 Federica Natali
Federica Natali Giulia Rancati
Giulia Rancati
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet. , 06 August 2019
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.00713
This article is part of the Research Topic Model Organisms: A Precious Resource for Understanding of the Molecular Mechanisms Underlying Human Physiology and Disease View all 14 articles
The mutator phenotype hypothesis was postulated almost 40 years ago to reconcile the observation that while cancer cells display widespread mutational burden, acquisition of mutations in non-transformed cells is a rare event. Moreover, it also suggested that cancer evolution could be fostered by increased genome instability. Given the evolutionary conservation throughout the tree of life and the genetic tractability of model organisms, yeast and bacterial species pioneered studies to dissect the functions of genes required for genome maintenance (caretaker genes) or for cell growth control (gatekeeper genes). In this review, we first provide an overview of what we learned from model organisms about the roles of these genes and the genome instability that arises as a consequence of their dysregulation. We then discuss our current understanding of how mutator phenotypes shape the evolution of bacteria and yeast species. We end by bringing clinical evidence that lessons learned from single-cell organisms can be applied to tumor evolution.
The mutator phenotype was proposed by Loeb almost 40 years ago to reconcile the observations that while cancer cells display widespread DNA and chromosomal changes (Alexandrov et al., 2013; Loeb, 2016), the rate of spontaneous mutation in somatic cells is low (Milholland et al., 2017). This hypothesis also suggested that an increased genome instability favors cancer evolution. Indeed, by reshuffling cancer cell genomes, the mutator phenotype generates cell-to-cell heterogeneity, which is the presence of cells with different genotypes and phenotypes within a population (Loeb, 2016). Since clonal competition within a tumor mass favors the expansion of fitter cells, the presence of cells with different phenotypes within a cancer sample increases the likelihood that some of them might be more aggressive and ultimately leads to poor patient survival (Greaves, 2015; McGranahan and Swanton, 2017). Thanks to the ease with which they can be grown in the laboratory and the wealth of genetic resources, in the last decades, model organisms have been extensively used to identify and dissect the function of genes required for genome stability. For instance, some of the available yeast genome-wide libraries include the systematic knockout collection (Winzeler et al., 1999; Giaever et al., 2002), the green fluorescent protein-tagged collection (Huh et al., 2003), and loss-of-function alleles for essential genes (Ben‐Aroya et al., 2008; Breslow et al., 2008; Li et al., 2011). Though progress in our ability to perform high-throughput screens and to manipulate mammalian cells has greatly improved (Behan et al., 2019), it is still difficult to envision a close future in which large-scale screens performed in model organisms could be easily and cost-effectively reproduced in mammalian cells. A more effective way is to directly translate findings of interesting candidates from model organisms to mammalian cells. For instance, a recent screen in budding yeast showed that, contrary to what was previously thought, heterozygous mutations in gatekeeper genes can cause genome instability (Coelho et al., 2019). Introduction of these mutations in human orthologs triggered genome instability also in human cells (Coelho et al., 2019). Interestingly, only some of the identified genes have been previously found mutated in cancer cells, supporting the idea that findings in budding yeast hold great potential for cancer cell biology.
Studies on model organisms have been instrumental to understand mechanisms generating cell-to-cell heterogeneity and its consequence in evolutionary outcomes. While an important route to heterogeneity in yeast is sexual outbreeding (Long et al., 2015; Vazquez‐Garcia et al., 2017; Li et al., 2019), this point will not be discussed as evolution of cancer cells is comparable to evolution of asexually reproducing organisms. A route to heterogeneity common between single-cell model organisms and cancer cells involves the dysregulation of the genome integrity network. Initial identification of players of this network in yeast and bacteria was soon followed by the realization that cancer cells carry recurrent mutations in their respective human orthologs and provided ground for key findings. Some popular examples include the identification in cancer cells of cohesion mutations (Hill et al., 2016), of the links between microsatellite instability and the mismatch repair pathway (Kolodner and Marsischky, 1999), and of predicting novel therapeutic targets based on synthetic lethality (Ashworth et al., 2011; Beijersbergen et al., 2017). Though most of the core machinery required for genome maintenance and replication is conserved throughout evolution, there are important exceptions. The most notable differences include the lack of a well-defined nucleotide sequence of the origin of replication in human cells (Gerhardt et al., 2006) and the presence of the human DNA replication inhibitor, geminin (Symeonidou et al., 2012). Yeast cells also lack obvious orthologs of key human DNA repair enzymes, such as breast related cancer antigens 1/2 (BRCA1/2) or poly (ADP-ribose) polymerase (PARP) (Morales et al., 2014; Walsh, 2015). Given the therapeutic importance of some of these proteins, “humanized” yeast strains carrying cancer-associated mutations in BRCA genes have been generated (Guaragnella et al., 2014; Maresca et al., 2018) and used to screen for novel therapeutics.
Due to space constraints, we briefly summarize the role of key DNA repair genes and DNA replication genes. For a detailed overview of the field, please refer to the reviews by Kunkel and Erie (2015), Lujan et al. (2016), and Liu et al. (2017). For a summary of the genes discussed in this paragraph, please refer to Table 1.
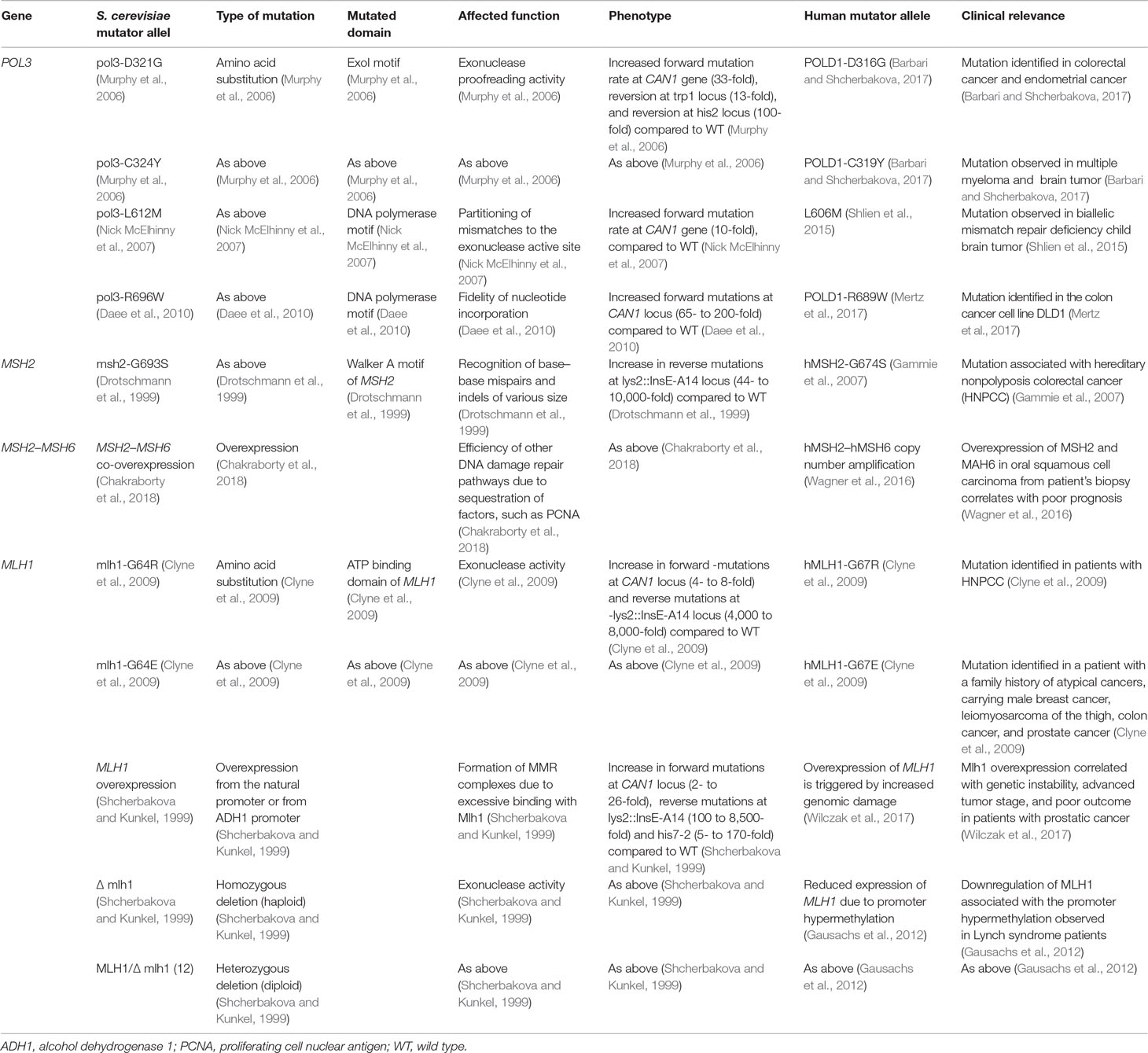
Table 1 The table reports a list of mutations discussed in the section “Cells losing balance” and details phenotypic consequences arising from such mutations.
