
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 18 September 2019
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.00867
This article is part of the Research Topic Advancing Genomics for Rare Disease Diagnosis and Therapy Development View all 29 articles
 Baoan Hong1,2,3,4,5,6†
Baoan Hong1,2,3,4,5,6† Kaifang Ma1,2,3,4†
Kaifang Ma1,2,3,4† Jingcheng Zhou1,2,3,4Jiufeng Zhang1,2,3,4
Jingcheng Zhou1,2,3,4Jiufeng Zhang1,2,3,4 Jiangyi Wang1,2,3,4Shengjie Liu1,2,3,4Zhongyuan Zhang1,2,3,4Lin Cai1,2,3,4*Ning Zhang5,6
Jiangyi Wang1,2,3,4Shengjie Liu1,2,3,4Zhongyuan Zhang1,2,3,4Lin Cai1,2,3,4*Ning Zhang5,6 Kan Gong1,2,3,4
Kan Gong1,2,3,4Von Hippel–Lindau (VHL) disease is a rare autosomal-dominant inherited tumor syndrome. We aimed to analyze the correlations between frequent VHL mutations and phenotypes in Chinese VHL families. We screened 540 patients from 187 unrelated Chinese VHL families for 19 frequent VHL mutations. The penetrance and mean age at onset for VHL-associated susceptible organs were calculated and compared. The overall survival of VHL patients was described with Kaplan–Meier curves. Among the 19 frequent germline mutations, there were four hotspot mutation sites (194, 481, 499, and 500). Missense mutations were the most common types of mutations (70.0%) followed by nonsense mutations (20.0%) and splicing mutations (10.0%). Due to the diversity of these mutations, the penetrance for each organ and the age at onset are distinct. Even in cases of similar mutations, variance in the penetrance and age at onset was observed. The mean age at death for the patients in this cohort was 42.4 ± 13.5 years, and variability was observed in the Kaplan–Meier curves. We present a precise summary of the phenotypes for the frequent VHL mutations in the largest Chinese VHL cohort, which provides valuable strategies for genetic counseling and clinical surveillance of VHL individuals.
Von Hippel–Lindau (VHL) disease (OMIM no. 193300) is an autosomal-dominant familial neoplastic condition that is caused by germline mutations in the VHL gene located on chromosome 3p25-26. This gene comprises three exons: exon 1 spans nucleotides 1–340 (codons 1–113), exon 2 spans nucleotides 341–463 (codons 114–154), and exon 3 spans nucleotides 464–642 (codons 155–213) (Latif et al., 1993; Nielsen et al., 2016; Varshney et al., 2017). Patients with VHL syndrome inherit a single mutant VHL allele from a parent and develop the disease when the second wild-type copy is deactivated or lost. Incidence of the VHL mutation is approximately 1 in 36,000 live births, and it is greater than 90% penetrant by age 65 years (Kim et al., 2010; Gossage et al., 2015). Common VHL-associated clinical manifestations include central nervous system hemangioblastoma (CHB), renal cell carcinoma or renal cyst (RCC), retinal angioma (RA), pancreatic tumor or cyst (PCT), pheochromocytoma and paragangliomas (PHEO), endolymphatic sac tumor, and epididymis or broad ligament cystadenoma (Supplementary Table 1; Lonser et al., 2003; Lonser et al., 2004; Butman et al., 2008; Chou et al., 2013; Launbjerg et al., 2017). Von Hippel–Lindau disease predisposes the affected individuals to the development of lesions in multiple systems with CHBs (25%–51%) and RCCs (13%–47%) as the major causes of mortality (Grubb et al., 2005; Wilding et al., 2012; Lonser et al., 2014). Individuals with a family history of VHL will be clinically diagnosed when he/she presents with VHL-associated tumors that include CHB, RA, or RCC (Chittiboina and Lonser, 2015). For patients who do not have a family history of VHL, two or more CHBs or RAs or one hemangioblastoma and a visceral tumor are required for a clinical diagnosis (Nielsen et al., 2016). Typically, genetic testing is the standard method to diagnose VHL disease. The detection of VHL mutations not only contributes to an early and precise diagnosis of at-risk individuals but also helps to elucidate the genotype–phenotype correlations within a given population.
A series of studies have reported genotype–phenotype correlations in VHL diseases from different research perspectives or within different ethnic backgrounds (Yoshida et al., 2000; Patocs et al., 2008; Gomy et al., 2010; Nordstrom-O’Brien et al., 2010; Vikkath et al., 2015). For example, a retrospective study that included 63 VHL patients from two large VHL kindreds (family 1: Y112H mutation and family 2: Y98H mutation) with pheochromocytoma/paraganglioma found that pheochromocytoma expressivity differed by genotype (Nielsen et al., 2011). Ong et al. (2007) evaluated the genotype–phenotype correlations in 573 VHL patients and confirmed that pheochromocytoma was linked to VHL missense mutations. Additionally, the age at onset for VHL syndrome was significantly earlier (P = 0.001) and the age-related risks of RA and RCC were higher (P = 0.022 and P = 0.0008, respectively) for individuals with nonsense or frameshift mutations compared to those with deletions. Importantly, the results of these studies provided valuable strategies for genetic counseling and clinical prophylactic surveillance for VHL family members.
Due to the rarity of VHL disease, studies on the correlations between the frequent mutations of the VHL gene and clinical phenotypes are relatively scarce, with the majority being case reports or studies involving a limited number of VHL patients or families. In clinical practice, there is an urgent need to identify the clinical symptoms and survival statistics for VHL patients based on their specific types of mutations. Therefore, an improved and precise understanding of specific genotype–phenotype correlations in VHL disease is essential for targeted monitoring and counseling. In this study, we screened for frequent mutations in the VHL gene across 187 unrelated Chinese VHL families and analyzed the genotype–phenotype correlations between frequent VHL mutations and clinical manifestations. This study improves our understanding of how frequent mutations of the VHL gene affect the age at onset for each susceptible organ and their impact on prognosis in a Chinese population and provides a more accurate resource for genetic counseling and the monitoring of VHL patients.
By May 31, 2018, 540 patients from 187 unrelated families had been diagnosed with VHL disease at the Peking University First Hospital, and all were screened in the present study. Any mutation site that appeared in two or more families was included in this study. Individuals who carried a VHL germline mutation or who met the clinical criteria were diagnosed with VHL disease (Wu et al., 2012). The evidence assessment for VHL disease includes a medical history, a physical examination by multidisciplinary teams, laboratory tests, and medical imaging of the abdomen, pelvis, brain, and spine (ultrasound, computed tomography, or magnetic resonance imaging). However, at least one patient from a family confirmed the presence of a VHL mutation through genetic testing.
