
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 18 October 2019
Sec. Cancer Genetics
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.01005
 Stavros Glentis1†
Stavros Glentis1† Alexandros C. Dimopoulos1
Alexandros C. Dimopoulos1 Konstantinos Rouskas1
Konstantinos Rouskas1 George Ntritsos2
George Ntritsos2 Evangelos Evangelou2,3
Evangelos Evangelou2,3 Steven A. Narod4,5Anne-Marie Mes-Masson6William D. Foulkes7,8,9Barbara Rivera7,8
Steven A. Narod4,5Anne-Marie Mes-Masson6William D. Foulkes7,8,9Barbara Rivera7,8 Patricia N. Tonin10,11,12
Patricia N. Tonin10,11,12 Jiannis Ragoussis7,13*
Jiannis Ragoussis7,13* Antigone S. Dimas1*
Antigone S. Dimas1*Approximately 10% of breast cancer (BC) cases are hereditary BC (HBC), with HBC most commonly encountered in the context of hereditary breast and ovarian cancer (HBOC) syndrome. Although thousands of loss-of-function (LoF) alleles in over 20 genes have been associated with HBC susceptibility, the genetic etiology of approximately 50% of cases remains unexplained, even when polygenic risk models are considered. We focused on one of the least-studied European populations and applied whole-exome sequencing (WES) to 52 individuals from 17 Greek HBOC families, in which at least one patient was negative for known HBC risk variants. Initial screening revealed pathogenic variants in known cancer genes, including BARD1:p.Trp91* detected in a cancer-free individual, and MEN1:p.Glu260Lys detected in a BC patient. Gene- and variant-based approaches were applied to exome data to identify candidate risk variants outside of known risk genes. Findings were verified in a collection of Canadian HBOC patients of European ancestry (FBRCAX), in an independent group of Canadian BC patients (CHUM-BC) and controls (CARTaGENE), as well as in individuals from The Cancer Genome Atlas (TCGA) and the UK Biobank (UKB). Rare LoF variants were uncovered in MDM1 and NBEAL1 in Greek and Canadian HBOC patients. We also report prioritized missense variants SETBP1:c.4129G > C and C7orf34:c.248C > T. These variants comprise promising candidates whose role in cancer pathogenicity needs to be explored further.
Breast cancer (BC) is the most common type of cancer in women worldwide, with almost 1.7 million new cases and 500,000 deaths annually (Ferlay et al., 2015). In Europe alone, there are 464,000 new cases and 131,000 deaths annually, but the age-standardized incidence rates across countries vary threefold (Ferlay et al., 2013). Greece, with 6,000 cases annually, displays one of the lowest BC rates among European populations. This, however, is likely to be an underestimate arising from under-reporting of cases (Ferlay et al., 2013). Although variation in incidence rates of BC is driven by both genetic and environmental factors (Garcia-Closas et al., 2014), approximately 10% of BC cases have a strong genetic basis (Lynch et al., 2008; King-Spohn and Pilarski, 2014; Terui-Kohbata and Yoshida, 2017). These cases, which show strong familial clustering, or are characterized by early onset of disease, are termed hereditary and are most commonly encountered in the context of hereditary breast and ovarian cancer (HBOC) syndrome (Lynch et al., 2008; Terui-Kohbata and Yoshida, 2017). This syndrome is primarily characterized by increased risk of BC and of ovarian cancer (OC), but also of other cancers, including pancreatic and prostate cancers (Solomon et al., 2012). BC can also manifest in other inherited cancer syndromes, including Cowden, Li-Fraumeni, Peutz-Jeghers, and hereditary gastric cancer syndromes (Rahman, 2014).
Disease-associated genetic variants located in two major predisposition genes, BRCA1 and BRCA2, account for approximately 25% of HBOC cases worldwide (Kast et al., 2016). Heterozygote individuals for pathogenic variants in BRCA1 have a 72% and 44% increased risk for BC and OC, respectively, by age 80 years. In the case of BRCA2, equivalent risks for BC and OC by age 80 years amount to 69% and 17% (Kuchenbaecker et al., 2017). Individuals harboring pathogenic variants in these genes also have increased risk for other malignancies, including melanoma, prostate, and pancreatic cancer, which suggests a broader role for these genes in cancer predisposition (Levy-Lahad and Friedman, 2007). Thousands of different loss-of-function (LoF) pathogenic variants have been identified in BRCA1 and BRCA2 across different populations, the majority of which are individually rare (Mcclellan and King, 2010). In the Greek population, BRCA1 and BRCA2 pathogenic variants account for 27.9% of HBOC cases nationally, with over half (58.5%) of these arising from six principal founder variants (Konstantopoulou et al., 2014).
In addition to pathogenic variants in BRCA1 and BRCA2, moderate- to high-penetrance LoF variants in at least 24 genes have been linked to BC susceptibility (Nielsen et al., 2016). However, of these, only ATM, CDH1, CHEK2, NF1, PALB2, PTEN, STK11, and TP53 are established risk genes, with the remaining candidates lacking reliable effect estimates to date (Easton et al., 2015). The majority of these genes have roles in biological pathways linked to genome maintenance, although other functions including cell adhesion (CDH1), RAS signaling (NF1), and PI3K/AKT/mTOR signaling (PTEN) are reported (Nielsen et al., 2016). Similar to pathogenic variants in BRCA1 and BRCA2, LoF alleles in these genes may also predispose to other forms of cancer (Solomon et al., 2012). In addition to BC arising from the effects of single highly penetrant variants, susceptibility to disease can also be explained by the presence of multiple, low-penetrance alleles. Population-level genome-wide association studies (GWAS) have highlighted at least 150 such BC susceptibility loci to date, which explain ∼18% of the familial relative risk (Michailidou et al., 2017).
Despite extensive research, approximately 50% of HBOC cases remain of unknown genetic etiology, even when polygenic risk scores are taken into consideration (Michailidou et al., 2017; Mavaddat et al., 2019). Applying whole-exome sequencing (WES) to HBOC families with unknown genetic etiology is an approach that has contributed to the identification of novel disease-predisposing variants (Thompson et al., 2012; Kiiski et al., 2014; Cybulski et al., 2015; Rivera et al., 2017). This strategy is expected to yield additional pathogenic variants, especially when applied to lesser-studied populations, such as that of Greece. Risk alleles segregating at extremely low frequencies in other populations may exist in slightly higher frequencies in Greek patients due to population-specific effects. In the present study, we applied WES to Greek HBOC families of unknown genetic etiology to identify novel BC susceptibility variants. Findings were followed up in a collection of Canadian HBOC patients of European ancestry (FBRCAX) (Cybulski et al., 2015; Rivera et al., 2017) and subsequently validated in an independent BC patient group from the Centre Hospitalier de l’Université de Montréal (CHUM-BC) and control population (CARTaGENE) (Awadalla et al., 2013). Findings were also explored in cancer patients from The Cancer Genome Atlas (TCGA) (www.cancergenome.nih.gov and the UK Biobank (UKB) (Sudlow et al., 2015). All data sets interrogated in the present study are summarized in Supplementary Table 1.
BC and OC patients were screened for known cancer risk variants at the molecular diagnostics laboratory at National Centre of Scientific Research (NCSR) Demokritos. Index patients were those individuals who developed BC or OC before the age of 40 years or had at least two first-degree relatives who were also diagnosed with cancer. Screening involved Sanger sequencing (exons and intron/exon boundaries) and multiplex ligation-dependent probe amplification (MLPA) analysis of BRCA1 and BRCA2. Patients who were negative for known disease-associated variants in BRCA1 and BRCA2, were further screened for single-nucleotide variants (SNVs) and indels in 94 cancer-predisposing genes on the Illumina TruSight Cancer Panel (Supplementary Note 1). Patients, who were negative for known causal variants and their informative relatives (including family members affected by any form of cancer, parents of index patients, and obligate carriers), were subjected to WES and comprised the Greek BC study (GRBC, 47 females and 5 males from 17 HBOC families). Of the selected individuals, 30 were BC and 3 were OC patients (Supplementary Table 2). Written informed consent was obtained from all individuals prior to genetic testing, and the study was approved by the bioethics committee of NCSR Demokritos (240/EH/11.3, updated Feb. 14, 2014) in agreement with the 1975 Helsinki statement.
