
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 04 January 2019
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2018 | https://doi.org/10.3389/fgene.2018.00680
 Ching-Yu Julius Chen1
Ching-Yu Julius Chen1 Tzu-Pin Lu2Lian-Yu Lin1Yen-Bin Liu1Li-Ting Ho1Hui-Chun Huang1Ling-Ping Lai1Juey-Jen Hwang1Shih-Fan Sherri Yeh3Cho-Kai Wu1
Tzu-Pin Lu2Lian-Yu Lin1Yen-Bin Liu1Li-Ting Ho1Hui-Chun Huang1Ling-Ping Lai1Juey-Jen Hwang1Shih-Fan Sherri Yeh3Cho-Kai Wu1 Jyh-Ming Jimmy Juang1*Charles Antzelevitch4,5,6
Jyh-Ming Jimmy Juang1*Charles Antzelevitch4,5,6Brugada syndrome (BrS) is a heritable disease that results in sudden cardiac death. In the exome/genomic era, certain reported pathogenic variants in some genetic diseases have been reclassified as benign owing to their high frequency in some ancestries. In the present study, we comprehensively reassessed all previously reported pathogenic variants of BrS. We collected all pathogenic variants of BrS reported in the Human Gene Mutation Database and ClinVar throughout April 2017. We compared the minor allele frequency (MAF) of each variant among different ancestries by searching public whole-genome and exome databases. After considering the maximum credible allele frequency, variants with a MAF ≥ 0.001 were considered to be of questionable pathogenicity. We also investigated the percentage of SCN5A variants with a MAF ≥ 0.001 in 124 BrS patients from the Han Chinese population. We collected a total of 440 BrS variants, of which 18 had a MAF ≥ 0.001. There was a greater percentage of non-SCN5A variants with a MAF ≥ 0.001 than of SCN5A variants (21.8 versus 1.6%, p < 0.0001). There were fewer frameshift and nonsense mutations than missense mutations (0.9 versus 5.6%, p = 0.032). Of the 18 variants, 14 (77.8%) were present only in the reference Asian population. In our cohort, we identified two SCN5A variants (p.A226V and p.V1340I) with MAFs ≥ 0.001 (0.45%). In conclusion, ancestral differences are important when considering the pathogenicity of BrS variants, especially in the case of missense variants and non-SCN5A variants, which may be pathogenic in some ancestries but only disease-predisposing in others.
Brugada syndrome (BrS) – a heritable arrhythmic disease responsible for sudden cardiac death (SCD) in patients with structurally normal hearts – was first reported by Brugada and Brugada (1992). It accounts for 4% of all sudden deaths and up to 20% of sudden deaths in patients without structural cardiac disease (Antzelevitch et al., 2005b), and has been identified as the same entity previously designated as sudden unexpected nocturnal death syndrome (SUNDS) (Vatta et al., 2002). Moreover, the prevalence of BrS is highest in the Asian population (Juang et al., 2015), which may be attributed to a distinct genetic background.
Numerous genes are associated with BrS, including SCN5A, SCN10A, SCN1B, SCN2B, SCN3B, KCNH2, KCND2, KCND3, KCNE3, KCNE5, KCNJ8, CACNA1C, CACNA2D1, CACNB2, ABCC9, HCN4, PKP2, SLMAP, TRPM4, RANGRF, GPD1L, FGF12, and SEMA3A (Fernandez-Falgueras et al., 2017); most encode proteins that control the transmembrane ion currents responsible for electrical impulses. The identification of pathogenic variants is crucial because it can help further investigation of the disease mechanism and facilitate family screening. To determine the pathogenicity of a variant, the American College of Medical Genetics and Genomics (ACMG) suggests taking into consideration the population allele frequency, the effect on protein structure, and the results of functional studies (Richards et al., 2015).
The expected allele frequency of a causative variant is related to the disease prevalence, the penetrance, and to the genetic contribution made by the causative variant to the disease (Whiffin et al., 2017). In other words, the prevalence of a monogenic disease is the summation of the contributions of all the pathogenic variants in a population. The expected allele frequency of each variant can vary across populations, depending on its contribution to the monogenic disease. Therefore, the pathogenicity of a variant with an allele frequency much higher than expected should be investigated carefully. The first universal genome-wide study regarding allele frequency was the 1000 Genomes Project (1000G). As the sample size increased in the Exome Aggregation Consortium (ExAC) and then the Genome Aggregation Database (gnomAD), it was expected that these studies would become more representative of the true population frequency. Ancestral differences in numerous variants were revealed, and some previously reported pathogenic variants have been reclassified as benign or disease-predisposing owing to their high frequencies in some ancestries (Manrai et al., 2016; Clemens et al., 2018). Although there have been functional in vitro studies of some of these variants, they could be considered disease-predisposing in high-frequency ancestries because the polymorphism of genes among ancestries is complex and may have protective effects.
Therefore, a local reference for population frequency is invaluable in determining the pathogenicity of a variant in a given ancestry. Current large-scale ancestry-specific databases for population frequency include the Taiwan Biobank (TWB), the integrative Japanese Genome Variation Database (iJGVD), and the NHLBI Grand Opportunity Exome Sequencing Project (ESP6500) for European Americans (EAs) and African Americans (AAs).
The present study was performed to investigate the ancestral differences in previously reported pathogenic variants of BrS, with the expectation that the pathogenicity of variants with relatively high allele frequencies in some ancestries should be reassessed. Because the prevalence of BrS is highest in the Asian population, we used our BrS cohort (TW-BrS registry) to validate the clinical impact of these variants, which were listed as disease-causing or pathogenic variants in the Human Gene Mutation Database (HGMD) and ClinVar.
We collected the pathogenic and likely pathogenic variants of BrS reported in ClinVar1 (Landrum et al., 2016), and the disease-causing mutations in HGMD - Professional2 (Stenson et al., 2012) throughout April 2017.
We compared the allele frequency of each variant among different ancestries by searching gnomAD version 2.0, TWB, and iJGVD. gnomAD was first released as ExAC in 2013 (Lek et al., 2016), and was expanded to 123,136 exomes and 15,496 genomes in 2017. TWB was released in 2014, and is composed of whole-genome sequencing data from 997 unrelated Han Chinese (HC), and genome-wide association study data from 16,036 unrelated HC (Fan et al., 2008). No subjects with BrS were enrolled in the TWB. iJGVD was first released in 2014 (Nagasaki et al., 2015), and consists of whole-genome sequencing data from 3,554 Japanese subjects (3KJPNv2) obtained in June 2018.