Clinical characteristics including date of birth, age at death, mutation type, clinical symptoms, and the age at onset were collected from family members or through medical records. The age at onset was defined as the age at which VHL-related symptoms or signs first appear. A follow-up was performed on all patients and asymptomatic carriers in the VHL families to determine the age at onset for six major VHL-related lesions: CHB, RA, RCC, PCT, PHEO, and GS (genital system including the epididymis or broad ligament). Follow-ups were carried out until May 31, 2018, or until patient death following the first presentation of the VHL-related clinical symptoms or a positive genetic diagnosis of VHL disease. The life span of the patient from birth until death or until the end of the follow-up period was used in the survival analysis.
Genetic testing was performed on at least one member from each family to confirm the diagnosis of VHL disease. Genomic DNA was extracted from peripheral blood samples that were obtained from members of the suspected families. Individuals who refused the genetic test or who died were excluded. The three exons and the flanking intronic sequences of the VHL gene were amplified by polymerase chain reaction, and direct sequencing was used to detect missense mutations, splicing mutations, and small indels. Large deletions and duplications were detected by multiplex ligation-dependent probe amplification (MLPA, P016-C2 kit, Amsterdam, the Netherlands) and verified by real-time quantitative polymerase chain reaction. The primers and reaction conditions used for the amplification were as previously described (Peng et al., 2017; Wang et al., 2018). The spectrum of VHL mutations was screened for frequent mutations and further analyzed to determine their correlations with phenotypes.
For each type of mutation, the mean age at onset of VHL-associated susceptible organs (CHB, RA, RCC, PCT, PHEO, and GS) and the mean age at death were calculated as the mean ± standard deviation. Statistical significance was determined by the Student t test or the LSD multiple-comparisons t test. The overall survival of VHL patients was described with a Kaplan–Meier curve. Statistical analysis was performed using SPSS 20.0 software (IBM-SPSS, Inc., Chicago, IL, USA), and P < 0.05 was considered to be statistically significant.
A total of 540 patients from 187 unrelated Chinese VHL families were included in our database, and 126 different types of VHL mutations were identified. Insertions, small deletions, and large deletions were detected in 61 families [61/187 (32.6%)]. Point mutations resulting in missense, nonsense, or splicing mutations were detected in 126 families [126/187 (67.4%)]. The distribution of germline point mutations was further analyzed and identified 19 germline mutation sites that appeared in two or more families. Of these 19 frequent germline mutations, 10 were located in exon 1, 3 were located in exon 2, 4 were located in exon 3, and 2 were located in intron 2 (Figure 1). The clinical characteristics and mutation frequencies for each of the relevant germline mutations in the 258 patients from 80 unrelated Chinese VHL families are shown in Table 1. The mean age of the 258 patients was 39.4 ± 15.5 years with a range of 3 to 74 years. Additionally, the mean age for each mutation group is listed in Table 1. As would be expected due to the consequences of frequent mutations, missense mutations were the most common mutation types in these families (70.0%) followed by nonsense mutations (20.0%) and splicing mutations (10.0%). Notably, there were four mutation hotspot sites at 194, 481, 499, and 500 that were separately distributed across 9, 10, 14, and 10 unrelated Chinese VHL families, respectively.
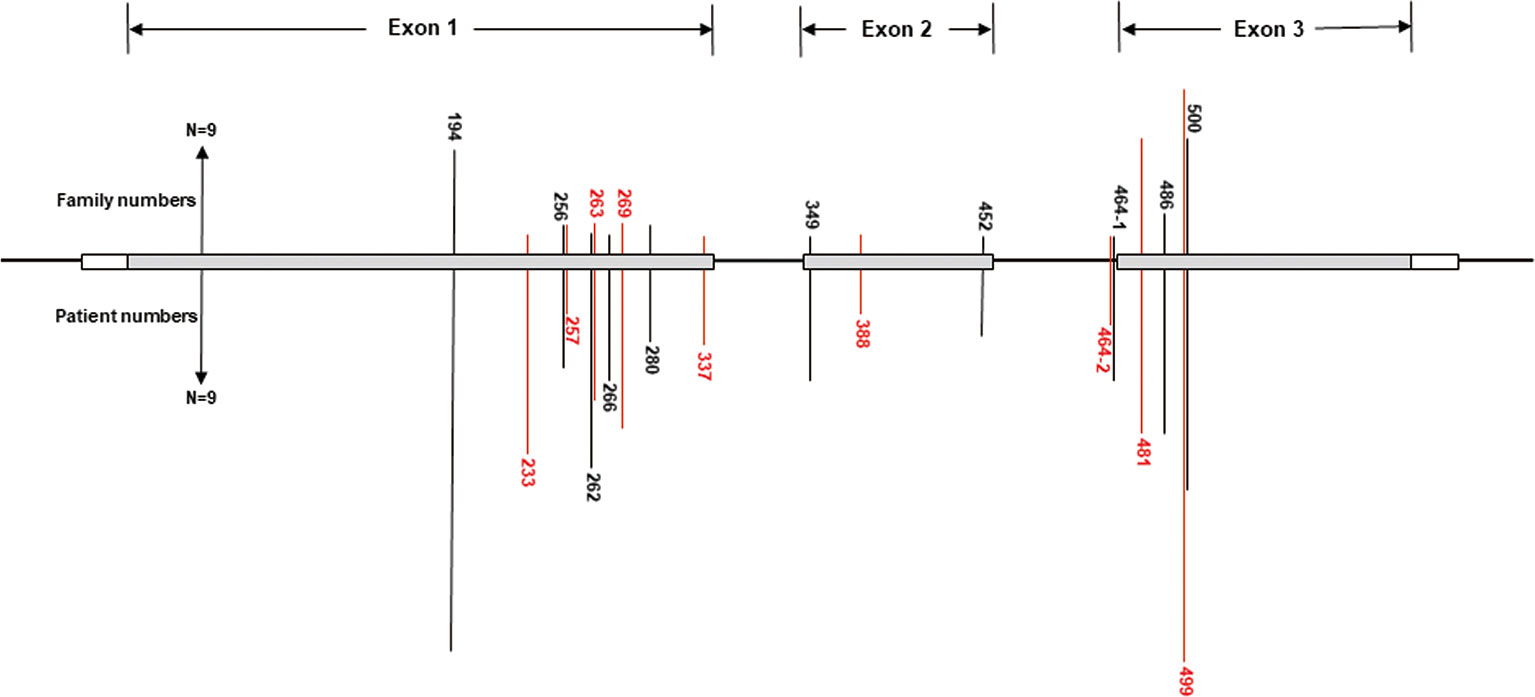
Figure 1 Distribution of the frequent germline mutation sites in VHL gene for 258 patients from 80 unrelated Chinese families with VHL disease. Of the 19 frequent germline mutations, 10 were located in exon 1, 3 were located in exon 2, 4 were located in exon 3, and 2 were located in intron 2. Notably, there were four hotspot mutation sites (194, 481, 499, and 500). The bars on the upper part represent mutational family numbers, and those on the lower part represent mutational patient numbers.
