 Sergey A. Krupenko
Sergey A. Krupenko David A. Horita
David A. Horita- 1Department of Nutrition, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 2Nutrition Research Institute, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
Folate (vitamin B9) is a common name for a group of coenzymes that function as carriers of chemical moieties called one-carbon groups in numerous biochemical reactions. The combination of these folate-dependent reactions constitutes one-carbon metabolism, the name synonymous to folate metabolism. Folate coenzymes and associated metabolic pathways are vital for cellular homeostasis due to their key roles in nucleic acid biosynthesis, DNA repair, methylation processes, amino acid biogenesis, and energy balance. Folate is an essential nutrient because humans are unable to synthesize this coenzyme and must obtain it from the diet. Insufficient folate intake can ultimately increase risk of certain diseases, most notably neural tube defects. More than 20 enzymes are known to participate in folate metabolism. Single-nucleotide polymorphisms (SNPs) in genes encoding for folate enzymes are associated with altered metabolism, changes in DNA methylation and modified risk for the development of human pathologies including cardiovascular diseases, birth defects, and cancer. ALDH1L1, one of the folate-metabolizing enzymes, serves a regulatory function in folate metabolism restricting the flux of one-carbon groups through biosynthetic processes. Numerous studies have established that ALDH1L1 is often silenced or strongly down-regulated in cancers. The loss of ALDH1L1 protein positively correlates with the occurrence of malignant tumors and tumor aggressiveness, hence the enzyme is viewed as a candidate tumor suppressor. ALDH1L1 has much higher frequency of non-synonymous exonic SNPs than most other genes for folate enzymes. Common SNPs at the polymorphic loci rs3796191, rs2886059, rs9282691, rs2276724, rs1127717, and rs4646750 in ALDH1L1 exons characterize more than 97% of Europeans while additional common variants are found in other ethnic populations. The effects of these SNPs on the enzyme is not clear but studies indicate that some coding and non-coding ALDH1L1 SNPs are associated with altered risk of certain cancer types and it is also likely that specific haplotypes define the metabolic response to dietary folate. This review discusses the role of ALDH1L1 in folate metabolism and etiology of diseases with the focus on non-synonymous coding ALDH1L1 SNPs and their effects on the enzyme structure/function, metabolic role and association with cancer.
Introduction: Folate Metabolism and Cellular Homeostasis
Folate (vitamin B9) is a common name for a group of coenzymes that function as carriers of chemical moieties called one-carbon groups (OCGs) in numerous biochemical reactions. The combination of these folate-dependent reactions constitutes one-carbon metabolism, the name synonymous to folate metabolism. The intracellular folate pool consists of several major coenzyme forms, including tetrahydrofolate (THF) and its derivatives differing by the oxidation state of conjugated OCG (Fox and Stover, 2008; Tibbetts and Appling, 2010). Folate coenzymes and associated metabolic pathways are vital for cellular homeostasis due to their key roles in nucleic acid biosynthesis, DNA repair, methylation processes, amino acid biogenesis, and energy balance (Blom et al., 2006; Fox and Stover, 2008; Tibbetts and Appling, 2010; Locasale, 2013; Fan et al., 2014; Ducker and Rabinowitz, 2017). Folate-dependent biochemical reactions underlying these processes include de novo purine and TMP biosynthesis, re-methylation of homocysteine to methionine linked to the production of the universal methyl donor S-adenosylmethionine, degradation of histidine and glycine, interconversion of serine and glycine, and the final step of carbon oxidation to CO2 linked with NADPH production (Tibbetts and Appling, 2010; Fan et al., 2014; Baggott and Tamura, 2015; Brosnan et al., 2015). Additional folate-dependent pathways include the clearance of formate (Brosnan et al., 2015) and the formylation of mitochondrial methionyl-tRNA, a process essential for translation initiation in eukaryotic mitochondria (Spencer and Spremulli, 2004; Tucker et al., 2011; Minton et al., 2018). Interestingly, a recent paper reported the direct involvement of one of folate coenzymes, 5,10-methylene-THF, in the methylation of mitochondrial tRNAs with the deficiency of this pathway likely being linked to defective oxidative phosphorylation in human cells (Morscher et al., 2018). This discovery not only extends the list of folate-dependent biochemical reactions and further underscores the indispensable role of the coenzyme but also emphasizes that precise molecular mechanisms underlying folate homeostasis are not completely understood.
Folate is an essential nutrient because humans are unable to synthesize this coenzyme and must obtain it from the diet (Cooper, 1986). Insufficient folate intake ultimately leads to deregulation of cellular homeostasis and is associated with increased risk of certain diseases, most notably neural tube defects (NTDs) (Rock et al., 2000; Fleming, 2001; Mitchell et al., 2004; Moat et al., 2004; Beaudin and Stover, 2007; Strickland et al., 2013; Newman and Maddocks, 2017). For example, periconceptional folate supplementation, in addition to preventing NTDs, has been associated with a significant reduction in the incidence of early spontaneous preterm births (Bukowski et al., 2009). Largely for NTD prevention, the FDA in 1996 approved a mandatory fortification of several types of grain foods in the US with a synthetic form of the vitamin, folic acid (FDA, 1996). The fortification resulted not only in a significant reduction of the incidence of NTDs in the US (Blom et al., 2006), but also improved folate status in the adult population (Jacques et al., 1999).
Folate Enzymes, Single-Nucleotide Polymorphisms and Diseases
More than 20 enzymes are known to participate in folate metabolism (Figure 1) (Fox and Stover, 2008; Tibbetts and Appling, 2010). They bring OCGs into folate pool, interconvert folate coenzymes, or use OCGs in biosynthetic reactions (Tibbetts and Appling, 2010). Of note, folate enzymes are highly compartmentalized in the cell, being localized to either cytoplasm or mitochondria (Tibbetts and Appling, 2010). Several cytoplasmic folate enzymes can also translocate to the nucleus to enable TMP biosynthesis at specific sites (MacFarlane et al., 2011; Anderson et al., 2012; Field et al., 2014; Field et al., 2015). The nucleus and cytoplasm exchange folate through a simple diffusion, but the mitochondrial membrane is not permeable to folate and shuttling requires a special transporter (Titus and Moran, 2000). Thus, mitochondrial folate metabolism is distinct from cytosolic and uses its own set of enzymes (Tibbetts and Appling, 2010). Several folate reactions in mitochondria parallel those in the cytoplasm; these are catalyzed by homologous enzymes which are products of different genes (Tibbetts and Appling, 2010; Strickland et al., 2011). Folate mitochondrial pathways (i) provide one-carbon groups (in the form of formate) for the cytosolic folate pool, where they are utilized for biosynthetic reactions (Tibbetts and Appling, 2010); (ii) generate NADPH (Fan et al., 2014), or (iii) serve specific mitochondrial functions (Tucker et al., 2011; Morscher et al., 2018; Tani et al., 2018).
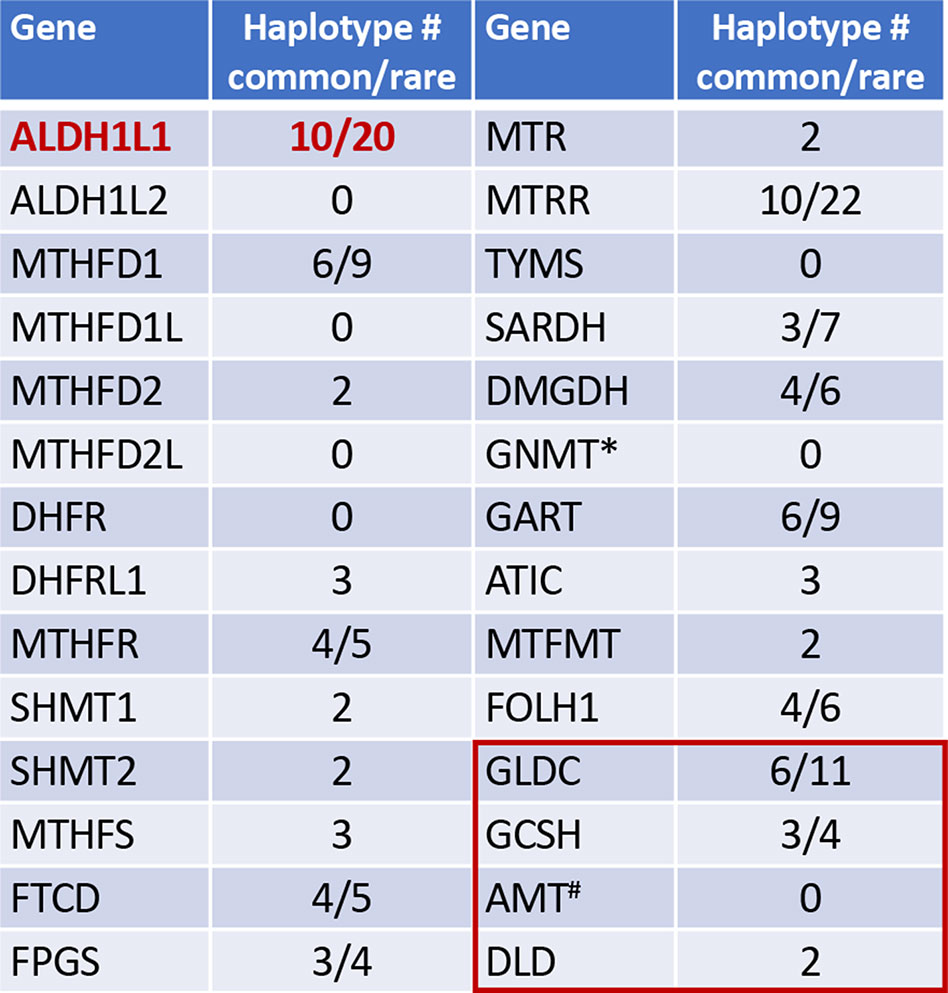
Figure 1 Numbers of common and rare haplotype alleles in genes of folate metabolism (human genome assembly GRCh37/hg19; rare haplotypes have frequency below 1%). *, GNMT is the enzyme regulated by folate. Red box indicates four enzymes of the mitochondrial glycine cleavage system, #, the folate dependent enzyme in glycine cleavage. Haplotypes were analyzed using UCSC Genome Browser (https://genome.ucsc.edu).
Changes in folate metabolism contribute to human pathologies (Stover, 2009), and recent studies underscore the role of several folate enzymes and associated pathways in NTDs and cancer (Jain et al., 2012; Narisawa et al., 2012; Momb et al., 2013; Nilsson et al., 2014; Pai et al., 2015; Piskounova et al., 2015; Ducker et al., 2016; Leung et al., 2017). Alterations in expression or activity of numerous enzymes of folate pathways can either enhance or impair folate metabolism. For example, the increased demand for nucleotides and methylation reactions in cancer cells commonly causes enhanced expression of folate enzymes to maintain the flux of folate-bound OCGs towards biosynthesis, thus supporting increased proliferation (Jain et al., 2012; Ducker and Rabinowitz, 2017; Rosenzweig et al., 2018). Accordingly, several of these enzymes were successfully targeted in cancer chemotherapy (Goldman et al., 2010; Visentin et al., 2012). Further links between the function of folate enzymes and onset of diseases have been clarified in studies using knockout mouse models. Thus, the loss of either MTHFD1L or the folate-dependent glycine cleavage (both localized to mitochondria) causes NTDs in mice (Momb et al., 2013; Pai et al., 2015). Another example is the knockout of folate-regulatory enzyme GNMT: the loss of this protein produces spontaneous tumors in the mouse liver (Martinez-Chantar et al., 2008). It has been also reported that the deficiency in the 10-formyl-THF synthetase activity of cytosolic trifunctional enzyme MTHFD1 is associated with increased incidence of congenital heart defects in mouse embryos (Christensen et al., 2015). Numerous studies also indicate strong gene-nutrient interactions in the folate metabolism regulation. For example, the loss of SHMT1 was insufficient to produce NTDs but caused exencephaly under conditions of maternal folate deficiency (Beaudin et al., 2011; Beaudin et al., 2012).
