 Tongjin Liu1†
Tongjin Liu1† Jinglei Wang1,2†
Jinglei Wang1,2† Chunhui Wu1
Chunhui Wu1 Youjun Zhang1*
Youjun Zhang1* Xiaohui Zhang1
Xiaohui Zhang1 Xiaoman Li1
Xiaoman Li1 Haiping Wang1Jiangping Song1
Haiping Wang1Jiangping Song1 Xixiang Li1*
Xixiang Li1*- 1Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Key Laboratory of Biology and Genetic Improvement of Horticultural Crops, Ministry of Agriculture, Beijing, China
- 2Institute of Vegetables Research, Zhejiang Academy of Agricultural Sciences, Hangzhou, China
Taproot skin color is a crucial visual and nutritional quality trait of radish, and purple skin is most attractive to consumers. However, the genetic mechanism underlying this character is unknown. Herein, F2 segregating populations were constructed to investigate radish genomic regions with purple skin genes. Segregation analysis suggested that pigment presence was controlled by one dominant gene, Rsps. A bulk segregant approach coupled to whole-genome sequencing (QTL-seq) and classical linkage mapping narrowed the Rsps location to a 238.51-kb region containing 18 genes. A gene in this region, designated RsMYB1.1 (an Arabidopsis PAP1 homolog), was a likely candidate gene because semiquantitative RT-PCR and quantitative real-time PCR revealed RsMYB1.1 expression in only purple-skinned genotypes, sequence variation was found between white- and purple-skinned radishes, and an InDel marker in this gene correctly predicted taproot skin color. Furthermore, four RsMYB1.1 homologs (RsMYB1.1-1.4) were found in “XYB36-2” radish. RsMYB1.1 and the previously mapped and cloned RsMYB1.4 (contributing to red skin) were located on different chromosomes and in different subclades of a phylogenetic tree; thus, they are different genes. These findings provide insight into the complex anthocyanin biosynthesis regulation in radish and information for molecular breeding to improve the anthocyanin content and appearance of radish taproots.
Introduction
Radish (Raphanus sativus, 2n = 2x = 18), a member of the Brassicaceae (Cruciferae) family, is an important root vegetable grown and consumed throughout the world. In addition to its root, the leaf, siliques, and seed oil of some cultivars are consumed in some regions. The taproots of radish exhibit significant variation in shape, size, color, and flavor. There are many different taproot skin colors, including purple, red, pink, white, green, and even black and yellow.
The color of agricultural products is an important visual quality trait that directly affects the choice behavior of consumers. For this reason, a number of studies have attempted to identify major genes that control the color of different vegetables by map-based cloning. In cucumber, two genes responsible for fruit color were identified by map-based cloning. A single-nucleotide insertion in APRR2 was responsible for a white immature fruit color (Liu H. et al., 2016), while a mutation in MYB36 resulted in the formation of a yellow-green peel mutant (Hao et al., 2018). A 4-bp insertion in a putative R3 MYB repressor, SIMYBATV, led to anthocyanin accumulation in tomato fruit (Cao et al., 2017). A previous study mapped a purple leaf gene in ornamental kale to a 44.8-kb interval, and a dihydroflavonol reductase-coding gene was identified as a candidate gene (Liu et al., 2017). Through map-based cloning, BnAPR2 was identified as a candidate gene responsible for the purple leaf trait in Brassica napus (Li et al., 2016). More recently, RsMYB1, an R2R3-MYB transcription factor, was identified in the red-skinned radish cultivar “Lian Yan No. 1” by map-based cloning (Yi et al., 2018). However, to date, no map-based cloning studies have been performed for the major genes responsible for the purple skin of radish taproots, which is an important visual and nutritional quality character.
In recent years, with an increase in people’s consumption level, red and purple radishes have gained popularity because their enriched glucosinolates and anthocyanins have health benefits (Park et al., 2011). Developing radish cultivars with different skin colors has become an important breeding objective, and illuminating the mechanism of skin color formation will significantly accelerate progress in skin color breeding. Previous studies have indicated that anthocyanin accumulation results in the red/purple skin of radish (Park et al., 2011; Muleke et al., 2017). The anthocyanin biosynthesis pathway is relatively conserved in plants and has been extensively studied (Park et al., 2011; Zhang Y. et al., 2015; Zhang et al., 2015; Chen et al., 2016). The regulation of this process is extremely complex, and some members of the MYB, basic helix–loop–helix (bHLH), WD40, lateral organ boundary domain (LBD), WRKY, and NAC transcription factor families have been reported to be involved in its positive or negative regulation (Johnson et al., 2002; Koes et al., 2005; Matsui et al., 2008; Rubin et al., 2009; Stracke et al., 2010; Zhou et al., 2015). Recently, most structural genes involved in a multi-enzymatic pathway that controls anthocyanin biosynthesis were reported in radish (Park et al., 2011; Chen et al., 2016; Muleke et al., 2017; Sun et al., 2018). However, only two transcription factors (RsMYB1 and RsTT8) that regulate anthocyanin accumulation in radish taproots identified by homology or map-based cloning have been reported (Lim et al., 2016; Lim et al., 2017; Yi et al., 2018).
Some early studies showed that a single locus or multiple loci may control root skin color and that red is dominant over white (Yarnell, 1956; He et al., 1997). He et al. (1997) indicated that red skin was probably controlled by three pairs of independent genes and that some genes controlling skin color likely affected flesh color inheritance (He et al., 1997). In contrast, Yi et al. (2018) reported that red skin was determined by a single dominant gene. These conflicting results most likely occur because the inheritance of radish root skin color is complex, and different inheritance mechanisms may be involved in different materials. The inheritance patterns and chromosomal locations of purple taproot skin genes in radish are still unclear. Therefore, the identification and cloning of major genes responsible for the purple skin of radish deserve further study.
In the present study, the radish inbred line CX16Q-25-2 (purple skin with white flesh) was crossed with the inbred line CX16Q-1-6-2 (white skin and flesh) to construct an F2 population, which was used for fine genetic mapping of the genes for purple skin. Furthermore, genes located within the mapping region were analyzed, a candidate gene was identified and named RsMYB1.1, and four RsMYB1.1 homologous genes distributed on three chromosomes of “XYB36-2” radish were identified and named RsMYB1.1-1.4 according to their chromosome position. These findings will facilitate elucidation of the molecular mechanism for purple skin formation in radish taproots.
Materials and Methods
Plant Materials
The white-skinned male radish inbred line CX16Q-1-6-2 (Figure 1A) was crossed with the purple-skinned female radish inbred line CX16Q-25-2 (Figure 1B) to generate the F1 generation. F2 populations (Figure 1C) were generated from self-pollination of a single F1 plant and further self-pollinated to generate the F2:3 population. All plants were grown at the Institute of Vegetables and Flowers (IVF), Chinese Academy of Agricultural Sciences (CAAS), Beijing, China.