We subsequently explored initial findings in 51 HBOC index patients of FBRCAX from Cybulski et al. (2015) and Rivera et al. (2017), who were negative for disease variants in BRCA1 and BRCA2, CHEK2, NBN, and PALB2. Exome capture, sequencing, and raw data analysis methods have been described previously (Cybulski et al., 2015) and are summarized in Supplementary Note 2. WES data from FBRCAX were screened to confirm absence of other known disease-associated variants (94 cancer genes; Illumina TruSight Cancer Panel).
Our study also included an independent group of female BC patients from Centre Hospitalier de l’Université de Montréal (CHUM-BC) (N = 512) who were diagnosed with invasive BC at <65 years of age. CHUM-BC patients were defined as French Canadian based on review of last names.
We also included a collection of cancer-free individuals of French-Canadian origin (N = 1,940: 970 women, 970 men) recruited through CARTaGENE. CARTaGENE individuals were born in Quebec, spoke French as their first language, and their parents and all four grandparents were born in Canada. They ranged in age from 45 to 65 years (average, 53 years), had no personal history of cancer, and no documented cancer cases in first-degree relatives.
Exome capture on gDNA was performed using the Ion Targetseq™ exome enrichment kit, and samples were sequenced on an Ion Proton (IP) platform (Thermo Fisher Scientific Inc., West Palm Beach, FL, USA) at the Genomics Facility of the Biomedical Sciences Research Center (BSRC) Alexander Fleming. Variants (SNVs and indels) were called from IP raw data using two software packages: the IP built-in Torrent Variant Caller (TVC v5.0) and the Genome Analyzer Toolkit (GATK) (Depristo et al., 2011). Copy number variants (CNVs) were called using eXome-Hidden Markov Model (XHMM) software, with default options (Fromer et al., 2012). Additional WES on an Illumina platform was performed for seven GRBC individuals to compare IP-generated data to data derived from the widely used Illumina platform, which employs different chemistry (Boland et al., 2013) (Supplementary Note 2).
Pairwise kinship analysis for all GRBC individuals was performed using vcftools (Danecek et al., 2011) to confirm reported pedigrees, to flag potential errors, and to reveal hidden genetic relationships. Principal component analysis (PCA) was carried out using EIGENSTRAT (Price et al., 2006) to determine the extent of population structure and to compare GRBC individuals with individuals from the 1,000 genome populations (Genomes Project et al., 2015).
To identify candidate HBOC susceptibility loci, we applied a gene-based shortlisting approach to the total of called variants in GRBC. Our approach focused on genes that harbored LoF variants (stop-gain, essential splice site, and frameshift), because these variant types are the most likely to have functional impact and are commonly linked to disease susceptibility (Richards et al., 2015). We limited our search to rare variants (minor allele frequency [MAF] ≤ 0.1% in both 1,000 genomes (Genomes Project et al., 2015) and gnomAD (Lek et al., 2016) global populations) that were present in at least two GRBC patients from a single family, where this was possible to be ascertained (Figure 1 and Supplementary Note 3). Shortlisted variants were inspected visually on the Integrative Genomics Viewer (IGV) to exclude possible false positives (Robinson et al., 2017). A subset of shortlisted variants was re-sequenced and experimentally validated using AmpliSeq (Thermo Fisher Scientific Inc., West Palm Beach, FL, USA) (Supplementary Note 4).
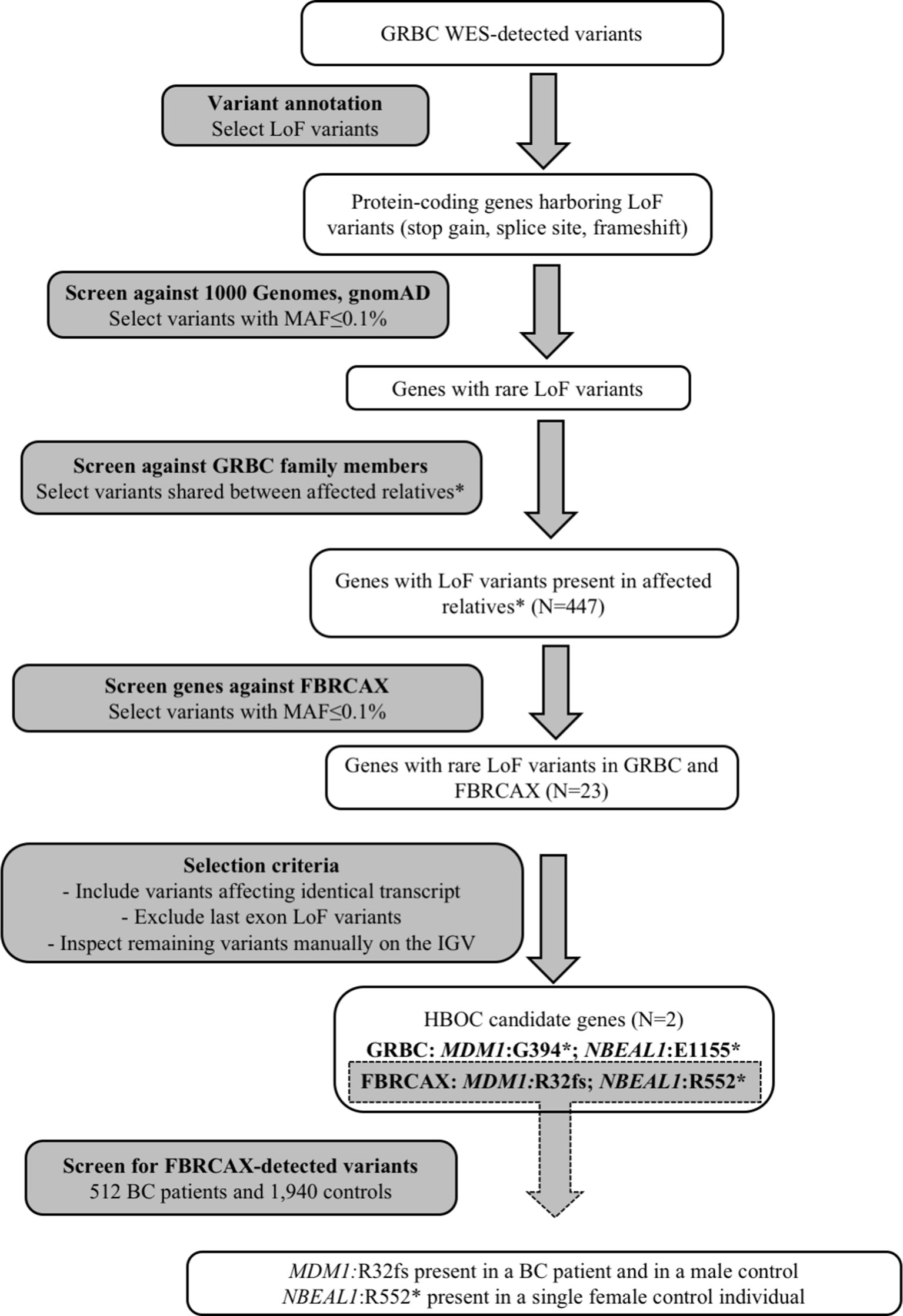
Figure 1 Gene-based prioritization workflow. *For families where this was possible to be ascertained.
Genes harboring shortlisted variants were considered candidate risk genes and were followed up in the FBRCAX collection of patients (N = 51). In FBRCAX, we examined the distribution of rare LoF variants in these prioritized genes. We focused on genes that harbored the same or a different rare LoF variant in at least one FBRCAX patient, restricting our analysis to variants mapping on the same transcript. Genes harboring at least one rare LoF variant in both GRBC and FBRCAX and meeting criteria pertaining to MAF, transcript, and position in gene were further shortlisted as likely risk candidates (Supplementary Note 3). Variants in FBRCAX were validated experimentally through Sanger sequencing (Supplementary Note 4). We further examined the presence of FBRCAX-detected candidate variants in female BC patients from CHUM-BC and in cancer-free individuals from CARTaGENE (Supplementary Note 5).
