 Xiaofang Xiao1†
Xiaofang Xiao1† Wen Zhu2†
Wen Zhu2† Bo Liao1,2*Junlin Xu1Changlong Gu1Binbin Ji2Yuhua Yao2
Bo Liao1,2*Junlin Xu1Changlong Gu1Binbin Ji2Yuhua Yao2 Lihong Peng3
Lihong Peng3 Jialiang Yang2,4*
Jialiang Yang2,4*- 1College of Information Science and Engineering, Hunan University, Changsha, China
- 2School of Mathematics and Statistics, Hainan Normal University, Haikou, China
- 3School of Computer Science, Hunan University of Technology, Zhuzhou, China
- 4Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY, United States
In recent years, it has been increasingly clear that long noncoding RNAs (lncRNAs) play critical roles in many biological processes associated with human diseases. Inferring potential lncRNA-disease associations is essential to reveal the secrets behind diseases, develop novel drugs, and optimize personalized treatments. However, biological experiments to validate lncRNA-disease associations are very time-consuming and costly. Thus, it is critical to develop effective computational models. In this study, we have proposed a method called BPLLDA to predict lncRNA-disease associations based on paths of fixed lengths in a heterogeneous lncRNA-disease association network. Specifically, BPLLDA first constructs a heterogeneous lncRNA-disease network by integrating the lncRNA-disease association network, the lncRNA functional similarity network, and the disease semantic similarity network. It then infers the probability of an lncRNA-disease association based on paths connecting them and their lengths in the network. Compared to existing methods, BPLLDA has a few advantages, including not demanding negative samples and the ability to predict associations related to novel lncRNAs or novel diseases. BPLLDA was applied to a canonical lncRNA-disease association database called LncRNADisease, together with two popular methods LRLSLDA and GrwLDA. The leave-one-out cross-validation areas under the receiver operating characteristic curve of BPLLDA are 0.87117, 0.82403, and 0.78528, respectively, for predicting overall associations, associations related to novel lncRNAs, and associations related to novel diseases, higher than those of the two compared methods. In addition, cervical cancer, glioma, and non-small-cell lung cancer were selected as case studies, for which the predicted top five lncRNA-disease associations were verified by recently published literature. In summary, BPLLDA exhibits good performances in predicting novel lncRNA-disease associations and associations related to novel lncRNAs and diseases. It may contribute to the understanding of lncRNA-associated diseases like certain cancers.
Introduction
It is known that there are about 20,000 protein-coding genes, consisting of less than 2% of the human genome (Bertone et al., 2004; Claverie, 2005). Most DNA regions in the human genome are either not transcribable or transcribed into noncoding RNAs (ncRNAs), which are deemed to be transcriptional noises in a long period of time. However, many recent studies have suggested that ncRNAs play key regulatory roles in many important biological processes such as cell proliferation (Esteller, 2011). Based on their sizes, ncRNAs can be divided into long ncRNAs (lncRNAs) (Pauli et al., 2011) and small ncRNAs such as microRNAs (miRNAs) (Farazi et al., 2013), transfer RNAs (tRNAs) (Birney et al., 2007), and Piwi-interacting RNAs (piRNAs) (Li et al., 2013). LncRNAs are ncRNAs of lengths greater than 200 nucleotides (Mercer et al., 2009; Mitchell Guttman et al., 2013). Compared to protein-coding, RNAs, lncRNAs are less conservative among species (Harrow et al., 2012; Cabili et al., 2016), and have a relatively low expression level, more tissue-specific patterns (Guttman et al., 2010), and longer but less exons (Chen, 2015). Recently, more and more lncRNAs have been identified in eukaryotes from nematodes to human beings due to the advancement in sequencing technologies and computational methods (Awan et al., 2017).
Previous studies have suggested that lncRNAs are critical in cell proliferation, cell differentiation, chromatin remodeling, genome splicing, epigenetic regulation, transcription, and many other important biological processes (Guttman et al., 2009). The dysregulation of lncRNAs has also been associated with the development of many diseases, including diabetes (Pasmant et al., 2011), cardiovascular diseases (Congrains et al., 2012), HIV (Zhang et al., 2013), neurological disorders (Johnson, 2012), and several cancers such as lung cancer (Ji et al., 2003; Zhang et al., 2003), breast cancer (Barsyte-Lovejoy et al., 2006; Gupta et al., 2010), and prostate cancer (Kok et al., 2002; Szell et al., 2008). As a result, it has become a hot topic recently to identify lncRNA-disease associations, and many important disease-associated lncRNAs have been discovered. For example, breast cancer metastasis patients have about 100 to 2,000 times higher HOTAIR expression than that of the healthy people, based on a quantitative PCR study (Gupta et al., 2010). HOTAIR is also related to metastasis and progression of other cancers, such as liver cancer (Hrdlickova et al., 2014), lung cancer (Li et al., 2014), colorectal cancer (Res, 2011; Maass et al., 2014), gastric cancer (Li et al., 2014; Liu et al., 2014), and so on. Therefore, HOTAIR is deemed to be a potential biomarker for cancers (Maass et al., 2014). In addition, the dysfunction of lncRNA H19 is found in several diseases, such as bladder cancer (Ariel et al., 2000). The downregulation of H19 also significantly reduces the clonogenic and anchored nondependent growth of breast cancer cells based on a knock-down study (Barsyte-Lovejoy et al., 2006).
Known lncRNA-disease associations have been stored in a few databases, including LncRNADisease (Chen et al., 2013), Lnc2Cancer (Ning et al., 2016), MNDR (Wang et al., 2013), and so on, which are the basis for predicting novel associations using efficient computational methods. The computational models to predict lncRNA-disease associations are generally divided into two categories including machine learning-based models and network-based models (Chen et al., 2017). Machine learning-based models usually train predictors from features based on training samples and test their performances based on cross-validation or independent data. For example, Chen et al. developed Laplacian Regularized Least Squares for LncRNA-Disease Association (LRLSLDA) for inferring candidates of disease-associated lncRNAs by applying a semisupervised learning framework (Chen and Yan, 2013). LRLSLDA assumes that similar diseases tend to correlate with functionally similar lncRNAs, and vice versa. Thus, known lncRNA-disease associations and lncRNA expression profiles are combined to prioritize disease-associated lncRNA candidates by LRLSLDA, which does not require negative samples (i.e., confirmed uncorrelated lncRNA-disease associations). However, LRLSLDA faces difficulty in optimizing the best model parameters. Zhao T. et al. (2015) proposed a naïve Bayesian classifier, which exploits various information related to cancer-associated lncRNAs, including regulome, genome, transcriptome, and multiomic data. As a result, 707 potential cancer-related lncRNAs were identified. However, this method requires negative samples, which are usually unknown. In contrast, network-based methods take the advantage of the lncRNA-disease association network, the disease similarity network, and the lncRNA similarity network to study the connectivity of lncRNAs and diseases. For instance, Sun et al. (2014) developed RWRlncD, which infers potential lncRNA-disease associations by a random walk with restart (RWR) on the lncRNA functional similarity network. However, the method cannot predict lncRNAs related to novel diseases (i.e., diseases with no known associated lncRNA). Gu et al. (2017) provided a global network random walk model for predicting lncRNA-disease associations (GrwLDA), which performs RWR on both lncRNA functional similarity network and disease similarity network. However, GrwLDA also faces a dilemma in optimizing model parameters.