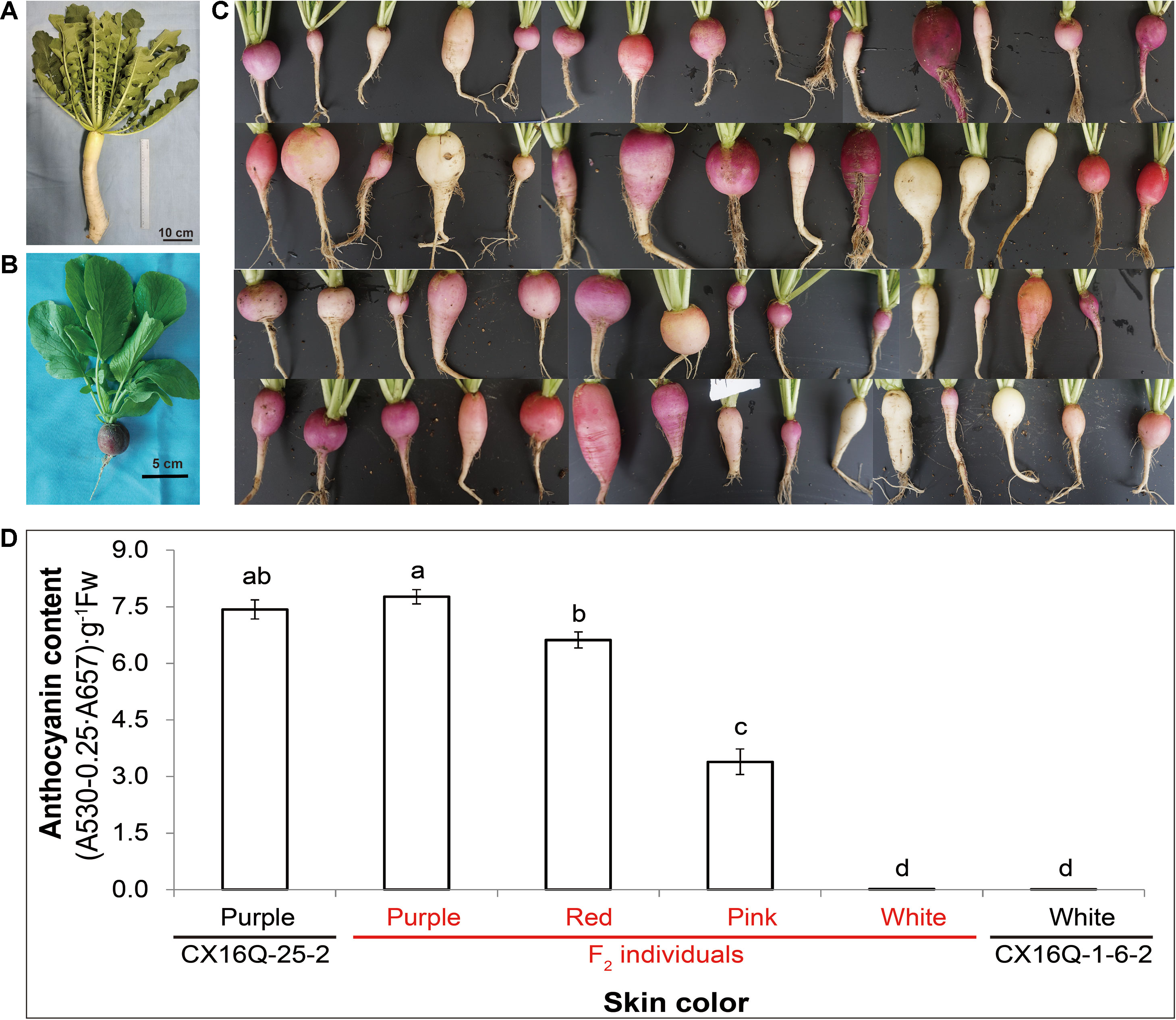
Figure 1 Skin color of the parental lines and their derived F2 population. (A) CX16Q-1-6-2; (B) CX16Q-25-2; (C) F2 individuals of CX16Q-25-2 × CX16Q-1-6-2 divided into purple skins with varying shades and white skins; (D) relative total anthocyanin content of the skin of taproots with different skin colors.
Phenotyping and DNA Extraction
Skin color was identified visually 40 days after planting. Young leaves of each plant were collected, freeze dried, and ground into a fine powder for DNA extraction. Genomic DNA was extracted from 100 mg of leaf powder of each plant using the CTAB method (Doyle and Doyle, 1987). The quality of the DNA was assessed by 1% agarose gel electrophoresis and a NanoPhotometer (Implen, CA, USA), and the concentrations were measured by a Qubit DNA Assay Kit in a Qubit 2.0 Fluorometer (Life Technologies, CA, USA).
Total Anthocyanin Content Measurements
Total anthocyanins were extracted from 0.5 g of finely ground taproot skin from CX16Q-25-2, CX16Q-1-6-2, and different F2 individuals (purple, red, pink, and white taproot skin) and evaluated by measuring the absorbance of the extract at 530 and 657 nm, as described by Chu et al. (2013). Anthocyanins were quantitated using the following formula: Q = (A530 − 0.25 × A657) × M−1, where Q is the anthocyanin content, A530 and A657 are the absorbance at the wavelengths 530 and 657 nm, respectively, and M is the weight of the sample. All samples were measured as triplicates in three independent biological replicates. Error bars indicate the standard error (SE) of the average of the anthocyanin content.
Bulk Segregant Approach Coupled to Whole-Genome Sequencing (QTL-seq) Analysis
For the QTL-seq analysis, a purple-skinned DNA pool (P-pool) and a white-skinned DNA pool (W-pool) were constructed by mixing equal amounts of DNA from 40 purple-skinned and 40 white-skinned F2 plants from the 2017 fall experiment, respectively. Four sequencing libraries, namely, P-pool, W-pool, and the two parental inbred lines (CX16Q-25-2 and CX16Q-1-6-2), were prepared for paired-end (150 bp) sequencing with the Illumina HiSeq platform. Clean reads were obtained by trimming adaptors of raw sequence reads (“.fastq” format), removing reads with ambiguous “N” nucleotides exceeding 5% of the read length, and eliminating reads containing a number of bases with a Q value below 5 exceeding 50% of the total length. High-quality clean reads were used for QTL-seq analysis and insertion and deletion (InDel) marker development (Gordon, 2009).
A QTL-seq pipeline (http://genome-e.ibrc.or.jp/home/bioinformatics-team/mutmap, developed by the Iwate Biotechnology Research Center, Japan) was used to identify candidate genomic regions. Briefly, the cleaned reads of CX16Q-25-2 were first aligned to the reference genome “XYB36-2” (Zhang et al., 2015b) using an inbuilt BWA aligner (Li and Durbin, 2009). The reference-guided assembly of the CX16Q-25-2 parent was developed by substituting the bases with high-confidence nucleotides called for the CX16Q-25-2 parent. Then, the reads from the P and W bulks were aligned to the developed assembly, and variants were called for both bulks. The single nucleotide polymorphism (SNP) index for both bulks was calculated at each SNP position as suggested by Hiroki et al. (2013). The ΔSNP index was obtained by subtracting the SNP index of the P-pool from that of the W-pool. The average ΔSNP index located in a certain genomic interval was calculated using a sliding window analysis with a 4-Mb window size and 20-kb increment. Statistical confidence intervals of the ΔSNP index were calculated under the null hypothesis of no quantitative trait locus (QTL) following the description by Takagi et al., and 95% confidence intervals of the ΔSNP index were used to identify genomic regions containing candidate major QTLs (Hiroki et al., 2013).
QTL Analyses With InDel Markers
QTL-seq analysis identified the major QTL for Rsps (purple skin). To fine map the Rsps gene, InDel markers were designed within the Rsps target region according to the polymorphism information of two resequenced parental inbred lines: CX16Q-25-2 and CX16Q-1-6-2. The short reads from parental inbred lines were aligned to the “XYB36-2” reference genome (Zhang et al., 2015b) with BWA software (Li and Durbin, 2009). InDel calling was performed by SAMtools software (Li and Durbin, 2009). Primer pairs were designed using Primer3 (Triinu and Maido, 2007), and specific primers were selected by alignment with the “XYB36-2” reference genome using electronic PCR (e-PCR) software (Schuler, 1997). Polymorphic markers were screened using the DNA of P-pool, W-pool, and the two parental inbred lines (CX16Q-25-2 and CX16Q-1-6-2) and then applied to the plants in the F2 population from the 2017 fall experiment.
PCR amplifications were performed in a 15-μl volume as described by Zhang et al. (2015a). The primers are listed in Supplementary Table 1. The amplified products were separated on an 8% polyacrylamide gel with 1× Tris/borate/EDTA (TBE) buffer at a constant voltage of 180 V for 1 h and then visualized with silver staining. The marker bands of individuals with the CX16Q-25-2, CX16Q-1-6-2, and F1 alleles were recorded as “a”, “b”, and “h”, respectively. JoinMap 4.0 was used to perform linkage analysis (Van Ooijen et al., 2002).
