
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet. , 02 October 2018
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2018 | https://doi.org/10.3389/fgene.2018.00436
This article is part of the Research Topic Model Organisms: A Precious Resource for Understanding of the Molecular Mechanisms Underlying Human Physiology and Disease View all 14 articles
 Anna Frappaolo1†
Anna Frappaolo1† Stefano Sechi1†
Stefano Sechi1† Tadahiro Kumagai2†
Tadahiro Kumagai2† Angela Karimpour-Ghahnavieh1
Angela Karimpour-Ghahnavieh1 Michael Tiemeyer2,3*
Michael Tiemeyer2,3* Maria Grazia Giansanti1*
Maria Grazia Giansanti1*Protein glycosylation, the enzymatic addition of N-linked or O-linked glycans to proteins, serves crucial functions in animal cells and requires the action of glycosyltransferases, glycosidases and nucleotide-sugar transporters, localized in the endoplasmic reticulum and Golgi apparatus. Congenital Disorders of Glycosylation (CDGs) comprise a family of multisystemic diseases caused by mutations in genes encoding proteins involved in glycosylation pathways. CDGs are classified into two large groups. Type I CDGs affect the synthesis of the dolichol-linked Glc3Man9GlcNac2 precursor of N-linked glycosylation or its transfer to acceptor proteins. Type II CDG (CDG-II) diseases impair either the trimming of the N-linked oligosaccharide, the addition of terminal glycans or the biosynthesis of O-linked oligosaccharides, which occur in the Golgi apparatus. So far, over 100 distinct forms of CDGs are known, with the majority of them characterized by neurological defects including mental retardation, seizures and hypotonia. Yet, it is unclear how defective glycosylation causes the pathology of CDGs. This issue can be only addressed by developing animal models of specific CDGs. Drosophila melanogaster is emerging as a highly suitable organism for analyzing glycan-dependent functions in the central nervous system (CNS) and the involvement of N-glycosylation in neuropathologies. In this review we illustrate recent work that highlights the genetic and neurobiologic advantages offered by D. melanogaster for dissecting glycosylation pathways and modeling CDG pathophysiology.
Protein glycosylation is one of the most frequent post-translational modifications in eukaryotes; approximately one fifth of all proteins in protein structural databases are glycosylated (Ohtsubo and Marth, 2006; Freeze and Ng, 2011; Khoury et al., 2011). The oligosaccharide moieties added to glycoproteins impact their structure and biological function by contributing to protein folding, stability, and transport to appropriate sub-cellular locations. Glycans also mediate cell–cell interactions, modulate signal transduction, and regulate molecular trafficking and endocytosis. The two main types of protein glycosylation are N-linked and O-linked glycosylation. The biosynthesis and elaboration of glycoprotein N-linked or O-linked glycans, require the coordinated action of hundreds of glycogenes, primarily glycosyltransferases and glycosidases, which are trafficked to specific locations within the endoplasmic reticulum (ER) and Golgi apparatus (Ohtsubo and Marth, 2006). Cytoplasmic and nuclear proteins are frequently modified with O-linked N-acetylglucosamine (GlcNAc) which regulates many biological processes but is beyond the scope of this review. N- and O-linked glycosylation of secreted and membrane protein starts in the ER or early cis-Golgi and is completed in later Golgi compartments. Major animal glycans contain ten monosacchararides: glucose (Glc), Galactose (Gal), N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), fucose (Fuc), mannose (Man), xylose (Xyl), glucuronic acid (GlcA), iduronic acid (IdoA), and sialic acid (SA, either as 5-N-acetylneuraminic acid, Neu5Ac, or as 5-N-acetylglycolylneuraminic acid, Neu5Gc).
The N-glycosylation pathway starts at the ER membrane where a precursor glycan is built upon a dolichol isoprenoid lipid (Figure 1). This precursor glycan, with the composition Glc3Man9GlcNAc2, is transferred en bloc onto asparagine residues located within glycosylation sequons of nascent polypeptide chains either co-translationally or shortly after translation by multi-subunit oligosaccharyltransferase (OST) complexes within the lumen of the ER (Li et al., 2008; Zielinska et al., 2010; Shwartz and Aebi, 2011). The transferred Glc3Man9GlcNAc2 glycan is subsequently trimmed by sequential action of ER glucosidases. Glc-trimming is an essential component of the folding process for most secretory pathway glycoproteins (Helenius and Aebi, 2004). Thus, human diseases that arise from altered biosynthesis or trimming of the N-linked precursor glycan, or ineffective transfer of the precursor to protein will impact the folding and stability of many glycoproteins and, consequently, manifest with multi-systemic and broadly severe clinical phenotypes.
