 Romina Burla1†
Romina Burla1† Mattia La Torre1†
Mattia La Torre1† Giorgia Zanetti1
Giorgia Zanetti1 Alex Bastianelli1
Alex Bastianelli1 Chiara Merigliano1,2
Chiara Merigliano1,2 Simona Del Giudice1
Simona Del Giudice1 Alessandro Vercelli3,4
Alessandro Vercelli3,4 Ferdinando Di Cunto3,4
Ferdinando Di Cunto3,4 Marina Boido3,4
Marina Boido3,4 Fiammetta Vernì1
Fiammetta Vernì1 Isabella Saggio1,2*
Isabella Saggio1,2*- 1Department of Biology and Biotechnology, Sapienza University of Rome, Rome, Italy
- 2Nanyang Technological University, Singapore, Singapore
- 3Neuroscience Institute Cavalieri Ottolenghi, Torino, Italy
- 4Department of Neuroscience, University of Torino, Piedmont, Italy
Epilepsy is a complex clinical condition characterized by repeated spontaneous seizures. Seizures have been linked to multiple drivers including DNA damage accumulation. Investigation of epilepsy physiopathology in humans imposes ethical and practical limitations, for this reason model systems are mostly preferred. Among animal models, mouse mutants are particularly valuable since they allow conjoint behavioral, organismal, and genetic analyses. Along with this, since aging has been associated with higher frequency of seizures, prematurely aging mice, simulating human progeroid diseases, offer a further useful modeling element as they recapitulate aging over a short time-window. Here we report on a mouse mutant with progeroid traits that displays repeated spontaneous seizures. Mutant mice were produced by reducing the expression of the gene Ft1 (AKTIP in humans). In vitro, AKTIP/Ft1 depletion causes telomere aberrations, DNA damage, and cell senescence. AKTIP/Ft1 interacts with lamins, which control nuclear architecture and DNA function. Premature aging defects of Ft1 mutant mice include skeletal alterations and lipodystrophy. The epileptic behavior of Ft1 mutant animals was age and sex linked. Seizures were observed in 18 mutant mice (23.6% of aged ≥ 21 weeks), at an average frequency of 2.33 events/mouse. Time distribution of seizures indicated non-random enrichment of seizures over the follow-up period, with 75% of seizures happening in consecutive weeks. The analysis of epileptic brains did not reveal overt brain morphological alterations or severe neurodegeneration, however, Ft1 reduction induced expression of the inflammatory markers IL-6 and TGF-β. Importantly, Ft1 mutant mice with concomitant genetic reduction of the guardian of the genome, p53, showed no seizures or inflammatory marker activation, implicating the DNA damage response into these phenotypes. This work adds insights into the connection among DNA damage, brain function, and aging. In addition, it further underscores the importance of model organisms for studying specific phenotypes, along with permitting the analysis of genetic interactions at the organismal level.
Introduction
Epilepsy comprises a family of disorders characterized by enduring predisposition to generate spontaneous seizures (Scharfman, 2007; Fisher et al., 2014). Seizures are underpinned by multiple mechanisms and their clinical outcome varies widely (Scharfman, 2007). Regardless of their outcome, seizures arise from disruption of mechanisms that create a balance between neuronal excitation and inhibition. Factors corrupting this balance result from alterations at different levels of brain function, from ion channels to receptors, and neuronal circuits (Stafstrom and Carmant, 2015). Epilepsies are often associated with morphological brain abnormalities (Bertram, 2013), but at least one-third have non-structural etiologies (Guerrini et al., 2014). In the last years, concomitantly with human population demographic changes, high incidence of epileptic disorders has been associated with aging, whose specific pathophysiology is under investigation (Leppik and Birnbaum, 2010).
Understanding the mechanistic path to disease is complex in humans due to ethical issues, unavailability of controls and high costs of human research. As a result, studies mostly rely on the use of models, including human 3D cultures and stem cell based systems (Riminucci et al., 2006; Simão et al., 2015), or, for organismal analyses, genetically engineered mice (Baraban, 2007; Saggio et al., 2014; Remoli et al., 2015; La Torre et al., 2018). One of the way in which epilepsy has been modeled in mice is via the inactivation of genes implicated in ion channels (Yu et al., 2006; Baraban, 2007; Glasscock et al., 2012), or of genes encoding for neurotransmitter receptors (Fonck et al., 2005). In addition, epileptic phenomena have been observed in mouse models of Alzheimer disease (Vogt et al., 2011; Ziyatdinova et al., 2011). However, no other genetic models of age related epilepsy have been yet described. Prematurely aging mouse mutants, which recapitulate aging traits over a short time-window (Blasco, 2005; Stewart et al., 2007), offer a specific advantage to model diseases caused or exacerbated by aging, including brain pathologies.
An emerging cause for brain disease and for the aging brain is DNA damage (Langie et al., 2017). DNA integrity poses a challenge for the nervous system as its development depends on a complex series of dynamic and adaptive processes associated to DNA damage (Mckinnon, 2013). Unrepaired DNA lesions have detrimental effects on the developing of a functional nervous system and neural progenitor cells rely on DNA repair systems during the developmental program. After completion of neurogenesis, DNA repair is still of paramount importance to safeguard the genome (Madabhushi et al., 2014), especially to protect the neurons against reactive oxygen species (Langie et al., 2017). DNA damage is also direct cause for cell senescence and for a related inflammatory response (Campisi, 2013; López-Otín et al., 2013).
Mouse mutants of DNA damage functions have opened the path to establish a link between DNA damage and the seizure phenotype (Shen et al., 2010; Bianchi et al., 2017). For example, the inactivation of XRCC1, a central factor in the DNA single strand break repair pathway, leads to profound neuropathology involving behavioral phenotypes consistent with epilepsy (Lee et al., 2009). Data based on studies in Drosophila suggest that nuclear architecture and lamins could play a role into epilepsy (Frost, 2016). However, the hierarchy and range of events connecting nuclear architecture, molecular DNA function to epileptic behavior is still to be dissected, along with the elements exacerbating this pathology in aging.
DNA damage affects genome near to randomly, but some chromosomal regions, such as telomeres, are more prone to DNA instability. In mammals, telomeric DNA is composed of double-stranded short tandem repeats of TTAGGG sequence forming higher-order DNA structures binding a specialized protein complex, known as shelterin (de Lange, 2005). We identified a telomere-associated protein named AKTIP in humans (and Ft1 in mouse), which interacts with the shelterin members TRF1 and TRF2 (Burla et al., 2015). AKTIP/Ft1 reduction causes telomere aberrations, DNA instability and cell senescence (Burla et al., 2015). In vivo, the genetic reduction of Ft1 causes premature aging defects including skeletal alterations and lipodystrophy (La Torre et al., 2018). AKTIP/Ft1 interacts with lamins (Burla et al., 2016a,b), pivotal elements for the control of nuclear architecture and DNA function, including DNA repair, replication and transcription (Dittmer and Misteli, 2011). Importantly, Ft1 mutant mice share similarities with lamin mutant animals, which are models of choice for human progeroid syndromes, linking the Ft1 model to premature aging and progeroid diseases (Burla et al., 2016a,b, 2018).
Here we report that Ft1 mutant mice are subjected to repeated seizures. We show that this trait is not linked to overt brain morphological alterations, but is age and inflammation linked. We also demonstrate that this phenotype is sensitive to the expression of the guardian of the genome p53, pointing to a role of DNA function in the seizure phenotype.