Single-nucleotide polymorphisms (SNPs) in genes encoding folate enzymes are associated with altered metabolism, changes in DNA methylation and modified risk for the development of human pathologies [reviewed in (Stover, 2011)] including cardiovascular diseases (Klerk et al., 2002), birth defects (Ou et al., 1996; Mills et al., 1999), and cancer (Sharp and Little, 2004; Lightfoot et al., 2010). The most investigated target in these studies was MTHFR (methylene-THF reductase) (Ueland et al., 2001; Hirschhorn et al., 2002; Klerk et al., 2002), which has two common SNPs in the coding region causing non-synonymous amino acid substitutions and creating enzyme variants with reduced activity (Frosst et al., 1995; Weisberg et al., 1998). Numerous SNPs in other key genes of folate pathways, including DHFR (dihydrofolate reductase) (Mishra et al., 2007), MTR (methionine synthase) (Harmon et al., 1999; Ma et al., 1999), TYMS (thymidylate synthase) (Pullarkat et al., 2001), and MTRR (methionine synthase reductase) (Wilson et al., 1999; Gaughan et al., 2001) were linked to human diseases. Of note, the effect of folate pathway gene polymorphisms on disease risk often depends on folate status (Friso et al., 2002; Ulrich et al., 2002; Philip et al., 2015).
ALDH1L1 Folate Regulatory Enzyme
ALDH1L1, one of the folate-metabolizing enzymes, converts 10-formyl-THF to THF with simultaneous production of NADPH from NADP+ (Krupenko, 2009). By oxidizing the formyl group to CO2, this reaction clears the OCG from the cell, thus restricting flux through biosynthetic processes (Figure 2). In this way, ALDH1L1 regulates one-carbon metabolism and serves a catabolic function (Krupenko and Oleinik, 2002; Anguera et al., 2006; Krupenko, 2009). ALDH1L1 is active as a tetramer and has a complex structure and catalytic mechanism (Figure 3). The ALDH1L1 gene originated from a natural fusion of three unrelated primordial genes (Strickland et al., 2011; Krupenko et al., 2015), and the resulting protein has a modular organization with three structurally and functionally distinct domains (Krupenko, 2009). The N-terminal folate binding/hydrolase domain structurally resembles methionine-tRNA formyltransferase (Schmitt et al., 1996; Chumanevich et al., 2004) and catalyzes the initial cleavage of the 10-formyl group from 10-formyl-THF (Krupenko et al., 1997a; Chumanevich et al., 2004). The C-terminal dehydrogenase domain forms the tetrameric core and is a structural and functional homolog of aldehyde dehydrogenases (ALDHs) (Krupenko et al., 1997b; Tsybovsky et al., 2007) [hence the assignment of ALDH1L1 to this superfamily of proteins (Marchitti et al., 2008)]. In humans, there are 19 genes encoding for aldehyde dehydrogenases (Marchitti et al., 2008; Koppaka et al., 2012). ALDHs catalyze NAD(P)+-dependent irreversible oxidation of a wide variety of endogenous and exogenous aldehydes to corresponding acids, display distinct substrate specificity, and are generally regarded as detoxification enzymes (Marchitti et al., 2008; Koppaka et al., 2012). The ALDH domain of ALDH1L1 shares about 49% of its amino acid sequence with ALDH1, has a typical ALDH fold and by itself catalyzes the oxidation of short-chain aldehydes to corresponding acid using strictly NADP+ (Krupenko, 2009). It is not clear whether ALDH1L1 is involved in the utilization of aldehyde substrates in vivo. As a part of the ALDH1L1 enzymatic machinery, this domain catalyzes the reduction of NADP+ and the oxidation of formyl group to CO2 (Krupenko et al., 1997b; Tsybovsky et al., 2007). The two catalytic domains communicate via the intermediate domain, which is a structural and functional homolog of acyl carrier proteins (Donato et al., 2007; Strickland et al., 2010). Its prosthetic group, 4′-phosphopantetheine (4′-PP), functions as a flexible arm reaching into the catalytic centers on the N- and C-terminal domains (Horita and Krupenko, 2017) and transporting the reaction intermediate (formyl) from one center to the other (Figure 3). The three domains of ALDH1L1 work in concert to enable the conversion of 10-formyl-THF to THF and NADPH production linked to the oxidation of formyl group to CO2. Thus, in the case of ALDH1L1 the recruitment of the folate-binding domain extended the substrate specificity of an aldehyde dehydrogenase. Of note, the ALDH family also includes ALDH1L2, the mitochondrial homolog of ALDH1L1 (Krupenko et al., 2010), which is the product of a separate gene [one of the 19 ALDH genes (Marchitti et al., 2008)].
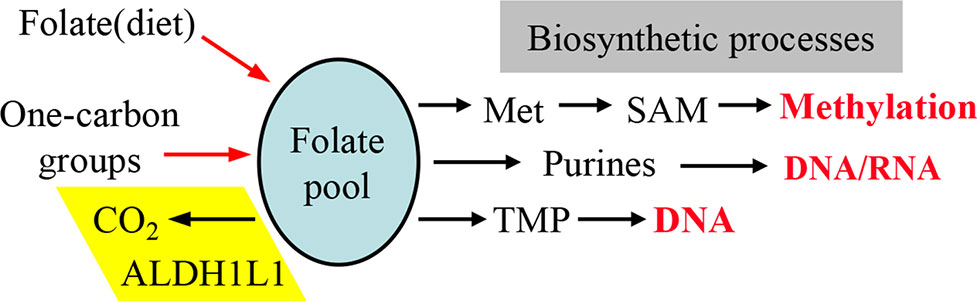
Figure 2 One-carbon groups (derived from amino acid oxidation or formate) enter the folate pool and are directed towards three biosynthetic pathways (methionine, purines and thymidylate synthesis). Note that the enzyme ALDH1L1 diverts these groups from biosynthetic pathways thus serving a catabolic function. Input of folate from diet is required to support the intracellular levels of the coenzyme. SAM, S-adenosylmethionine.
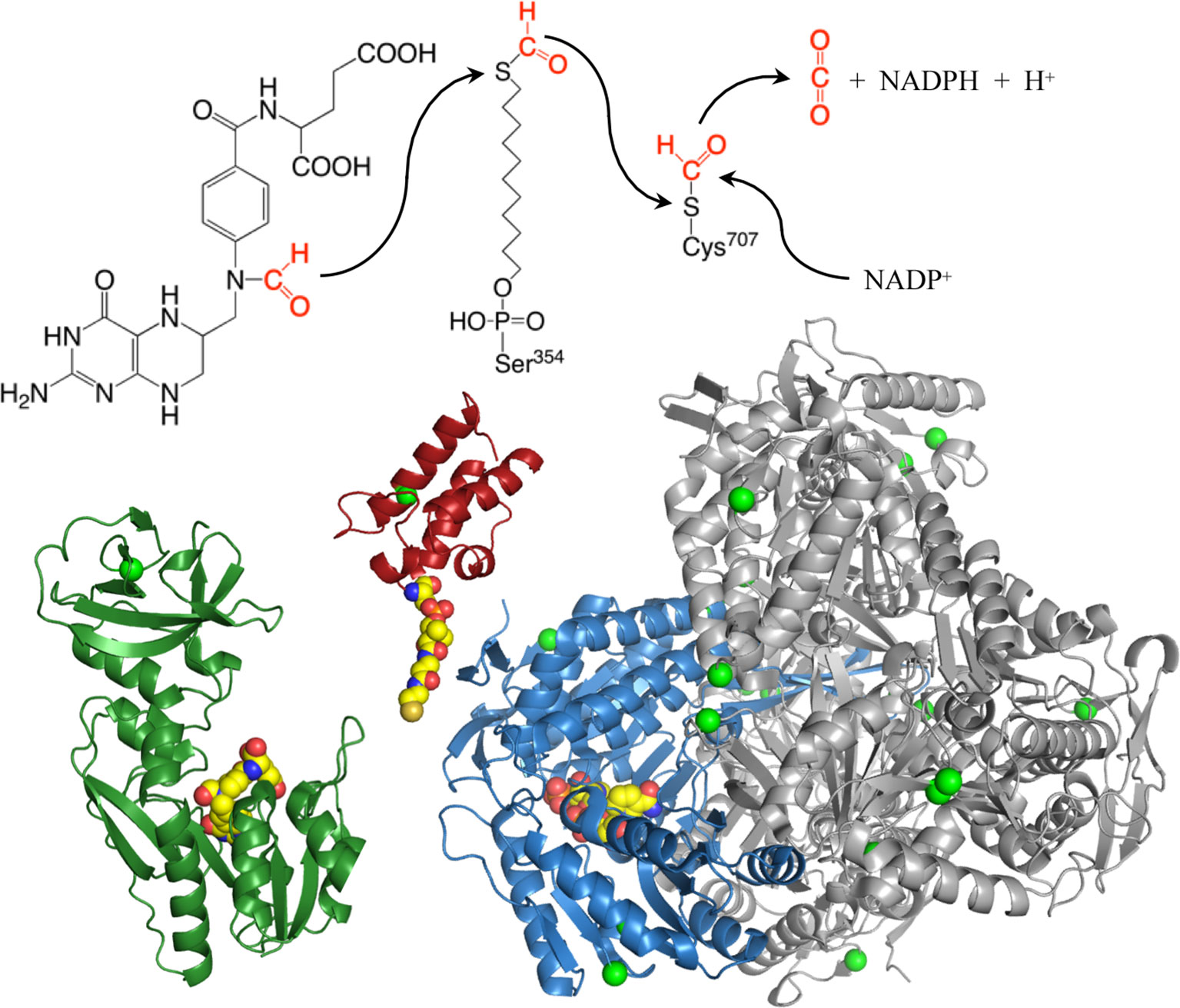
Figure 3 Structures of the N-terminal formyltransferase (dark green), central acyl-carrier (red, with manually added phosphopantetheinyl moiety), and C-terminal dehydrogenase (blue and gray) domains of ALDH1L1. Green spheres highlight positions of amino acids corresponding to exonic SNPs discussed in the text, other colored spheres show positions of 10-formyltetrahydrofolate (N-terminal domain), phosphopantetheine (intermediate domain), and NADP+ (C-terminal domain). Subunit A of the tetrameric dehydrogenase domain is blue, subunits B, C, and D are gray. The extended phosphopantetheine is critical to the reaction as both the formyl donor (10-formyltetrahydrofolate) and electron acceptor (NADP+) are located at the bottoms of clefts in the protein surface. PDB structures are: 4tt8 (N-terminal domain); 2cq8 (intermediate domain); and 2o2q (aldehyde dehydrogenase domain).