In this study, we have proposed a novel method BPLLDA to predict lncRNA-disease associations based on paths connecting them with limited lengths in a heterogeneous network. Specifically, BPLLDA first establishes a heterogeneous network consisting of the known lncRNA-disease association network, the disease similarity network, and the lncRNA similarity network. It then calculates the association between a disease and an lncRNA by the paths connecting them and their lengths. BPLLDA does not require negative samples and is capable of predicting novel diseases and novel lncRNAs.
Materials and Methods
lncRNA-Disease Associations
The lncRNA-disease association data were retrieved from the database LncRNADisease (Chen et al., 2013; Sun et al., 2014). After eliminating identical lncRNA-disease entries from distinct pieces of evidence, there were 352 experimentally confirmed lncRNA-disease associations, containing 156 lncRNAs and 190 diseases (see Supplementary Figure 1 and Supplementary Tables 2, 3). We summarize some basic characteristics (e.g., the average degree) of the dataset in Table 1. We then established the lncRNA-disease association network, whose adjacency matrix is denoted by LD. That is, LD(i, j) is set to 1 if lncRNA l(i) is associated with disease d(j), and 0 if otherwise. Before presenting the details of BPLLDA, we first introduced two important notations, namely, disease semantic similarity and lncRNA functional similarity.

Table 1. The basic characteristics of the lncRNA-disease association dataset.
Disease Semantic Similarity
The Disease Ontology (DO) is an open source ontology of human diseases (http://www.disease-ontology.org/). The terms in DO are diseases or disease-correlated concepts, which are organized in a directed acyclic graph (DAG). On the basis of Disease Ontology, Li et al. (2011) provided an R package called DOSim to calculate the disease semantic similarity, and we adopted this method in this study. Specifically, we used a symmetric matrix SS to record semantic similarity values among diseases, in which SS(i, j) represents semantic similarity between disease d(i) and d(j) as calculated by DOSim. We plot the distribution of SS in Figure 1A. There are overall 36100 (190 × 190) values, among which 21148 values (58.58%) are 0 s.
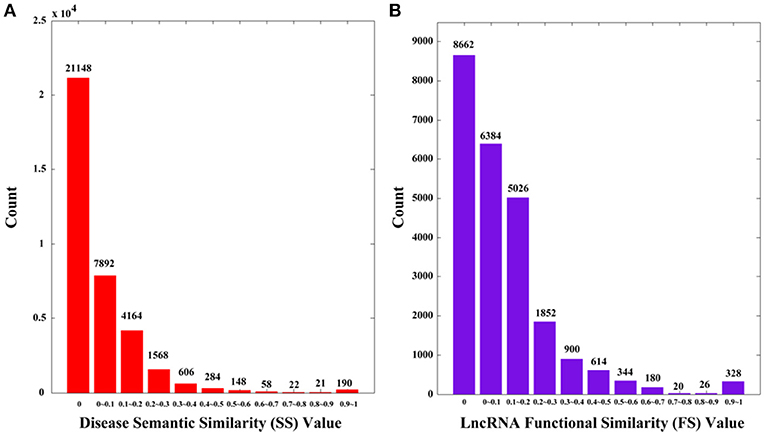
Figure 1. The distributions of disease semantic and lncRNA functional similarity. (A) Disease semantic similarity (SS) distribution. (B) lncRNA functional similarity (FS) distribution. The x-axis indicates the intervals of similarity values and the y-axis indicates the numbers of values in the interval. The actual values are also marked above the histograms.
lncRNA Functional Similarity
We adopted a similar method to Sun et al. for measuring the functional similarity between two lncRNAs (Wang et al., 2010; Sun et al., 2014). Specifically, suppose lncRNA l(i) is associated with a disease set Di = {dik| 1 ≤ k ≤ m} and lncRNA l(j) is associated with Dj = {djl| 1 ≤ l ≤ n}. The method first calculates the semantic similarity between a disease, say di1, and a disease group, say Dj, as
Then, the functional similarity between l(i) and l(j) is calculated as
It is clear that the lncRNA functional similarity matrix FS is symmetric. Similarly, we plot the distribution of FS in Figure 1B. There are 24336 (156 × 156) values, among which 8662 (35.59%) are 0 s.
Gaussian Interaction Profile Kernel Similarity for lncRNAs
There are many zeros in FS due to the fact that lncRNA-disease associations are rather incomplete. To avoid such scenario, we introduced the Gaussian interaction profile kernel similarity between lncRNA l(i) and l(i) as
where IP(l(i)) and IP(l(j)) are the vectors in the ith and jth row of the lncRNA-disease association matrix LD. The parameter γl is a regulation parameter of the kernel bandwidth with , where ln is the number of all lncRNAs studied and is usually set to 1 according to van Laarhoven et al. (2011).
Gaussian Interaction Profile Kernel Similarity for Diseases
Similarly, we defined the Gaussian interaction profile kernel similarity for diseases as
with , where IP(d(i)) and IP(d(i)) are the binary vectors in the ith and jth column of the adjacency matrix LD and dn is the numbers of diseases. Clearly, GD is also symmetric.
Integrated Similarity Between lncRNAs and Between Diseases
We integrated disease semantic similarity (lncRNA functional similarity) with the Gaussian interaction profile kernel similarity for diseases (lncRNAs) as follows:
where NS is the set of diseases with no sematic similarity with any other disease, and NF is the set of lncRNAs with no functional similarity with any other lncRNAs. By definition, DS and LS are symmetric. We plot the distributions of DS and LS in Figure 2, in which the numbers of 0 s are greatly reduced compared to SS and FS.
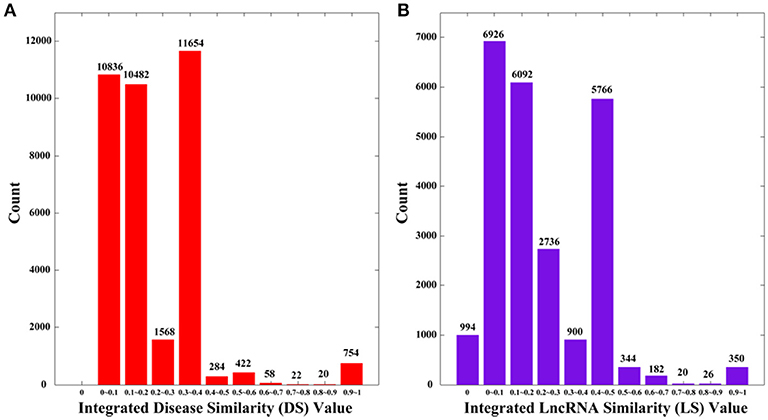
Figure 2. The distributions of integrated similarities. (A) Distribution of the integrated similarity for diseases (DS). (B) Distribution of the integrated similarity for lncRNAs (LS). The x-axis indicates the intervals of similarity values and the y-axis indicates the numbers of values in the interval. The actual values are also marked above the histograms.
