 Xiao-Tun Ren1
Xiao-Tun Ren1 Xiao-Hui Wang1Chang-Hong Ding1
Xiao-Hui Wang1Chang-Hong Ding1 Xiang Shen2Hao Zhang2Wei-Hua Zhang1Jiu-Wei Li1Chang-Hong Ren1
Xiang Shen2Hao Zhang2Wei-Hua Zhang1Jiu-Wei Li1Chang-Hong Ren1 Fang Fang1*
Fang Fang1*- 1Department of Neurology, National Centre for Children's Health, Beijing Children's Hospital, Capital Medical University, Beijing, China
- 2Running Gene Inc., Beijing, China
Neuronal Ceroid Lipofuscinoses (NCLs) are progressive degenerative diseases mainly affect brain and retina. They are characterized by accumulation of autofluorescent storage material, mitochondrial ATPase subunit C, or sphingolipid activator proteins A and D in lysosomes of most cells. Heterogenous storage material in NCLs is not completely disease-specific. Most of CLN proteins and their natural substrates are not well-characterized. Studies have suggested variants of Late-Infantile NCLs (LINCLs) include the major type CLN2 and minor types CLN5, CLN6, CLN7, and CLN8. Therefore, combination of clinical and molecular analysis has become a more effective diagnosis method. We studied 4 late-infantile NCL siblings characterized by seizures, ataxia as early symptoms, followed by progressive regression in intelligence and behavior, but mutations are located in different genes. Symptoms and progression of 4 types of LINCLs are compared. Pathology of LINCLs is also discussed. We performed Nest-Generation Sequencing on these phenotypically similar families. Three novel variants c.1551+1insTGAT in TPP1, c.244G>T in CLN6, c.554-5A>G in MFSD8 were identified. Potential outcome of the mutations in structure and function of proteins are studied. In addition, we observed some common and unique clinical features of Chinese LINCL patient as compared with those of Western patients, which greatly improved our understanding of the LINCLs.
Introduction
NCLs (Neuronal Ceroid Lipofuscinoses) are characterized by accumulation of lysosomal autofluorescent storage material and progressive neurodegeneration (Oishi et al., 1999; Getty and Pearce, 2011; Blom et al., 2013; Kollmann et al., 2013; Mink et al., 2013; Sands, 2013; Patino et al., 2014). The most common clinical features of NCLs include epileptic seizures, progressive regression of intelligence, loss of motor function, retinal degeneration, and premature death (Haltia and Goebel, 2013; Mink et al., 2013; Warrier et al., 2013; De Silva et al., 2015). NCL is one of the most frequent classes of childhood-onset neurodegenerative diseases with prevalence around 0.5–8 per 100,000 live births varying by the regions (Oishi et al., 1999; Getty and Pearce, 2011; Cotman et al., 2013; Haltia and Goebel, 2013; Beltran et al., 2018). So far there are 13 genes identified as candidate genes of NCLs, i.e., CLN1/PPT1, CLN2/TPP1, CLN3, CLN4/DNAJC5, CLN5, CLN6, CLN7/MFSD8, CLN8, CLN10/CTSD, CLN11/GRN, CLN12/ATP13A2, CLN13/CTSF, and CLN14/KCTD7 (Kollmann et al., 2013; Warrier et al., 2013). Most human NCLs follow autosomal recessive model except that caused by CLN4/DNAJC5 gene which presents autosomal dominant form (Haltia and Goebel, 2013; Kollmann et al., 2013; Warrier et al., 2013). However, the storage material of NCLs is not disease-specific and the function of most CLN protein has not been well classified. It has been difficult to diagnose merely based on clinical findings. Enzyme assay is used to help diagnose NCLs with mutations in enzyme-coding genes, CLN1/PPT1, CLN2/TPP1, CLN10/CTSD with deficiency of respective enzymes (Kamate et al., 2012; Mole and Williams, 2013). Another enzyme-coding genes, CLN13/CTSF, as a novel candidate gene identified in year 2013 is also presumably to cause impairment in enzyme cathepsin F (Mole and Williams, 2013; Schulz et al., 2013), but no supportive experiment due to few cases. Merely based on clinical presentations, NCL could not be easily distinguished from other diseases including Leber's Hereditary Optic Neuropathy (LHON), the symptoms of which include sezures, regression, ataxia, and vision impairment (Fang et al., 2017). Due to limitations of other methods, sequencing has emerged as an effective diagnosis method.
By onset ages, NCLs are classified into congenital, infantile, late-infantile, juvenile, and adult NCLs. Late-Infantile NCLs (LINCLs) include the classic CLN2 disease and variant CLN5, CLN6, CLN7, and CLN8 disease (Getty and Pearce, 2011; Mole and Williams, 2013; Schulz et al., 2013; Warrier et al., 2013; Patino et al., 2014). In this study, we present four families with CLN2, CLN5, CLN6, and CLN7 disease, respectively.
CLN2 disease caused by mutations in CLN2 gene which encodes the tripeptidyl peptidase 1, a lysosomal serine protease that removes tripeptides from N-terminus of peptides (Kollmann et al., 2013). Mitochondrial ATP-synthase subunit C, a significant component of storage material in LINCL has been demonstrated as one of the substrates of TPP1 (Ezaki et al., 2000). Deficiency of TPP1 activity results in accumulation of mitochondrial ATP-synthase subunit C, which may be the pathology of LINCL.
CLN5 gene encodes a soluble polypeptide which predominantly colocalizes with lysosomal-associated membrane protein-1 (LAMP1). Mutations in CLN5 may cause retention in the ER/Golgi (Isosomppi et al., 2002; Lebrun et al., 2009; Schmiedt et al., 2010). Since CLN5 is a highly glycosylated protein, it may play an essential role as a sensor in trafficking or integrity of lysosomes (Kollmann et al., 2013).
CLN6 gene encodes a highly conserved membrane protein that exclusively resides in endoplasmic reticulum (ER) (Heine et al., 2004; Kollmann et al., 2013). However, the exact function of CLN6 is still unknown. Previous experiment shows that loss of CLN6 activity may affect lysosomal degradation of Arylsulfatase A (Heine et al., 2004). This finding may indicate CLN6 could play a role in degradation involving ER. Besides, CLN6 was also proved to interact with Collapsin Response Mediator Protein-2 (CRMP-2). This interaction probably affects maturation and integrity of axonal outgrowth thus contribute to neuronal dysfunction of LINCL patients (Benedict et al., 2009).