Candidate Gene Identification
To amplify the candidate gene, the DNA sequence was extracted from the radish genome (Zhang et al., 2015b), and a specific PCR primer was designed using Primer3web (Untergasser et al., 2012) (see Supplementary Table 1). PCR amplification was performed in a total volume of 50 µl using KOD-Plus-Neo polymerase (Toyobo, Osaka, Japan) according to the manufacturer’s specifications with the two-step cycle procedure (Liu T. et al., 2016). The PCR products were subjected to Sanger sequencing using an ABI 3730 instrument (Applied Biosystems, CA, USA).
RNA Extraction, Semiquantitative RT-PCR, and Quantitative Real-Time PCR (qPCR) of the Candidate Gene
Total RNA was extracted from the taproot skin of CX16Q-25-2, CX16Q-1-6-2, and different F2 individuals (purple, red, pink, and whitetaproot skin), treated with DNase and then reverse transcribed into cDNA as previously described (Liu T. et al., 2016). The specific primer was designed using Primer3web (Untergasser et al., 2012) and is shown in Supplementary Table 1. Semiquantitative RT-PCR for the candidate gene was carried out between CX16Q-25-2 and CX16Q-1-6-2 in a total volume of 50 µl using KOD-Plus-Neo polymerase with a T100 thermal cycler (Bio-Rad, Hercules, CA, USA). The following cycling parameters were used: 2 min of predenaturation at 94°C, followed by 35 cycles of denaturation (98°C, 10 s), annealing (63°C, 30 s), and extension (68°C, 30 s). Ten microliters of PCR product was then separated on a 2% agarose gel stained with GoldView. To investigate the pigment-controlling role of the candidate gene, qPCR analysis was performed in the taproot skin of parental lines and different F2 individuals (purple, red, pink, and white). The cDNA was diluted fivefold as templates for qPCR using SuperReal PreMix Plus (TIANGEN BIOTECH CO., LTD, Beijing, China). The reaction volume was 20 μl, including 0.6 μl of forward and reverse primers at 10 mM each, 10 μl of 2 × SuperReal PreMix Plus, 2.0 μl of the cDNA sample, 2.0 μl of 50 × ROX Reference Dye, and 4.8 μl of ddH2O. The thermal cycling profile was 95°C for 15 min; 40 cycles of 95°C for 10 s, 60°C for 30 s; then 95°C for 15 s, 60°C for 1 min, and ramping to 95°C for 15 s. Three independent biological and technical replicates were performed. Data were analyzed using StepOne™ Software v.2.0 (Applied Biosystems). Radish glyceraldehyde 3-phosphate dehydrogenase (RsGADPH) transcripts were used as a control for semiquantitative RT-PCR and an internal reference for qPCR (Liu et al., 2019).
Search for Homologs of the Candidate Gene
The predicted protein sequence of the candidate gene was used to search for corresponding homologs by BLASTP with previously published radish genome data (Zhang et al., 2015). Related protein sequences of other species were obtained from the NCBI database (http://www.ncbi.nlm.nih.gov/). DNAMAN8 software (Lynnon, Quebec, Canada) was used to align sequences with the default parameters, and MEGA5 software was used to generate a phylogenetic tree according to the predicted protein sequences (Tamura et al., 2011).
Results
Inheritance of the Purple Skin of Radish Taproots
To determine the inheritance of purple skin in radish, purple-skinned (CX16Q-25-2) and white-skinned (CX16Q-1-6-2) radish inbred lines were used to generate F1 and F2 populations (Figure 1). The fleshy taproot skin color of all F1 individuals was purple (Table 1). The 557 F2 individuals exhibited complex skin color segregation including white, pink, red, and purple of different shades. The total anthocyanin content of the parental lines and different F2 radish roots are significantly different (Figure 1D). This result indicated that the genetics of purple taproot skin were complex, with multiple factors involved. However, the segregation was consistent with a 3:1 Mendelian segregation ratio for pigmented (pink, red, and purple with different shades) and white roots, which indicated that pigment presence was controlled by a single dominant gene, designated Rsps (purple skin). The present study was mainly focused on the identification of this dominant gene.

Table 1 Segregation of taproot skin color in the F1 and F2 populations.
Preliminary Mapping of Rsps by QTL-seq
The two parental lines (CX16Q-25-2 and CX16Q-1-6-2) and two extreme pools (purple-skinned pool (P-pool) and white-skinned pool (W-pool)) from the F2 population were paired-end (150 bp) sequenced with an Illumina HiSeq platform. In total, 5.6, 5.9, 23.4, and 30.0 Gb of clean reads from CX16Q-25-2 (11.2× depth coverage), CX16Q-1-6-2 (11.8× depth coverage), P-pool (46.8× depth coverage), and W-pool (60× depth coverage) were generated, respectively. These short reads were aligned to the “XYB36-2” reference genome, and 1,330,575, 1,130,541, 2,406,284, and 2,452,285 SNPs were identified between CX16Q-25-2 and “XYB36-2,” CX16Q-1-6-2, and “XYB36-2,” P-pool and “XYB36-2,” W-pool and “XYB36-2,” respectively. Subsequent QTL-seq analysis identified only one genomic region from 31.45 to 33.00 Mb on “XYB36-2” radish chromosome 2 that had a Δ(SNP index) value that was significantly different from 0 at the 95% confidence level (Figure 2A). These results indicated that there was a major QTL controlling purple skin in the 31.45- to 33.00-Mb region on chromosome 2 in radish (Figure 2A), which was designated Rsps.
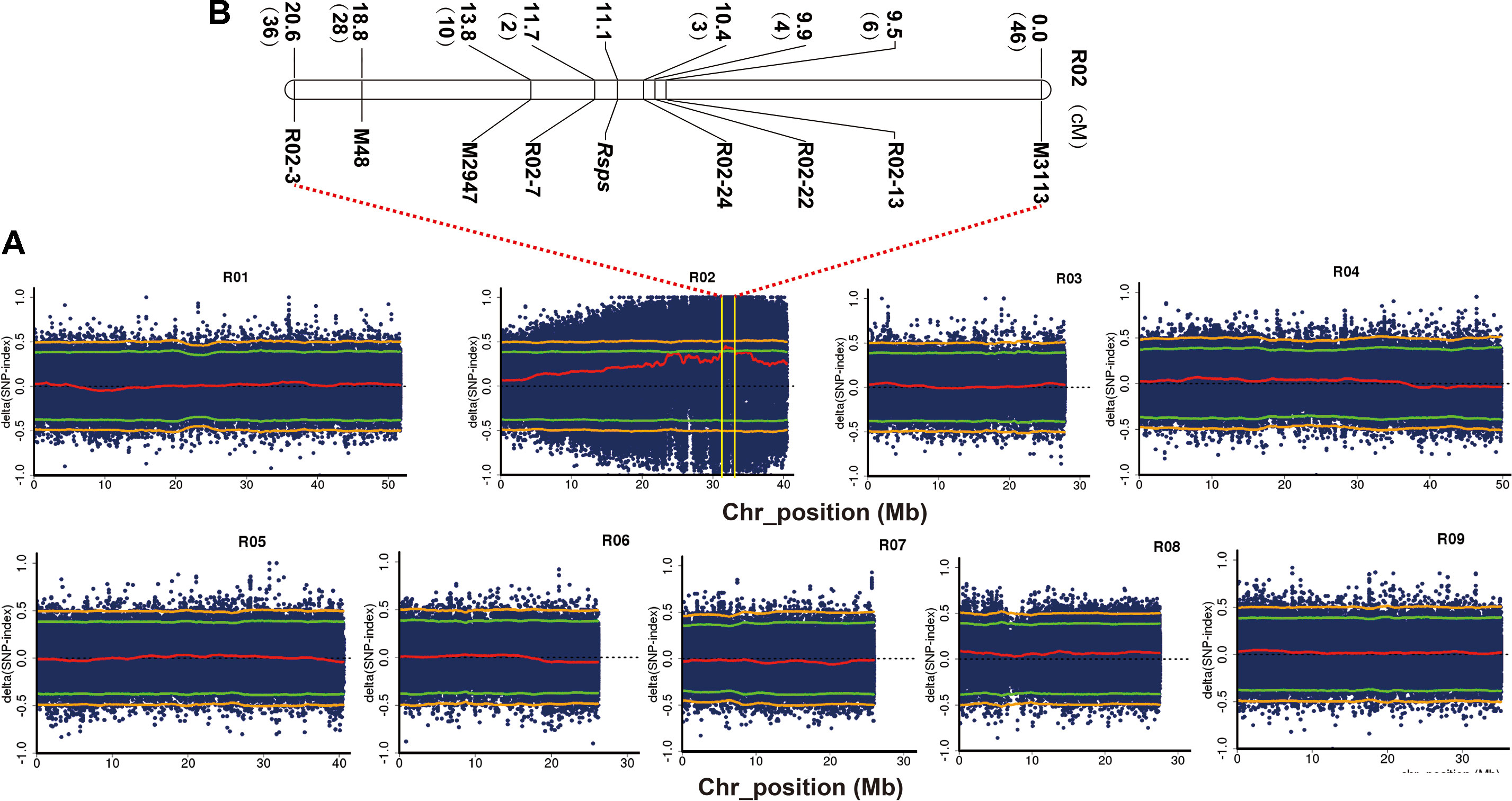
Figure 2 Combined QTL-seq and traditional linkage analysis to map a purple skin locus (Rsps) in radish. (A) Δ(SNP index) plot of the purple and white bulks with confidence intervals under the null hypothesis of no QTLs (green, p < 0.05; orange, p < 0.01); (B) genetic linkage analysis with InDel markers confirming the position of Rsps. The numbers in brackets refer to the number of recombinants of each marker.
