
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
TECHNOLOGY AND CODE article
Front. Genet. , 04 July 2019
Sec. Systems Biology Archive
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.00633
This article is part of the Research Topic User-Friendly Tools Applied to Genetics or Systems Biology View all 16 articles
 Thiago Castanheira Merigueti1
Thiago Castanheira Merigueti1 Marcia Weber Carneiro4
Marcia Weber Carneiro4 Ana Paula D’A. Carvalho-Assef3
Ana Paula D’A. Carvalho-Assef3 Floriano Paes Silva-Jr2*
Floriano Paes Silva-Jr2* Fabricio Alves Barbosa da Silva1*
Fabricio Alves Barbosa da Silva1*Background: Healthcare-associated infections (HAIs) are a serious public health problem. They can be associated with morbidity and mortality and are responsible for the increase in patient hospitalization. Antimicrobial resistance among pathogens causing HAI has increased at alarming levels. In this paper, a robust method for analyzing genome-scale metabolic networks of bacteria is proposed in order to identify potential therapeutic targets, along with its corresponding web implementation, dubbed FindTargetsWEB. The proposed method assumes that every metabolic network presents fragile genes whose blockade will impair one or more metabolic functions, such as biomass accumulation. FindTargetsWEB automates the process of identification of such fragile genes using flux balance analysis (FBA), flux variability analysis (FVA), extended Systems Biology Markup Language (SBML) file parsing, and queries to three public repositories, i.e., KEGG, UniProt, and DrugBank. The web application was developed in Python using COBRApy and Django.
Results: The proposed method was demonstrated to be robust enough to process even non-curated, incomplete, or imprecise metabolic networks, in addition to integrated host-pathogen models. A list of potential therapeutic targets and their putative inhibitors was generated as a result of the analysis of Pseudomonas aeruginosa metabolic networks available in the literature and a curated version of the metabolic network of a multidrug-resistant P. aeruginosa strain belonging to a clone endemic in Brazil (P. aeruginosa ST277). Genome-scale metabolic networks of other gram-positive and gram-negative bacteria, such as Staphylococcus aureus, Klebsiella pneumoniae, and Haemophilus influenzae, were also analyzed using FindTargetsWEB. Multiple potential targets have been found using the proposed method in all metabolic networks, including some overlapping between two or more pathogens. Among the potential targets, several have been previously reported in the literature as targets for antimicrobial development, and many targets have approved drugs. Despite similarities in the metabolic network structure for closely related bacteria, we show that the method is able to selectively identify targets in pathogenic versus non-pathogenic organisms.
Conclusions: This new computational system can give insights into the identification of new candidate therapeutic targets for pathogenic bacteria and discovery of new antimicrobial drugs through genome-scale metabolic network analysis and heterogeneous data integration, even for non-curated or incomplete networks.
Healthcare-associated infections (HAIs), previously called hospital infections, are a serious public health problem and can develop either as a direct result of medical or surgical treatment or from being in contact with a healthcare setting. These infections include central line-associated bloodstream infections, catheter-associated urinary tract infections, ventilator-associated pneumonia (VAP), and surgical site infections. Among the pathogens related to HAI, the group of bacteria is the one that stands out. More than 2 million HAIs occur each year in the USA (Stone et al., 2005), with 50–60% being caused by antimicrobial resistant bacteria. In 2014, the World Health Organization (WHO) published the report “Antimicrobial resistance: global report on surveillance” (WHO, 2014) warning of the growing increase in antimicrobial resistance in the world. Antimicrobial resistance among hospital pathogens has increased at alarming levels, both in developed and developing countries. It is estimated that there will be a worldwide spread of untreatable infections both inside and outside hospitals. According to a bulletin published in 2017 by WHO (WHO, 2017), there are 12 major antibiotic-resistant bacteria that deserve attention and urgently need more research and development (R&D) of new and effective antibiotic treatments. Gram-negative bacteria are the most involved in HAI (carbapenem-resistant Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacteriaceae family), and R&D on new antibiotics against these is considered to be of critical priority (WHO, 2017). In humans, P. aeruginosa is an opportunistic pathogen that causes severe infections in immunocompromised individuals. This pathogen is the main cause of morbi-mortality in patients with cystic fibrosis (Kerr and Snelling, 2009) and is a major cause of VAP.
Given the potential severity of multidrug-resistant bacteria and the lack of treatment options, the identification and implementation of effective strategies to prevent such infections are urgent priorities.
The integration of mathematical, statistical, and computational methods for biological data analysis to enable the discovery of new therapeutic targets for any bacteria is extremely relevant. The combination of bioinformatics, system modeling, and heterogeneous data integration can be a powerful tool for this purpose.
Several strategies have been proposed to search for drug targets from genome-scale models of bacterial metabolism. More often, essential genes are identified from single virtual knockouts where flux balance analysis (FBA) (Orth et al., 2010) is used to assess if this gene deletion is able to halt a selected function of bacterial metabolism. Usually, such function is biomass production (Rienksma et al., 2014). Other criteria can be combined to prioritize genes among candidate drug targets, such as existence of druggable pockets (Kozakov et al., 2015) or specificity to the bacteria as compared to the host proteins.
The construction of genome-scale metabolic network is a laborious endeavor. It combines automated steps with manual curation. The most used protocol, proposed by Thiele and Palsson (2010), lists a total of 94 steps. Nevertheless, the process is error-prone, and normally the resulting network may correctly predict some phenomena while disregarding others, which are less relevant to the study related to the reconstructed metabolic network.
The BioCyc database (Caspi et al., 2015) classifies pathway/genome databases (PGDB), each containing the full genome and predicted metabolic network of one organism, into three tiers. Tier 1 corresponds to PGDBs that have received at least 1 year of manual curation and are updated continuously. Tier 2 includes PGDBs that have received moderate (less than a year) amounts of review and are usually not updated on an ongoing basis. Finally, Tier 3 refers to PGDBs that were created computationally and received no subsequent manual review or updating.
In this work, the same classification for genome-scale metabolic network models is adopted. The focus here is on metabolic network models that can be classified as Tier 2 and Tier 3, according to the BioCyc database classification. In this manuscript, draft metabolic reconstructions are considered Tier 3 models. Published curated metabolic models are classified as Tier 2, unless the model is identified in the literature as Tier 1.
Herein, a method for analyzing genome-scale metabolic networks of bacteria is proposed in order to identify potential therapeutic targets, along with its corresponding web implementation, dubbed FindTargetsWEB. The proposed method is computationally efficient, user-friendly, and robust to errors in reconstructed genome-scale metabolic networks, which are more frequent in Tier 3 (draft) metabolic networks. The web interface of the application is straightforward, and results are sent directly to an email address informed by the user. To demonstrate the flexibility of FindTargetsWEB, 10 genomic-scale metabolic networks of bacterial strains are analyzed in this paper. Nine of the 10 networks are available in the literature, all classified as Tier 2 models in this work: P. aeruginosa PAO1—version 2008 (Oberhardt et al., 2008), P. aeruginosa PAO1—version 2017 (Bartell et al., 2017), P. aeruginosa PA14 (Bartell et al., 2017), Klebsiella pneumoniae (Liao et al., 2011), Haemophilus influenzae (Schilling and Palsson, 2000), a host-pathogen genome-scale reconstruction based on the Mycobacterium tuberculosis metabolic network (Bordbar et al., 2010), Staphylococcus aureus (Becker and Palsson, 2005), and Pseudomonas putida (Puchałka et al., 2008). Results are also presented for two metabolic networks of P. aeruginosa CCBH4851, which is a multi-drug resistant strain belonging to a clone endemic in Brazil (P. aeruginosa ST277) (Silveira et al., 2014). Both reconstructions of P. aeruginosa CCBH4851 were made by our group. One reconstruction can be classified as Tier 3, and the other is the corresponding curated version, classified as Tier 2.
The web application proposed in this work combines FBA, flux variability analysis (FVA) (Gudmundsson and Thiele, 2010), extended Systems Biology Markup Language (SBML) parsing, and heterogeneous data integration in order to identify the most promising therapeutic targets. All SBML files processed in this work are available as Supplementary Material. The underlying hypothesis related to FVA is that reactions which the maximum flux is equal to the minimum flux (i.e., flux range equal to zero), given the optimal biomass production, are less robust to potential perturbations. Indeed, a high rigidity for a given reaction flux (i.e., flux range equal to zero) may indicate that the flux through this reaction is crucial for sustaining optimal growth, while a lower rigidity (i.e., flux range greater than zero) indicates that there might be alternate pathways to carry the reaction flux (Oberhardt et al., 2010). Flux ranges fell into three categories: i) inflexible fluxes (flux range equal to zero), ii) fluxes with bounded flexibility (flux range greater than zero, but bounded), and iii) infinitely flexible fluxes (flux range greater than zero, unbounded). The FVA analysis carried out by FindTargetsWEB aims to identify potential targets associated with inflexible fluxes, i.e., flux range equal to zero. The genome-scale metabolic network analysis is combined with several queries to multiple public repositories, such as KEGG (Ogata et al., 1999), UniProt (UniProt, 2018), and DrugBank (Wishart et al., 2008), to assess the druggability and toxicology of potential targets. FindTargetsWEB has identified potential targets for all networks. Several of the potential targets have been described in the literature. Other targets are candidates for future experimental investigation.