CLN7/MFSD8 encodes a lysosomal membrane protein called Major Facilitator Superfamily Domain-containing protein 8 (MFSD8). This protein is ubiquitously expressed with several splicing variants. It could transport small solutes by electrochemical gradients (Siintola et al., 2007). However, the specific substrates of CLN7 require further investigation.
In the present study, we found three novel mutations of LINCLs. They are likely pathogenic by analyzing their functional consequences and correlation with the phenotypes. In addition, we also observed some common and unique clinical features of Chinese LINCL patient as compared with those of Western patients, which may improve understanding of the LINCLs.
Case Presentation
Eight patients including four probands were born in four healthy non-consanguineous Chinese families with normal pregnancy and perinatal history. Pedigrees of four families were presented in Figure 1A. All four families possess unremarkable family history. Typically, unsteady gait was observed between age 3–5 as the initial symptom. Only in Family one, seizures were observed as the first symptom at slightly earlier ages from 8 months to 3 years old. Regression in cognition and behavior were then observed in all affected children. Ataxia, seizures were also presented in all patients. Low vision was found in a few patients at this stage. At later stage, most patients lost the ability to sit, stand or walk unaided, they also lost their vision. Two patients in family one died at age seven, one patient in family three died at age 16 (for a comparison of all patients see Figure 1B).
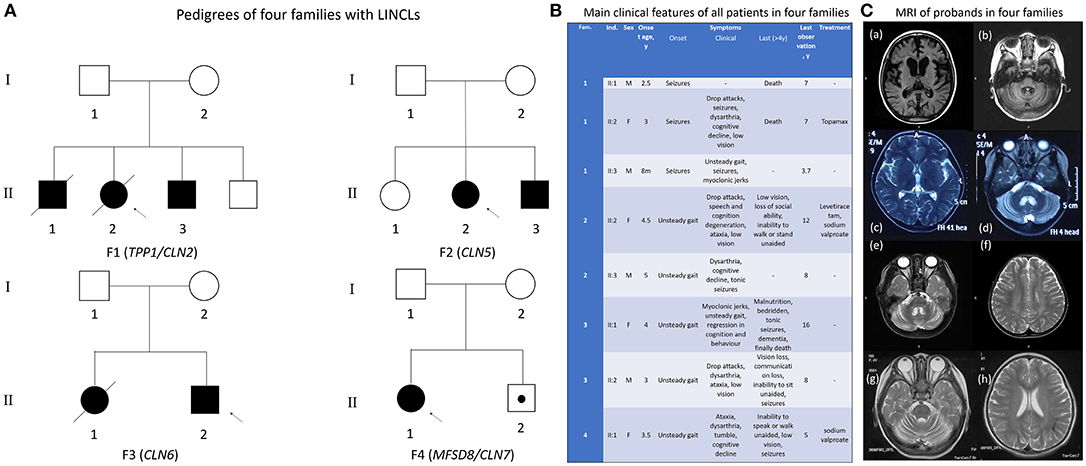
Figure 1. Clinical information of all patients in four families. (A) Pedigrees of the four families. Proband (F1 II:2) and proband's elder brother (F1 II:1) in family 1 and proband's elder sister (F3 II:1) in family 3 passed away. The little brother of proband in family 4 (F4 II:2) was just 1 year old and did not reach the general onset age, 3–4.5 years old. However, he was identified with the same pathogenic gene as his sister, the proband of family 4. (B) Overview of the main clinical features of all patients. Clinical course of all patients is nearly identical except for the earlier onset age and different initial symptom in family 1. (C) MRI images from 4 probands demonstrating severe cerebellar atrophy in all patients. Atrophy in cerebrum and brain stem was observed in proband 1 of family 1 (a, b). Abnormal myelination in white matter was found in proband 3 of family 3.
Cerebellar atrophy was confirmed by MRI imaging in all four probands. Proband in family 1 also had very significant cerebral atrophy accompanied by atrophy of brain stem, while proband in family three had abnormal myelination (Figure 1C). Only symptomatic treatment was used for patients, whereas no obvious improvement was observed. Proteolytic activity of TPP1 was completely lost in proband 1 (individual II:2 of family 1). The precise information of patients was collected in Table S1. All phenotypes of patients was standardized as HPO terms.
Proband 1 (II:2 of family 1) presented general epileptic discharges accompanied by burst-suppression. For proband 2 (II:1 of family 2), EEG background rhythm was slow, general medium to high amplitude slow waves, transient, or continuous slow spike-and-waves was observed. EEG of proband 3 (II:2 of family 3) showed frequent spike-and-wave and slow spike-and-wave discharged during the sleep period in right central electrode. For proband 4 (II:1 of family 4), massive epileptic discharges were noticed.
Since nearly all the probands have siblings who presents similar phenotypes, Whole-Exome Sequencing was performed to investigate the molecular genetic basis of the disease in these family. Sanger sequencing was performed to confirm the identified mutations.
Informed consents for genetic analyses were obtained from the children's parents. The study was approved by the ethics committee of Beijing Children's Hospital. Written informed consent was obtained from the patients' parents for the publication of this report and any accompanying images.
Methods
Reference Sequence
All positions in the mutated genes are annotated to reference sequence, namely RefSeq NM_000391, Ensembl ENST00000299427 for TPP/CLN2; NM_006493, Ensembl ENST00000377453 for CLN5; NM_017882, Ensembl ENST00000249806 for CLN6, NM_152778, Ensembl ENST00000296468 for MFSD/CLN7 in this publication. Whole Exome Sequence data were mapped and aligned to Human Genome Build GRCh37/hg19.
Next-Generation Sequencing
Proband DNA was sequenced to discover the causal gene. DNA was isolated from peripheral blood using DNA Isolation Kit (Bioteke, AU1802). One microgram of genomic DNA was fragmented into 200–300 bp length by Covaris Acoustic System. The DNA fragments were then processed by end-repairing, A-tailing and adaptor ligation, a 4-cycle pre-capture PCR amplification, targeted sequences capture. Captured DNA fragments were eluted and amplified by 15 cycle post capture PCR. The final products were sequenced with 150-bp paired-end reads on Illumina HiSeq X platform according to the standard manual. The raw data produced on HiSeq X were filtered and aligned against the human reference genome (hg19) using the BWA Aligner (http://bio-bwa.sourceforge.net/). The quality recalibration was performed using GATK Base Recalibrator(Genome Analysis ToolKit) (www.broadinstitute.org/gatk). The single-nucleotide polymorphisms (SNPs) and small insertions or deletions (indel) were called by GATK Unified Genotyper (Genome Analysis ToolKit) (www.broadinstitute.org/gatk). Variants were annotated using ANNOVAR (annovar.openbioinformatics.org/en/latest/).