According to the clinical genetic interpretation method proposed by Whiffin et al. (2017), in addition to considering the disease prevalence and penetrance, it is also necessary to consider the allelic contribution made by each pathogenic variant. Accordingly, the maximal credible allele frequency of a pathogenic variant is as follows:
Brugada syndrome has the highest prevalence (∼0.12%) in East Asian populations (Mizusawa and Wilde, 2012; Juang et al., 2015), with a genetic yield rate of approximately 30–35% (Juang and Horie, 2016), and the contribution of a single variant is no more than 1% (Kapplinger et al., 2010). Moreover, BrS-associated variants may have a penetrance of approximately 16–32.7% according to familial co-segregation analysis (Priori et al., 2000; Hong et al., 2004). Therefore, the allele frequency of a pathogenic variant of BrS is expected to be lower than 0.000075, implying that the pathogenicity of any variants with allele frequencies higher than this value may require careful interpretation. However, in vitro studies have revealed that some variants are associated with functional alterations (Matsusue et al., 2016). Such cases should be treated as disease predisposition alleles rather than totally excluding their pathogenic role. Thus, we used a conservative cut-off of 0.001, at least 10 times higher than the frequency mentioned above, which may have prevented the exclusion of some true pathogenic variants from the subsequent phenotype-driven analysis due to too strict a cut-off. The cut-off set by us in the present study was not very strict, and was reasonable as the first step for the reclassification of previously reported pathogenic variants of BrS across different populations.
The present study complied with governmental laws and regulations and was carried out in accordance with the Good Practice guidelines provided by Research Ethics Committee B of the National Taiwan University Hospital. All subjects provided written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by Research Ethics Committee B of the National Taiwan University Hospital. We enrolled 124 consecutive unrelated BrS patients from the Han Chinese population of Taiwan between 1998 and 2017 (SADS-TW BrS registry) (Wu et al., 2018). The majority (89.4%) of the patients were male, and their mean age was 44.7 years old. The diagnosis of BrS was in accordance with the expert consensus of the Heart Rhythm Society (HRS), the European Heart Rhythm Association (EHRA), and the Asia-Pacific Heart Rhythm Society (APHRS) (Antzelevitch et al., 2005a; Priori et al., 2013). The Brugada electrocardiographic (ECG) patterns are composed of three types as follows: type 1 ECG has coved-type ST-segment elevation with J point elevation >0.2 mV, followed by a negative T wave; type 2 ECG has saddle-back ST-segment elevation with J point elevation >0.2 mV, followed by a gradually descending ST-segment elevation >0.1 mV and a positive or biphasic T wave; and type 3 ECG has either a saddle-back or coved appearance but with ST-segment elevation <0.1 mV. Only spontaneous or drug-induced type 1 ECG is diagnostic, while type 2 or 3 ECG is suspicious and should undergo drug provocation test by sodium channel blocker. The demographic data were recorded, including gender, age, initial presentation, family history of sudden cardiac death or BrS and electrocardiographic parameters.
With permission from patients, 10 ml of peripheral venous blood was used for genetic analysis. We used deoxyribonucleic acid (DNA) extraction kit (Qiagen company) to extract DNA from the buffy coat of patients’ peripheral blood. SCN5A variants were screened by Sanger’s sequencing. We investigated the prevalence and clinical characteristics of the patients with these reclassified variants in our 124 consecutive unrelated BrS cohort.
Chi-square or Fisher’s exact tests were used to compare categorical variables. A two-tailed p-value < 0.05 was considered statistically significant. Statistical analysis was performed using IBM SPSS Statistics for Windows (version 22.0) (Armonk, NY, United States).
Four-hundred and five disease-causing mutations of BrS were reported in HGMD, and 45 pathogenic and 23 likely pathogenic variants were reported in ClinVar. There were 440 variants in total, with 33 overlapping variants. A total of 385 (87.5%) variants were in the SCN5A gene, followed by the CACNA1C, SCN10A, and TRPM4 genes (Figure 1). Twenty-three (6.0%) SCN5A variants were in the voltage-sensing domain (VSD), 52 (13.5%) were in the pore-forming domain, 53 (13.8%) were in other transmembrane domains, 114 (29.6%) were in the extracellular domain, and 143 (37.1%) were in the cytoplasmic domain.
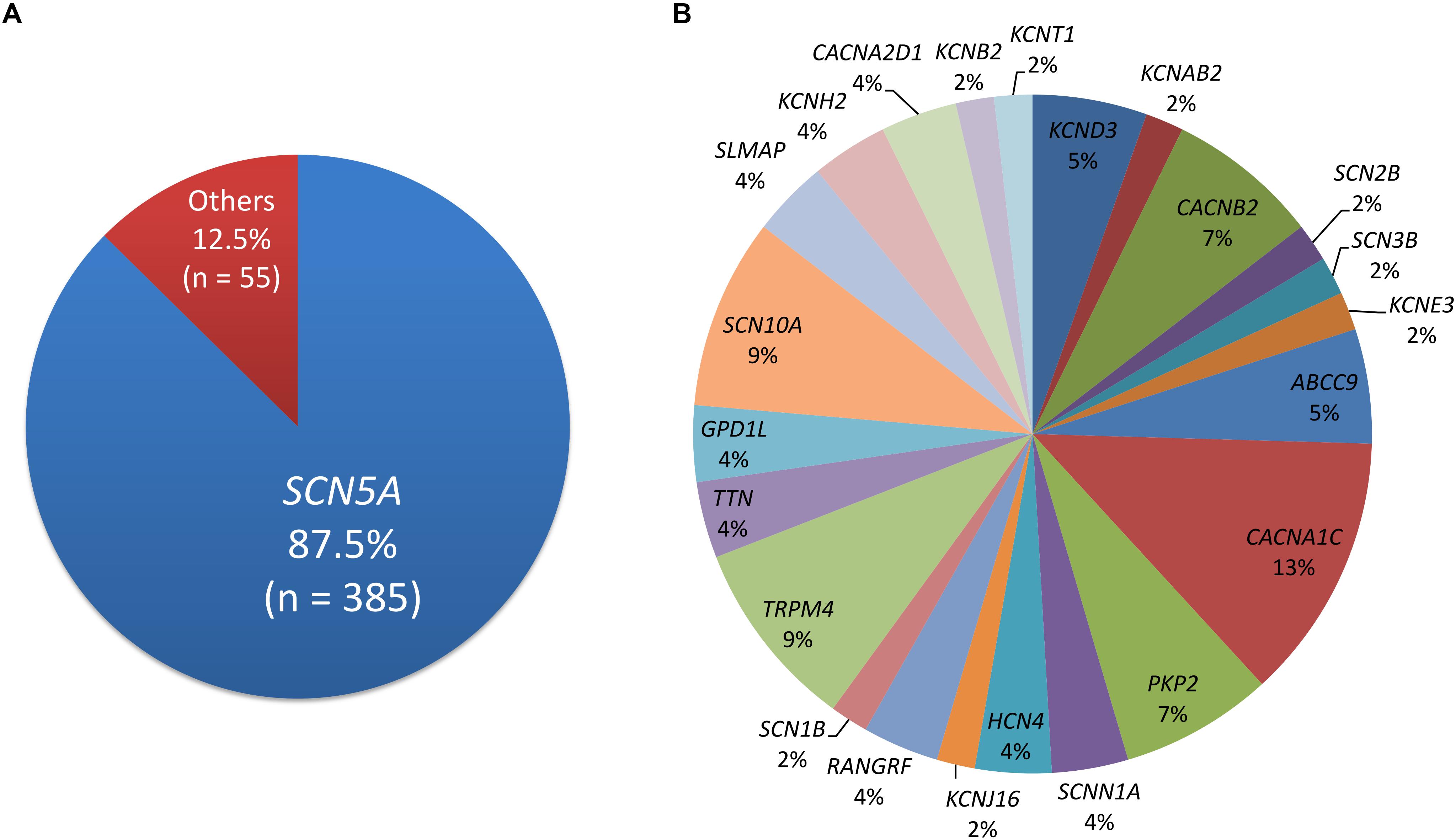
Figure 1. The distribution of pathogenic and likely pathogenic variants. (A) SCN5A accounts for 87.5%. (B) The remaining genes.