To evaluate the possible involvement of shortlisted genes in general cancer susceptibility, we explored large publicly available data sets and performed burden analysis. LoF variant cumulative frequencies were compared across non–Finnish European cancer patients from TCGA (NFE-TCGA), and NFE individuals from the Exome Aggregation Consortium (ExAC) (Lek et al., 2016) after excluding TCGA patients (NFE-ExAC-nonTCGA) (Supplementary Note 6). We applied the sequence kernel association test (SKAT) (p < 0.05) (Wu et al., 2011) with inclusion of variants with MAF ≤ 0.1% in both groups. Last-exon variants as well variants with MAF ≥ 0.1% in either 1000 Genomes or gnomAD were excluded.
To complement the gene-based variant prioritization strategy, we also applied three variant-based prioritization approaches to identify candidate BC risk variants in GRBC. Through these approaches, in addition to LoF variants, we also examined in-frame indels, missense, and stop-loss variants (Supplementary Note 7 and Supplementary Figure 1). The three approaches addressed: a) variants that mapped in any of 1,580 genes with a role in cancer (cancer gene variants [CGV]; Supplementary Note 8, and Supplementary Table 3), b) identical variants shared across unrelated patients, or different variants mapping to the same gene in unrelated patients (shared variants/genes in unrelated [SVGU]), and c) ultra-rare variants (MAF < 0.01%) present in a single patient or family (family-specific variants [FSV]). We subsequently considered variants prioritized by one, two or all three of the above approaches, and that fulfilled criteria for MAF, predicted pathogenicity as predicted by in silico tools, and presence in affected relatives (Supplementary Note 7). Following visual inspection on the IGV and experimental validation of a subset of variants (Supplementary Note 4), we explored the presence of shortlisted variants in FBRCAX.
To further evaluate the variants prioritized through the above approach, we explored their presence across independent groups of cancer patients and cancer-free individuals. We examined the following groups: a) NFE-TCGA cancer patients, b) NFE-ExAC-nonTCGA cancer-free individuals, c) UKB cancer patients of European ancestry (EUR-UKB cancer patients), and d) UKB cancer-free individuals of European ancestry (EUR-UKB cancer free individuals). In the case of EUR-UKB cancer patients, we considered: i) all cancer patients, and ii) female BC patients (Supplementary Note 6).
To investigate the possible role of variants shortlisted through the above approach in general cancer susceptibility, we used Fisher’s exact test (FET) (two-sided p < 0.05) to compare allele frequencies across: a) NFE-TCGA (cancer patients) versus NFE-ExAC-nonTCGA (cancer-free individuals), b) EUR-UKB cancer patients versus EUR-UKB cancer-free individuals, and c) EUR-UKB female BC patients versus EUR-UKB female cancer-free individuals (Supplementary Note 9).
We report a mean coverage depth of 154× for targeted sequenced regions (range, 71–210×, Supplementary Tables 4 and 5). On average, 33,048 SNVs, 1,212 indels, and 30 CNVs were detected per GRBC individual (Supplementary Table 6). For IP-sequenced samples, we report an average overlap of 90.1% for SNVs called using TVC versus GATK (Supplementary Table 7). Comparison across sequencing platforms (IP vs. Illumina) revealed average overlaps of 85.7% (SNVs) and 48.8% (indels) (Supplementary Note 10 and Supplementary Tables 8 and 9). Kinship analysis confirmed patient-reported relationships in 16 out of 17 pedigrees. A single BC patient (F14S01) was found to be genetically unrelated to her reported mother and sister (relatedness ϕ = −0.019 and −0.015, respectively) and to all other GRBC individuals and was excluded from subsequent analyses. PCA revealed that GRBC unrelated patients map close to populations of European ancestry, particularly to the Iberian (IBS) and Tuscan (TSI) 1000 Genomes populations (Genomes Project et al., 2015) (Figure 2). Although this finding derives from coding variants only, it is in line with work reporting genetic similarity between Greek and Italian subpopulations (Stamatoyannopoulos et al., 2017).
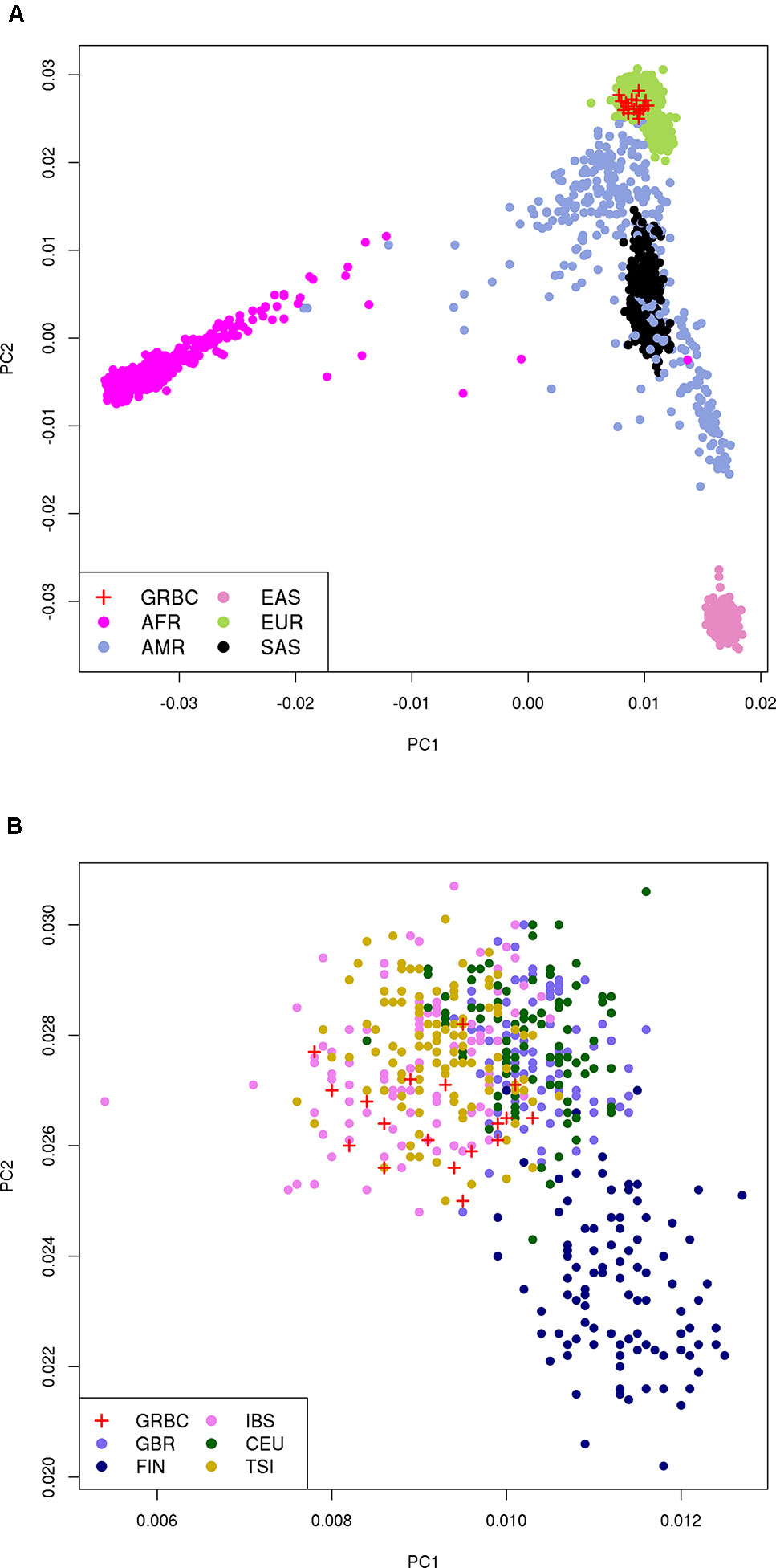
Figure 2 Principal component analysis (PCA) on 27,666 coding variant positions reveals that GRBC patients (one patient per family) map close to: (A) the EUR superpopulation, and (B) the IBS and TSI populations of 1000 Genomes. Abbreviations: GRBC, Greek Breast Cancer study; AFR, African; AMR, Admixed American; EAS, East Asian; EUR, European; SAS, South Asian; CEU, Central European; TSI, Tuscan; FIN, Finnish; GBR, British; IBS, Iberian.