BPLLDA
The general workflow of BPLLDA is illustrated in Figure 3, in which a heterogeneous network is first constructed with nodes denoting lncRNAs or diseases. For any two diseases d(i) and d(j), the weight of the edge between them is defined to be
where T is a threshold value to avoid all diseases being connected (You et al., 2017). Similarly, the weight of the edge between two lncRNAs l(i) and l(j) is
The weight of an edge between an lncRNA l(i) and a disease d(j) is LD(l(i), d(j)), that is, the weight is 1 if they are associated and 0 if otherwise. We tuned T from 0.1 to 0.5 with interval 0.1 by a leave-one-out cross-validation (LOOCV) process and finally chose T to be 0.2.
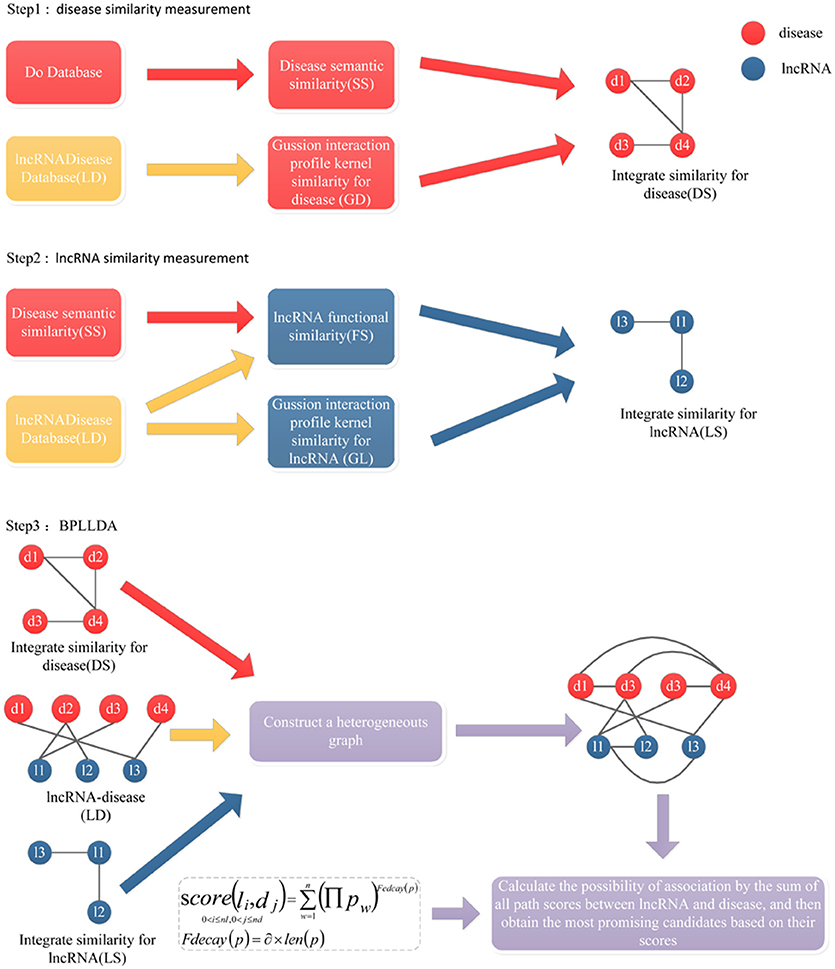
Figure 3. The flowchart of BPLLDA. It consists of three steps: (1) disease similarity measurement, (2) lncRNA similarity measurement, and (3) the BPLLDA algorithm.
For a given lncRNA node l(i) and a disease node d(j), we performed a depth-first search (Hopcroft and Tarjan, 1974) to identify all noncyclic paths between them. To avoid long paths, we restricted the maximum number of edges in the path to be τ. Similarly, we performed an LOOCV search for τ being 1 to 4 and decided τ to be 3. Intuitively, l(i) and d(j) tend to be associated if there are many paths with high edge weights connecting them. Therefore, a score measuring their association confidence can be defined using the paths together with a decay function Fdecay(pw):
where p = {p1, p2, …, pn} is the set of paths connecting l(i) and d(j), and ∏pw denotes the product of the weights of all edges in the path pw. Generally speaking, long paths will have little contribution to the total score. So the decay function Fdecay(p) is denoted as
where the decay factor α is set to 2.26 based on a previous study (Ba-Alawi et al., 2016; You et al., 2017) and len(pw) is the length of the path pw. Clearly, the higher the score(l(i), d(j)), the more likely that l(i) and d(j) will be associated.
Analysis of the Computational Complexity
We analyzed the time complexity and space complexity of BPLLDA. Recall that there are m diseases and n lncRNAs with m > n. The algorithm mainly consists of two steps. First, a heterogeneous network was constructed, for which two matrices were established. So the time complexity and space complexity are O(m2) respectively in this step. Then, BPLLDA infers the probability of an lncRNA-disease association based on paths with limited lengths in the network. We performed a depth-first search to identify all noncyclic paths between nodes and the time complexity is O((m + n)2) on each node. Because there are m diseases, the time complexity is O(m3) in this step. And the space complexity is O(mn) because we need to only save the prediction result. In summary, the time complexity and space complexity are at most O(m3) and O(m2), respectively, for BPLLDA.
Results and Discussions
Performance of BPLLDA in Predicting lncRNA-Disease Associations
We applied BPLLDA to a known lncRNA-disease association data LD, together with two popular methods GrwLDA (Gu et al., 2017) and LRLSLDA (Chen and Yan, 2013). The reason why we selected the two methods for comparison is that they can both predict novel lncRNAs and novel diseases. Specifically, two LOOCV methods namely global LOOCV and local LOOCV were adopted to evaluate their performances. Global LOOCV sets each experimentally confirmed lncRNA-disease association as a test sample once, but local LOOCV sets all associations of an lncRNA or those of a disease as test samples once. Other known lncRNA-disease associations are considered as training samples. The performances of the methods were evaluated by the area under the receiver operating characteristic (ROC) curve (AUC).
As a result, we plotted the global LOOCV ROC curves and their associated AUCs of BPLLDA, GrwLDA, and LRLSLDA, respectively, in Figure 4. BPLLDA has an AUC of 0.87117, and outperformed LRLSLDA (0.81952) and GrwLDA (0.78246). Similarly, we plotted the local LOOCV ROC curves and AUCs of the three methods on novel lncRNAs in Figure 5. As can be seen, BPLLDA has an AUC of 0.82403, about 8 and 18% higher than that of LRLSLDA (0.76542) and GrwLDA (0.69817), respectively. Finally, the AUC of BPLLDA (0.78528) in predicting novel diseases is significantly higher than that of LRLSLDA (0.65812) with an increase of 19% and GrwLDA (0.65802) with an increase of 20% (see Figure 6). In summary, our method is better than LRLSLDA and GrwLDA in both lncRNA-disease association prediction and prediction related to novel lnRNAs and diseases.