Of the 440 previously reported BrS mutations, 18 (4.1%) variants had an allele frequency ≥0.001 in at least one ancestry (Table 1): 17 from HGMD and 1 from ClinVar. There was no significant difference in the percentages of variants found in these two databases (HGMD 4.2% versus ClinVar 1.5%, p = 0.4915). All of the original studies used internal controls with 50–700 healthy subjects; four of them used two controls (2: internal controls and ESP6500; 2: internal controls and 1000G). None of the original studies used greater than 3 controls. Eleven of the 18 variants (61.1%) in the previous 13 studies used the Caucasian population as healthy controls. These variants included 6 SCN5A and 12 non-SCN5A variants. There were more previously reported non-SCN5A variants with allele frequencies ≥0.001 than SCN5A variants (21.8 versus 1.6%, p < 0.0001). Among the SCN5A variants with allele frequencies ≥0.001, one was located in the VSD, one in the pore-forming domain, one in another transmembrane domain, and the other three variants were in the cytoplasmic domain (Figure 2). The variants with allele frequencies <0.001 across all ancestries are listed in Supplementary Table 1.
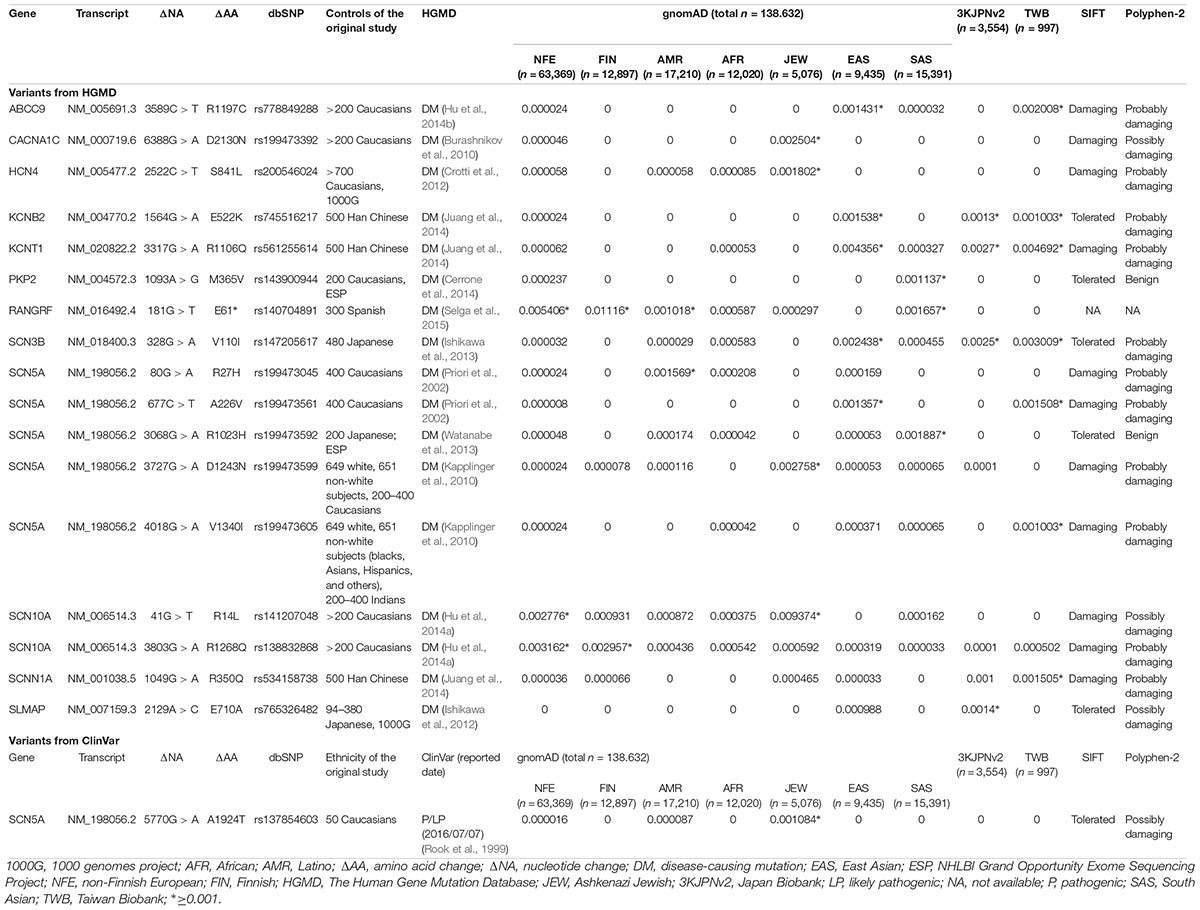
Table 1. The 18 of the 440 reported pathogenic variants in Brugada syndrome showed allele frequencies ≥0.001 in at least one ancestry.