QTL Analyses With InDel Markers
To verify the accuracy of QTL-seq in the detection of Rsps and narrow down its location, traditional QTL analysis was carried out with 352 F2 plants. Three polymorphic InDel primer pairs were identified from markers developed in our previous studies (Supplementary Table 1). In addition, a total of 674 pairs of InDel primers were developed in the preliminary mapped regions with potential variation of >4 bp (Supplementary Table 2). Of the pairs, 52 were synthesized and used to screen polymorphic markers between CX16Q-25-2 and CX16Q-1-6-2 and between P-pool and W-pool, and five markers were determined to be useful (Supplementary Table 1). Then, these eight polymorphic markers were applied to the F2 populations for QTL mapping. The Rsps locus was localized to a 1.3-cM interval between R02-7 and R02-24, flanking the gene at genetic distances of 0.6 cM and 0.7 cM, and this region matched that from QTL-seq (Figure 2B). Based on the marker locations of R02-7 and R02-24 in the reference genome of “XYB36-2,” the 238.51-kb interval (31584238–31822749 bp) on chromosome 2 harbors the candidate gene for Rsps.
Candidate Gene Prediction for Purple Taproot Skin in Radish
There were 18 genes within the 238.51-kb candidate region in the “XYB36-2” reference genome (Table 2) (http://brassicadb.org/brad/datasets/pub/Genomes/Raphanus_sativus/) (Zhang et al., 2015b). Based on the functional annotations, Rsa10008423 was predicted to be a member of the MYB family of transcription factors. Blast alignment results showed that the coding sequence (CDS) identity of Rsa10008423 with the Arabidopsis PRODUCTION OF ANTHOCYANIN PIGMENT1 (AtPAP1, AT1G56650) gene was up to 82.40% (Supplementary Figure1A), and the predicted encoded proteins had 77.51% sequence identity (Supplementary Figure 1B). AtPAP1 was reported to be involved in regulating the expression of anthocyanin biosynthetic genes (Borevitz et al., 2000). Rsa10008423 is a homologous gene of AtPAP1 in radish; therefore, Rsa10008423 was designated as RsMYB1.1.
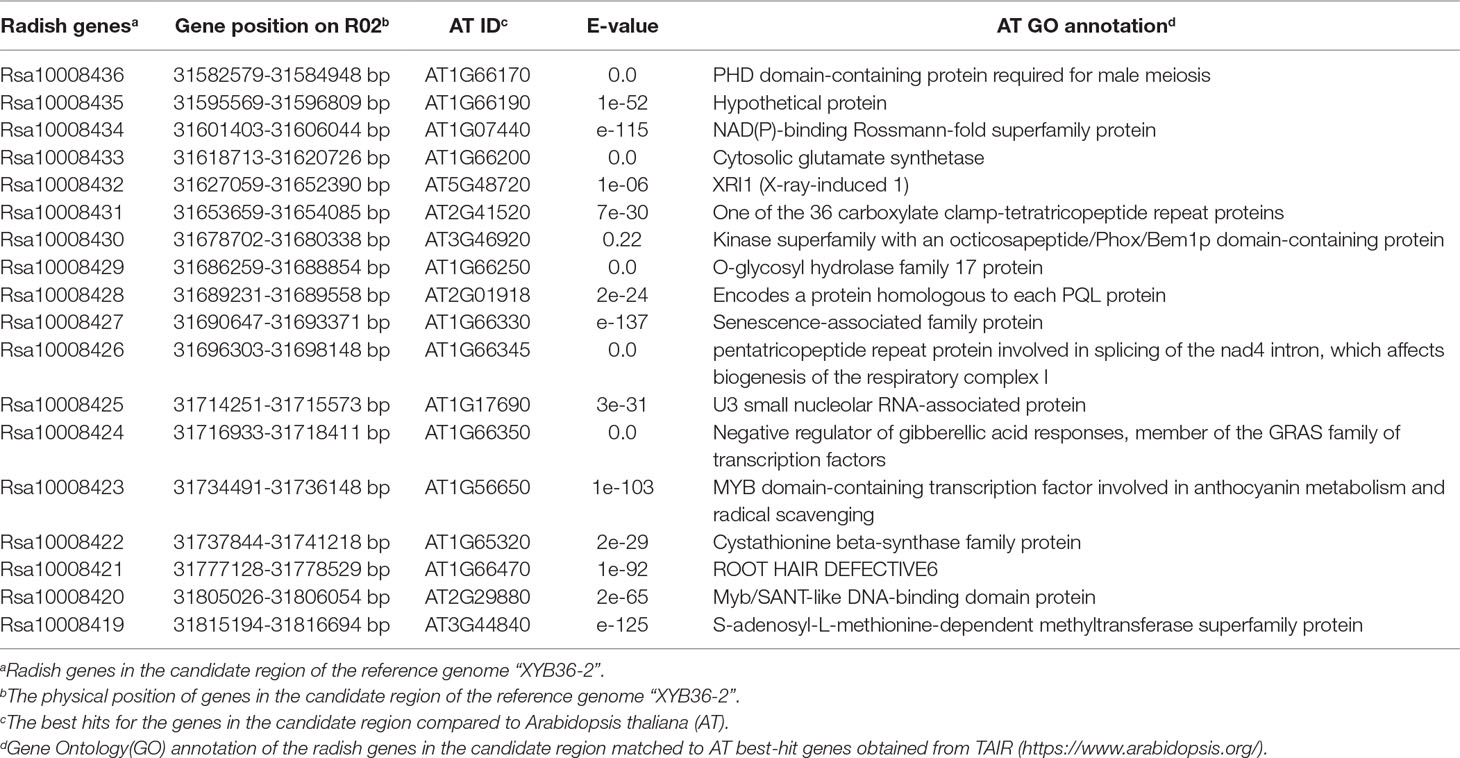
Table 2 Annotation of radish genes in the candidate region.
Analysis of the Candidate Gene
We investigated the expression patterns of RsMYB1.1 with semiquantitative RT-PCR in two parental lines to analyze whether the expression level of RsMYB1.1 may contribute to the variance in taproot skin color (Figure 3). The RsMYB1.1 transcript was detected only in the taproot skin of purple-skinned radish (Figure 3A).

Figure 3 Analysis of candidate genes. (A) Expression patterns of RsMYB1.1 with semiquantitative RT-PCR in the two parental lines; (B) alignment of RsMYB1.1 sequences of white and purple radishes; the positions of the forward and reverse primer pairs designed according to the parental sequence variation used to genotype the F2 individuals are shown with arrows; (C) genotypes of F2 individuals for PCR markers; (D) genotypes of F2:3 individuals for PCR markers. M, size marker.