Results
Seizures in Ft1 Mutant Mice
Mice with reduced levels of Ft1 were generated using the knock out first (kof) strategy based on the insertion into the target gene (referred as kof allele) of the βgeok cassette (Testa et al., 2004), which traps and truncates Ft1 nascent transcript reducing the expression of the targeted gene (La Torre et al., 2018). Previous observations of Ft1kof/kof mice revealed that mutant animals display significant reduction in body weight and lifespan, compared to controls. Twenty-one percent of the animals show a severe body size reduction and early post-natal death (we refer to these mice as severely affected Ft1kof/kof mice, abbreviated with SA Ft1kof/kof or SA mutant mice). The leftovers, non SA Ft1kof/kof mice have a mild phenotype, with a median survival of 113 weeks, allowing adult phenotype observation (La Torre et al., 2018).
To investigate mutant mouse behavior, we worked on a cohort of animals aged ≥ 21 weeks non SA Ft1kof/kof. We recorded spontaneous behavioral abnormalities in non SA Ft1kof/kof mice including episodes of motor tremors and convulsions, fast runs, jumps, and excessive salivation (Figure 1 and Supplementary Movies 1–3). Dissection of video recordings indicated that non SA Ft1kof/kof displayed sudden movements followed by facial twitching, violent hind limbs shaking and falling, back arching, short jerks in muscles of the hind limbs and forelimbs extension, Straub tail and incontinence, followed by short jerks and fast breathing (Figure 1A and Supplementary Movies 1–3). Eventually, animals stood up and returned to normal activity; after few minutes switched to post-ictal phase characterized by short periods of complete immobility, interrupted by short intervals of movement (Supplementary Movies 4–6). Recording showed moving rhythmical up-down or left-right, stroking mouth with forepaws, in a repetitive motion, and appearing to be chewing and grooming (Figure 1A and Supplementary Movies 4–6). Temporal evaluation of the data indicated that non SA Ft1kof/kof mice display a behavioral repertoire corresponding in quality and duration to epileptic phenotypes (Figure 1B), as described for other mouse models (Chabrol et al., 2010; Robie et al., 2017).
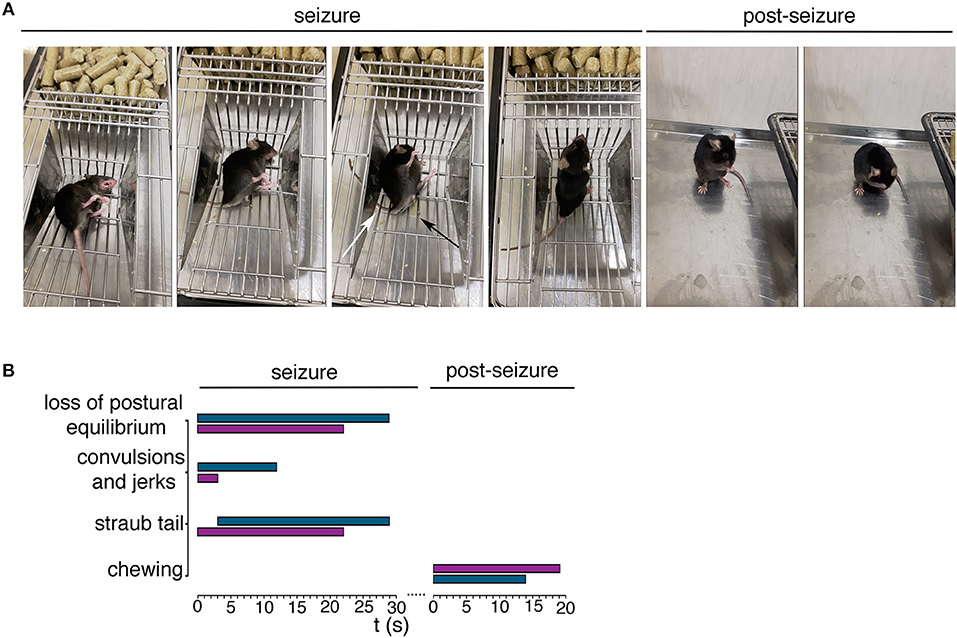
Figure 1. Ft1kof/kof mice exhibit seizures. (A) Representative movie frames from non SA Ft1kof/kof mice recording during and post seizure. Starting from left of the seizure panel group: loss of postural equilibrium, arching of the back, Straub tail (third picture, white arrow) and incontinence (third picture, black arrow), recovery of the postural equilibrium. Frames from the movie also show motor automatisms in the post seizure panel group, as chewing (first picture) and grooming (second picture). (B) Progression of behavior of two Ft1kof/kof mice, during and post seizure events, each represented by a horizontal bar. The length of each bar indicates the duration of the relative motor type. In turquoise seizure analysis of mouse ID #13489, and in purple the same analysis of mouse ID #16247.
Over a total of 260 animals (including Ft1+/+, Ft1+/kof, and Ft1kof/kof), 18 Ft1kof/kof exhibited at least one seizure manifestation (Figures 2A,B). No seizure episodes were observed in heterozygote Ft1+/kof or wt mice (Figure 2A). The frequency of seizures reached a maximum of one seizure episode per week. Of 18 seizure positive mutants, 7 exhibited one seizure-like manifestation during their follow-up period, while the leftover displayed more than one seizure. On average, we observed 2.33 ± 0.14 seizures during the entire follow-up period in the seizure positive mutants (Figure 2B). Time distribution of seizures for each mutant animal experiencing more than one seizure indicated non-random enrichment of seizures over the follow-up period, with 75% of seizures happening in consecutive weeks (Figure 2C).
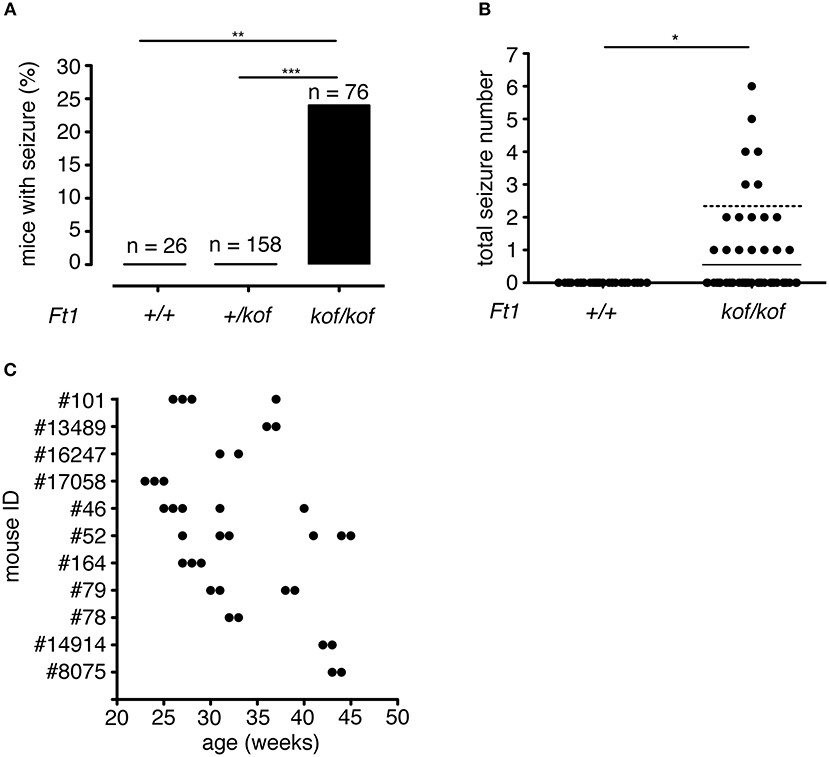
Figure 2. Seizure frequency in Ft1kof/kof mice. (A) Percentages of mice that exhibited seizure; no seizures were observed in wt (**p < 0.01 χ2 test) or in heterozygous Ft1+/kof mice (***p < 0.001 χ2 test). (B) Total number of seizure experienced by wt and Ft1kof/kof mice, each dot represents one mouse. Line indicates average seizure number considering all Ft1kof/kof; dashed line indicates average seizure number (2.33 ± 0.14) considering Ft1kof/kof which experienced seizures (*p < 0.05 Student's t-test). (C) Temporal seizure distribution for each Ft1kof/kof mouse which experienced more than one seizure. Each dot represents a seizure episode. n, total number of analyzed mice.