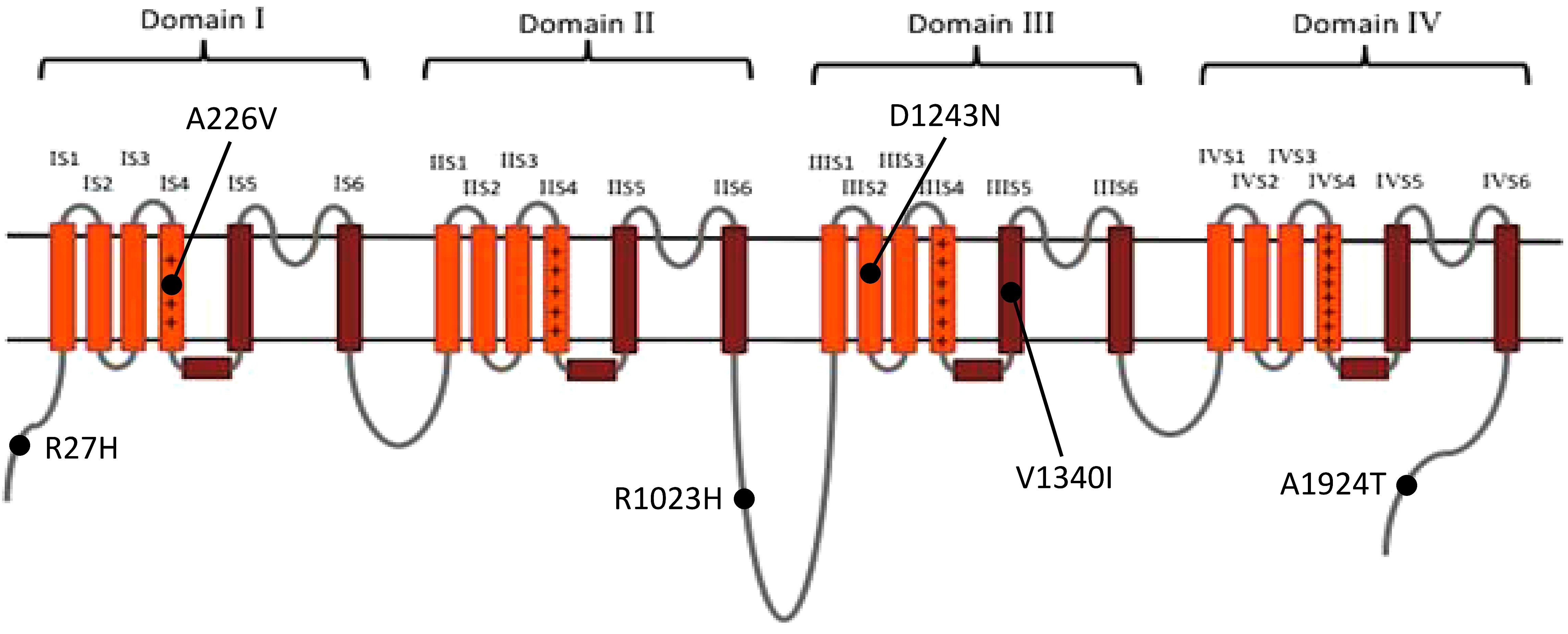
Figure 2. The distribution of the previously reported SCN5A variants with allele frequency ≥0.001 in at least one ancestry. Brown: pore-forming domain; +: voltage-sensing domain. Data from gnomAD, iJGVD, and Taiwan Biobank.
With regard to ancestral differences, 14 (77.8%) variants had allele frequencies ≥0.001 only in Asian population, including all the non-SCN5A variants except one RANGRF variant and two SCN10A variants. The allele frequencies of RANGRF p.E61∗ were ≥0.001 across all ancestries except for African, whereas the allele frequencies of SCN5A p.R27H were ≥0.001 only in American ancestries. These findings demonstrate that ancestral differences impact genetic predisposition in BrS.
Five variants had allele frequencies ≥0.001 in 1000G or ESP, but they were lower in gnomAD (Supplementary Table 2). Because a larger database is more representative of the population frequency, the results from gnomAD should be more reliable than those reported previously.
Regarding the types of mutations, there were 40 nonsense (9.1%), 73 frameshift (16.6%), 23 splice site (9.1%), and 304 missense (69.1%) mutations among the previously reported 440 BrS mutations. The percentages of the 18 variants with minor allele frequency >0.001 in at least one ancestry within each type of mutation were: 0% (0/73) in the frameshift mutations, 2.5% (1/40) in the nonsense mutations, 0% (0/23) in the splice site mutations, and 5.6% (17/304) in the missense mutations. There was a significantly lower percentage of radical mutations (frameshift plus nonsense) than missense mutations (0.9 versus 5.6%, p = 0.032).
In our SADS-TW BrS registry cohort, for the variants with minor allele frequencies ≥0.001, two SCN5A variants (p.A226V and p.V1340I) (0.45%) were identified in 5 patients (4%) (Table 1). Of those patients, 4 carried p.A226V and one had p.V1340I. The clinical characteristics of the patients are shown in Table 2. Among the three patients carrying SCN5A p.A226V underwent the electrophysiological examinations, ventricular tachycardia/fibrillation was induced in two patients. In the other hand, we also found 10 pathogenic or likely pathogenic SCN5A variants with minor allele frequencies <0.001 in 10 patients with BrS (8.1%), for which the pathogenicity was determined according to the American College of Medical Genetics and Genomics (ACMG) criteria (Richards et al., 2015).