DNA replication of the lagging and the leading strands depends on the activity of two high-fidelity DNA polymerases, DNA polymerases δ (Polδ) and ε (Polε), respectively (Maslowska et al., 2018). The faithfulness of the process relies on the accuracy of nucleotide incorporation coupled with the 3′–5′ exonuclease proofreading activity (Pavlov and Shcherbakova, 2010). Indeed, biochemical assays showed that while purified human Polδ catalyzes one base substitution error every 22,000 nucleotides, the error rate decreased at least 10-fold in presence of a functional proofreading domain (Schmitt et al., 2009). Mouse models lacking a functional Polδ proofreading activity develop spontaneous cancers at high frequency (Goldsby et al., 2002), confirming in vivo the importance of the domain for genome stability and cancer formation. Moreover, germline mutations in the proofreading domain of Polδ and Polε have been identified in a number of families with increased susceptibility to colorectal adenomas and carcinomas (Palles et al., 2013; Heitzer and Tomlinson, 2014). Accordingly with an inability to repair mispaired bases inserted during DNA replication, tumors from affected patients increased the rate of base substitution mutations while maintaining microsatellite stability (Palles et al., 2013). Mutations in the proofreading activity of S. cerevisiae that mimic mutations in tumors resulted in a mutator phenotype and elevated spontaneous base substitution rates (Murphy et al., 2006; Nick McElhinny et al., 2007).
However, Polδ and Polε mutations outside of the proofreading domain were also mapped in sporadic cancers and cancer cell lines (Briggs and Tomlinson, 2013). Introduction of one of such variant, pol3-R696W (human POLD1-R689W), in heterozygosity in S. cerevisiae increased 30-fold the rate of forward mutations. At a biochemical level, pol3-R696W was shown to be an error-prone DNA polymerase with an increased nucleotide misinsertion rate and a specific mutational pattern (Daee et al., 2010) that is consistent with the one observed in colorectal cancer lines bearing the POLD1-R689W variant (Mertz et al., 2017). Collectively, these observations suggest that mutations affecting both the polymerase and the 3’–5’ exonuclease domains confer a mutator phenotype that can be translated from bacteria and yeast to human cells.
The mismatch repair (MMR) pathway is a conserved surveillance system that recognizes and resolves misincorporated bases (Fishel, 2015). In prokaryotes, the MMR machinery is relatively simple and involves proteins detecting DNA mismatches (MutS), processing the damage (MutH), and bridging these two proteins together (MutL) (Fukui, 2010). While mutations in mutS and mutL human orthologs were found in the germline of patients with hereditary nonpolyposis colorectal cancer/Lynch syndrome (HNPCC/LS), and other cancer-predisposing Lynch variant syndromes (Lynch and de la Chapelle, 2003; Morales-Burgos et al., 2008; Wimmer and Kratz, 2010), they also somatically occur in up to 15% of sporadic colorectal, gastric, or endometrial carcinomas (Boland et al., 1998). Experimentally, engineered mice lacking functional MMR proteins are genomically unstable and predisposed to spontaneous cancer onset (Lee et al., 2016). Modeling cancer-related MMR mutations in yeast has been instrumental to dissect the consequences on cellular physiology of a non-functional mismatch repair pathway. For instance, mimicking MMR mutations found in HNPCC (Kurzawski et al., 2002) in yeast cells caused an increase in the rate of spontaneous (Drotschmann et al., 1999) and forward mutations (Clyne et al., 2009). Moreover, consistent with the observation that human cancer cell lines with dysregulation in the expression of the MMR proteins are genomically unstable (Ryan et al., 2017; Wilczak et al., 2017), tinkering with the expression levels of the yeast orthologs in S. cerevisiae resulted in significant increase of repeats’ instability and forward mutations (Shcherbakova and Kunkel, 1999; Chakraborty et al., 2018).
While maintenance of genome stability is key for reproductive success of prokaryotes and eukaryotes, laboratory and clinical evidence suggests that tinkering with such pathways favors cellular adaptation and population expansion during exposure to challenges. Below we discuss some such evidence.
