 Wentao Cai
Wentao Cai Cong Li
Cong Li Shuli Liu
Shuli Liu Chenghao Zhou
Chenghao Zhou Hongwei Yin
Hongwei Yin Jiuzhou Song
Jiuzhou Song Qin Zhang1
Qin Zhang1 Shengli Zhang
Shengli Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 30 July 2018
Sec. Livestock Genomics
Volume 9 - 2018 | https://doi.org/10.3389/fgene.2018.00281
Long non-coding RNAs (lncRNAs) have emerged as a novel class of regulatory molecules involved in various biological processes. However, their role in milk performance is unknown. Here, whole transcriptome RNA sequencing was used to generate the lncRNA transcriptome profiles in mammary tissue samples from 6 Chinese Holstein cows with 3 extremely high and 3 low milk protein percentage phenotypes. In this study, 6,450 lncRNA transcripts were identified through 5 stringent steps and filtration by coding potential. In total, 31 lncRNAs and 18 novel genes were identified to be differentially expressed in high milk protein samples (HP) relative to low milk protein samples (LP), respectively. Differentially expressed lncRNAs were selected to predict target genes through bioinformatics analysis, followed by the integration of differentially expressed mRNA data, gene function, gene ontology (GO) and pathway, genome wide association study (GWAS) and quantitative trait locus (QTL) information, as well as network analysis to further characterize potential interactions. Several lncRNAs were found (such as XLOC_059976) that could be used as candidate markers for milk protein content prediction. This is the first study to perform global expression profiling of lncRNAs and mRNAs related to milk protein traits in dairy cows. These results provide important information and insights into the synthesis of milk proteins, and potential targets for the future improvement of milk quality.
Milk proteins are among the most important nutrients for human, therefore, these proteins could serve as valuable indices to evaluate the quality of milk. The amount and composition of proteins in milk are largely determined by the genetic factors of the cows (Auldist et al., 2004). Although some causal genes and mutations for milk yield and composition have been identified using QTL mapping, candidate gene analysis, GWAS or NGS technologies in dairy cows (Georges et al., 1995; Andersson, 2009; Canovas et al., 2010; Wickramasinghe et al., 2011), the synthesis and secretion of milk proteins involve complex processes, and need to be thoroughly examined. It has been demonstrated that the milk production traits are under strong epigenetic regulation (Singh et al., 2012). Mechanisms of epigenetic gene regulation function through the modulation of chromatin structure, and may either repress or enhance gene expression. It has been reported that milk proteins are in fact influenced by DNA methylation (Nguyen et al., 2014; Liu et al., 2017), histone modifications, such as acetylation, ubiquitination, and phosphorylation (Wang et al., 2017), and microRNAs (Li R. et al., 2016; Wang et al., 2016). However, a new epigenetic component – long non-coding RNAs (lncRNAs), which were recently demonstrated to be critical gene regulators of cellular metabolism (Zhao and Lin, 2015), are yet to be thoroughly examined.
Long non-coding RNAs (lncRNAs), which are at least 200 bases in length, and do not possess protein-coding capabilities (Rinn and Chang, 2012), have received much attention in the past decade. It has been shown that some lncRNAs regulate the expression of genes in close proximity (cis-acting) or at a distance (trans-acting) in the genome via different mechanisms, including the modification of promoter activities by nucleosome repositioning (Rinn et al., 2007; Zhao et al., 2008), histone modification (Pandey et al., 2008; Khalil et al., 2009), DNA methylation (Lai and Shiekhattar, 2014; Merry et al., 2015), activating/gathering/transporting of accessory proteins, epigenetic silencing, and repression (Ponting et al., 2009; Kornienko et al., 2013). Some lncRNAs are believed to be precursor molecules that are processed into small RNAs, while others have been demonstrated to function as intact, long molecules that participate in a range of regulatory roles (Cesana et al., 2011). Increasing evidence supports the notion that lncRNAs are associated with developmental, metabolic and immunological regulation, as well as adaptations and phenotypic variation of complex traits in domestic animals (Qu and Adelson, 2012; Weikard et al., 2013; Zhou et al., 2014; He et al., 2015; Ren et al., 2016; Tong et al., 2017). Recent advances in sequencing technologies have opened a new horizon for the identification and annotation of this class of RNAs in many species (Esteve-Codina et al., 2011; Li T. et al., 2012). Although lncRNAs have emerged as a novel class of regulatory molecules involved in various biological processes, their expression profile and role in the regulation of milk protein synthesis in dairy cattle remains unknown.
In the current study, we performed whole transcriptome ssRNA-seq to examine the mammary tissue transcriptome and identify lncRNAs from Chinese Holstein cows with extremely high and low milk protein levels during peak lactation. Our study seeks to systematically identify lncRNAs potentially involved in milk protein synthesis, reveal expression profiles of lncRNAs in Chinese Holstein mammary glands and provide the resources to effectively explore their functional roles in the improvement of milk quality.
All procedures pertaining to the handling of experimental animals were conducted in accordance with and approved by the Animal Welfare Committee of China Agricultural University (Permit Number: DK996). All efforts were made to minimize discomfort and suffering.
Based on the DHI data, 6 multiparous and healthy, mastitis-free Chinese Holstein cows with 3 extremely high and 3 low phenotypic values for milk protein percentage (high ≥ 3.5% and low ≤ 3.0%) at approximately 60 days postpartum (peak lactation) were selected for the study (Supplementary Table S1). The cows selected in this study were the same with our previous cows in peak lactation (Li C. et al., 2016). The cows at the Beijing Sanyuan Dairy Farm Center (Beijing, China) were maintained in free stall housing, and were fed a total mixed ration (TMR) with ad libitum access to water. Mammary samples were collected from the 6 cows via biopsies, as previously described (Li C. et al., 2016). All tissue samples (∼500 mg) were snap-frozen in liquid nitrogen and stored at -80°C for further testing.
Total RNA was extracted from each mammary tissue sample using TRIzol (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s protocol with an average concentration 1,018.5 ng/μl (Supplementary Table S2). RNA quantity and quality were assessed using the Qubit® RNA Assay Kit in a Qubit® 2.0 Flurometer (Life Technologies, Camarillo, CA, United States) and by 1% agarose gel electrophoresis along with the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, United States), respectively. All 6 samples had an RIN value greater than 7.0 (Supplementary Table S2). Different from previous study (Li C. et al., 2016), libraries were constructed using ribosomal RNA removal methods. Ribosomal RNA of the 6 RNA samples was removed using the Epicentre Ribo-zero® rRNA Removal Kit (Epicentre, United States). Strand-specific sequencing libraries were constructed following a previously described protocol (Borodina et al., 2011). Finally, the libraries were sequenced on an Illumina Hiseq 2500 platform, which generated 125 bp paired-end reads. The sequencing data have been submitted to the NCBI Sequence Read Archive (SRA), and are accessible through the accession number PRJNA416150.