That ALDH1L1 serves a regulatory role was determined by several reports that demonstrated the effect of the enzyme on folate and purine pools and on methylation (Champion et al., 1994; Oleinik et al., 2005; Anguera et al., 2006; Oleinik et al., 2006; Hoeferlin et al., 2011). ALDH1L1 is also a key component of the formate degradation pathway, which converts toxic formate to neutral CO2, through 10-formyltetrahydrofolate as an intermediate (Strickland et al., 2011). In the cell, formate is directly produced not only from the degradation of 3-methyl-branched fatty acids and the shortening of 2-hydroxy long chain fatty acids (Casteels et al., 2007) but also from the oxidation of methanol present in juices and alcoholic beverages (Hang and Woodams, 2010) and from metabolism of artificial sweetener aspartame (Choudhary and Pretorius, 2017). The first step of the formate degradation pathway, the incorporation of formate into the folate pool, is catalyzed by MTHFD1 and the second rate-limiting step releasing CO2 is catalyzed by ALDH1L1 (Neymeyer et al., 1997). It appears that the ALDH1L1-dependent pathway is the only pathway in humans to metabolize formate, and it is more prominent for the clearance of lower, physiological doses of formate (Cook et al., 2001). In further support of this role, decreased expression of ALDH1L1 was observed in cobalamin-deficient rats, likely as a mechanism to divert formate towards methyl group production (MacMillan et al., 2018). ALDH1L1 was also highlighted as a pan-astrocyte marker (Cahoy et al., 2008), but its importance for the astrocyte function is not clear. Interestingly, decreased levels of ALDH1L1 in cerebrospinal fluid were linked to neonatal hydrocephalus in a rat model (Cains et al., 2009). Further studies of this model suggested a role for the enzyme in cerebral folate transport and regulation of folate availability in the brain (Naz et al., 2016; Jimenez et al., 2019). In line with such function, it has been also demonstrated that ALDH1L1 protects folate from degradation in zebrafish embryos, which is a defense mechanism against oxidative stress (Chang et al., 2014; Hsiao et al., 2014). Furthermore, the protective effect of ALDH1L1 on THF degradation has been recently observed in cancer cells (Zheng et al., 2018). These studies provide experimental support for the hypothesis that ALDH1L1 serves as folate depot (Krupenko and Krupenko, 2018).
Evidence That ALDH1L1 Is a Candidate Tumor Suppressor
ALDH1L1 is most abundant in liver, kidney and pancreas comprising about 1% of total cytosolic protein in hepatocytes (Krupenko, 2009). However, it is not a housekeeping gene and its expression is tissue-specific with some tissues lacking this protein expression (Krupenko and Oleinik, 2002). Furthermore, the enzyme is tightly regulated during mouse brain development (Anthony and Heintz, 2007) and during the progression of NIH3T3 cells through the cell cycle (Khan et al., 2018). In both cases, ALDH1L1 protein is dramatically decreased in proliferating cells but elevated in non-proliferating/resting cells. During mouse brain development, ALDH1L1 expression is likely controlled by transcriptional regulation (Anthony and Heintz, 2007) while in NIH3T3 cells it is rapidly degraded through the ubiquitin-proteasome pathway during the transition from G0/G1 to S-phase (Khan et al., 2018). Because the enzyme limits proliferation by diverting OCGs from biosynthetic to catabolic pathways, its down-regulation could be one of the mechanisms to maintain proliferative state.
In line with its antiproliferative function, ALDH1L1 is often silenced or strongly down-regulated in cancer cell lines and malignant tumors [reviewed in (Krupenko and Krupenko, 2018; Krupenko and Krupenko, 2019)]. This is in strict contrast to other folate enzymes, which are commonly up-regulated in cancer (Jain et al., 2012; Ducker and Rabinowitz, 2017). Several studies have established that the silencing of ALDH1L1 in human cancers is driven by gene methylation (Oleinik et al., 2011; Dmitriev et al., 2012; Senchenko et al., 2013; Dmitriev et al., 2014; Beniaminov et al., 2018). Methylation takes place in the CpG island, which includes 96 CpG base pairs and covers the promoter, first exon and the part of the first intron in ALDH1L1 (Oleinik et al., 2011; Beniaminov et al., 2018). Remarkably, a microarray-based global gene expression profiling of approximately 42,000 genes has found that ALDH1L1 was one of the most down-regulated proteins in primary hepatocellular carcinomas and in liver metastases (Tackels-Horne et al., 2001). Analysis of gene expression profiles across 33 human cancer types using The Cancer Genome Atlas (TCGA) data indicated that ALDH1L1 is more strongly down-regulated in late-stage cancers (Li et al., 2017). Overall, the loss of ALDH1L1 protein positively correlates with the occurrence of malignant tumors and tumor aggressiveness [reviewed in (Krupenko and Krupenko, 2018; Krupenko and Krupenko, 2019)], hence the suggestion that the enzyme is a candidate tumor suppressor (Senchenko et al., 2013).
SNPs in ALDH1L1 and Their Association With Pathologies
ALDH1L1 is located on the minus strand of chromosome 3, spans about 94 thousand nucleotides and may harbor numerous SNPs. Several reports have investigated the functional role of some of these SNPs as well as their associations with diseases. For example, genome-wide association studies (GWAS) revealed that SNPs in ALDH1L1 are associated with serine to glycine ratio in serum (Dharuri et al., 2013) thus supporting the role of the enzyme as metabolic regulator. Another GWAS analysis identified an association between rs1107366, located about 3800 nucleotides upstream of the ALDH1L1 transcription start site, and glycine to serine ratios (Xie et al., 2013). This study also indicated that the rs1107366-linked glycine to serine ratio is associated with insulin sensitivity but not with type 2 diabetes. ALDH1L1 SNPs were also associated with NTDs in Dutch and Chinese Han populations (Franke et al., 2009; Wu et al., 2016).
An interesting study evaluated the effect of two intronic ALDH1L1 SNPs, rs2276731 and rs2002287, on genome-wide DNA methylation as well as site-specific methylation in normal breast tissues from healthy women (Song et al., 2016). This study identified 57 CpG sites in human genome that were differentially methylated depending on SNPs in six genes of folate metabolism. The strongest association for differential methylation at these sites were with the ALDH1L1 SNPs. Furthermore, rs2276731 was also associated with a significantly higher global DNA methylation as well as with differential methylation of CpGs within ALDH1L1 itself. Of note, for both ALDH1L1 SNPs, the pattern of differentially methylated sites was different between whites and blacks (Song et al., 2016). Importantly, a modifying effect on breast cancer incidence of these ALDH1L1 SNPs has also been reported (Stevens et al., 2007). Here, however, these SNPs have opposite effects: the rs2276731 allele was associated with increased risk whereas the rs2002287 allele was associated with decreased risk of breast cancer.
The rs2276731 SNP could also have a role in the host-gut microbiome interaction. This has been suggested from the 16S rRNA-based analysis of the gut microbiome in 1,126 twin pairs, which thought to calculate the heritability of specific components of the gut microbiota and to find associations between the abundance of specific microbes and host gene alleles (Goodrich et al., 2016). The study identified an association between the host gene ALDH1L1 (via rs2276731) and the bacteria SHA-98 [unclassified genus of the order SHA-98, phylum Firmicutes (Goodrich et al., 2014)]. It further suggested that this association is linked to the metabolism of formate (as discussed above, ALDH1L1 is a key component of the formate clearance). In addition to the sources listed in the previous section, formate is also a fermentation product which acts as a major interspecies electron carrier promoting syntrophy (Goodrich et al., 2016). Of note, it has been shown that urinary formate excretion significantly correlated with blood pressure (Holmes et al., 2008). Since a SNP in ALDH1L1 was associated with incident ischemic stroke (Williams et al., 2014), the enzyme might link formate metabolism with the risk of cardiovascular diseases.
Interestingly, ALDH1L1 has much higher frequency of non-synonymous exonic SNPs than most other genes for folate enzymes (Figure 1). Such SNPs cause amino acid substitutions, could affect the enzyme function, and thus could be relevant to the role of the enzyme in cancer. Curiously, a highly similar mitochondrial homolog, ALDH1L2, which is a product of a separate gene resulted from gene duplication (Krupenko et al., 2010; Strickland et al., 2011; Krupenko et al., 2015), does not have common SNPs (Figure 1). SNPs in ALDH1L1 are common but their effect on metabolism and the etiology of cancer disease is not well understood. Notably, the frequency of exonic SNPs in this gene is highly different between ethnic populations [Figure 4; analyzed using UCSC genome browser (Mangan et al., 2014)]. While common SNPs at the polymorphic loci rs3796191, rs2886059, rs9282691, rs2276724, rs1127717 and rs4646750 in ALDH1L1 exons characterize more than 97% of Europeans, additional common variants are found in African, Hispanic, and Chinese populations (Figure 4). Several studies indicated that coding SNPs in ALDH1L1 are associated with altered risk of certain cancer types. Thus, ALDH1L1 rs1127717 was associated with the increased risk of hepatocellular carcinoma in Chinese population (1500 cancer patients and 1500 controls were enrolled in this study) (Zhang et al., 2015). Another SNP, rs2276724, could be associated with the post-operative survival of patients with hepatitis B-related hepatocellular carcinoma (Zhu et al., 2017). This study indicates that the effect of the SNP is associated with the expression level of ALDH1L1 mRNA and also depends on the p53 status. An elevated risk of non-Hodgkin lymphoma (NHL) was observed among carriers of the G allele at ALDH1L1 Ex21+31 (p.D793G; rs1127717) (Lee et al., 2007; Lim et al., 2007; Suthandiram et al., 2015). Furthermore, the protective effect of methionine on NHL was associated with ALDH1L1 SNPs (Lim et al., 2007; Li et al., 2013) suggesting gene-nutrient interactions. Importantly, four exonic SNPs shown in Figure 4 are associated with leukocyte telomere length (Pusceddu et al., 2017), implicating these polymorphisms in cancer (Sarek et al., 2015; Zhu et al., 2016). Of note, studies investigating ALDH1L1 SNPs as a risk factor for prostate and renal cancers did not find any associations (Stevens et al., 2008; Gibson et al., 2011), which could suggest the cancer type-specific role of the SNPs. Additionally, the overall effect of ALDH1L1 SNPs is likely ethnicity-specific (Marini et al., 2016; Wu et al., 2016) and could also be modified by the folate status.