In our effort to identify novel HBOC risk loci, we first sought to ascertain whether known risk variants were present in GRBC non-index patients, or in unaffected individuals. A private stop-gain variant (not reported in public databases to date) in exon three of BARD1 (ENST00000260947:c.273G > A, p.Trp91*) was detected in a single unaffected female individual (F14S03; age, > 60 years). Of note, the variant was not present in the carrier’s daughter, who developed BC at 37 years of age. BARD1 encodes a ligase that forms a heterodimer with BRCA1 and is involved in DNA repair (Shakya et al., 2008). Accumulating evidence suggests that LoF variants in BARD1 may elevate the risk for BC (Castera et al., 2018). We also detected an established pathogenic variant for multiple endocrine neoplasia type 1 (MEN-1) syndrome, in MEN1 (ENST00000337652:c.778G > A, p.Glu260Lys, rs104894268) (Teh et al., 1998) in a non-index patient, diagnosed with BC and primary parathyroid hyperplasia at 50 years. Given that MEN-1 syndrome predisposes to BC and to parathyroid adenomas (Dreijerink et al., 2014), this variant may have contributed to the development of both malignancies. However, it was not detected in the carrier’s niece who developed BC at 39 years of age, suggesting that disease risk in these two related patients may have different underlying genetic etiologies.
Gene- and variant-based prioritization approaches yielded a total of 468 (mapping in 447 genes) and 1,844 variants, respectively. To experimentally validate shortlisted variants, we re-sequenced a subset of 148 SNVs and confirmed the presence of 140 (validation rate 94.6%, Supplementary Note 4 and Supplementary Table 10).
Gene-based prioritized LoF variants were present in GRBC index patients and in at least one affected relative. Of the 447 genes harboring these variants, two (MDM1 and NBEAL1) also harbored at least one rare LoF variant in FBRCAX (Figure 1 and Table 1). LoF variants in MDM1 and NBEAL1 mapped on the same transcript in GRBC and FBRCAX, did not reside in the last exon, and their presence was confirmed experimentally through AmpliSeq (GRBC) and Sanger sequencing (FBRCAX). For GRBC cases where family pedigrees were available, we confirmed that the reported LoF variant (for both MDM1 and NBEAL1) was present in all related patients examined. Burden analysis did not reveal LoF variant enrichment for either gene in NFE-TCGA cancer patients (Supplementary Table 11), but this likely arises in part from the fact that TCGA includes patients with different cancer types.
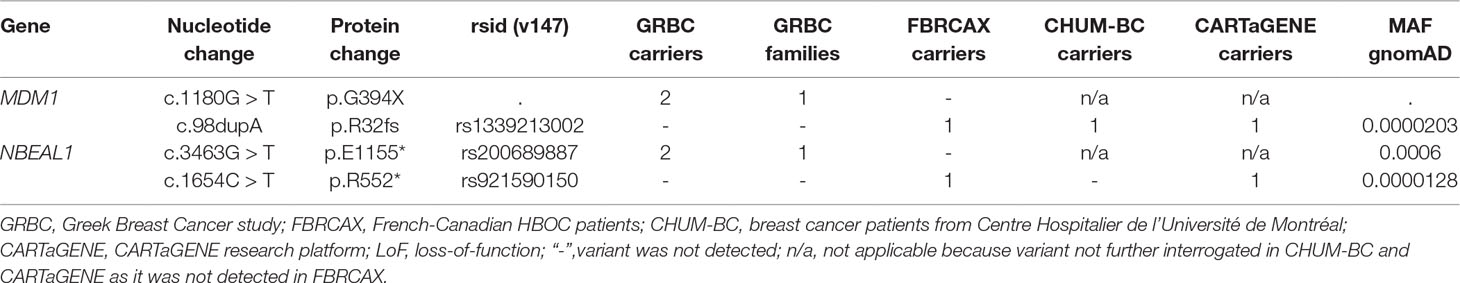
Table 1 Genes with rare LoF variants in individuals from GRBC, FBRCAX, CHUM-BC, and CARTaGENE.
In the case of MDM1, variants MDM1:p.G394* and MDM1:p.R32fs were initially detected in GRBC and FBRCAX, respectively. Notably, MDM1:p.R32fs was also detected in a CHUM-BC patient. These variants localize on opposite ends of the same functional domain (Figure 3). Stop-gain variant MDM1:p.G394* maps on a splice site and may thus exert additional functional effects. MDM1:p.R32fs (gnomAD MAF = 0.002%) is adjacent to a position where two other distinct LoF variants, that affect the same amino acid, have been reported (gnomAD cumulative MAF = 0.0016%), but whose phenotypic impact remains to be described. MDM1:p.G394* was detected in a GRBC mother-daughter pair, diagnosed, respectively, with bilateral BC (at 46 and 56 years) and BC at 44 years. This variant was absent in all other GRBC individuals and has not been reported in public databases to date. MDM1:p.R32fs was present in two French-Canadian BC patients, one each from FBRCAX (out of 51 patients) and CHUM-BC (out of 512 patients). The MDM1 carrier in FBRCAX developed BC at the age of 47 years and had a strong family history of cancer, including five cases of BC and one case each of esophageal and bladder cancer. The MDM1 carrier from CHUM-BC, who was negative for BRCA1, BRCA2, and PALB2 pathogenic variants, developed BC at the age of 51 years. For this individual there is no information on cancer family history. MDM1:p.R32fs, was also detected in a single French-Canadian male individual from CARTaGENE (of 1,924 individuals) (Supplementary Note 4) who was cancer-free at the age of 65 years. MDM1 is an evolutionary conserved gene encoding a protein that binds and stabilizes microtubules, and acts as a negative regulator of centriole duplication (Van De Mark et al., 2015). It is expressed in multiple tissues, including breast, and is predicted to be intolerant to homozygous/double heterozygous LoF variants (ExAC pRec score = 0.96, LoF_Z score = 2.15) (Lek et al., 2016). At the somatic mutation level, MDM1 point mutations and copy number alterations (CNA) are present in 3% of 817 BC tumors of the TCGA data set (Supplementary Figure 2). These mutations tend to co-occur with BRCA2 mutations (p = 0.002) (Supplementary Table 12).