The resulting directional 125 bp paired-end reads were assessed for quality using FastQC. Clean reads were obtained by removing contaminating adapter molecules, reads containing poly-N, and low-quality reads (parameter -q 36 -p 90) in the raw data using the Fastx_toolkit (0.0.13). All downstream analyses were based on the high quality clean data. Sequencing reads in the FASTQ format were aligned to the bovine genome (UMD 3.1), and novel splice junctions were automatically determined using TopHat2 (version 1.4.1) (Kim et al., 2013). The default parameters were used for the analysis, except for the “-G” option together with Gene Transfer Format (GTF) file of Ensembl gene annotation (UMD3.1.85) and “–library-type = fr-firststrand”. The sampled alignment data were then fed to Cufflinks, Scripture, Stringtie and Transcomb to assemble aligned reads into transcripts (Guttman et al., 2010; Trapnell et al., 2012; Pertea et al., 2015; Liu et al., 2016). Cufflinks (version 2.02) was run with “min-frags-per-transfrag = 0”, “–library-type = fr-firststrand” and “–mask-file = ncRNA.gtf”), the ncRNA.gtf contains all the known rRNA, tRNA, snRNA and snoRNA annotations in the bovine genome. Scripture (beta2) was run with default parameters (with an omission of the “-pairedEnd” option), StringTie (version 1.0.1) was run with the parameters (-f 0.01 -c 0.01). Transcomb (V1.0) was run using the default parameters, except for “-s first”, “-l 200” and “-e 50”.
All transcripts identified using Cufflinks, Scripture, Stringtie and Transcomb were independently matched and guided by the Ensembl gene models of Cuffcompare. The transcripts with the same start, end position and exon–intron boundary, which were supported by at least two assembly programs or occurred in at least two samples, were extracted as stringent transcripts. The novel transcripts were then filtered and assembled in order to obtain putative transcripts, as previously reported (Cabili et al., 2011; Pauli et al., 2012), according to the following steps:
Step (1) Transcripts that were likely to be assembly artifacts or PCR run-on fragments according to class code annotated by Cuffcompare were removed. Among the different classes, only those annotated by ‘i’, ‘u’ and ‘x’ were retained, which represent novel intergenic, intronic, and cis-antisense transcripts, respectively. Transcripts with class code “=” annotated by Cuffcompare were considered as known genes.
Step (2) To avoid incomplete assemble and too many splicing events, transcripts with length ≥ 200 nt, exon ≥ 2 were retained.
Step (3) FPKM ≥ 0.3 were retained. Extremely low expression was generally considered to be transcriptional noise.
Step (4) Maximum ORF lengths of less than 120 amino acids (360 nt) were obtained by TransDecoder (3.0.1)1.
Step (5) Transcripts with predicted protein-coding potential were removed (protein-coding potential criteria: CPC score > 0, PLEK score > 0, and CNCI score > 0).
Step (6) All transcripts were translated into amino acid sequences through all three reading frames to remove transcripts that contain known protein domains. HMMER (Finn et al., 2011) was used to identify any known protein domain by searching against the Pfam database (Pfam 30.0) (Finn et al., 2008). Transcripts with significant Pfam hits were excluded.
Finally, those without coding potential made up the candidate sets of lncRNAs. In addition, transcripts with long ORF lengths (more than 120 amino acids) or having protein-coding potential were considered as novel protein-coding genes.
PhyloFit was applied to compute phylogenetic models for conserved and non-conserved regions among species. The model and HMM transition parameters were entered into phastCons in order to compute a set of conservation scores of lncRNAs and coding genes (Siepel et al., 2005). PhastCons scores were downloaded from the UCSC database. To assign a conservation score to a transcript, the median phastCons score for the concatenated exonic regions of each transcript model was calculated. The conservation score was compared among the protein-coding sequences, lncRNAs, and 5000 random genomic sequences. To generate a set of random sequences, the non-gap genome region was initially taken as the full set, and coding exons from all known and computational gene models were excluded. Next, the sequences with lengths identical to those of the lncRNAs under investigation were randomly drawn. The lncRNAs were annotated to the NONCODE database using BLASTN with p-value < 1e-06 (McGinnis and Madden, 2004).
The expression levels of known genes, novel genes, and lncRNAs were calculated in fragments per kilo-base of exon per 106 mapped fragments (FPKM) using Cuffdiff, which provides statistical routines for determining differentially expressed known genes, novel genes and lncRNAs through the use of a model based on the negative binomial distribution (Trapnell et al., 2012). Genes or lncRNAs with a Benjamini-Hochberg (BH) adjusted p-value < 0.05 were designated as differentially expressed. Hierarchical clustering was performed to visualize the expression patterns of differentially expressed lncRNAs among the samples.
Gene ontology enrichment analysis of DEGs or lncRNA target genes were performed in DAVID database (Huang et al., 2008). BH method was used to adjust significant p-values. GO terms with p-value < 0.05 were considered to be significantly enriched. The Ingenuity Pathway Analysis (IPA) with fisher exact test method was used to test the statistical pathways enrichment of lncRNA correlated genes (Krämer et al., 2013). Pathways with -log2(p-value) > 1.3 were considered to be significantly enriched.
To explore whether some lncRNAs were precursors of miRNAs, miRNAs published in miRBase (Release 21)2 were aligned to the sequences of lncRNAs in order to find the known miRNA precursors using BLASTN with 100% match. Prediction of secondary structures for the lncRNA transcripts was made using the Vienna RNA package in the RNAfold program. The prediction software miRanda was applied to predict the interaction between miRNAs and lncRNAs with score > 160, energy < -15.
The cis role of lncRNAs was defined as those exerting effects on neighboring target genes (Guil and Esteller, 2012). Coding genes located in both 10 and 100 Kb upstream and downstream of lncRNAs were checked using in-house Perl scripts. The trans-acting correlation of lncRNA and mRNA was used to identify each other through the expression level (Derrien et al., 2012). The expressed correlations between lncRNAs and coding genes were calculated using the Pearson method with p-value < 0.05. The QTL information of milk protein traits were extracted from the AnimalQTLdb.3 The 972 significant SNPs associated with milk protein traits were collected from 12 previous GWAS studies (Supplementary Table S3). The functional analysis was conducted based on their positions on chromosome using our in-house Perl scripts. Circos and Cytoscape softwares were used to plot integrated results, and candidate lncRNA-mRNA network, respectively (Shannon et al., 2003; Krzywinski et al., 2009).
RNA samples, the same sources and concentrations with library preparation (Supplementary Table S2), were reverse-transcribed to cDNA using the PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa) according to the manufacturer’s instructions. Primers for qRT-PCR were designed using Primer 5.0, their specificity and complementarity were assessed using NCBI BLAST algorithm. QRT-PCR was run in triplicate using the LightCycler® 480 SYBR Green I Master Kit (Roche). A reaction volume of 15 μL containing 7.5 μL of SYBR Green PCR Master Mix, 0.75 μL of forward and reverse primers (10 mM), 2 μL of cDNA, and 4.75 μL of double-distilled water. The qRT-PCR reaction conditions were: denaturation at 95°C for 10 min, 45 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 10 s. A final extension of 72°C for 6 min was included. The results were normalized to GAPDH and MARVELD1 expression to obtain ΔCt values (Saremi et al., 2012; Li C. et al., 2016). Fold changes in expression were calculated using the 2-ΔΔCt method. Differences in gene expression levels between HP samples and LP samples were analyzed using a Student’s t-test, with p-value < 0.05 considered to be statistically significant.
After quality trimming and adaptor removal of the Illumina reads, an average of 128 million clean reads (range: 120 – 137 million) for each sample were obtained by ssRNA-seq (Supplementary Table S4). An average of 91.55% (range: 91.10–92.02%) of the reads were mapped to the bovine genome (Ensembl UMD3.1) using Tophat2. Of these, 83.22% (range: 82.01–84.41%) were uniquely mapped reads and 8.33% (range: 7.17–9.44%) were multi-mapped reads (Supplementary Table S4). The proportion of reads aligning to mRNAs, miscRNAs, ncRNAs, precursor RNAs, pseudogenes, rRNAs and tRNAs are presented in Supplementary Figure S1. It was observed that most reads were matched with mRNAs (51% - 58%), whereas between 33 and 39% of the reads were mapped outside of annotated loci, which potentially harbored the promising lncRNA transcripts.