Several clinical isolates and natural populations of pathogenic bacteria and fungi were reported to have an enhanced mutation rate mostly mapped to defects in the methyl-directed mismatch repair system (Oliver et al., 2000; Bjorkholm et al., 2001; Chopra et al., 2003; Wang et al., 2013; Healey et al., 2016), suggesting that a mutator phenotype could be selected in fluctuating or hostile environments, such as the presence of drugs or adaptation to new ecological niches. Experimental studies have supported this idea. For instance, MSH2-defective Cryptococcus neoformans, Candida glabrata, and Cryptococcus deuterogattii strains increased mutation rates and underwent rapid adaptation to antifungal drugs (Healey et al., 2016; Billmyre et al., 2017; Boyce et al., 2017). Similarly, hypermutator Staphylococcus aureus bacteria strains impaired in the DNA mismatch repair pathway developed vancomycin resistance more rapidly than control strains (Schaaff et al., 2002). Moreover, mutant strains defective in DNA repair and characterized by increased mutation rates outcompeted wild-type strains and were fixed in 6 out of 12 E. coli populations in the Long-Term Evolution Experiment (Tenaillon et al., 2016). A link between increased mutation rates and adaptability comes also from observations that impairment in the activity of DNA repair pathways was often found to co-segregate with mutations conferring antibiotic resistance (Gould et al., 2007). At the theoretical level, a mutator phenotype potentially endows populations with a higher adaptability by generating cell-to-cell heterogeneity and a pool of allelic variants on which selection could select upon (Figure 1). Accordingly, mutator msh2Δ S. cerevisiae strains acquired resistance to the toxic arginine analog canavanine up to 20-fold faster than wild type (Bowers et al., 1999). Adaptive mutations encompassed single-nucleotide misincorporations and deletions of the canavanine influx pump gene (Sokolsky and Alani, 2000). Since such mutations are edited by the MMR pathway, these observations suggest that crippling with DNA replication or repair pathways could generate beneficial allelic variants. However, mutators with no direct effect on cellular fitness in asexually evolving unicellular organisms could sweep in a population if they are linked to beneficial mutations, a process called mutator hitchhiking. A large body of evidence coming from theoretical and experimental studies in both bacteria and yeast showed that the probability of hitchhiking has been linked to the population size and the fitness effects of beneficial mutations in complex-to-predict scenarios (Taddei et al., 1997; Notley-McRobb et al., 2002; Shaver et al., 2002; Wahl et al., 2002; Andre and Godelle, 2006; Thompson et al., 2006; Gerrish et al., 2007; Gentile et al., 2011; Raynes et al., 2014; Bui et al., 2015; Good and Desai, 2016). For instance, large population sizes are known to increase clonal interference, which has been shown to either delay or enhance fixation of mutators in different conditions (Raynes et al., 2014; Good and Desai, 2016). However, in populations at local fitness minima or experiencing fluctuating environments, mutators can hitchhike to a higher frequency when linked to strong beneficial mutations. It comes, therefore, as no surprise that mutator multidrug-resistant bacterial strains are a common feature of chronic infections, like cystic fibrosis or urinary tract infections (Oliver et al., 2000; Labat et al., 2005; Feliziani et al., 2010; Macia et al., 2014), where bacterial strains endure pulses of antibiotic treatments. In the laboratory, coupling of cycles of antibiotic or carbon source selection with mutagenesis increased the percentage of strains carrying mutations in mismatch repair genes up to 50–100% (Mao et al., 1997), further supporting the notion that fluctuating environments positively select for mutator strains. However, since the vast majority of mutations are detrimental, genome instability in populations at their fitness peaks comes with a cost (Figure 1). Indeed, a general role for asexual pathogenic mutators in the emergence of drug resistance is still being debated, possibly because mutators are selected against once beneficial mutations have been acquired. For instance, mutS mutant S. aureus laboratory strains characterized by a 78-fold increased mutation frequency did not increase the rate of adaptation to vancomycin (O’Neill and Chopra, 2002). Also, MSH2 mutations in C. glabrata clades were shown to be present as polymorphisms within different natural populations that were equally sensitive or resistant to antifungal drugs (Carrete et al., 2018). At the same time, C. glabrata clinical isolates carrying MSH2 mutations did not show increased resistance to azole or echinocandin (Singh et al., 2018). Lastly, an in vivo model for chronic bone infection in the rat showed that MSH2 mutant S. aureus strains carried a decreased fitness and did not acquire antibiotic resistance (Daurel et al., 2007). The reported discrepancies on the effect of mutators on evolving populations of asexual singe-cell organisms could be linked to clonal interference, mutation effects, or population fitness or could be the result of the negative selection that mutators face once beneficial mutations have been acquired. Indeed, laboratory evidence showed that while mutator strains were initially selected for, both bacteria and yeast mutants experienced reduced transmission and recolonization abilities as well as rapid fitness decline upon prolonged passaging (Giraud et al., 2001; Trindade et al., 2010). These observations suggest that mutator strains could be counterselected once adaptation to the novel environment is achieved. Accordingly, mathematical modeling of E. coli population dynamics showed a sharp decline in the frequency of mutator strains once adaptation was achieved (Taddei et al., 1997). At a molecular level, experimental evolution correlated fitness drop of evolving strains with acquisition of detrimental mutations in genes required for optimal fitness (Andersson and Hughes, 1996; Funchain et al., 2000), suggesting that the mutational load of mutator strains could become a selective pressure itself. Accordingly, decreased cellular fitness after prolonged passaging of msh2Δ mutator S. cerevisiae strains in non-challenging environments was followed by restoration of genome stability by increasing the buffering ability of heat shock proteins (McDonald et al., 2012). Alternatively, restoration of genome stability arose either by acquisition of antimutator suppressor alleles or by replacing the mutator alleles with functional ones through horizontal gene transfer (Denamur et al., 2000; Wielgoss et al., 2013). Taken together, all of this evidence suggests that while the mutator phenotype is initially selected for during adaptation, it could be selected against once adaptive mutations are fixed.
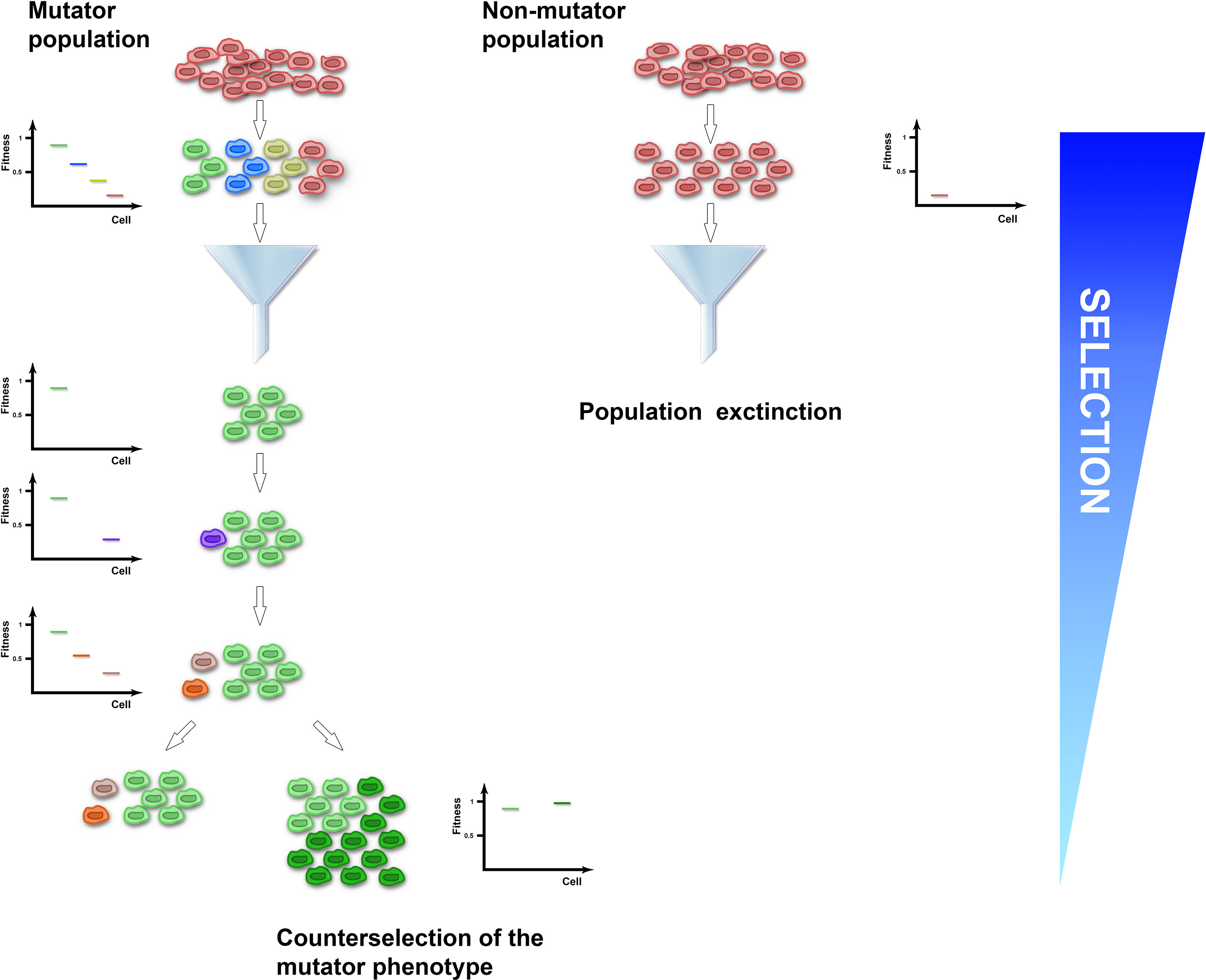
Figure 1 The mutator population (left) experiences enhanced genome instability and acquires cell-to-cell heterogeneity, while the non-mutator population (right) expands clonally. Upon application of selection, the mutator-induced phenotypic variation increases the probability of the population to have cells with a selective advantage (green cells) that could be fixed. Conversely, the clonal non-mutator population has higher probability of becoming extinct (red cells). Once adaptive mutations have been fixed and the population reaches a local optimum, acquisition of additional variation is detrimental and selected against (purple, orange, and grey cells). To increase adaptation in non-selective conditions, the mutator population can evolve a suppressor of the genome instability phenotype (dark green cells).