Some of the main requirements related to the implementation of the general method described in this work, dubbed FindTargetsWEB, were ease of use, availability, robustness, and performance. After careful consideration, Python was selected as the implementation language. Python is a high-level, interpreted, scripted, imperative, object-oriented, dynamic, and strongly typed programming language created by Van Rossum and Drake (2003). Its many advantages favor the fulfillment of the main requirements of the application. Another advantage is the availability of the COBRApy package. COnstraint-Based Reconstruction and Analysis Toolbox (COBRA) (Hyduke et al. 2011) methods are widely used for genome-scale modeling of metabolic networks in prokaryotes and eukaryotes. The COBRA Toolbox for MATLAB is a leading software package for analyzing metabolism on a genomic scale. On the other hand, COBRApy (Ebrahim et al., 2013) is a Python module that provides support for basic COBRA methods. COBRApy is designed in an object-oriented way, which facilitates the representation of the complex biological processes of metabolism. COBRApy does not require MATLAB to work; however, it includes an interface to the COBRA Toolbox for MATLAB to facilitate the use of legacy codes. To improve performance, COBRApy includes parallel processing support for computationally intensive processes. FindTargetsWEB is implemented as a web application. Therefore, the user only needs a web browser to access the system. The system interface is intuitive: the user needs to provide the SBML file describing the metabolic network reconstruction, the organism species associated with the metabolic network reconstruction, which defines a filter to KEGG queries, and information such as name and e-mail address (Figure 1). It should be emphasized that the FindTargetsWEB list of analyzable species is easily expandable and can include both gram-negative bacteria, gram-positive bacteria, and bacteria that cannot be classified as either gram-positive or gram-negative. In the following screen, the user decides if he/she wants to analyze the network using the FBA method alone or a combination of the FBA+FVA methods (Figure 2). The FBA+FVA method pinpoints reactions and associated genes in which knockout completely stops (zeroes) biomass generation and has an FVA range of zero. Therefore, the FBA+FVA method is more restrictive than the FBA-only option. It should be highlighted that the targets found by the FBA+FVA method compose a proper subset of the set of targets found by the FBA-only method. Robustness is provided by the design of the method itself, as described in the following paragraphs.
Figure 1 FindTargetsWEB user interface—SBML file input.
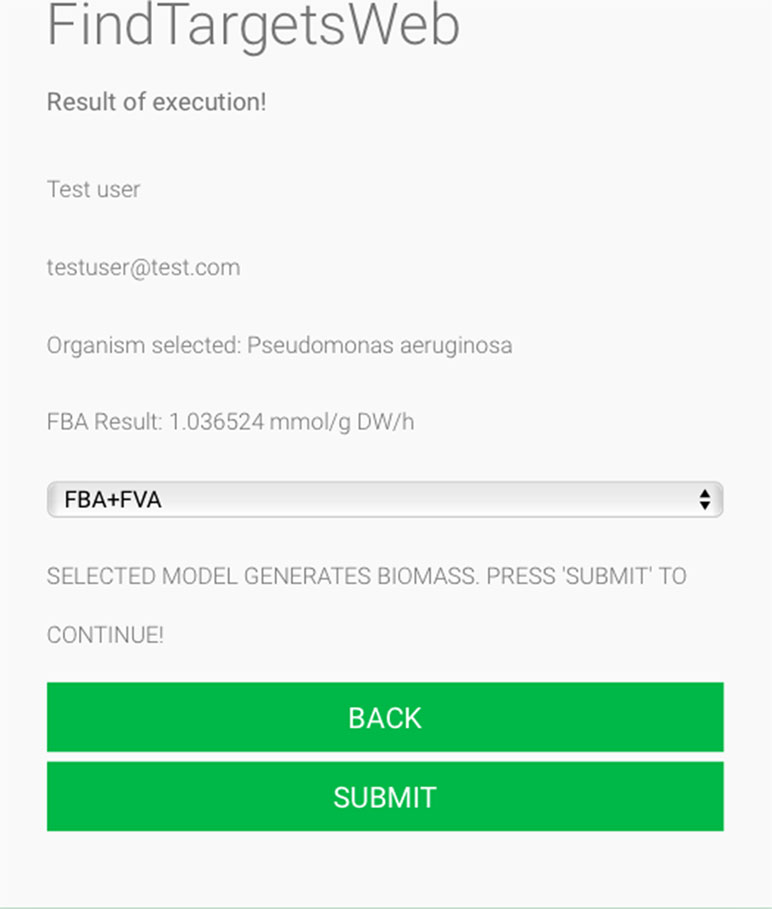
Figure 2 FindTargetsWEB user interface—choice of analysis method.
Target identification is carried out through a computational workflow that runs the metabolic network analysis and pinpoints genes whose virtual knockout interrupts the generation of biomass. Therefore, the minimum level of curation required for a metabolic network model to be processed by FindTargetsWEB is to have a biomass reaction flux greater than zero. The list of potential targets is filtered using FVA (if the user decides to do so), and the workflow retrieves possible inhibitors for the identified genes, verify if such inhibitors are available as approved drugs, and evaluate their toxicity to humans by querying several repositories.
The workflow was implemented using the Python programming language, version 3.6.3, and the COBRApy framework version 0.9.0. This framework has the necessary methods for reading the SBML (Hucka et al., 2015) file that describes the genome-scale metabolic network of the bacterium under analysis. The solver used for FBA and FVA analysis is GLPK (https://www.gnu.org/software/glpk/), which is the COBRApy default solver that is easily deployable on Linux platforms. The system is deployed in an Ubuntu v18.04 server with 64GB RAM. Prior to processing, when needed, SBML files were converted to the SBML level 3 format using the command cobra.io.sbml3.write_sbml_model from COBRA. The SBML files processed in this manuscript were retrieved from the BioModels repository (Glont et al., 2017) or directly from the supplementary material of the associated reference. The main steps of the method are described below. The whole method is depicted in Figure 3.
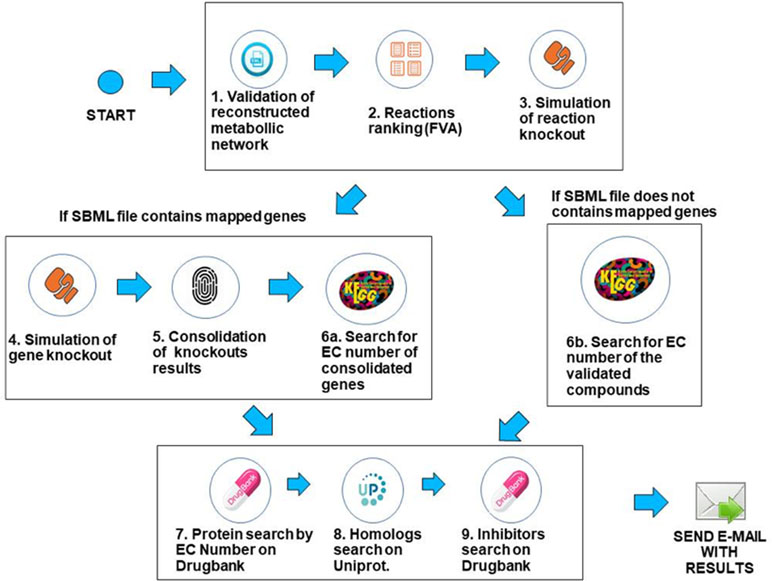
Figure 3 Description of the computational workflow.
1. Validation of the SBML file describing the genome-scale metabolic network—In this step, the system first creates a table containing gene/reaction/metabolite data obtained from the SBML file and then checks if the metabolic network reconstruction generates biomass. This is done through the FBA method, considering the biomass reaction as the target for maximization. If the biomass value is zero, the system outputs an error to the user and halts processing. If the maximum flux of the biomass reaction is greater than zero, the workflow proceeds to the next step.
2. Use of FVA to filter reactions—After validating the metabolic network, reactions are filtered using the FVA method, if the user has decided to analyze the metabolic network using a combination of the FBA+FVA methods. The objective is to consider, in the following processing steps, those reactions which the range of possible flux values is equal to zero, given the optimal biomass generation value determined in the previous step. The underlying assumption is that reactions with a range equal to zero are less robust, i.e., more susceptible to perturbations, as stated in the introduction. Note that the FVA method can be implemented in a computationally efficient way (Gudmundsson and Thiele, 2010), and the cost of FVA analysis on the overall execution time of FindTargetsWEB is negligible.