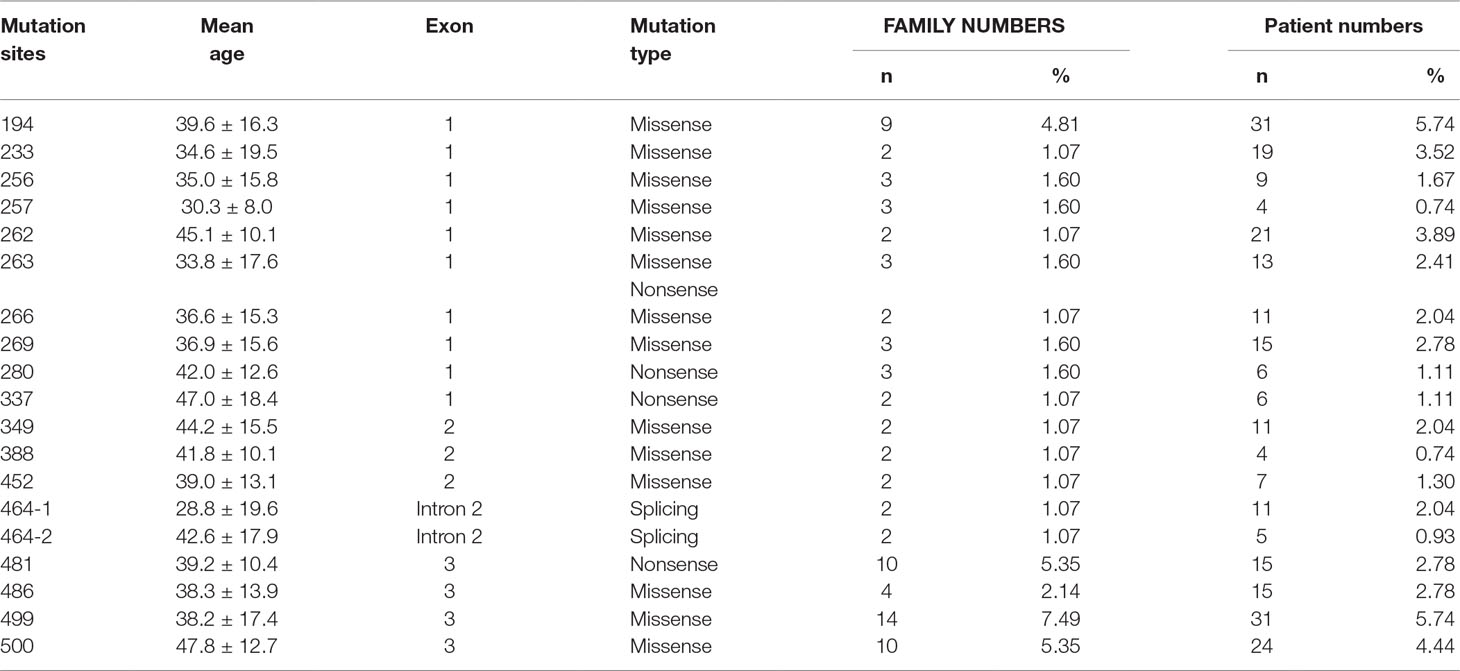
Table 1 Clinical characteristics and mutation frequency of relevant frequent germline mutations of 258 patients from 80 unrelated Chinese VHL families.
In this study, we analyzed and compared the mean age at onset for six major VHL-related lesions (CHB, RA, RCC, PCT, PHEO, and GS) in 19 frequent germline mutations (Table 2). Due to diversity in the types of mutations, the penetrance for each organ and the age at onset are not the same. For CHB, both the c.481C > T p.Arg161stop (group 16) and c.486C > G p.Cys162Trp (group 17) mutations had a high penetrance of approximately 80.0% (12/15). The mean age at onset of CHB for the c.481C > T p.Arg161stop (group 16) mutation was 27.4 ± 9.4 years (range = 14–40 years), while for the c.486C > G p.Cys162Trp (group 17) mutation, it was 31.4 ± 10.0 years (range = 12–49 years). For RCC, the mean ages at onset for the c.269A > T p.Asn90Ile (group 8) and c.486C > G p.Cys162Trp (group 17) mutations were 41.5 ± 15.5 years and 41.8 ± 10.3 years, while the penetrance was 26.7% (4/15) and 53.3% (8/15), respectively. Variation in the penetrance and age at onset exists even when the types of mutations are similar. There were two types of missense mutations in group 1 located in the 194 mutation site, c.194C > T p.Ser65Leu and c.194C > G p.Ser65Trp, but the clinical phenotypes were different between these two mutational subgroups. Six major VHL-related lesions (CHB, RA, RCC, PCT, PHEO, and GS) were observed in the c.194C > T p.Ser65Leu mutational subgroup, while only three VHL lesions (CHB, RCC, and PCT) presented in the c.194C > G p.Ser65Trp mutational subgroup. The mean age at onset for the common VHL lesions in the c.194C > T p.Ser65Leu mutational subgroup was older than that of the c.194C > G p.Ser65Trp mutational subgroup (Figures 2A–D). Three mutations were related to codon 88 (Trp) in groups 5 and 6. Both c.262T > C p.Trp88Arg and c.263G > C p.Trp88Ser were missense mutations, while c.263G > A p.Trp88Stop resulted in a nonsense mutation. Additionally, the VHL lesions associated with these three mutations were not the same. A comparison was made between the mean age at onset for CHB in these three subgroups and found that the c.262T > C p.Trp88Arg mutational group was older than that of the c.263G > C p.Trp88Ser mutational subgroup (P = 0.0152) and the c.263G > A p.Trp88Stop mutational subgroup (P = 0.0232) (Figure 2E). However, the CHB-associated age at onset for the c.263G > A p.Trp88Stop mutational subgroup was younger than the c.263G > C p.Trp88Ser mutational subgroup, but the difference was not significant (P = 0.481) (Figure 2E). In groups 18 and 19, the c.499C > T p.Arg167Trp and c.500G > A p.Arg167Gln mutations were located in codon 167 (Arg). There were differences in the penetrance and age at onset for six major VHL-related lesions (CHB, RA, RCC, PCT, PHEO, and GS) between these two mutation types, but the difference was not statistically significant (P > 0.05) (Figures 2F–I).