Seizures of Ft1 Mutant Mice are Age and Gender Linked
To analyze the distribution of seizures during aging, we subdivided mouse lifespan in three major intervals: young (3 ≤ weeks ≤ 20), juvenile (21 ≤ weeks ≤ 60), and adult (61 ≤ weeks ≤ 100). We monitored, for each age interval, the fraction of mice exhibiting seizures for the first time (Figure 3A). None of young non SA Ft1kof/kof mice (n = 189) displayed seizures, while seizures were observed in juvenile and adult mice, in 16 out of 68 and 2 out of 8, respectively.
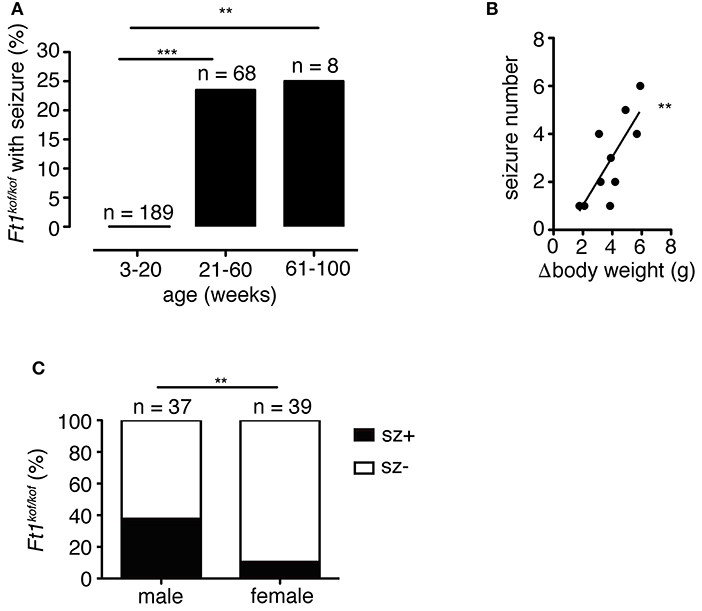
Figure 3. Seizures in Ft1kof/kof mice are age, body weight and sex-linked. (A) Percentage of Ft1kof/kof mice exhibiting first seizure clustered in three age intervals (young: 3 ≤ weeks ≤ 20; juvenile: 21 ≤ weeks ≤ 60; adult: 61 ≤ weeks ≤ 100). No seizures were observed in young animals (**p < 0.01 and ***p < 0.001 in χ2 test). (B) Correlation between the severity of growth defect and seizure frequency (r2 = 0.60; **p < 0.01 in Pearson's R test). Each dot represents data from an individual animal: body weight differences (Δbody weight) were obtained subtracting average body weight of Ft1kof/kof to the average weight of age and sex-matched wt group. (C) Gender related differences in seizure behavior (**p < 0.01 in χ2 test). n, total number of analyzed mice.
We previously reported that non SA Ft1kof/kof mice display growth defects, which start within the juvenile interval and become prominent through aging (La Torre et al., 2018). We then asked whether the seizure phenotype was paralleled by age-associated growth impairment. We considered the difference between the average body weight of wt (n = 6) and sex-matched seizure positive mutant mice (n = 8) (Δbody weight), and plotted it against the number of seizures observed in each mutant animal to generate a regression curve (Figure 3B). The two variables were linked by positive correlation (r2 = 0.60; **p < 0.01 in Pearson's R test).
Non SA Ft1kof/kof male mice were previously shown to display a stronger phenotype as compared to Ft1kof/kof females (La Torre et al., 2018). We thus decided to explore gender differences in the seizure trait. We analyzed a cohort of non SA Ft1kof/kof animals aged ≥ 21 weeks including 37 males and 39 females. Seizure positive mutants were 37.8% among males and 10.3% among non SA Ft1kof/kof female mice (Figure 3C).
In order to verify whether aging would induce variations in Ft1 expression we monitored two cohorts of Ft1+/+ mice by QPCR. Results indicate that there are no significant variation in Ft1 expression with aging (Supplementary Figure 1A). In addition, the human counterpart of Ft1, AKTIP, is also expressed in brain cells (Supplementary Figure 1B).
Altogether these results indicate that Ft1 reduction causes seizures, and that this phenotype parallels overall organismal alterations characteristic of non SA Ft1kof/kof mice including age dependent growth impairment and sex-linked defects (La Torre et al., 2018).
Structure of Ft1 Mutant Brain
To define whether Ft1 function impacted on brain structure we firstly measured skull and cranial length of non SA Ft1kof/kof mice by X-ray analysis (Figure 4). Mutant and age- and sex-matched wt animals were monitored at 2, 4, 8, and 13 months. Length measures revealed a continuous increase during postnatal development in mutant mice as in controls, with a mild alteration induced by Ft1 reduction at the age of 8 weeks (*p < 0.05 in Student's t-test) (Figures 4A–D). We then investigated brain morphology by histological analysis. We firstly evaluated the hippocampus, as hippocampal defects are often cause of epilepsy. Nissl-staining of coronal sections of Ft1kof/kof brains did not highlight overt alterations of hippocampal organization or gross lesions, such as cell loss, structural deformation or scars (Figures 5A–B). By immunofluorescence, we further analyzed hippocampus cytoarchitecture. Semi-quantitative analysis of Parvalbumin-positive GABAergic interneurons showed a mild reduction in Ft1kof/kof mice as compared to controls (Figure 5C and Supplementary Figure 2). Then we evaluated the somatosensory cortex cytoarchitecture: by Nissl-staining and MAP2 immunofluorescence, we did not detect evident alterations, including cortical thinning and layering defects in Ft1kof/kof brain compared to wt (Figures 6A–C). The density of Parvalbumin-positive GABAergic interneurons in the cortex was non-significantly affected by Ft1 reduction (Figure 6D and Supplementary Figure 3). Moreover, we did not observe any evident macroscopic alterations in the whole brain structure, as for example shrinkage of specific cerebral regions, ventricle enlargement, corpus callosum thinning (data not shown), or in brain volume (wt 177.95 mm3, Ft1kof/kof 189.46 mm3; Supplementary Figure 4). Overall, these analyses did not reveal robust differences between non SA Ft1kof/kof and control mice, thus in principle excluding the presence of severe brain degenerative processes.