Method of Mapping, Genotype, SNP Calling, and Indel Calling
Image analysis and base calling were performed using the Illumina Pipeline. BWA Aligner (http://bio-bwa.sourceforge.net/) was used to align clean reads to human reference genome (hg19), the parameters were set as default. The alignment result was then passed to GATK to identify the breakpoints, the parameters were set as “mismatch Fraction = 0.05, lod = 5, masReadsF or Realignment = 30,000, maxReadsInRam = 1,000,000.”
We selected variations obtained from exome sequencing with minor allele frequencies <0.05 in any of the following databases (dbSNP, Hapmap, 1000 Genomes Project). Effects of single-nucleotide variants (SNVs) were predicted by SIFT, Polyphen-2, and MutationTaster programs. All variants were interpreted according to ACMG standards and categorized to be pathogenic, likely pathogenic, variants of unknown clinical significance (VUS), likely benign, and benign. We further compared the rest of the deleterious variations in the patients with their unaffected parents and investigated the function of all identified genes according to the published reports and OMIM database.
Sanger Sequencing
The candidate causal genes discovered via WES were then confirmed by Sanger sequencing and co-segregation analyses among the family were also conducted. The primers were designed using Primer Premier 5.0 (Premier Biosoft) and PCR was carried out to amplify the fragments covering the mutated sites. The PCR products were further purified with Zymoclean PCR purification Kit and then sequenced by ABI 3730 DNA Sequencer (Applied Biosystems, Foster City, CA, United States). Sanger sequencing results were analyzed by Chromas Lite v2.01 (Technelysium Pty Ltd., Tewantin, QLD, Australia).
Result
Variants Identified by Whole Exome Sequencing
Next-Generation Sequencing was carried out in exome of probands. All four probands were identified with mutations in TPP1/CLN2, CLN5, CLN6, and MFSD8/CLN7, respectively (Table 1, Figure S1). Among these seven mutations, c.1551+1insTGAT of TPP1 gene, c.244G>T of CLN6 and c.554-5A>G of MFSD8 gene are novel mutations that haven't been reported before. According to ACMG guidelines, mutation c.1551+1insTGAT of TPP1 was interpreted as pathogenic since this mutation is a null variant (PVS1), with extremely low frequency (PM2), and the phenotype of the patient is specific for the disease that related to the gene (PP4). Mutation c.244G>T of CLN6 can be classified as likely pathogenic since it is absent from controls (PM2), was detected in trans with a recently reported pathogenic variant c.892G>A (PM3) co-segregated in multiple affected family members (PP1), predicted to be deleterious by multiple lines of computational evidence (PP3) i.e., predicted to be damaging by SIFT with score 0, deleterious by PROVEAN with score −5.92, probably damaging by PolyPhen-2 with score 1.00, disease causing by MutationTaster with score >0.99, the phenotype of the patient was also specific for disease CLN6 (PP4). Intronic variant c.554-5A>G of MFSD8 was predicted to be VUS (variants of uncertain significance) according to ACMG standard, since this variant is absent from controls (PM2), detected in trans with a pathogenic variant c.1444C>T (PM3), and the phenotype of patient was similar to CLN7 (PP4).
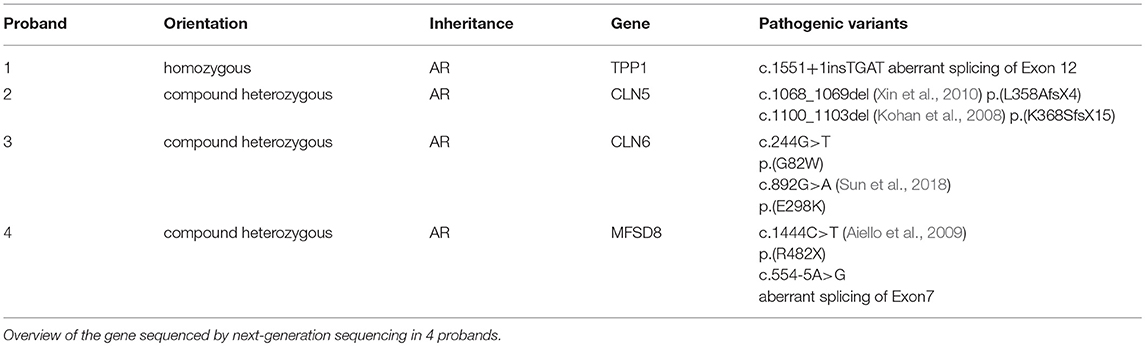
Table 1. Gene sequencing result of 4 patients with NCL.
Sanger Sequencing
All variants identified in Next-Generation Sequencing were then confirmed in other family members by Sanger Sequencing. Both parents of probands are the carriers of one of the two mutations, respectively. Other patients in the family also carry the same mutation as the proband. All results are shown in Figure S2.
Discussion
NCLs are a group of neurological diseases without typical clinical symptoms. The symptoms of NCLs could not be well-distinguished from other neurological diseases especially at early stage. Traditional enzymatic activity detection can only determine certain types of NCLs, i.e., CLN1, CLN2, and CLN10. Other types of NCLs cannot be well-diagnosed until gene sequencing was introduced (Patino et al., 2014). Here we performed Whole-Exome Sequencing in four Chinese siblings with LINCLs, mutations in CLN genes were identified in all families including 3 novel mutations. Genetic test especially Whole-Exome Sequencing is now a suggestive tool in diagnosis of rare disease and is accepted and recommended by more clinicians now (Jin et al., 2018; Shen, 2018).
Pathogenesis of Novel Mutations
In this study, 3 novel mutations in TPP1/CLN2, CLN6, and CLN7 were found, respectively. According to ACMG guidelines, mutation c.1551+1insTGAT in TPP1/CLN2 was characterized as pathogenic, mutation c.244G>T (G82W) in CLN6 was interpreted as likely pathogenic and mutation c.554-5A>G was classified as VUS.
For mutation c.1551+1insTGAT, in silico analysis suggests that this variant is a four-nucleotide-insertion in exonic region (Figure 2) It is probably not a splicing variant but an insertion variant which affects all the downstream sequence. Insertion of these 4 nucleotides would cause nonsense variant p.V518X. Resulted protein will be truncated after the residue D517. Residue from 518 to 563 would be missing. The highly conserved Ca-binding loop in sedolisin family (aa517-547) (Wlodawer et al., 2001, 2003) would be destroyed. Ca2+ is the cofactor of enzyme TPP1 and it was demonstrated necessary for the autocatalysis of the precursor TPP1 into mature form (Kuizon et al., 2010). Destruction of Ca-binding loop would disrupt the Ca2+ binding and sequentially hinder the autocatalysis of precursor TPP1. Another vital residue W542 which involves in tripeptidyl peptidase activity and autocatalytic activity of TPP1 (Kuizon et al., 2010) is also obliterated when the protein is truncated after D517.