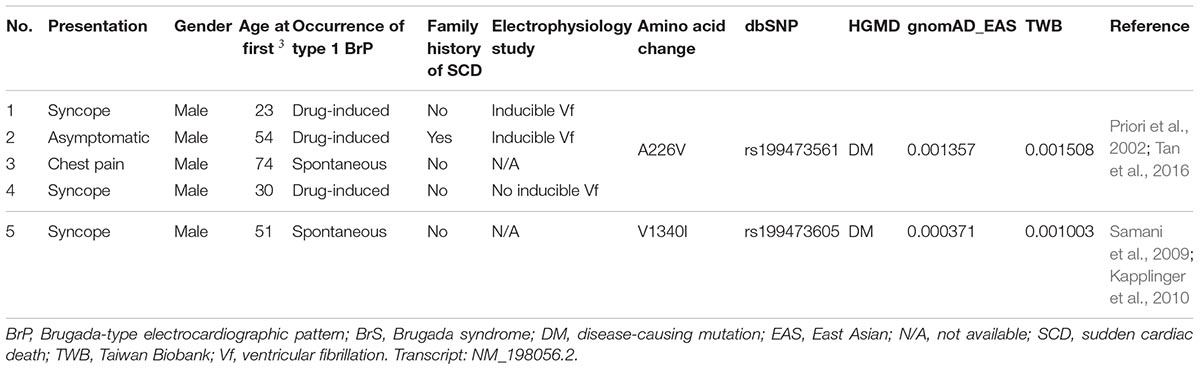
Table 2. Clinical characteristics of the patients (SADS-TW BrS registry) carrying previously reported pathogenic variants in HGMD with allele frequencies ≥0.001 in at least one ancestry.
In dealing with a heritable disease, the identification of the causative genetic variant is important for familial consultation, because it can aid risk stratification in a family and eliminate the anxiety of unaffected members. In the era of precision medicine, misinterpretation of variants may lead to incorrect genetic diagnoses and ineffective treatment strategies.
Brugada syndrome is characterized by the loss of the epicardial action potential dome in the right ventricular outflow tract (Antzelevitch, 2001); thus, variants that alter channel function are potential pathogenic variants. Given that BrS is a rare disease, the allele frequency of each pathogenic variant would be expected to be lower than its prevalence. That is, a variant with a functional alteration but a relatively high allele frequency may not be pathogenic by itself, or may only increase susceptibility to the disease.
With the development of next-generation sequencing (NGS), population frequency studies can be conducted faster and on a larger scale, and the pathogenicity of variants can be evaluated on the basis of ancestry-specific references. For example, the SCN5A p.R1193Q substitution, which accelerates the inactivation of sodium channels (Vatta et al., 2002), is now considered to be a disease predisposition allele rather than a causative mutation in Asian populations; its allele frequency in this ancestry is as high as 0.05 (Matsusue et al., 2016), which is much higher than the disease prevalence rate. Another example involves SCN5A c.2893C > T (p.R965C), which exhibits major differences in allele frequency among ancestries (Figure 3). These findings support the importance of establishing a large, population-specific database of allele frequency, which could be the best reference for determining the rarity of variants.
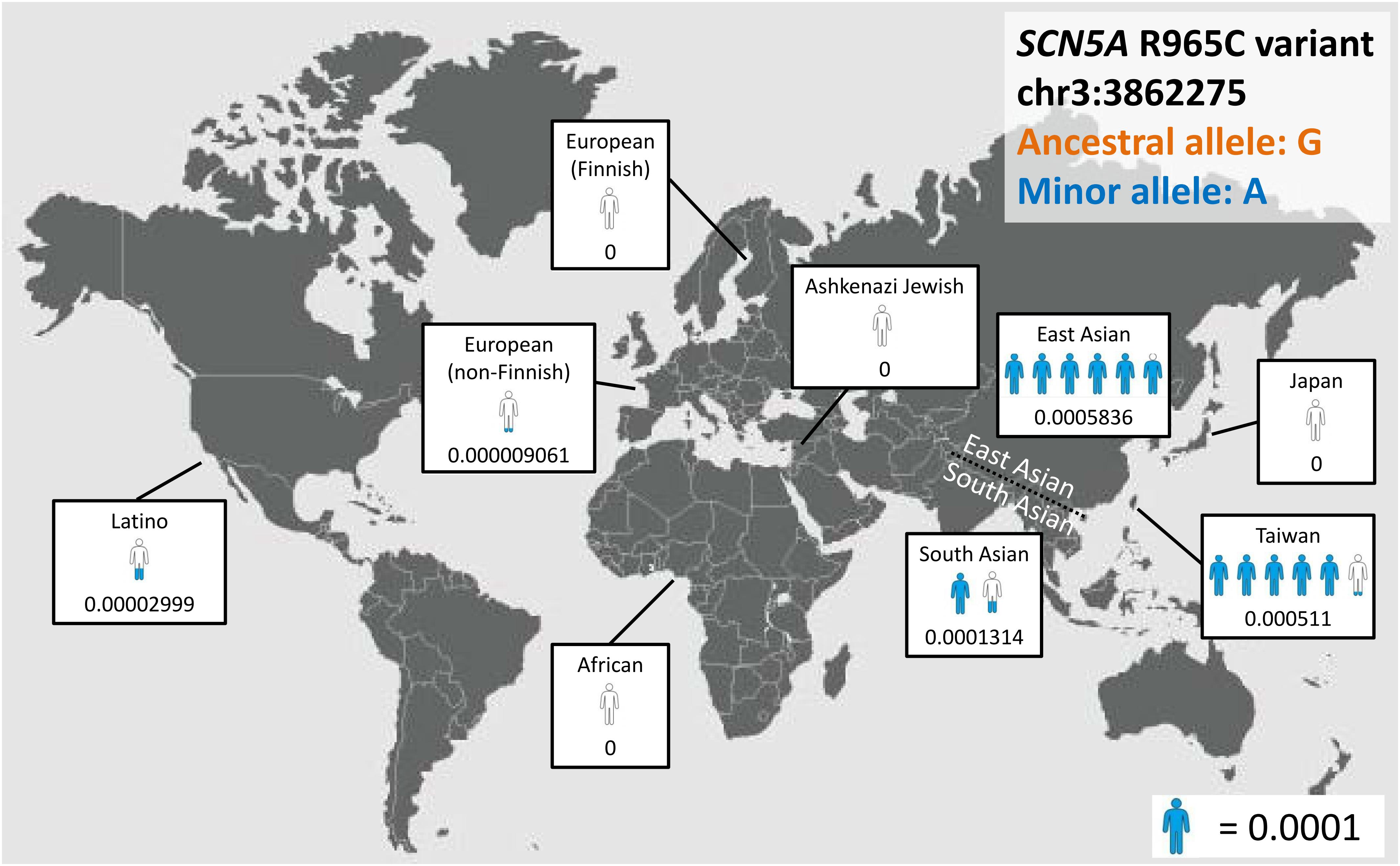
Figure 3. SCN5A, c.2893C > T, p.R965C as an example of ancestral differences in allele frequency.