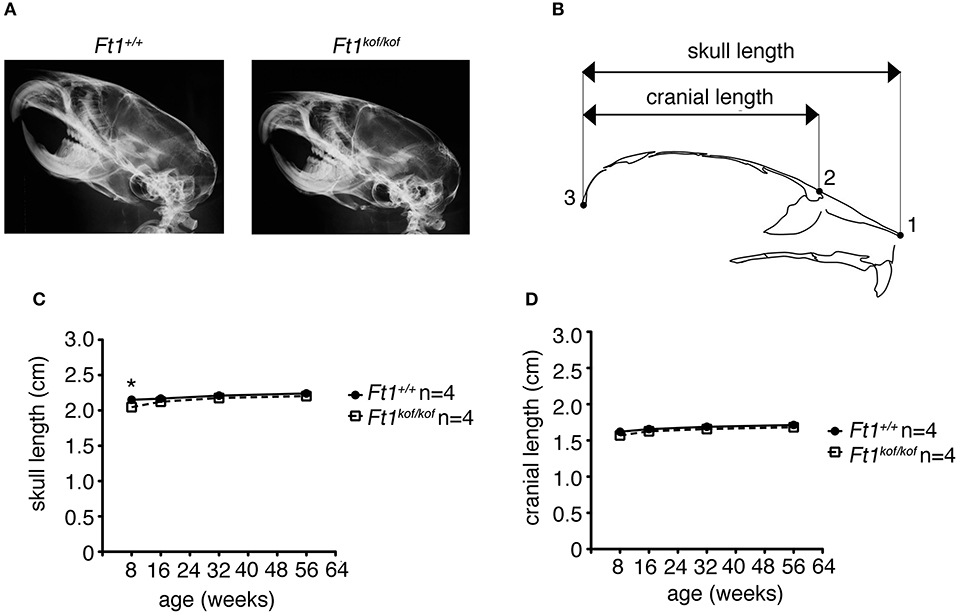
Figure 4. Ft1 reduction does not affect skull development. (A) X-ray analysis of craniofacial skeleton of wt and Ft1kof/kof mice. (B) Schematic view of mouse skull and description of landmarks used for the anterior-posterior craniofacial skeleton length analysis. (C,D) Skull and cranium length analysis of Ft1kof/kof animals and age-matched wt controls. Curves are not overall significantly different in Student's t-test (p = 0.225), the difference of the skull length between Ft1kof/kof and Ft1+/+ animals is significantly different at the age of 8 weeks (*p < 0.05 in Student's t-test). n, total number of analyzed mice.
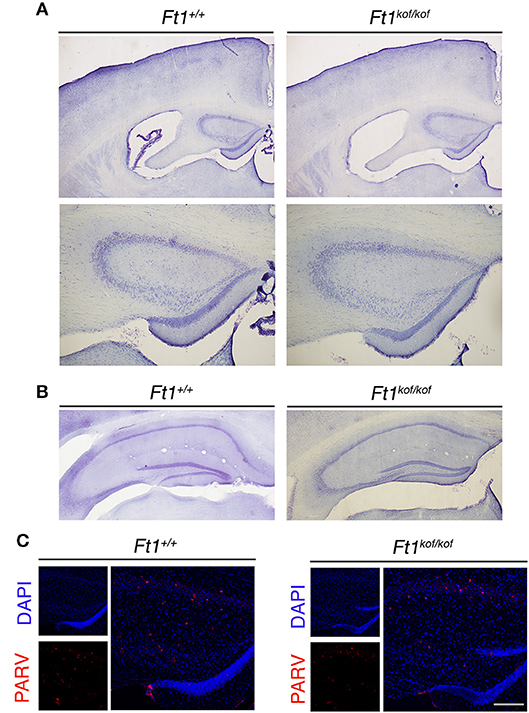
Figure 5. Ft1 reduction does not overtly affect hippocampal organization. (A) Nissl-stained coronal sections of Ft1+/+ and Ft1kof/kof mouse brains (upper panels), and higher magnification of the anterior hippocampus (bottom panels), at about Bregma −1.22 mm. (B) Nissl-stained hippocampus of Ft1+/+ and Ft1kof/kof mice, at about Bregma −2.18 mm. (C) Confocal images showing parvalbumin-positive interneurons (PARV, in red) at the hippocampal level; cell nuclei are labeled by DAPI staining (in blue). Scale bars: (A) 500 μm (upper panel), (A) 200 μm (bottom panel), (B) 400 μm, (C) (merge) 160 μm, (C) (DAPI/PARV) 200 μm.
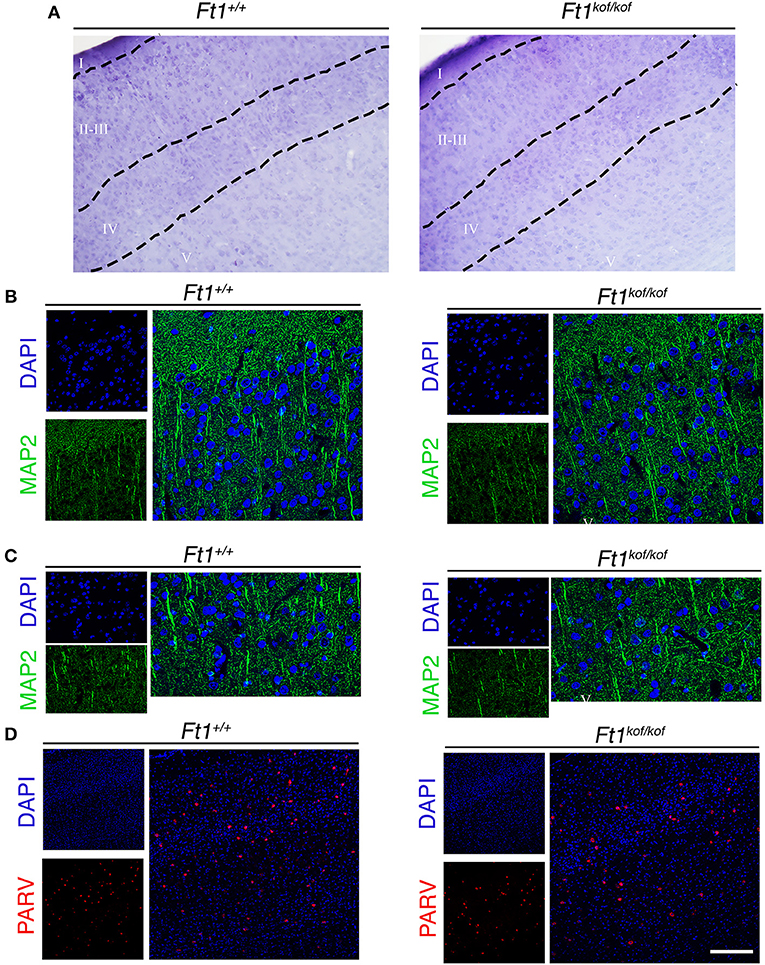
Figure 6. Ft1 reduction does not affect somatosensory cortex cytoarchitecture. (A) Nissl-stained coronal sections, showing somatosensory cortex of Ft1+/+ and Ft1kof/kof mice brains. (B) MAP2-positive neurons (green) located in the cortical supragranular layers I-II-III and (C) in the infragranular layer V; cell nuclei are labeled by DAPI staining (in blue). (D) Confocal images showing parvalbumin-positive interneurons (PARV, in red) and DAPI-positive nuclei (in blue). Scale bars: (A) 100 μm, (B,C) 55 μm, (D) 160 μm.
p53-sensitive Inflammatory Response in Ft1 Mutant Brain
To investigate on molecular drivers for the seizure phenotype observed in Ft1kof/kof mice we reasoned on studies in aging and progeroid models which link DNA damage to systemic inflammation and neurodegeneration (Campisi, 2013; López-Otín et al., 2013). Given the progeroid traits observed in non SA Ft1kof/kof mice and the implication of Ft1 in telomere metabolism and DNA function we decided to monitor the level of two canonical, interrelated, inflammatory cytokines: IL-6 and TGF-β. To get a full picture also on the connection with DNA damage, we explored this aspect in non SA Ft1kof/kof mice, expressing normal levels of the guardian of the genome p53 and in mice defective for p53 expression due to heterozygous p53 inactivation (Ft1kof/kof; p53+/ko). QPCR analysis showed significant higher levels of both IL-6 and TGF-β in Ft1kof/kof brains as compared to age matched control mice (Figures 7A,B; **p < 0.01). Indeed, in Ft1kof/kof; p53+/ko mutant mice IL-6 and TGF-β activation was reversed. These results suggest that the activation of the DNA damage response pivotal player, p53, is a crucial event in the organismal response to Ft1 reduction (Figures 7A,B). As expected, p53 reduction did not impact on Ft1 expression, which remained significantly reduced in Ft1kof/kof; p53+/ko as in Ft1kof/kof animals (Figure 7C).