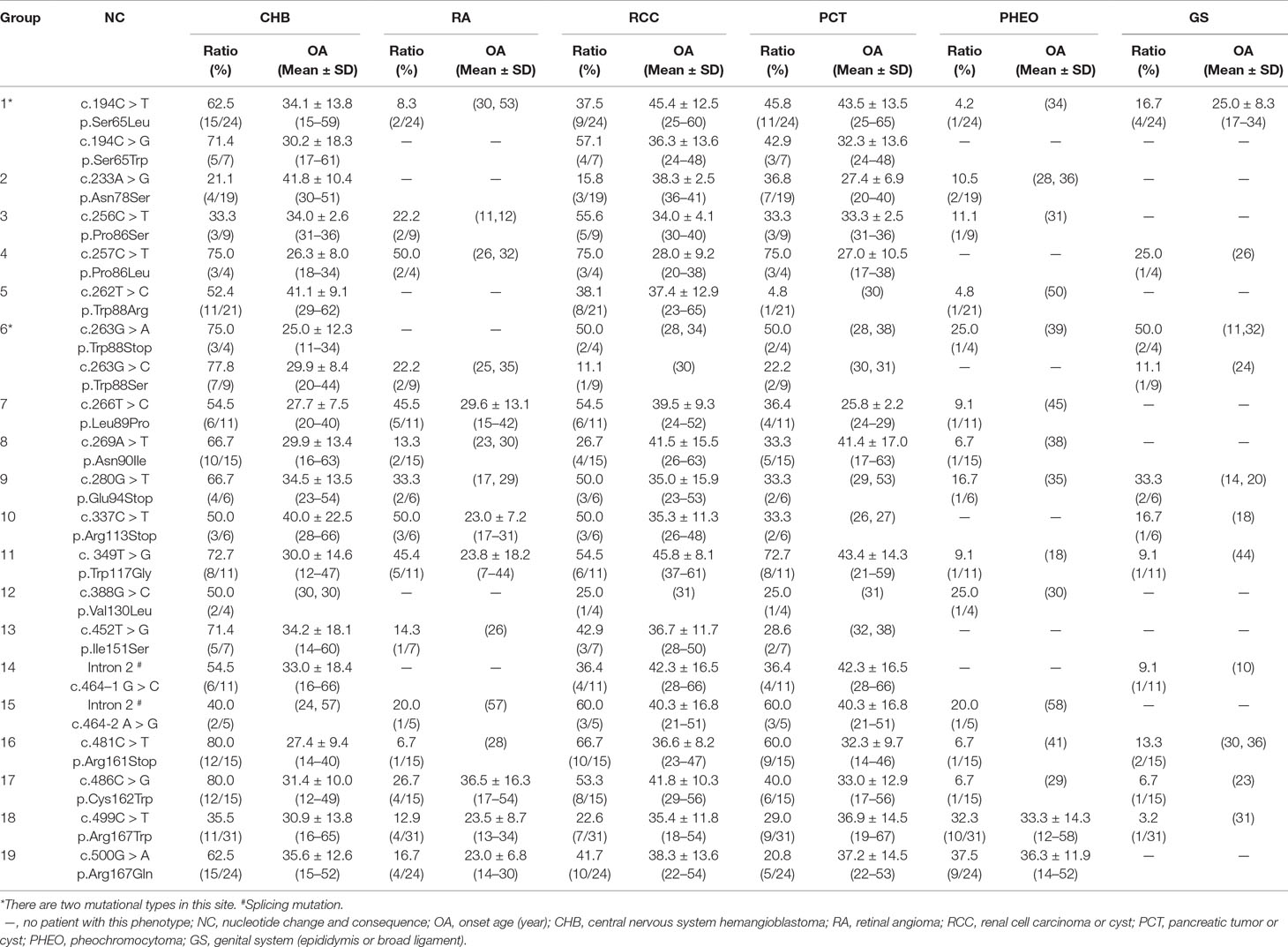
Table 2 Frequent VHL germline mutations and related phenotypes in Chinese patients with VHL disease.
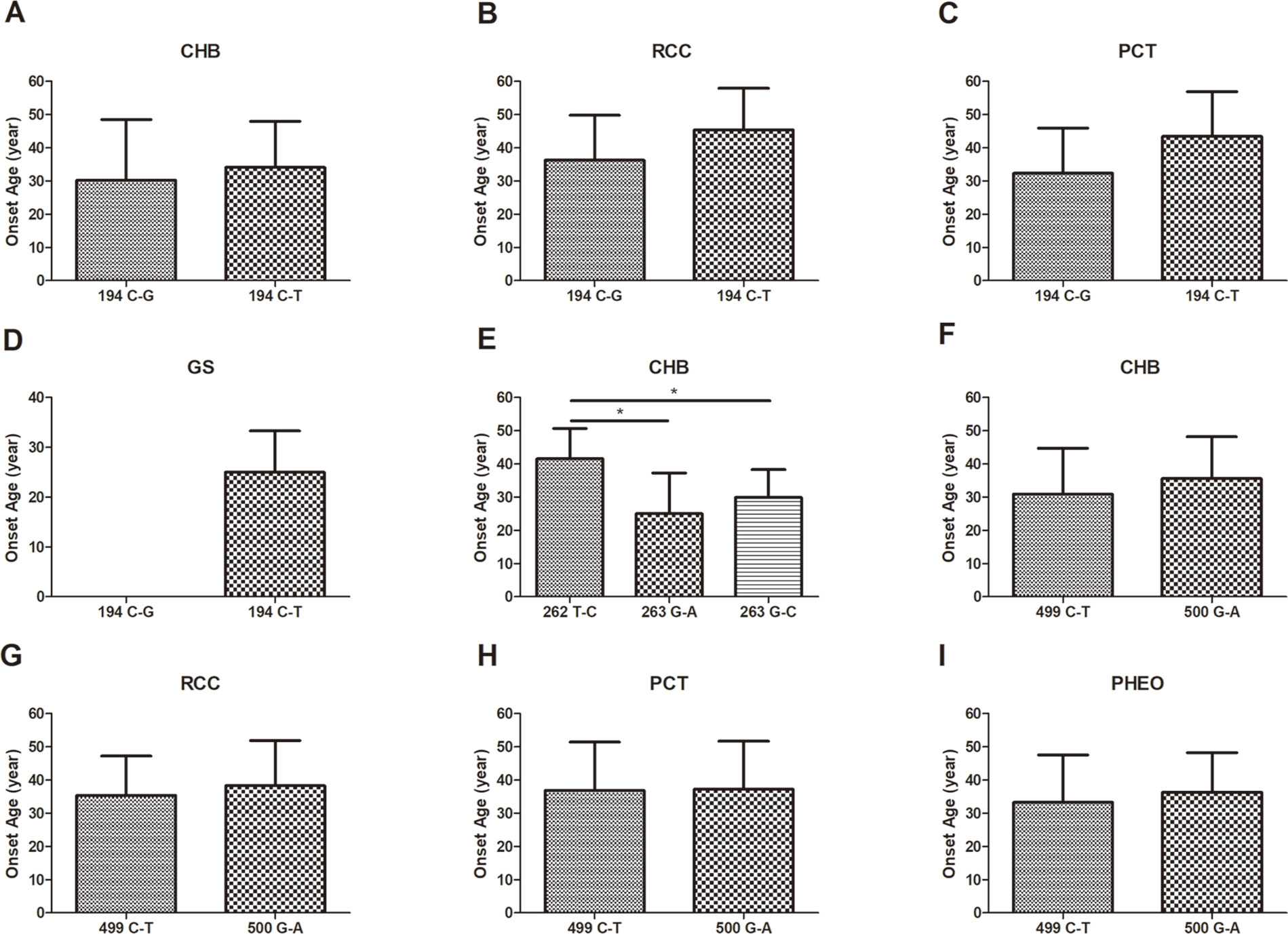
Figure 2 The mean age at onset for the common VHL lesions in patients with different germline mutations. The mean age at onset for the common VHL lesions in the c.194C > T p.Ser65Leu mutational subgroup and the c.194C > G p.Ser65Trp mutational subgroup (A–D). Comparison of the mean age at onset for CHB among c.262T > C p.Trp88Arg, c.263G > A p.Trp88Stop, and c.263G > C p.Trp88Ser mutations (E). Differences in the mean age at onset for the four major VHL lesions between the c.499C > T p.Arg167Trp and c.500G > A p.Arg167Gln mutations (F–I, P > 0.05). *P < 0.05.