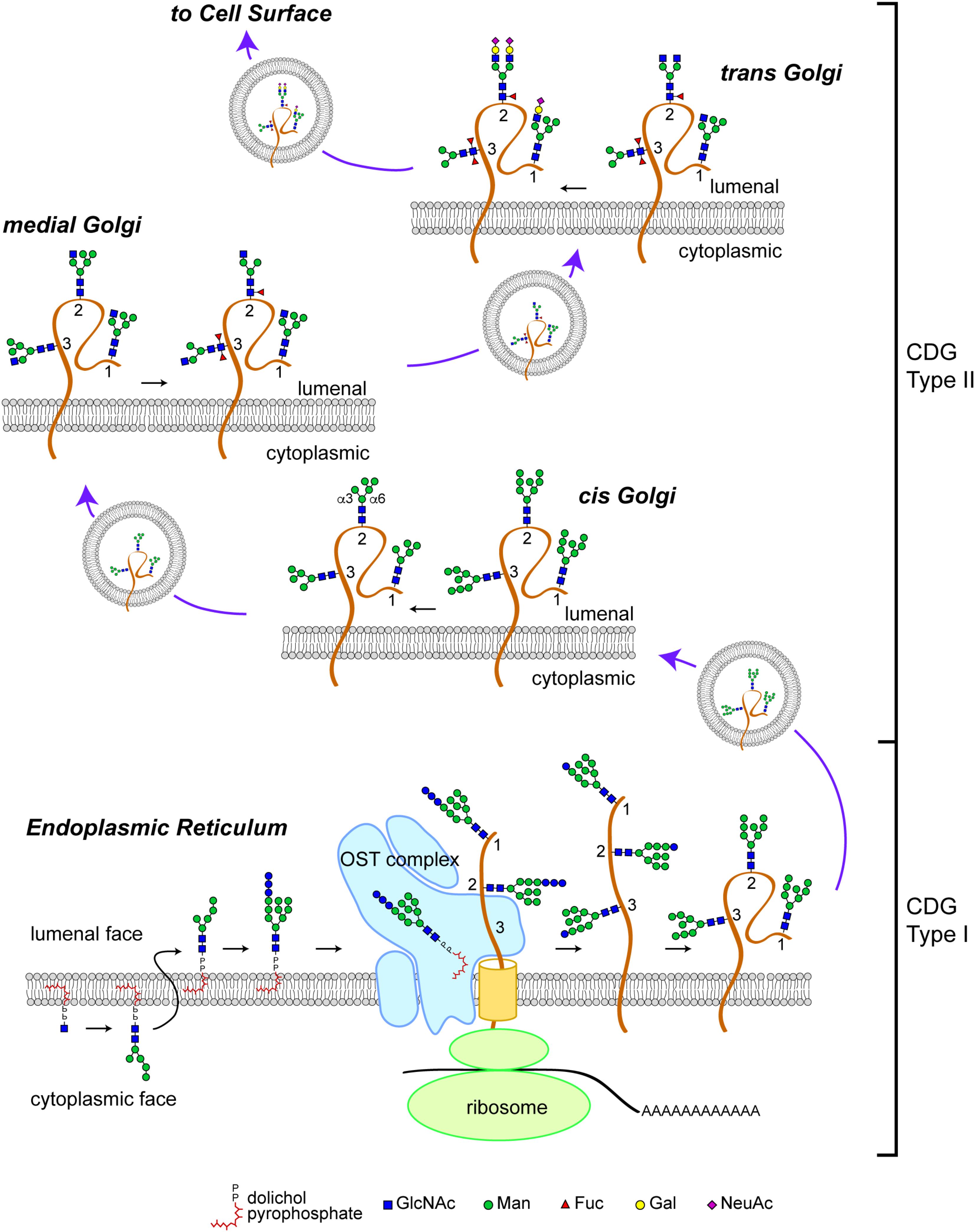
FIGURE 1. N-linked glycosylation pathway. Biosynthesis of the N-linked precursor glycan begins on the cytoplasmic face of the ER where a GlcNAc residue is added in a pyrophosphate linkage to dolichol, an isoprenoid lipid. The GlcNAc-P-P-Dol is extended to form Man5GlcNAc2-P-P-Dol which is then flipped so that the glycan moiety is within the lumen of the ER. Further extension produces a Glc3Man9GlcNAc2-P-P-Dol that is a substrate for the oligosaccharyltransferase (OST) complex, which transfers the precursor glycan en bloc to a nascent polypeptide. This figure depicts the glycosylation of a glycoprotein (brown) with 3 N-linked glycosylation sites (labeled 1, 2, and 3). Once transfered to protein, the glycan precursor is trimmed of its Glc residues during folding as part of the calnexin/calreticulin quality control cycle. CDG Type I mutations affect the biosynthesis of the precursor glycan, its transfer to protein, and early trimming steps. Once successfully folded, glycoproteins bearing high-Man glycans are transported to the Golgi apparatus where Man trimming occurs. In the early cis Golgi, high-Man glycans can be trimmed to Man5GlcNAc2 by complete removal of Man residues on the α3 arm and partial removal of Man residues on the α6 arm. In the medial Golgi, the first committed step toward production of a complex glycan is taken; GlcNAcT1 adds a GlcNAc to the α3 Man residue to form a hybrid type glycan (site 1 retains this structure). The GlcNAc-extended Man5GlcNAc2 glycan can be core fucosylated by the addition of a Fuc residue to the internal GlcNAc (site 2). In Drosophila and other arthropods, a second Fuc residue can be added (site 3). Additional Man trimming by Golgi mannosidases provide substrates for branching in the medial and trans Golgi (site 2). Subsequent extention with Gal and capping with sialic acid (shown here, as N-acetylneuraminic acid, NeuAc) completes the maturation of complex N-linked glycans. Hybrid glycans can also be extended on the α3 arm (site 1). The abundance of hybrid and complex glycans is reduced in Drosophila compared to vertebrate species due to the presence of an hexosaminidase that removes the GlcNAc added by GlcNAcT1, thereby blocking additional branching/extension and producing a paucimanose glycan (site 3). CDG Type II mutations impact the availability of substrates and the activity of enzymes that process N-glycans in the Golgi apparatus. Graphical representation of monosaccharide residues and glycan structures is consistent with the Symbol Nomenclature For Glycans (SNFG), which has been broadly adopted by the glycobiology community (Varki et al., 2015).
Glycoproteins arrive at the cis-Golgi carrying high-Man glycans (Figure 1). Mannose trimming in the cis-Golgi by Golgi α-mannosidases removes Man residues to generate the Man5GlcNAc2 intermediate. In medial Golgi compartments, Man5GlcNAc2 is the substrate for GlcNAcT-1, a glycosyltransferase that transfers a GlcNAc residue to a terminal Man residue on the α3-arm of the Man5GlcNAc2 structure, thereby initiating the synthesis of hybrid and complex N-linked glycans (Stanley, 2011; Moremen et al., 2012). The product of GlcNAcT-1 is also a substrate for core fucosylation, the addition of one or more Fuc residues (depending on the species) to the most proximal core GlcNAc residues attached to Asn. The α3-arm initiated by GlcNAT-1 can be extended with Gal, Sia and/or other residues, resulting in the production of hybrid structures. Removal of the remaining Man residues from the α6-arm allows branching with additional GlcNAc residues catalyzed by specific GlcNAcT enzymes and subsequent extension to generate fully elaborated multiantennary complex glycans in late medial and trans Golgi compartments (Stanley, 2011). The vectorial nature of N-glycan processing is facilitated by enzyme specificity and by the spatial distribution of processing steps across the Golgi apparatus. Therefore, human diseases that impact N-glycan fine structure may arise from genes that encode for processing enzymes or for proteins that regulate Golgi architecture and trafficking. Such diseases may be characterized by relatively restricted phenotypes associated with altered function, half-life, or targeting of specific glycoproteins.