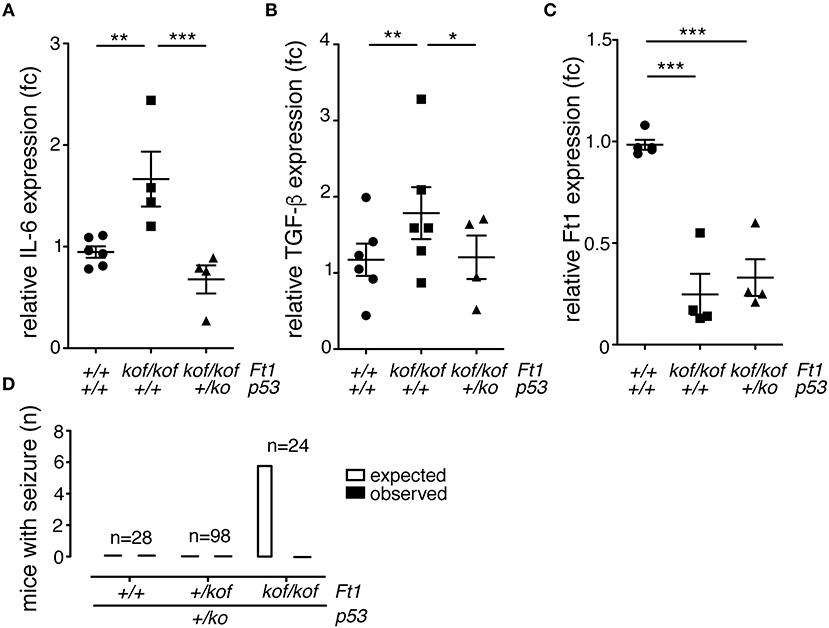
Figure 7. p53 reduction reverses inflammation and seizures in Ft1kof/kof mice. (A–C) QPCR expression quantification of IL-6, TGF-β and Ft1 in Ft1kof/kof; Ft1+/kof Ft1kof/kof in a wt or p53+/ko background (*p < 0.05; **p < 0.01; ***p < 0.001 Student's t-test). (D) Expected versus observed number of mice with seizures in Ft1kof/kof; Ft1+/kof Ft1kof/kof in a wt or p53+/ko background. Expected seizure number was calculated considering the seizure frequency in Ft1kof/kof age matched cohort (see Figure 2A). To note that none of Ft1kof/kof mice with the p53+/ko mutation displayed seizures. n, total number of analyzed mice.
Given the activation of the inflammatory cytokines and its rescue by p53 reduction, we decided to explore the link between p53, as an element directly associated with DNA function, to the seizure phenotype of Ft1kof/kof mice. To this purpose, we analyzed three cohorts of ≥ 21 weeks mice including Ft1+/+;p53+/ko, Ft1+/kof;p53+/ko and Ft1kof/kof;p53+/ko mice, as compared to genetically and age matched controls. None of the mice with the p53+/ko mutation displayed seizures, suggesting on one side that p53 reduction alone does not cause seizures, and, on the other side, that it rescues the seizure phenotype of our Ft1 mutant mice (Figure 7D).
Altogether these results establish a link among Ft1, p53, inflammatory parameters and seizure behavior.
Discussion
Epilepsy is a complex disorder codified by the concept of predisposition to generate spontaneous seizures. Studies to interpret seizure etiology are complex in humans, indeed many open questions remain about its causes and mechanisms. An important tool to understand and interpret epileptic phenomena comes from model systems. Mutant mice in particular have been instrumental for the identification of genes and gene-determined molecular processes generating brain dysfunction and epileptic phenotypes (Fonck et al., 2005; Yu et al., 2006; Baraban, 2007; Löscher, 2011; Glasscock et al., 2012). In this work we report on the epileptic behavior of a mouse mutant obtained by genetically reducing the expression of the telomeric gene Ft1 (AKTIP in humans) and present data supportive of a link between DNA damage and epileptic behavior.
As a consequence to its biological function, AKTIP/Ft1 mutant cells display DNA replication defects, DNA and telomere damage, along with cell senescence (Burla et al., 2015, 2016a). AKTIP/Ft1 is linked to nuclear architecture through the biochemical interaction occurring with A- and B-type lamins, the main components of the nucleoskeleton, which, in turn, is a pivotal element for the control of DNA function (Dittmer and Misteli, 2011; Burla et al., 2018). Mice with reduced Ft1 expression display a segmental phenotype including reduced subcutaneous fat, growth and skeletal defects. These traits partly recapitulate the premature aging phenotypes observed in progeroid mice generated by mutations of lamins or DNA maintenance genes (Blasco, 2005; Stewart et al., 2007; Burla et al., 2018), and are exacerbated in adult individuals as compared to juvenile. Given the connection of AKTIP/Ft1 with both lamins and DNA function, Ft1 mutant mice represent an attractive model for investigating how these connections may impact on the organismal phenotype and on different tissues and organs.
We report here that Ft1 mutant mice exhibit spontaneous seizures. Digital movies document the seizure manifestation highlighting typical traits, as loss of postural equilibrium, limb jerks, and convulsions, as in other epileptic mouse models (Yang et al., 2007; Minkeviciene et al., 2009; Chabrol et al., 2010; Glasscock et al., 2012; Simeone et al., 2018). Seizures were observed after the 20th week of age suggesting age-dependent degenerative changes. Seizures also correlated with growth impairment, which, in Ft1kof/kof mice, is exacerbated with aging (La Torre et al., 2018). The frequency of seizures was higher in male mice, in line with the overall impact of Ft1 reduction observed in our model (La Torre et al., 2018).
Along with behavioral descriptions, the importance of mouse models resides in the fact that they allow investigating upstream events to behavioral phenotypes, which, in humans, is complex to explore. We thus exploited these animals to analyze the pathophysiological path to seizures in an attempt to contribute to establish experimentally proven links between DNA function and brain alterations.
The analysis of brain morphology and cytoarchitecture in non SA epileptic Ft1kof/kof mice did not highlight overt defects nor macroscopic neurodegeneration. These results were obtained by analyzing restricted brain areas and cell subtypes, and we do not exclude the possibility of other or more subtle brain alterations escaping our analysis. However, given the data, we hypothesized that molecular, rather than overt structural aberrations would underpin the epileptic behavior. This interpretation would be in line with the fact that many types of epilepsy have been associated with molecular alterations, rather than with macroscopic brain structure defects (Guerrini et al., 2014).
On the basis of this hypothesis, we investigated on putative molecular culprits for the seizure phenotype. Telomeres and cell senescence have been implicated in disease and aging. In particular, the secretion of a panel of inflammatory signals (Senescence Associated Secretory Phenotype, SASP) is considered a pivotal element in the alteration of tissue homeostasis contributing to pathological status (Campisi et al., 2001; Campisi, 2013). Given the function of AKTIP/Ft1 in telomere protection along with its importance in preventing cell senescence and DNA damage (Burla et al., 2015; La Torre et al., 2018), we hypothesized that in Ft1 mutant mice accumulation of damage and consequent senescence would provoke inflammatory cytokine production, which, in turn, could be implicated into the epileptic phenotype.
We thus measured inflammatory factors in the brain along with exploring the implication of DNA function in inflammation. In the brain of Ft1 mutant mice we detected the activation of IL-6 and TGF-β. Both these factors are related with inflammation (Coppe et al., 2010). IL-6 activation is assigned to the SASP group of factors linked to cell senescence and has been defined as a senescence biomarker in mice (Coppe et al., 2010; Hudgins et al., 2018). The cytokine TGF-β, in association with IL-6, is a further inflammatory stimulus (Sanjabi et al., 2009). Altogether these data suggested a connection between seizures and inflammation in the brain of Ft1kof/kof mice.