3. Simulation of reaction knockout—In this step, single reaction knockouts are performed. The process is done by zeroing the maximum and minimum reaction flux constraints and running FBA again, for each reaction in the network. If biomass generation is zeroed when knocking out a given reaction, its information is stored in a list for further processing. If gene IDs are available in the SBML file, the workflow proceeds to step 4. Otherwise, it jumps directly to step 6b.
4. Simulation of gene knockout—In this step, the system performs single knockouts for each gene described in the model, where the COBRApy framework queries the reactions that are linked to the selected gene and zeroes the minimum and maximum value of each reaction bound to the gene, taking into account gene-protein-reaction (GPR) relations. In the same way as the previous step, if the value of the generation of biomass has zeroed, the corresponding gene information is stored in a second list. It is worth noting that one gene can be associated with more than one reaction, and one reaction may require the expression of several genes.
5. Consolidation/unification of knockouts results—In this step, both lists generated in the previous steps are unified, i.e., the list of reactions generated in step 3 and the gene list generated in step 4. In order to a gene to be included in the final list, it should be included in the list of step 4 and be associated with at least one reaction stored in step 3 (see Algorithm 1). These are the candidate genes that the workflow is going to consider in the following steps. It should be highlighted that the final list is filtered according to the FVA processing performed in step 2, if the option FBA+FVA is selected by the user.
Algorithm 1: Consolidation of knockout results (SBML with mapped genes)
Input: List of genes from knocked-out reactions/list of knocked-out genes
Output: Unified list of target genes in a text file
1: procedure UnificationTargetsList(targetGeneListFromReact, targetGeneList) 2: read targetGeneListFromReact 3: read targetGeneList 4: open file “targetgenes.txt” 5: for all targetgene in targetGeneListFromReact do 6: if targetgene in targetGeneList then 7: write targetgene in file “targetgenes.txt” 8: end if 9: end for 10: close file “targetgenes.txt” 11: end procedure
6a. Search for EC numbers of consolidated genes. In this step, the system queries the KEGG repository to obtain the EC number of each gene included in the final gene list obtained in the previous step (file “targetgenes.txt”). KEGG (Kyoto Encyclopedia of Genes and Genomes) is a knowledge base for systematic analysis of gene functions, linking genomic information with higher order functional information (Ogata et al., 1999). This step is important because drug retrieval in DrugBank requires the associated EC number. The result of this step is a list of EC numbers associated to their respective genes. The workflow then proceeds to step 7.
6b. Search for EC number using reaction information. If gene IDs are not available in the SBML file, which may be the case in draft (Tier 3) metabolic network models, EC numbers are retrieved from KEGG based on reaction information. This step is particularly important for incomplete metabolic reconstructions that do not include GPR relations and is directly related to the application’s requirement of robustness to incompleteness on metabolic network data. The KEGG search is performed using all the compounds involved in the corresponding reaction. See Algorithm 2 for a detailed description of the processing related to this step. It is worth emphasizing that this step is executed only for incomplete descriptions of genome-scale metabolic networks. The complexity of Algorithm 2 is O(C), where C is the number of compounds included in the SBML file.
Algorithm 2: Search for EC numbers using reaction information (SBML without mapped genes)
Input: List of chemical compounds of reaction
Output: List of EC numbers found
1: procedure alternativeStepToGetECNumberWithoutGenes(listCompoundFromSBML) 2: # file with all compounds in SBML. 3: read listCompoundFromSBML 4: 5: # Instance of biomodels python module 6: k <- KEGG instance 7: 8: # setting timeout in seconds 9: k.timeout <- 200000 10: 11: # All compounds in SBML file. Ex.: 2 A –> B 12: for all compound in listCompoundFromSBML do 13: 14: # find all stoichiometric values with regex method in compound. Ex.: A –> B 15: compound_no_stoich <- remove all stoichiometric values in compound 16: param_splt <- empty 17: 18: # Verify if reaction is reversible or irreversible 19: if “< = >“ in compound_no_stoich then 20: param_splt <- “< = >“ 21: else 22: param_splt <- “–>“ 23: end if 24: 25: # Separate compounds in reactant and product. Ex.: compound_splt = [‘A’, ‘B’] 26: compound_splt <- compound_no_stoich.split(param_splt) 27: 28: # If compound belongs to a transport reaction (influx or eflux), jump to next iteration 29: if compound_splt.length < 2 then 30: continue 31: end if 32: 33: list_ec_number_0 <- initialize empty list 34: list_ec_number_1 <- initialize empty list 35: 36: # Start iterating compound_splt list with reactant and product 37: for (x = 0,1) do 38: 39: # Get reactant or product in this variable 40: item_compound <- compound_splt[x] without spaces 41: list_id_cpd_KEGG <- initialize empty list 42: 43: # If reactant or product contains “+”, find ID in KEGG for components. 44: # Else, find ID in KEGG for only one component. 45: if “ + “ in item_compound then 46: item_compound_splt <- item_compound.split(“ + “) 47: for all cpd_item in item_compound_splt do 48: # find id compound in KEGG for cpd_item 49: # and insert in ids list 50: result_id_cpd <- k.find(“compound”, cpd_item) 51: insert result_id_cpd in list_id_cpd_KEGG 52: end for 53: else 54: result_id_cpd <- k.find(“compound”, item_compound) 55: insert item_compound in list_id_cpd_KEGG 56: end if 57: 58: # Here, all list_id_cpd_KEGG are concatenated 59: # found to search the reaction in KEGG. 60: # In Python, if list_id_cpd_KEGG length is less than 2, 61: # don’t put the “+” in end of string. 62: str_item_compound_in_cpd <- list_id_cpd_KEGG concat with “+” 63: 64: # find all reactions in KEGG with IDs of compounds 65: result_link_reactions_cpd <- k.link(“reaction”, str_item_compound_in_cpd) 66: 67: # All results of result_link_reactions_cpd are inserted here 68: set_id_reaction_KEGG <- insert all reactions found in KEGG. 69: 70: # find all EC numbers in KEGG with reactions IDs 71: # in set_id_reaction_KEGG and insert in result_list_ec 72: result_list_ec = k.link(“enzyme”, set_id_reaction_KEGG) 73: if x = 0 then 74: insert result_list_ec in list_ec_number_0 75: else 76: insert result_list_ec in list_ec_number_1 77: end if 78: 79: end for 80: 81: list_ec_number_intersect <- initialize empty list 82: txt_file <- initialize txt file 83: 84: # Starts to iterate the list of ECs to identify intersections 85: # If found, related EC numbers are written in a text file 86: for all ec_number_0 in list_ec_number_0 do 87: if ec_number_0 in list_ec_number_1 then 88: record ec_number_0 in a txt_file 89: end if 90: end for 91: 92: end for 93: 94: end procedure
7. Search for EC numbers on DrugBank—With the EC numbers obtained in the previous steps, the system queries the DrugBank repository to verify if this database has any record of the listed EC numbers. The DrugBank database is a repository that combines detailed drug data with comprehensive drug target information (Wishart et al., 2008). If an exact match is found, the system retrieves the values of the name of the protein, organism, and UniProt ID.
When executing this query, the protein retrieved can be mapped in another organism, distinct from the target bacterium. Thus, the next step (step 8) is necessary to confirm whether the protein retrieved has a homologue in the target bacterium. Clearly, exact matches are also possible. In any case, the retrieved data is validated in the next step.
8. Search for homologues on UniProt—Finally, the system searches for sequence similarity between the proteins described by UniProt IDs retrieved in the last step and the proteins encoded by the genome of the target bacterium using the BLAST (basic local alignment search tool) (Altschul et al., 1990) application deployed in the UniProt server. If there is a hit (i.e., sequence similarity above 30%), all corresponding data concerning the homologue found is stored.
In this step, the homology between the target protein and human proteins is also considered. If the sequence similarity with a human protein is greater than the similarity with the target bacterium, the protein under analysis is discarded, since the inhibition of that protein could be harmful to the host. Otherwise, several data are stored, such as metabolic pathway, function, and catalytic activity, among others. This step of the workflow is the most time-consuming, since BLAST is executed for all proteins identified in the previous step.
9. Search for existing inhibitors—The last step is to query the DrugBank repository, using the stored UniProt IDs, in order to retrieve known inhibitors, if available. After this last step, the system generates spreadsheets containing all results that are sent to the user in a compressed file.
This method presents as results candidate genes that, when knocked-out, will cease the biomass production of the microorganism. Candidate genes must be associated with potential drug targets in DrugBank, and their sequence similarity to human proteins is also checked. The application then identifies available ligands, most often inhibitors, to the selected genes.