In contrast to N-linked glycosylation, O-linked glycosylation does not rely on a precursor core that is transferred en bloc to the nascent polypeptide. Instead, O-glycosylation is initiated on folding or folded proteins and involves the formation of a glycosidic linkage between serine or threonine and GalNAc, GlcNAc, Man, Glc, Xyl, or Fuc residues (Stanley, 2011). Some O-glycans are specifically elaborated on well-defined protein domains and contribute to protein folding, stability, protease sensitivity, and protein function. The biosynthesis of O-glycans in the secretory pathway is initiated in the early cis-Golgi or in a transitional compartment that retains characteristics of the ER and is completed in subsequent processing steps distributed across the Golgi apparatus. Therefore, human diseases arising from genes that regulate Golgi dynamics may impact both N-linked and O-linked glycoprotein glycosylation.
Congenital disorders of glycosylation (CDGs) are inborn errors in protein and lipid glycosylation that arise from mutations in genes controlling steps in glycan addition (Freeze and Ng, 2011; Jaeken, 2013). More than 100 distinct forms of CDGs have been discovered, many of which display multisystemic defects including severe neurological impairment, highlighting the important role of regulated glycosylation in central nervous system (CNS) functions (Barone et al., 2014; Freeze et al., 2015). Because 1–2% of the genome encodes for glyco-enzymes and glycan transporters, it is likely that many other CDGs remain to be discovered (Freeze et al., 2015). CDGs have been traditionally divided into two large groups (Goreta et al., 2012; Freeze et al., 2014). Type I CDGs impair the synthesis of the dolichol pyrophosphate oligosaccharide precursor of N-linked glycoproteins or its transfer to acceptor proteins (Freeze and Ng, 2011) resulting in decreased efficiency of protein N-glycosylation. Type II CDGs (CDG-II) are characterized by defects in the processing of N-linked glycans or the biosynthesis of O-linked oligosaccharides (Freeze and Ng, 2011; Goreta et al., 2012; Freeze et al., 2014). Although most CDGs exhibit neurological impairment (Freeze et al., 2015), there are no comprehensive studies, aimed at elucidating the molecular mechanisms that link defective glycosylation to the neuropathological aspects of the disease. Animal models that faithfully recapitulate the pathological aspects of the disease, including the neurological defects, provide a valuable resource to study the molecular mechanisms underlying pathology in CDGs. In this review we focus our attention on the advantages offered by the use of Drosophila melanogaster for understanding and modeling the glycobiology of CDGs.
Drosophila models offer many advantages for studying CDGs as well as other human diseases (Moulton and Letsou, 2016). Fundamental biological processes are highly conserved between Drosophila and humans; approximately 75% of human-disease related genes have a homolog in Drosophila (Reiter et al., 2001; Chien et al., 2002). Moreover the genome of D. melanogaster is far less complex than the human genome and exhibits fewer gene duplications (Hartl, 2000). All of these characteristics, and its extraordinary repertoire of readily available genetic tools, have combined to make Drosophila a valuable, emerging model system for investigating glycan-dependent functions in vivo and for understanding the link between CDG neuropathology and glycan changes (Katoh and Tiemeyer, 2013; Scott and Panin, 2014). Such studies are extremely challenging in vertebrates due to the complexity of the nervous system and the redundancy of glycosylation pathways and enzymes (Freeze et al., 2014; Scott and Panin, 2014). Drosophila combines the advantages of a well-characterized glycome and the availability of electrophysiological and behavioral assays to test neurological impairment in the whole organism (Gatto and Broadie, 2011; Dani et al., 2014; Scott and Panin, 2014). Moreover, larval neuromuscular junction (NMJ) synapses use ionotropic glutamate receptors (GluRs), providing an excellent model system for excitatory synapses in the mammalian CNS.
Various analytic techniques have revealed that the most abundant N-linked glycans on Drosophila glycoproteins are of the high-Man or paucimannosidic type. However, hybrid and complex glycans are also present, although they represent a lower fraction of the total glycan profile when compared with vertebrates (Katoh and Tiemeyer, 2013). The relative paucity of complex glycans in Drosophila is a result of an arthropod-specific glycan processing enzyme encoded by a gene named Fused Lobes (Fdl) that removes the GlcNAc residue added by GlcNAcT-1, thereby blocking further glycan elaboration (Figures 1, 2). The presence of Fdl in the secretory pathway means that Drosophila glycan profiles are skewed away from the highly abundant complex profiles found in most vertebrates. Nonetheless, the glycans that escape the activity of Fdl in Drosophila are readily processed to complexity, indicating common logic underlies glycan maturation in vertebrates and Drosophila. The resulting low content of complex glycans also provides a benefit for this system because it generates a simpler profile to analyze and a larger dynamic range for detecting shifts induced by mutations. Additionally, unlike mammalian organisms, the Drosophila genome contains only a single sialyltransferase (DSiaT), which greatly simplifies in vivo analysis of glycoprotein sialylation (Aoki et al., 2007; Koles et al., 2007; Repnikova et al., 2010).