To further investigate on the causative cascade of pathologic events occurring in mice from Ft1 mutation to seizures, we generated Ft1 mutant mice with a heterozygous ko mutation in p53. p53 is a pivotal element in the DNA damage response and we have demonstrated that p53 is activated in AKTIP reduced primary human cells, which contributes to blocking cell proliferation and inducing senescence (Burla et al., 2015; La Torre et al., 2018). Interestingly p53 loss was shown to rescue aging traits in vivo, by releasing DNA damage-induced checkpoints and cell senescence (Varela et al., 2005). In addition, p53 regulated senescence has been defined as a pivotal element in generating the SASP phenotype. Consistently, reduction of p53 expression rescues SASP related aging traits (Baar et al., 2017). In Ft1 mutant mice, p53 reduction not only reversed back the expression of IL-6 and TGF-β, but also rescued the seizure phenotype. These results, taken together, induce to speculate that the path to seizures generated by Ft1 reduction could start with DNA damage, including telomere dysfunction, followed by p53 activation, cell senescence, and SASP. The latter could eventually induce brain seizures, through a mechanism that remains to be elucidated.
This interpretation of the data is interesting for investigating on seizure causative events in basal physiological conditions, but also in aging. In fact, aging is characterized by the exacerbation of the alteration of the biological pathways which we have taken into consideration, including senescence, genomic instability and telomere fragility (López-Otín et al., 2013). These factors act on aging in a cell intrinsic way and through extrinsic mechanisms (Lopez-Otin et al., 2016), as secretion of SASP factors (Coppe et al., 2010) and chronic inflammation, profoundly altering tissue microenvironment (Fulop et al., 2017), which, we would suggest, could impact also on brain function.
In conclusion, this work adds new insights into the connection among DNA damage, brain function, and inflammation. In addition, it further underscores the importance of model organisms for studying molecular path to specific phenotypes, along with permitting the study of genetic interactions at the organismal level. These aspects are even more relevant in aging and brain research studies for which work in humans is inaccessible due to time, ethical and sample accessibility issues.
Materials and Methods
Mice and Ethical Statements
Mice were maintained and bred in a 12 h light/12 h dark cycle, in a pathogen free unit of the animal house at Biology and Biotechnology Department, Sapienza University. Animals were housed and treated in accordance with protocol 355/2017-PR approved by the Italian Ministry of Health. Animals carrying the knockout first mutations in the Ft1 gene (Ft1 kof) were obtained as previously described (La Torre et al., 2018). In order to obtain Ft1 and p53 mutant animals Ft1+/kof were crossed with p53+/ko animals [kindly provided by G. Piaggio and S. Soddu IFO, Italy; (Jacks et al., 1994)]; subsequently doubly heterozygous mice were intercrossed to obtain the desired genotypes. Offspring were weaned at 3 weeks and tail biopsies were used for genotyping. When needed, mice were anesthetized by intramuscular Zoletil 20 (Virbac S.A., France), or euthanized by asphyxiation with carbon dioxide or cervical dislocation.
Genotyping
Mice were genotyped as previously described (La Torre et al., 2018). Briefly genomic DNA was extracted from tail biopsies digested 50°C with a proteinase K/SDS solution using the blackPREP Rodent Tail DNA Kit (Analytik Jena, Jena, Germany) following manufacturer's instructions.
Mice were PCR genotyped using the following primers:
Ft1 E4 F: 5′-GTGAAGCAGAAGCTGCCAGGAGT−3′;
Ft1 E6 R: 5′-AGCTCACCCGAGGTGGGATCAA−3′;
p53-X6 F: 5′-AGCGTGGTGGTACCTTATGAGC−3′;
p53-Neo19 F: 5′-GCTATCAGGACATAGCGTTGGC−3′;
p53-X7 R: 5′-GGATGGTGGTATACTCAGAGCC−3′
Seizure Observation
Seizures were observed during routine mouse handling. Where indicated seizure and post-seizure events were video recorded. The movies were then analyzed for specific behavioral signs as previously described (Chabrol et al., 2010).
X-ray and Cephalometric Measurements
X-ray images were taken using Faxitron MX-20 (Faxitron X-ray Corp.) at 24 kV for 6 s; images captured with Medical Imaging Film HM Plus (Ferrania). Skull length and cranial length were measured by Image J software as previously described (Rueden et al., 2017).
Histological Analysis
Eight month-old mice were euthanized and brains were removed and postfixed in 4% PFA for 2 h at 4°C. Samples were then immersed in a solution containing 30% sucrose in phosphate buffer 0.1 M at 4°C for cryoprotection, embedded in cryostat medium (Killik; Bio-Optica, Milan, Italy), and cut on the cryostat (HM 550; Microm) in serial transverse 50 μm thick sections. Some brain sections (one section every 600 μm) were Nissl-stained to evaluate the gross cerebral morphology: briefly sections were mounted on 2% gelatin-coated Superfrost slides and air-dried overnight; slides were hydrated in distilled water for 1 min before staining in 0.1% Cresyl violet acetate (Sigma Aldrich, St. Louis, MO) for 10 min, dehydrated in an ascending series of ethanol, cleared in xylene and cover-slipped with Eukitt (Bioptica, Milan, Italy). Sections were examined at a Nikon Eclipse 80i microscope Equipped with a Nikon DS-Fi1 camera. Brain volume of wt and Ft1kof/kof mice was calculated considering Bregma 2.10 mm to Bregma −2.54 mm segments in Nissl-stained serial sections reconstructed by Neurolucida software (MicroBrightField, Williston, VT, USA) and the volume (expressed in mm3) was obtained by NeuroExplorer software (MicroBrightField). For immunofluorescence, brain sections were permeabilized with in PBS 0.3% Triton X-100 at RT on a tilting plate for 20 min; then, to block unspecific binding of the antibody, sections were incubated for 30 min at RT with 0.3% Triton X-100 and 10% normal donkey serum or normal goat serum (Sigma-Aldrich) in PBS (pH 7.4). Sections were then incubated at 4°C overnight with the following antibodies: 1:7,500 anti-Parvalbumin (rabbit; Swant); 1:200 anti-MAP2 (mouse; Chemicon). Then, sections were incubated with appropriate fluorochrome-conjugated secondary antibodies, for 1h at RT: 1:400 Alexa Fluor 647, goat anti mouse (Jackson ImmunoResearch Laboratories); 1:200 cyanine 3-conjugated secondary antibody, donkey anti-rabbit (Jackson ImmunoResearch Laboratories); 1:200 cyanine 2-conjugated secondary antibody, donkey anti-mouse (Jackson ImmunoResearch Laboratories). Finally sections were incubated with 4,6 Diamino-2 phenyindole Dilactate (DAPI; Sigma Aldrich) in PBS 1:50 for 3 min. Samples were washed and coverslips were mounted with the anti-fade mounting medium Mowiol. For double staining and 3D reconstructions, slides were examined with a Leica TCS SP5 confocal laser scanning microscope.
Cell Culture
Human foreskin primary fibroblasts (HPFs), HeLa (ATCC CCL-2) and 293T (ATCC CRL-11268) cells were cultured in DMEM with 10% FBS. SH-SY5Y (ATCC CRL-2266) cells were cultured in EMEM supplemented with 10% FBS.