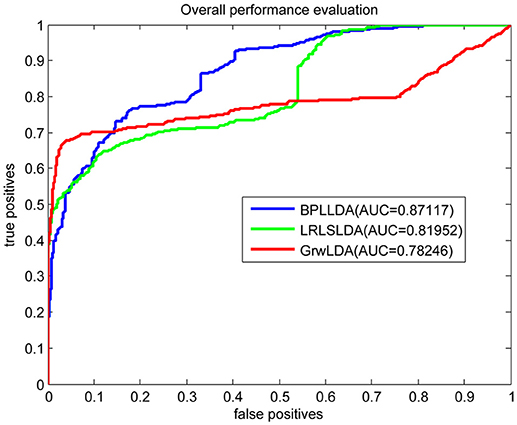
Figure 4. Performance evaluation of BPLLDA, LRLSLDA, and GrwLDA in predicting lncRNA-disease associations by global LOOCV.
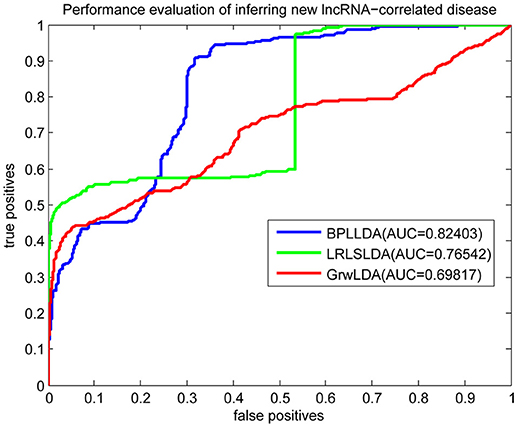
Figure 5. Performance evaluation of BPLLDA, LRLSLDA, and GrwLDA in predicting novel lncRNA-associated diseases.
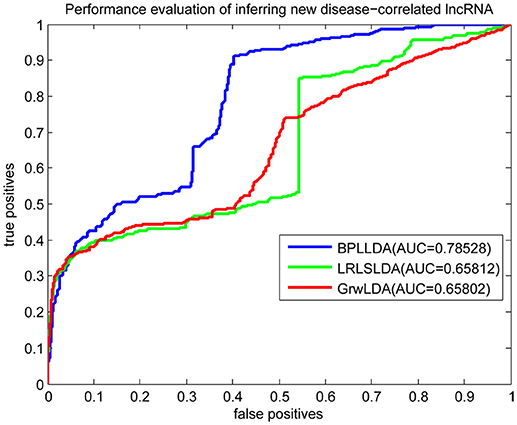
Figure 6. Performance evaluation of BPLLDA, LRLSLDA, and GrwLDA in predicting novel disease-associated lncRNAs.
Meanwhile, we list in Table 2 the precision versus the prediction scores in the global LOOCV. In general, the higher the score, the more likely the disease is related to the lncRNAs. The association confidence is greater than 0.9 when the prediction score is larger than 21.58.

Table 2. Precision of BPLLDA on global LOOCV.
Effects of Parameters
There are two model parameters in BPLLDA, including the maximum path length L and the weight threshold T. We tested the effects of these parameters on AUCs for LOOCV with L (L = 2, 3, 4) and T (T = 0.2, 0.4, 0.5), and we list the results in Table 3. As can be seen, the parameter L has significant effects on the performance of BPLLDA, and the best AUC is achieved at L = 3. In contrast, T has only minor effects on the performance of our method. To further illustrate this, we fixed L to be 3, and let T vary from 0.1 to 0.5 with interval 0.1 (see Table 4). The AUC values are between 0.85568 and 0.87117, only about 2% difference.
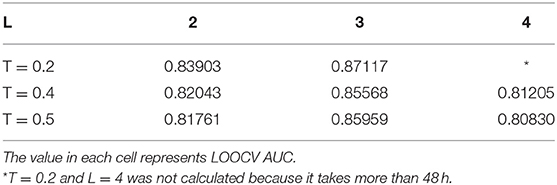
Table 3. Tuning two model parameters: the maximum path length L and the weight threshold T by LOOCV.
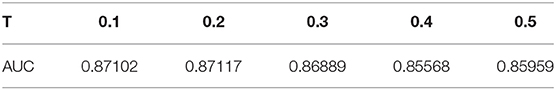
Table 4. The effects of T on AUC when fixing L = 3.
Effects of Gaussian Interaction Profile Kernel Similarity for lncRNAs and Diseases
Disease similarity and lncRNA similarity are calculated by integrating disease semantic similarity, lncRNA functional similarity, as well as the Gaussian interaction profile kernel similarity for lncRNAs and diseases. We tested the effects of the Gaussian interaction profile kernel similarity for lncRNAs and diseases on LOOCV with L = 3 and T = 0.2 with four settings: (1) without using both the Gaussian interaction profile kernel similarity for lncRNAs and diseases; (2) only using the Gaussian interaction profile kernel similarity for lncRNAs; (3) only using the Gaussian interaction profile kernel similarity for diseases; (4) using both the Gaussian interaction profile kernel similarity for lncRNAs and diseases. The results are summarized in Table 5. As can be seen, the two similarities indeed have a significant influence on the LOOCV AUC. The best AUC (0.87117) was achieved when both similarities were adopted into our model.

Table 5. The effects of the Gaussian interaction profile kernel similarity for lncRNAs and diseases on LOOCV.
Case Studies on Predicted lncRNA-Disease Associations
It is known that lncRNAs play critical roles in the development of many diseases. To further evaluate the ability of BPLLDA in inferring novel lncRNA-disease associations, we used all known lncRNA-disease associations in LD as training data and assessed the potential of predicted associations by our model. The novel lncRNA-disease associations were ranked according to the predicted score of BPLLDA. To validate the predictions, the newest LncRNADisease database was used, which curated 1766 distinct known lncRNA-disease associations among 888 lncRNAs and 328 diseases. Specifically, we listed the top five lncRNAs associated with three diseases, including cervical cancer, glioma, and non-small-cell lung cancer (NSCLC), respectively, in Table 6 and the paths of cervical cancer in Supplementary Table 1. For a better view, we also plotted the associations of the three diseases and their top 10 predicted lncRNAs in Figure 7.
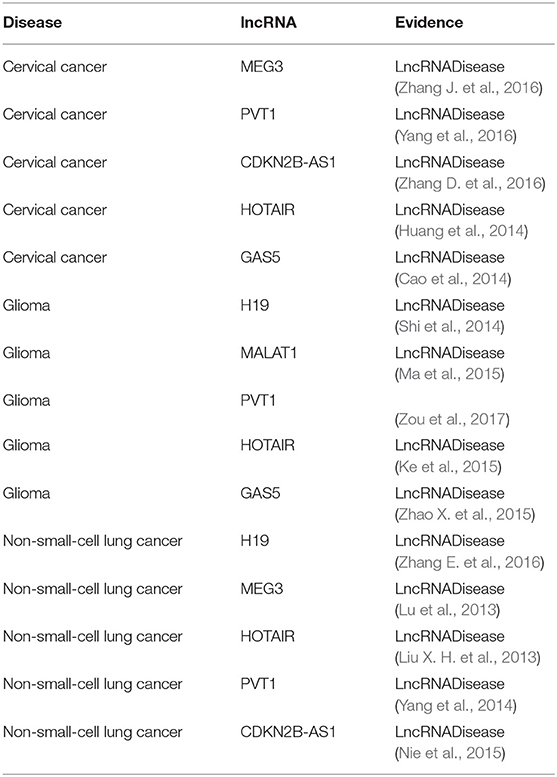
Table 6. The top five lncRNA candidates predicted for cervical cancer, glioma, and non-small-cell lung cancer.