In the present study, we identified 18 variants with minor allele frequencies ≥0.001 in at least one ancestry, indicating that they may be common variants in these populations. In the poly-ancestry population databases, the number of participants in gnomAD is larger than that in 1000G (poly-ancestry) and ESP6500 (European ancestry and African Americans only) whereas in the population-specific databases, TWB (Han Chinese) and iJGVD (Japanese) are probably larger than most internal controls from an individual institution. As a result, the allele frequency in gnomAD, TWB and iJGVD were more approximate to the true allele frequencies of each ancestry. Importantly, 14 (77.8%) of the variants had allele frequencies ≥0.001 only in Asian population. This may imply the distinct genetic background of Asian BrS patients and a different pathogenicity of the same variants in BrS patients across populations. Some variants with definite effects on transmembrane currents, or those predicted to be deleterious by in silico analysis, may be reclassified as disease predisposition alleles; however, the pathogenicity of other variants should be subjected to further careful investigation. Since most of these studies used Caucasian population as healthy controls, the MAF in other ancestries could not be known. Therefore, a large database collected from all ancestries is crucial, and variants with allele frequencies <0.001 across all ancestries (European, African, American, and Asian) are much more likely to be pathogenic.
Our study revealed that significantly more previously reported non-SCN5A variants than SCN5A variants should be reclassified. This finding is consistent with the findings of Le Scouarnec et al. (2015), who showed that there was a higher percentage of BrS cases with rare functional variants in SCN5A than in non-SCN5A genes, compared with internal controls. There are several possible explanations. First, there have been many comprehensive in vitro electrophysiological functional studies of SCN5A. The authors of all such studies investigated each variant repeatedly and rigorously, and were therefore able to exclude any variant for which pathogenicity could not be demonstrated. However, studies regarding other genes have usually been small-scale and of limited scope, and may not have excluded some benign variants. Second, SCN5A plays an important role in cardiac development and function; thus, non-synonymous variants that alter protein function may directly enhance disease development. Thus, non-SCN5A variants should be interpreted carefully.
The substitution of the amino acids that constitute the pore-forming domain of SCN5A may alter its gating function; therefore, this domain should be more highly conserved in evolution across different ancestries than other regions. Alterations to the pore function may be less well tolerated than changes in other regions, which could explain the consistently low allele frequencies of pore-forming domain variants among the databases examined.
Regarding the types of mutations, only 0.9% of the nonsense and frameshift mutations had allele frequencies ≥0.001 in at least one ancestry (European, African, American, or Asian), a significantly lower percentage than that of the missense mutations (5.6%). This difference may imply that BrS-associated nonsense and frameshift mutations are much more likely to be pathogenic, which may be attributed to critical alterations in protein function in these types of mutations.
We found that 4% of the patients with BrS in our cohort carried SCN5A variants with minor allele frequencies ≥0.001. Of these patients, four had p.A226V and one had p.V1340I. SCN5A p.A226V is located in the VSD, and p.V1340I is located in the pore-forming domain; both reduce the sodium current in patch-clamp studies (Samani et al., 2009; Tan et al., 2016). However, in a Singaporean family reported by Tan et al., the father carrying p.A226V did not show type 1 Brugada ECG after flecainide infusion. This suggested that the pathogenicity of p.A226V may be questionable in the East Asian population, demonstrating that considering ancestral difference and clinical data is as important as functional studies when we interpret the pathogenicity of a genetic variant.
There are several possible reasons why a variant that causes functional alterations in vitro could be a disease predisposition allele in at least some ancestries. First, because protein–protein interactions are complicated in living cells, the effects of variants in transfected cells may not always persist in vivo, where a gene that does not encode an ion channel may nevertheless alter the current by interacting with that channel (Cerrone et al., 2014). Second, a pathogenic variant may be masked by another variant (Marangoni et al., 2011), and an ancestry with an abundance of such protective variants could be resistant to this pathogenic variant. Third, deoxyribonucleic acid (DNA) methylation is known to regulate gene expression and is highly divergent among ancestries (Fraser et al., 2012). Therefore, functional studies are essential, and ancestral differences should be taken into account when judging the pathogenicity of a novel variant.
The present study has limitations. First, more evidences emerged now that the inheritance underlying BrS may be complex with multiple genes and possibly some environmental factors contributing to the phenotype, and some common variants may also be contributive (Bezzina et al., 2013; Abriel, 2015; Behr et al., 2015). Therefore, we may not be able to exclude possible contribution to pathogenicity of a variant in polygenic model solely because of its high allele frequency. Second, because electrocardiograms are generally not available in the public control databases, it is possible that some asymptomatic individuals carrying pathogenic BrS-associated variants might be enrolled as “apparently healthy” controls in the public databases. However, considering Brugada syndrome is a rare disease with the prevalence of 12/10,000 (Southeast Asia populations) and 5/10,000 (Caucasians) in the general populations, and healthy controls are usually enrolled randomly from a general population, the expected contribution from the minor allele of BrS-associated variants to the public control databases could be very low (0–0.06%). Third, although TWB and iJGVD are the population-specific references for Han Chinese and Japanese populations, respectively, the number of the samples in the two references are relatively small compared with gnomAD. Therefore, the representativity for the two populations and the power of excluding benign variants may also be a concern. Fourth, if a potentially pathogenic BrS-related variant is “protected” by other genes or by epigenetic effects, its pathogenic phenotype may be attenuated. As a result, its allele frequency in a population may be relatively high. However, this variant may remain pathogenic in other populations or segments of the population; for example, for one gender or in certain ancestries. In such a case, the variant would have been considered to have “questionable pathogenicity” in our study and would require periodic re-evaluation.
In conclusion, the allele frequencies of BrS variants differ significantly with ancestry. This finding should be taken into consideration before judging a variant as pathogenic, particularly in the case of non-SCN5A variants, which may be pathogenic in some ancestries but only disease-predisposing in others. Besides, in-house controls and small-scale databases of population frequency have less power to exclude benign variants; large-scale super-controls, such as gnomAD and ancestry-specific databases, could be used to address this issue.
J-MJ and CA contributed to the conception and design of the study. Y-BL, L-TH, and H-CH collected the data. T-PL and L-PL contributed to the data analysis. C-YC, S-FY, and C-KW wrote the manuscript. L-YL and J-JH supervised the whole project. All authors read and approved the final manuscript.