To identify lncRNAs related to milk protein synthesis, we employed a computational approach using different filter criteria (Figure 1A). First, 5,291,989, 589,031, 2,320,598, and 572,551 transcripts in the 6 samples were obtained using Cufflinks, Scripture, StringTie and Transcomb, respectively, which includes all possible RNA types like protein-coding genes, novel genes, lncRNAs and pseudogenes (Supplementary Figure S2). After merging the 4 assemblers, a total of 1,926,647 transcripts were obtained that were detected by at least 2 assemblers in a given sample or were identified in at least 2 individual samples by the same assembler (details in Materials and Methods). Of these, 1,219,607 transcripts with class codes ‘i’, ‘u’, ‘x’, representing ilncRNAs, lincRNAs, and lncNATs were retained, respectively. Next, transcripts that were shorter than 200 bp in total length and possessed only one single exon, with a FPKM < 0.3, as well as those predicted to harbor maximum lengths of ORFs longer than 120 amino acids were filtered. Transcripts with a CPC score > 0, a CNCI score > 0, a PLEK score > 0 or overlapping with the Pfam database were then excluded. Finally, a total of 6,450 lncRNA transcripts in 5,256 lncRNA loci were identified among the 6 Chinese Holstein cows through our computational pipeline (Supplementary Table S5).
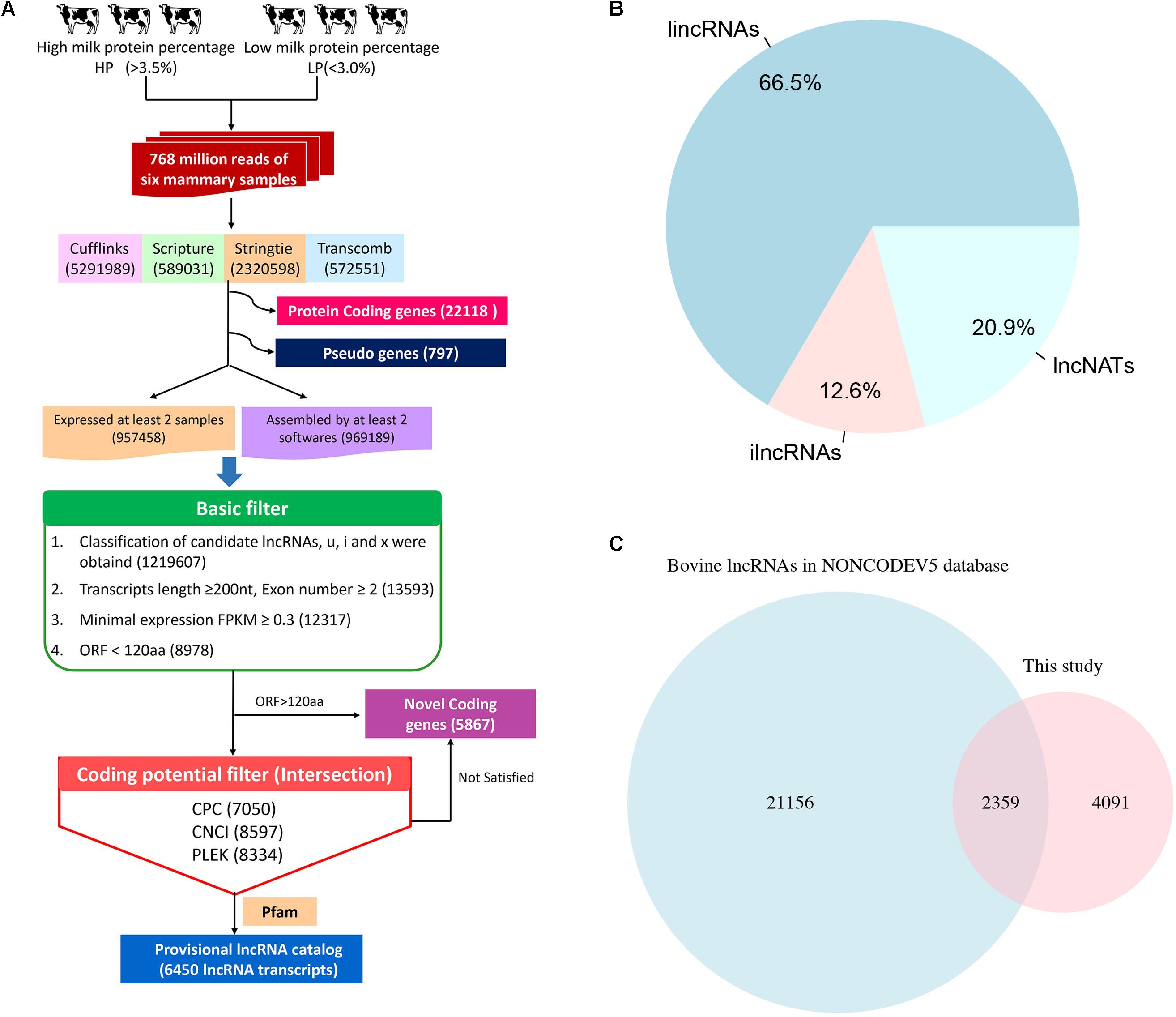
FIGURE 1. The computational pipeline used to identify lncRNA genes from RNA-seq data. (A) The pipeline of lncRNA identification. (B) The category of lncRNAs. (C) The result of lncRNAs overlapping with NONCODE Database.
Among the 6,450 identified lncRNA transcripts, 4,292 and 810 were lincRNAs and ilncRNAs, respectively, while 1,348 lncRNAs flanked a protein-coding gene in a divergent orientation (lncNATs) (Figure 1B). The genomic coordinates of the identified lncRNA transcripts are provided in Supplementary Table S4. Using the BLAST algorithm, a total of 2,359 lncRNAs were found in bovine NONCODE v5.0 database (Figure 1C). In addition, we detected 5,867 novel mRNA transcripts in 4093 loci without any Ensemble annotation (Supplementary Table S6).
The characteristics of expression, length, exon number, structure and conservation of sequence of the newly obtained 6,450 lncRNA transcripts, 5,867 novel mRNA transcripts, 22,118 known mRNAs and 797 pseudogenes are shown in Figure 2. Most of the detected lncRNAs were expressed at low levels (Figure 2A). The size of lncRNAs was notably smaller than that of protein-coding transcripts, novel genes and pseudogenes (Figures 2B,C). The length of the ORFs of protein-coding RNAs (543.6 AAs) was significantly longer than those observed in lncRNAs (77.1 AAs), novel genes (153.6 AAs) and pseudogenes (266.8 AAs) (Figure 2D). However, approximately 4% of ORF lengths of protein-coding RNAs were observed to be shorter than 120 AAs. LncRNAs and pseudogenes exhibited lower GC contents with 47.9 and 49.2%, respectively, as compared to that of protein-coding RNAs (52.5%) and novel genes (50.2%) (Figure 2E). PhastCons score demonstrated that lncRNAs were less conserved than protein-coding regions but were much more conserved than random size-matched intergenic regions (Figure 2F). In addition, a search of the sequences from 6,450 lncRNAs against the lncRNAs from 5 other mammalian species (Human, Mouse, Rat, Pig, and Chicken) in the NONCODE v5.0 database was conducted. The conservation of lncRNA sequences across the species was limited (Supplementary Table S7).