Intra- and inter-tumor heterogeneity has been observed in early (Jamal-Hanjani et al., 2017) as well as in advanced stages of tumor progression (Caswell and Swanton, 2017; Jamal-Hanjani et al., 2017). Its presence suggests that cancer proceeds through a branched evolutionary pathway (Nowell, 1976; McGranahan and Swanton, 2017). Specifically, single-cell–derived clones carrying different genomes, epigenomes, and karyotypes compete in a non-linear model that favors the expansion and the coexistence of clones containing distinct beneficial mutations under challenging environments (Merlo et al., 2006). The presence of cell-to-cell heterogeneity promotes cancer progression (Gerlinger et al., 2012; Loeb, 2016; McGranahan and Swanton, 2017) by potentially increasing the number of clones with penetrant driver mutations (Jamal-Hanjani et al., 2017), with resistance to drugs or poor environmental conditions (Scalerandi et al., 1999; Calcagno et al., 2010), or immune to interaction with host immune cells (Seliger, 2005).
Cancer cell populations evolve as asexually reproducing organisms and can be modeled as bacteria or mating-type locked laboratory yeast strains. As discussed above, studies on mutator populations in these model organisms indicate that while a mutator phenotype can initially promote adaptation to a variety of selective pressures, it has detrimental effects once adaptive mutations have been fixed. Does this dynamic also occur during cancer progression? We would like to propose this to be the case. In recent years, the tumultuous advances of deep-sequencing technologies increased our ability to perform and analyze large single-cell sequencing data sets (Gerlinger et al., 2012). By looking at mutations present in cancer cells in spatially distinct regions at different stages of cancer progression, a few lessons have emerged. First, consistent with a positive contribution of genome instability to cancer development and evolution in response to challenges, cancer cells display a high level of intra- and inter-tumor heterogeneity (McGranahan and Swanton, 2017). Experimentally, several mouse models of genome instability display an increased spontaneous incidence of cancer onset (Liu et al., 2007; Weaver et al., 2007) and increased tumor relapse when challenged by oncogene withdrawal (Sotillo et al., 2010). This suggests that mutator phenotype could increase aggressiveness or drug resistance of cancer cells. Second, early evolution stages of different types of cancers display genome instability. Thanks to the long latency and frequent biopsies patients are subjected to, one of the cancer types that undergoes the most frequent longitudinal sampling is Barrett’s esophagus. This neoplastic lesion frequently gives rise to esophageal adenocarcinoma and is associated with a high level of genomic instability (Reid et al., 2001). Consistent with the idea that mutator phenotype is an enabling characteristic of tumor development, heterogeneity of premalignant Barrett’s esophagus populations is a prognostic marker that correlates with increased probability of esophageal adenocarcinoma development (Maley et al., 2006). Another line of evidence that a mutator phenotype is an early event comes from clinical evidence that mutations in mismatch repair genes and genome instability occur early in HNPCC and colon cancer evolution. For instance, microsatellite instability was found in premalignant adenomas (Shibata et al., 1996), consistent with the idea that mutations in mismatch repair genes occur prior to hallmark mutation markers for colon cancer (Huang et al., 1996). Lastly, mathematical modeling favors a positive contribution of mutator phenotype in early events of cancer progression leading to rapid tumor growth (Beckman, 2009). Therefore, similarly to yeast and bacteria adaptation to hostile environments, the mutator phenotype can facilitate early stages as well as later stages of cancer evolution. Consistently, it was recently shown that metastatic cells have higher mutations rates than non-metastatic cancer cells (Bertucci et al., 2019). However, extreme genomic instability was reported to have a negative effect on tumor growth, leading to massive cancer cell death (Kops et al., 2004; Janssen et al., 2009). Similarly to what observed in model organisms, it was proposed that excessive mutational burden decreased cellular fitness as cells cannot tolerate high levels of genome instability (Komarova and Wodarz, 2004). Accordingly, clinical evidence suggests that high levels of chromosomal instability are a marker for better prognosis than intermediate ones in non-small-cell lung carcinoma (Jamal-Hanjani et al., 2015). Similar observations have been made in other epithelial tumors, such as ovarian and squamous non-small-cell lung cancer and gastric adenocarcinoma (Birkbak et al., 2011). Taken together, all of this clinical evidence suggests that cancer cells, pretty much like mutator yeasts, can evolve adaptive mechanisms to decrease the rate of genome instability once fitter and more aggressive cancer clones have emerged. This view is also supported by recent studies showing that at different stages of tumor progression, cancer cells exhibit distinct types of genome instability (Nik-Zainal et al., 2012). For instance, sequencing of spatially distant clear-cell renal carcinoma masses within patients showed that, while the bulk of the primary tumor was stable and diploid, cells from metastatic regions derived from a tetraploid intermediate and were genomically unstable (Gerlinger et al., 2012). Moreover, phylogenetic reconstruction of breast cancer tissues carrying BRCA mutations showed that while early mutations during cancer development were consistent with patients’ germline mutations, late-stage genome instability had a significantly different mutational pattern consistent with localized hypermutation with specific base substation (Nik-Zainal et al., 2012). Taken together, all of this clinical evidence suggests that different types of genome instability of tumor cells can be selected to better adapt to cycles of selective and non-selective environments as well as different selective pressures.
As speculated above, to better adapt to a variety of different selective and non-selective environments, cancer cells could tinker with their genome instability to either generate cell-to-cell heterogeneity or stabilize fitter clones or change their mutational landscape. Since different types of mutations could allow cells to differently hike the fitness landscape (Pavelka et al., 2010), the ability of cancer cells to switch between different mutational patterns could equip them with different “gears” to successfully adapt to challenges. Therefore, to successfully eradicate cancer cells, strategies to curb their incredible genome plasticity should be found. Given the similarity in the evolution of mutator phenotypes between single-cell model organisms and cancer cells, we predict that dissecting the molecular mechanisms that allow yeast or bacteria to fine-tune their genome instability will pinpoint targets to curb cancer genome plasticity.
FN and GR conceived and wrote the manuscript.
This study is funded by the NRF Investigatorship (ref. no. NRF-NRFI05-2019-0008) awarded to GR.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Sebastien Teissier and Realite Design for assisting with Figure 1.