Financial support for this research was provided partially through grants NTUH 106-S3469, NTUH106-S3458, NTUH 105-012, NTUH 106-018, NTUH 105-S2995, UN 103-018, and UN104-001 from National Taiwan University Hospital, National Taiwan University, NSC 101-2314-B-002-168-MY2, NSC 101-2314-B-002-173-MY2, NSC 103-2314-B-002-148, MOST 104-2314-B-002-193-MY3, MOST 106-2314-B-002-047-MY3, MOST 106-2314-B-002-134-MY2, MOST 106-2314-B-002-206, MOST 107-2314-B-002-009, and MOST 107-2314-B-002-261-MY3 from the Ministry of Science and Technology, Taiwan Health Foundation, and National Institutes of Health grant # HL47678 (CA).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are sincerely grateful to the staff of the Sixth Core Lab, Department of Medical Research, National Taiwan University Hospital for technical support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00680/full#supplementary-material
Abriel, H. (2015). Genetic background of Brugada syndrome is more complex than what we would like it to be! Cardiovasc. Res. 106, 351–352. doi: 10.1093/cvr/cvv135
Antzelevitch, C. (2001). The Brugada syndrome: ionic basis and arrhythmia mechanisms. J. Cardiovasc. Electrophysiol. 12, 268–272. doi: 10.1046/j.1540-8167.2001.00268.x
Antzelevitch, C., Brugada, P., Borggrefe, M., Brugada, J., Brugada, R., Corrado, D., et al. (2005a). Brugada syndrome: report of the second consensus conference. Heart Rhythm 2, 429–440. doi: 10.1016/j.hrthm.2005.01.005
Antzelevitch, C., Brugada, P., Borggrefe, M., Brugada, J., Brugada, R., Corrado, D., et al. (2005b). Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 111, 659–670. doi: 10.1161/01.CIR.0000152479.54298.51
Behr, E. R., Savio-Galimberti, E., Barc, J., Holst, A. G., Petropoulou, E., Prins, B. P., et al. (2015). Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc. Res. 106, 520–529. doi: 10.1093/cvr/cvv042
Bezzina, C. R., Barc, J., Mizusawa, Y., Remme, C. A., Gourraud, J. B., Simonet, F., et al. (2013). Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 45, 1044–1049. doi: 10.1038/ng.2712
Brugada, P., and Brugada, J. (1992). Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 20, 1391–1396. doi: 10.1016/0735-1097(92)90253-J
Burashnikov, E., Pfeiffer, R., Barajas-Martinez, H., Delpon, E., Hu, D., Desai, M., et al. (2010). Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7, 1872–1882. doi: 10.1016/j.hrthm.2010.08.026
Cerrone, M., Lin, X., Zhang, M., Agullo-Pascual, E., Pfenniger, A., Chkourko Gusky, H., et al. (2014). Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 129, 1092–1103. doi: 10.1161/CIRCULATIONAHA.113.003077
Clemens, D. J., Lentino, A. R., Kapplinger, J. D., Ye, D., Zhou, W., Tester, D. J., et al. (2018). Using the genome aggregation database, computational pathogenicity prediction tools, and patch clamp heterologous expression studies to demote previously published long QT syndrome type 1 mutations from pathogenic to benign. Heart Rhythm 15, 555–561. doi: 10.1016/j.hrthm.2017.11.032
Crotti, L., Marcou, C. A., Tester, D. J., Castelletti, S., Giudicessi, J. R., Torchio, M., et al. (2012). Spectrum and prevalence of mutations involving BrS1- through BrS12-susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: implications for genetic testing. J. Am. Coll. Cardiol. 60, 1410–1418. doi: 10.1016/j.jacc.2012.04.037
Fan, C. T., Lin, J. C., and Lee, C. H. (2008). Taiwan Biobank: a project aiming to aid Taiwan’s transition into a biomedical island. Pharmacogenomics 9, 235–246. doi: 10.2217/14622416.9.2.235
Fernandez-Falgueras, A., Sarquella-Brugada, G., Brugada, J., Brugada, R., and Campuzano, O. (2017). Cardiac channelopathies and sudden death: recent clinical and genetic advances. Biology 6:E7. doi: 10.3390/biology6010007
Fraser, H. B., Lam, L. L., Neumann, S. M., and Kobor, M. S. (2012). Population-specificity of human DNA methylation. Genome Biol. 13:R8. doi: 10.1186/gb-2012-13-2-r8
Hong, K., Brugada, J., Oliva, A., Berruezo-Sanchez, A., Potenza, D., Pollevick, G. D., et al. (2004). Value of electrocardiographic parameters and ajmaline test in the diagnosis of Brugada syndrome caused by SCN5A mutations. Circulation 110, 3023–3027. doi: 10.1161/01.CIR.0000144299.17008.07
Hu, D., Barajas-Martinez, H., Pfeiffer, R., Dezi, F., Pfeiffer, J., Buch, T., et al. (2014a). Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J. Am. Coll. Cardiol. 64, 66–79. doi: 10.1016/j.jacc.2014.04.032
Hu, D., Barajas-Martinez, H., Terzic, A., Park, S., Pfeiffer, R., Burashnikov, E., et al. (2014b). ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int. J. Cardiol. 171, 431–442. doi: 10.1016/j.ijcard.2013.12.084
Ishikawa, T., Sato, A., Marcou, C. A., Tester, D. J., Ackerman, M. J., Crotti, L., et al. (2012). A novel disease gene for Brugada syndrome: sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ. Arrhythm Electrophysiol. 5, 1098–1107. doi: 10.1161/CIRCEP.111.969972
Ishikawa, T., Takahashi, N., Ohno, S., Sakurada, H., Nakamura, K., On, Y. K., et al. (2013). Novel SCN3B mutation associated with brugada syndrome affects intracellular trafficking and function of Nav1.5. Circ. J. 77, 959–967. doi: 10.1253/circj.CJ-12-0995
Juang, J. J., and Horie, M. (2016). Genetics of Brugada syndrome. J. Arrhythm 32, 418–425. doi: 10.1016/j.joa.2016.07.012
Juang, J. M., Chen, C. Y., Chen, Y. H., Wu, I. C., Hsu, C. C., Chen, L. N., et al. (2015). Prevalence and prognosis of Brugada electrocardiogram patterns in an elderly Han Chinese population: a nation-wide community-based study (HALST cohort). Europace 17(Suppl. 2), ii54–ii62. doi: 10.1093/europace/euv141