Results of FindTargetsWEB’s analysis are sent to the user as a compressed file, to the e-mail address informed at the start of execution. Five spreadsheets are included in the compressed file:
- 08-filter_ECNumbers_DrugBank—This spreadsheet contains the EC number of putative targets, along with product, organism name, UniProt ID, and DrugBank ID
- 11- hits_Uniprot—This spreadsheet contains additional information related to UniProt queries, such as percentage of sequence similarity, BLAST e-value, gene name, pathway, function, and catalytic activity.
- 13-list_inhibitors_per_target—This spreadsheet lists all inhibitors found for all targets. Included information are drug name, drug group (e.g. experimental, approved, investigational), and drug action.
- 14-list_inhibitors_approved—This spreadsheet lists all inhibitors with approved drugs found for all targets. Included information are drug name, drug group (approved), and drug action.
- model_data—This spreadsheet lists data related to the input SBML file, such as gene IDs and associated reactions. The complete information of which reactions are associated with each gene in the metabolic network model is included in this file.
- summary_results—This spreadsheet contains a summary of data included in the previous files. Included fields are EC numbers, product, organism name, gene name, pathway, function, catalytic activity, drug name, drug group, and drug action.
In this section, analysis results for several strains of P. aeruginosa, K. pneumoniae, H. influenzae, S. aureus, P. putida, and a host-pathogen genome-scale reconstruction based on the M. tuberculosis metabolic network are presented. It should be highlighted that FindTargetsWEB can carry out analysis for other bacterial species, as indicated by the list box on the initial web page of the application. Indeed, even this list can be easily expanded to include additional species of interest, through a user request to FindTargetsWEB support team.
To evaluate the accuracy of results for several metabolic networks, initially, the analysis of four metabolic networks of P. aeruginosa is discussed. A survey of the literature is also presented to confirm the feasibility of the candidate genes as antibacterial drug targets. Gene function and related pathways are also considered in the evaluation of results.
The four metabolic networks of P. aeruginosa strains analyzed by FindTargetWEB were: PAO1 version 2008—iMO1056 (BioModels ID 1507180020) (Oberhardt et al., 2008), PAO1 version 2017—iPAE1146 (Bartell et al., 2017), PA14—iPAU1129 (Bartell et al., 2017), and a curated version (Tier 2) of the metabolic network of P. aeruginosa CCBH4851 (Silveira et al., 2014). The SBML level 3 file describing the Tier 2 P. aeruginosa CCBH4851 network is available as supplementary material, as well as the SBML files of the other networks considered in this paper. It is worth noting that each metabolic network model presents a different value for the growth rate after validation of biomass generation by FBA; for PA01 version 2008, the growth rate corresponds to 1.047929 h-1; PA01 version 2017 has a growth rate of 15.509635 h-1; for the PA14 model, the growth rate is 15.508373 h-1, and the Tier 2 CCBH4851 model has a growth rate of 1.036524 h-1. Differences in growth rate among metabolic network models are due to the distinct biomass equations, as well as variation in the number of genes, reactions, and metabolites in each of the metabolic network models.
It should be mentioned that the growth rates associated with the PA14 and PAO1-2017 (Bartell et al., 2017) models depart by far from the observed growth rates of P. aeruginosa spp., which may vary between 0.3 and 0.8 h-1, depending on cultivation conditions (Brown, 1957) (Seto and Noda, 1982) (Yang et al., 2008). Nevertheless, FindTargetsWEB can still process those networks. The only requirement is to have a growth rate greater than zero.
In this subsection, common targets for all Tier 2 P. aeruginosa networks are listed. The metabolic network models of P. aeruginosa analyzed in this subsection are described at Oberhardt et al., 2008 (PAO1) and Bartell et al., 2017 (PAO1 and PA14). The P. aeruginosa CCBH4851 metabolic network is being modeled by our group and represents a bacterium found in a catheter of a patient hospitalized at the Brazilian state of Goiás (Silveira et al., 2014). It is worth highlighting that the Bartell et al. (2017) networks focused on modeling virulence factors. Due to this fact, the biomass equation received less attention and the growth rate is not inside the range observed for Pseudomonas spp. Nevertheless, the workflow was able to process both networks and found several targets common to other metabolic reconstructions. The number of unique targets found in each network, for both FAB+FVA and FBA-only methods, are listed in Table 1. The spreadsheet detailing all targets found is available as supplementary material.

Table 1 Number of unique targets found in the Tier 2 metabolic networks of P. aeruginosa.
For the FBA-only method, 25 targets are common to all four networks. For the FBA+FVA method, 11 targets are common to all four networks.
It is important to highlight some of the genes identified as common targets for all four metabolic network models of P. aeruginosa (Table 2). The murA (EC 2.5.1.7) and murB (EC 1.3.1.98) genes encode enzymes involved in bacterial cell wall synthesis and have been identified as essential in both Pseudomonas spp. and Escherichia coli (Benson et al., 1996). The folP gene product (EC 2.5.1.15) is important for folic acid biosynthesis, which is fundamental for bacterial growth and reproduction (Dallas et al., 1992). The folA gene product (EC 1.5.1.3) is related to the biosynthesis of cofactors, being an important intermediary of folate metabolism. It is considered the key enzyme of this process and essential for microbial growth (Myllykallio et al., 2003). Another target worth mentioning is the aroE gene (EC 1.1.1.25), which has been described as a potential therapeutic target of both P. putida and E. coli (Peek et al., 2014).
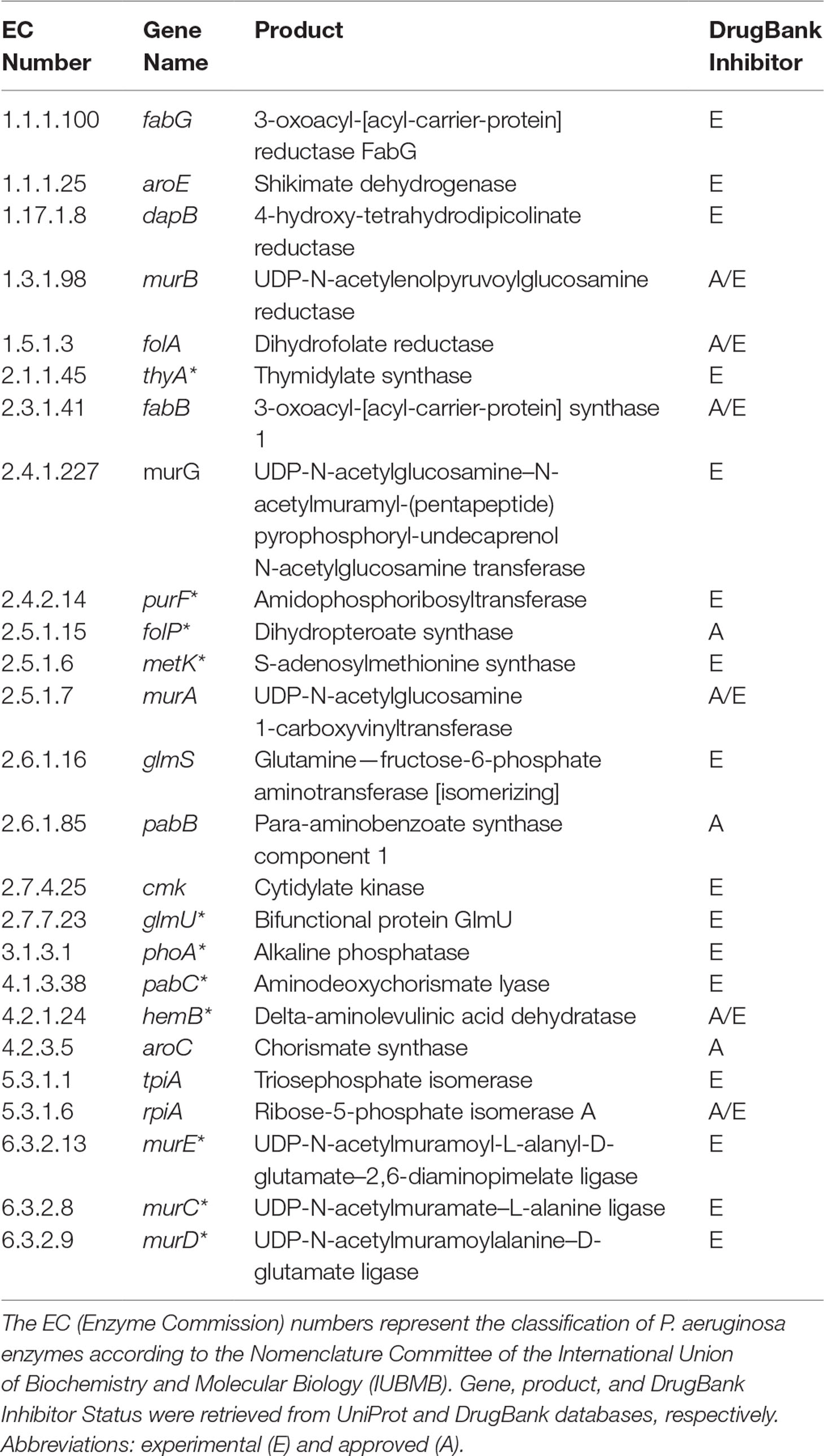
Table 2 Potential targets common to all Tier 2 P. aeruginosa metabolic network models. Common targets identified by both FBA-only and FBA+FVA methods are marked with asterisks (*). The other targets were identified by the FBA-only method but not by the FBA+FVA method.