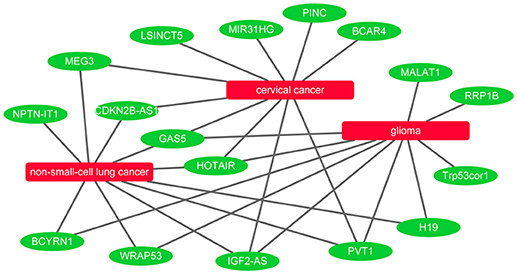
Figure 7. Network view of the top 10 predicted lncRNAs for cervical cancer, glioma, and non-small-cell lung cancer.
Cervical cancer is a cancer in the cervix and its early symptoms are hard to uncover. As the second common cancer among women all over the world, cervical cancer causes numerous incidents of death in developing countries (Forouzanfar et al., 2011). It was reported that there are approximately 500,000 novel cases of cervical cancer diagnosed annually (Tewari et al., 2014). Therefore, there is an urgent need to explore its biological mechanisms and develop effective treatment strategies. Interestingly, all of the top five novel cervical cancer-associated lncRNAs predicted by BPLLDA were confirmed by the newest updates of the LncRNADisease database. For example, the top predicted lncRNA, MEG3, can inhibit tumor growth in cervical cancer by regulating miR-21-5p, which is regarded as a tumor suppressor (Zhang J. et al., 2016). Serum PVT1 can accurately differentiate patients with cervical cancer from healthy controls (Yang et al., 2016). The high expression of HOTAIR is involved in cervical cancer progression and may be a potential target for diagnosis and gene therapy (Huang et al., 2014).
Glioma is considered to be the most common malignant tumor in the central nervous system and is characterized by aggressive blood vessel formation (Khasraw et al., 2010). Despite the continuous improvement of various treatments, including surgery, radiotherapy, and chemotherapy, the overall survival of patients with glioma is only about 12–14 months after diagnosis (Wang et al., 2015). The poor treatment effect is mainly due to the prominent tumor angiogenesis. Similarly, BPLLDA achieved good performance in predicting glioma-associated lncRNAs as all top five predicted lncRNAs were confirmed by the newest LncRNADisease database and literature. For example, it was shown that H19 regulates the development of glioma by deriving miR-675 and offers an essential clue to understanding the key role of the lncRNA-miRNA functional network in glioma (Shi et al., 2014). The expression level of lncRNA MALAT1 is significantly correlated with the overall survival of patients with glioma and can be used as a convictive prognostic biomarker for patients with glioma (Ma et al., 2015). In addition, Gas5 inhibits tumor malignancy by downregulating miR-222, which may be a promising treatment for glioma (Zhao X. et al., 2015).
NSCLC, including adenocarcinoma and squamous cell carcinoma, is a predominant form of lung cancer (Siegel et al., 2012). Despite the progress in clinical and experimental oncology, the prognosis remains difficult. More and more evidence indicates that ncRNAs could take part in the pathogenesis of NSCLC. Similarly, the top five NSCLC-correlated lncRNA candidates predicted by BPLLDA were validated by literature. For example, HOTAIR is significantly upregulated in NSCLC tissues and partly regulates cell invasion and metastasis of NSCLC by HOXA5 downregulation (Liu X. H. et al., 2013). So, HOTAIR is a potential therapeutic target for NSCLC intervention. In addition, patients with NSCLC with high PVT1 expression have a significantly lower overall survival rate than those with low PVT1 expression (Yang et al., 2014). Finally, the expression of CDKN2B-AS1 (ANRIL) might damage cell proliferation and leads to cell apoptosis in vitro and in vivo (Nie et al., 2015), which is linked to the survival of patients with NSCLC.
Case Studies on Predicted Novel Diseases and Novel lncRNAs
To test the ability of BPLLDA in predicting novel disease-associated lncRNAs, all known lncRNA-disease associations correlated with a disease were eliminated. We selected two diseases: colorectal cancer and breast cancer (see Table 7). As can be seen, all top five predicted lncRNAs associated with colorectal cancer were confirmed by the newest LncRNADisease database, whereas four of the top five lncRNAs associated with breast cancer were also validated by the database or literature.
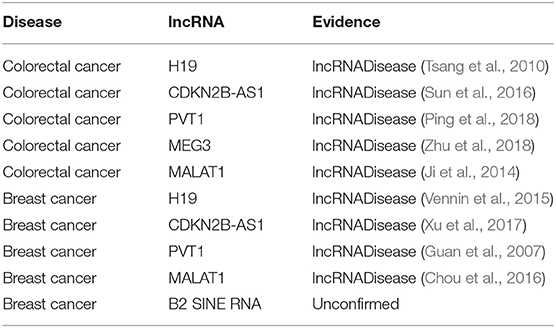
Table 7. The top five novel disease-correlated lncRNA candidates predicted for colorectal cancer and breast cancer.
Similarly, to test the ability of BPLLDA in predicting novel lncRNA-associated diseases, all known lncRNA-disease associations correlated with an lncRNA were removed. As two case studies, we selected two lncRNAs, H19, and HOTAIR (see Table 8). In both cases, four of the top five associated diseases were validated by the database and literature. In summary, BPLLDA achieves favorable performances in predicting novel disease-associated lncRNAs and novel lncRNA-associated diseases.
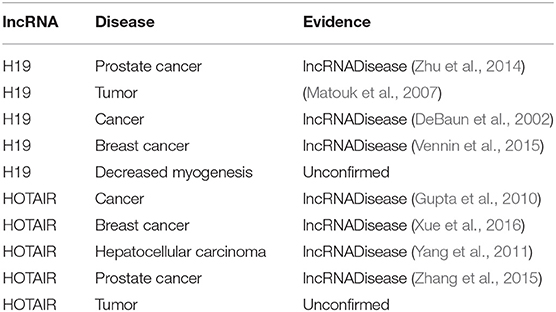
Table 8. The top five novel disease-correlated lncRNA candidates predicted for H19 and HOTAIR.
Conclusions
Many studies have demonstrated that lncRNAs are essential in many physiological processes related to human diseases. They could be important biomarkers for the diagnosis, prognosis, and treatment of these diseases. However, the biological experiments to validate lncRNA-disease associations are not only time consuming but also costly, which promotes the need for developing computational prediction models. In this study, we proposed BPLLDA, a novel computational method to predict lncRNA-disease associations based on simple paths with limited lengths in a heterogeneous network consisting of the lncRNA similarity network, the disease similarity network, and the lncRNA-disease association network. BPLLDA outperforms two compared methods in prediction accuracy, and most top predicted novel lncRNA-disease associations were validated by literature. However, there are a few limitations of BPLLDA. First, available experimentally validated lncRNA-disease associations are rather incomplete. Secondly, lncRNA similarity is computed on the basis of known lncRNA-disease associations. There is a problem of sparseness in the disease semantic similarity and lncRNA functional similarity, which is remedied by integrating the Gaussian interaction profile kernel similarity for diseases and lncRNAs, respectively. So, BPLLDA may result in biased predictions. Finally, the distance-decay function in BPLLDA is relatively simple and could be improved by machine learning methods.