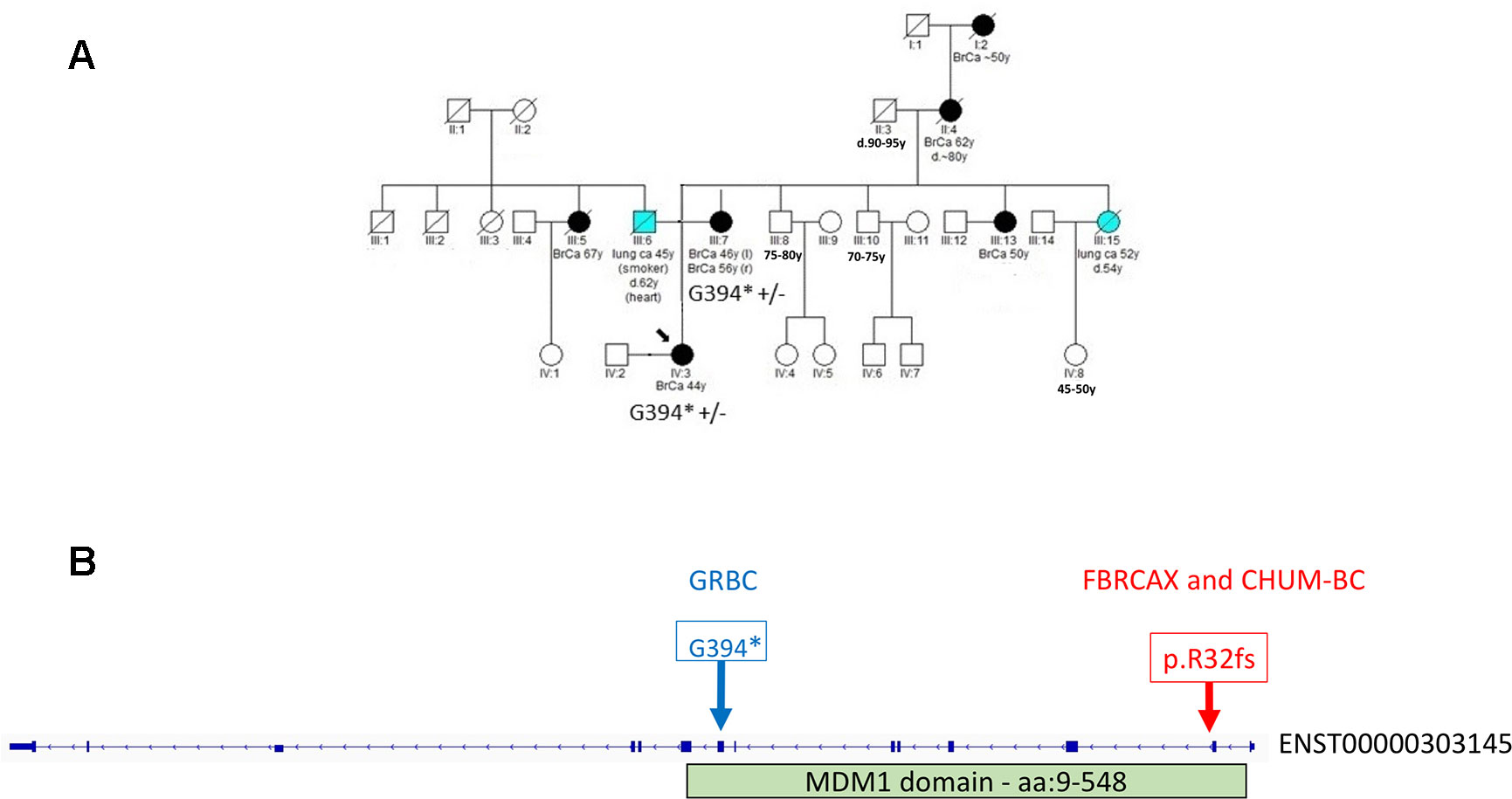
Figure 3 (A) Segregation of MDM1:G394* in GRBC pedigree F25. Both mother (diagnosed with bilateral BC at 46 and 56 years of age) and daughter (diagnosed with BC at 44 years of age) were heterozygous carriers of the MDM1 stop-gain variant. (B) Genomic positions of MDM1 LoF variants detected in Greek (GRBC, MDM1:G394*) and French Canadian (FBRCAX and CHUM-BC, MDM1:p.R32fs) patients. Variants were experimentally validated using Ampliseq (GRBC) and Sanger sequencing (FBRCAX). CHUM-BC variants were detected using the iPLEX MassARRAY.
In the case of NBEAL1, variants NBEAL1:p.E1155* and NBEAL1:p.R552* were detected in GRBC and FBRCAX, respectively (Figure 4). NBEAL1:p.E1155* (gnomAD MAF = 0.06%) was present in two GRBC affected sisters, diagnosed with BC at 59 and 60 years. In FBRCAX, NBEAL1:p.R552* (gnomAD MAF = 0.001%) was present in a single patient (of 51 patients), who developed BC at the age of 23 years. NBEAL1:p.R552* was also detected in a single female individual from CARTaGENE (of 1,919 individuals) who was cancer-free at the age of 48 years. NBEAL1 encodes a protein that possesses two BEACH domains, suggesting a role in vesicle trafficking, membrane dynamics, and receptor signaling (Chen et al., 2004). It is expressed in multiple tissues, including breast, and is predicted to be intolerant to homozygous/double heterozygous LoF variants (ExAC pRec score = 0.96, LoF_Z score = 2.15) (Lek et al., 2016). At the somatic mutation level, NBEAL1 point mutations and CNA are present in 2.3% of 817 BC tumors of the TCGA data set (Supplementary Figure 2). These mutations tend to co-occur with BRCA2 and TP53 mutations and are mutually exclusive with BRCA1 mutations, although these results were not statistically significant (Supplementary Table 12).
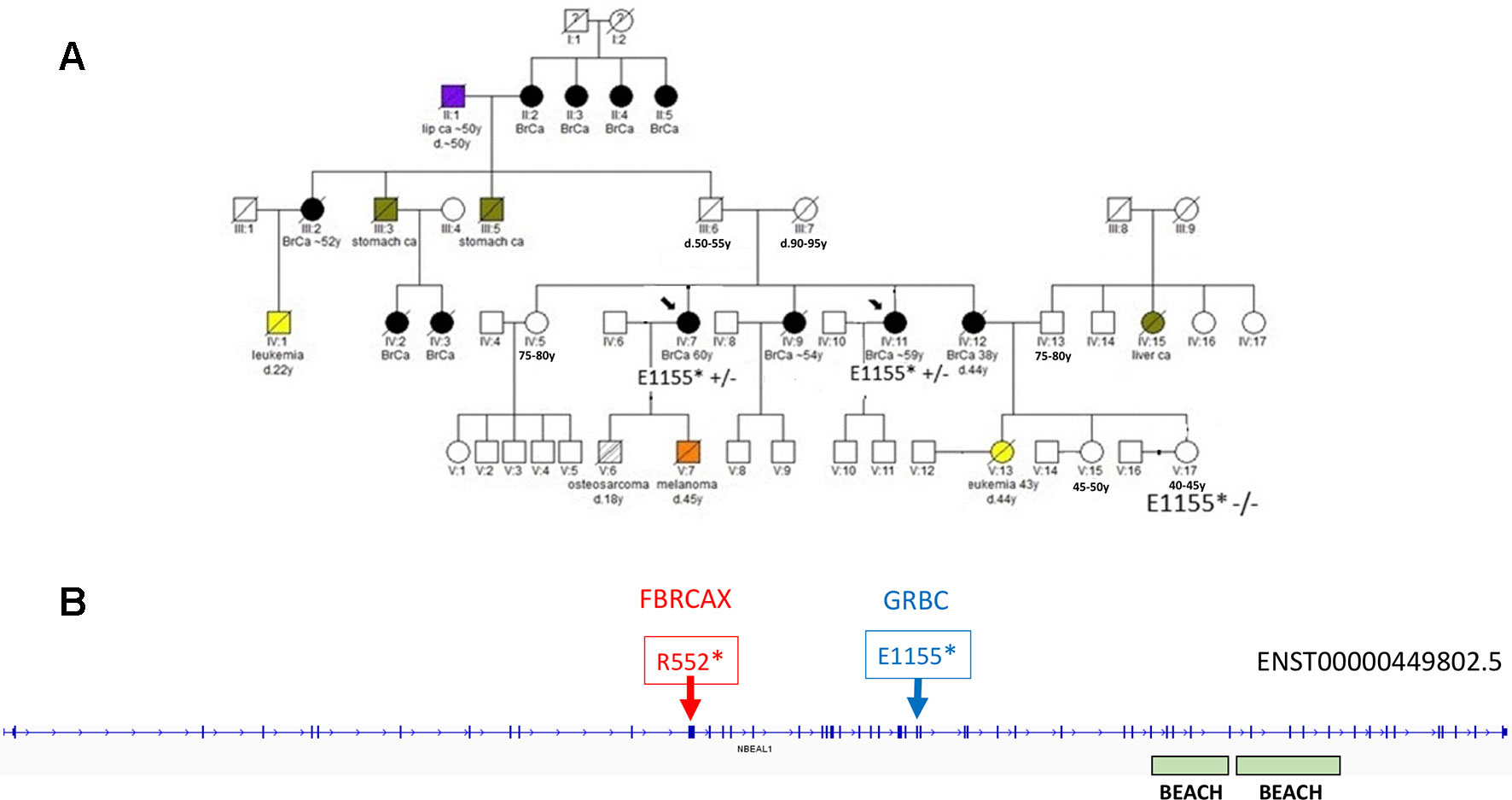
Figure 4 (A) Segregation of NBEAL1:E1155* in GRBC pedigree F22. Two sisters (diagnosed with BC at 59 and 60 years) were heterozygous carriers of the NBEAL1 stop-gain variant. Their niece, who was cancer-free at 40 years of age, was not a carrier of the variant. (B) Genomic positions of NBEAL1 LoF variants detected in GRBC (NBEAL1:E1155*) and FBRCAX (NBEAL1:p.R552*) patients. Variants were experimentally validated using Ampliseq (GRBC) and Sanger sequencing (FBRCAX).