Table 3 shows common targets with approved drugs. It is worth mentioning that several approved drugs have been identified; some of them are potential candidates for drug repositioning. Another relevant remark is the fact that most targets are also associated with experimental drugs.
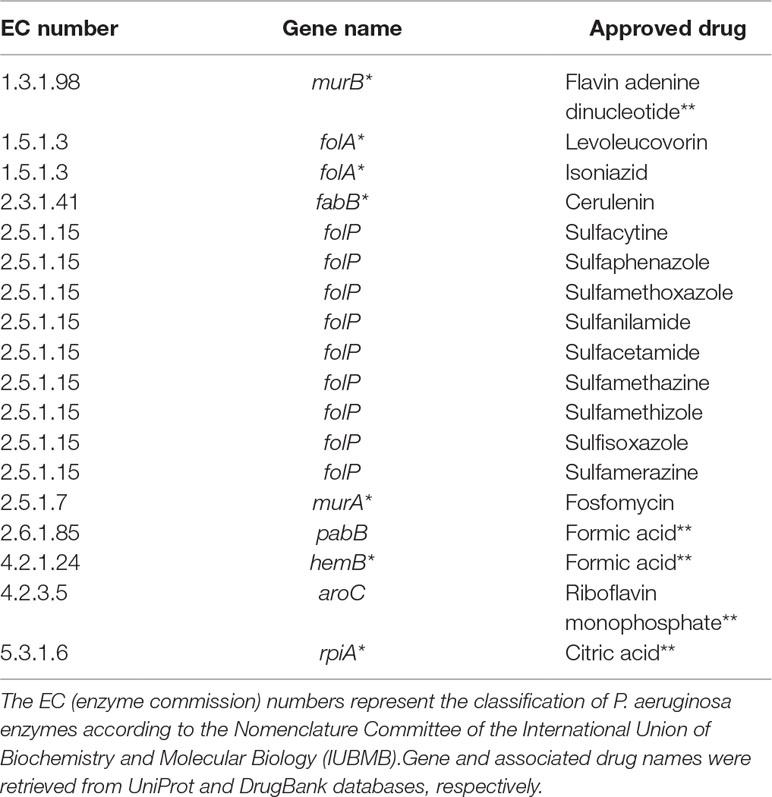
Table 3 Putative targets with approved drugs common to all Tier 2 metabolic network models of P. aeruginosa. Targets marked with asterisks are also associated with drugs in the experimental stage. Drugs marked with double asterisks are most probably artifacts inherited from DrugBank.
Another noteworthy observation is that a considerable number of approved drugs in Table 3 are most probably artifacts from the DrugBank database. For instance, flavin adenine dinucleotide (FAD), listed as an approved drug related to gene murB, is in fact approved for use in Japan under the trade name adeflavin as an ophthalmic treatment for vitamin B2 deficiency, it is just a cofactor for the product of gene murB, the enzyme UDP-N-acetylenolpyruvoylglucosamine reductase. All similar cases are highlighted with double asterisks in Table 3. This observation only reinforces a known limitation of all computational methods relying on databases at least partially annotated using automated workflows.
Considering the curated version of the metabolic network of multidrug-resistant strain P. aeruginosa CCBH4851, 17 unique targets were identified using the FBA+FVA method, while the FBA-only method returned 50 unique potential targets. Among those results, it is important to highlight four potential targets: asd, ispE, fabA, and dapA. Both asd and dapA are involved in the L-lysine biosynthesis via DAP pathway, which synthesizes L-lysine from aspartate and pyruvate. In bacteria, the lysine biosynthesis pathway yields the important metabolites meso-2,6-diaminopimelate (meso-DAP) and lysine. Lysine is utilized for protein synthesis in bacteria and forms part of the peptidoglycan cross-link structure in the cell wall of most gram-positive species, whilst meso-DAP is the peptidoglycan cross-linking moiety in the cell wall of gram-negative bacteria (Dogovski et al., 2012). This pathway is utilized by most bacteria, some archaea, some fungi, some algae, and plants (Liu et al., 2010b), and therefore are suitable candidates for therapeutic targets. Only experimental drugs are available to both targets.
ispE encodes a cytoplasmic kinase of the MEP pathway that is involved in the biosynthesis of the isoprenoids used by many gram-negative bacteria (including P. aeruginosa) (Heuston et al., 2012). Because isoprenoids are involved in a wide variety of vital biological functions, the seven enzymes without close human homologs that participate in their metabolism (encoded by dxr, ispC, ispD, ispE, ispF, ispG, ispH genes) are favorable candidate drug targets and several inhibitors have been already reported (Masini and Hirsch, 2014). Specifically for ispE, only experimental drugs are available.
fabA participates in fatty acid synthesis (FAS) processes, which includes also fabB, fabD, fabI, and fabH. The proteins encoded by these genes have an essential role during the synthesis of bacterial phospholipid membranes, lipopolysaccharide (LPS), and lipoproteins, thus representing attractive targets due to the structural differences between the human and bacterial proteins and the essentiality of FAS (Zhang et al., 2006; Leibundgut et al., 2008). Only experimental drugs are available to this target.
All four potential targets described above are reported to be overexpressed in K. pneumoniae when the pathogen is exposed to polymyxin B (Ramos et al., 2018), which is considered as a “last resort” antibiotic for infections caused by Carbapenem-resistant Enterobacteriaceae. Indeed, it has been shown that P. aeruginosa CCBH4851 is sensible only to polymyxin B (Silveira et al., 2014). This observation can be of interest in a combination therapy perspective when dealing with resistant P. aeruginosa infections, possibly acting synergistically with other drugs. An interesting observation is that the same target may be associated with similar reactions in both Tier 2 P. aeruginosa CCBH4851 and K. pneumoniae. For instance, asd is associated with the aspartate-semialdehyde dehydrogenase reaction in both metabolic networks, but reactants, products, and directionality differ. On the other hand, reactions associated with fabA differ in both metabolic network models. The gene fabA is associated to 13 reactions in K. pneumoniae and nine reactions in Tier 2 P. aeruginosa CCBH4851.
Another interesting observation is that the above targets have been identified by the FBA-only method. Only dapA is included in FBA+FVA results. One possible inference from this fact is that dapA should be prioritized over the other targets. Nevertheless, it also highlights the importance of considering both methods when looking for new potential targets.
A fifth target worth mentioning is algC, which encodes a highly reversible phosphoryltransferase. The phosphomannomutase activity produces a precursor for alginate polymerization; the alginate layer causes a mucoid phenotype and provides a protective barrier against host immune defenses and antibiotics. It is involved in core LPS biosynthesis due to its phosphoglucomutase activity and is essential for rhamnolipid production, an exoproduct correlated with pathogenicity (Olvera et al., 1999). It is also required for biofilm production (Davies and Geesey, 1995). This particular target was identified using the FBA-only method. Only experimental drugs are available to algC.
To evaluate the robustness of FindTargetsWEB regarding Tier 3 networks, which generally are networks generated automatically without manual curation, FindTargetsWEB processed a preliminary version of the metabolic network model of P. aeruginosa CCBH4851, which precedes the Tier 2 network described previously. This network is the only one in this paper which was processed using step 6b (algorithm 2) of the overall method. The growth rate of the Tier 3 version of the P. aeruginosa CCBH4851 network is 1.757 h-1, which is less consistent to the biology of P. aeruginosa spp. than the growth rate obtained by the Tier 2 version of the network. The processing of this Tier 3 network generated 32 targets in the FBA+FVA analysis, and 48 targets using the FBA-only method. It is remarkable that this less curated version of P. aeruginosa CCBH4851 network generated more potential targets in the FBA+FVA analysis than the corresponding Tier 2 network.
Among targets identified using the FVA+FBA method, 10 targets are common between the Tier 2 and Tier 3 networks. For the FBA-only analysis, 21 targets are common between the two versions. It is worth mentioning that many targets found in Tier 2 networks are present in the analysis of the CCBH4851 Tier 3 network, which corroborates the relevance of the targets found even in draft versions of metabolic networks. This comparison also highlights the importance of careful curation of automatically generated metabolic networks. For instance, from the targets discussed in the previous subsection, only dapA is present as a potential target in the Tier 3 network.