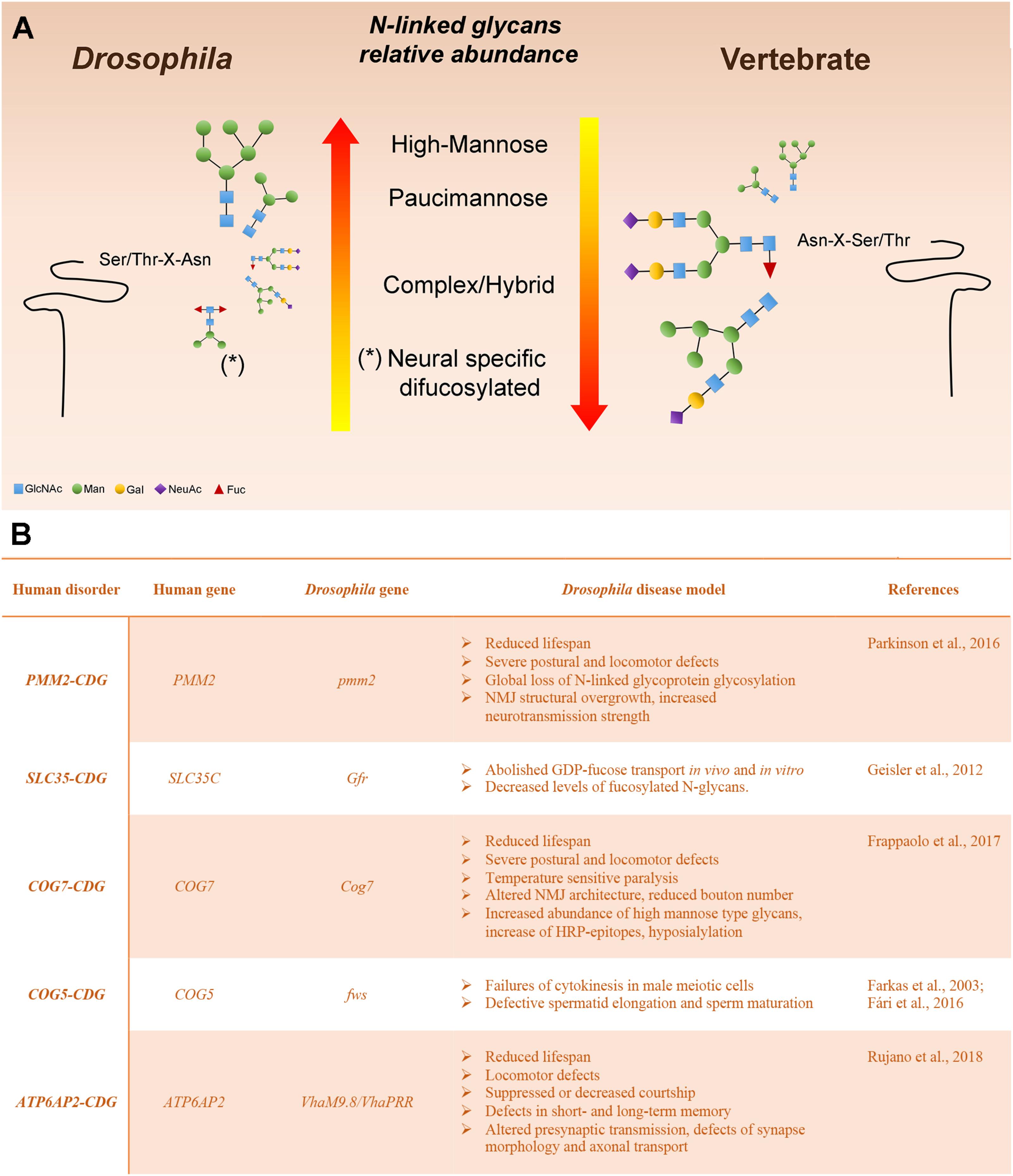
FIGURE 2. Drosophila melanogaster as a model system to study glycoprotein N-glycosylation (A) Representation of Drosophila and vertebrate N-glycome characteristics. N-linked glycans are scaled proportionally to their relative abundance. The major reason for the high-mannose and pauci-mannose dominance in the Drosophila N-linked glycan profile is the existence of an arthropod-specific, N-acetylhexosaminidase known as Fused lobes (Fdl), which converts the precursor for complex glycans (GlcNAc1Man3−5GlcNAc2-Protein) to a paucimannose structure (Man3−5GlcNAc2-Protein) that cannot be extended further. Glycoprotein glycans that escape Fdl are fully capable of being processed into complex structures. (B) Human glycosylation disorders and phenotypic characteristics of the Drosophila model.
In this section we describe Drosophila mutants that offer functional models for characterized human CDGs (Figure 2). We discuss the phenotypic characteristics that recapitulate the pathological aspects of the human disease and the translational impact of modeling the CDG in this organism. Many other Drosophila mutants have been shown to impact glycoprotein or glycolipid glycosylation and it is likely that the impact of many others is underappreciated (Seppo et al., 2003; Baas et al., 2011; Daenzer et al., 2016; Jumbo-Lucioni et al., 2016). But here, we focus on those mutations that have immediate parallels with human type I and II CDGs.
The most prevalent CDG is known as CDG-1a or PMM2-CDG, accounting for around 80% of all diagnosed cases. PMM2-CDG is inherited as an autosomal-recessive trait resulting from mutations in human PMM2, which encodes phosphomannomutase-2, that converts mannose-6-phosphate to mannose-1-phosphate, the precursor of GDP-mannose (Freeze et al., 2014, 2015). Because GDP-mannose is the donor for the addition of the first 5 Man residues to the dolichol-linked precursor, synthesis of N-linked glycans is impacted, as is O-mannosylation. The defect results in hypoglycosylation of many types of glycoproteins including serum glycoproteins, plasma membrane glycoproteins, and lysosomal enzymes. Pediatric patients suffering with CDG-Ia present with variable clinical features that affect nearly all systems and include failure to thrive, hypotonia, psychomotor retardation, ataxia, dysmorphia and coagulopathy (Freeze, 2009; Grünewald, 2009; Jaeken, 2013). Most adult CDG-Ia patients are wheelchair bound and display peripheral neuropathy and mental retardation (Grünewald, 2009).
Overall, the Drosophila PMM2 protein displays 56% amino acid identity with human PMM2 (Parkinson et al., 2016). Parkinson et al. (2016) generated Drosophila pmm2 mutants. Similar to CDG-1a patients, pmm2 mutants displayed uncoordinated movement and reduced lifespan. Analysis of the N-linked glycome of the pmm2-null mutant larval body demonstrated a global suppression of N-linked glycosylation. Furthermore, the N-linked glycome of adult heads with neurally targeted pmm2 RNAi revealed increased abundance of pauci-mannose glycans. Analysis of the larval NMJs revealed altered glycan composition within the heavily glycosylated synaptomatrix which correlated with striking NMJ structural overgrowth and increased neurotransmission strength. Since NMJ synaptogenesis requires trans-synaptic Wnt/Wingless signaling, which in turn depends on expression of Dally-like protein (Dlp, a heparan sulfate proteoglycan) and matrix metalloproteinases (MMPs). Because knockdown of PMM2 resulted in loss of MMP2, reduced synaptic levels of Wingless, Dlp co-receptor and downstream trans-synaptic signaling, the authors propose that the matrix metalloproteome and Wnt signaling pathway might provide potential new targets for developing CDG-1a treatments.