Figure 2. Prediction result of c.1551+1insTGAT by NetGene Server 2 (Brunak et al., 1991; Hebsgaard et al., 1996) and Softberry. (A) According to the result of NetGene Server 2, this variant could not alter the donor splice site but insert 4 nucleotides before the exon and intron border. (B) Result of Softberry (http://www.softberry.com) presents the same result that this variant is an insert variant rather than splicing variant.
Mutation c.244G>T (G82W) in CLN6 is a missense mutation which changed the nonpolar negative amino acid Glycine into a nonpolar neutral amino acid. This mutation was predicted as deleterious mutation by PolyPhen2, SIFT, PROVEAN, and MutationTaster. This result indicates that this alteration may harm the proper folding of protein and consequently affect the protein function. In addition, protein CLN6 is a transmembrane protein. The mutated residue is located on the second transmembrane domain (Figure 3A). Although the exact function and interaction of this residue is still unclear, alteration of this residue may change the anchor of CLN6 protein in lysosomal membrane. This G82 residue is highly conservative among various species (Figure 3B). It indicates that this residue is functionally important.

Figure 3. The location and conservation of residue G82. (A) The residue is located in the second transmembrane domain of CLN6 protein according to the previous studies. (B) This residue is highly conserved in various species, which indicates the importance of this residue.
Mutation c.554-5A>G, it is not a variation in canonical splice sites thus cannot be simply classified as null variant. GT/AG mRNA processing rule is valid in almost all eukaryotes including the wild type MFSD8 sequence (Figure 4). The mutation c.554-5A>G changes the normal intronic site “aa” into another splice acceptor recognition sequence “ag” (Figure 4), which may influence the normal splicing. This hypothesis is further strengthened when predicted by Human Splicing Finder (Figure 5).

Figure 4. Sequences mutated MFSD8 and wildtype MFSD8. Mutation c.554-5A>G altered the “aa” sequence into an intronic splice acceptor site “ag”. This might induce splice from after the mutated “ag” site and induce four nucleotides insertion (TAAG) before the real exon thus altered all the downstream sequence.
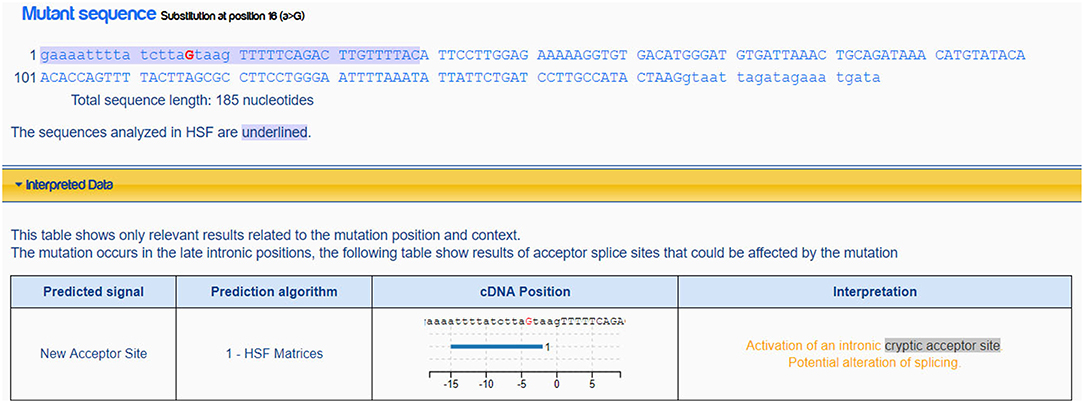
Figure 5. Prediction result of mutation c.554-5>G in MFSD8. This mutation would active an intronic cryptic acceptor site and potentially alter the splicing according to Human Splicing Finder Desmet et al. (2009).
Phenotype Study of LINCL Patients
Although CLN2, CLN5, CLN6, and CLN7 are all LINCLs, their symptoms and onset ages are slightly different in previous reports. Comparing to CLN2, clinical course of CLN5 is milder and slower, the onset age is significantly later, age of death is also significantly delayed. Onset age of visual loss in CLN5 is also significantly later than any other types of LINCLs. Reported age of death of CLN5 is around age 15 and most patients were still alive when reports were published. CLN6 was first reported as the NCL that presented similar clinical course to CLN2. The development of CLN6 was slightly slower than CLN2. Time for ambulation loss and death varied a lot. Seizures appear at the early stage in most CLN6 patients. Development of CLN7 is more severe than CLN2 as most of the patients lost ambulation within 2 years after onset, but the age of death varied from 6.5 to 18 years old. According to the previous reports, clinical information of LINCLs was summarized in Table 2 (Santavuori et al., 1982, 1991; Eva et al., 1988; Taratuto et al., 1995; Gao et al., 2002; Steinfeld et al., 2002; Sharp et al., 2003; Topcu et al., 2004; Siintola et al., 2007; Cismondi et al., 2008; Kohan et al., 2008; Aiello et al., 2009; Al-Muhaizea et al., 2009; Cannelli et al., 2009; Kousi et al., 2009; Stogmann et al., 2009; Xin et al., 2010; Perez-Poyato et al., 2012; Guerreiro et al., 2013; Patino et al., 2014; Canafoglia et al., 2015; Sato et al., 2016).

Table 2. Clinical information of reported cases of CLN2, CLN5, CLN6, and CLN7 before.
Table 3 above is the clinical information of Chinese CLN2, CLN5, CLN6, and CLN7 patients mentioned in this study. The initial symptoms in this study are all involved in muscle system and motor function. The onset ages of visual loss are all slightly later than most of the reported cases. The time for becoming bedridden and death is in the range of other reported cases.

Table 3. Clinical information of Chinese CLN2, CLN5, CLN6, and CLN7 patients in this study.