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Campbell, P. J., Stratton, M. R. (2013). Deciphering signatures of mutational processes operative in human cancer. Cell. Rep. 3 (1), 246–259. doi: 10.1016/j.celrep.2012.12.008
Andersson, D. I., Hughes, D. (1996). Muller’s ratchet decreases fitness of a DNA-based microbe. Proc. Natl. Acad. Sci. U.S.A. 93 (2), 906–907. doi: 10.1073/pnas.93.2.906
Andre, J. B., Godelle, B. (2006). The evolution of mutation rate in finite asexual populations. Genetics 172 (1), 611–626. doi: 10.1534/genetics.105.046680
Ashworth, A., Lord, C. J., Reis-Filho, J. S. (2011). Genetic interactions in cancer progression and treatment. Cell. 145 (1), 30–38. doi: 10.1016/j.cell.2011.03.020
Barbari, S. R., Shcherbakova, P. V. (2017). Replicative DNA polymerase defects in human cancers: consequences, mechanisms, and implications for therapy. DNA Repair (Amst.) 56, 16–25. doi: 10.1016/j.dnarep.2017.06.003
Beckman, R. A. (2009). Mutator mutations enhance tumorigenic efficiency across fitness landscapes. PLoS One 4 (6), e5860. doi: 10.1371/journal.pone.0005860
Behan, F. M., Iorio, F., Picco, G., Goncalves, E., Beaver, C. M., Migliardi, G., et al. (2019). Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568 (7753), 511–516. doi: 10.1038/s41586-019-1103-9
Beijersbergen, R. L., Wessels, L. F. A., Bernards, R. (2017). Synthetic lethality in cancer therapeutics. Annu. Rev. Cancer Biol. 1 (1), 141–161. doi: 10.1146/annurev-cancerbio-042016-073434
Ben-Aroya, S., Coombes, C., Kwok, T., O’Donnell, K. A., Boeke, J. D., Hieter, P. (2008). Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol. Cell 30 (2), 248–258. doi: 10.1016/j.molcel.2008.02.021
Bertucci, F., Ng, C. K. Y., Patsouris, A., Droin, N., Piscuoglio, S., Carbuccia, N., et al. (2019). Genomic characterization of metastatic breast cancers. Nature 569 (7757), 560–564. doi: 10.1038/s41586-019-1056-z
Billmyre, R. B., Clancey, S. A., Heitman, J. (2017). Natural mismatch repair mutations mediate phenotypic diversity and drug resistance in Cryptococcus deuterogattii.Elife 6, e28802. doi: 10.7554/eLife.28802
Birkbak, N. J., Eklund, A. C., Li, Q., McClelland, S. E., Endesfelder, D., Tan, P., et al. (2011). Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 71 (10), 3447–3452. doi: 10.1158/0008-5472.CAN-10-3667
Bjorkholm, B., Sjolund, M., Falk, P. G., Berg, O. G., Engstrand, L., Andersson, D. I. (2001). Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 98 (25), 14607–14612. doi: 10.1073/pnas.241517298
Boland, C. R., Thibodeau, S. N., Hamilton, S. R., Sidransky, D., Eshleman, J. R., Burt, R. W., et al. (1998). A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 58 (22), 5248–5257.
Bowers, J., Sokolsky, T., Quach, T., Alani, E. (1999). A mutation in the MSH6 subunit of the Saccharomyces cerevisiae MSH2–MSH6 complex disrupts mismatch recognition. J. Biol. Chem. 274 (23), 16115–16125. doi: 10.1074/jbc.274.23.16115
Boyce, K. J., Wang, Y., Verma, S., Shakya, V. P. S., Xue, C., Idnurm, A. (2017). Mismatch repair of DNA replication errors contributes to microevolution in the pathogenic fungus Cryptococcus neoformans. MBio 8 (3), e00595–17. doi: 10.1128/mBio.00595-17
Breslow, D. K., Cameron, D. M., Collins, S. R., Schuldiner, M., Stewart-Ornstein, J., Newman, H. W., et al. (2008). A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods 5 (8), 711–718. doi: 10.1038/nmeth.1234
Briggs, S., Tomlinson, I. (2013). Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J. Pathol. 230 (2), 148–153. doi: 10.1002/path.4185
Bui, D. T., Dine, E., Anderson, J. B., Aquadro, C. F., Alani, E. E. (2015). A genetic incompatibility accelerates adaptation in yeast. PLoS Genet. 11 (7), e1005407. doi: 10.1371/journal.pgen.1005407
Calcagno, A. M., Salcido, C. D., Gillet, J. P., Wu, C. P., Fostel, J. M., Mumau, M. D., et al. (2010). Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J. Natl. Cancer Inst. 102 (21), 1637–1652. doi: 10.1093/jnci/djq361
Carrete, L., Ksiezopolska, E., Pegueroles, C., Gomez-Molero, E., Saus, E., Iraola-Guzman, S., et al. (2018). Patterns of genomic variation in the opportunistic pathogen Candida glabrata suggest the existence of mating and a secondary association with humans. Curr. Biol. 28 (1), 15–27 e17. doi: 10.1016/j.cub.2017.11.027
Caswell, D. R., Swanton, C. (2017). The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med. 15 (1), 133. doi: 10.1186/s12916-017-0900-y
Chakraborty, U., Dinh, T. A., Alani, E. (2018). Genomic instability promoted by overexpression of mismatch repair factors in yeast: a model for understanding cancer progression. Genetics 209 (2), 439–456. doi: 10.1534/genetics.118.300923
Chopra, I., O’Neill, A. J., Miller, K. (2003). The role of mutators in the emergence of antibiotic-resistant bacteria. Drug Resist. Updat. 6 (3), 137–145. doi: 10.1016/S1368-7646(03)00041-4
Clyne, M., Offman, J., Shanley, S., Virgo, J. D., Radulovic, M., Wang, Y., et al. (2009). The G67E mutation in hMLH1 is associated with an unusual presentation of Lynch syndrome. Br. J. Cancer 100 (2), 376–380. doi: 10.1038/sj.bjc.6604860
Coelho, M. C., Pinto, R. M., Murray, A. W. (2019). Heterozygous mutations cause genetic instability in a yeast model of cancer evolution. Nature 566 (7743), 275–278. doi: 10.1038/s41586-019-0887-y
Daee, D. L., Mertz, T. M., Shcherbakova, P. V. (2010). A cancer-associated DNA polymerase delta variant modeled in yeast causes a catastrophic increase in genomic instability. Proc. Natl. Acad. Sci. U.S.A. 107 (1), 157–162. doi: 10.1073/pnas.0907526106
Daurel, C., Prunier, A. L., Chau, F., Garry, L., Leclercq, R., Fantin, B. (2007). Role of hypermutability on bacterial fitness and emergence of resistance in experimental osteomyelitis due to Staphylococcus aureus. FEMS Immunol. Med. Microbiol. 51 (2), 344–349. doi: 10.1111/j.1574-695X.2007.00310.x
Denamur, E., Lecointre, G., Darlu, P., Tenaillon, O., Acquaviva, C., Sayada, C., et al. (2000). Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell. 103 (5), 711–721. doi: 10.1016/S0092-8674(00)00175-6
Drotschmann, K., Clark, A. B., Tran, H. T., Resnick, M. A., Gordenin, D. A., Kunkel, T. A. (1999). Mutator phenotypes of yeast strains heterozygous for mutations in the MSH2 gene. Proc. Natl. Acad. Sci. U.S.A. 96 (6), 2970–2975. doi: 10.1073/pnas.96.6.2970
Feliziani, S., Lujan, A. M., Moyano, A. J., Sola, C., Bocco, J. L., Montanaro, P., et al. (2010). Mucoidy, quorum sensing, mismatch repair and antibiotic resistance in Pseudomonas aeruginosa from cystic fibrosis chronic airways infections. PLoS One 5 (9), e12669. doi: 10.1371/journal.pone.0012669
Fishel, R. (2015). Mismatch repair. J. Biol. Chem. 290 (44), 26395–26403. doi: 10.1074/jbc.R115.660142
Fukui, K. (2010). DNA mismatch repair in eukaryotes and bacteria. J. Nucleic Acids 2010, 260512. doi: 10.4061/2010/260512
Funchain, P., Yeung, A., Lee Stewart, J., Lin, R., Slupska, M. M., Miller, J. H. (2000). The consequences of growth of a mutator strain of Escherichia coli as measured by loss of function among multiple gene targets and loss of fitness. Genetics 154 (3), 959–970.