Also known as CDG-IIc and leukocyte adhesion deficiency type II syndrome (LAD II), this disorder is caused by mutations in a GDP-Fuc transporter (GFR). SLC35-CDG patients show craniofacial dysmorphism, severe retardation and chronic infections with unusually high leukocytosis. Importantly neutrophils of these patients lack the ability to synthesize the fucosylated glycan sialyl-Lewis X, a ligand of the selectin family of cell adhesion molecules, that is necessary for their recruitment to infection sites (Yakubenia et al., 2008). Several mutations in SLC35 nucleotide sugar transporters have been identified in Drosophila, including the ER Fuc transporter (Efr), fringe connection (frc), slalom (sll) and neurally altered carbohydrate (nac). In nac1 mutant, a conserved serine at position 29 of the Golgi GFR is replaced by a leucine, which abolishes GDP-Fuc transport in vivo and in vitro. Mass spectrometry and HPLC analysis demonstrated reduced core α1,3-and α1,6-fucosylation in nac1 (Geisler et al., 2012). While Lewis-type glycans have not yet been identified in Drosophila, the commonality of altered fucosylation in SLC35 CDG-IIc and in select Drosophila mutants provides opportunities to investigate the regulation of protein fucosylation in a whole organism.
The eight-subunit Conserved Oligomeric Golgi (COG) complex is a Golgi tether required for intra Golgi trafficking of vesicles that recycle Golgi resident proteins and is essential for proper localization of Golgi-localized glycosylation enzymes including glycosyltransferases (Ungar et al., 2002; Kranz et al., 2007; Miller and Ungar, 2012; Willett et al., 2013; Climer et al., 2015). Mutations in the genes encoding human COG1, COG2, and COG4–COG8 are associated with monogenic forms of inherited, autosomal recessive, CDGs-II (Freeze and Ng, 2011; Climer et al., 2015). Common features of patients carrying mutations in COG proteins (COG-CDG) are feeding problems and developmental defects, including microcephaly and growth retardation associated with dysmorphic features, hypotonia and cerebral atrophy (Wu et al., 2004; Spaapen et al., 2005; Foulquier et al., 2006; Kranz et al., 2007; Morava et al., 2007; Ng et al., 2007; Paesold-Burda et al., 2009; Reynders et al., 2009; Zeevaert et al., 2009; Lübbehusen et al., 2010; Fung et al., 2012; Kodera et al., 2015). COG7–CDG patients had the highest mortality within the first year of life and presented with dysmorphic facial features, generalized hypotonia, skeletal anomalies, hepatomegaly, progressive jaundice, cardiac insufficiency, microcephaly, and severe epilepsy (Wu et al., 2004; Spaapen et al., 2005; Morava et al., 2007; Ng et al., 2007; Zeevaert et al., 2009). Defects in COG proteins have been linked to glycosylation alterations in mammalian cultured cells and in COG–CDG patients, including hyposialylation of serum proteins, abnormal synthesis of N- and O-linked glycans and altered glycolipid glycosylation (Kingsley et al., 1986; Suvorova et al., 2002; Wu et al., 2004; Spaapen et al., 2005; Morava et al., 2007; Ng et al., 2007; Zeevaert et al., 2009; Struwe and Reinhold, 2012).
Analysis of phenotypes associated with mutations in Drosophila homologs of human COG complex members highlights both the value and also the limitations of modeling COG-complex disorders in this organism. The Drosophila homolog of human COG5, Four way stop (Fws), is not essential for adult survival but is required for male fertility. Mutations in fws impair spermatocyte cytokinesis, acroblast structure and elongation and individualization of differentiating spermatids (Farkas et al., 2003; Fári et al., 2016). Thus, the Drosophila COG5 mutant presents less severe involvement than the presently known human COG-complex CDGs. On the other hand, loss of COG7 in COG7–CDG patients and in Drosophila mutants results in reduced life span and severe psychomotor defects (Frappaolo et al., 2017). Analysis of N-glycans from heads of Drosophila Cog7 mutants, revealed increased abundance of high-Man type glycans compared to wild type, accompanied by a disproportionate increase of the Man5GlcNAc2 glycan, which is the precursor for all complex glycans. Additionally, a substantial increase in the abundance of a family of neural-specific, difucosylated N-glycans known as HRP-epitopes, was detected. However, not all N-glycans were increased in Cog7 mutants. A single sialylated N-glycan was detected among the glycans harvested from adult heads and quantification relative to standard indicated that it was decreased in two mutant allelic combinations compared to wild type (Frappaolo et al., 2017). Moreover, like DSiaT mutants, Cog7 mutant flies exhibit temperature sensitive (TS) paralysis, coordination defects, and altered architecture of larval NMJ. Thus the phenotypic characteristics of our Drosophila COG7–CDG model closely parallel the pathological characteristics of COG7–CDG patients including N-linked glycome defects with hyposialylation. Ongoing analysis of the COG protein interactome is beginning to highlight molecular hierarchies and trafficking paradigms that may underlie altered protein glycosylation (Belloni et al., 2012; Miller et al., 2012; Willett et al., 2013; Hong and Lev, 2014; Climer et al., 2015; Bailey Blackburn et al., 2016; Frappaolo et al., 2017; Sechi et al., 2017).
The multi-subunit vacuolar-type proton ATPase (V-ATP-ase) is a highly conserved proton pump, which acidifies intracellular compartments and is essential for endocytosis and vesicular trafficking. Rujano et al. (2018) identified missense mutations in the extracellular domain of the accessory V-ATPase subunit ATP6AP2 that cause a novel glycosylation disorder associated with hepatopathy, immunodeficiency, cutis laxa, muscular hypotonia, dysmorphic features, and psychomotor impairment. Analysis of ATP6AP2-CDG patients’ serum proteins revealed hypoglycosylation, a defect that could be recapitulated by ATP6AP2 deficiency in the mouse. Null alleles of the Drosophila ortholog of ATP6AP2 cause an early lethal phenotype in Drosophila. The introduction of an ATP6AP2 transgene carrying the p.L98S mutation in the background of Drosophila ATP6AP2 null mutants, reduced viability and affected the developing optic lobes in larval brains by expanding the pool of optic lobe neuroblasts, a phenotype associated with altered Notch signaling (Vaccari et al., 2010). In agreement with the role of V-ATPase-mediated acidification in autophagic degradation (Mauvezin et al., 2015), p.L98S mutation leads to lipid accumulation and autophagic dysregulation in the liver-like fat body, associated with defects of lysosomal acidification and mTOR signaling. Thus the Drosophila ATP6AP2-CDG model has allowed the elucidation of molecular mechanisms underlying pathological aspects of the human disease.