Kaplan–Meier curves were used to describe the survival of patients with different VHL mutations, and the results are presented in Figure 3A. This analysis identified a variety of Kaplan–Meier curves for different frequent VHL germline mutations. The mutation sites 256 and 257, 262 and 263, and 499 and 500 were located in codons 86, 88, and 167, respectively, and grouped together for the analysis. Of the 258 patients from the 80 unrelated Chinese VHL families, 59 died of VHL-related diseases, such as CHB [71.2% (42/59)], RCC [25.4% (15/59)], and PCT-related complications [3.4% (2/59)]. The mean age at death for this cohort was 42.4 ± 13.5 years (range = 16–68 years). Additionally, we summarized the mean age at death for each of the frequent VHL germline mutation groups, and no deaths were observed in the groups with mutation sites 337, 388, and 464-2 (Figure 3B). Differences in the risk of VHL-related death across the different frequent VHL germline mutations were not statistically significant.
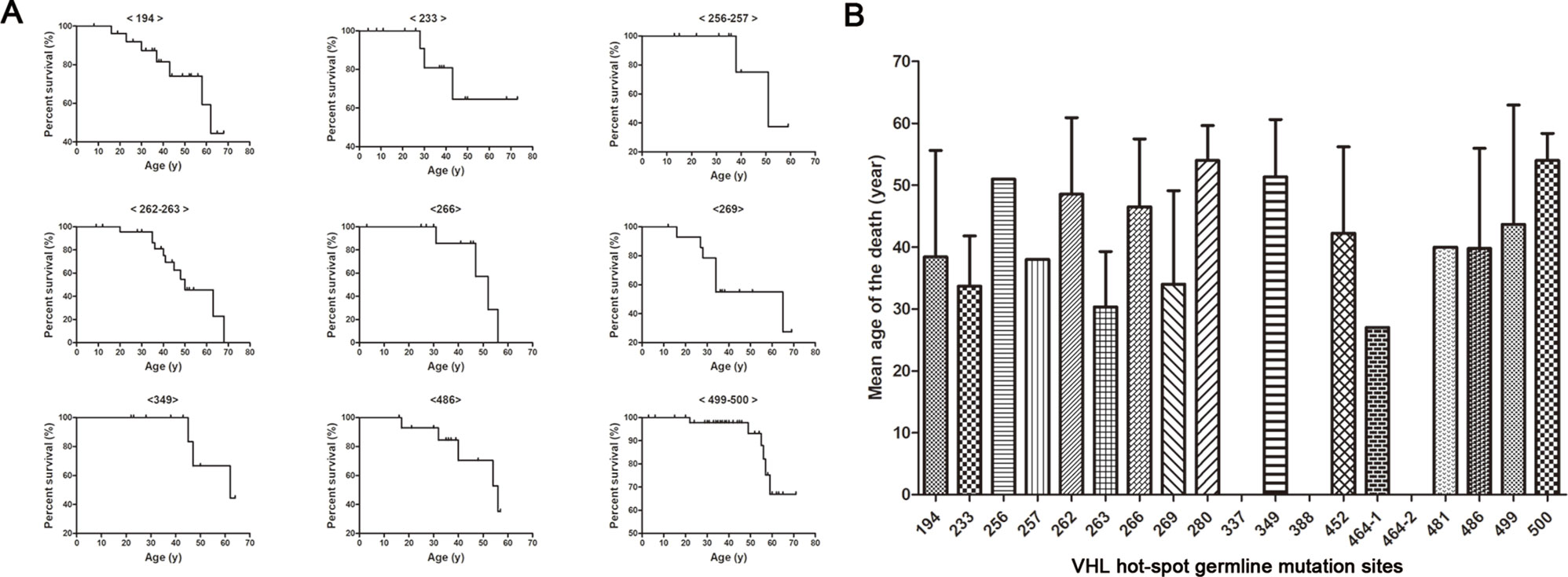
Figure 3 Von Hippel–Lindau frequent germline mutations and survival. Kaplan–Meier curves were used to describe the survival of patients with different VHL mutations (A). Variability was observed in the mean age at death for the different frequent VHL germline mutations, but no death was observed in groups with mutation sites 337, 388 and 464-2 (B).
A comprehensive understanding of the correlations between genotype and phenotype for hereditary diseases is critical to the clinical management and scientific analysis of their pathogenesis. Changes in specific genotypes can lead to alterations in protein expression patterns that result in their corresponding phenotypes. Elucidating these correlations may provide insight into the molecular pathogenesis of the individual manifestations of VHL syndrome. Screening for mutations in the VHL gene helps to clarify the diagnosis of asymptomatic first-degree relatives, thereby improving patient outcomes through early disease surveillance. To date, the studies on genotype–phenotype correlations have provided clinicians with tools that help predict the VHL disease processes in individual patients. Hence, it is important to increase the sample size and analyze the correlations between genotypes and phenotypes for different ethnic groups.
In this study, we analyzed the correlations between frequent mutations in the VHL gene and clinical phenotypes in the largest Chinese VHL cohort to date. In total, we screened 540 patients from 187 unrelated VHL families and identified 126 different VHL mutations. Furthermore, we identified 19 frequent mutations and four mutation hotspots and further investigated the genotype–phenotype correlations. Notably, patients or families with the VHL disease have a range of different phenotypes. A variety of factors may contribute to this diversity of phenotypes, including the type of VHL mutation, the site of the mutation, and ethnic background.