In addition to gene-based prioritization, 1,844 variants were prioritized through a variant-based shortlisting strategy (Supplementary Table 10). Variants were prioritized because they fulfilled at least one of the following criteria: a) they localized in cancer genes (CGV); b) they were shared between unrelated patients, or mapped to the same gene in unrelated patients (SVGU) or c) they were FSV. Importantly, shortlisted variants met criteria pertaining to MAF, predicted pathogenicity and presence in affected relatives (Supplementary Figure 1). To further shortlist candidates, we explored their presence in FBRCAX, TCGA, and UKB and report that: a) 284 were detected in FBRCAX patients, b) 97 were enriched (FET, p < 0.05) in NFE-TCGA patients versus NFE-ExAC-nonTCGA cancer-free individuals, c) 20 were enriched (FET, p < 0.05) in EUR-UKB cancer patients versus EUR-UKB cancer-free individuals, and d) 19 were enriched (FET p < 0.05) in EUR-UKB female BC patients versus EUR-UKB female cancer-free individuals (Supplementary Note 9 and Supplementary Table 13).
Two GRBC-prioritized missense variants, SETBP1:c.4129G > C and C7orf34:c.248C > T, were present in FBRCAX and were also enriched in cancer patients from both NFE-TCGA and EUR-UKB (Table 2). SETBP1 encodes a protein that binds to SET, a nuclear oncogene involved in DNA replication (Piazza et al., 2013). Although SETBP1:c.4129G > C missense variant is predicted to have a damaging effect by only one of seven prediction tools (LRT score = 0.0001619), it was shortlisted through two strategies (CGV and SVGU), was present in two GRBC-affected sisters (diagnosed with BC at 41 and 44 years of age) and in a single FBRCAX patient. Additionally, this variant was enriched in NFE-TCGA cancer patients (OR = 1.25, 95% CI = 1.05–1.49) and in EUR-UKB female BC patients (OR = 1.13, 95% CI = 1.01–1.26). C7orf34:c.248C > T maps to a protein-coding gene of unknown function and is predicted to have a damaging effect by four of seven prediction tools (SIFT; Polyphen; MutationTaster; CADD). This variant was present in a GRBC mother–daughter pair (diagnosed at 60 and 47 years of age, respectively) in an unrelated GRBC patient (diagnosed at 37 years of age) and in a single FBRCAX patient. The variant was enriched in cancer patients from both NFE-TCGA (OR = 1.46, 95% CI = 1.02–2.07) and EUR-UKB cancer patients (OR = 1.14, 95% CI = 1.02–1.27).
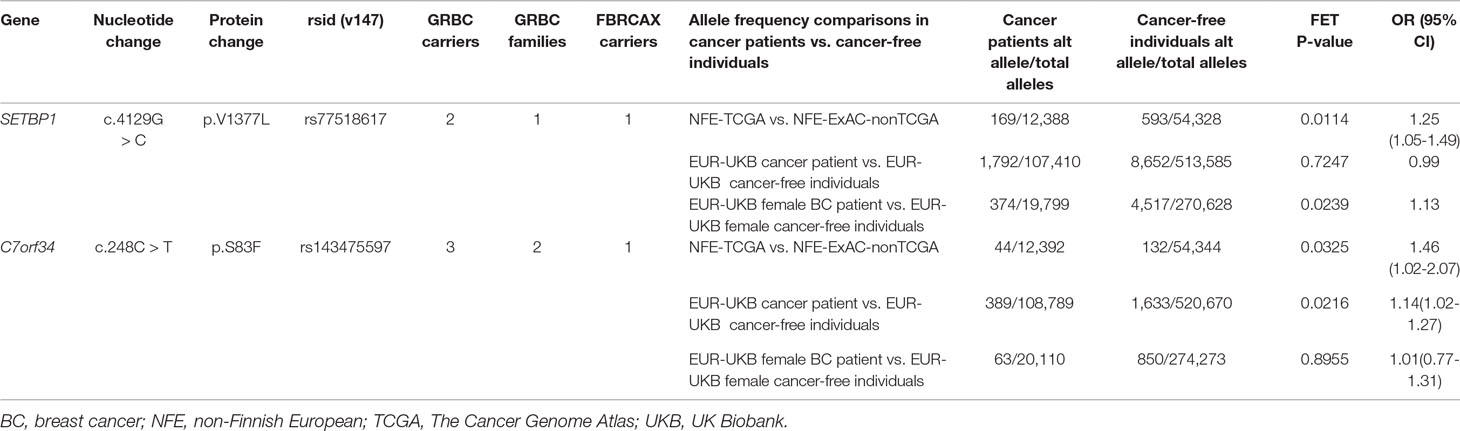
Table 2 Genetic variants in GRBC and FBRCAX enriched in cancer patients from TCGA and UKB.
Associating new genes to BC susceptibility has been challenging. Although numerous sequencing-based studies, focusing primarily on candidate genes, have aimed to identify new risk candidates, over 50% HBOC cases remain of unexplained etiology, even after polygenic risk is accounted for. This is largely due to the extreme rarity of pathogenic variants, prompting some groups to suggest family-specific risk variant models for undiagnosed HBOC cases (Lynch et al., 2013). In the present study, following initial screening of patients to exclude individuals with known BC risk variants, we interrogated the whole exome of 52 Greek individuals, from 17 HBOC families for risk-associated variants. Although this is a modest-sized study, it is an important first contribution to the field, given that the Greek population is relatively understudied. In fact, a consequence of this, which is also a limitation of the present study, is the lack of allele frequency information for the Greek population, which would enable direct comparison of frequencies between Greek BC patients and cancer-free individuals. A catalogue of genetic variation is yet to be generated for the Greek population. However, given the genetic proximity of GRBC to European populations (Figure 2), our rationale was that risk alleles segregating at extremely low frequencies in other populations may exist in slightly higher frequencies in Greek individuals due to population-specific effects. We, therefore, sought to validate GRBC-shortlisted variants in two European ancestry French-Canadian HBOC patient groups (FBRCAX and CHUM-BC), and in TCGA and UKB cancer patients. Data sets used were from distinct studies, and thus differences in assays and data processing exist (Supplementary Notes 2, 5, and 6). However, given our conservative approach, which included strict filtering criteria, we do not expect the recorded differences to affect the robustness of reported findings. In fact, it is more likely that true positive findings may have been excluded as a result of the stringent approach that was applied. Given that our study focused on coding regions only, was of relatively small size, and results could not be evaluated against local allele frequencies, polygenic risk scores were not assigned in order of avoid biased or inaccurate results.