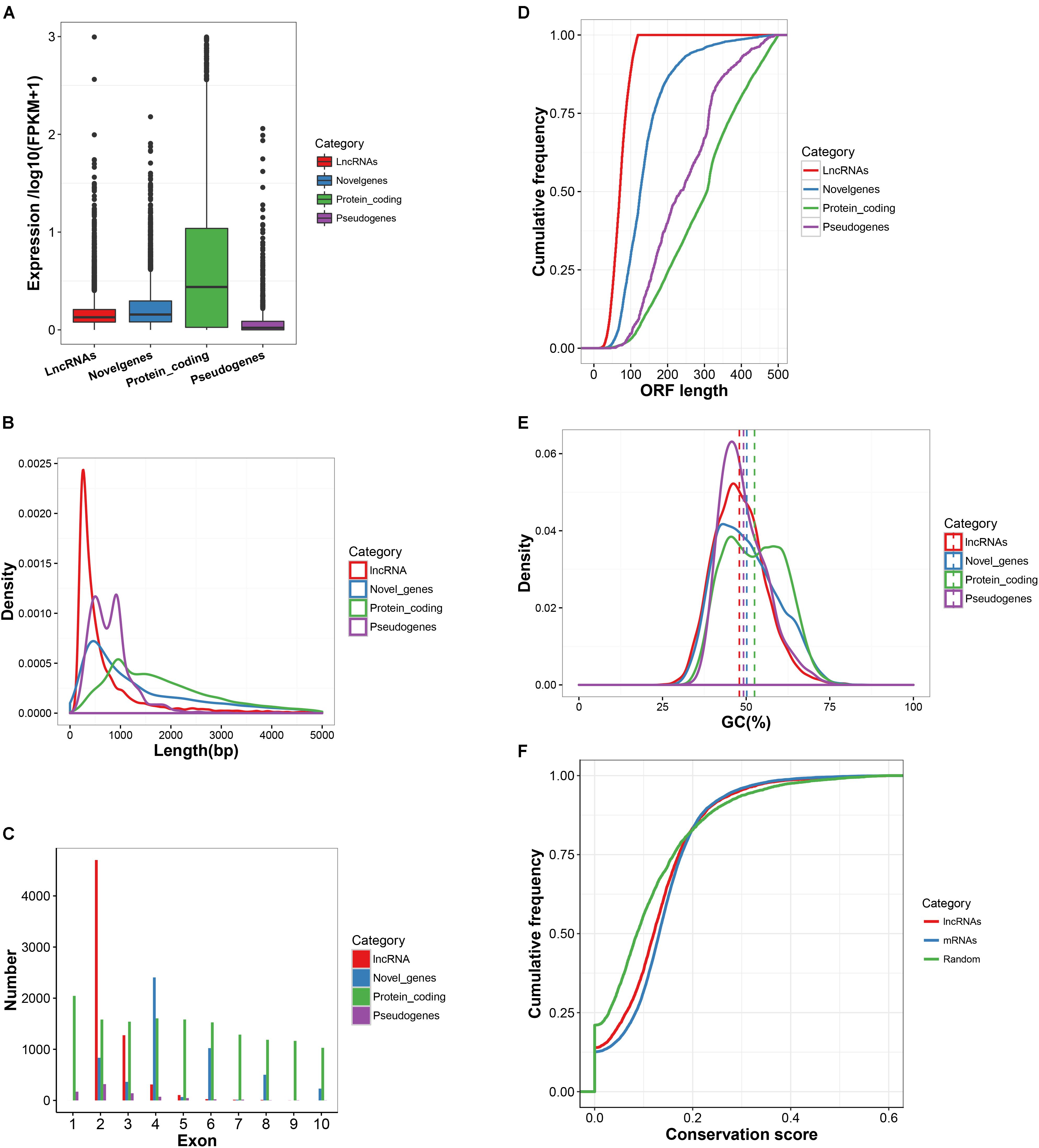
FIGURE 2. Basic features of lncRNAs. (A) Expression level of lncRNA transcripts, marked by red box. (B) Length distribution of lncRNA transcripts, marked by red line in the Figure. (C) The number of exons for lncRNA transcripts. As marked by a red bar line in the Figure, on the average there are 2.39 exons per transcript. (D) The ORF length of lncRNA transcripts compared to protein-coding genes, novel genes and pseudo genes. (E) The GC content of lncRNAs transcripts compared to protein-coding genes, novel genes and pseudo genes. (F) LncRNA sequence conservation measured by phastCons scores. lncRNAs show intermediate sequence conservation as compared to random region and protein-coding genes.
A total of 31 lncRNA genes were differentially expressed in HP relative to LP, including 15 up-regulated and 16 down-regulated lncRNAs (Figure 3A and Supplementary Table S8a, q-value < 0.05). Meanwhile, 18 significantly dysregulated novel genes were identified, including 6 up-regulated and 12 down-regulated novel genes (Figure 3A and Supplementary Table S8b, q-value < 0.05). Among the 31 differentially expressed lncRNAs, 8 lncRNAs and 10 lncRNAs were only expressed in either the HP or LP group, respectively (Table 1). The other 13 lncRNAs, with 5 being up-regulated and 8 down-regulated are listed in Table 2. Cluster analysis of differentially expressed lncRNAs are depicted in a heatmap (Figure 3B).
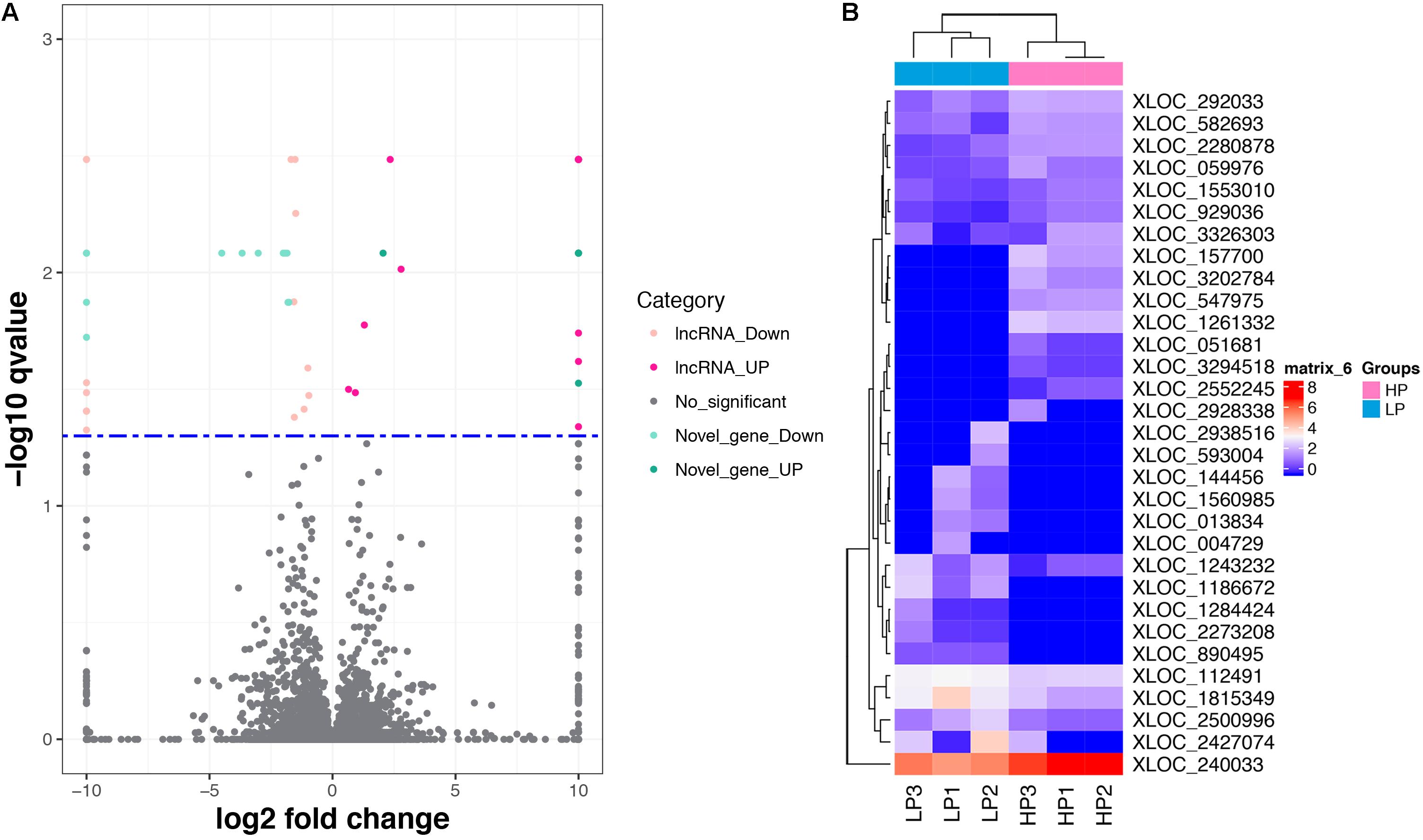
FIGURE 3. The differentially expressed lncRNAs and mRNAs. (A) Volcano plot displaying differentially expressed lncRNAs and mRNAs within two different comparison groups. The red and orange dots represent the differentially expressed lncRNAs (q < 0.05); the light blue and blue dots represent the differential expressed mRNAs (q < 0.05); the gray dots represent transcripts whose expression levels did not reach statistical significance (q > 0.05). (B) Cluster analysis of differentially expressed lncRNAs.