Metabolic networks of bacteria other than P. aeruginosa were also processed using FindTargetsWEB. In the previous subsections, results for P. aeruginosa metabolic network models were presented, but it is also possible to analyze networks of other species of bacteria. In this subsection, FindTargetsWEB results for a metabolic network reconstruction of K. pneumoniae MGH78578—iYL1228 (BioModels ID 1507180054) (Liao et al., 2011) and H. influenzae—iCS400 (BioModels ID 1507180053) (Schilling and Palsson, 2000) are presented (Table 4).

Table 4 List of EC numbers, product, and DrugBank inhibitor status for putative targets for metabolic network models of K. pneumoniae and H. influenzae. All targets listed in this table are included in the results of both FBA+FVA and FBA-only methods.
For K. pneumoniae, a total of 45 unique potential targets were found using the FBA+FVA method and also 45 for the FBA-only method. Some of the more representative targets are listed in Table 4 (complete results are available as Supplementary Material).
Several targets identified in Table 4 are worth mentioning. For instance, the cytoplasmic enzyme encoded by lpxA gene is involved in the initial steps of lipid A production through the Raetz pathway. As stated in the previous subsection, fabA, fabB, and fabF participate in FAS processes and represent attractive targets due to the structural differences between the human and bacterial proteins and the essentiality of FAS. The cytoplasmic protein N-acetylglutamate (NAG) kinase (encoded by argB), which promotes phosphorylation of NAG in a rate-limiting step of bacterial L-arginine production, occurs through acetylated intermediates, unlike mammals which use non-acetylated intermediates, and for this reason, it was previously considered a candidate drug target (Marcos et al., 2010). Indeed, Ramos et al. (2018) identified several potential targets found by FindTargetsWEB as priority targets for K. pneumoniae. Examples are dapD, lpxA, fabA, fabB, tmk, murE, and murD. Their analysis included a reconstruction of the metabolic network model of K. pneumoniae Kp13 and an essentiality analysis based on literature search. A target prioritization pipeline was proposed that takes into account gene essentiality, topological measures, literature information, and gene expression data. It is worth noting that neither FBA nor FVA were used in their analysis.
For the metabolic network model of H. influenzae, 16 unique potential targets were found by FindTargetsWEB for both FBA+FVA and FBA-only methods (Table 4). Complete results are available as Supplementary Material.
It is worth mentioning that the genes folA, tmk, kdsB, metG, thrS, and guaA were identified as essential for H. influenzae growth and survival by Akerley and colleagues (2002), using a high-density transposon mutagenesis strategy. Another relevant observation is the presence of potential targets common to K. pneumoniae (tmk) and P. aeruginosa (folA). Both methods, FBA+FVA and FBA-only, generate exactly the same results. Therefore, the FVA ranges for all targets in Table 4 are equal to zero.
FindTargetsWEB is also capable of processing integrated metabolic network models. The analysis presented in this subsection used a host-pathogen genome-scale reconstruction, iAB-AMØ-1410-Mt-661 (BIOMODELS ID 1011090001), which integrates a cell-specific alveolar macrophage model, iAB-AMØ-1410, from the global human metabolic reconstruction, with an M. tuberculosis H37Rv model, iNJ661 (Bordbar et al., 2010). The integrated host-pathogen network enables simulation of the metabolic changes during infection.
A total of 35 unique potential targets was identified by FindTargetsWEB on the integrated model by both the FBA+FVA and FBA-only methods (complete results are available as Supplementary Material). Several potential targets found by FindTargetsWEB in the host-pathogen integrated model have been previously reported in the literature as essential to M. tuberculosis survival (Bordbar et al., 2010; Sassetti and Rubin, 2003). Examples are nrdE, mmaA2, mmaA3, aroQ, and ahcY, from which only mmaA2 and mmaA3 have approved drugs. It is worth highlighting that the selection of potential targets of FindTargetsWEB depends not only on network analysis, but also on data retrieved from DrugBank and additional filters, such as a low level of similarity with human proteins.
None of the results presented in the previous subsections include gram-positive bacteria. To emphasize FindTargetsWEB flexibility, in this subsection, we present results from the metabolic network model analysis of a gram-positive pathogen. S. aureus is a pathogenic gram-positive bacterium that causes a variety of disease conditions both in hospital settings and in the community at large. The metabolic model iSB619 (BIOMODELS ID 1507180070) (Becker and Palsson, 2005), reconstructed from the strain N315, was processed using FindTargetsWEB. Complete results for both FBA-only and FVA+FBA are available as Supplementary Material.
A total of 27 unique potential targets were generated using the FBA-only method. The FBA+FVA analysis returned 22 unique targets. Some potential targets are common to gram-negative bacteria (such as murB, aroC), while others such as mvaA (locus tag SA2333 for the N315 strain, SAOUHSC_02859 for the NCTC8325 strain), tkt (SA1177, SAOUHSC_01337), and dfrA (SA1259, SAOUHSC_01434) are defined as essential for S. aureus in both minimal and rich medias (Becker and Palsson, 2005). Regarding the metabolic network models analyzed in this manuscript, the potential targets mvaA, tkt, and dfrA only appear in the S. aureus metabolic network model.
The pseudomonads include a diverse set of bacteria whose metabolic versatility and genetic plasticity have enabled their survival in a broad range of environments. Many members of this family are able to either degrade toxic compounds or to efficiently produce high value compounds and are therefore of interest for both bioremediation and bulk chemical production. P. putida is a representative of those industrially relevant pseudomonads. In this subsection, an analysis of the metabolic network model of the P. putida KT2440 (Puchałka et al., 2008), named iJP815 (BIOMODELS ID 1507180044), is compared to the previous analysis of a pathogenic member of the family, P. aeruginosa. Complete results for the analysis of the P. putida metabolic network model is available as supplementary material.
A first comparison between P. putida e P. aeruginosa metabolic network models is the number of potential targets. The analysis of the metabolic network model of P. putida returned a comparable number of potential targets: 52 for FBA-only, 50 for the FBA+FVA method (see Table 1). Indeed, the size of the metabolic network model iJP815 is comparable with other P. aeruginosa metabolic networks: 824 intracellular and 62 extracellular metabolites connected by 877 reactions. Other interesting observation is that some targets present in the multidrug-resistant P. aeruginosa CCBH4851 are absent in P. putida, despite the comparable number of potential targets. Remarkable examples are asd, ispE, fabA, dapA, and algC. Indeed, from the 25 targets common to all Tier 2 P. aeruginosa metabolic network model displayed in Table 2 (FBA-only method), only 18 are also potential targets for the P. putida KT2440 metabolic network model.
Several advantages of the proposed method can be highlighted: first the robustness of the system, which can identify potential targets even for draft (Tier 3) networks, pointing out that such metabolic network models are very common and are the only models available for some organisms. The system is deployed as a web application and is asynchronous: the user is notified when results are available. The performance of the system is optimized, since the COBRApy framework can make use of multiple cores available in the host machine, and it is able to process the metabolic network of various bacteria, as described in the previous section. The only requirement is the availability of an SBML level 3 file describing the corresponding genome-scale metabolic network. The user interface is straightforward (see Figures 1 and 2), and the user should only provide a name, an e-mail address, and the corresponding SBML file. The user should also indicate the species of bacterium associated with the metabolic network model. FindTargetsWEB is a highly flexible tool, capable of processing genome-scale metabolic network models of gram-negative bacteria, gram-positive bacteria, bacteria not classified as either gram-positive or gram-negative, and even integrated host-pathogen genome-scale metabolic network models.
Other proposals for the analysis of metabolic networks at genomic scale are available in the literature. Chavali et al. (2012) used FBA and FVA for identification of potential targets, but their application does not propose any drugs for the targets found neither describes the potential targets in detail. The procedure reported in (Oberhardt et al., 2010) describes a processing similar to the one proposed in this work up to the EC number mapping step, and then uses graphical tools to identify the potential targets for E. coli and Bacillus subtilis, without pinpointing any potential drug. Ramos et al. (2018) propose a method to identify drug targets in metabolic network model of K. pneumoniae. However, their method is not automated, and it was not applied to other species of bacteria. None of these works go as far as FindTargetsWEB, which can process metabolic network models of several species of bacteria, identify potential targets, confirm homology with the analyzed gene, and identify all available drugs for the potential target in a fully automated manner.