Different ethnic backgrounds are associated with diverse phenotypes. Several studies about Western and Japanese populations highlighted the differences in the spectrum of VHL germline mutations (Maher et al., 1996; Patel et al., 2000). The mutation hotspots of the VHL gene that are already known include Leu178, Cys162, Arg167, Asn78, Pro86, and Tyr98 and have a frequency of approximately 3% to 17% (Stebbins et al., 1999). However, the common mutations are varied across different ethnic groups. Hwang et al. (2014) reported that Glu70Lys was a high-frequency VHL germline mutation in the Korean population, with nine unrelated patients [16.4% (9/55)] who had the same amino-acid alteration at codon 70 (Glu70Lys) and exhibited VHL type 1 phenotypes. However, in our cohort, the high-frequency mutations included Ser65 (4.81%), Arg161 (5.35%), and Arg167 (12.84%). Thus, the spectrum of VHL mutations varies in countries that have different ethnic backgrounds. Patients from different ethnic backgrounds that have the same VHL germline mutation may also develop distinct phenotypes. For example, Yoshida et al. (2000) found four mutations (Arg113Stop, Gln132Stop, Leu158Val, and Cys162Tyr) in Japanese families with the VHL type 2 phenotype, whereas Crossey et al. (1994) reported that these mutations were associated with the VHL type 1 phenotype in Western populations. Similarly, c.500G > A p.Arg167Gln is a hotspot mutation in many populations. Studies in Western populations showed that the mutation of c.500G > A p.Arg167Gln was associated with RCC and renal cysts, indicating that this mutation was associated with the VHL type 1 phenotype (Hes et al., 2007; Ciotti et al., 2009). However, according to our database, we found that this mutation was also related to PHEO (37.5%, 9 of 24), which indicated that the phenotype of the c.500G > A p.Arg167Gln mutation also differs in different ethnic backgrounds.
Different mutations may produce remarkably diverse phenotypes. For example, in group 1, two missense mutations occurred in the 194 mutation site, c.194C > T p.Ser65Leu and c.194C > G p.Ser65Trp. Intriguingly, the six major VHL-related lesions (CHB, RA, RCC, PCT, PHEO, and GS) were observed in the c.194C > T p.Ser65Leu mutational subgroup, while only three VHL lesions (CHB, RCC, and PCT) presented in the c.194C > G p.Ser65Trp mutational subgroup. Therefore, it indicates that missense mutations of different nucleotides in the same codon have potential effects on the clinical manifestation. The much lower number of patients in c.194C > G p.Ser65Trp mutational subgroup may limit the observation of the six major VHL-related lesions. This finding needs to be corroborated in larger cohorts. Bradley et al. (1999) also reported on the phenotypes of two distinct missense mutations in the same codon of the VHL gene (c.334T > A p.Tyr112Asn and c.334T > C p.Tyr112His). Thirteen patients were found with the c.334T > A p.Tyr112Asn mutation, seven of whom had RCC, and one of these patients had a pheochromocytoma, which suggests that this type of mutation causes the VHL type 1 phenotype, as most of the patients presented with RCC. Conversely, the c.334T > C p.Tyr112His mutation was associated with the VHL type 2A phenotype, as every affected individual in two families (22 patients) had PHEO but did not have RCC. Thus, different amino-acid changes at the same position may have different effects on the stability of the VHL protein, resulting in distinct clinical phenotypes.
Family members with the same mutation in VHL can also display different phenotypes. Mete et al. (2014) evaluated the clinical presentation of 49 family members from three generations of a Turkish family and identified the VHL p.A149S mutation. All of the patients were diagnosed with VHL syndrome type 2B, while nine patients were diagnosed with a pheochromocytoma, and one patient was diagnosed with a lumbar spinal hemangioblastoma and a pancreatic neuroendocrine tumor without pheochromocytoma. In our study, group 5 represented the c.262T > C p.Trp88Arg mutation that included 21 patients from 2 families (Figure 4 and Table 3). Variability was observed in the mean age at onset for CHB and RCC, which were 41.1 ± 9.1 (range = 29–62) and 37.4 ± 12.9 (range = 23–65), respectively. Moreover, the penetrance of VHL lesions was also distinct. Taken together, this phenotypic variability suggests that other factors or molecular mechanisms may affect the phenotypes caused by specific mutations, such as environmental factors or telomere length (Ning et al., 2014; Wang et al., 2017).
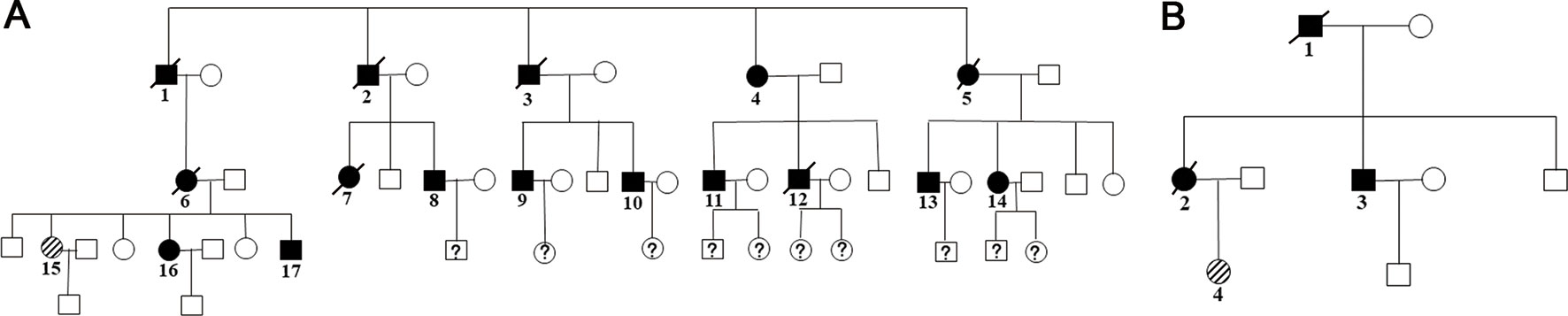
Figure 4 Two family pedigrees of group 5. □ and ○ indicate normal males and females, ▪ and • represent males and females with VHL mutation, and patient 15 in family 1 (A) and patient 4 in family 2 (B) were the probands.
Table 3 Characteristics of genotype and phenotype in group 5.
A classification of VHL diseases was proposed that was based on the patient’s preference for PHEO development. For example, the VHL type 1 phenotype has a low risk of PHEOs compared to the VHL type 2 phenotype, which is associated with PHEOs (Ong et al., 2007). The VHL type 2 phenotype is further classified into type 2A (hemangioblastoma and PHEO, but rarely RCC), type 2B (hemangioblastoma, PHEO, and RCC), and type 2C (PHEO only). However, during follow-up, the VHL disease shows characteristics of phenotypic variability, and new clinical manifestations may appear during the patient’s lifetime. Recently, Liu et al. (2018) reported genotype–phenotype correlations in VHL disease based on the alteration of a HIF-α binding site in the VHL protein. Lee et al. (2016) analyzed the genotype–phenotype correlations of VHL syndrome in Korean families and concluded that missense mutations in the Hypoxia-inducible factor-α (HIF-α) binding site elevate the age-specific risk for CHB. The studies cited above linked mutations in the VHL gene, protein binding sites, and phenotypic diversity to provide insights into the genotype–phenotype correlations based on amino-acid changes in the HIF-α binding site. In this study, we provide a precise summary of the penetrance and overall survival for each of the frequent mutations in the VHL gene within the largest Chinese VHL cohort. Our findings provide a more precise and individualized dataset that can be used in genetic counseling and research of the disease pathogenesis.