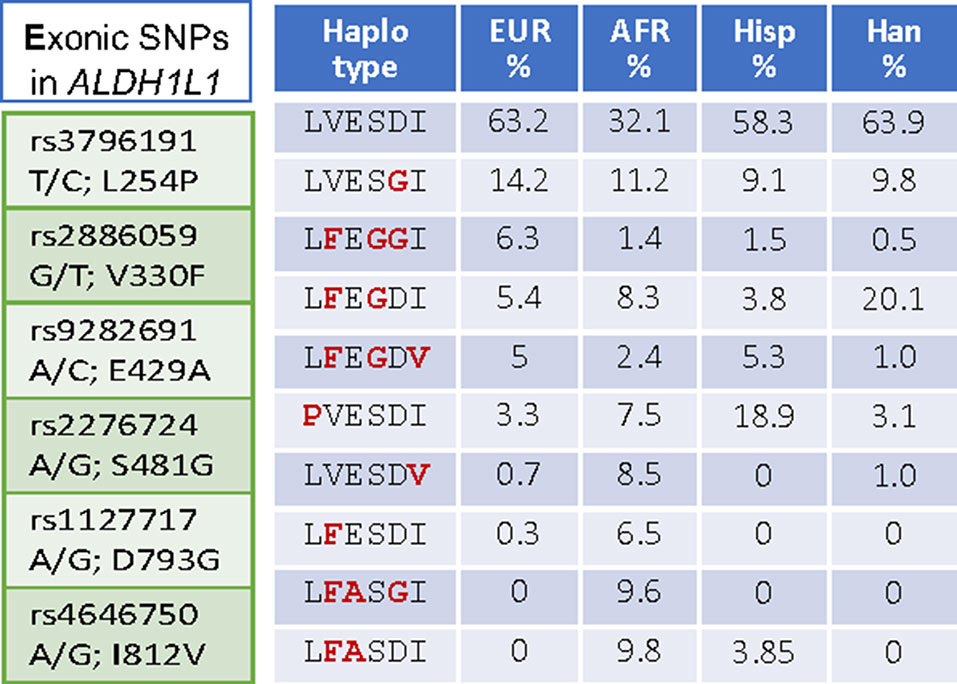
Figure 4 Left panel, SNPs in the exonic region causing non-synonymous amino acid substitutions are common in ALDH1L1. Right panel, SNP-associated haplotypes are markedly different between ethnic populations.
Potential Impact of ALDH1L1 Exonic SNPs
The substitution of a single amino acid residue in the protein structure, caused by a SNP, could be mute or could cause significant alterations in protein properties. For example, one of the exonic SNPs in MTHFR, C677T, results in the A222V amino acid change in the FAD-binding catalytic domain of the enzyme. This substitution produces a less thermostable protein with reduced catalytic activity (Frosst et al., 1995). Another common exonic SNP in MTHFR, A1298C (Weisberg et al., 1998), exists in strong linkage disequilibrium with C677T (Stover, 2011) and results in the E429A enzyme variant. The effect on the enzyme activity of this substitution, which is in the regulatory domain of the protein, is less clear. Initial report indicated that this substitution decreases the enzyme activity though to a lesser extent than the A222V substitution (Weisberg et al., 1998). A later study of purified recombinant human MTHFR concluded that the E429A protein has biochemical properties that are indistinguishable from the wild-type enzyme (Yamada et al., 2001). In vivo, however, MTHFR is phosphorylated at multiple residues (Yamada et al., 2005), and both the A222V and E429A mutations are predicted to disrupt phosphorylation of neighboring Ser residues (Shahzad et al., 2013). Notably, the recently solved crystal structure of human MTHFR links the enzyme’s phosphorylation state to its sensitivity to inhibition by S-adenosylmethionine (Froese et al., 2018).
Amino acid substitutions associated with common exonic ALDH1L1 SNPs are found in each of the functional domains (Figures 3 and 4) but their effect on protein properties have not been studied. Analysis of the crystal structures of the ALDH1L1 domains identifies potential important structural roles for residues mutated by these polymorphisms. For example, Ser481 is an α-helix N-cap and its side chain makes a hydrogen bond with Gln549 in a different subunit, suggesting a role in protein oligomerization and stability. Two other residues affected by ALDH1L1 SNPs, Asp793 and Ile812 (changed to Gly and Val, respectively) are strictly conserved through all species. Interestingly, these residues are adjacent on parallel β-strands and form backbone hydrogen bonds (Figure 5). This can be interpreted as a role in supporting protein conformation and stability. Of note, the co-occurrence of both SNPs is not found, suggesting that it perhaps would have too damaging a structural effect if both residues are changed. Our previous studies indicate that point mutations in the ALDH1L1 aldehyde dehydrogenase domain can significantly alter the protein conformation, with some of them impairing the protein’s stability (Tsybovsky et al., 2007; Tsybovsky and Krupenko, 2011; Tsybovsky et al., 2013). Furthermore, a long-range communication between the aldehyde dehydrogenase catalytic center and the NADP+-binding domain, observed previously (Tsybovsky and Krupenko, 2011), could transduce the effect of an amino acid substitution to distant domains with an unpredictable effect. In line with this notion, the structure of MTHFR suggests a long-range influence of S-adenosylmethionine binding in the regulatory domain of the enzyme on the catalytic domain some 300 amino acids away (Froese et al., 2018).
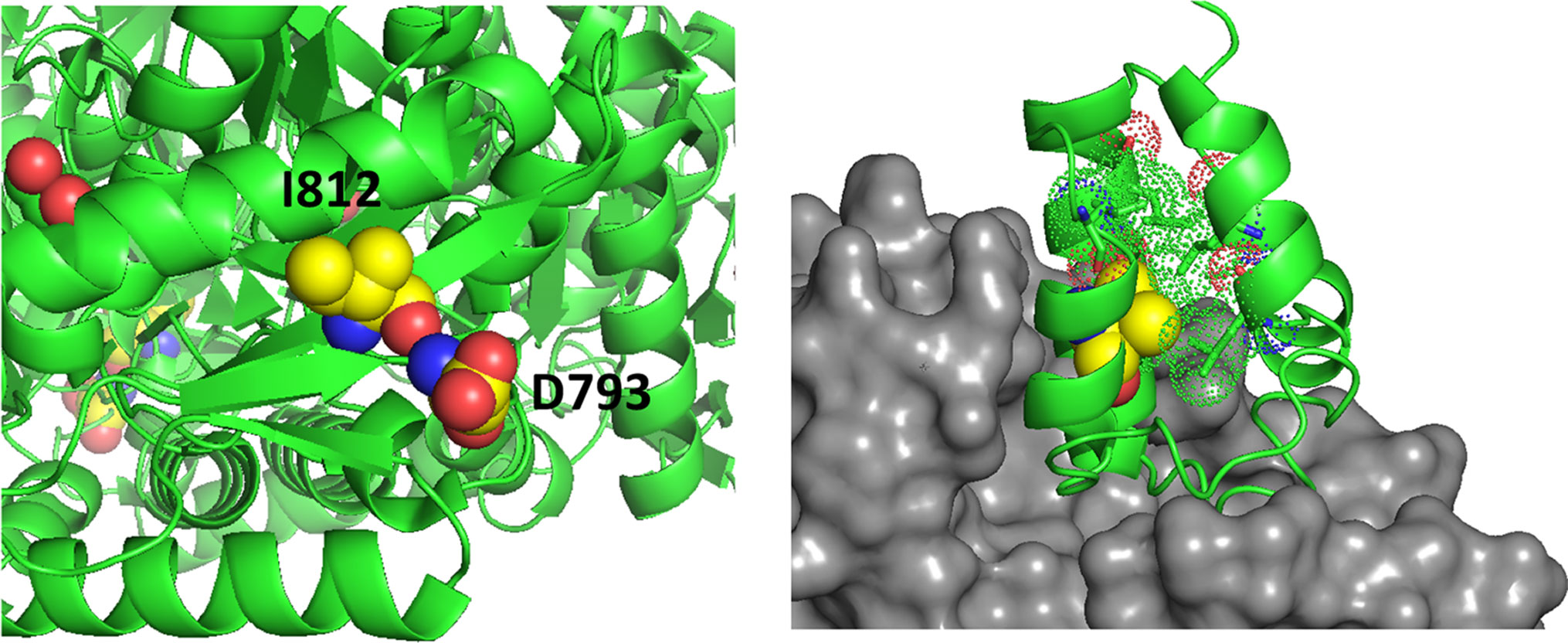
Figure 5 Left panel, D793 and I812 are adjacent on parallel β-strands making backbone hydrogen bonds. Right panel, the structure of a phosphopantetheinyl transferase (gray surface) in complex with an ACP (acyl carrier protein) domain (green ribbon) shows that initial modification of the ACP domain serine (spheres) requires substantial access to the ACP surface. ACP helices 1 and 2 and the connecting loop lie on the surface of the transferase. The side chain of V330 (yellow spheres) packs in the interior of the ACP domain helical bundle. The substitution with Phe (rs2886059) will clash with surrounding residues (dots), likely causing a shift of the helix which contacts the transferase domain (gray surface) and interfering with binding.
The SNP rs2886059 produces the V330F substitution in the intermediate domain of ALDH1L1, close to the modification site where the prosthetic group is attached (Figure 5). This substitution introduces a bulky side-chain in the core of the intermediate domain helical bundle which could interfere with the binding of phosphopantetheine transferase (PPTase) (Bunkoczi et al., 2007). PPTase appends the 4′-phosphopantetheinyl moiety to a serine in the intermediate domain and converts inactive apo-ALDH1L1 into active holo-ALDH1L1 (Strickland et al., 2010). Conformational changes associated with other SNPs could interfere with PPTase binding or hinder the ability of the intermediate domain to shuttle reactant between the catalytic domains. The SNP rs3796191 creates the L254P amino acid substitution in the C-terminal lobe of the N-terminal folate binding domain of ALDH1L1. In the structurally homologous enzyme, MTFMT, this sub-domain is responsible for the binding of methionyl-tRNA (Schmitt et al., 1996) but the role of this part of the ALDH1L1 molecule in the enzyme’s function is not clear. It perhaps serves to properly align the folate-binding and the intermediate domains for the acceptance of the formyl group by the 4′-PP arm. Replacement of Leu with Pro will alter and restrict backbone conformation and loop flexibility, and perhaps cause a misalignment between the N-terminal and intermediate domains, impeding access to the folate-binding pocket. In fact, the role of this sub-domain for the proper ALDH1L1 function, likely through the proper orientation of the functional domains, has been demonstrated (Reuland et al., 2006).
Finally, as in the case with MTHFR, coding SNPs can affect ALDH1L1 stability and degradation rate. Towards this end, we have recently demonstrated that ALDH1L1 can be rapidly degraded through the ubiquitin-proteasome pathway (Khan et al., 2018). It is known that protein variants associated with non-synonymous SNPs can be differently degraded by the ubiquitin-proteasome pathway (Siegel et al., 2001; Bandiera et al., 2005). These findings raise the question of whether amino acid substitutions caused by coding SNPs will affect the ALDH1L1 degradation, which would affect the protein function as the proliferation regulator.
Concluding Remarks
While the phenomenon of ALDH1L1 silencing/down-regulation in cancer is now well recognized (Krupenko and Krupenko, 2018; Krupenko and Krupenko, 2019), the effects of exonic SNPs on the protein function in tumorigenesis and tumor progression are not clear. It is also not known whether this gene is involved in tumor initiation or whether its loss provides selective advantage for tumor progression at later stages. The high prevalence of exonic SNPs causing non-synonymous amino acid substitutions in ALDH1L1 raises the question of how these SNPs affect cellular metabolism and proliferation regulated by ALDH1L1. If ALDH1L1 polymorphic variants have altered activity or stability/half-life, they are likely to cause the imbalance of intracellular reduced folate pools with a consequent effect on de novo purine biosynthesis and amino acid metabolism. Overall, ALDH1L1-dependent metabolic reprogramming associated with functional exonic SNPs could be an important contributor to disease etiology with a more profound effect in populations with certain ALDH1L1 haplotypes (Figure 6). With regard to gene-diet interactions, the effect of dietary folate on the ALDH1L1 regulatory role is not clear, and the impact of functional SNPs is yet to be investigated. The understanding of how haplotype-specific effects are modified by folate supplementation could empower precision nutrition approach in disease prevention/treatment. Finally, since ALDH1L1 is involved in formate clearance, it could be an important component of the methanol detoxification pathway (Tephly, 1991). In this regard, it will be interesting to learn whether individuals with different ALDH1L1 haplotypes have a different susceptibility to methanol toxicity.