Author Contributions
JY and BL: conceived the concept of the work and designed the experiments; XX, JX, BJ and YY: performed the literature search; XX, WZ, CG, and LP: collected and analyzed the data; XX and JY: wrote the paper, and all authors have approved the manuscript.
Funding
This work was supported by National Nature Science Foundation of China (Grant Nos. 61863010, 61873076, 61370171, 61300128, 61472127, 11171369, 61272395, 61572178, 61672214, and 61702054) and the Natural Science Foundation of Hunan, China (Grant Nos. 2018JJ2461 and 2018JJ3568).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00411/full#supplementary-material
References
Ariel, I., Sughayer, M., Fellig, Y., Pizov, G., Ayesh, S., Podeh, D., et al. (2000). The imprinted H19 gene is a marker of early recurrence in human bladder carcinoma. Mol. Pathol. 53:320. doi: 10.1136/mp.53.6.320
Awan, H. M., Shah, A., Rashid, F., and Shan, G. (2017). Primate-specific long non-coding RNAs and MicroRNAs. Genomics Proteomics Bioinform. 15, 187–195. doi: 10.1016/j.gpb.2017.04.002
Ba-Alawi, W., Soufan, O., Essack, M., Kalnis, P., and Bajic, V. B. (2016). DASPfind: new efficient method to predict drug-target interactions. J. Cheminform. 8:15. doi: 10.1186/s13321-016-0128-4
Barsyte-Lovejoy, D., Lau, S. K., Boutros, P. C., Khosravi, F., Jurisica, I., Andrulis, I. L., et al. (2006). The c-Myc oncogene directly induces the H19 Noncoding RNA by Allele-Specific binding to potentiate tumorigenesis. Cancer Res. 66:5330. doi: 10.1158/0008-5472.CAN-06-0037
Bertone, P., Stolc, V., Royce, T. E., Rozowsky, J. S., Urban, A. E., Zhu, X., et al. (2004). Global identification of human transcribed sequences with genome tiling arrays. Science 306, 2242–2246. doi: 10.1126/science.1103388
Birney, E., Stamatoyannopoulos, J. A., Dutta, A., Guigó, R., Gingeras, T. R., Margulies, E. H., et al. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816. doi: 10.1038/nature05874
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazonvega, B., Regev, A., et al. (2016). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25:1915. doi: 10.1101/gad.17446611
Cao, S., Liu, W., Li, F., Zhao, W., and Qin, C. (2014). Decreased expression of lncRNA GAS5 predicts a poor prognosis in cervical cancer. Int. J. Clin. Exp. Pathol. 7, 6776–6783.
Chen, G., Wang, Z., Wang, D., Qiu, C., Liu, M., Chen, X., et al. (2013). LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 41, D983–D986. doi: 10.1093/nar/gks1099
Chen, X. (2015). Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci. Rep. 5:13186. doi: 10.1038/srep13186
Chen, X., Yan, C. C., Zhang, X., and You, Z. H. (2017). Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief Bioinform 18, 558–576. doi: 10.1093/bib/bbw060
Chen, X., and Yan, G. Y. (2013). Novel human lncRNA–disease association inference based on lncRNA expression profiles. Bioinformatics 29, 2617–2624. doi: 10.1093/bioinformatics/btt426
Chou, J., Wang, B., Zheng, T., Li, X., Zheng, L., Hu, J., et al. (2016). MALAT1 induced migration and invasion of human breast cancer cells by competitively binding miR-1 with cdc42. Biochem Biophys. Res. Commun. 472, 262–269. doi: 10.1016/j.bbrc.2016.02.102
Claverie, J. M. (2005). Fewer genes, more noncoding RNA. Science 309, 1529–1530. doi: 10.1126/science.1116800
Congrains, A., Kamide, K., Oguro, R., Yasuda, O., Miyata, K., Yamamoto, E., et al. (2012). Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B ? Atherosclerosis 220, 449–455. doi: 10.1016/j.atherosclerosis.2011.11.017
DeBaun, M. R., Niemitz, E. L., Mcneil, D. E., Brandenburg, S. A., Lee, M. P., and Feinberg, A. P. (2002). Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. Am. J. Hum. Genetics 70, 604–611. doi: 10.1086/338934
Esteller, M. (2011). Non-coding RNAs in human disease. Nat. Rev. Genetics 12, 861–874. doi: 10.1038/nrg3074
Farazi, T. A., Hoell, J. I., Morozov, P., and Tuschl, T. (2013). MicroRNAs in human cancer. Adv. Exp. Med. Biol. 774, 1–20. doi: 10.1007/978-94-007-5590-1_1
Forouzanfar, M. H., Foreman, K. J., Delossantos, A. M., Lozano, R., Lopez, A. D., Murray, C. J. L., et al. (2011). Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. Lancet 378, 1461–1484. doi: 10.1016/S0140-6736(11)61351-2
Gu, C., Liao, B., Li, X., Cai, L., Li, Z., Li, K., et al. (2017). Global network random walk for predicting potential human lncRNA-disease associations. Sci. Rep. 7:12442. doi: 10.1038/s41598-017-12763-z
Guan, Y., Kuo, W. L., Stilwell, J. L., Takano, H., Lapuk, A. V., Fridlyand, J., et al. (2007). Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin. Cancer Res. 13, 5745–5755. doi: 10.1158/1078-0432.CCR-06-2882
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., et al. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076. doi: 10.1038/nature08975
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D., et al. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227. doi: 10.1038/nature07672
Guttman, M., Garber, M., Levin, J. Z., Donaghey, J., Robinson, J., Adiconis, X., et al. (2010). Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 28, 503–510. doi: 10.1038/nbt.1633
Harrow, J., Frankish, A., Gonzalez, J. M., Tapanari, E., Diekhans, M., Kokocinski, F., et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. doi: 10.1101/gr.135350.111
Hopcroft, J., and Tarjan, R. (1974). Efficient planarity testing. J. Acm 21, 549–568. doi: 10.1145/321850.321852
Hrdlickova, B., de Almeida, R. C., Borek, Z., and Withoff, S. (2014). Genetic variation in the non-coding genome: involvement of micro-RNAs and long non-coding RNAs in disease. BBA Mol. Basis Dis. 1842, 1910–1922. doi: 10.1016/j.bbadis.2014.03.011
Huang, L., Liao, L. M., Liu, A. W., Wu, J. B., Cheng, X. L., Lin, J. X., et al. (2014). Overexpression of long noncoding RNA HOTAIR predicts a poor prognosis in patients with cervical cancer. Arch. Gynecol. Obstetrics 290, 717–723. doi: 10.1007/s00404-014-3236-2
Ji, P., Diederichs, S., Wang, W., Böing, S., Metzger, R., Schneider, P. M., et al. (2003). MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 22, 6087–6097. doi: 10.1038/sj.onc.1206928
Ji, Q., Zhang, L., Liu, X., Zhou, L., Wang, W., Han, Z., et al. (2014). Long non-coding RNA MALAT1 promotes tumour growth and metastasis in colorectal cancer through binding to SFPQ and releasing oncogene PTBP2 from SFPQ|[sol]|PTBP2 complex. Br. J. Cancer 111, 736–748. doi: 10.1038/bjc.2014.383
Johnson, R. (2012). Long non-coding RNAs in Huntington's disease neurodegeneration. Neurobiol. Dis. 46, 245–254. doi: 10.1016/j.nbd.2011.12.006
Ke, J., Yao, Y. L., Zheng, J., Wang, P., Liu, Y. H., Ma, J., et al. (2015). Knockdown of long non-coding RNA HOTAIR inhibits malignant biological behaviors of human glioma cells via modulation of miR-326. Oncotarget 6, 21934–21949. doi: 10.18632/oncotarget.4290
Khasraw, M., Ameratunga, M. S., Grant, R., Wheeler, H., and Pavlakis, N. (2010). Antiangiogenic therapy for high-grade glioma. Cochrane Database Syst Rev 9:CD008218. doi: 10.1002/14651858.CD008218.pub3
Kok, J. B. D., Verhaegh, G. W., Roelofs, R. W., Hessels, D., Kiemeney, L. A., Aalders, T. W., et al. (2002). DD3PCA3, a very sensitive and specific marker to detect prostate tumors. Cancer Res. 62, 2695–2698.