RNA Extraction and QPCR Analysis
Cells were lysed using the TRIzol reagent (Invitrogen). Brains were removed from euthanized mice and frozen in liquid nitrogen. RNA was extracted using the TRIzol reagent (Invitrogen) according to manufacturer. After DNaseI treatment (Invitrogen) RNA from cells and brains was reverse transcribed into cDNA with oligo d(T) primer and OMNISCRIPT RT KIT (Qiagen). QPCRs were performed as described (Piersanti et al., 2015) using the following primers:
Ft1 E3 F: AACCAGTCCTCCACGAAGTGCA;
Ft1 E3 R: TAGGGCTTCGCTATGGGTAGAGCA;
Ft1 E6 F: CCGTCTTTCACCCACTAGTTGAT;
Ft1 E6 R: TTGCGAACGCTCTTTTCACA;
mGAPDH F: GTGGCAAAGTGGAGATTGTTGCC;
mGAPDH R: TGTGCCGTTGAATTTGCCGT;
IL-6 F: CTCTGGGAAATCGTGGAAAT;
IL-6 R: CCAGTTTGGTAGCATCCATC
TGF-β F: CCCTATATTTGGAGCCTGGA;
TGF-β R: CTTGCGACCCACGTAGTAGA;
AKTIP F: TCCACGCTTGGTGTTCGAT;
AKTIP R: TCACCTGAGGTGGGATCAACT
GAPDH F: TGGGCTACACTGAGCACCAG
GAPDH R: GGGTGTCGCTGTTGAAGTCA
Statistics
χ2 test was applied for comparisons of the mouse cohorts. Independent data sets were analyzed with the Student's t-test (unpaired, two-tailed). Correlation analyses were performed via Pearson's R test.
Ethics Statement
This study was carried out in accordance with the recommendations of the European Directive 2010/63/EU on the protection of animals used for scientific purposes. The protocol was approved by the Italian Ministry of Health (Ministero della Salute) with protocol number 355/2017-PR.
Author Contributions
RB, MLT, FDC, AV, and FV contributed to the design of the experiments and to the writing of the manuscript. MLT, GZ, AB, SDG, MB, and CM performed the experiments and analyzed the data. IS designed the experiments, analyzed the data and wrote the manuscript.
Funding
This work has been supported by Sapienza Ateneo grants to FV, IS, MLT, and RB; by grants EU FP7 Brainvectors (n. 286071), Telethon GEP15033 and PRF 2016-67 to IS. This work is in the memory of P. Bianco.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been supported by Sapienza Ateneo grants to FV, IS, ML, and RB; by grants EU FP7 Brainvectors (n. 286071), Telethon GEP15033 and PRF 2016-67 to IS. This work is in the memory of P. Bianco.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00581/full#supplementary-material
References
Baar, M. P., Brandt, R. M. C., Putavet, D. A., Klein, J. D. D., Derks, K. W. J., Bourgeois, B. R. M., et al. (2017). Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147 e116. doi: 10.1016/j.cell.2017.02.031
Baraban, S. C. (2007). Emerging epilepsy models: insights from mice, flies, worms and fish. Curr. Opin. Neurol. 20, 164–168. doi: 10.1097/WCO.0b013e328042bae0
Bertram, E. H. (2013). Neuronal circuits in epilepsy: do they matter? Exp. Neurol. 244, 67–74. doi: 10.1016/j.expneurol.2012.01.028
Bianchi, F. T., Tocco, C., Pallavicini, G., Liu, Y., Vernì, F., Merigliano, C., et al. (2017). Citron kinase deficiency leads to chromosomal instability and TP53-sensitive microcephaly. Cell Rep. 18, 1674–1686. doi: 10.1016/j.celrep.2017.01.054
Blasco, M. A. (2005). Mice with bad ends: mouse models for the study of telomeres and telomerase in cancer and aging. EMBO J. 24, 1095–1103. doi: 10.1038/sj.emboj.7600598
Burla, R., Carcuro, M., Raffa, G. D., Galati, A., Raimondo, D., Rizzo, A., et al. (2015). AKTIP/Ft1, a new shelterin-interacting factor required for telomere maintenance. PLoS Genet. 11:e1005167. doi: 10.1371/journal.pgen.1005167
Burla, R., Carcuro, M., Torre, M. L., Fratini, F., Crescenzi, M., D'apice, M. R., et al. (2016a). The telomeric protein AKTIP interacts with A- and B-type lamins and is involved in regulation of cellular senescence. Open Biol. 6:160103. doi: 10.1098/rsob.160103
Burla, R., La Torre, M., Merigliano, C., Verni, F., and Saggio, I. (2018). Genomic instability and DNA replication defects in progeroid syndromes. Nucleus 9, 368–379. doi: 10.1080/19491034.2018.1476793
Burla, R., La Torre, M., and Saggio, I. (2016b). Mammalian telomeres and their partnership with lamins. Nucleus 7, 187–202. doi: 10.1080/19491034.2016.1179409
Campisi, J. (2013). Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705. doi: 10.1146/annurev-physiol-030212-183653
Campisi, J., Kim, S. H., Lim, C. S., and Rubio, M. (2001). Cellular senescence, cancer and aging: the telomere connection. Exp. Gerontol. 36, 1619–1637. doi: 10.1016/S0531-5565(01)00160-7
Chabrol, E., Navarro, V., Provenzano, G., Cohen, I., Dinocourt, C., Rivaud-Péchoux, S., et al. (2010). Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain 133, 2749–2762. doi: 10.1093/brain/awq171
Coppe, J. P., Desprez, P. Y., Krtolica, A., and Campisi, J. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118. doi: 10.1146/annurev-pathol-121808-102144
de Lange, T. (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100–2110. doi: 10.1101/gad.1346005
Dittmer, T. A., and Misteli, T. (2011). The lamin protein family. Genome Biol. 12:222. doi: 10.1186/gb-2011-12-5-222
Fisher, R. S., Acevedo, C., Arzimanoglou, A., Bogacz, A., Cross, J. H., Elger, C. E., et al. (2014). ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55, 475–482. doi: 10.1111/epi.12550
Fonck, C., Cohen, B. N., Nashmi, R., Whiteaker, P., Wagenaar, D. A., Rodrigues-Pinguet, N., et al. (2005). Novel seizure phenotype and sleep disruptions in knock-in mice with hypersensitive alpha 4* nicotinic receptors. J. Neurosci. 25, 11396–11411. doi: 10.1523/JNEUROSCI.3597-05.2005
Frost, B. (2016). Alzheimer's disease: an acquired neurodegenerative laminopathy. Nucleus 7, 275–283. doi: 10.1080/19491034.2016.1183859
Fulop, T., Larbi, A., Dupuis, G., Le Page, A., Frost, E. H., Cohen, A. A., et al. (2017). Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front. Immunol. 8:1960. doi: 10.3389/fimmu.2017.01960.