The current study had several limitations. Von Hippel–Lindau disease is rare, the size of this cohort is relatively small, and the follow-up durations are not sufficiently long, which may influence the correlation analysis between the frequent mutations in the VHL gene and the clinical phenotypes. Prospective, large-scale, and long-term follow-up studies are needed to further validate these results. Ultimately, elucidating the genotype–phenotype correlations of VHL disease will help to predict the risk of developing the diverse range of VHL-related phenotypes and their prognoses for individuals with VHL.
The involvement of human participants in this study was based on the Declaration of Helsinki. The ethics of this study were reviewed and approved by the Institutional Ethics Committee of Peking University First Hospital. Informed consent to use clinical data was received from each patient or from their legal guardian.
Conceptualization: BH. Data curation: KM, JCZ, JFZ, JW, and SL. Formal analysis: BH. Funding acquisition: KG. Methodology: ZZ and LC. Project administration and supervision: KG and NZ. Original draft writing: BH.
This work was supported by the National Natural Science Foundation of China (grant 81572506), the Special Health Development Research Project of Capital (grant 2016-2-4074), and the Fundamental Research Funds for the Central Universities (grant BMU2018JI002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We sincerely thank all the patients and the families in this study for their collaboration and support: Dingfang Bu, Medical Experiment Center, Peking University First Hospital, for his technical assistance and guidance for this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00867/full#supplementary-material
Bradley, J. F., Collins, D. L., Schimke, R. N., Parrott, H. N., Rothberg, P. G. (1999). Two distinct phenotypes caused by two different missense mutations in the same codon of the VHL gene. Am. J. Med. Genet. 87, 163–167. doi: 10.1002/(SICI)1096-8628(19991119)87:2<163::AID-AJMG7>3.0.CO;2-A
Butman, J. A., Linehan, W. M., Lonser, R. R. (2008). Neurologic manifestations of von Hippel–Lindau disease. JAMA 300, 1334–1342. doi: 10.1001/jama.300.11.1334
Chittiboina, P., Lonser, R. R. (2015). Von Hippel–Lindau disease. Handb. Clin. Neurol. 132, 139–156. doi: 10.1016/B978-0-444-62702-5.00010-X
Chou, A., Toon, C., Pickett, J., Gill, A. J. (2013). Von Hippel–Lindau syndrome. Front. Horm. Res. 41, 30–49. doi: 10.1159/000345668
Ciotti, P., Garuti, A., Gulli, R., Ballestrero, A., Bellone, E., Mandich, P. (2009). Germline mutations in the von Hippel–Lindau gene in Italian patients. Eur. J. Med. Genet. 52, 311–314. doi: 10.1016/j.ejmg.2009.05.007
Crossey, P. A., Foster, K., Richards, F. M., Phipps, M. E., Latif, F., Tory, K., et al. (1994). Molecular genetic investigations of the mechanism of tumourigenesis in von Hippel–Lindau disease: analysis of allele loss in VHL tumours. Hum. Genet. 93, 53–58. doi: 10.1007/BF00218913
Gomy, I., Molfetta, G. A., de Andrade, B. E., Ferreira, C. A., Zanette, D. L., Casali-da-Rocha, J. C., et al. (2010). Clinical and molecular characterization of Brazilian families with von Hippel–Lindau disease: a need for delineating genotype–phenotype correlation. Fam. Cancer 9, 635–642. doi: 10.1007/s10689-010-9357-2
Gossage, L., Eisen, T., Maher, E. R. (2015). VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 15, 55–64. doi: 10.1038/nrc3844
Grubb, R. R., Choyke, P. L., Pinto, P. A., Linehan, W. M., Walther, M. M. (2005). Management of von Hippel–Lindau–associated kidney cancer. Nat. Clin. Pract. Urol. 2, 248–255. doi: 10.1038/ncpuro0179
Hes, F. J., van der Luijt, R. B., Janssen, A. L., Zewald, R. A., de Jong, G. J., Lenders, J. W., et al. (2007). Frequency of Von Hippel–Lindau germline mutations in classic and non-classic Von Hippel-Lindau disease identified by DNA sequencing, Southern blot analysis and multiplex ligation-dependent probe amplification. Clin. Genet. 72, 122–129. doi: 10.1111/j.1399-0004.2007.00827.x
Hwang, S., Ku, C. R., Lee, J. I., Hur, K. Y., Lee, M. S., Lee, C. H., et al. (2014). Germline mutation of Glu70Lys is highly frequent in Korean patients with von Hippel–Lindau (VHL) disease. J. Hum. Genet. 59, 488–493. doi: 10.1038/jhg.2014.61
Kim, J. J., Rini, B. I., Hansel, D. E. (2010). Von Hippel Lindau syndrome. Adv. Exp. Med. Biol. 685, 228–249. doi: 10.1007/978-1-4419-6448-9_22
Latif, F., Tory, K., Gnarra, J., Yao, M., Duh, F. M., Orcutt, M. L., et al. (1993). Identification of the von Hippel–Lindau disease tumor suppressor gene. Science 260, 1317–1320. doi: 10.1126/science.8493574
Launbjerg, K., Bache, I., Galanakis, M., Bisgaard, M. L., Binderup, M. (2017). Von Hippel–Lindau development in children and adolescents. Am. J. Med. Genet. A 173, 2381–2394. doi: 10.1002/ajmg.a.38324
Lee, J. S., Lee, J. H., Lee, K. E., Kim, J. H., Hong, J. M., Ra, E. K., et al. (2016). Genotype–phenotype analysis of von Hippel–Lindau syndrome in Korean families: HIF-alpha binding site missense mutations elevate age-specific risk for CNS hemangioblastoma. BMC Med. Genet. 17, 48. doi: 10.1186/s12881-016-0306-2
Liu, S. J., Wang, J. Y., Peng, S. H., Li, T., Ning, X. H., Hong, B. A., et al. (2018). Genotype and phenotype correlation in von Hippel-Lindau disease based on alteration of the HIF-alpha binding site in VHL protein. Genet. Med. 20 (10), 1266–1273. doi: 10.1038/gim.2017.261