Motor function disruption then progress to intellectual function (language and cognition disorders) are predominant in the clinical course of the Chinese patients in current study. Visual system is last affected. The disease course of patients in our study is particularly consistent. Another independent report of a Chinese CLN6 patient with homozygous mutation c.892G>A (p.E298K) presented a patient with uncoordinated movements and seizures at 1.5 years, then slow response and developmental milestones were observed. Visual loss was not observed when the boy was 5 years old at the last observation (Sun et al., 2018). Other studies on CLN2 and CLN5 also presented that visual decline is never the first symptom in Chinese LINCL patients (Chang et al., 2012; Ge et al., 2018). Normal vision was found in all three Chinese CLN5 patients in the study of Ge and his colleagues (Ge et al., 2018). These studies and ours suggest that the Chinese LINCL patients may have a consistent clinical course that is slightly different from the western LINCL patients. Expanded study on Chinese patients may shed more light on the observed difference.
Follow Up and Treatments
There is no cure in NCLs, and the treatments are limited to palliative care (Getty and Pearce, 2011). In this study, we used Topiramate to treat the CLN2 patient, Levetiracetam and sodium valproate to treat CLN5 patient, sodium valproate to treat CLN7 patient. These treatments did not generate desired outcome. We then reviewed the development of novel treatment, enzyme replacement therapy, stem cell transplantation and gene therapy.
Enzyme replacement therapy has been reported to be a more effective and safe way to treat with strong improvement observed in murine and canine models (Katz et al., 2014; Lu et al., 2015). A phase half clinical trial of the intracerebroventricular enzyme in CLN2 patients proved the safety of enzyme replacement treatment. Significant improvement of motor-language function was reported after treatment (https://www.clinicaltrials.gov/ct2/show/results/NCT01907087).
Stem cell transplantation is also considered to treat NCLs. However, hematopoietic stem cell transplantation did not perform ideally, only transient effect was observed in a few experiments (Lonnqvist et al., 2001; Yuza et al., 2005). Transplantation of neural stem cell performed better in murine model (Tamaki et al., 2009). Whereas in human, it did not change the neurological function or attenuates seizures (Selden et al., 2013).
Gene therapy was also tested. Human enzyme gene such as PPT1 or TPP1 was integrated into Adeno-Associated Virus 2 (AAV2) of AAV2/5 vector and intracranial injected into murine models or canine CLN2 models. Recombinant AAV-PPT1 successfully lowered the accumulation of autofluorescent material, increased brain mass, slowed neurodegeneration and protected behavioral functions (Griffey et al., 2004, 2006; Roberts et al., 2012; Katz et al., 2015). Safety trial for gene therapy in human is still underway (https://www.clinicaltrials.gov/ct2/show/NCT00151216).
Individual II:2 of family 4 still has not reached the onset age. Disease symptoms are not presented. These novel therapies may give positive effect on this patient as all therapies performed better at the presymptomatic stage. It certainly requires genetic test to identify causal mutation carrier in order to perform any treatment before any symptoms observed.
Conclusion
This study described four Chinese LINCL siblings who were diagnosed by WES. The patients of these four families had similar disease courses started from motor regression or seizures to cognition regression and visual loss but carried mutations in different genes i.e. CLN2, CLN5, CLN6, and CLN7. The clinical features of LINCLs in these four Chinese siblings were not significantly different from those of Western patients. However, all Chinese LINCL patients in this study presented similar clinical course despite the affected genes. We assumed it as an ethnic specific clinical course according to our observation. Expanded sample size will be helpful to investigation of phenotype-genotype correlation. Besides, a platform for better communication, data and diagnostic experience sharing between Chinese and international clinicians is also required for further investigation (Jia and Shi, 2017).
Moreover, three mutations that detected in this study are novel mutations, and two of them occurred in intronic regions. These findings expanded the variant diversity of LINCLs.
Ethics Statement
This study was carried out is approved by Capital Medical University Beijing Children's Hospital Ethics Committee. The protocol was approved by the Capital Medical University Beijing Children's Hospital Ethics Committee. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Consent for Publication
The patient's parents gave written informed consent to studies and publication of clinical information, images and sequencing data.
Author Contributions
X-TR and X-HW designed the study. X-TR, X-HW, C-HD, W-HZ, J-WL, C-HR, and FF collected the clinical information of all patients. X-TR, X-HW, and C-HD collected the follow-up and prognosis information of all patients. XS and HZ performed the Next-Generation and Sanger Sequencing. X-TR, X-HW, XS, and HZ wrote the manuscript. X-TR, XS, HZ, and C-HD revised the manuscript. All authors listed have made a substantial, direct and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
XS and HZ were employed by company Running Gene Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to all of the family members for their participation in the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00370/full#supplementary-material
Figure S1. Next-Generation Sequencing results of 4 probands. (a) Proband 1 (Individual II:2 of family 1) is homozygous of mutation c.1551+1insTGAT. (b,c) Proband 2 (Individual II:2 of family 2) carries 2 mutations in CLN5 gene, c.1068_1069del (b) and c.1100_1103del (c). (d,e) Proband 3 (Individual II:2 of family 3) carries 2 mutations in CLN6 gene, namely c.244G>T (d) and c.892G>A (e). (f,g) Proband 4 (Individual II:1 of family 4) carries 2 mutations in MFSD8 gene, c.1444C>T (f) and c.554-5A>G (g).
Figure S2. Sanger Sequencing results of 4 probands. (A) Individual II:2 and II:3 of family 1 are the homozygous of mutation c.1551+1insTGAT. Their parents are all carriers of this mutation and don't have the disease or any symptoms, which is corresponding to the inheritance pattern. (B,C) Individual II:2 and II:3 are the compound heterozygous of mutations c.1068_1069del and c.1100_1103del and meanwhile suffered from LINCL. Their father (I:1) and sister (II:1) are the carriers of mutation c.1100_1103del, their mother is the carrier of mutation c.1068_1069del. Their parent and sister are all healthy with no regression which also corresponds to the inheritance pattern. (D,E) Individual II:1 and II:2 of family 3 are the compound heterozygous of mutation c.244G>T and c.892G>A, and these mutations are inherited from their mother and father. Their mother is the carrier of mutation c.224G>T and their father carries mutation c.892G>A. The two carrier parents are health with no disease symptom, while 2 heterozygous children are the patients of CLN6. (F,G) Individual II:1 and II:2 are compound heterozygous of mutations c.1444C>T and c.554-5A>G and their parents are the carriers of these mutations, mother with c.1444C>T and father with c.554-5A>G. Two parents don't have disease, proband individual II:1 of family 4 is the patient of NCL, which is true to the heritance pattern. However, another heterozygous, individual II:2 of family 4, is asymptomatic. This is because is individual is too young and doesn't reach the onset age of CLN7.
Table S1. The precise information of patients.