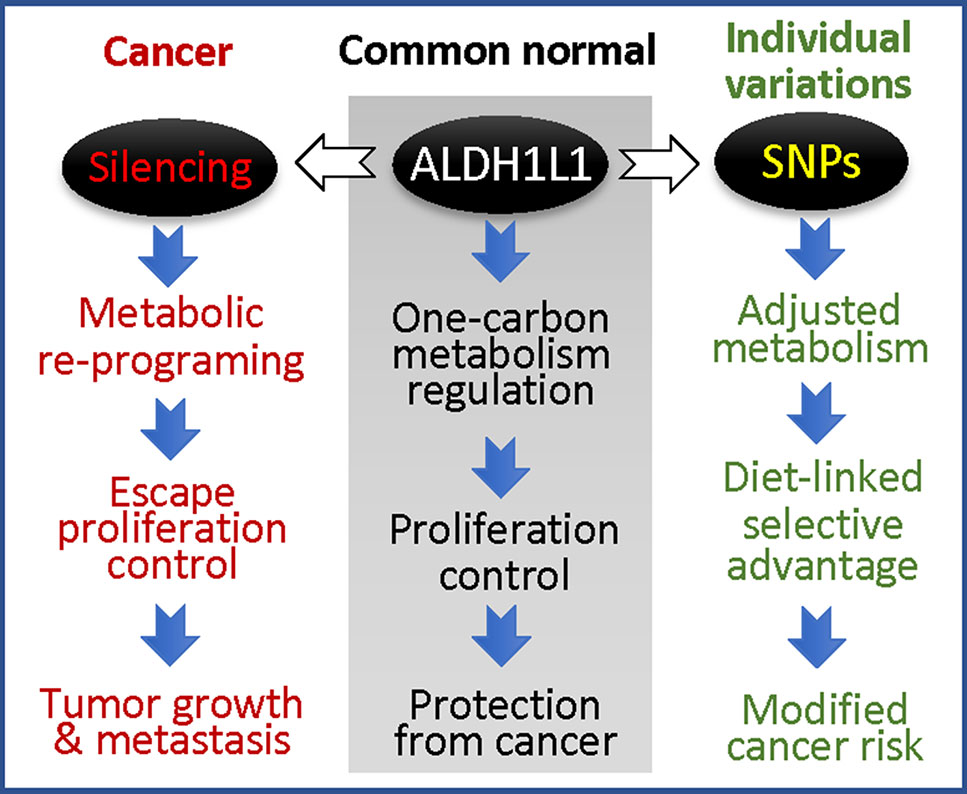
Figure 6 ALDH1L1 is a main regulator of folate metabolism, and its gene is commonly silenced in cancer (the loss of the protein is linked to accelerated proliferation and tumor progression); coding SNPs in this gene are likely to modify cancer risk.
Author Contributions
SK conceived the project, performed analysis of ALDH1L1 gene for coding SNPs and wrote the manuscript. DH performed structural analysis of ALDH1L1 variants and participated in data analysis and manuscript writing.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SK was supported by the National Institute of Health grants DK054388, CA095030 and DK117854.
References
Anderson, D. D., Woeller, C. F., Chiang, E. P., Shane, B., Stover, P. J. (2012). Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. J. Biol. Chem. 287 (10), 7051–7062. doi: 10.1074/jbc.M111.333120
Anguera, M. C., Field, M. S., Perry, C., Ghandour, H., Chiang, E. P., Selhub, J., et al. (2006). Regulation of folate-mediated one-carbon metabolism by 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 281 (27), 18335–18342. doi: 10.1074/jbc.M510623200
Anthony, T. E., Heintz, N. (2007). The folate metabolic enzyme ALDH1L1 is restricted to the midline of the early CNS, suggesting a role in human neural tube defects. J. Comp. Neurol. 500 (2), 368–383. doi: 10.1002/cne.21179
Baggott, J. E., Tamura, T. (2015). folate-dependent purine nucleotide biosynthesis in humans. Adv. Nutr. 6 (5), 564–571. doi: 10.3945/an.115.008300.
Bandiera, S., Weidlich, S., Harth, V., Broede, P., Ko, Y., Friedberg, T. (2005). Proteasomal degradation of human CYP1B1: effect of the Asn453Ser polymorphism on the post-translational regulation of CYP1B1 expression. Mol. Pharmacol. 67 (2), 435–443. doi: 10.1124/mol.104.006056
Beaudin, A. E., Stover, P. J. (2007). Folate-mediated one-carbon metabolism and neural tube defects: balancing genome synthesis and gene expression. Birth Defects Res. C Embryo Today 81 (3), 183–203. doi: 10.1002/bdrc.20100
Beaudin, A. E., Abarinov, E. V., Noden, D. M., Perry, C. A., Chu, S., Stabler, S. P., et al. (2011). Shmt1 and de novo thymidylate biosynthesis underlie folate-responsive neural tube defects in mice. Am. J. Clin. Nutr. 93 (4), 789–798. doi: 10.3945/ajcn.110.002766
Beaudin, A. E., Abarinov, E. V., Malysheva, O., Perry, C. A., Caudill, M., Stover, P. J. (2012). Dietary folate, but not choline, modifies neural tube defect risk in Shmt1 knockout mice. Am. J. Clin. Nutr. 95 (1), 109–114. doi: 10.3945/ajcn.111.020305
Beniaminov, A. D., Puzanov, G. A., Krasnov, G. S., Kaluzhny, D. N., Kazubskaya, T. P., Braga, E. A., et al. (2018). Deep sequencing revealed a cpg methylation pattern associated with aldh1l1 suppression in breast cancer. Front. Genet. 9, 169. doi: 10.3389/fgene.2018.00169
Blom, H. J., Shaw, G. M., Heijer, M., Finnell, R. H. (2006). Neural tube defects and folate: case far from closed. Nat. Rev. Neurosci. 7 (9), 724–731. doi: 10.1038/nrn1986
Brosnan, M. E., MacMillan, L., Stevens, J. R., Brosnan, J. T. (2015). Division of labour: how does folate metabolism partition between one-carbon metabolism and amino acid oxidation? Biochem. J. 472 (2), 135–146. doi: 10.1042/BJ20150837
Bukowski, R., Malone, F. D., Porter, F. T., Nyberg, D. A., Comstock, C. H., Hankins, G. D., et al. (2009). Preconceptional folate supplementation and the risk of spontaneous preterm birth: a cohort study. PLoS Med. 6 (5), e1000061. doi: 10.1371/journal.pmed.1000061
Bunkoczi, G., Pasta, S., Joshi, A., Wu, X., Kavanagh, K. L., Smith, S., et al. (2007). Mechanism and substrate recognition of human holo ACP synthase. Chem. Biol. 14 (11), 1243–1253. doi: 10.1016/j.chembiol.2007.10.013
Cahoy, J. D., Emery, B., Kaushal, A., Foo, L. C., Zamanian, J. L., Christopherson, K. S., et al. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28 (1), 264–278. doi: 10.1523/JNEUROSCI.4178-07.2008
Cains, S., Shepherd, A., Nabiuni, M., Owen-Lynch, P. J., Miyan, J. (2009). Addressing a folate imbalance in fetal cerebrospinal fluid can decrease the incidence of congenital hydrocephalus. J. Neuropathol. Exp. Neurol. 68 (4), 404–416. doi: 10.1097/NEN.0b013e31819e64a7
Casteels, M., Sniekers, M., Fraccascia, P., Mannaerts, G. P., Van Veldhoven, P. P. (2007). The role of 2-hydroxyacyl-CoA lyase, a thiamin pyrophosphate-dependent enzyme, in the peroxisomal metabolism of 3-methyl-branched fatty acids and 2-hydroxy straight-chain fatty acids. Biochem. Soc. Trans. 35 (Pt 5), 876–880. doi: 10.1042/BST0350876
Champion, K. M., Cook, R. J., Tollaksen, S. L., Giometti, C. S. (1994). Identification of a heritable deficiency of the folate-dependent enzyme 10-formyltetrahydrofolate dehydrogenase in mice. Proc. Natl. Acad. Sci. U. S. A. 91 (24), 11338–11342. doi: 10.1073/pnas.91.24.11338
Chang, W. N., Lee, G. H., Kao, T. T., Lin, C. Y., Hsiao, T. H., Tsai, J. N., et al. (2014). Knocking down 10-Formyltetrahydrofolate dehydrogenase increased oxidative stress and impeded zebrafish embryogenesis by obstructing morphogenetic movement. Biochim. Biophys. Acta 1840 (7), 2340–2350. doi: 10.1016/j.bbagen.2014.04.009
Choudhary, A. K., Pretorius, E. (2017). Revisiting the safety of aspartame. Nutr. Rev. 75 (9), 718–730. doi: 10.1093/nutrit/nux035
Christensen, K. E., Deng, L., Bahous, R. H., Jerome-Majewska, L. A., Rozen, R. (2015). MTHFD1 formyltetrahydrofolate synthetase deficiency, a model for the MTHFD1 R653Q variant, leads to congenital heart defects in mice. Birth Defects Res. A Clin. Mol. Teratol. 103 (12), 1031–1038. doi: 10.1002/bdra.23451
Chumanevich, A. A., Krupenko, S. A., Davies, C. (2004). The crystal structure of the hydrolase domain of 10-formyltetrahydrofolate dehydrogenase: mechanism of hydrolysis and its interplay with the dehydrogenase domain. J. Biol. Chem. 279 (14), 14355–14364. doi: 10.1074/jbc.M313934200
Cook, R. J., Champion, K. M., Giometti, C. S. (2001). Methanol toxicity and formate oxidation in NEUT2 mice. Arch. Biochem. Biophys. 393 (2), 192–198. doi: 10.1006/abbi.2001.2485
Cooper, B. A. (1986). “Folate nutrition in man and animals,” in Folates and pterins. Eds. Blakley, R. L., Whitehead, V. M. (New York: John Wiley & Sons, Inc)
Dharuri, H., Henneman, P., Demirkan, A., van Klinken, J. B., Mook-Kanamori, D. O., Wang-Sattler, R., et al. (2013). Automated workflow-based exploitation of pathway databases provides new insights into genetic associations of metabolite profiles. BMC Genomics 14, 865. doi: 10.1186/1471-2164-14-865
Dmitriev, A. A., Kashuba, V. I., Haraldson, K., Senchenko, V. N., Pavlova, T. V., Kudryavtseva, A. V., et al. (2012). Genetic and epigenetic analysis of non-small cell lung cancer with NotI-microarrays. Epigenetics 7 (5), 502–513. doi: 10.4161/epi.19801
Dmitriev, A. A., Rudenko, E. E., Kudryavtseva, A. V., Krasnov, G. S., Gordiyuk, V. V., Melnikova, N. V., et al. (2014). Epigenetic alterations of chromosome 3 revealed by NotI-microarrays in clear cell renal cell carcinoma. Biomed. Res. Int. 2014, 735292. doi: 10.1155/2014/735292
Donato, H., Krupenko, N. I., Tsybovsky, Y., Krupenko, S. A. (2007). 10-formyltetrahydrofolate dehydrogenase requires a 4'-phosphopantetheine prosthetic group for catalysis. J. Biol. Chem. 282 (47), 34159–34166. doi: 10.1074/jbc.M707627200
Ducker, G. S., Rabinowitz, J. D. (2017). One-carbon metabolism in health and disease. Cell Metab. 25 (1), 27–42. doi: 10.1016/j.cmet.2016.08.009
Ducker, G. S., Chen, L., Morscher, R. J., Ghergurovich, J. M., Esposito, M., Teng, X., et al. (2016). Reversal of cytosolic one-carbon flux compensates for loss of the mitochondrial folate pathway. Cell Metab. 23 (6), 1140–1153. doi: 10.1016/j.cmet.2016.04.016