Lonser, R. R., Butman, J. A., Huntoon, K., Asthagiri, A. R., Wu, T., Bakhtian, K. D., et al. (2014). Prospective natural history study of central nervous system hemangioblastomas in von Hippel–Lindau disease. J. Neurosurg. 120, 1055–1062. doi: 10.3171/2014.1.JNS131431
Lonser, R. R., Glenn, G. M., Walther, M., Chew, E. Y., Libutti, S. K., Linehan, W. M., et al. (2003). Von Hippel–Lindau disease. Lancet 361, 2059–2067. doi: 10.1016/S0140-6736(03)13643-4
Lonser, R. R., Kim, H. J., Butman, J. A., Vortmeyer, A. O., Choo, D. I., Oldfield, E. H. (2004). Tumors of the endolymphatic sac in von Hippel–Lindau disease. N. Engl. J. Med. 350, 2481–2486. doi: 10.1056/NEJMoa040666
Maher, E. R., Webster, A. R., Richards, F. M., Green, J. S., Crossey, P. A., Payne, S. J., et al. (1996). Phenotypic expression in von Hippel–Lindau disease: correlations with germline VHL gene mutations. J. Med. Genet. 33, 328–332. doi: 10.1136/jmg.33.4.328
Mete, T., Berker, D., Yilmaz, E., Ozgen, G., Yalcin, Y., Tuna, M., et al. (2014). Clinical presentation of Von Hippel Lindau syndrome type 2B associated with VHL p.A149S mutation in a large Turkish family. Endocrine 45, 128–135. doi: 10.1007/s12020-013-9982-2
Nielsen, S. M., Rhodes, L., Blanco, I., Chung, W. K., Eng, C., Maher, E. R., et al. (2016). Von Hippel–Lindau disease: genetics and role of genetic counseling in a multiple neoplasia syndrome. J. Clin. Oncol. 34, 2172–2181. doi: 10.1200/JCO.2015.65.6140
Nielsen, S. M., Rubinstein, W. S., Thull, D. L., Armstrong, M. J., Feingold, E., Stang, M. T., et al. (2011). Genotype–phenotype correlations of pheochromocytoma in two large von Hippel–Lindau (VHL) type 2A kindreds with different missense mutations. Am. J. Med. Genet. A 155A, 168–173. doi: 10.1002/ajmg.a.33760
Ning, X. H., Zhang, N., Li, T., Wu, P. J., Wang, X., Li, X. Y., et al. (2014). Telomere shortening is associated with genetic anticipation in Chinese Von Hippel–Lindau disease families. Cancer Res. 74, 3802–3809. doi: 10.1158/0008-5472.CAN-14-0024
Nordstrom-O’Brien, M., van der Luijt, R. B., van Rooijen, E., van den Ouweland, A. M., Majoor-Krakauer, D. F., Lolkema, M. P., et al. (2010). Genetic analysis of von Hippel–Lindau disease. Hum. Mutat. 31, 521–537. doi: 10.1002/humu.21219
Ong, K. R., Woodward, E. R., Killick, P., Lim, C., Macdonald, F., Maher, E. R. (2007). Genotype–phenotype correlations in von Hippel–Lindau disease. Hum. Mutat. 28, 143–149. doi: 10.1002/humu.20385
Patel, R. J., Appukuttan, B., Ott, S., Wang, X., Stout, J. T. (2000). DNA-based diagnosis of the von Hippel–Lindau syndrome. Am. J. Ophthalmol. 129, 258–260. doi: 10.1016/S0002-9394(99)00328-1
Patocs, A., Gergics, P., Balogh, K., Toth, M., Fazakas, F., Liko, I., et al. (2008). Ser80Ile mutation and a concurrent Pro25Leu variant of the VHL gene in an extended Hungarian von Hippel–Lindau family. BMC Med. Genet. 9, 29. doi: 10.1186/1471-2350-9-29
Peng, S., Shepard, M. J., Wang, J., Li, T., Ning, X., Cai, L., et al. (2017). Genotype–phenotype correlations in Chinese von Hippel–Lindau disease patients. Oncotarget 8, 38456–38465. doi: 10.18632/oncotarget.16594
Stebbins, C. E., Kaelin, W. J., Pavletich, N. P. (1999). Structure of the VHL–ElonginC–ElonginB complex: implications for VHL tumor suppressor function. Science 284, 455–461. doi: 10.1126/science.284.5413.455
Varshney, N., Kebede, A. A., Owusu-Dapaah, H., Lather, J., Kaushik, M., Bhullar, J. S. (2017). A review of von Hippel–Lindau syndrome. J. Kidney Cancer VHL 4, 20–29. doi: 10.15586/jkcvhl.2017.88
Vikkath, N., Valiyaveedan, S., Nampoothiri, S., Radhakrishnan, N., Pillai, G. S., Nair, V., et al. (2015). Genotype–phenotype analysis of von Hippel–Lindau syndrome in fifteen Indian families. Fam. Cancer 14, 585–594. doi: 10.1007/s10689-015-9806-z
Wang, J. Y., Peng, S. H., Li, T., Ning, X. H., Liu, S. J., Hong, B. A., et al. (2018). Risk factors for survival in patients with von Hippel–Lindau disease. J. Med. Genet. 55, 322–328. doi: 10.1136/jmedgenet-2017-104995
Wang, J. Y., Peng, S. H., Ning, X. H., Li, T., Liu, S. J., Liu, J. Y., et al. (2017). Shorter telomere length increases age-related tumor risks in von Hippel–Lindau disease patients. Cancer Med. 6, 2131–2141. doi: 10.1002/cam4.1134
Wilding, A., Ingham, S. L., Lalloo, F., Clancy, T., Huson, S. M., Moran, A., et al. (2012). Life expectancy in hereditary cancer predisposing diseases: an observational study. J. Med. Genet. 49, 264–269. doi: 10.1136/jmedgenet-2011-100562
Wu, P., Zhang, N., Wang, X., Ning, X., Li, T., Bu, D., et al. (2012). Family history of von Hippel–Lindau disease was uncommon in Chinese patients: suggesting the higher frequency of de novo mutations in VHL gene in these patients. J. Hum. Genet. 57, 238–243. doi: 10.1038/jhg.2012.10
Keywords: von Hippel–Lindau disease, VHL mutation, genotype–phenotype correlation, onset age, survival
Citation: Hong B, Ma K, Zhou J, Zhang J, Wang J, Liu S, Zhang Z, Cai L, Zhang N and Gong K (2019) Frequent Mutations of VHL Gene and the Clinical Phenotypes in the Largest Chinese Cohort With Von Hippel–Lindau Disease. Front. Genet. 10:867. doi: 10.3389/fgene.2019.00867
Received: 05 September 2018; Accepted: 20 August 2019;
Published: 18 September 2019.
Edited by:
Weida Tong, National Center for Toxicological Research (FDA), United StatesReviewed by:
Asma Sultana, King Saud University Medical City, Saudi ArabiaCopyright © 2019 Hong, Ma, Zhou, Zhang, Wang, Liu, Zhang, Cai, Zhang and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Cai, ZHJjYWlsaW5AMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.