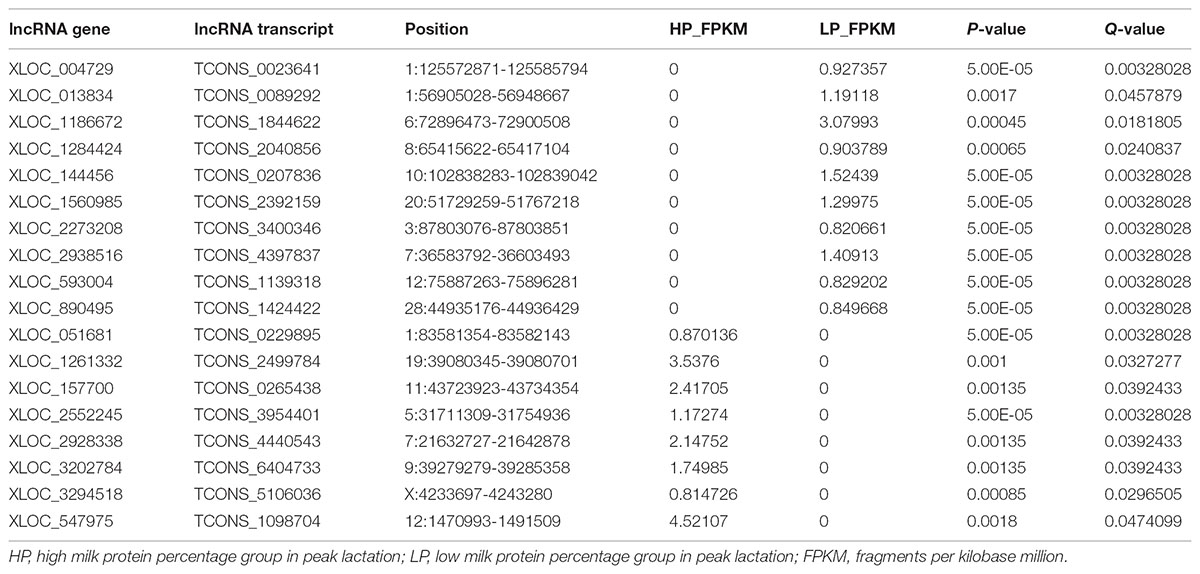
TABLE 1. The differential lncRNAs specifically expressed in HP group or LP group.
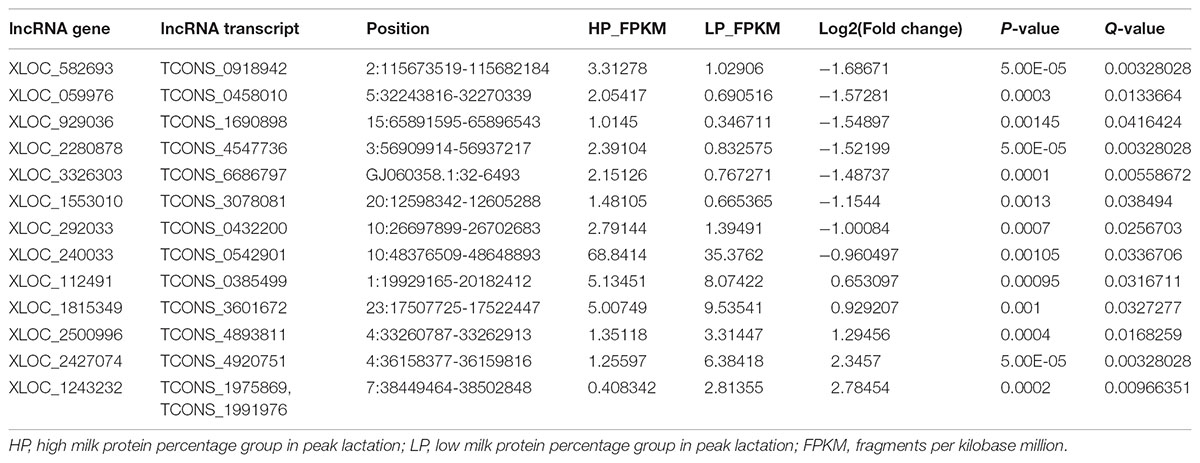
TABLE 2. The differential lncRNAs expressed in both HP and LP group.
To validate the RNA-seq results, 10 differentially expressed mRNAs (ANXA2, TRIB3, ATF4, IDH1, LARP4B, GALE, HSPA8, ERBB2, CYP1A1 and ENPP5) and 6 lncRNAs (XLOC_582693, XLOC_0599776, XLOC_929036, XLOC_2280878, XLOC_1815349 and XLOC_2500996) were randomly selected for qRT-PCR analysis. In Figure 4, the relative fold changes in expression detected by qRT-PCR are presented, and were found to be consistent with the RNA-seq data (R = 0.931 and 0.873). This indicated that our transcript identification and abundance estimation were highly reliable.