Regarding the options to identify potential targets, i.e., FBA+FVA and FBA-only, one can conclude that the FBA+FVA method represents a way to prioritize the targets identified by the FBA-only method, since the set of targets identified by FBA+FVA is a proper subset of the set of targets identified by FBA-only. However, as stated in the detailed description of the targets of the Results section, potential targets that are associated with the FBA-only method and do not appear as results of the FBA+FVA method should not be disconsidered. Many important targets described in the literature have a FVA range greater than zero, and a careful analysis of both sets of potential targets is advised.
Several of the approved drugs identified by FindTargetsWEB are already used against P. aeruginosa and other bacteria and can be effective against non multidrug-resistant strains. As expected, for the multidrug-resistant strain, most of the approved drugs are not effective. For instance, it is known that P. aeruginosa can be resistant to both to trimethoprim and sulfamethoxazole (see Table 3) due to the MexAB-OprM multidrug efflux system (Köhler et al., 1996). Nevertheless, FindTargetsWEB also pinpoints a large number of experimental drugs that can be effective. Actually, most of the targets identified by FindTargetsWEB for all strains are associated to experimental drugs and may represent new therapeutic options. Clearly, additional in vitro and in vivo testings are needed in order to confirm the experimental drugs as new therapeutic options.
Additional information provided by FindTargetsWEB can also be considered in the definition of new strategies to fight multidrug-resistant bacteria. Information such as pathway, target function, and catalytic activity can be considered in order to devise a multi-target strategy, which can be very effective in some scenarios. As an example of a multi-target strategy, in bacteremia caused by P. aeruginosa, the combination of efflux pump inhibitors and iron chelators has been proposed to control the infection process in view of the overexpression of the MexAB-OprM efflux system during iron deprivation (Liu et al., 2010a). Indeed, several targets in the analysis of results for P. aeruginosa are related to different cellular functions. Targeting several cellular functions and processes at the same time can be a more promising strategy than considering only one isolated target. For instance, it is known that inhibiting bacterial growth can accelerate the process of biofilm formation (Xu et al., 2013). Therefore, the pathogen can form a biofilm before it is eliminated. Multi-target therapies are already commonplace in treating bacteria infections, and the wealth of information provided by FindTargetsWEB can be used to define new multi-target treatments not considered before. For instance, algC (P. aeruginosa CCBH4851, PA14, and PAO1-2017 metabolic networks) is both essential to metabolic growth and biofilm formation, according to the FUNCTION field returned by FindTargetsWEB and literature sources (Davies and Geesey, 1995). Therefore, a targeting strategy based on other genes may consider also targeting algC to prevent biofilm formation.
FindTargetsWEB is a user-friendly web application that combines bioinformatics and systems biology, providing insights of new therapeutic targets for multidrug-resistant bacteria, increasing the available therapeutic options. By identifying more effectively potential targets along with candidate active compounds for posterior experimental confirmation, this tool prevents exhaustive bacterial drug screening. Importantly, FindTargetWEB can also be applied to the study of other bacteria due to the flexibility proposed by computational modeling, serving as a base for other relevant studies. In addition, it will serve as a starting point for the creation of even more complete applications in a web environment, such as one capable of processing integrated computational models and retrieving data from more databases.
Project name: FindTargetsWEB
Project home page: http://pseudomonas.procc.fiocruz.br/FindTargetsWEB
Operating system: e.g. Web-based, Platform independent
Programming language: Python 3.6
Other requirements: An updated web browser (e.g. Google Chrome, Mozilla Firefox, Apple Safari, Microsoft Edge)
License: Not Applicable
Any restriction to use by non-academics: Not Applicable
The user must provide a SBML level 3 file describing the metabolic network reconstruction and an e-mail address to which the results will be forwarded.
All datasets generated for this study are included in the manuscript and the supplementary files.
TCM, FPSJ, and FABS designed the system. TCM was the main programmer. MWC and ADC-A tested the system and evaluated its correctness. All authors have equally participated in the writing of this paper.
The authors would like to thank CAPES, FAPERJ, CNPq and FIOCRUZ (INOVA-FIOCRUZ VPPCB-007-FIO-18-2-29) for financial support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00633/full#supplementary-material
Akerley, B. J., Rubin, E. J., Novick, V. L., Amaya, K., Judson, N., Mekalanos, J. J. (2002). A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc. Natl. Acad. Sci. 99 (2), 966–971. doi: 10.1073/pnas.012602299
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215 (3), 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bartell, J. A., Blazier, A. S., Yen, P., Thøgersen, J. C., Jelsbak, L., Goldberg, J. B., et al. (2017). Reconstruction of the metabolic network of Pseudomonas aeruginosa to interrogate virulence factor synthesis. Nat. Commun 8, 14631. doi: 10.1038/ncomms14631
Becker, S. A., Palsson, BØ. (2005). Genome-scale reconstruction of the metabolic network in Staphylococcus aureus N315: an initial draft to the two-dimensional annotation. BMC Microbiol. 5 (1), 8. doi: 10.1186/1471-2180-5-8
Benson, T. E., Walsh, C. T., Hogle, J. M. (1996). The structure of the substrate-free form of MurB, an essential enzyme for the synthesis of bacterial cell walls. Structure 4 (1), 47–54. doi: 10.1016/S0969-2126(96)00008-1
Bordbar, A., Lewis, N. E., Schellenberger, J., Palsson, BØ., Jamshidi, N. (2010). Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol. Syst. Biol. 6 (1), 422. doi: 10.1038/msb.2010.68
Brown, A. D. (1957). Some general properties of a psychrophilic pseudomonad: the effects of temperature on some of these properties and the utilization of glucose by this organism and Pseudomonas aeruginosa Microbiology 17 (3), 640–648. doi: 10.1099/00221287-17-3-640
Caspi, R., Billington, R., Ferrer, L., Foerster, H., Fulcher, C. A., Keseler, I. M., et al. (2015). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 44 (D1), D471–D480. doi: 10.1093/nar/gkv1164
Chavali, A. K., D’Auria, K. M., Hewlett, E. L., Pearson, R. D., Papin, J. A. (2012). A metabolic network approach for the identification and prioritization of antimicrobial drug targets. Trends Microbiol. 20, 113–123. doi: 10.1016/j.tim.2011.12.004
Dallas, W. S., Gowen, J. E., Ray, P. H., Cox, M. J., Dev, I. K. (1992). Cloning, sequencing, and enhanced expression of the dihydropteroate synthase gene of Escherichia coli MC4100. J. Bacteriol. 174 (18), 5961–5970. doi: 10.1128/jb.174.18.5961-5970.1992
Davies, D. G., Geesey, G. G. (1995). Regulation of the alginate biosynthesis gene algC in Pseudomonas aeruginosa during biofilm development in continuous culture. Appl. Environ. Microbiol. 61 (3), 860–867.