The DNA sequences of the two parental lines and the CDS of RsMYB1.1 in CX16Q-25-2 were cloned, sequenced, and analyzed to investigate their sequence differences (Figure 3B). The alignment of RsMYB1.1 DNA and CDSs revealed multiple mutations (Figure 3B). The DNA sequence is 1,610-bp long, with a 750-bp predicted open reading frame encoding a polypeptide of 249 amino acids. RsMYB1.1 consists of three exons and two introns, and the first intron (which consists of 769 nucleotides) is significantly longer (8.5 times) than the second intron and represents 47.8% of the total length of the gene. The first and second exons are highly conserved between the two parental lines, while six SNP mutations were found in the third exon. In addition, two SNP mutations were found in the second intron. More notably, the first intron exhibited more SNP and InDel mutations between the two parental lines. Accordingly, an InDel marker was designed in this region, as shown in Figure 3B. Forty-six pigmented-skinned (pink, red, and purple with different shades) and 22 white-skinned radishes were selected from the F2 population to determine the consistency between these marker genotypes and the taproot skin color phenotypes. Furthermore, the present InDel marker was validated in 17 randomly selected pigmented- and white-skinned radishes from the F2:3 population. The results showed that all taproots were predicted correctly (Figures 3C, D).
The expression levels of RsMYB1.1 in the taproot skin of both parents and F2 individuals with purple, red, pink, and white skin were measured by qPCR. RsMYB1.1 had significantly higher expression levels in pigmented-skinned individuals than in white taproot skin (Figure 4). Among pigmented-skinned individuals, purple-skinned plants had the significantly highest expression levels of RsMYB1.1, red-skinned plants had the second highest levels, and pink-skinned plants had the significantly lowest expression. The expression trend of RsMYB1.1 in the taproot skin of parents and F2 individuals correlated with their anthocyanin accumulation (Figures 1 and 4). These results suggest that RsMYB1.1 may be a candidate gene controlling purple skin in CX16Q-25-2 radish.
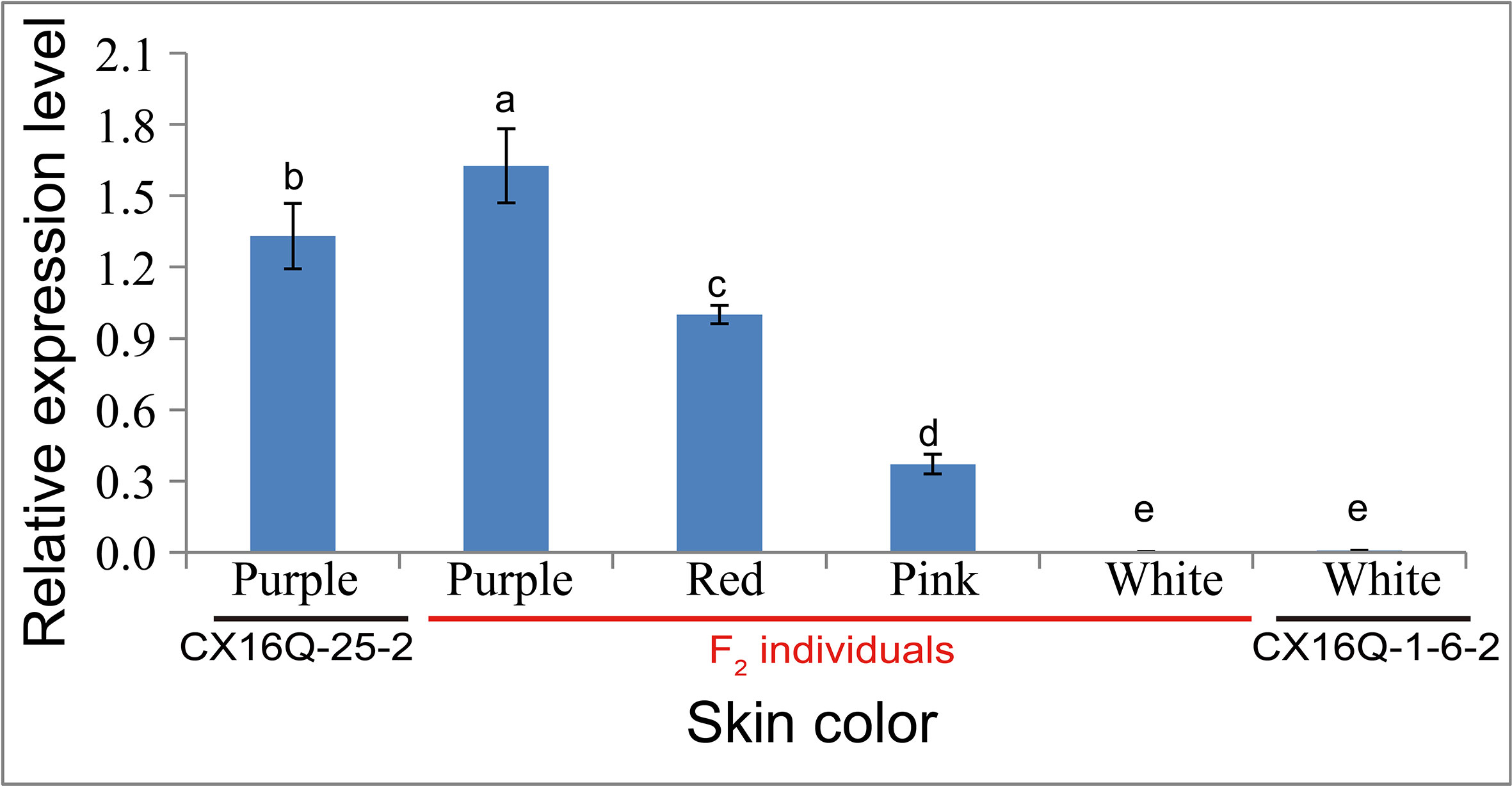
Figure 4 Relative expression of RsMYB1.1 in the skin of taproots with different skin colors detected by quantitative real-time PCR analysis. (a-e) indicate that the expression values were significantly different.
MYB1 Genes Were Expanded in the Radish Genome
Four and three genes were identified by homology searches in “XYB36-2” (Zhang et al., 2015b) and “WK10039” (Jeong et al., 2016) protein datasets, respectively, with the deduced protein sequence of RsMYB1.1 from CX16Q-25-2 as a reference. All of these homologous genes were best matched to Arabidopsis PRODUCTION OF ANTHOCYANIN PIGMENT1 (AtPAP1, AT1G56650) in The Arabidopsis Information Resource (TAIR) database (https://www.arabidopsis.org/). In a phylogenetic tree of anthocyanin-regulated R2R3-MYB proteins from Arabidopsis and radish, four “XYB36-2” and three “WK10039” homologous genes and the published radish gene MYB1 all fell in the AtPAP1 subgroup (Figure 5A). Sequence alignments showed 96.79%, 99.60%, and 100% identity at the amino acid level between Rsa10034073 and Rs388430, Rsa10008423 and Rs094840, and Rsa10042324 and Rs278810, respectively (Supplementary Figure 2), indicating that these three gene pairs were most likely the same gene in different radish cultivars. In addition, Rsa10033919 was identified in the “XYB36-2” radish genome but not in the “WK10039” genome (Figures 5B, C). This gene falls within the same subclade as the previously reported MYB1 genes from the “Xinlimei” (green skin with red flesh) and “Bordeaux” (red skin with red flesh) radish cultivars and red-skinned “Lian Yan No. 1” radish progeny, indicating that Rsa10033919 is a homolog of RsMYB1. Therefore, according to their chromosome locations in the “XYB36-2” genome, Rsa10008423 (Rs094840), Rsa10042324 (Rs278810), Rsa10033919, and Rsa10034073 (Rs388430) were designated RsMYB1.1, RsMYB1.2, RsMYB1.3, and RsMYB1.4, respectively.