Gammie, A. E., Erdeniz, N., Beaver, J., Devlin, B., Nanji, A., Rose, M. D. (2007). Functional characterization of pathogenic human MSH2 missense mutations in Saccharomyces cerevisiae. Genetics 177 (2), 707–721. doi: 10.1534/genetics.107.071084
Gausachs, M., Mur, P., Corral, J., Pineda, M., González, S., Benito, L., et al. (2012). MLH1 promoter hypermethylation in the analytical algorithm of Lynch syndrome: a cost-effectiveness study. Eur. J. Hum. Genet. 20 (7), 762–768. doi: 10.1038/ejhg.2011.277
Gentile, C. F., Yu, S. C., Serrano, S. A., Gerrish, P. J., Sniegowski, P. D. (2011). Competition between high- and higher-mutating strains of Escherichia coli. Biol. Lett. 7 (3), 422–424. doi: 10.1098/rsbl.2010.1036
Gerhardt, J., Jafar, S., Spindler, M. P., Ott, E., Schepers, A. (2006). Identification of new human origins of DNA replication by an origin-trapping assay. Mol. Cell. Biol. 26 (20), 7731–7746. doi: 10.1128/MCB.01392-06
Gerlinger, M., Rowan, A. J., Horswell, S., Math, M., Larkin, J., Endesfelder, D., et al. (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366 (10), 883–892. doi: 10.1056/NEJMoa1113205
Gerrish, P. J., Colato, A., Perelson, A. S., Sniegowski, P. D. (2007). Complete genetic linkage can subvert natural selecti.on. Proc. Natl. Acad. Sci. U.S.A. 104 (15), 6266–6271. doi: 10.1073/pnas.0607280104
Giaever, G., Chu, A. M., Ni, L., Connelly, C., Riles, L., Veronneau, S., et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418 (6896), 387–391. doi: 10.1038/nature00935
Giraud, A., Matic, I., Tenaillon, O., Clara, A., Radman, M., Fons, M., et al. (2001). Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science 291 (5513), 2606–2608. doi: 10.1126/science.1056421
Goldsby, R. E., Hays, L. E., Chen, X., Olmsted, E. A., Slayton, W. B., Spangrude, G. J., et al. (2002). High incidence of epithelial cancers in mice deficient for DNA polymerase delta proofreading. Proc. Natl. Acad. Sci. U.S.A. 99 (24), 15560–15565. doi: 10.1073/pnas.232340999
Good, B. H., Desai, M. M. (2016). Evolution of mutation rates in rapidly adapting asexual populations. Genetics 204 (3), 1249–1266. doi: 10.1534/genetics.116.193565
Gould, C. V., Sniegowski, P. D., Shchepetov, M., Metlay, J. P., Weiser, J. N. (2007). Identifying mutator phenotypes among fluoroquinolone-resistant strains of Streptococcus pneumoniae using fluctuation analysis. Antimicrob. Agents Chemother. 51 (9), 3225–3229. doi: 10.1128/AAC.00336-07
Greaves, M. (2015). Evolutionary determinants of cancer. Cancer Discov. 5 (8), 806–820. doi: 10.1158/2159-8290.CD-15-0439
Guaragnella, N., Palermo, V., Galli, A., Moro, L., Mazzoni, C., Giannattasio, S. (2014). The expanding role of yeast in cancer research and diagnosis: insights into the function of the oncosuppressors p53 and BRCA1/2. FEMS Yeast Res. 14 (1), 2–16. doi: 10.1111/1567-1364.12094
Healey, K. R., Zhao, Y., Perez, W. B., Lockhart, S. R., Sobel, J. D., Farmakiotis, D., et al. (2016). Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat. Commun. 7, 11128. doi: 10.1038/ncomms11128
Heitzer, E., Tomlinson, I. (2014). Replicative DNA polymerase mutations in cancer. Curr. Opin. Genet. Dev. 24, 107–113. doi: 10.1016/j.gde.2013.12.005
Hill, V. K., Kim, J. S., Waldman, T. (2016). Cohesin mutations in human cancer. Biochim. Biophys. Acta 1866 (1), 1–11. doi: 10.1016/j.bbcan.2016.05.002
Huang, J., Papadopoulos, N., McKinley, A. J., Farrington, S. M., Curtis, L. J., Wyllie, A. H., et al. (1996). APC mutations in colorectal tumors with mismatch repair deficiency. Proc. Natl. Acad. Sci. U.S.A. 93 (17), 9049–9054. doi: 10.1073/pnas.93.17.9049
Huh, W. K., Falvo, J. V., Gerke, L. C., Carroll, A. S., Howson, R. W., Weissman, J. S., et al. (2003). Global analysis of protein localization in budding yeast. Nature 425 (6959), 686–691. doi: 10.1038/nature02026
Jamal-Hanjani, M., A’Hern, R., Birkbak, N. J., Gorman, P., Gronroos, E., Ngang, S., et al. (2015). Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: a prospective validation cohort study from the TACT trial. Ann. Oncol. 26 (7), 1340–1346. doi: 10.1093/annonc/mdv178
Jamal-Hanjani, M., Wilson, G. A., McGranahan, N., Birkbak, N. J., Watkins, T. B. K., Veeriah, S., et al. (2017). Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 376 (22), 2109–2121. doi: 10.1056/NEJMoa1616288
Janssen, A., Kops, G. J., Medema, R. H. (2009). Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc. Natl. Acad. Sci. U.S.A. 106 (45), 19108–19113. doi: 10.1073/pnas.0904343106
Kolodner, R. D., Marsischky, G. T. (1999). Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 9 (1), 89–96. doi: 10.1016/S0959-437X(99)80013-6
Komarova, N. L., Wodarz, D. (2004). The optimal rate of chromosome loss for the inactivation of tumor suppressor genes in cancer. Proc. Natl. Acad. Sci. U.S.A. 101 (18), 7017–7021. doi: 10.1073/pnas.0401943101
Kops, G. J., Foltz, D. R., Cleveland, D. W. (2004). Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. U.S.A. 101 (23), 8699–8704. doi: 10.1073/pnas.0401142101
Kunkel, T. A., Erie, D. A. (2015). Eukaryotic mismatch repair in relation to DNA replication. Annu. Rev. Genet. 49, 291–313. doi: 10.1146/annurev-genet-112414-054722
Kurzawski, G., Suchy, J., Kladny, J., Safranow, K., Jakubowska, A., Elsakov, P., et al. (2002). Germline MSH2 and MLH1 mutational spectrum in HNPCC families from Poland and the Baltic states. J. Med. Genet. 39 (10), E65. doi: 10.1136/jmg.39.10.e65
Labat, F., Pradillon, O., Garry, L., Peuchmaur, M., Fantin, B., Denamur, E. (2005). Mutator phenotype confers advantage in Escherichia coli chronic urinary tract infection pathogenesis. FEMS Immunol. Med. Microbiol. 44 (3), 317–321. doi: 10.1016/j.femsim.2005.01.003
Lee, K., Tosti, E., Edelmann, W. (2016). Mouse models of DNA mismatch repair in cancer research. DNA Repair (Amst.) 38, 140–146. doi: 10.1016/j.dnarep.2015.11.015
Li, J., Vazquez-Garcia, I., Persson, K., Gonzalez, A., Yue, J. X., Barre, B., et al. (2019). Shared molecular targets confer resistance over short and long evolutionary timescales. Mol. Biol. Evol. 36 (4), 691–708. doi: 10.1093/molbev/msz006
Li, Z., Vizeacoumar, F. J., Bahr, S., Li, J., Warringer, J., Vizeacoumar, F. S., et al. (2011). Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat. Biotechnol. 29 (4), 361–367. doi: 10.1038/nbt.1832
Liu, D., Keijzers, G., Rasmussen, L. J. (2017). DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. 773, 174–187. doi: 10.1016/j.mrrev.2017.07.001
Liu, X., Holstege, H., van der Gulden, H., Treur-Mulder, M., Zevenhoven, J., Velds, A., et al. (2007). Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc. Natl. Acad. Sci. U.S.A. 104 (29), 12111–12116. doi: 10.1073/pnas.0702969104
Loeb, L. A. (2016). Human cancers express a mutator phenotype: hypothesis, origin, and consequences. Cancer Res. 76 (8), 2057–2059. doi: 10.1158/0008-5472.CAN-16-0794