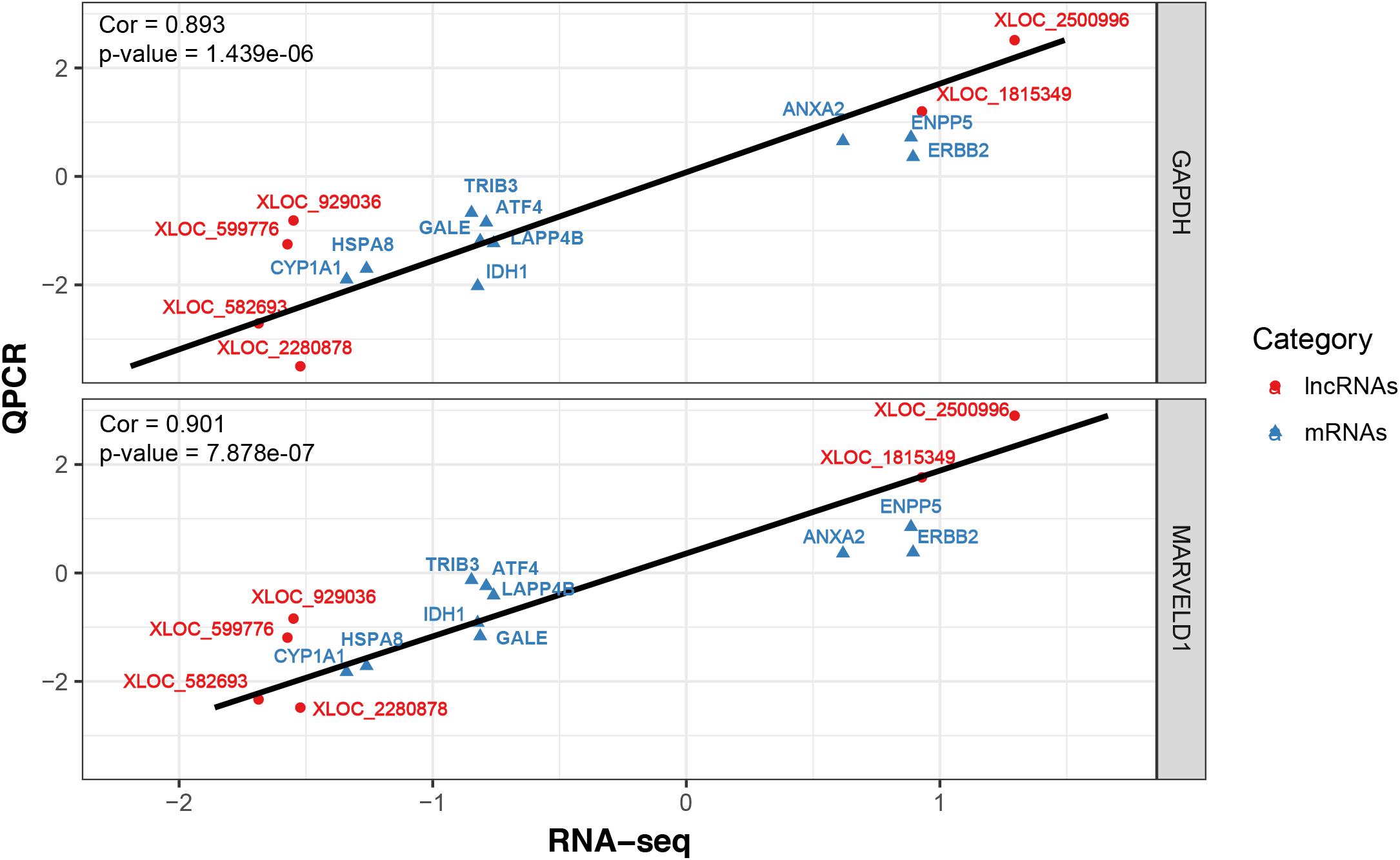
FIGURE 4. Correlations of gene expression level of 10 differentially expressed genes and 6 lncRNAs between high and low milk protein percentage using RNA-Seq and qRT-PCR. The x- and y-axis show the log2 (ratio of mRNA levels) measured by RNA-seq and qRT-PCR, respectively. GAPDH and MARVELD1 gene were used as housekeeping internal control. The red and blue dots represent the differentially expressed genes and lncRNAs, respectively.
To determine whether lncRNAs are in fact precursors of miRNAs, we compared the lncRNA sequences and miRNA sequences that obtained from miRbase, and found that 13 lncRNAs harbored 8 complete miRNA precursors (Supplementary Table S9). Prediction of the secondary structures of lncRNA transcripts indicated that some lncRNAs contained a stable hairpin structure for miRNA precursors. For example, XLOC_1223728 harbored bta-mir-2887-2 (Supplementary Figure S3). To investigate whether the identified lncRNAs were targeted by miRNAs, we analyzed the 6,450 lncRNA transcripts using miRanda. A total of 4,972 lncRNA transcripts were predicted to be targeted by 788 bovine miRNAs (Supplementary Table S10a). Of these, 206 lncRNAs were targeted by miR-15a, miR-486, miR-135, miR-101a, miR-152 and miR-139, which were reported to involve in milk protein synthesis (Supplementary Table S10b). Interestingly, one differentially expressed lncRNA (XLOC_059976) was predicted to be targeted by miR-139 and miR-152, which implied XLOC_059976 could act as a regulatory factor for the process of milk protein synthesis.
To understand the possible biological roles of the lncRNAs in bovine milk protein synthesis, we investigated co-expression patterns of lncRNA genes and protein-coding genes in these 6 mammary gland samples. Trans-acting correlations of expression between lncRNAs and protein-coding genes were examined first. We found 5,251 lncRNAs were significantly correlated with 18,227 mRNAs (p-value < 0.05), including 31 differentially expressed lncRNAs that significantly correlated with 11,161 mRNAs (p-value < 0.05, Supplementary Table S11).
A search for protein-coding genes within 10 kb upstream/downstream and 100 k upstream/downstream of the lncRNAs were conducted. It was revealed that 3,369 protein-coding genes resided within a range of 10 kb of 3,065 lncRNAs, as well 9,656 protein-coding genes resided within a range of 100 kb of 4,535 lncRNAs (Supplementary Table S12a). Of particular interest is the observation that 7 lncRNA transcripts (TCONS_4241670, TCONS_0484807, TCONS_0484809, TCONS_1820871, TCONS_1859003, TCONS_1845219, TCONS_5606159) were detected in close proximity to casein genes such as CSN1S1, CSN1S2, CSN2 and CSN3. Furthermore, TCONS_0484809 (XLOC_061135), an intronic lncRNA, is located in the first intron of the CSN3 gene, indicating that milk protein synthesis may be regulated by the action of lncRNAs on neighboring protein-coding genes (Supplementary Figure S4).
Interestingly, we found the expression in both 10 and 100 kb upstream/downstream genes of lncRNAs were observed to be higher than random protein genes. Moreover, the closer the neighboring genes were to lncRNAs, the higher the co-expression would be (10k > 100k, Supplementary Figure S5). Within the 100 kb upstream and downstream of lncRNAs, 979 neighboring protein-coding genes were detected to be significantly correlated with 867 lncRNAs (Supplementary Table S12b). GO analysis demonstrated these 979 neighboring genes were involved in regulation of transcription, mammary gland branching involved in thelarche, negative regulation of transcription from RNA polymerase II promoter, positive regulation of tyrosine phosphorylation of Stat5 protein and peptide catabolic process (Supplementary Table S13a). IPA showed that these target genes of lncRNAs were significantly enriched in 141 pathways, many pathways were associated with the process of protein synthesis, such as EIF2 signaling, JAK/Stat signaling, GnRH signaling, PRL signaling, mTOR signaling and growth hormone signaling (Supplementary Table S13b). In addition, we observed seven differentially expressed protein-coding genes were in close proximity of 5 differentially expressed lncRNA genes. In summary, 6 lncRNA-mRNA gene pairs were regulated in the same direction, including 5 down-down pairs and 1 up-up pair, and 1 pair inversely. Three target genes (ENSBTAG00000032428, CCDC184 and ASB8) for XLOC_059976 were regulated in the same direction, and one target gene (PRR15L) for XLOC_1261332 was regulated inversely (Supplementary Table S12c).