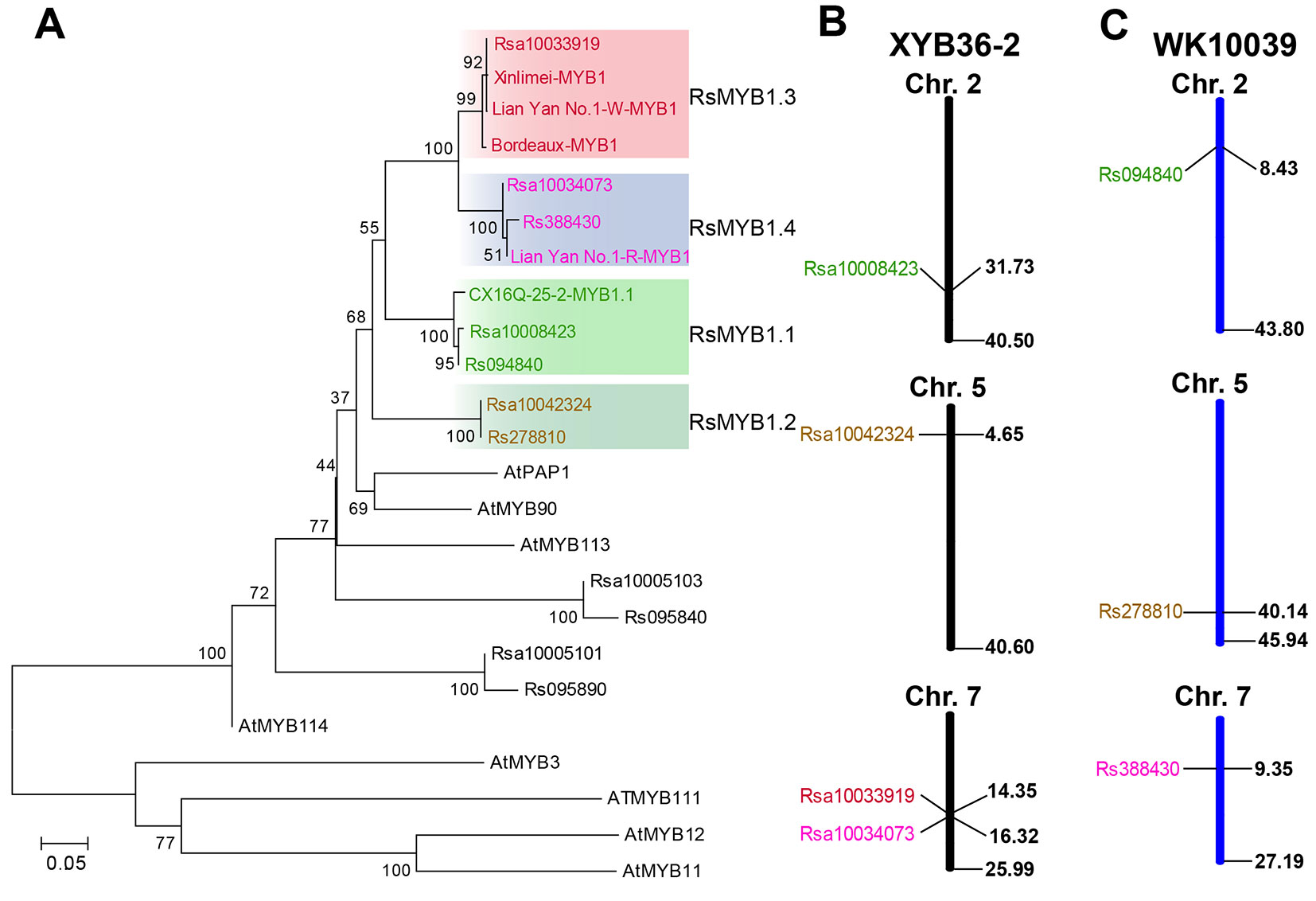
Figure 5 Phylogenetic tree of RsMYB1.1 homologous genes in radish and anthocyanin R2R3-MYB regulators in Arabidopsis based on their amino acid sequences. (A) The chromosome locations of these genes in the “XYB36-2” (B) and “WK10039” (C) radish genomes. GenBank accession numbers are as follows: AtMYB11 (NP_191820), AtMYB12 (NP_182268.1), AtMYB75 (NP_176057.1), AtMYB90 (NM_105310), AtMYB111 (NP_199744), AtMYB114 (NP_176812), and Bordeaux-MYB1 (AKM95888.1).
Discussion
Fleshy taproot skin color is an important visual and nutritional quality trait of radish that influences radish production and consumption. The identification and cloning of major genes responsible for the skin color of radish will promote effectiveness in the breeding process. To date, possible inheritance patterns of radish taproot skin color have been reported by numerous studies (Yarnell, 1956; He et al., 1997). In different hybrid combinations, green root skin might be controlled by one or two pairs of independent genes or two pairs of linked genes (He et al., 1997). Previous research also indicated that a single locus or multiple loci may control red root skin, and red is dominant over white (Yarnell, 1956; He et al., 1997; Yi et al., 2018). In the present study, a cross between purple (CX16Q-25-2) and white (CX16Q-1-6-2) inbred lines produced F2 plants with complex skin color segregation, which was consistent with a previous report that the genetics of purple coloration are complex and involve multiple factors (He et al., 1997). However, because the segregation of pigmented and white roots was consistent with a 3:1 Mendelian segregation ratio, pigment presence was controlled by a single dominant gene. This hypothesis was also verified by subsequent QTL-seq analysis results with two extreme pools (purple- and white-skinned pools) in which only one QTL region was detected. The results are consistent with the finding of a recent study that a single genetic factor distinguishes between red- and white-skinned progenies derived from the Chinese commercial F1 cultivar “Lian Yan No. 1” (red skin with white flesh) (Yi et al., 2018). Therefore, our present study mainly focused on the identification of candidate genes located in this QTL region.
We used QTL-seq combined with classical methods to rapidly identify a major QTL, Rsps, which was localized to a 238.51-kb physical interval on chromosome 2 (Figure 3). Only 18 genes are located in this interval, based on the “XYB36-2” reference genome (Table 2). The present study indicated that combined QTL-seq, traditional linkage analysis, and high-quality whole-genome re-sequencing can significantly promote progress in gene mapping and subsequent cloning.
The taproot skins of some radish cultivars are purple and red because of the accumulation of a large concentration of anthocyanins (Park et al., 2011; Yi et al., 2018). The biosynthesis of anthocyanins is regulated by some transcription factors, especially members of the MYB gene family. Among the 18 genes located in the 238.51-kb candidate region of Rsps (Table 2), the expression pattern (Figures 3A and 4), gene structure (Figure 3B), segregation analysis (Figures 3C, D), and phylogenetic analysis (Figure 5A) of RsaMYB1.1 suggested that it could be a candidate gene for Rsps. RsaMYB1.1 shared 77.51% amino acid sequence identity with Arabidopsis AtPAP1 and was expressed in purple-skinned radish plants but not in white-skinned ones. There are multiple mutations in RsaMYB1.1 between the two parental lines, and the marker designed according to a variable locus separated the purple-skinned and white-skinned radishes correctly (Figures 3C, D). Therefore, it is reasonable to postulate that RsaMYB1.1 is the candidate gene for purple taproot skin in radish. However, further functional research is needed to validate the present findings. The MBW complex composed of R2R3-MYB, basic helix–loop–helix, and WD40 is highly conserved in plants involved in the regulation of anthocyanin biosynthesis (Koes et al., 2005). Among MBW complex components, R2R3-MYB plays a more important role because it is thought to directly bind to the promoter sequences of anthocyanin biosynthesis genes and activate their expression (Hichri et al., 2011). Numerous studies have reported that MYB homologs are involved in regulating the biosynthesis of anthocyanin in many plant species, including radish (Koes et al., 2005). A subgroup of Arabidopsis R2R3-MYBs including AtPAP1 (AtMYB75), AtPAP2 (AtMYB90), AtMYB114, and AtMYB113 demonstrates a high degree of amino acid conservation and is involved in anthocyanin regulation (Allan et al., 2008). MdMYBA, MdMYB1, and MdMYB10 are reportedly responsible for apple anthocyanin accumulation (Takos et al., 2006; Ban et al., 2007; Espley et al., 2007). BoMYB2 is involved in controlling anthocyanin biosynthesis in purple cauliflower (Chiu and Li, 2012). SmMYB1 is reported to participate in the regulation of anthocyanin biosynthesis in the peel of eggplant (Zhang et al., 2014). In radish, RsMYB1 is reported to function in the activation of anthocyanin biosynthesis (Lim et al., 2016; Yi et al., 2018). Therefore, it is not surprising that an AtPAP1 homologous gene was identified as the candidate gene responsible for the purple skin of CX16Q-25-2 radish.