Juang, J. M., Lu, T. P., Lai, L. C., Ho, C. C., Liu, Y. B., Tsai, C. T., et al. (2014). Disease-targeted sequencing of ion channel genes identifies de novo mutations in patients with non-familial Brugada syndrome. Sci. Rep. 4:6733. doi: 10.1038/srep06733
Kapplinger, J. D., Tester, D. J., Alders, M., Benito, B., Berthet, M., Brugada, J., et al. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7, 33–46. doi: 10.1016/j.hrthm.2009.09.069
Landrum, M. J., Lee, J. M., Benson, M., Brown, G., Chao, C., Chitipiralla, S., et al. (2016). ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868. doi: 10.1093/nar/gkv1222
Le Scouarnec, S., Karakachoff, M., Gourraud, J. B., Lindenbaum, P., Bonnaud, S., Portero, V., et al. (2015). Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum. Mol. Genet. 24, 2757–2763. doi: 10.1093/hmg/ddv036
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Manrai, A. K., Funke, B. H., Rehm, H. L., Olesen, M. S., Maron, B. A., Szolovits, P., et al. (2016). Genetic misdiagnoses and the potential for health disparities. N. Engl. J. Med. 375, 655–665. doi: 10.1056/NEJMsa1507092
Marangoni, S., Di Resta, C., Rocchetti, M., Barile, L., Rizzetto, R., Summa, A., et al. (2011). A Brugada syndrome mutation (p.S216L) and its modulation by p.H558R polymorphism: standard and dynamic characterization. Cardiovasc. Res. 91, 606–616. doi: 10.1093/cvr/cvr142
Matsusue, A., Yuasa, I., Umetsu, K., Nakayashiki, N., Dewa, K., Nishimukai, H., et al. (2016). The global distribution of the p.R1193Q polymorphism in the SCN5A gene. Leg Med. 19, 72–76. doi: 10.1016/j.legalmed.2015.07.010
Mizusawa, Y., and Wilde, A. A. (2012). Brugada syndrome. Circ. Arrhythm Electrophysiol. 5, 606–616. doi: 10.1161/CIRCEP.111.964577
Nagasaki, M., Yasuda, J., Katsuoka, F., Nariai, N., Kojima, K., Kawai, Y., et al. (2015). Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat. Commun. 6:8018. doi: 10.1038/ncomms9018
Priori, S. G., Napolitano, C., Gasparini, M., Pappone, C., Della Bella, P., Brignole, M., et al. (2000). Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: a prospective evaluation of 52 families. Circulation 102, 2509–2515. doi: 10.1161/01.CIR.102.20.2509
Priori, S. G., Napolitano, C., Gasparini, M., Pappone, C., Della Bella, P., Giordano, U., et al. (2002). Natural history of Brugada syndrome: insights for risk stratification and management. Circulation 105, 1342–1347. doi: 10.1161/hc1102.105288
Priori, S. G., Wilde, A. A., Horie, M., Cho, Y., Behr, E. R., Berul, C., et al. (2013). HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 10, 1932–1963. doi: 10.1016/j.hrthm.2013.05.014
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rook, M. B., Bezzina Alshinawi, C., Groenewegen, W. A., van Gelder, I. C., van Ginneken, A. C., Jongsma, H. J., et al. (1999). Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc. Res. 44, 507–517. doi: 10.1016/S0008-6363(99)00350-8
Samani, K., Wu, G., Ai, T., Shuraih, M., Mathuria, N. S., Li, Z., et al. (2009). A novel SCN5A mutation V1340I in Brugada syndrome augmenting arrhythmias during febrile illness. Heart Rhythm 6, 1318–1326. doi: 10.1016/j.hrthm.2009.05.016
Selga, E., Campuzano, O., Pinsach-Abuin, M. L., Perez-Serra, A., Mademont-Soler, I., Riuro, H., et al. (2015). Comprehensive genetic characterization of a spanish brugada syndrome cohort. PLoS One 10:e0132888. doi: 10.1371/journal.pone.0132888
Stenson, P. D., Ball, E. V., Mort, M., Phillips, A. D., Shaw, K., and Cooper, D. N. (2012). The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr. Protoc. Bioinformatics 39, 1.13.1–1.13.20. doi: 10.1002/0471250953.bi0113s39
Tan, B. Y., Yong, R. Y., Barajas-Martinez, H., Dumaine, R., Chew, Y. X., Wasan, P. S., et al. (2016). A Brugada syndrome proband with compound heterozygote SCN5A mutations identified from a Chinese family in Singapore. Europace 18, 897–904. doi: 10.1093/europace/euv058
Vatta, M., Dumaine, R., Varghese, G., Richard, T. A., Shimizu, W., Aihara, N., et al. (2002). Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum. Mol. Genet. 11, 337–345. doi: 10.1093/hmg/11.3.337
Watanabe, H., Ohkubo, K., Watanabe, I., Matsuyama, T. A., Ishibashi-Ueda, H., Yagihara, N., et al. (2013). SCN5A mutation associated with ventricular fibrillation, early repolarization, and concealed myocardial abnormalities. Int. J. Cardiol. 165, e21–e23. doi: 10.1016/j.ijcard.2012.10.074
Whiffin, N., Minikel, E., Walsh, R., O’Donnell-Luria, A. H., Karczewski, K., Ing, A. Y., et al. (2017). Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 19, 1151–1158. doi: 10.1038/gim.2017.26
Keywords: Brugada syndrome, sudden cardiac death, inherited cardiac arrhythmia syndrome, allele frequency, pathogenic variants
Citation: Chen C-YJ, Lu T-P, Lin L-Y, Liu Y-B, Ho L-T, Huang H-C, Lai L-P, Hwang J-J, Yeh S-FS, Wu C-K, Juang J-MJ and Antzelevitch C (2019) Impact of Ancestral Differences and Reassessment of the Classification of Previously Reported Pathogenic Variants in Patients With Brugada Syndrome in the Genomic Era: A SADS-TW BrS Registry. Front. Genet. 9:680. doi: 10.3389/fgene.2018.00680
Received: 14 September 2018; Accepted: 07 December 2018;
Published: 04 January 2019.
Edited by:
M. Z. A. Bhuiyan, Lausanne University Hospital (CHUV), SwitzerlandReviewed by:
Apichai Khong, Chulalongkorn University, ThailandCopyright © 2019 Chen, Lu, Lin, Liu, Ho, Huang, Lai, Hwang, Yeh, Wu, Juang and Antzelevitch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jyh-Ming Jimmy Juang, amptanVhbmdAbnR1LmVkdS50dw== orcid.org/0000-0003-4767-7636
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.