To better understand the relationship between lncRNAs and milk protein traits, we selectively analyzed the 2,868 lncRNA-mRNA pairs in which both lncRNAs and their neighboring or expression correlated genes were differentially expressed between HP and LP groups. At the same time, the selected target genes should be associated with milk protein metabolism based on reported gene function or their GO and pathway results, and the differentially expressed lncRNAs and their target DEGs should be closed to milk protein QTLs or 972 milk protein SNPs from GWAS studies (Supplementary Table S3). The integrated plot is presented in Figure 5. It was observed that the majority of QTLs and significant SNPs associated with milk proteins are located in chromosomes 6 and 20. Several differentially expressed lncRNAs and mRNAs were also detected in these regions. According to the integrated study, 30 lncRNAs potentially regulated 34 genes that were involved in milk protein synthesis (Supplementary Table S14). For example, IGFBP2, a significantly dysregulated milk protein gene (Li C. et al., 2016), the expression of IGFBP2 was significantly correlated with XLOC_1186672, XLOC_1284424, XLOC_1243232, and XLOC_2273208 simultaneously. EIF2S2, significantly correlated with XLOC_929036, initiated mRNA translation and directly induced protein synthesis. Additional genes are listed in Table 3. In addition, we believed that we were able to account for all known target genes involved in milk protein synthesis through various pathways (Figure 6).
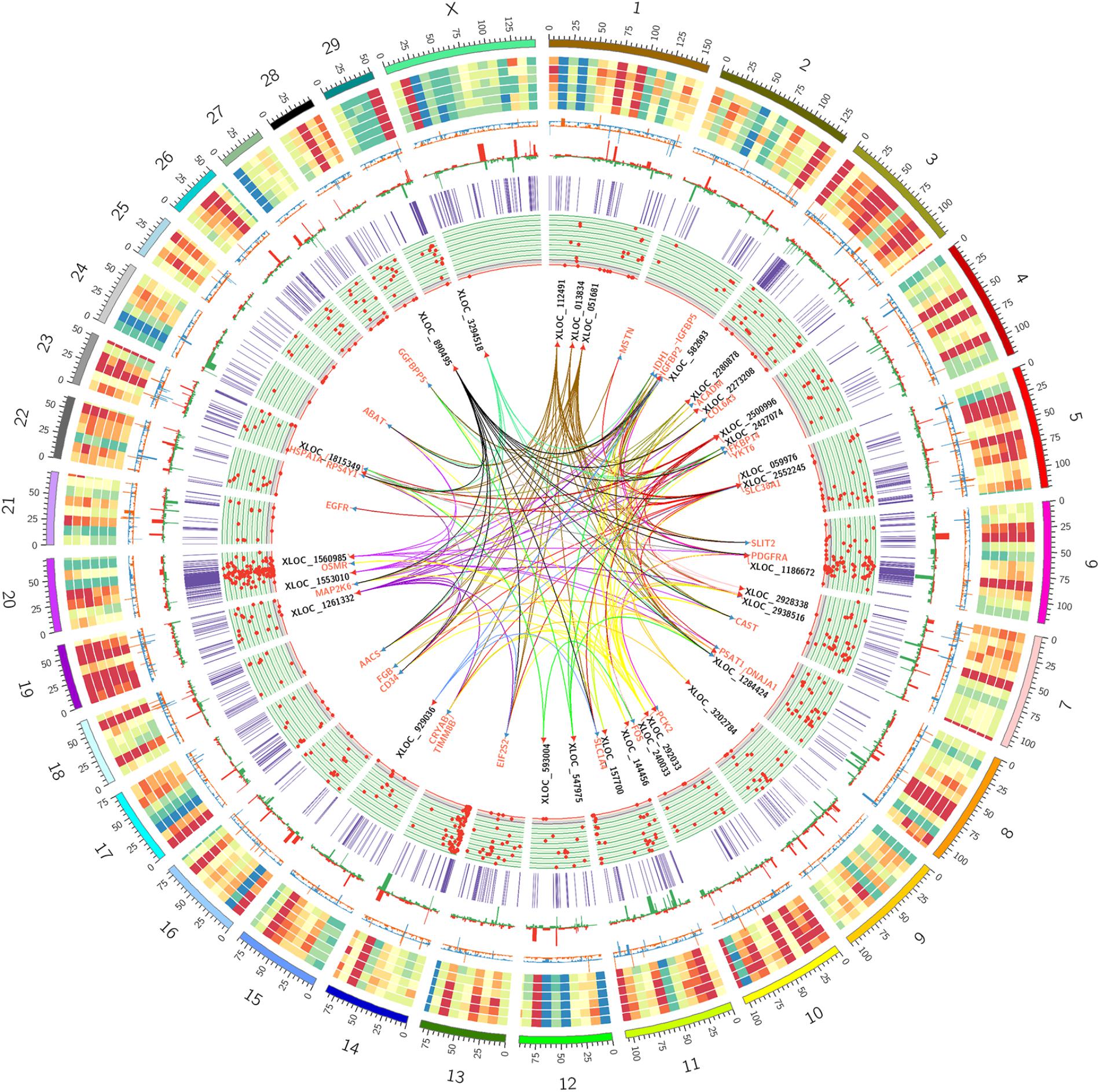
FIGURE 5. Circos plot of the RNA-seq data, QTL databases and GWAS data. From outside to inside: chromosome number, reads coverage of six samples (three LP samples and three HP samples), mRNAs relative change between HP and LP (orange represents upregulation, blue represents downregulation), lncRNAs relative change between HP and LP (red represents upregulation, green represents downregulation), the location of milk protein QTLs from animal QTL database, the location of significant SNPs on milk protein traits from several previous research, the names of promising lncRNAs and mRNAs pairs associate with milk protein and the cis/trans target correlation between promising lncRNAs and mRNAs associate with milk protein.