Li, G., Zhang, H., Wan, X., Yang, X., Zhu, C., Wang, A., et al. (2014). Long noncoding RNA plays a key role in metastasis and prognosis of hepatocellular carcinoma. Biomed. Res. Int. 2014:780521. doi: 10.1155/2014/780521
Li, J., Gong, B., Chen, X., Liu, T., Wu, C., Zhang, F., et al. (2011). DOSim: an R package for similarity between diseases based on Disease Ontology. BMC Bioinformatics 12:266. doi: 10.1186/1471-2105-12-266
Li, X. Z., Roy, C. K., Moore, M. J., and Zamore, P. D. (2013). Defining piRNA primary transcripts. Cell Cycle 12, 1657–1658. doi: 10.4161/cc.24989
Liu, X. H., Liu, Z. L., Sun, M., Liu, J., Wang, Z. X., and De, W. (2013). The long non-coding RNA HOTAIR indicates a poor prognosis and promotes metastasis in non-small cell lung cancer. BMC Cancer 13:464. doi: 10.1186/1471-2407-13-464
Liu, X. H., Sun, M., Nie, F. Q., Ge, Y. B., Zhang, E. B., Yin, D. D., et al. (2014). Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol. Cancer 13:92. doi: 10.1186/1476-4598-13-92
Lu, K. H., Li, W., Liu, X. H., Sun, M., Zhang, M. L., Wu, W. Q., et al. (2013). Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer 13:461. doi: 10.1186/1471-2407-13-461
Ma, K. X., Wang, H. J., Li, X. R., Li, T., Su, G., Yang, P., et al. (2015). Long noncoding RNA MALAT1 associates with the malignant status and poor prognosis in glioma. Tumor Biology 36, 3355–3359. doi: 10.1007/s13277-014-2969-7
Maass, P. G., Luft, F. C., and Bähring, S. (2014). Long non-coding RNA in health and disease. J. Mol. Med. 92, 337–346. doi: 10.1007/s00109-014-1131-8
Matouk, I. J., Degroot, N., Mezan, S., Ayesh, S., Abu-Lail, R., Hochberg, A., et al. (2007). The H19 Non-coding RNA is essential for human tumor growth. PLoS ONE 2:e845. doi: 10.1371/journal.pone.0000845
Mercer, T. R., Dinger, M. E., and Mattick, J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159. doi: 10.1038/nrg2521
Mitchell Guttman, P. R., Nicholas, T., Ingolia, J. S., and Weissman, E. S., (2013). Ribosome profiling provides evidence that large Noncoding RNAs do not encode proteins. Cell 154, 240–251. doi: 10.1016/j.cell.2013.06.009
Nie, F. Q., Sun, M., Yang, J. S., Xie, M., Xu, T. P., Xia, R., et al. (2015). Long Noncoding RNA ANRIL promotes non–small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol. Cancer Therapeutics 14, 268–277. doi: 10.1158/1535-7163.MCT-14-0492
Ning, S., Zhang, J., Wang, P., Zhi, H., Wang, J., Liu, Y., et al. (2016). Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 44(Database issue), D980–D985. doi: 10.1093/nar/gkv1094
Pasmant, E., Sabbagh, A., Vidaud, M., and Bièche, I. (2011). ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 25:444–448. doi: 10.1096/fj.10-172452
Pauli, A., Rinn, J. L., and Schier, A. F. (2011). Non-coding RNAs as regulators of embryogenesis. Nat. Rev. Genet. 12, 136–149. doi: 10.1038/nrg2904
Ping, G., Xiong, W., Zhang, L., Li, Y., Zhang, Y., and Zhao, Y. (2018). Silencing long noncoding RNA PVT1 inhibits tumorigenesis and cisplatin resistance of colorectal cancer. Am. J. Transl. Res. 10, 138–149.