Gene- and variant-based prioritization strategies were applied to shortlist and validate GRBC-detected candidate variants. Using the former strategy, we identified LoF variants that put forth MDM1 and NBEAL1 as putative HBOC risk genes. MDM1 encodes a nuclear protein that binds to and stabilizes microtubules. This protein localizes to centrioles and negatively regulates their duplication in dividing and in differentiating multi-ciliated cells (Van De Mark et al., 2015). Pathogenic variants in genes involved in the formation and function of centrosomes, such as MDM1, have been linked to ciliopathies, a group of diseases affecting the cilia (Hildebrandt et al., 2011). Ciliopathies have an established connection with inherited cancer syndromes, including Von Hippel–Lindau disease (VHL) (Hildebrandt et al., 2011). NBEAL1 encodes one of nine human BEACH domain-containing proteins (BDCPs) (Cullinane et al., 2013). BDCPs play a role in molecular mechanisms including vesicular transport, apoptosis and receptor signaling (Cullinane et al., 2013). Variants in these genes have been associated with diseases such as BC, prostate cancer, multiple myeloma, Chediak–Higashi syndrome, and autism (Cullinane et al., 2013). NBEAL1 is upregulated in gliomas (Chen et al., 2004) and comprises the least studied of human BDCPs.
The latter strategy yielded two missense SNVs, SETBP1:c.4129G > C and C7orf34:c.248C > T, detected in both GRBC and FBRCAX, that were also enriched in cancer patients of European ancestry in TCGA and UKB. Considering their relatively high frequencies (gnomAD, 0.8% and 0.2%, respectively), the reported candidates are likely to be associated with a very small increment to disease risk, if any. Whereas the function of C7orf34 has not been described to date, SETBP1 encodes a protein that contains numerous motifs, three nuclear localization signals, and binds to the SET oncogene involved in DNA replication (Piazza et al., 2013). Germline pathogenic variants in SETBP1 cause Schinzel–Giedion syndrome, a severely debilitating condition that predisposes to neuroepithelial tumors (Lehman et al., 2008). Additionally, somatic mutations in this gene have been associated with atypical chronic myeloid leukemia (Piazza et al., 2013).
We also report two additional noteworthy findings. A private stop-gain variant was detected in the third of eleven exons of BARD1 (c.273G > A) in a cancer-free mother (> 60 years old) of a HBOC patient (diagnosed at 37 years of age). Although there is accumulating evidence linking variants in BARD1 to BC risk (Castera et al., 2018), LoF variants in this gene have also been reported in cancer-free individuals (https://whi.color.com). We, therefore, cannot yet ascertain whether the above variant contributes to disease risk. We also report an instance where a pathogenic variant in the established cancer gene MEN1 (c.778G > A) is present in an affected family member, but absent from an affected second-degree relative, implying possible differential genetic causality for disease within a single pedigree. These findings highlight the high levels of genetic heterogeneity in HBOC syndrome and emphasize the need for careful interpretation of pedigrees and of the impact of genetic variants.
Studies focusing on whole exomes, and more recently on whole genomes, rather than candidate genes only, are currently fueling the discovery of novel associations between genes and disease. Whole-genome sequencing studies, which also interrogate non-coding regions of the genome, are expected to uncover new variants that influence disease risk for HBOC syndrome and multiple other diseases. In addition, recent technological advances, which address underlying structural variation in the genome, are starting to reveal the presence of genetic variants that have remained largely undetected using short read sequencing technologies and that are likely to contribute to disease. However, until these advances are applied at a large scale, the findings presented here can be used to inform lists of candidate variants and genes to be screened in additional cancer patients worldwide and to be taken forward to functional studies.
The datasets generated for this study can be found in the European Nucleotide Archive accession number PRJEB31704.
Informed consent was obtained from all individuals prior to genetic testing and the study was approved by the bioethics committee of NCSR Demokritos (240/EH/11.3, updated Feb. 14, 2014) in agreement with the 1975 Helsinki statement.
ASD and JR conceived the study and were in charge of overall direction and planning. SG and KR carried out lab work. ACD and SG performed bioinformatic analyses. SG, ACD, EE, and GN performed statistical analyses. SN and A-MM-M ascertained cases, and provided patient samples. BR, WF, and PT contributed to interpretation of results from FBRCAX. SG and ASD wrote the manuscript. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
This study was supported by funds from NSRF 2007-2013 grant SYNERGASIA II (SYN11_10_19) and the Stavros Niarchos Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Drs Drakoulis Yannoukakos, Rena Konstantopoulou, and Florentia Fostira from NCSR Demokritos, as well as the patients and their families who took part in this study. The authors would also like to thank Dr Vagelis Harokopos for performing NGS and Drs Pantelis Hatzis and Martin Reczko for helpful suggestions. Tissue banking was supported by the Réseau de recherche sur le cancer of the Fond de recherche Québec-Santé, associated with the Canadian Tissue Repository Network (CTRNet).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01005/full#supplementary-material
Awadalla, P., Boileau, C., Payette, Y., Idaghdour, Y., Goulet, J. P., Knoppers, B., et al. (2013). Cohort profile of the CARTaGENE study: quebec’s population-based biobank for public health and personalized genomics. Int. J. Epidemiol. 42, 1285–1299. doi: 10.1093/ije/dys160
Boland, J. F., Chung, C. C., Roberson, D., Mitchell, J., Zhang, X., Im, K. M., et al. (2013). The new sequencer on the block: comparison of life technology’s proton sequencer to an Illumina HiSeq for whole-exome sequencing. Hum. Genet. 132, 1153–1163. doi: 10.1007/s00439-013-1321-4
Castera, L., Harter, V., Muller, E., Krieger, S., Goardon, N., Ricou, A., et al. (2018). Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. doi: 10.1038/s41436-018-0005-9
Chen, J., Lu, Y., Xu, J., Huang, Y., Cheng, H., Hu, G., et al. (2004). Identification and characterization of NBEAL1, a novel human neurobeachin-like 1 protein gene from fetal brain, which is up regulated in glioma. Brain Res. Mol. Brain Res. 125, 147–155. doi: 10.1016/j.molbrainres.2004.02.022
Cullinane, A. R., Schaffer, A. A., Huizing, M. (2013). The BEACH is hot: a LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic 14, 749–766. doi: 10.1111/tra.12069
Cybulski, C., Carrot-Zhang, J., Kluzniak, W., Rivera, B., Kashyap, A., Wokolorczyk, D., et al.. (2015). Germline RECQL mutations are associated with breast cancer susceptibility. Nat. Genet. 47, 643–646. doi: 10.1038/ng.3284
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., Depristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
De La Vega, F. M., Bustamante, C. D. (2018). Polygenic risk scores: a biased prediction? Genome Med. 10, 100. doi: 10.1186/s13073-018-0610-x
Depristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Dreijerink, K. M., Goudet, P., Burgess, J. R., Valk, G. D. International Breast Cancer In M.E.N.S.G. (2014). Breast-cancer predisposition in multiple endocrine neoplasia type 1. N. Engl. J. Med. 371, 583–584. doi: 10.1056/NEJMc1406028