Many impactful studies utilizing model systems (Zebrafish, flies, C. elegans, mice, etc.,) have enhanced our understanding of the underlying biochemical and phenotypic consequences of altered glycan biosynthesis associated with human CDG subtypes. Clinical phenotypes of human CDGs have parallels in these model systems. For the growing subset of CDGs modeled in Drosophila, specific phenotypes related to neural function, lifespan, viability, and glycomic diversity are replicated across these highly divergent species. This phenotypic reproducibility across species should not be surprising since the core biosynthetic pathways for protein glycosylation as well as the basic mechanisms that regulate Golgi trafficking are shared across broad swaths of evolutionary space. This conservation will allow mechanistic questions to be effectively answered in CDG models. One of these key questions is whether phenotypes arise from altered glycosylation of broad sets of glycoproteins or whether aberrant glycosylation of small subsets of glycoproteins can be linked to underlying pathologies. Once candidate proteins, whose glycosylation is altered in a given CDG, are identified by cutting edge glycoproteomics, the genetic tools offered by model systems such as Drosophila will allow unprecedented targeted investigations of the cell- and tissue-specific impacts of glycosylation deficiencies.
All authors edited and critically revised the manuscript.
The Research in MG lab was supported by a grant from Associazione Italiana per la Ricerca sul Cancro (AIRC), grant IG 2017, Id 20779 and a grant from Fondazione Telethon Italy (grant number GEP 14076). Work in the MT laboratory was supported by a grant from the NIGMS/NIH, P41GM103490.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor and author MG declared their involvement as co-editors in the Research Topic, and confirm the absence of any other collaboration.
Aoki, K., Perlman, M., Lim, J. M., Cantu, R., Wells, L., and Tiemeyer, M. (2007). Dynamic developmental elaboration of N-linked glycan complexity in the Drosophila melanogaster embryo. J. Biol. Chem. 282, 9127–9142. doi: 10.1074/jbc.M606711200
Baas, S., Sharrow, M., Kotu, V., Middleton, M., Nguyen, K., Flanagan-Steet, H., et al. (2011). Sugar-free frosting, a homolog of SAD kinase, drives neural-specific glycan expression in the Drosophila embryo. Development 138, 553–563. doi: 10.1242/dev.055376
Bailey Blackburn, J. B., Pokrovskaya, I., Fisher, P., Ungar, D., and Lupashin, V. V. (2016). COG complex complexities: detailed characterization of a complete set of HEK293T cells lacking individual COG subunits. Front. Cell Dev. Biol. 4:23. doi: 10.3389/fcell.2016.00023
Barone, R., Fiumara, A., and Jaeken, J. (2014). Congenital disorders of glycosylation with emphasis on cerebellar involvement. Semin. Neurol. 34, 357–366. doi: 10.1055/s-0034-1387197
Belloni, G., Sechi, S., Riparbelli, M. G., Fuller, M. T., Callaini, G., and Giansanti, M. G. (2012). Mutations in Cog7 affect Golgi structure, meiotic cytokinesis and sperm development during Drosophila spermatogenesis. J. Cell Sci. 125, 5441–5452. doi: 10.1242/jcs.108878
Chien, S., Reiter, L. T., Bier, E., and Gribskov, M. (2002). Homophila: human disease gene cognates in Drosophila. Nucleic Acid Res. 30, 149–151. doi: 10.1093/nar/30.1.149
Climer, L. K., Dobretsov, M., and Lupashin, V. (2015). Defects in the COG complex and COG-related trafficking regulators affect neuronal Golgi function. Front. Neurosci. 9:405. doi: 10.3389/fnins.2015.00405
Daenzer, J. M., Jumbo-Lucioni, P. P., Hopson, M. L., Garza, K. R., Ryan, E. L., and Fridovich-Keil, J. L. (2016). Acute and long-term outcomes in a Drosophila melanogaster model of classic galactosemia occur independently of galactose-1-phosphate accumulation. Dis. Model Mech. 9, 1375–1382. doi: 10.1242/dmm.022988
Dani, N., Zhu, H., and Broadie, K. (2014). Two protein N-acetylgalactosaminyl transferases regulate synaptic plasticity by activity-dependent regulation of integrin signaling. J. Neurosci. 34, 13047–13065. doi: 10.1523/JNEUROSCI.1484-14.2014
Fári, K., Takács, S., Ungár, D., and Sinka, R. (2016). The role of acroblast formation during Drosophila spermatogenesis. Biol. Open 5, 1102–1110. doi: 10.1242/bio.018275
Farkas, R. M., Giansanti, M. G., Gatti, M., and Fuller, M. T. (2003). The Drosophila Cog5 homologue is required for cytokinesis, cell elongation, and assembly of specialized Golgi architecture during spermatogenesis. Mol. Biol. Cell 14, 190–200. doi: 10.1091/mbc.e02-06-0343
Foulquier, F., Vasile, E., Schollen, E., Callewaert, N., Raemaekers, T., Quelhas, D., et al. (2006). Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc. Natl. Acad. Sci. U.S.A. 103, 3764–3769. doi: 10.1073/pnas.0507685103
Frappaolo, A., Sechi, S., Kumagai, T., Robinson, S., Fraschini, R., Karimpour-Ghahnavieh, A., et al. (2017). COG7 deficiency in Drosophila generates multifaceted developmental, behavioral and protein glycosylation phenotypes. J. Cell Sci. 130, 3637–3649. doi: 10.1242/jcs.209049
Freeze, H. H. (2009). Towards a therapy for phosphomannomutase 2 deficiency, the defect in CDG-Ia patients. Biochim. Biophys. Acta 1792, 835–840. doi: 10.1016/j.bbadis.2009.01.004