Fan, J., Ye, J., Kamphorst, J. J., Shlomi, T., Thompson, C. B., Rabinowitz, J. D. (2014). Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510 (7504), 298–302. doi: 10.1038/nature13236
FDA. (1996). Final rule. Food standards: amendment of standards of identity for en- riched grain products to require addition of folic acid. Final rule. Fed. Regist. 61, 8781– 8797
Field, M. S., Kamynina, E., Agunloye, O. C., Liebenthal, R. P., Lamarre, S. G., Brosnan, M. E., et al. (2014). Nuclear enrichment of folate cofactors and methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) protect de novo thymidylate biosynthesis during folate deficiency. J. Biol. Chem. 289 (43), 29642–29650. doi: 10.1074/jbc.M114.599589
Field, M. S., Kamynina, E., Watkins, D., Rosenblatt, D. S., Stover, P. J. (2015). Human mutations in methylenetetrahydrofolate dehydrogenase 1 impair nuclear de novo thymidylate biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 112 (2), 400–405. doi: 10.1073/pnas.1414555112
Fleming, A. (2001). The role of folate in the prevention of neural tube defects: human and animal studies. Nutr. Rev. 59 (8 Pt 2), S13–20; discussion S21–3. doi: 10.1111/j.1753-4887.2001.tb05497.x
Fox, J. T., Stover, P. J. (2008). Folate-mediated one-carbon metabolism. Vitam. Horm. 79, 1–44. doi: 10.1016/S0083-6729(08)00401-9
Franke, B., Vermeulen, S. H., Steegers-Theunissen, R. P., Coenen, M. J., Schijvenaars, M. M., Scheffer, H., et al. (2009). An association study of 45 folate-related genes in spina bifida: Involvement of cubilin (CUBN) and tRNA aspartic acid methyltransferase 1 (TRDMT1). Birth Defects Res. A Clin. Mol. Teratol. 85 (3), 216–226. doi: 10.1002/bdra.20556
Friso, S., Choi, S. W., Girelli, D., Mason, J. B., Dolnikowski, G. G., Bagley, P. J., et al. (2002). A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc. Natl. Acad. Sci. U. S. A. 99 (8), 5606–5611. doi: 10.1073/pnas.062066299
Froese, D. S., Kopec, J., Rembeza, E., Bezerra, G. A., Oberholzer, A. E., Suormala, T., et al. (2018). Structural basis for the regulation of human 5,10-methylenetetrahydrofolate reductase by phosphorylation and S-adenosylmethionine inhibition. Nat. Commun. 9 (1), 2261. doi: 10.1038/s41467-018-04735-2
Frosst, P., Blom, H. J., Milos, R., Goyette, P., Sheppard, C. A., Matthews, R. G., et al. (1995). A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 10 (1), 111–113. doi: 10.1038/ng0595-111
Gaughan, D. J., Kluijtmans, L. A., Barbaux, S., McMaster, D., Young, I. S., Yarnell, J. W., et al. (2001). The methionine synthase reductase (MTRR) A66G polymorphism is a novel genetic determinant of plasma homocysteine concentrations. Atherosclerosis 157 (2), 451–456. doi: 10.1016/S0021-9150(00)00739-5
Gibson, T. M., Brennan, P., Han, S., Karami, S., Zaridze, D., Janout, V., et al. (2011). Comprehensive evaluation of one-carbon metabolism pathway gene variants and renal cell cancer risk. PLoS One 6 (10), e26165. doi: 10.1371/journal.pone.0026165
Goldman, I. D., Chattopadhyay, S., Zhao, R., Moran, R. (2010). The antifolates: evolution, new agents in the clinic, and how targeting delivery via specific membrane transporters is driving the development of a next generation of folate analogs. Curr. Opin. Investig. Drugs 11 (12), 1409–1423
Goodrich, J. K., Waters, J. L., Poole, A. C., Sutter, J. L., Koren, O., Blekhman, R., et al. (2014). Human genetics shape the gut microbiome. Cell 159 (4), 789–799. doi: 10.1016/j.cell.2014.09.053
Goodrich, J. K., Davenport, E. R., Beaumont, M., Jackson, M. A., Knight, R., Ober, C., et al. (2016). Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19 (5), 731–743. doi: 10.1016/j.chom.2016.04.017
Hang, Y. D., Woodams, E. E. (2010). Influence of apple cultivar and juice pasteurization on hard cider and eau-de-vie methanol content. Bioresour. Technol. 101 (4), 1396–1398. doi: 10.1016/j.biortech.2009.09.069
Harmon, D. L., Shields, D. C., Woodside, J. V., McMaster, D., Yarnell, J. W., Young, I. S., et al. (1999). Methionine synthase D919G polymorphism is a significant but modest determinant of circulating homocysteine concentrations. Genet. Epidemiol. 17 (4), 298–309. doi: 10.1002/(SICI)1098-2272(199911)17:4<298::AID-GEPI5>3.0.CO;2-V
Hirschhorn, J. N., Lohmueller, K., Byrne, E., Hirschhorn, K. (2002). A comprehensive review of genetic association studies. Genet. Med. 4 (2), 45–61. doi: 10.1097/00125817-200203000-00002
Hoeferlin, L. A., Oleinik, N. V., Krupenko, N. I., Krupenko, S. A. (2011). Activation of p21-dependent G1/G2 arrest in the absence of DNA damage as an antiapoptotic response to metabolic stress. Genes Cancer 2 (9), 889–899. doi: 10.1177/1947601911432495
Holmes, E., Loo, R. L., Stamler, J., Bictash, M., Yap, I. K., Chan, Q., et al. (2008). Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 453 (7193), 396–400. doi: 10.1038/nature06882
Horita, D. A., Krupenko, S. A. (2017). Modeling of interactions between functional domains of ALDH1L1. Chem. Biol. Interact. 276, 23–30. doi: 10.1016/j.cbi.2017.04.011
Hsiao, T. H., Lin, C. J., Chung, Y. S., Lee, G. H., Kao, T. T., Chang, W. N., et al. (2014). Ethanol-induced upregulation of 10-formyltetrahydrofolate dehydrogenase helps relieve ethanol-induced oxidative stress. Mol. Cell Biol. 34 (3), 498–509. doi: 10.1128/MCB.01427-13
Jacques, P. F., Selhub, J., Bostom, A. G., Wilson, P. W., Rosenberg, I. H. (1999). The effect of folic acid fortification on plasma folate and total homocysteine concentrations. N. Engl. J. Med. 340 (19), 1449–1454. doi: 10.1056/NEJM199905133401901
Jain, M., Nilsson, R., Sharma, S., Madhusudhan, N., Kitami, T., Souza, A. L., et al. (2012). Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336 (6084), 1040–1044. doi: 10.1126/science.1218595
Jimenez, A. R., Naz, N., Miyan, J. A. (2019). Altered folate binding protein expression and folate delivery are associated with congenital hydrocephalus in the hydrocephalic Texas rat. J. Cereb. Blood Flow Metab. 39 (10), 2061–2073. doi: 10.1177/0271678X18776226
Khan, Q. A., Pediaditakis, P., Malakhau, Y., Esmaeilniakooshkghazi, A., Ashkavand, Z., Sereda, V., et al. (2018). CHIP E3 ligase mediates proteasomal degradation of the proliferation regulatory protein ALDH1L1 during the transition of NIH3T3 fibroblasts from G0/G1 to S-phase. PLoS One 13 (7): e0199699. doi: 10.1371/journal.pone.0199699
Klerk, M., Verhoef, P., Clarke, R., Blom, H. J., Kok, F. J., Schouten, E. G., et al. (2002). MTHFR 677C–>T polymorphism and risk of coronary heart disease: a meta-analysis. JAMA 288 (16), 2023–2031. doi: 10.1001/jama.288.16.2023
Koppaka, V., Thompson, D. C., Chen, Y., Ellermann, M., Nicolaou, K. C., Juvonen, R. O., et al. (2012). Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 64 (3), 520–539. doi: 10.1124/pr.111.005538
Krupenko, S. A. (2009). FDH: an aldehyde dehydrogenase fusion enzyme in folate metabolism. Chem. Biol. Interact. 178 (1–3), 84–93. doi: 10.1016/j.cbi.2008.09.007
Krupenko, S. A., Krupenko, N. I. (2018). ALDH1L1 and ALDH1L2 folate regulatory enzymes in cancer. Adv. Exp. Med. Biol. 1032, 127–143. doi: 10.1007/978-3-319-98788-0_10
Krupenko, S. A., Krupenko, N. I. (2019). Loss of ALDH1L1 folate enzyme confers a selective metabolic advantage for tumor progression. Chem. Biol. Interact. 302, 149–155. doi: 10.1016/j.cbi.2019.02.013
Krupenko, S. A., Oleinik, N. V. (2002). 10-formyltetrahydrofolate dehydrogenase, one of the major folate enzymes, is down-regulated in tumor tissues and possesses suppressor effects on cancer cells. Cell Growth Differ. 13 (5), 227–236
Krupenko, S. A., Wagner, C., Cook, R. J. (1997a). Domain structure of rat 10-formyltetrahydrofolate dehydrogenase. Resolution of the amino-terminal domain as 10-formyltetrahydrofolate hydrolase. J. Biol. Chem. 272 (15), 10273–10278. doi: 10.1074/jbc.272.15.10273.