A previous study suggested that the taproot color of radish was determined by the ratio of pelargonidin and cyanidin, which are responsible for the red and purple coloration, respectively, and the H locus was proposed to be involved in the biosynthesis of pigments from pelargonidin to cyanidin (Yi et al., 2018). However, our present research did not detect a QTL region responsible for this trait. The most likely reason for this lack of detection was that the purple or red skin trait in the present F2 population was easily affected by environmental conditions. The F2 population generated by red- and purple-skinned radish inbred lines may be ideal for mapping the H locus.
Four RsMYB1.1 homologs were identified in the “XYB36-2” radish genome and named RsMYB1.1, RsMYB1.2, RsMYB1.3, and RsMYB1.4 (Figure 5B). RsMYB1.1 was first reported in the present study and may function in the regulation of anthocyanin biosynthesis in radish taproot skin. The function of RsMYB1.2 is still unknown and requires further research. In the phylogenetic analysis, the RsMYB1.3 subclade included the previously reported RsMYB1 gene from the “Bordeaux” homozygous F3 progeny (Lim et al., 2016) and “Xinlimei” (Liu et al., 2019), both of which have red flesh. These results indicate that RsMYB1.3 plays an important role in the regulation of anthocyanin biosynthesis in radish taproot flesh. More recently, another RsMYB1gene was identified in the red-skinned radish cultivar “Lianyan No. 1” by map-based cloning (Yi et al., 2018). This gene falls in the RsMYB1.4 subclade with Rsa10034073 from “XYB36-2” and Rs388430 from “WK10039”. RsMYB1.4 and RsMYB1.1 are located on different chromosomes and fall into different subclades, indicating that they are two different genes, but they might have the same function in the regulation of anthocyanin biosynthesis in radish taproot skin. The identification of different genes previously in “Lianyan No. 1” and in CX16Q-25-2 in the present study may be attributable to the fact that different cultivars were used in mapping candidate genes. Further research is needed to confirm the role of these genes, which would enrich the research on anthocyanin biosynthesis regulation in radish.
Data Availability
Sequences data generated in this study are available on the Sequence Read Archive http://www.ncbi.nlm.nih.gov/Traces/sra/, at accession PRJNA563272.
Author Contributions
XXL conceived and supervised the study. TL, JW, and CW analyzed the data and drafted the manuscript. TL, XXL, HW, and JS performed the experiments. JW, TL, and XML helped with mapping population construction and sample collection. XXL, YZ, XZ, and JW revised the manuscript. All authors have read and approved the manuscript.
Funding
This work was supported by a grant from the National Key Research and Development Program of China (2016YFD0100204-2), the National Natural Science Foundation of China (31801858), the China Postdoctoral Science Foundation Funded Project (2017M620971), and the Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-XTCX2016016-4-4, CAAS-XTCX2016001-5-2, and CAAS-XTCX2016017).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00808/full#supplementary-material
Supplementary Table 1 | The primers used in the present study.
Supplementary Table 2 | InDel primers developed in the preliminary mapping regions with potential variation of >4 bp.
Supplementary Figure 1 | Alignment of radish Rsa10008423 with the Arabidopsis PAP1 (MYB75, AT1G56650) gene coding sequence (a) and their deduced amino acid sequences (b).
Supplementary Figure 2 | Alignment of radish Rsa10034073 and Rs388430 (a), Rsa10008423 and Rs094840 (b), and Rsa10042324 and Rs278810 (c) based on their deduced amino acid sequences.
References
Allan, A. C., Hellens, R. P., Laing, W. A. (2008). MYB transcription factors that colour our fruit. Trends Plant Sci. 13, 99–102. doi: 10.1016/j.tplants.2007.11.012
Ban, Y., Honda, C., Hatsuyama, Y., Igarashi, M., Bessho, H., Moriguchi, T. (2007). Isolation and functional analysis of a MYB transcription factor gene that is a key regulator for the development of red coloration in apple skin. Plant Cell Physiol. 48 (7), 958–970. doi: 10.1093/pcp/pcm066
Borevitz, J. O., Xia, Y., Blount, J., Dixon, R. A., Lamb, C. (2000). Activation tagging identifies a conserved MYB regulator of phenylpropanoid biosynthesis. Plant Cell 12, 2383–2393. doi: 10.1105/tpc.12.12.2383
Cao, X., Qiu, Z., Wang, X., Giang, T., Liu, X., Wang, J., et al. (2017). A putative R3 MYB repressor is the candidate gene underlying atroviolacium, a locus for anthocyanin pigmentation in tomato fruit. J. Exp. Bot. 68, 5745–5758. doi: 10.1093/jxb/erx382
Chen, F. B., Xing, C. Y., Huo, S. P., Cao, C. L., Yao, Q. L., Fang, P. (2016). Red pigment content and expression of genes related to anthocyanin biosynthesis inradishes (Raphanus sativus L.) with different colored flesh. J. Agr. Sci. 8, 126–135. doi: 10.5539/jas.v8n8p126
Chiu, L. W., Li, L. (2012). Characterization of the regulatory network of BoMYB2 in controlling anthocyanin biosynthesis in purple cauliflower. Planta 236, 1153–1164. doi: 10.1007/s00425-012-1665-3
Chu, H., Jeong, J. C., Kim, W. J., Chung, D. M., Jeon, H. K., Ahn, Y. O., et al. (2013). Expression of the sweet potato R2R3-type IbMYB1a gene induces anthocyanin accumulation in Arabidopsis. Physiol. Plantarum. 148 (2), 189–199. doi: 10.1111/j.1399-3054.2012.01706.x
Doyle, J., Doyle, J. (1987). Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem. Bull. 19, 11–15.
Espley, R. V., Hellens, R. P., Putterill, J., Stevenson, D. E., Kutty-Amma, S., Allan, A. C. (2007). Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10 . Plant J. 49 (3), 414–427. doi: 10.1111/j.1365-313X.2006.02964.x
Gordon, A. (2009). Fastx-toolkit. http://hannonlab.cshl.edu/fastx_toolkit/.