Freeze, H. H., Chong, J. X., Bamshad, M. J., and Ng, B. G. (2014). Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am. J. Hum. Genet. 94, 161–175. doi: 10.1016/j.ajhg.2013.10.024
Freeze, H. H., Eklund, E. A., Ng, B. G., and Patterson, M. C. (2015). Neurological aspects of human glycosylation disorders. Annu. Rev. Neurosci. 38, 105–125. doi: 10.1146/annurev-neuro-071714-034019
Freeze, H. H., and Ng, B. G. (2011). Golgi glycosylation and human inherited diseases. Cold Spring Harb. Perspect. Biol. 3:a005371. doi: 10.1101/cshperspect.a005371
Fung, C. W., Matthijs, G., Sturiale, L., Garozzo, D., Wong, K. Y., Wong, R., et al. (2012). COG5-CDG with a mild neurohepatic presentation. JIMD Rep. 3, 67–70. doi: 10.1007/8904_2011_61
Gatto, C. L., and Broadie, K. (2011). Fragile X mental retardation protein is required for programmed cell death and clearance of developmentally-transient peptidergic neurons. Dev. Biol. 356, 291–317. doi: 10.1016/j.ydbio.2011.05.001
Geisler, C., Kotu, V., Sharrow, M., Rendić, D., Pöltl, G., Tiemeyer, M., et al. (2012). The Drosophila neurally altered carbohydrate mutant has a defective Golgi GDP-fucose transporter. J. Biol. Chem. 287, 29599–29609. doi: 10.1074/jbc.M112.379313
Goreta, S. S., Dabelic, S., and Dumic, J. (2012). Insights into complexity of congenital disorders of glycosylation. Biochem. Med. 22, 156–170. doi: 10.11613/BM.2012.019
Grünewald, S. (2009). The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia). Biochim. Biophys. Acta 1792, 827–834. doi: 10.1016/j.bbadis.2009.01.003
Hartl, D. L. (2000). Molecular melodies in high and low C. Nat. Rev. Genet. 1, 145–149. doi: 10.1038/35038580
Helenius, A., and Aebi, M. (2004). Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752
Hong, W., and Lev, S. (2014). Tethering the assembly of SNARE complexes. Trends Cell Biol. 24, 35–43. doi: 10.1016/j.tcb.2013.09.006
Jaeken, J. (2013). Congenital disorders of glycosylation. Handb. Clin. Neurol. 113, 1737–1743. doi: 10.1016/B978-0-444-59565-2.00044-7
Jumbo-Lucioni, P. P., Parkinson, W. M., Kopke, D. L., and Broadie, K. (2016). Coordinated movement, neuromuscular synaptogenesis and trans-synaptic signaling defects in Drosophila galactosemia models. Hum. Mol. Genet. 25, 3699–3714. doi: 10.1093/hmg/ddw217
Katoh, T., and Tiemeyer, M. (2013). The N’s and O’s of Drosophila glycoprotein glycobiology. Glycoconj. J. 30, 57–66. doi: 10.1007/s10719-012-9442-x
Khoury, G. A., Baliban, R. C., and Floudas, C. A. (2011). Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci. Rep. 1:90. doi: 10.1038/srep00090
Kingsley, D. M., Kozarsky, K. F., Segal, M., and Krieger, M. (1986). Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol. 102, 1576–1585. doi: 10.1083/jcb.102.5.1576
Kodera, H., Ando, N., Yuasa, I., Wada, Y., Tsurusaki, Y., Nakashima, M., et al. (2015). Mutations in COG2 encoding a subunit of the conserved oligomeric golgi complex cause a congenital disorder of glycosylation. Clin. Genet. 87, 455–460. doi: 10.1111/cge.12417
Koles, K., Lim, J. M., Aoki, K., Porterfield, M., Tiemeyer, M., Wells, L., et al. (2007). Identification of N-glycosylated proteins from the central nervous system of Drosophila melanogaster. Glycobiology 17, 1388–1403. doi: 10.1093/glycob/cwm097
Kranz, C., Ng, B. G., Sun, L., Sharma, V., Eklund, E. A., Miura, Y., et al. (2007). COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum. Mol. Genet. 16, 731–741. doi: 10.1093/hmg/ddm028
Li, H., Chavan, M., Schindelin, H., Lennarz, W. J., and Li, H. (2008). Structure of the oligosaccharyl transferase complex at 12 A resolution. Structure 16, 432–440. doi: 10.1016/j.str.2007.12.013
Lübbehusen, J., Thiel, C., Rind, N., Ungar, D., Prinsen, B. H., de Koning, T. J., et al. (2010). Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Hum. Mol. Genet. 19, 3623–3633. doi: 10.1093/hmg/ddq278
Mauvezin, C., Nagy, P., Juhász, G., and Neufeld, T. P. (2015). Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 6:7007. doi: 10.1038/ncomms8007
Miller, V. J., Sharma, P., Kudlyk, T. A., Frost, L., Rofe, A. P., Watson, I. J., et al. (2012). Molecular insights into vesicle tethering at the Golgi by the conserved oligomeric Golgi (COG) complex and the golgin TATA element modulatory factor (TMF). J. Biol. Chem. 288, 4229–4240. doi: 10.1074/jbc.M112.426767
Miller, V. J., and Ungar, D. (2012). Re’COG’nition at the Golgi. Traffic 13, 891–897. doi: 10.1111/j.1600-0854.2012.01338.x
Morava, E., Zeevaert, R., Korsch, E., Huijben, K., Wopereis, S., Matthijs, G., et al. (2007). A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur. J. Hum. Genet. 15, 638–645. doi: 10.1038/sj.ejhg.5201813
Moremen, K. W., Tiemeyer, M., and Nairn, A. V. (2012). Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462. doi: 10.1038/nrm3383
Moulton, M. J., and Letsou, A. (2016). Modeling congenital disease and inborn errors of development in Drosophila melanogaster. Dis. Model Mech. 9, 253–269. doi: 10.1242/dmm.023564
Ng, B. G., Kranz, C., Hagebeuk, E. E., Duran, M., Abeling, N. G., Wuyts, B., et al. (2007). Molecular and clinical characterization of a Moroccan Cog7 deficient patient. Mol. Genet. Metab. 91, 201–214. doi: 10.1016/j.ymgme.2007.02.011
Ohtsubo, K., and Marth, J. D. (2006). Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867. doi: 10.1016/j.cell.2006.08.019