Long, A., Liti, G., Luptak, A., Tenaillon, O. (2015). Elucidating the molecular architecture of adaptation via evolve and resequence experiments. Nat. Rev. Genet. 16 (10), 567–582. doi: 10.1038/nrg3937
Lujan, S. A., Williams, J. S., Kunkel, T. A. (2016). DNA polymerases divide the labor of genome replication. Trends Cell. Biol. 26 (9), 640–654. doi: 10.1016/j.tcb.2016.04.012
Lynch, H. T., de la Chapelle, A. (2003). Hereditary colorectal cancer. N. Engl. J. Med. 348 (10), 919–932. doi: 10.1056/NEJMra012242
Macia, M. D., Rojo-Molinero, E., Oliver, A. (2014). Antimicrobial susceptibility testing in biofilm-growing bacteria. Clin. Microbiol. Infect. 20 (10), 981–990. doi: 10.1111/1469-0691.12651
Maley, C. C., Galipeau, P. C., Finley, J. C., Wongsurawat, V. J., Li, X., Sanchez, C. A., et al. (2006). Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat. Genet. 38 (4), 468–473. doi: 10.1038/ng1768
Mao, E. F., Lane, L., Lee, J., Miller, J. H. (1997). Proliferation of mutators in A cell population. J. Bacteriol. 179 (2), 417–422. doi: 10.1128/jb.179.2.417-422.1997
Maresca, L., Lodovichi, S., Lorenzoni, A., Cervelli, T., Monaco, R., Spugnesi, L., et al. (2018). Functional interaction between BRCA1 and DNA repair in yeast may uncover a role of RAD50, RAD51, MRE11A, and MSH6 somatic variants in cancer development. Front. Genet. 9, 397. doi: 10.3389/fgene.2018.00397
Maslowska, K. H., Makiela-Dzbenska, K., Mo, J. Y., Fijalkowska, I. J., Schaaper, R. M. (2018). High-accuracy lagging-strand DNA replication mediated by DNA polymerase dissociation. Proc. Natl. Acad. Sci. U.S.A. 115 (16), 4212–4217. doi: 10.1073/pnas.1720353115
McDonald, M. J., Hsieh, Y. Y., Yu, Y. H., Chang, S. L., Leu, J. Y. (2012). The evolution of low mutation rates in experimental mutator populations of Saccharomyces cerevisiae. Curr. Biol. 22 (13), 1235–1240. doi: 10.1016/j.cub.2012.04.056
McGranahan, N., Swanton, C. (2017). Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 168 (4), 613–628. doi: 10.1016/j.cell.2017.01.018
Merlo, L. M., Pepper, J. W., Reid, B. J., Maley, C. C. (2006). Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 6 (12), 924–935. doi: 10.1038/nrc2013
Mertz, T. M., Baranovskiy, A. G., Wang, J., Tahirov, T. H., Shcherbakova, P. V. (2017). Nucleotide selectivity defect and mutator phenotype conferred by a colon cancer–associated DNA polymerase delta mutation in human cells. Oncogene 36 (31), 4427–4433. doi: 10.1038/onc.2017.22
Milholland, B., Dong, X., Zhang, L., Hao, X., Suh, Y., Vijg, J. (2017). Differences between germline and somatic mutation rates in humans and mice. Nat. Commun. 8, 15183. doi: 10.1038/ncomms15183
Morales-Burgos, A., Sanchez, J. L., Figueroa, L. D., De Jesus-Monge, W. E., Cruz-Correa, M. R., Gonzalez-Keelan, C., et al. (2008). MSH-2 and MLH-1 protein expression in Muir Torre syndrome–related and sporadic sebaceous neoplasms. P. R. Health Sci. J. 27 (4), 322–327.
Morales, J., Li, L., Fattah, F. J., Dong, Y., Bey, E. A., Patel, M., et al. (2014). Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene. Expr. 24 (1), 15–28. doi: 10.1615/CritRevEukaryotGeneExpr.2013006875
Murphy, K., Darmawan, H., Schultz, A., Fidalgo da Silva, E., Reha-Krantz, L. J. (2006). A method to select for mutator DNA polymerase deltas in Saccharomyces cerevisiae. Genome 49 (4), 403–410. doi: 10.1139/g05-106
Nick McElhinny, S. A., Stith, C. M., Burgers, P. M., Kunkel, T. A. (2007). Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase delta. J. Biol. Chem. 282 (4), 2324–2332. doi: 10.1074/jbc.M609591200
Nik-Zainal, S., Van Loo, P., Wedge, D. C., Alexandrov, L. B., Greenman, C. D., Lau, K. W., et al. (2012). The life history of 21 breast cancers. Cell. 149 (5), 994–1007. doi: 10.1016/j.cell.2012.04.023
Notley-McRobb, L., Seeto, S., Ferenci, T. (2002). Enrichment and elimination of mutY mutators in Escherichia coli populations. Genetics 162 (3), 1055–1062.
Nowell, P. C. (1976). The clonal evolution of tumor cell populations. Science 194 (4260), 23–28. doi: 10.1126/science.959840
O’Neill, A. J., Chopra, I. (2002). Insertional inactivation of mutS in Staphylococcus aureus reveals potential for elevated mutation frequencies, although the prevalence of mutators in clinical isolates is low. J. Antimicrob. Chemother. 50 (2), 161–169. doi: 10.1093/jac/dkf118
Oliver, A., Canton, R., Campo, P., Baquero, F., Blazquez, J. (2000). High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288 (5469), 1251–1254. doi: 10.1126/science.288.5469.1251
Palles, C., Cazier, J. B., Howarth, K. M., Domingo, E., Jones, A. M., Broderick, P., et al. (2013). Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 45 (2), 136–144. doi: 10.1038/ng.2503
Pavelka, N., Rancati, G., Li, R. (2010). Dr Jekyll and Mr Hyde: role of aneuploidy in cellular adaptation and cancer. Curr. Opin. Cell Biol. 22 (6), 809–815. doi: 10.1016/j.ceb.2010.06.003
Pavlov, Y. I., Shcherbakova, P. V. (2010). DNA polymerases at the eukaryotic fork-20 years later. Mutat. Res. 685 (1-2), 45–53. doi: 10.1016/j.mrfmmm.2009.08.002
Raynes, Y., Halstead, A. L., Sniegowski, P. D. (2014). The effect of population bottlenecks on mutation rate evolution in asexual populations. J. Evol. Biol. 27 (1), 161–169. doi: 10.1111/jeb.12284
Reid, B. J., Prevo, L. J., Galipeau, P. C., Sanchez, C. A., Longton, G., Levine, D. S., et al. (2001). Predictors of progression in Barrett’s esophagus II: baseline 17p (p53) loss of heterozygosity identifies a patient subset at increased risk for neoplastic progression. Am. J. Gastroenterol. 96 (10), 2839–2848. doi: 10.1111/j.1572-0241.2001.04236.x
Ryan, E., Sheahan, K., Creavin, B., Mohan, H. M., Winter, D. C. (2017). The current value of determining the mismatch repair status of colorectal cancer: a rationale for routine testing. Crit. Rev. Oncol. Hematol. 116, 38–57. doi: 10.1016/j.critrevonc.2017.05.006
Scalerandi, M., Romano, A., Pescarmona, G. P., Delsanto, P. P., Condat, C. A. (1999). Nutrient competition as a determinant for cancer growth. Phys. Rev. E 59 (2), 2206–2217. doi: 10.1103/PhysRevE.59.2206
Schaaff, F., Reipert, A., Bierbaum, G. (2002). An elevated mutation frequency favors development of vancomycin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 46 (11), 3540–3548. doi: 10.1128/AAC.46.11.3540-3548.2002
Schmitt, M. W., Matsumoto, Y., Loeb, L. A. (2009). High fidelity and lesion bypass capability of human DNA polymerase delta. Biochimie 91 (9), 1163–1172. doi: 10.1016/j.biochi.2009.06.007
Seliger, B. (2005). Strategies of tumor immune evasion. BioDrugs 19 (6), 347–354. doi: 10.2165/00063030-200519060-00002
Shaver, A. C., Dombrowski, P. G., Sweeney, J. Y., Treis, T., Zappala, R. M., Sniegowski, P. D. (2002). Fitness evolution and the rise of mutator alleles in experimental Escherichia coli populations. Genetics 162 (2), 557–566.