Glasscock, E., Qian, J., Kole, M. J., and Noebels, J. L. (2012). Transcompartmental reversal of single fibre hyperexcitability in juxtaparanodal Kv1.1-deficient vagus nerve axons by activation of nodal KCNQ channels. J. Physiol. 590, 3913–3926. doi: 10.1113/jphysiol.2012.235606
Guerrini, R., Marini, C., and Mantegazza, M. (2014). Genetic epilepsy syndromes without structural brain abnormalities: clinical features and experimental models. Neurotherapeutics 11, 269–285. doi: 10.1007/s13311-014-0267-0
Hudgins, A. D., Tazearslan, C., Tare, A., Zhu, Y., Huffman, D., and Suh, Y. (2018). Age- and tissue-specific expression of senescence biomarkers in mice. Front. Genet. 9:59. doi: 10.3389/fgene.2018.00059
Jacks, T., Remington, L., Williams, B. O., Schmitt, E. M., Halachmi, S., Bronson, R. T., et al. (1994). Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4, 1–7. doi: 10.1016/S0960-9822(00)00002-6
La Torre, M., Merigliano, C., Burla, R., Mottini, C., Zanetti, G., Del Giudice, S., et al. (2018). Mice with reduced expression of the telomere-associated protein Ft1 develop p53-sensitive progeroid traits. Aging Cell, 17:e12730. doi: 10.1111/acel.12730
Langie, S. A., Cameron, K. M., Ficz, G., Oxley, D., Tomaszewski, B., Gorniak, J. P., et al. (2017). The ageing brain: effects on DNA repair and DNA methylation in mice. Genes 8:E75 doi: 10.3390/genes8020075
Lee, Y., Katyal, S., Li, Y., El-Khamisy, S. F., Russell, H. R., Caldecott, K. W., et al. (2009). The genesis of cerebellar interneurons and the prevention of neural DNA damage require XRCC1. Nat. Neurosci. 12, 973–980. doi: 10.1038/nn.2375
Leppik, I. E., and Birnbaum, A. K. (2010). Epilepsy in the elderly. Ann. N. Y. Acad. Sci. 1184, 208–224. doi: 10.1111/j.1749-6632.2009.05113.x
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Lopez-Otin, C., Galluzzi, L., Freije, J. M. P., Madeo, F., and Kroemer, G. (2016). Metabolic control of longevity. Cell 166, 802–821. doi: 10.1016/j.cell.2016.07.031
Löscher, W. (2011). Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 20, 359–368. doi: 10.1016/j.seizure.2011.01.003
Madabhushi, R., Pan, L., and Tsai, L. H. (2014). DNA damage and its links to neurodegeneration. Neuron 83, 266–282. doi: 10.1016/j.neuron.2014.06.034
Mckinnon, P. J. (2013). Maintaining genome stability in the nervous system. Nat. Neurosci. 16, 1523–1529. doi: 10.1038/nn.3537
Minkeviciene, R., Rheims, S., Dobszay, M. B., Zilberter, M., Hartikainen, J., Fulop, L., et al. (2009). Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 29, 3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009
Piersanti, S., Burla, R., Licursi, V., Brito, C., La Torre, M., Alves, P. M., et al. (2015). Transcriptional response of human neurospheres to helper-dependent CAV-2 vectors involves the modulation of DNA damage response, microtubule and centromere gene groups. PLoS ONE 10:e0133607. doi: 10.1371/journal.pone.0133607
Remoli, C., Michienzi, S., Sacchetti, B., Consiglio, A. D., Cersosimo, S., Spica, E., et al. (2015). Osteoblast-specific expression of the fibrous dysplasia (FD)-causing mutation Gsalpha(R201C) produces a high bone mass phenotype but does not reproduce FD in the mouse. J. Bone Miner. Res. 30, 1030–1043. doi: 10.1002/jbmr.2425
Riminucci, M., Saggio, I., Robey, P. G., and Bianco, P. (2006). Fibrous dysplasia as a stem cell disease. J. Bone Miner. Res. 21 (Suppl. 2), P125–131. doi: 10.1359/jbmr.06s224
Robie, A. A., Seagraves, K. M., Egnor, S. E., and Branson, K. (2017). Machine vision methods for analyzing social interactions. J. Exp. Biol. 220, 25–34. doi: 10.1242/jeb.142281
Rueden, C. T., Schindelin, J., Hiner, M. C., Dezonia, B. E., Walter, A. E., Arena, E. T., et al. (2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18:529. doi: 10.1186/s12859-017-1934-z
Saggio, I., Remoli, C., Spica, E., Cersosimo, S., Sacchetti, B., Robey, P. G., et al. (2014). Constitutive expression of Gsalpha(R201C) in mice produces a heritable, direct replica of human fibrous dysplasia bone pathology and demonstrates its natural history. J. Bone Miner. Res. 29, 2357–2368. doi: 10.1002/jbmr.2267
Sanjabi, S., Zenewicz, L. A., Kamanaka, M., and Flavell, R. A. (2009). Anti-inflammatory and pro-inflammatory roles of TGF-beta, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 9, 447–453. doi: 10.1016/j.coph.2009.04.008
Scharfman, H. E. (2007). The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep. 7, 348–354. doi: 10.1007/s11910-007-0053-z
Shen, J., Gilmore, E. C., Marshall, C. A., Haddadin, M., Reynolds, J. J., Eyaid, W., et al. (2010). Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat. Genet. 42, 245–249. doi: 10.1038/ng.526
Simão, D., Pinto, C., Piersanti, S., Weston, A., Peddie, C. J., Bastos, A. E., et al. (2015). Modeling human neural functionality in vitro: three-dimensional culture for dopaminergic differentiation. Tissue Eng. Part A 21, 654–668. doi: 10.1089/ten.tea.2014.0079
Simeone, K. A., Hallgren, J., Bockman, C. S., Aggarwal, A., Kansal, V., Netzel, L., et al. (2018). Respiratory dysfunction progresses with age in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia 59, 345–357. doi: 10.1111/epi.13971
Stafstrom, C. E., and Carmant, L. (2015). Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb. Perspect. Med. 5:a022426. doi: 10.1101/cshperspect.a022426
Stewart, C. L., Kozlov, S., Fong, L. G., and Young, S. G. (2007). Mouse models of the laminopathies. Exp. Cell Res. 313, 2144–2156. doi: 10.1016/j.yexcr.2007.03.026
Testa, G., Schaft, J., Van Der Hoeven, F., Glaser, S., Anastassiadis, K., Zhang, Y., et al. (2004). A reliable lacZ expression reporter cassette for multipurpose, knockout-first alleles. Genesis 38, 151–158. doi: 10.1002/gene.20012
Varela, I., Cadiñanos, J., Pendás, A. M., Gutiérrez-Fernández, A., Folgueras, A. R., Sánchez, L. M., et al. (2005). Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 437, 564–568. doi: 10.1038/nature04019
Vogt, D. L., Thomas, D., Galvan, V., Bredesen, D. E., Lamb, B. T., and Pimplikar, S. W. (2011). Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol. Aging 32, 1725–1729. doi: 10.1016/j.neurobiolaging.2009.09.002
Yang, Y., Mahaffey, C. L., Bérubé, N., Maddatu, T. P., Cox, G. A., and Frankel, W. N. (2007). Complex seizure disorder caused by Brunol4 deficiency in mice. PLoS Genet. 3:e124. doi: 10.1371/journal.pgen.0030124
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. doi: 10.1038/nn1754
Ziyatdinova, S., Gurevicius, K., Kutchiashvili, N., Bolkvadze, T., Nissinen, J., Tanila, H., et al. (2011). Spontaneous epileptiform discharges in a mouse model of Alzheimer's disease are suppressed by antiepileptic drugs that block sodium channels. Epilepsy Res. 94, 75–85. doi: 10.1016/j.eplepsyres.2011.01.003
Keywords: aging, epilepsy, DNA damage, p53, DNA repair
Citation: Burla R, La Torre M, Zanetti G, Bastianelli A, Merigliano C, Del Giudice S, Vercelli A, Di Cunto F, Boido M, Vernì F and Saggio I (2018) p53-Sensitive Epileptic Behavior and Inflammation in Ft1 Hypomorphic Mice. Front. Genet. 9:581. doi: 10.3389/fgene.2018.00581
Received: 29 August 2018; Accepted: 08 November 2018;
Published: 27 November 2018.
Edited by:
Maria Grazia Giansanti, Consiglio Nazionale Delle Ricerche (CNR), ItalyReviewed by:
Maria Rosaria D'Apice, Consiglio Nazionale Delle Ricerche (CNR), ItalyCristina Capanni, Istituto di Genetica Molecolare (IGM), Italy
Copyright © 2018 Burla, La Torre, Zanetti, Bastianelli, Merigliano, Del Giudice, Vercelli, Di Cunto, Boido, Vernì and Saggio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabella Saggio, aXNhYmVsbGEuc2FnZ2lvQHVuaXJvbWExLml0
†These authors share first authorship