References
Aiello, C., Terracciano, A., Simonati, A., Discepoli, G., Cannelli, N., Claps, D., et al. (2009). Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum. Mutat. 30, E530–E540. doi: 10.1002/humu.20975
Al-Muhaizea, M. A., Al-Hassnan, Z. N., and Chedrawi, A. (2009). Variant late infantile neuronal ceroid lipofuscinosis (CLN6 gene) in Saudi Arabia. Pediatr. Neurol. 41, 74–76. doi: 10.1016/j.pediatrneurol.2009.01.012
Beltran, L., Valenzuela, G. R., Loos, M., Vargas, R., Lizama, R., Spinsanti, P., et al. (2018). Late-onset childhood neuronal ceroid lipofuscinosis: early clinical and electroencephalographic markers. Epilepsy Res. 144, 49–52. doi: 10.1016/j.eplepsyres.2018.05.005
Benedict, J. W., Getty, A. L., Wishart, T. M., Gillingwater, T. H., and Pearce, D. A. (2009). Protein product of CLN6 gene responsible for variant late-onset infantile neuronal ceroid lipofuscinosis interacts with CRMP-2. J. Neurosci. Res. 87, 2157–2166. doi: 10.1002/jnr.22032
Blom, T., Schmiedt, M. L., Wong, A. M., Kyttala, A., Soronen, J., Jauhiainen, M., et al. (2013). Exacerbated neuronal ceroid lipofuscinosis phenotype in Cln1/5 double-knockout mice. Dis. Model. Mech. 6, 342–357. doi: 10.1242/dmm.010140
Brunak, S., Engelbrecht, J., and Knudsen, S. (1991). Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol. 220, 49–65.
Canafoglia, L., Gilioli, I., Invernizzi, F., Sofia, V., Fugnanesi, V., Morbin, M., et al. (2015). Electroclinical spectrum of the neuronal ceroid lipofuscinoses associated with CLN6 mutations. Neurology 85, 316–324. doi: 10.1212/WNL.0000000000001784
Cannelli, N., Garavaglia, B., Simonati, A., Aiello, C., Barzaghi, C., Pezzini, F., et al. (2009). Variant late infantile ceroid lipofuscinoses associated with novel mutations in CLN6. Biochem. Biophys. Res. Commun. 379, 892–897. doi: 10.1016/j.bbrc.2008.12.159
Chang, X., Huang, Y., Meng, H., Jiang, Y., Wu, Y., Xiong, H., et al. (2012). Clinical study in Chinese patients with late-infantile form neuronal ceroid lipofuscinoses. Brain Dev. 34, 739–745. doi: 10.1016/j.braindev.2011.12.005
Cismondi, I. A., Kohan, R., Ghio, A., Ramirez, A. M., and Halac, I. N. (2008). Gene symbol: CLN6. Disease: neuronal ceroid lipofuscinosis, late Infantile. Hum. Genet. 124:324.
Cotman, S. L., Karaa, A., Staropoli, J. F., and Sims, K. B. (2013). Neuronal ceroid lipofuscinosis: impact of recent genetic advances and expansion of the clinicopathologic spectrum. Curr. Neurol. Neurosci. Rep. 13:366. doi: 10.1007/s11910-013-0366-z
De Silva, B., Adams, J., and Lee, S. Y. (2015). Proteolytic processing of the neuronal ceroid lipofuscinosis related lysosomal protein CLN5. Exp. Cell Res. 338, 45–53. doi: 10.1016/j.yexcr.2015.08.021
Desmet, F. O., Hamroun, D., Lalande, M., Collod-Beroud, G., Claustres, M., and Beroud, C. (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37:e67. doi: 10.1093/nar/gkp215
Eva, A., Jacob, J. C., Frederick, A., Stirling, C., Leonhard, W., Berkovic, S. F., et al. (1988). The Newfoundland aggregate of neuronal ceroid-lipofuscinosis. Am. J. Med. Genet. 31, 111–116. doi: 10.1002/ajmg.1320310615
Ezaki, J., Takeda-Ezaki, M., and Kominami, E. (2000). Tripeptidyl peptidase I, the late infantile neuronal ceroid lipofuscinosis gene product, initiates the lysosomal degradation of subunit c of ATP synthase. J. Biochem. 128, 509–516. doi: 10.1093/oxfordjournals.jbchem.a022781
Fang, F., Liu, Z., Fang, H., Wu, J., Shen, D., Sun, S., et al. (2017). The clinical and genetic characteristics in children with mitochondrial disease in China. Sci. China Life Sci. 60, 746–757. doi: 10.1007/s11427-017-9080-y
Gao, H., Boustany, R. M., Espinola, J. A., Cotman, S. L., Srinidhi, L., Antonellis, K. A., et al. (2002). Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am. J. Hum. Genet. 70, 324–335. doi: 10.1086/338190
Ge, L., Li, H. Y., Hai, Y., Min, L., Xing, L., Min, J., et al. (2018). Novel mutations in CLN5 of Chinese patients with neuronal ceroid Lipofuscinosis. J. Child Neurol. 33, 837–850. doi: 10.1177/0883073818789024
Getty, A. L., and Pearce, D. A. (2011). Interactions of the proteins of neuronal ceroid lipofuscinosis: clues to function. Cell. Mol. Life Sci. 68, 453–474. doi: 10.1007/s00018-010-0468-6
Griffey, M., Bible, E., Vogler, C., Levy, B., Gupta, P., Cooper, J., et al. (2004). Adeno-associated virus 2-mediated gene therapy decreases autofluorescent storage material and increases brain mass in a murine model of infantile neuronal ceroid lipofuscinosis. Neurobiol. Dis. 16, 360–369. doi: 10.1016/j.nbd.2004.03.005
Griffey, M. A., Wozniak, D., Wong, M., Bible, E., Johnson, K., Rothman, S. M., et al. (2006). CNS-directed AAV2-mediated gene therapy ameliorates functional deficits in a murine model of infantile neuronal ceroid lipofuscinosis. Mol. Ther. 13, 538–547. doi: 10.1016/j.ymthe.2005.11.008
Guerreiro, R., Bras, J. T., Vieira, M., Warrier, V., Agrawal, S., Stewart, H., et al. (2013). CLN6 disease caused by the same mutation originating in Pakistan has varying pathology. Eur. J. Paediatr. Neurol. 17, 657–660. doi: 10.1016/j.ejpn.2013.04.011
Haltia, M., and Goebel, H. H. (2013). The neuronal ceroid-lipofuscinoses: a historical introduction. Biochim. Biophys. Acta 1832, 1795–1800. doi: 10.1016/j.bbadis.2012.08.012
Hebsgaard, S. M., Korning, P. G., Tolstrup, N., Engelbrecht, J., Rouze, P., and Brunak, S (1996). Splice site prediction in Arabidopsis thaliana DNA by combining local and global sequence information. Nucleic Acids Res. 24, 3439–3452.