Dogovski, C., Atkinson, S. C., Dommaraju, S. R., Downton, M., Hor, L., Moore, S., et al. (2012). Enzymology of bacterial lysine biosynthesis. Biochemistry 1, 225–262. doi: 10.5772/34121
Ebrahim, A., Lerman, J. A., Palsson, BØ, Hyduke, D. R. (2013). COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 7, 74–79. doi: 10.1186/1752-0509-7-74
Glont, M., Nguyen, T. V. N., Graesslin, M., Hälke, R., Ali, R., Schramm, J., et al. (2017). BioModels: expanding horizons to include more modelling approaches and formats. Nucleic Acids Res. 46 (D1), D1248–D1253. doi: 10.1093/nar/gkx1023
Gudmundsson, S., Thiele, I. (2010). Computationally efficient flux variability analysis. BMC Bioinf. 11(1), 489. doi: 10.1186/1471-2105-11-489
Heuston, S., Begley, M., Gahan, C. G., Hill, C. (2012). Isoprenoid biosynthesis in bacterial pathogens. Microbiology 158 (6), 1389–1401. doi: 10.1099/mic.0.051599-0
Hucka, M., Bergmann, F. T., Hoops, S., Keating, S. M., Sahle, S., Schaff, J. C., et al. (2015). The Systems Biology Markup Language (SBML): language specification for level 3 version 1 core. J. Integrat. Bioinf. 12 (2), 382–549. doi: 10.1515/jib-2015-266
Hyduke, D., Schellenberger, J., Que, R., Fleming, R., Thiele, I., Orth J., et al. (2011). COBRA Toolbox 2.0. Protoc. Exch. 22. doi: 10.1038/protex.2011.234
Kerr, K. G., Snelling, A. M. (2009). Pseudomonas aeruginosa: a formidable and ever-present adversary. J. Hosp. Infect. 73, 338–344. doi: 10.1016/j.jhin.2009.04.020
Köhler, T., Kok, M., Michea-Hamzehpour, M., Plesiat, P., Gotoh, N., Nishino, T., et al. (1996). Multidrug efflux in intrinsic resistance to trimethoprim and sulfamethoxazole in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 40 (10), 2288–2290. doi: 10.1128/AAC.40.10.2288
Kozakov, D., Hall, D. R., Napoleon, R. L., Yueh, C., Whitty, A., Vajda, S. (2015). New frontiers in druggability. J. Med. Chem. 58 (23), 9063–9088. doi: 10.1021/acs.jmedchem.5b00586
Leibundgut, M., Maier, T., Jenni, S., Ban, N. (2008). The multienzyme architecture of eukaryotic fatty acid synthases. Curr. Opin. Struct. Biol. 18 (6), 714–725. doi: 10.1016/j.sbi.2008.09.008
Liao, Y.-C., Huang, T.-W., Chen, F.-C., Charusanti, P., Hong, J. S. J., Chang, H.-Y., et al. (2011). An experimentally validated genome-scale metabolic reconstruction of Klebsiella pneumoniae MGH 78578, iYL1228. J. Bacteriol. 193, 1710–1717. doi: 10.1128/JB.01218-10
Liu, Y., Yang, L., Molin, S. (2010a). Synergistic activities of an efflux pump inhibitor and iron chelators against Pseudomonas aeruginosa growth and biofilm formation. Antimicrob. Agents Chemother. 54, 3960–3963. doi: 10.1128/AAC.00463-10
Liu, Y., White, R. H., Whitman, W. B. (2010b). Methanococci use the diaminopimelate aminotransferase (DapL) pathway for lysine biosynthesis. J. Bacteriol. 192 (13), 3304–3310. doi: 10.1128/JB.00172-10
Marcos, E., Ramon, C., Ivet, B. (2010). On the conservation of the slow conformational dynamics within the amino acid kinase family: NAGK the paradigm. PLoS Computat. Biol 6 (4), e1000738. doi: 10.1371/journal.pcbi.1000738
Masini, T., Hirsch, A. K. (2014). Development of inhibitors of the 2 C-Methyl-d-erythritol 4-phosphate (MEP) pathway enzymes as potential anti-infective agents. J. Med. Chem. 57 (23), 9740–9763. doi: 10.1021/jm5010978
Myllykallio, H., Leduc, D., Filee, J., Liebl, U. (2003). Life without dihydrofolate reductase FolA. Trends Microbiol. 11, 220–223. doi: 10.1016/S0966-842X(03)00101-X
Oberhardt, M. A., Puchałka, J., Fryer, K. E., Martins Dos Santos, V. A. P., Papin, J. A. (2008). Genome-scale metabolic network analysis of the opportunistic pathogen Pseudomonas aeruginosa PAO1. J. Bacteriol. 190, 2790–2803. doi: 10.1128/JB.01583-07
Oberhardt, M. A., Goldberg, J. B., Hogardt, M., Papin, J. A. (2010). Metabolic network analysis of Pseudomonas aeruginosa during chronic cystic fibrosis lung infection. J. Bacteriol. 192 (20), 5534–5548. doi: 10.1128/JB.00900-10
Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., Kanehisa, M. (1999). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34. doi: 10.1093/nar/27.1.29
Olvera, C., Goldberg, J. B., Sánchez, R., Soberón-Chávez, G. (1999). The Pseudomonas aeruginosa algC gene product participates in rhamnolipid biosynthesis. FEMS Microbiol. Lett. 179 (1), 85–90. doi: 10.1111/j.1574-6968.1999.tb08712.x
Orth, J. D., Thiele, I., Palsson, BØ (2010). What is flux balance analysis? Nat. Biotechnol. 28, 245–248. doi: 10.1038/nbt.1614
Peek, J., Shi, T., Christendat, D. (2014). Identification of novel polyphenolic inhibitors of shikimate dehydrogenase (AroE). J. Biomol. Screen 19 (7), 1090–1098. doi: 10.1177/1087057114527127
Puchałka, J., Oberhardt, M. A., Godinho, M., Bielecka, A., Regenhardt, D., Timmis, K. N., et al. (2008). Genome-scale reconstruction and analysis of the Pseudomonas putida KT2440 metabolic network facilitates applications in biotechnology. PLoS Computat. Biol. 4 (10), e1000210. doi: 10.1371/journal.pcbi.1000210
Ramos, P. I. P., Do Porto, D. F., Lanzarotti, E., Sosa, E. J., Burguener, G., Pardo, A. M., et al. (2018). An integrative, multi-omics approach towards the prioritization of Klebsiella pneumoniae drug targets. Sci. Rep. 8 (1), 10755. doi: 10.1038/s41598-018-28916-7
Rienksma, R. A., Suarez-Diez, M., Spina, L., Schaap, P. J., Martins Dos Santos, V. A. P. (2014). Systems-level modeling of mycobacterial metabolism for the identification of new (multi-)drug targets. Semin. Immunol. 26, 610–622. doi: 10.1016/j.smim.2014.09.013
Sassetti, C. M., Rubin, E. J. (2003). Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 100, 12989–12994. doi: 10.1073/pnas.2134250100
Schilling, C. H., Palsson, BØ. (2000). Assessment of the metabolic capabilities of Haemophilus influenzae Rd through a genome-scale pathway analysis. J. Theor. Biol. 203, 249–283. doi: 10.1006/jtbi.2000.1088
Seto, M., Noda, M. (1982). Growth rate, biomass production and carbon balance of Pseudomonas aeruginosa at pH extremes in a carbon-limited medium. Jap. J. Limnol. 43(4), 263–271. doi: 10.3739/rikusui.43.263
Silva, F. A. B., Medeiros Filho, F., Merigueti, T. C., Giannini, T., Brum, R., de Faria, L. M., et al., (2018). “Computational Modeling of Multidrug-Resistant Bacteria,” in Theoretical and Applied Aspects of Systems Biology. Eds. Silva, F. A. B., Carels, N., Silva-Jr, F., Cham, Switzerland: Springer International Publishing AG, 195–220. doi: 10.1007/978-3-319-74974-7_11
Silveira, M., Albano, R., Asensi, M., Assef, A. P. C. (2014). The draft genome sequence of multidrug-resistant Pseudomonas aeruginosa strain CCBH4851, a nosocomial isolate belonging to clone SP (ST277) that is prevalent in Brazil. Mem. Inst. Oswaldo Cruz 190, 1086–1087. doi: 10.1590/0074-0276140336
Stone, P. W., Braccia, D., Larson, E. (2005). Systematic review of economic analyses of health care-associated infections. Am. J. Infect. Control 33, 501–509. doi: 10.1016/j.ajic.2005.04.246
Thiele, I., and Palsson, BØ. (2010). A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121. doi: 10.1038/nprot.2009.203
UniProt Consortium (2018). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 46 (5), 2699. doi: 10.1093/nar/gky092
Van Rossum, G., Drake, F. L. (2003). An introduction to Python. Bristol: Network Theory Ltd. 115 pp.
WHO (World Health Organization) (2014). Antimicrobial resistance: global report on surveillance. Geneva: World Health Organization.
WHO (World Health Organization) (2017). Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. Geneva: World Health Organization.
Wishart, D. S., Knox, C., Guo, A. C., Cheng, D., Shrivastava, S., Tzur, D., et al. (2008). DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 36, D901–D906. doi: 10.1093/nar/gkm958
Xu, Z., Fang, X., Wood, T. K. (2013). Huang ZJ. A system-level approach for investigating Pseudomonas aeruginosa biofilm formation. PLoS One 8 (2), e57050. doi: 10.1371/journal.pone.0057050
Yang, L., Haagensen, J. A., Jelsbak, L., Johansen, H. K., Sternberg, C., Høiby, N., et al. (2008). In situ growth rates and biofilm development of Pseudomonas aeruginosa populations in chronic lung infections. J. Bacteriol. 190 (8), 2767–2776. doi: 10.1128/JB.01581-07
Keywords: systems biology, flux balance analysis, metabolic network, COBRA analysis, Python (programming language)
Citation: Merigueti TC, Carneiro MW, Carvalho-Assef APD’A, Silva-Jr FP and Silva FAB (2019) FindTargetsWEB: A User-Friendly Tool for Identification of Potential Therapeutic Targets in Metabolic Networks of Bacteria. Front. Genet. 10:633. doi: 10.3389/fgene.2019.00633
Received: 27 March 2019; Accepted: 17 June 2019;
Published: 04 July 2019.
Edited by:
Helder Nakaya, University of São Paulo, BrazilReviewed by:
Priyanka Baloni, Institute for Systems Biology (ISB), United StatesCopyright © 2019 Merigueti, Carneiro, Carvalho-Assef, Silva-Jr and Silva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Floriano Paes Silva-Jr, Zmxvcmlhbm9AaW9jLmZpb2NydXouYnI=; Fabricio Alves Barbosa da Silva, ZmFicmljaW8uc2lsdmFAZmlvY3J1ei5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.