Hao, N., Du, Y., Li, H., Wang, C., Wang, C., Gong, S., et al. (2018). CsMYB36 is involved in the formation of yellow green peel in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 131, 1659–1669. doi: 10.1007/s00122-018-3105-7
He, Q., Zhao, S., Shi, H., An, Z., Lang, F., Wang, S. (1997). Preliminary study on skin color inheritance of Chinese radish. J. Shandong Agric. Sci. 2, 4–9. doi: 10.14083/j.issn.1001-4942.1997.02.001
Hichri, I., Barrieu, F., Bogs, J., Kappel, C., Delrot, S., Lauvergeat, V. (2011). Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 62, 2465–2483. doi: 10.1093/jxb/erq442
Hiroki, T., Akira, A., Kentaro, Y., Shunichi, K., Satoshi, N., Chikako, M., et al. (2013). QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74, 174–183. doi: 10.1111/tpj.12105
Jeong, Y. M., Kim, N., Ahn, B. O., Oh, M., Chung, W. H., Chung, H., et al. (2016). Elucidating the triplicated ancestral genome structure of radish based on chromosome-level comparison with the Brassica genomes. Theor. Appl. Genet. 129, 1357–1372. doi: 10.1007/s00122-016-2708-0
Johnson, C. S., Kolevski, B., Smyth, D. R. (2002). Transparent testa GLABRA2, a trichome and seed coat development gene of Arabidopsis, encodes a WRKY transcription factor. Plant Cell 14, 1359–1375. doi: 10.1105/tpc.001404
Koes, R., Verweij, W., Quattrocchio, F. (2005). Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 10, 236–242. doi: 10.1016/j.tplants.2005.03.002
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Zhu, L., Yuan, G., Heng, S., Yi, B., Ma, C., et al. (2016). Fine mapping and candidate gene analysis of an anthocyanin-rich gene, BnaA.PL1, conferring purple leaves in Brassica napus L. Mol. Genet. Genomics 291, 1523–1534. doi: 10.1007/s00438-016-1199-7
Lim, S. H., Kim, D. H., Kim, J. K., Lee, J. Y., Ha, S. H. (2017). A radish basic helix–loop–helix transcription factor, RsTT8 acts a positive regulator for anthocyanin biosynthesis. Front. Plant Sci. 8, 1917. doi: 10.3389/fpls.2017.01917
Lim, S. H., Song, J. H., Kim, D. H., Kim, J. K., Lee, J. Y., Kim, Y. M., et al. (2016). Activation of anthocyanin biosynthesis by expression of the radish R2R3-MYB transcription factor gene RsMYB1. Plant Cell Rep. 35, 641–653. doi: 10.1007/s00299-015-1909-3
Liu, H., Jiao, J., Liang, X., Liu, J., Meng, H., Chen, S., et al. (2016). Map-based cloning, identification and characterization of the w gene controlling white immature fruit color in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 129, 1247–1256. doi: 10.1007/s00122-016-2700-8
Liu, T., Zhang, X., Yang, H., Agerbirk, N., Yang, Q., Wang, H., et al. (2016). Aromatic glucosinolate biosynthesis pathway in Barbarea vulgaris and its response to Plutella xylostella infestation. Front. Plant Sci. 7, 83. doi: 10.3389/fpls.2016.00083
Liu, T., Zhang, Y., Zhang, X., Sun, Y., Wang, H., Song, J., et al. (2019). Transcriptome analyses reveal key genes involved in skin color changes of ‘Xinlimei’ radish taproot. Plant Physiol. Biochem. 139, 528–539. doi: 10.1016/j.plaphy.2019.04.006
Liu, X., Gao, B., Han, F., Fang, Z., Yang, L., Zhuang, M., et al. (2017). Genetics and fine mapping of a purple leaf gene, BoPr, in ornamental kale (Brassica oleracea L. var. acephala). BMC Genomics 18, 230. doi: 10.1186/s12864-017-3613-x
Matsui, K., Umemura, Y., Ohme-Takagi, M. (2008). AtMYBL2, a protein with a single MYB domain, acts as a negative regulator of anthocyanin biosynthesis in Arabidopsis. Plant J. 55, 954–967. doi: 10.1111/j.1365-313X.2008.03565.x
Muleke, E. M., Fan, L., Wang, Y., Xu, L., Zhu, X., Zhang, W., et al. (2017). Coordinated regulation of anthocyanin biosynthesis genes confers varied phenotypic and spatial–temporal anthocyanin accumulation in radish (Raphanus sativus L.). Front. Plant Sci. 8: 1243 doi: 10.3389/fpls.2017.01243
Park, N. I., Xu, H., Li, X., Jang, I. H., Park, S., Ahn, G. H., et al. (2011). Anthocyanin accumulation and expression of anthocyanin biosynthetic genes in radish (Raphanus sativus). J. Agric. Food Chem. 59, 6034–6039. doi: 10.1021/jf200824c
Rubin, G., Tohge, T., Matsuda, F., Saito, K., Scheible, W. R. (2009). Members of the LBD family of transcription factors repress anthocyanin synthesis and affect additional nitrogen responses in Arabidopsis. Plant Cell 21, 3567–3584. doi: 10.1105/tpc.109.067041
Schuler, G. D. (1997). Sequence mapping by electronic PCR. Genome Res. 7, 541. doi: 10.1101/gr.7.5.541
Stracke, R., Jahns, O., Keck, M., Tohge, T., Niehaus, K., Fernie, A. R., et al. (2010). Analysis of Production of flavonol glycosides-dependent flavonol glycoside accumulation in Arabidopsis thaliana plants reveals MYB11-, MYB12- and MYB111-independent flavonol glycoside accumulation. New Phytol. 188, 985–1000. doi: 10.1111/j.1469-8137.2010.03421.x
Sun, Y., Wang, J., Qiu, Y., Liu, T., Song, J., Li, X. (2018). Identification of ‘Xinlimei’ radish candidate genes associated with anthocyanin biosynthesis based on a transcriptome analysis. Gene 657, 81–91. doi: 10.1016/j.gene.2018.03.001
Takos, A. M., Jaffé, F. W., Jacob, S. R., Bogs, J., Robinson, S. P., Walker, A. R. (2006). Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 142 (3), 1216–1232. doi: 10.1104/pp.106.088104
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Triinu, K., Maido, R. (2007). Enhancements and modifications of primer design program Primer3 . Bioinformatics 23, 1289–1291. doi: 10.1093/bioinformatics/btm091
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115. doi: 10.1093/nar/gks596
Van Ooijen, J. W., Boer, M. P., Jansen, R. C., Maliepaard, C. (2002). MapQTl4.0, software for the calculation of QTL positions on genetic maps. Plant Res. Int. Wagenigen.
Yarnell, S. H. (1956). Cytogenetics of the vegetable crops. II. Crucifers. Bot. Rev. 22, 81–166. doi: 10.1007/BF02872468
Yi, G., Kim, J. S., Park, J. E., Shin, H., Yu, S. H., Park, S., et al. (2018). MYB1 transcription factor is a candidate responsible for red root skin in radish (Raphanus sativus L.). PLoS One 13, e0204241. doi: 10.1371/journal.pone.0204241
Zhang, X., Liu, T., Wei, X., Qiu, Y., Song, J., Wang, H., et al. (2015a). Expression patterns, molecular markers and genetic diversity of insect-susceptible and resistant Barbarea genotypes by comparative transcriptome analysis. BMC Genomics 16, 486. doi: 10.1186/s12864-015-1609-y
Zhang, X., Yue, Z., Mei, S., Qiu, Y., Yang, X., Chen, X., et al. (2015b). A de novo genome of a Chinese radish cultivar. Hortic. Plant J. 1, 155–164. doi: 10.16420/j.issn.2095-9885.2016-0028
Zhang, Y., Hu, Z., Chu, G., Huang, C., Tian, S., Zhao, Z., et al. (2014). Anthocyanin accumulation and molecular analysis of anthocyanin biosynthesis-associated genes in eggplant (Solanum melongena L.). J. Agric. Food Chem. 62, 2906–2912. doi: 10.1021/jf404574c
Zhang, Y., Hu, Z., Zhu, M., Zhu, Z., Wang, Z., Tian, S., et al. (2015). Anthocyanin accumulation and molecular analysis of correlated genes in purple kohlrabi (Brassica oleracea var. gongylodes L.). J. Agric. Food Chem. 63, 4160–4169. doi: 10.1021/acs.jafc.5b00473
Keywords: radish, purple taproot skin, inheritance, QTL-seq, candidate gene
Citation: Liu T, Wang J, Wu C, Zhang Y, Zhang X, Li X, Wang H, Song J and Li X (2019) Combined QTL-Seq and Traditional Linkage Analysis to Identify Candidate Genes for Purple Skin of Radish Fleshy Taproots. Front. Genet. 10:808. doi: 10.3389/fgene.2019.00808
Received: 18 March 2019; Accepted: 02 August 2019;
Published: 20 September 2019.
Edited by:
Reyazul Rouf Mir, Sher-e-Kashmir University of Agricultural Sciences and Technology of Kashmir, IndiaReviewed by:
Bilal A. Padder, Sher-e-Kashmir University of Agricultural Sciences and Technology, IndiaZhisheng Xu, Nanjing Agricultural University, China
Ru Gang Yu, Huaibei Normal University, China
Copyright © 2019 Liu, Wang, Wu, Zhang, Zhang, Li, Wang, Song and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youjun Zhang, emhhbmd5b3VqdW5AY2Fhcy5jbg==; Xixiang Li, bGl4aXhpYW5nQGNhYXMuY24=
†These authors have contributed equally to this work.