). Familial skewed X-chromosome inactivation linked to a component of the cohesin complex, SA2. J. Hum. Genet. 56 (5), 390–397. doi: 10.1038/jhg.2011.25
Ross, J. L., Scott, C., Marttila, P., Kowal, K., Nass, A., Papenhausen, P., et al. (2001). Phenotypes Associated with SHOX Deficiency. J. Clin. Endocrinol. Metab. 86 (12), 5674–5680. doi: 10.1210/jcem.86.12.8125
Sabherwal, N., Bangs, F., Roth, R., Weiss, B., Jantz, K., Tiecke, E., et al. (2007). Long-range conserved non-coding SHOX sequences regulate expression in developing chicken limb and are associated with short stature phenotypes in human patients. Hum. Mol. Genet. 16 (2), 210–222. doi: 10.1093/hmg/ddl470
Sankaran, V. G., Ulirsch, J. C., Tchaikovskii, V., Ludwig, L. S., Wakabayashi, A., Kadirvel, S., et al. (2015). X-linked macrocytic dyserythropoietic anemia in females with an ALAS2 mutation. J. Clin. Invest. 125 (4), 1665–1669. doi: 10.1172/JCI78619
Seki, A., Jinno, T., Suzuki, E., Takayama, S., Ogata, T., Fukami, M. (2014). Skeletal deformity associated with SHOX deficiency. Clin. Pediatr. Endocrinol. 23 (3), 65–72. doi: 10.1297/cpe.23.65
Shears, D. J., Guillen-Navarro, E., Sempere-Miralles, M., Domingo-Jimenez, R., Scambler, P. J., Winter, R. M. (2002). Pseudodominant inheritance of Langer mesomelic dysplasia caused by a SHOX homeobox missense mutation. Am. J. Med. Genet. 110 (2), 153–157. doi: 10.1002/ajmg.10421
Skuplik, I., Benito-Sanz, S., Rosin, J. M., Bobick, B. E., Heath, K. E., Cobb, J. (2018). Identification of a limb enhancer that is removed by pathogenic deletions downstream of the SHOX gene. Sci. Rep. 8 (1), 14292. doi: 10.1038/s41598-018-32565-1
Sun, Y. X., Zhang, Y. X., Zhang, D., Xu, C. M., Chen, S. C., Zhang, J. Y., et al. (2017). XCI-escaping gene KDM5C contributes to ovarian development via downregulating miR-320a. Hum. Genet. 136 (2), 227–239. doi: 10.1007/s00439-016-1752-9
Suzuki, E., Shima, H., Toki, M., Hanew, K., Matsubara, K., Kurahashi, H., et al. (2016). Complex X-chromosomal rearrangements in two women with ovarian dysfunction: implications of chromothripsis/chromoanasynthesis-dependent and -independent origins of complex genomic alterations. Cytogenet. Genome Res. 150 (2), 86–92. doi: 10.1159/000455026
Torres, R. J., Puig, J. G. (2017). Skewed X inactivation in Lesch-Nyhan disease carrier females. J. Hum. Genet. 62 (12), 1079–1083. doi: 10.1038/jhg.2017.88
Verdin, H., Fernandez-Minan, A., Benito-Sanz, S., Janssens, S., Callewaert, B., De Waele, K., et al. (2015). Profiling of conserved non-coding elements upstream of SHOX and functional characterisation of the SHOX cis-regulatory landscape. Sci. Rep. 5, 17667. doi: 10.1038/srep17667
Keywords: SHOX gene enhancer, Leri-Weill dyschondrosteosis, skewed X-chromosome inactivation (XCI), clinical heterogeneity, HUMARA assay, escaping X-chromosome inactivation (XCI)
Citation: Sun Y, Luo Y, Qian Y, Chen M, Wang L, Li H, Zou Y and Dong M (2019) Heterozygous Deletion of the SHOX Gene Enhancer in two Females With Clinical Heterogeneity Associating With Skewed XCI and Escaping XCI. Front. Genet. 10:1086. doi: 10.3389/fgene.2019.01086
Received: 06 June 2019; Accepted: 09 October 2019;
Published: 06 November 2019.
Edited by:
Yueqiu Tan, Central South University, ChinaReviewed by:
Tsutomu Ogata, Hamamatsu University School of Medicine, JapanGudrun Rappold, Heidelberg University, Germany
Liming Tao, Broad Institute, United States
Copyright © 2019 Sun, Luo, Qian, Chen, Wang, Li, Zou and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minyue Dong, ZG9uZ215QHpqdS5lZHUuY24=