Heine, C., Koch, B., Storch, S., Kohlschutter, A., Palmer, D. N., and Braulke, T. (2004). Defective endoplasmic reticulum-resident membrane protein CLN6 affects lysosomal degradation of endocytosed arylsulfatase A. J. Biol. Chem. 279, 22347–22352. doi: 10.1074/jbc.M400643200
Isosomppi, J., Vesa, J., Jalanko, A., and Peltonen, L. (2002). Lysosomal localization of the neuronal ceroid lipofuscinosis CLN5 protein. Hum. Mol. Genet. 11, 885–891. doi: 10.1093/hmg/11.8.885
Jia, J., and Shi, T. (2017). Towards efficiency in rare disease research: what is distinctive and important? Sci. China Life Sci. 60, 686–691. doi: 10.1007/s11427-017-9099-3
Jin, Y., Zhang, L., Ning, B., Hong, H., Xiao, W., Tong, W., et al. (2018). Application of genome analysis strategies in the clinical testing for pediatric diseases. Pediatr. Investig. 2, 72–81. doi: 10.1002/ped4.12044
Kamate, M., Prashanth, G. P., and Hattiholi, V. (2012). Clinico-investigative profile of infantile and late-infantile neuronal ceroid lipofuscinoses. Neurol. India 60, 316–320. doi: 10.4103/0028-3886.98524
Katz, M. L., Coates, J. R., Sibigtroth, C. M., Taylor, J. D., Carpentier, M., Young, W. M., et al. (2014). Enzyme replacement therapy attenuates disease progression in a canine model of late-infantile neuronal ceroid lipofuscinosis (CLN2 disease). J. Neurosci. Res. 92, 1591–1598. doi: 10.1002/jnr.23423
Katz, M. L., Tecedor, L., Chen, Y., Williamson, B. G., Lysenko, E., Wininger, F. A., et al. (2015). AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Sci. Transl. Med. 7:313ra180. doi: 10.1126/scitranslmed.aac6191
Kohan, R., Cannelli, N., Aiello, C., Santorelli, F., Cismondi, A., Mila, M., et al. (2008). Novel human pathological mutations. Hum. Genet. 123, 537–555. doi: 10.1007/s00439-008-0503-y
Kollmann, K., Uusi-Rauva, K., Scifo, E., Tyynela, J., Jalanko, A., and Braulke, T. (2013). Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim. Biophys. Acta 1832, 1866–1881. doi: 10.1016/j.bbadis.2013.01.019
Kousi, M., Siintola, E., Dvorakova, L., Vlaskova, H., Turnbull, J., Topcu, M., et al. (2009). Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain 132, 810–819. doi: 10.1093/brain/awn366
Kuizon, S., Dimaiuta, K., Walus, M., Jenkins, E. C Jr., Kuizon, M., Kida, E., et al. (2010). A. critical tryptophan and Ca2+ in activation and catalysis of TPPI, the enzyme deficient in classic late-infantile neuronal ceroid lipofuscinosis. PLoS ONE 5:e(11929). doi: 10.1371/journal.pone.0011929
Lebrun, A. H., Storch, S., Ruschendorf, F., Schmiedt, M. L., Kyttala, A., Mole, S. E., et al. (2009). Retention of lysosomal protein CLN5 in the endoplasmic reticulum causes neuronal ceroid lipofuscinosis in Asian sibship. Hum. Mutat. 30, E651–E661. doi: 10.1002/humu.21010
Lonnqvist, T., Vanhanen, S. L., Vettenranta, K., Autti, T., Rapola, J., Santavuori, P., et al. (2001). Hematopoietic stem cell transplantation in infantile neuronal ceroid lipofuscinosis. Neurology 57, 1411–1416. doi: 10.1212/WNL.57.8.1411
Lu, J. Y., Nelvagal, H. R., Wang, L., Birnbaum, S. G., Cooper, J. D., and Hofmann, S. L. (2015). Intrathecal enzyme replacement therapy improves motor function and survival in a preclinical mouse model of infantile neuronal ceroid lipofuscinosis. Mol. Genet. Metab. 116, 98–105. doi: 10.1016/j.ymgme.2015.05.005
Mink, J. W., Augustine, E. F., Adams, H. R., Marshall, F. J., and Kwon, J. M. (2013). Classification and natural history of the neuronal ceroid lipofuscinoses. J. Child Neurol. 28, 1101–1105. doi: 10.1177/0883073813494268
Mole, S. E., and Williams, R. E. (2013). “Neuronal ceroid-lipofuscinoses,” in GeneReviews®, eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, and A. Amemiya, A (Seattle, WA: University of Washington).