Paesold-Burda, P., Maag, C., Troxler, H., Foulquier, F., Kleinert, P., Schnabel, S., et al. (2009). Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum. Mol. Genet. 18, 4350–4356. doi: 10.1093/hmg/ddp389
Parkinson, W. M., Dookwah, M., Dear, M. L., Gatto, C. L., Aoki, K., Tiemeyer, M., et al. (2016). Synaptic roles for phosphomannomutase type 2 in a new Drosophila congenital disorder of glycosylation disease model. Dis. Model Mech. 9, 513–527. doi: 10.1242/dmm.022939
Reiter, L. T., Potocki, L., Chien, S., Gribskov, M., and Bier, E. (2001). A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 11, 1114–1125. doi: 10.1101/gr.169101
Repnikova, E., Koles, K., Nakamura, M., Pitts, J., Li, H., Ambavane, A., et al. (2010). Sialyltransferase regulates nervous system function in Drosophila. J. Neurosci. 30, 6466–6476. doi: 10.1523/JNEUROSCI.5253-09.2010
Reynders, E., Foulquier, F., Leão Teles, E., Quelhas, D., Morelle, W., Rabouille, C., et al. (2009). Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum. Mol. Genet. 18, 3244–3256. doi: 10.1093/hmg/ddp262
Rujano, M. A., Cannata Serio, M., Panasyuk, G., Péanne, R., Reunert, J., Rymen, D., et al. (2018). Mutations in the X-linked ATP6AP2 cause a glycosylation disorder with autophagic defects. J. Exp. Med. 214, 3707–3729. doi: 10.1084/jem.20170453
Scott, H., and Panin, V. M. (2014). N-glycosylation in regulation of the nervous system. Adv. Neurobiol. 9, 367–394. doi: 10.1007/978-1-4939-1154-7_17
Sechi, S., Frappaolo, A., Fraschini, R., Capalbo, L., Gottardo, M., Belloni, G., et al. (2017). Rab1 interacts with GOLPH3 and controls Golgi structure and contractile ring constriction during cytokinesis in Drosophila melanogaster. Open Biol. 7:160257. doi: 10.1098/rsob.160257
Seppo, A., Matani, P., Sharrow, M., and Tiemeyer, M. (2003). Induction of neuron-specific glycosylation by Tollo/Toll-8, a Drosophila toll-like receptor expressed in non-neural cells. Development 130, 1439–1448. doi: 10.1242/dev.00347
Shwartz, F., and Aebi, M. (2011). Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 21, 576–582. doi: 10.1016/j.sbi.2011.08.005
Spaapen, L. J., Bakker, J. A., van der Meer, S. B., Sijstermans, H. J., Steet, R. A., Wevers, R. A., et al. (2005). Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. J. Inherit. Metab. Dis. 28, 707–714. doi: 10.1007/s10545-005-0015-z
Stanley, P. (2011). Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 3:a005199. doi: 10.1101/cshperspect.a005199
Struwe, W. B., and Reinhold, V. N. (2012). The conserved oligomeric Golgi complex is required for fucosylation of N-glycans in Caenorhabditis elegans. Glycobiology 22, 863–875. doi: 10.1093/glycob/cws053
Suvorova, E. S., Duden, R., and Lupashin, V. V. (2002). The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J. Cell Biol. 157, 631–643. doi: 10.1083/jcb.200111081
Ungar, D., Oka, T., Brittle, E. E., Vasile, E., Lupashin, V. V., Chatterton, J. E., et al. (2002). Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 157, 405–415. doi: 10.1083/jcb.200202016
Vaccari, T., Duchi, S., Cortese, K., Tacchetti, C., and Bilder, D. (2010). The vacuolar ATPase is required for physiological as well as pathological activation of the Notch receptor. Development 137, 1825–1832. doi: 10.1242/dev.045484
Varki, A., Cummings, R. D., Aebi, M., Packer, N. H., Seeberger, P. H., Esko, J. D., et al. (2015). Symbol nomenclature for graphical representations of glycans. Glycobiology 25, 1323–1324. doi: 10.1093/glycob/cwv091
Willett, R., Ungar, D., and Lupashin, V. (2013). The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem. Cell Biol. 140, 271–283. doi: 10.1007/s00418-013-1117-6
Wu, X., Steet, R. A., Bohorov, O., Bakker, J., Newell, J., Krieger, M., et al. (2004). Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 10, 518–523. doi: 10.1038/nm1041
Yakubenia, S., Frommhold, D., Schölch, D., Hellbusch, C. C., Körner, C., Petri, B., et al. (2008). Leukocyte trafficking in a mouse model for leukocyte adhesion deficiency II/congenital disorder of glycosylation IIc. Blood 112, 1472–1481. doi: 10.1182/blood-2008-01-132035
Zeevaert, R., Foulquier, F., Cheillan, D., Cloix, I., Guffon, N., Sturiale, L., et al. (2009). A new mutation in COG7 extends the spectrum of COG subunit deficiencies. Eur. J. Med. Genet. 52, 303–305. doi: 10.1016/j.ejmg.2009.06.006
Keywords: Drosophila, glycosylation, congenital disorders, Golgi, model organism
Citation: Frappaolo A, Sechi S, Kumagai T, Karimpour-Ghahnavieh A, Tiemeyer M and Giansanti MG (2018) Modeling Congenital Disorders of N-Linked Glycoprotein Glycosylation in Drosophila melanogaster. Front. Genet. 9:436. doi: 10.3389/fgene.2018.00436
Received: 10 July 2018; Accepted: 14 September 2018;
Published: 02 October 2018.
Edited by:
Roberta Fraschini, Università degli Studi Milano-Bicocca, ItalyReviewed by:
Vladimir Lupashin, University of Arkansas for Medical Sciences, United StatesCopyright © 2018 Frappaolo, Sechi, Kumagai, Karimpour-Ghahnavieh, Tiemeyer and Giansanti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Tiemeyer, bXRpZW1leWVyQGNjcmMudWdhLmVkdQ== Maria Grazia Giansanti, bWFyaWFncmF6aWEuZ2lhbnNhbnRpQHVuaXJvbWExLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.