Shcherbakova, P. V., Kunkel, T. A. (1999). Mutator phenotypes conferred by MLH1 overexpression and by heterozygosity for mlh1 mutations. Mol. Cell. Biol. 19 (4), 3177–3183. doi: 10.1128/MCB.19.4.3177
Shibata, D., Navidi, W., Salovaara, R., Li, Z.-H., Aaltonen, L. A. (1996). Somatic microsatellite mutations as molecular tumor clocks. Nat. Med. 2 (6), 676–681. doi: 10.1038/nm0696-676
Shlien, A., Campbell, B. B., de Borja, R., Alexandrov, L. B., Merico, D., Wedge, D., et al. (2015). Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 47 (3), 257–262. doi: 10.1038/ng.3202
Singh, A., Healey, K. R., Yadav, P., Upadhyaya, G., Sachdeva, N., Sarma, S., et al. (2018). Absence of azole or echinocandin resistance in Candida glabrata isolates in India despite background prevalence of strains with defects in the DNA mismatch repair pathway. Antimicrob. Agents Chemother. 62 (6), e00195–18. doi: 10.1128/AAC.00195-18
Sokolsky, T., Alani, E. (2000). EXO1 and MSH6 are high-copy suppressors of conditional mutations in the MSH2 mismatch repair gene of Saccharomyces cerevisiae. Genetics 155 (2), 589–599.
Sotillo, R., Schvartzman, J. M., Socci, N. D., Benezra, R. (2010). Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 464 (7287), 436–440. doi: 10.1038/nature08803
Symeonidou, I. E., Taraviras, S., Lygerou, Z. (2012). Control over DNA replication in time and space. FEBS Lett. 586 (18), 2803–2812. doi: 10.1016/j.febslet.2012.07.042
Taddei, F., Radman, M., Maynard-Smith, J., Toupance, B., Gouyon, P. H., Godelle, B. (1997). Role of mutator alleles in adaptive evolution. Nature 387 (6634), 700–702. doi: 10.1038/42696
Tenaillon, O., Barrick, J. E., Ribeck, N., Deatherage, D. E., Blanchard, J. L., Dasgupta, A., et al. (2016). Tempo and mode of genome evolution in a 50,000-generation experiment. Nature 536 (7615), 165–170. doi: 10.1038/nature18959
Thompson, D. A., Desai, M. M., Murray, A. W. (2006). Ploidy controls the success of mutators and nature of mutations during budding yeast evolution. Curr. Biol. 16 (16), 1581–1590. doi: 10.1016/j.cub.2006.06.070
Trindade, S., Perfeito, L., Gordo, I. (2010). Rate and effects of spontaneous mutations that affect fitness in mutator Escherichia coli. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 365 (1544), 1177–1186. doi: 10.1098/rstb.2009.0287
Vazquez-Garcia, I., Salinas, F., Li, J., Fischer, A., Barre, B., Hallin, J., et al. (2017). Clonal heterogeneity influences the fate of new adaptive mutations. Cell. Rep. 21 (3), 732–744. doi: 10.1016/j.celrep.2017.09.046
Wagner, V. P., Webber, L. P., Salvadori, G., Meurer, L., Fonseca, F. P., Castilho, R. M., et al. (2016). Overexpression of MutSalpha complex proteins predicts poor prognosis in oral squamous cell carcinoma. Medicine (Baltimore) 95 (22), e3725. doi: 10.1097/MD.0000000000003725
Wahl, L. M., Gerrish, P. J., Saika-Voivod, I. (2002). Evaluating the impact of population bottlenecks in experimental evolution. Genetics 162 (2), 961–971.
Walsh, C. S. (2015). Two decades beyond BRCA1/2: homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol. Oncol. 137 (2), 343–350. doi: 10.1016/j.ygyno.2015.02.017
Wang, S., Wang, Y., Shen, J., Wu, Y., Wu, C. (2013). Polymorphic mutation frequencies in clinical isolates of Staphylococcus aureus: the role of weak mutators in the development of fluoroquinolone resistance. FEMS Microbiol. Lett. 341 (1), 13–17. doi: 10.1111/1574-6968.12085
Weaver, B. A., Silk, A. D., Montagna, C., Verdier-Pinard, P., Cleveland, D. W. (2007). Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 11 (1), 25–36. doi: 10.1016/j.ccr.2006.12.003
Wielgoss, S., Barrick, J. E., Tenaillon, O., Wiser, M. J., Dittmar, W. J., Cruveiller, S., et al. (2013). Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load. Proc. Natl. Acad. Sci. U.S.A. 110 (1), 222–227. doi: 10.1073/pnas.1219574110
Wilczak, W., Rashed, S., Hube-Magg, C., Kluth, M., Simon, R., Buscheck, F., et al. (2017). Up-regulation of mismatch repair genes MSH6, PMS2 and MLH1 parallels development of genetic instability and is linked to tumor aggressiveness and early PSA recurrence in prostate cancer. Carcinogenesis 38 (1), 19–27. doi: 10.1093/carcin/bgw116
Wimmer, K., Kratz, C. P. (2010). Constitutional mismatch repair-deficiency syndrome. Haematologica 95 (5), 699–701. doi: 10.3324/haematol.2009.021626
Keywords: mutator phenotype, cell-to-cell heterogeneity, adaptation, selective pressure, asexually reproducing organisms
Citation: Natali F and Rancati G (2019) The Mutator Phenotype: Adapting Microbial Evolution to Cancer Biology. Front. Genet. 10:713. doi: 10.3389/fgene.2019.00713
Received: 15 October 2018; Accepted: 05 July 2019;
Published: 06 August 2019.
Edited by:
Roberta Fraschini, University of Milano-Bicocca, ItalyReviewed by:
Michael McDonald, Monash University, AustraliaCopyright © 2019 Natali and Rancati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulia Rancati, Z2l1bGlhLnJhbmNhdGlAaW1iLmEtc3Rhci5lZHUuc2c=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.