Oishi, K., Ida, H., Kurosawa, K., and Eto, Y. (1999). Clinical and molecular analysis of Japanese patients with neuronal ceroid lipofuscinosis. Mol. Genet. Metab. 66, 344–348. doi: 10.1006/mgme.1999.2835
Patino, L. C., Battu, R., Ortega-Recalde, O., Nallathambi, J., Anandula, V. R., Renukaradhya, U., et al. (2014). Exome sequencing is an efficient tool for variant late-infantile neuronal ceroid lipofuscinosis molecular diagnosis. PLoS ONE 9:e109576. doi: 10.1371/journal.pone.0109576
Perez-Poyato, M. S., Mila-Recasens, M., Ferrer-Abizanda, I., Cusi-Sanchez, V., Vazquez-Lopez, M., Camino-Leon, R., et al. (2012). [Neuronal ceroid lipofuscinosis: diagnostic algorithm and clinical description of the Finnish (CLN5) and Turkish (CLN7) variants late infantile]. Rev. Neurol. 54, 544–550. doi: 10.33588/rn.5409.2011657
Roberts, M. S., Macauley, S. L., Wong, A. M., Yilmas, D., Hohm, S., Cooper, J. D., et al. (2012). Combination small molecule PPT1 mimetic and CNS-directed gene therapy as a treatment for infantile neuronal ceroid lipofuscinosis. J. Inherit. Metab. Dis. 35, 847–857. doi: 10.1007/s10545-011-9446-x
Sands, M. S. (2013). Considerations for the treatment of infantile neuronal ceroid lipofuscinosis (infantile Batten disease). J. Child Neurol. 28, 1151–1158. doi: 10.1177/0883073813495960
Santavuori, P., Rapola, J., Nuutila, A., Raininko, R., Lappi, M., Launes, J., et al. (1991). The spectrum of Jansky-Bielschowsky disease. Neuropediatrics 22, 92–96. doi: 10.1055/s-2008-1071423
Santavuori, P., Rapola, J., Sainio, K., and Raitta, C. (1982). A variant of Jansky-Bielschowsky disease. Neuropediatrics 13, 135–141. doi: 10.1055/s-2008-1059612
Sato, R., Inui, T., Endo, W., Okubo, Y., Takezawa, Y., Anzai, M., et al. (2016). First Japanese variant of late infantile neuronal ceroid lipofuscinosis caused by novel CLN6 mutations. Brain Dev. 38, 852–856. doi: 10.1016/j.braindev.2016.04.007
Schmiedt, M. L., Bessa, C., Heine, C., Ribeiro, M. G., Jalanko, A., and Kyttala, A. (2010). The neuronal ceroid lipofuscinosis protein CLN5: new insights into cellular maturation, transport, and consequences of mutations. Hum. Mutat. 31, 356–365. doi: 10.1002/humu.21195
Schulz, A., Kohlschutter, A., Mink, J., Simonati, A., and Williams, R. (2013). NCL diseases—clinical perspectives. Biochim. Biophys. Acta 1832, 1801–1806. doi: 10.1016/j.bbadis.2013.04.008
Selden, N. R., Al-Uzri, A., Huhn, S. L., Koch, T. K., Sikora, D. M., Nguyen-Driver, M. D., et al. (2013). Central nervous system stem cell transplantation for children with neuronal ceroid lipofuscinosis. J. Neurosurg. Pediatr. 11, 643–652. doi: 10.3171/2013.3.PEDS12397
Sharp, J. D., Wheeler, R. B., Parker, K. A., Gardiner, R. M., Williams, R. E., and Mole, S. E. (2003). Spectrum of CLN6 mutations in variant late infantile neuronal ceroid lipofuscinosis. Hum. Mutat. 22, 35–42. doi: 10.1002/humu.10227
Shen, Y. (2018). Next-generation sequencing based molecular testing is an equalizer for diagnostic service of rare genetic disorders in China. Pediatr. Investig. 2, 96–97. doi: 10.1002/ped4.12036
Siintola, E., Topcu, M., Aula, N., Lohi, H., Minassian, B. A., Paterson, A. D., et al. (2007). The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am. J. Hum. Genet. 81, 136–146. doi: 10.1086/518902
Steinfeld, R., Heim, P., Von Gregory, H., Meyer, K., Ullrich, K., Goebel, H. H., et al. (2002). Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. Am. J. Med. Genet. 112, 347–354. doi: 10.1002/ajmg.10660
Stogmann, E., El Tawil, S., Wagenstaller, J., Gaber, A., Edris, S., Abdelhady, A., et al. (2009). A. novel mutation in the MFSD8 gene in late infantile neuronal ceroid lipofuscinosis. Neurogenetics 10, 73–77. doi: 10.1007/s10048-008-0153-1
Sun, G., Yao, F., Tian, Z., Ma, T., and Yang, Z. (2018). A. first CLN6 variant case of late infantile neuronal ceroid lipofuscinosis caused by a homozygous mutation in a boy from China: a case report. BMC Med. Genet. 19:177. doi: 10.1186/s12881-018-0690-x
Tamaki, S. J., Jacobs, Y., Dohse, M., Capela, A., Cooper, J. D., Reitsma, M., et al. (2009). Neuroprotection of host cells by human central nervous system stem cells in a mouse model of infantile neuronal ceroid lipofuscinosis. Cell Stem Cell 5, 310–319. doi: 10.1016/j.stem.2009.05.022
Taratuto, A. L., Saccoliti, M., Sevlever, G., Ruggieri, V., Arroyo, H., Herrero, M., et al. (1995). Childhood neuronal ceroid-lipofuscinoses in Argentina. Am. J. Med. Genet. 57, 144–149. doi: 10.1002/ajmg.1320570207
Topcu, M., Tan, H., Yalnizoglu, D., Usubutun, A., Saatci, I., Aynaci, M., et al. (2004). Evaluation of 36 patients from Turkey with neuronal ceroid lipofuscinosis: clinical, neurophysiological, neuroradiological, and histopathologic studies. Turk. J. Pediatr. 46, 1–10.
Warrier, V., Vieira, M., and Mole, S. E. (2013). Genetic basis and phenotypic correlations of the neuronal ceroid lipofusinoses. Biochim. Biophys. Acta 1832, 1827–1830. doi: 10.1016/j.bbadis.2013.03.017
Wlodawer, A., Li, M., Dauter, Z., Gustchina, A., Uchida, K., Oyama, H., et al. (2001). Carboxyl proteinase from Pseudomonas defines a novel family of subtilisin-like enzymes. Nat. Struct. Biol. 8, 442–446. doi: 10.1038/87610
Wlodawer, A., Li, M., Gustchina, A., Oyama, H., Dunn, B. M., and Oda, K. (2003). Structural and enzymatic properties of the sedolisin family of serine-carboxyl peptidases. Acta Biochim. Pol. 50, 81–102.
Xin, W., Mullen, T. E., Kiely, R., Min, J., Feng, X., Cao, Y., et al. (2010). CLN5 mutations are frequent in juvenile and late-onset non-Finnish patients with NCL. Neurology 74, 565–571. doi: 10.1212/WNL.0b013e3181cff70d
Keywords: Next-Generation Sequencing (NGS), Neuronal Ceroid Lipofuscinosis (NCL), late-infantile, CLN2, CLN5, CLN6, CLN7
Citation: Ren X-T, Wang X-H, Ding C-H, Shen X, Zhang H, Zhang W-H, Li J-W, Ren C-H and Fang F (2019) Next-Generation Sequencing Analysis Reveals Novel Pathogenic Variants in Four Chinese Siblings With Late-Infantile Neuronal Ceroid Lipofuscinosis. Front. Genet. 10:370. doi: 10.3389/fgene.2019.00370
Received: 08 November 2018; Accepted: 08 April 2019;
Published: 25 April 2019.
Edited by:
Tieliu Shi, East China Normal University, ChinaReviewed by:
Theodora Katsila, National Hellenic Research Foundation, GreeceFan Jin, Zhejiang University, China
Copyright © 2019 Ren, Wang, Ding, Shen, Zhang, Zhang, Li, Ren and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Fang, MTM5MTAxNTAzODlAMTYzLmNvbQ==