Krupenko, S. A., Wagner, C., Cook, R. J. (1997b). Expression, purification, and properties of the aldehyde dehydrogenase homologous carboxyl-terminal domain of rat 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 272 (15), 10266–10272. doi: 10.1074/jbc.272.15.10266
Krupenko, N. I., Dubard, M. E., Strickland, K. C., Moxley, K. M., Oleinik, N. V., Krupenko, S. A. (2010). ALDH1L2 is the mitochondrial homolog of 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 285 (30), 23056–23063. doi: 10.1074/jbc.M110.128843
Krupenko, N. I., Holmes, R. S., Tsybovsky, Y., Krupenko, S. A. (2015). Aldehyde dehydrogenase homologous folate enzymes: evolutionary switch between cytoplasmic and mitochondrial localization. Chem. Biol. Interact. 234, 12–17. doi: 10.1016/j.cbi.2014.12.022
Lee, K. M., Lan, Q., Kricker, A., Purdue, M. P., Grulich, A. E., Vajdic, C. M., et al. (2007). One-carbon metabolism gene polymorphisms and risk of non-Hodgkin lymphoma in Australia. Hum. Genet. 122 (5), 525–533. doi: 10.1007/s00439-007-0431-2
Leung, K. Y., Pai, Y. J., Chen, Q., Santos, C., Calvani, E., Sudiwala, S., et al. (2017). Partitioning of one-carbon units in folate and methionine metabolism is essential for neural tube closure. Cell Rep. 21 (7), 1795–1808. doi: 10.1016/j.celrep.2017.10.072
Li, Q., Lan, Q., Zhang, Y., Bassig, B. A., Holford, T. R., Leaderer, B., et al. (2013). Role of one-carbon metabolizing pathway genes and gene-nutrient interaction in the risk of non-Hodgkin lymphoma. Cancer Causes Control 24 (10), 1875–1884. doi: 10.1007/s10552-013-0264-3
Li, M., Sun, Q., Wang, X. (2017). Transcriptional landscape of human cancers. Oncotarget 8 (21), 34534–34551. doi: 10.18632/oncotarget.15837
Lightfoot, T. J., Johnston, W. T., Painter, D., Simpson, J., Roman, E., Skibola, C. F., et al. (2010). Genetic variation in the folate metabolic pathway and risk of childhood leukemia. Blood 115 (19), 3923–3929. doi: 10.1182/blood-2009-10-249722
Lim, U., Wang, S. S., Hartge, P., Cozen, W., Kelemen, L. E., Chanock, S., et al. (2007). Gene-nutrient interactions among determinants of folate and one-carbon metabolism on the risk of non-Hodgkin lymphoma: NCI-SEER case-control study. Blood 109 (7), 3050–3059. doi: 10.1182/blood-2006-07-034330
Locasale, J. W. (2013). Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer 13 (8), 572–583. doi: 10.1038/nrc3557
Ma, J., Stampfer, M. J., Christensen, B., Giovannucci, E., Hunter, D. J., Chen, J., et al. (1999). A polymorphism of the methionine synthase gene: association with plasma folate, vitamin B12, homocyst(e)ine, and colorectal cancer risk. Cancer Epidemiol. Biomarkers Prev. 8 (9), 825–829
MacFarlane, A. J., Anderson, D. D., Flodby, P., Perry, C. A., Allen, R. H., Stabler, S. P., et al. (2011). Nuclear localization of de novo thymidylate biosynthesis pathway is required to prevent uracil accumulation in DNA. J. Biol. Chem. 286 (51), 44015–44022. doi: 10.1074/jbc.M111.307629
MacMillan, L., Tingley, G., Young, S. K., Clow, K. A., Randell, E. W., Brosnan, M. E., et al. (2018). Cobalamin deficiency results in increased production of formate secondary to decreased mitochondrial oxidation of one-carbon units in rats. J. Nutr. 148 (3), 358–363. doi: 10.1093/jn/nxx057
Mangan, M. E., Williams, J. M., Kuhn, R. M., Lathe, W. C. 3rd (2014). The UCSC genome browser: what every molecular biologist should know. Curr. Protoc. Mol. Biol. 10719 9, 1–36. doi: 10.1002/0471142727.mb1909s107
Marchitti, S. A., Brocker, C., Stagos, D., Vasiliou, V. (2008). Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert. Opin. Drug Metab. Toxicol. 4 (6), 697–720. doi: 10.1517/17425255.4.6.697
Marini, N. J., Yang, W., Asrani, K., Witte, J. S., Rine, J., Lammer, E. J., et al. (2016). Sequence variation in folate pathway genes and risks of human cleft lip with or without cleft palate. Am. J. Med. Genet. A 170 (11), 2777–2787. doi: 10.1002/ajmg.a.37874
Martinez-Chantar, M. L., Vazquez-Chantada, M., Ariz, U., Martinez, N., Varela, M., Luka, Z., et al. (2008). Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology 47 (4), 1191–1199. doi: 10.1002/hep.22159
Mills, J. L., Kirke, P. N., Molloy, A. M., Burke, H., Conley, M. R., Lee, Y. J., et al. (1999). Methylenetetrahydrofolate reductase thermolabile variant and oral clefts. Am. J. Med. Genet. 86 (1), 71–74. doi: 10.1002/(SICI)1096-8628(19990903)86:1<71::AID-AJMG14>3.0.CO;2-Y
Minton, D. R., Nam, M., McLaughlin, D. J., Shin, J., Bayraktar, E. C., Alvarez, S. W., et al. (2018). Serine catabolism by shmt2 is required for proper mitochondrial translation initiation and maintenance of formylmethionyl-tRNAs. Mol. Cell 69 (4), 610–621 e5. doi: 10.1016/j.molcel.2018.01.024
Mishra, P. J., Humeniuk, R., Mishra, P. J., Longo-Sorbello, G. S., Banerjee, D., Bertino, J. R. (2007). A miR-24 microRNA binding-site polymorphism in dihydrofolate reductase gene leads to methotrexate resistance. Proc. Natl. Acad. Sci. U. S. A. 104 (33), 13513–13518. doi: 10.1073/pnas.0706217104
Mitchell, L. E., Adzick, N. S., Melchionne, J., Pasquariello, P. S., Sutton, L. N., Whitehead, A. S. (2004). Spina bifida. Lancet 364 (9448), 1885–1895. doi: 10.1016/S0140-6736(04)17445-X
Moat, S. J., Lang, D., McDowell, I. F., Clarke, Z. L., Madhavan, A. K., Lewis, M. J., et al. (2004). Folate, homocysteine, endothelial function and cardiovascular disease. J. Nutr. Biochem. 15 (2), 64–79. doi: 10.1016/j.jnutbio.2003.08.010
Momb, J., Lewandowski, J. P., Bryant, J. D., Fitch, R., Surman, D. R., Vokes, S. A., et al. (2013). Deletion of Mthfd1l causes embryonic lethality and neural tube and craniofacial defects in mice. Proc. Natl. Acad. Sci. U. S. A. 110 (2), 549–554. doi: 10.1073/pnas.1211199110
Morscher, R. J., Ducker, G. S., Li, S. H., Mayer, J. A., Gitai, Z., Sperl, W., et al. (2018). Mitochondrial translation requires folate-dependent tRNA methylation. Nature 554 (7690), 128–132. doi: 10.1038/nature25460
Narisawa, A., Komatsuzaki, S., Kikuchi, A., Niihori, T., Aoki, Y., Fujiwara, K., et al. (2012). : Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Hum. Mol. Genet. 21 (7), 1496–1503. doi: 10.1093/hmg/ddr585
Naz, N., Jimenez, A. R., Sanjuan-Vilaplana, A., Gurney, M., Miyan, J. (2016). Neonatal hydrocephalus is a result of a block in folate handling and metabolism involving 10-formyltetrahydrofolate dehydrogenase. J. Neurochem. 138 (4), 610–623. doi: 10.1111/jnc.13686
Newman, A. C., Maddocks, O. D. K. (2017). One-carbon metabolism in cancer. Br. J. Cancer 116 (12), 1499–1504. doi: 10.1038/bjc.2017.118.
Neymeyer, V., Tephly, T. R., Miller, M. W. (1997). Folate and 10-formyltetrahydrofolate dehydrogenase (FDH) expression in the central nervous system of the mature rat. Brain Res. 766 (1-2), 195–204. doi: 10.1016/S0006-8993(97)00528-3.
Nilsson, R., Jain, M., Madhusudhan, N., Sheppard, N. G., Strittmatter, L., Kampf, C., et al. (2014). Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 5, 3128. doi: 10.1038/ncomms4128.
Oleinik, N. V., Krupenko, N. I., Priest, D. G., Krupenko, S. A. (2005). Cancer cells activate p53 in response to 10-formyltetrahydrofolate dehydrogenase expression. Biochem. J. 391 (Pt 3), 503–511. doi: 10.1042/BJ20050533
Oleinik, N. V., Krupenko, N. I., Reuland, S. N., Krupenko, S. A. (2006). Leucovorin-induced resistance against FDH growth suppressor effects occurs through DHFR up-regulation. Biochem. Pharmacol. 72 (2), 256–266. doi: 10.1016/j.bcp.2006.04.005
Oleinik, N. V., Krupenko, N. I., Krupenko, S. A. (2011). Epigenetic silencing of ALDH1L1, a metabolic regulator of cellular proliferation, in cancers. Genes Cancer 2 (2), 130–139. doi: 10.1177/1947601911405841
Ou, C. Y., Stevenson, R. E., Brown, V. K., Schwartz, C. E., Allen, W. P., Khoury, M. J., et al. (1996). 5,10 Methylenetetrahydrofolate reductase genetic polymorphism as a risk factor for neural tube defects. Am. J. Med. Genet. 63 (4), 610–614. doi: 10.1002/(SICI)1096-8628(19960628)63:4<610::AID-AJMG15>3.0.CO;2-L
Pai, Y. J., Leung, K. Y., Savery, D., Hutchin, T., Prunty, H., Heales, S., et al. (2015). Glycine decarboxylase deficiency causes neural tube defects and features of non-ketotic hyperglycinemia in mice. Nat. Commun. 6, 6388. doi: 10.1038/ncomms7388
Philip, D., Buch, A., Moorthy, D., Scott, T. M., Parnell, L. D., Lai, C. Q., et al. (2015). Dihydrofolate reductase 19-bp deletion polymorphism modifies the association of folate status with memory in a cross-sectional multi-ethnic study of adults. Am. J. Clin. Nutr. 102 (5), 1279–1288. doi: 10.3945/ajcn.115.111054
Piskounova, E., Agathocleous, M., Murphy, M. M., Hu, Z., Huddlestun, S. E., Zhao, Z., et al. (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. doi: 10.1038/nature15726
Pullarkat, S. T., Stoehlmacher, J., Ghaderi, V., Xiong, Y. P., Ingles, S. A., Sherrod, A., et al. (2001). Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharmacogenomics J. 1 (1), 65–70. doi: 10.1038/sj.tpj.6500012
Pusceddu, I., Herrmann, M., Kirsch, S. H., Werner, C., Hubner, U., Bodis, M., et al. (2017). One-carbon metabolites and telomere length in a prospective and randomized study of B- and/or D-vitamin supplementation. Eur. J. Nutr. 56 (5), 1887–1898. doi: 10.1007/s00394-016-1231-z
Reuland, S. N., Vlasov, A. P., Krupenko, S. A. (2006). Modular organization of FDH: exploring the basis of hydrolase catalysis. Protein Sci. 15 (5), 1076–1084. doi: 10.1110/ps.052062806
Rock, C. L., Lampe, J. W., Patterson, R. E. (2000). Nutrition, genetics, and risks of cancer. Annu. Rev. Public Health 21, 47–64. doi: 10.1146/annurev.publhealth.21.1.47
Rosenzweig, A., Blenis, J., Gomes, A. P. (2018). Beyond the warburg effect: how do cancer cells regulate one-carbon metabolism? Front. Cell Dev. Biol. 6, 90. doi: 10.3389/fcell.2018.00090