Res, C. (2011). Correction: long Noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 71, 6320–6326. doi: 10.1158/0008-5472.CAN-11-1021
Shi, Y., Wang, Y., Luan, W., Wang, P., Tao, T., Zhang, J., et al. (2014). Long Non-coding RNA H19 promotes glioma cell invasion by deriving miR-675. PLoS ONE 9:e86295. doi: 10.1371/journal.pone.0086295
Siegel, R., Naishadham, D., and Jemal, A. (2012). Cancer statistics, 2012. Ca A Cancer J Clinicians 63:11. doi: 10.3322/caac.21166
Sun, J., Shi, H., Wang, Z., Zhang, C., Liu, L., Wang, L., et al. (2014). Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Mol. Biosyst. 10, 2074–2081. doi: 10.1039/C3MB70608G
Sun, Y., Zheng, Z. P., Li, H., Zhang, H. Q., and Ma, F. Q. (2016). ANRIL is associated with the survival rate of patients with colorectal cancer, and affects cell migration and invasion in vitro. Mol. Med. Rep. 14, 1714–1720. doi: 10.3892/mmr.2016.5409
Széll, M., Bata-Csörgo, Z., and Kemény, L. (2008). The enigmatic world of mRNA-like ncRNAs: their role in human evolution and in human diseases. Semin. Cancer Biol. 18, 141–148. doi: 10.1016/j.semcancer.2008.01.007
Tewari, K. S., Sill, M. W., Long, H. J., Penson, R. T., Huang, H., Ramondetta, L. M., et al. (2014). Improved survival with bevacizumab in advanced cervical cancer. N. Engl. J. Med. 370, 734–743. doi: 10.1056/NEJMoa1309748
Tsang, W. P., Ng, E. K., Ng, S. S., Jin, H., Yu, J., Sung, J. J., et al. (2010). Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 31, 350–358. doi: 10.1093/carcin/bgp181
van Laarhoven, T., Nabuurs, S. B., and Marchiori, E. (2011). Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 27, 3036–3043. doi: 10.1093/bioinformatics/btr500
Vennin, C., Spruyt, N., Dahmani, F., Julien, S., Bertucci, F., Finetti, P., et al. (2015). H19 non coding RNA-derived miR-675 enhances tumorigenesis and metastasis of breast cancer cells by downregulating c-Cbl and Cbl-b. Oncotarget 6, 29209–29223. doi: 10.18632/oncotarget.4976
Wang, D., Wang, J., Lu, M., Song, F., and Cui, Q. (2010). Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 26, 1644–1650. doi: 10.1093/bioinformatics/btq241
Wang, J., Su, H. K., Zhao, H. F., Chen, Z. P., and To, S. S. T. (2015). Progress in the application of molecular biomarkers in gliomas. Biochem. Biophys. Res. Commun. 465, 1–4. doi: 10.1016/j.bbrc.2015.07.148
Wang, Y., Chen, L., Chen, B., Li, X., Kang, J., Fan, K., et al. (2013). Mammalian ncRNA-disease repository: a global view of ncRNA-mediated disease network. Cell Death Dis. 4:e765. doi: 10.1038/cddis.2013.292
Xu, S. T., Xu, J. H., Zheng, Z. R., Zhao, Q. Q., Zeng, X. S., Cheng, S. X., et al. (2017). Long non-coding RNA ANRIL promotes carcinogenesis via sponging miR-199a in triple-negative breast cancer. Biomed. Pharmacother. 96, 14–21. doi: 10.1016/j.biopha.2017.09.107
Xue, X., Yang, Y. A., Zhang, A., Fong, K., Kim, J., Song, B., et al. (2016). LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene 35, 2746–2755. doi: 10.1038/onc.2015.340
Yang, J. P., Yang, X. J., Xiao, L., and Wang, Y. (2016). Long noncoding RNA PVT1 as a novel serum biomarker for detection of cervical cancer. Eur. Rev. Med. Pharmacol. Sci. 20, 3980–3986.
Yang, Y. R., Zang, S. Z., Zhong, C. L., Li, Y. X., Zhao, S. S., and Feng, X. J. (2014). Increased expression of the lncRNA PVT1 promotes tumorigenesis in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 7, 6929–6935.
Yang, Z., Zhou, L., Wu, L. M., Lai, M. C., Xie, H. Y., Zhang, F., et al. (2011). Overexpression of long Non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann. Surg. Oncol. 18, 1243–1250. doi: 10.1245/s10434-011-1581-y
You, Z. H., Huang, Z. A., Zhu, Z., Yan, G. Y., Li, Z. W., Wen, Z., et al. (2017). PBMDA: a novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 13:e1005455. doi: 10.1371/journal.pcbi.1005455
Zhang, A., Zhao, J. C., Kim, J., Fong, K. W., Yang, Y. A., Chakravarti, D., et al. (2015). LncRNA HOTAIR enhances the androgen-receptor-mediated transcriptional program and drives castration-resistant prostate cancer. Cell Rep. 13, 209–221. doi: 10.1016/j.celrep.2015.08.069
Zhang, D., Sun, G., Zhang, H., Tian, J., and Li, Y. (2016). Long non-coding RNA ANRIL indicates a poor prognosis of cervical cancer and promotes carcinogenesis via PI3K/Akt pathways. Biomed. Pharmacother. 85, 511–516. doi: 10.1016/j.biopha.2016.11.058
Zhang, E., Li, W., Yin, D., Wei, D., Zhu, L., Sun, S., et al. (2016). c-Myc-regulated long non-coding RNA H19 indicates a poor prognosis and affects cell proliferation in non-small-cell lung cancer. Tumor Biol. 37, 4007–4015. doi: 10.1007/s13277-015-4185-5
Zhang, J., Yao, T., Wang, Y., Yu, J., Liu, Y., and Lin, Z. (2016). Long noncoding RNA MEG3 is downregulated in cervical cancer and affects cell proliferation and apoptosis by regulating miR-21. Cancer Biol. Ther. 17, 104–113. doi: 10.1080/15384047.2015.1108496
Zhang, Q., Chen, C. Y., Yedavalli, V. S. R. K., and Jeang, K. T. (2013). NEAT1 Long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. Mbio 4, 00596–00512. doi: 10.1128/mBio.00596-12
Zhang, X., Zhou, Y., Mehta, K. R., Danila, D. C., Scolavino, S., Johnson, S. R., et al. (2003). A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metab. 88, 5119–5126. doi: 10.1210/jc.2003-030222
Zhao, T., Xu, J., Liu, L., Bai, J., Xu, C., Xiao, Y., et al. (2015). Identification of cancer-related lncRNAs through integrating genome, regulome and transcriptome features. Mol. Biosyst. 11, 126–136. doi: 10.1039/c4mb00478g
Zhao, X., Wang, P., Liu, J., Zheng, J., Liu, Y., Chen, J., et al. (2015). Gas5 exerts tumor-suppressive functions in human glioma cells by targeting miR-222. Mol. Ther. 23, 1899–1911. doi: 10.1038/mt.2015.170
Zhu, M., Chen, Q., Liu, X., Sun, Q., Zhao, X., Deng, R., et al. (2014). lncRNA H19/miR-675 axis represses prostate cancer metastasis by targeting TGFBI. FEBS J. 281, 3766–3775. doi: 10.1111/febs.12902
Zhu, Y., Chen, P., Gao, Y., Na, T., Zhang, Y., Cai, J., et al. (2018). MEG3 activated by Vitamin D inhibits colorectal cancer cells proliferation and migration via regulating clusterin. Ebiomedicine 30, 148–157. doi: 10.1016/j.ebiom.2018.03.032
Keywords: disease similarity, lncRNA similarity, path with limited length, Gaussian interaction profile kernel similarity, leave-one-out cross validation, ROC curve
Citation: Xiao X, Zhu W, Liao B, Xu J, Gu C, Ji B, Yao Y, Peng L and Yang J (2018) BPLLDA: Predicting lncRNA-Disease Associations Based on Simple Paths With Limited Lengths in a Heterogeneous Network. Front. Genet. 9:411. doi: 10.3389/fgene.2018.00411
Received: 01 July 2018; Accepted: 05 September 2018;
Published: 16 October 2018.
Edited by:
Tao Zeng, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences (CAS), ChinaReviewed by:
Jianbo Pan, Johns Hopkins Medicine, United StatesXianwen Ren, Peking University, China
Copyright © 2018 Xiao, Zhu, Liao, Xu, Gu, Ji, Yao, Peng and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Liao, ZHJhZ29uYndAMTYzLmNvbQ==
Jialiang Yang, amlhbGlhbmcueWFuZ0Btc3NtLmVkdQ==
†These authors have contributed equally to this work