Easton, D. F., Pharoah, P. D., Antoniou, A. C., Tischkowitz, M., Tavtigian, S. V., Nathanson, K. L., et al. (2015). Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 372, 2243–2257. doi: 10.1056/NEJMsr1501341
Ferlay, J., Soerjomataram, I., Dikshit, R., Eser, S., Mathers, C., Rebelo, M., et al. (2015). Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–E386. doi: 10.1002/ijc.29210
Ferlay, J., Steliarova-Foucher, E., Lortet-Tieulent, J., Rosso, S., Coebergh, J. W., Comber, H., et al. (2013). Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur. J. Cancer 49, 1374–1403. doi: 10.1016/j.ejca.2012.12.027
Fromer, M., Moran, J. L., Chambert, K., Banks, E., Bergen, S. E., Ruderfer, D., et al. (2012). Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 91, 597–607. doi: 10.1016/j.ajhg.2012.08.005
Garcia-Closas, M., Gunsoy, N. B., Chatterjee, N. (2014). Combined associations of genetic and environmental risk factors: implications for prevention of breast cancer. J. Natl. Cancer Inst. 106. doi: 10.1093/jnci/dju305
Genomes Project, C., Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Hildebrandt, F., Benzing, T., Katsanis, N. (2011). Ciliopathies. N. Engl. J. Med. 364, 1533–1543. doi: 10.1056/NEJMra1010172
Kast, K., Rhiem, K., Wappenschmidt, B., Hahnen, E., Hauke, J., Bluemcke, B., et al. (2016). Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J. Med. Genet. 53, 465–471. doi: 10.1136/jmedgenet-2015-103672
Kiiski, J. I., Pelttari, L. M., Khan, S., Freysteinsdottir, E. S., Reynisdottir, I., Hart, S. N., et al. (2014). Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc. Natl. Acad. Sci. U. S. A. 111, 15172–15177. doi: 10.1073/pnas.1407909111
King-Spohn, K., Pilarski, R. (2014). Beyond BRCA1 and BRCA2. Curr. Probl. Cancer 38, 235–248. doi: 10.1016/j.currproblcancer.2014.10.004
Konstantopoulou, I., Tsitlaidou, M., Fostira, F., Pertesi, M., Stavropoulou, A. V., Triantafyllidou, O., et al.. (2014). High prevalence of BRCA1 founder mutations in Greek breast/ovarian families. Clin. Genet. 85, 36–42. doi: 10.1111/cge.12274
Kuchenbaecker, K. B., Hopper, J. L., Barnes, D. R., Phillips, K. A., Mooij, T. M., Roos-Blom, M. J., et al. (2017). Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 317, 2402–2416. doi: 10.1001/jama.2017.7112
Lehman, A. M., Mcfadden, D., Pugash, D., Sangha, K., Gibson, W. T., Patel, M. S. (2008). Schinzel-Giedion syndrome: report of splenopancreatic fusion and proposed diagnostic criteria. Am. J. Med. Genet. A 146A, 1299–1306. doi: 10.1002/ajmg.a.32277
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Levy-Lahad, E., Friedman, E. (2007). Cancer risks among BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 96, 11–15. doi: 10.1038/sj.bjc.6603535
Lynch, H., Wen, H., Kim, Y. C., Snyder, C., Kinarsky, Y., Chen, P. X., et al. (2013). Can unknown predisposition in familial breast cancer be family-specific? Breast J. 19, 520–528. doi: 10.1111/tbj.12145
Lynch, H. T., Silva, E., Snyder, C., Lynch, J. F. (2008). Hereditary breast cancer: part I. Diagnosing hereditary breast cancer syndromes. Breast J. 14, 3–13. doi: 10.1111/j.1524-4741.2007.00515.x
Mavaddat, N., Michailidou, K., Dennis, J., Lush, M., Fachal, L., Lee, A., et al. (2019). Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 104, 21–34. doi: 10.1016/j.ajhg.2018.11.002
Mcclellan, J., King, M. C. (2010). Genetic heterogeneity in human disease. Cell 141, 210–217. doi: 10.1016/j.cell.2010.03.032
Michailidou, K., Lindstrom, S., Dennis, J., Beesley, J., Hui, S., Kar, S., et al. (2017). Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94. doi: 10.1038/nature24284
Nielsen, F. C., Van Overeem Hansen, T., Sorensen, C. S. (2016). Hereditary breast and ovarian cancer: new genes in confined pathways. Nat. Rev. Cancer 16, 599–612. doi: 10.1038/nrc.2016.72
Piazza, R., Valletta, S., Winkelmann, N., Redaelli, S., Spinelli, R., Pirola, A., et al. (2013). Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat. Genet. 45, 18–24. doi: 10.1038/ng.2495
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, et al. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi: 10.1038/ng1847
Rahman, N. (2014). Realizing the promise of cancer predisposition genes. Nature 505, 302–308. doi: 10.1038/nature12981
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rivera, B., Di Iorio, M., Frankum, J., Nadaf, J., Fahiminiya, S., Arcand, S. L., et al. (2017). Functionally null RAD51D missense mutation associates strongly with ovarian carcinoma. Cancer Res. 77, 4517–4529. doi: 10.1158/0008-5472.CAN-17-0190
Robinson, J. T., Thorvaldsdottir, H., Wenger, A. M., Zehir, A., Mesirov, J. P. (2017). Variant review with the integrative genomics viewer. Cancer Res. 77, e31–e34. doi: 10.1158/0008-5472.CAN-17-0337
Shakya, R., Szabolcs, M., Mccarthy, E., Ospina, E., Basso, K., Nandula, S., et al. (2008). The basal-like mammary carcinomas induced by Brca1 or Bard1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc. Natl. Acad. Sci. U. S. A. 105, 7040–7045. doi: 10.1073/pnas.0711032105
Solomon, S., Das, S., Brand, R., Whitcomb, D. C. (2012). Inherited pancreatic cancer syndromes. Cancer J. 18, 485–491. doi: 10.1097/PPO.0b013e318278c4a6
Stamatoyannopoulos, G., Bose, A., Teodosiadis, A., Tsetsos, F., Plantinga, A., Psatha, N., et al. (2017). Genetics of the peloponnesean populations and the theory of extinction of the medieval peloponnesean Greeks. Eur. J. Hum. Genet. 25, 637–645. doi: 10.1038/ejhg.2017.18
Sudlow, C., Gallacher, J., Allen, N., Beral, V., Burton, P., Danesh, J., et al. (2015). UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779. doi: 10.1371/journal.pmed.1001779
Teh, B. T., Esapa, C. T., Houlston, R., Grandell, U., Farnebo, F., Nordenskjold, M., et al. (1998). A family with isolated hyperparathyroidism segregating a missense MEN1 mutation and showing loss of the wild-type alleles in the parathyroid tumors. Am. J. Hum. Genet. 63, 1544–1549. doi: 10.1086/302097
Terui-Kohbata, H., Yoshida, M. (2017). Current condition of genetic medicine for hereditary breast cancer. Mol. Clin. Oncol. 7, 98–102. doi: 10.3892/mco.2017.1260
Thompson, E. R., Doyle, M. A., Ryland, G. L., Rowley, S. M., Choong, D. Y., Tothill, R. W., et al-. (2012). Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 8, e1002894. doi: 10.1371/journal.pgen.1002894
Van De Mark, D., Kong, D., Loncarek, J., Stearns, T. (2015). MDM1 is a microtubule-binding protein that negatively regulates centriole duplication. Mol. Biol. Cell 26, 3788–3802. doi: 10.1091/mbc.E15-04-0235
Keywords: hereditary breast cancer, exome sequencing, Greek population, candidate risk variants, MDM1, NBEAL1
Citation: Glentis S, Dimopoulos AC, Rouskas K, Ntritsos G, Evangelou E, Narod SA, Mes-Masson A-M, Foulkes WD, Rivera B, Tonin PN, Ragoussis J and Dimas AS (2019) Exome Sequencing in BRCA1- and BRCA2-Negative Greek Families Identifies MDM1 and NBEAL1 as Candidate Risk Genes for Hereditary Breast Cancer. Front. Genet. 10:1005. doi: 10.3389/fgene.2019.01005
Received: 01 March 2019; Accepted: 20 September 2019;
Published: 18 October 2019.
Edited by:
John Frederick Pearson, University of Otago, New ZealandReviewed by:
Chiara Vardabasso, Gotham Therapeutics, United StatesCopyright © 2019 Glentis, Dimopoulos, Rouskas, Ntritsos, Evangelou, Narod, Mes-Masson, Foulkes, Rivera, Tonin, Ragoussis and Dimas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antigone S. Dimas, ZGltYXNAZmxlbWluZy5ncg==; Jiannis Ragoussis, aW9hbm5pcy5yYWdvdXNzaXNAbWNnaWxsLmNh
†Present Adress: Stavros Glentis, Division of Pediatric Hematology/Oncology, First Department of Pediatrics, University of Athens, Aghia Sophia Children’s Hospital, Athens, Greece
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.