Sarek, G., Marzec, P., Margalef, P., Boulton, S. J. (2015). Molecular basis of telomere dysfunction in human genetic diseases. Nat. Struct. Mol. Biol. 22 (11), 867–874. doi: 10.1038/nsmb.3093
Schmitt, E., Blanquet, S., Mechulam, Y. (1996). Structure of crystalline escherichia coli methionyl-tRNA(f)met formyltransferase: comparison with glycinamide ribonucleotide formyltransferase. EMBO J. 15 (17), 4749–4758. doi: 10.1002/j.1460-2075.1996.tb00852.x
Senchenko, V. N., Kisseljova, N. P., Ivanova, T. A., Dmitriev, A. A., Krasnov, G. S., Kudryavtseva, A. V., et al. (2013). Novel tumor suppressor candidates on chromosome 3 revealed by NotI-microarrays in cervical cancer. Epigenetics 8 (4), 409–420. doi: 10.4161/epi.24233
Shahzad, K., Hai, A., Ahmed, A., Kizilbash, N., Alruwaili, J. (2013). A structured-based model for the decreased activity of Ala222Val and Glu429Ala Methylenetetrahydrofolate Reductase (MTHFR) Mutants. Bioinformation 9 (18), 929–936. doi: 10.6026/97320630009929
Sharp, L., Little, J. (2004). Polymorphisms in genes involved in folate metabolism and colorectal neoplasia: a HuGE review. Am. J. Epidemiol. 159 (5), 423–443. doi: 10.1093/aje/kwh066
Siegel, D., Anwar, A., Winski, S. L., Kepa, J. K., Zolman, K. L., Ross, D. (2001). Rapid polyubiquitination and proteasomal degradation of a mutant form of NAD(P)H:quinone oxidoreductase 1. Mol. Pharmacol. 59 (2), 263–268. doi: 10.1124/mol.59.2.263
Song, M. A., Brasky, T. M., Marian, C., Weng, D. Y., Taslim, C., Llanos, A. A., et al. (2016). Genetic variation in one-carbon metabolism in relation to genome-wide DNA methylation in breast tissue from heathy women. Carcinogenesis 37 (5), 471–480. doi: 10.1093/carcin/bgw030
Spencer, A. C., Spremulli, L. L. (2004). Interaction of mitochondrial initiation factor 2 with mitochondrial fMet-tRNA. Nucleic Acids Res. 32 (18), 5464–5470. doi: 10.1093/nar/gkh886
Stevens, V. L., McCullough, M. L., Pavluck, A. L., Talbot, J. T., Feigelson, H. S., Thun, M. J., et al. (2007). Association of polymorphisms in one-carbon metabolism genes and postmenopausal breast cancer incidence. Cancer Epidemiol. Biomarkers Prev. 16 (6), 1140–1147. doi: 10.1158/1055-9965.EPI-06-1037
Stevens, V. L., Rodriguez, C., Sun, J., Talbot, J. T., Thun, M. J., Calle, E. E. (2008). No association of single nucleotide polymorphisms in one-carbon metabolism genes with prostate cancer risk. Cancer Epidemiol. Biomarkers Prev. 17 (12), 3612–3614. doi: 10.1158/1055-9965.EPI-08-0789
Stover, P. J. (2009). One-carbon metabolism-genome interactions in folate-associated pathologies. J. Nutr. 139 (12), 2402–2405. doi: 10.3945/jn.109.113670
Stover, P. J. (2011). Polymorphisms in 1-carbon metabolism, epigenetics and folate-related pathologies. J. Nutrigenet. Nutrigenomics 4 (5), 293–305. doi: 10.1159/000334586
Strickland, K. C., Hoeferlin, L. A., Oleinik, N. V., Krupenko, N. I., Krupenko, S. A. (2010). Acyl carrier protein-specific 4'-phosphopantetheinyl transferase activates 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 285 (3), 1627–1633. doi: 10.1074/jbc.M109.080556
Strickland, K. C., Holmes, R. S., Oleinik, N. V., Krupenko, N. I., Krupenko, S. A. (2011). Phylogeny and evolution of aldehyde dehydrogenase-homologous folate enzymes. Chem. Biol. Interact. 191 (1-3), 122–128. doi: 10.1016/j.cbi.2010.12.025
Strickland, K. C., Krupenko, N. I., Krupenko, S. A. (2013). Molecular mechanisms underlying the potentially adverse effects of folate. Clin. Chem. Lab. Med. 51 (3), 607–616. doi: 10.1515/cclm-2012-0561
Suthandiram, S., Gan, G. G., Mohd Zain, S., Bee, P. C., Lian, L. H., Chang, K. M., et al. (2015). Genetic polymorphisms in the one-carbon metabolism pathway genes and susceptibility to non-Hodgkin lymphoma. Tumour Biol. 36 (3), 1819–1834. doi: 10.1007/s13277-014-2785-0
Tackels-Horne, D., Goodman, M. D., Williams, A. J., Wilson, D. J., Eskandari, T., Vogt, L. M., et al. (2001). Identification of differentially expressed genes in hepatocellular carcinoma and metastatic liver tumors by oligonucleotide expression profiling. Cancer 92 (2), 395–405. doi: 10.1002/1097-0142(20010715)92:2<395::AID-CNCR1335>3.0.CO;2-U
Tani, H., Ohnishi, S., Shitara, H., Mito, T., Yamaguchi, M., Yonekawa, H., et al. (2018). Mice deficient in the Shmt2 gene have mitochondrial respiration defects and are embryonic lethal. Sci. Rep. 8 (1), 425. doi: 10.1038/s41598-017-18828-3
Tephly, T. R. (1991). The toxicity of methanol. Life Sci. 48 (11), 1031–1041. doi: 10.1016/0024-3205(91)90504-5
Tibbetts, A. S., Appling, D. R. (2010). Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30, 57–81. doi: 10.1146/annurev.nutr.012809.104810
Titus, S. A., Moran, R. G. (2000). Retrovirally mediated complementation of the glyB phenotype. Cloning of a human gene encoding the carrier for entry of folates into mitochondria. J. Biol. Chem. 275 (47), 36811–36817. doi: 10.1074/jbc.M005163200
Tsybovsky, Y., Krupenko, S. A. (2011). Conserved catalytic residues of the ALDH1L1 aldehyde dehydrogenase domain control binding and discharging of the coenzyme. J. Biol. Chem. 286 (26), 23357–23367. doi: 10.1074/jbc.M111.221069
Tsybovsky, Y., Donato, H., Krupenko, N. I., Davies, C., Krupenko, S. A. (2007). Crystal structures of the carboxyl terminal domain of rat 10-formyltetrahydrofolate dehydrogenase: implications for the catalytic mechanism of aldehyde dehydrogenases. Biochemistry 46 (11), 2917–2929. doi: 10.1021/bi0619573
Tsybovsky, Y., Malakhau, Y., Strickland, K. C., Krupenko, S. A. (2013). The mechanism of discrimination between oxidized and reduced coenzyme in the aldehyde dehydrogenase domain of Aldh1l1. Chem. Biol. Interact. 202 (1–3), 62–69. doi: 10.1016/j.cbi.2012.12.015
Tucker, E. J., Hershman, S. G., Kohrer, C., Belcher-Timme, C. A., Patel, J., Goldberger, O. A., et al. (2011). Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab. 14 (3), 428–434. doi: 10.1016/j.cmet.2011.07.010
Ueland, P. M., Hustad, S., Schneede, J., Refsum, H., Vollset, S. E. (2001). Biological and clinical implications of the MTHFR C677T polymorphism. Trends Pharmacol. Sci. 22 (4), 195–201. doi: 10.1016/S0165-6147(00)01675-8
Ulrich, C. M., Bigler, J., Bostick, R., Fosdick, L., Potter, J. D. (2002). Thymidylate synthase promoter polymorphism, interaction with folate intake, and risk of colorectal adenomas. Cancer Res. 62 (12), 3361–3364
Visentin, M., Zhao, R., Goldman, I. D. (2012). The antifolates. Hematol. Oncol. Clin. North Am. 26 (3), 629–48, ix. doi: 10.1016/j.hoc.2012.02.002
Weisberg, I., Tran, P., Christensen, B., Sibani, S., Rozen, R. (1998). A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol. Genet. Metab. 64 (3), 169–172. doi: 10.1006/mgme.1998.2714
Williams, S. R., Yang, Q., Chen, F., Liu, X., Keene, K. L., Jacques, P., et al. (2014). : Genome-wide meta-analysis of homocysteine and methionine metabolism identifies five one carbon metabolism loci and a novel association of ALDH1L1 with ischemic stroke. PLoS Genet. 10 (3), e1004214. doi: 10.1371/journal.pgen.1004214
Wilson, A., Platt, R., Wu, Q., Leclerc, D., Christensen, B., Yang, H., et al. (1999). A common variant in methionine synthase reductase combined with low cobalamin (vitamin B12) increases risk for spina bifida. Mol. Genet. Metab. 67 (4), 317–323. doi: 10.1006/mgme.1999.2879
Wu, L., Lu, X., Guo, J., Zhang, T., Wang, F., Bao, Y. (2016). Association between ALDH1L1 gene polymorphism and neural tube defects in the Chinese Han population. Neurol. Sci. 37 (7), 1049–1054. doi: 10.1007/s10072-016-2527-8
Xie, W., Wood, A. R., Lyssenko, V., Weedon, M. N., Knowles, J. W., Alkayyali, S., et al. (2013). Genetic variants associated with glycine metabolism and their role in insulin sensitivity and type 2 diabetes. Diabetes 62 (6), 2141–2150. doi: 10.2337/db12-0876
Yamada, K., Chen, Z., Rozen, R., Matthews, R. G. (2001). Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc. Natl. Acad. Sci. U. S. A. 98 (26), 14853–14858. doi: 10.1073/pnas.261469998
Yamada, K., Strahler, J. R., Andrews, P. C., Matthews, R. G. (2005). Regulation of human methylenetetrahydrofolate reductase by phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 102 (30), 10454–10459. doi: 10.1073/pnas.0504786102
Zhang, H., Liu, C., Han, Y. C., Ma, Z., Zhang, H., Ma, Y., et al. (2015). Genetic variations in the one-carbon metabolism pathway genes and susceptibility to hepatocellular carcinoma risk: a case-control study. Tumour Biol. 36 (2), 997–1002. doi: 10.1007/s13277-014-2725-z
Zheng, Y., Lin, T. Y., Lee, G., Paddock, M. N., Momb, J., Cheng, Z., et al. (2018). Mitochondrial one-carbon pathway supports cytosolic folate integrity in cancer cells. Cell 175 (6), 1546–1560 e17. doi: 10.1016/j.cell.2018.09.041
Zhu, X., Han, W., Xue, W., Zou, Y., Xie, C., Du, J., et al. (2016). The association between telomere length and cancer risk in population studies. Sci. Rep. 6, 22243. doi: 10.1038/srep22243
Keywords: folate metabolism, ALDH1L1, candidate tumor suppressor, SNPs, human diseases
Citation: Krupenko SA and Horita DA (2019) The Role of Single-Nucleotide Polymorphisms in the Function of Candidate Tumor Suppressor ALDH1L1. Front. Genet. 10:1013. doi: 10.3389/fgene.2019.01013
Received: 04 December 2018; Accepted: 23 September 2019;
Published: 30 October 2019.
Edited by:
Manlio Vinciguerra, International Clinical Research Center (FNUSA-ICRC), CzechiaReviewed by:
Nicholas Daniel Edward Greene, University College London, United KingdomKaori Kimura-Kataoka, Shimane University, Japan
Copyright © 2019 Krupenko and Horita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergey A. Krupenko, c2VyZ2V5X2tydXBlbmtvQHVuYy5lZHU=