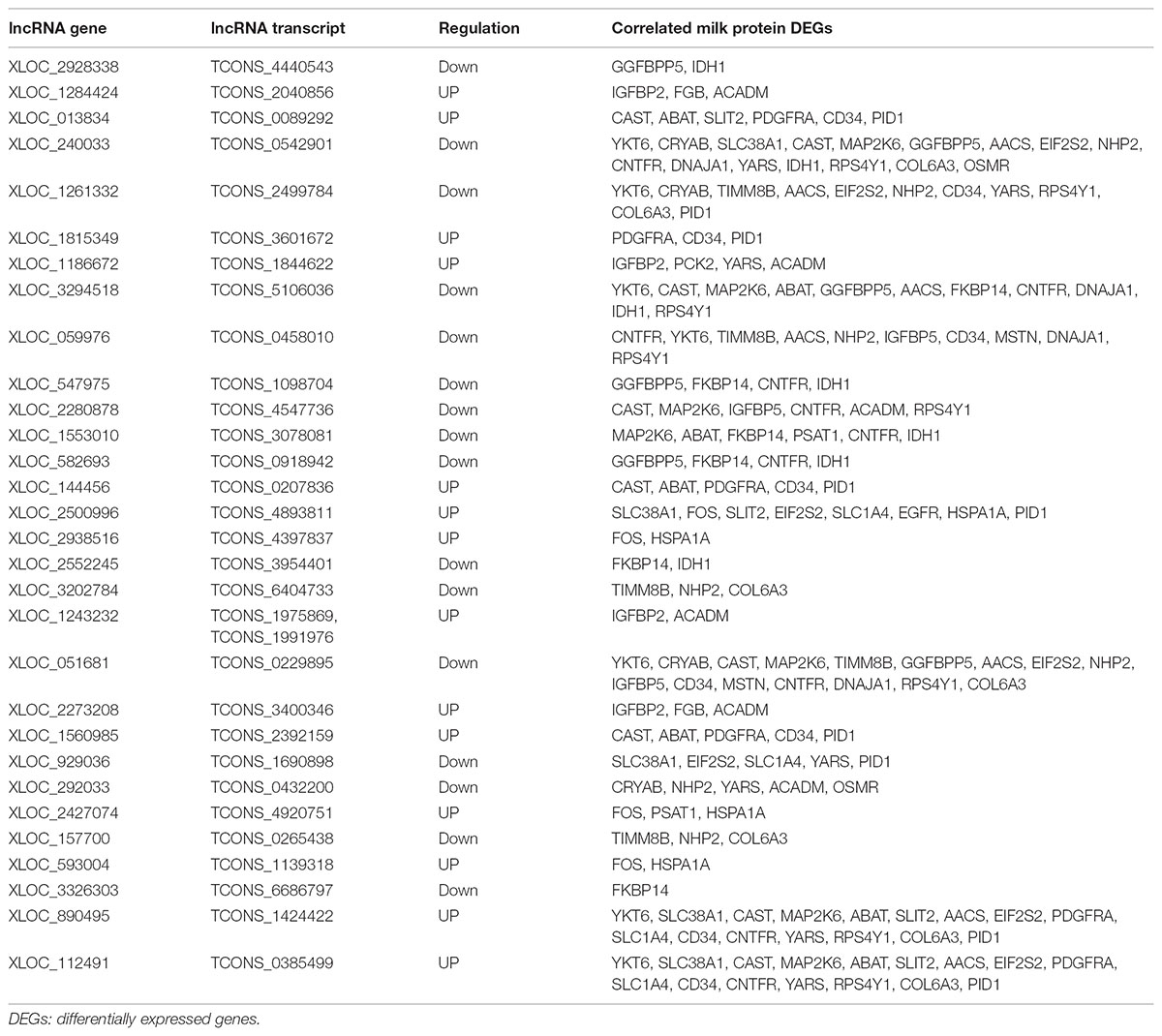
TABLE 3. The differentially expressed lncRNAs with their potential target genes related to milk protein synthesis.
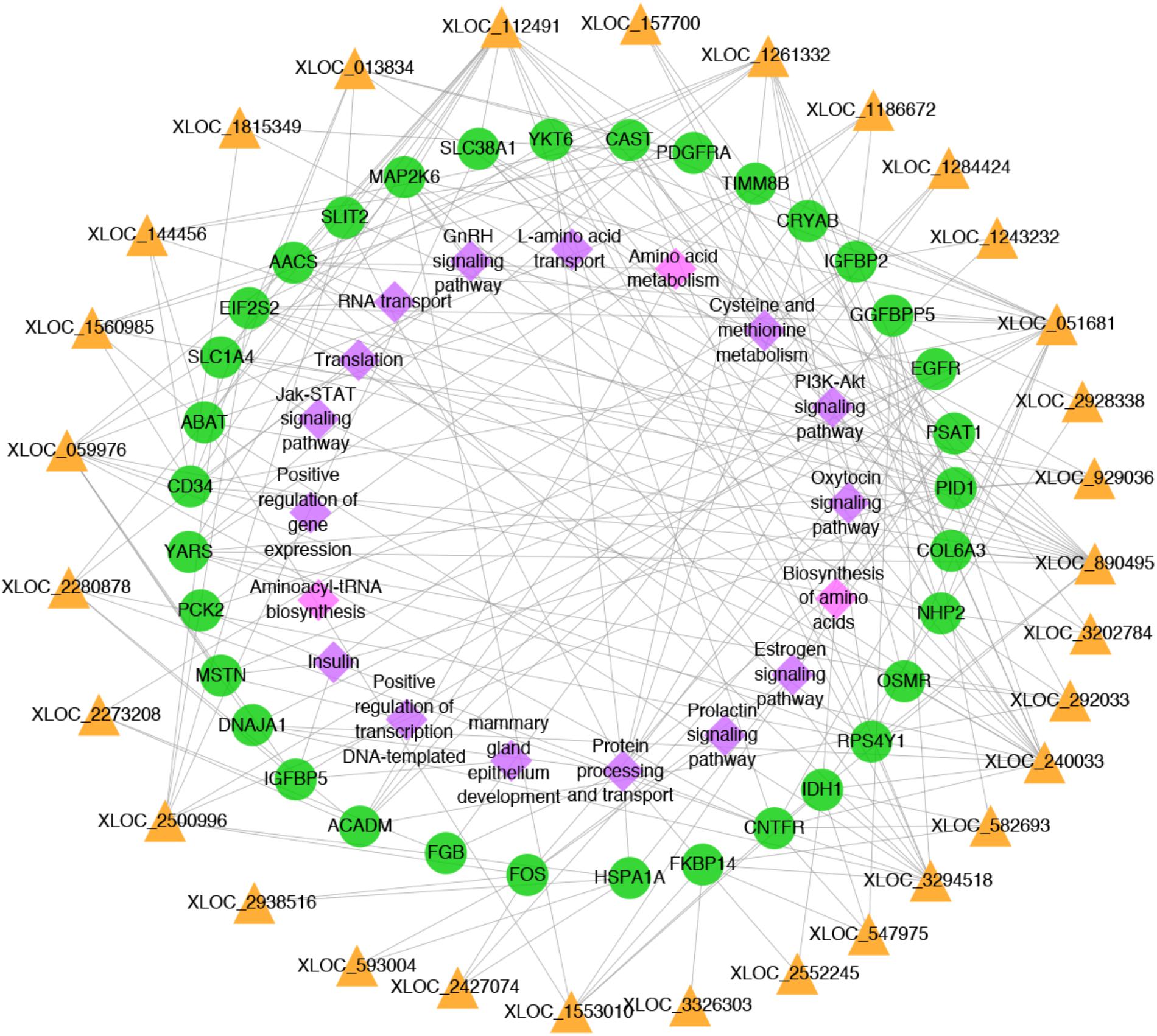
FIGURE 6. Network plot of candidate lncRNAs, mRNAs and pathways. The yellow triangles, green circles and pink diamonds represent lncRNAs, mRNAs and pathways, respectively.
Notably, lncRNA (XLOC_059976) had a strong positive correlation (r2= 0.947) with the expression of CNTFR, which was identified as a candidate gene for milk protein traits in our previous study (Li C. et al., 2016). To confirm whether XLOC_059976 has coding potential, a search for a Kozak consensus sequence was conducted. However, the sequence (gcc)gccRccAUGG was not observed in XLOC_059976, indicating that there was no efficient and reliable Kozak sequence for initiation of translation (Kozak, 1986). Interestingly, a specific interaction between XLOC_059976 and the upstream region of CNTFR was found using the ‘LncTar’ algorithm, which indicated XLOC_059976 could bind to an upstream site of CNTFR (Supplementary Figure S6). Validation of the result by qRT-PCR revealed that both XLOC_059976 and CNTFR were expressed at high levels in the HP group (Supplementary Figure S7). Therefore, it is possible that XLOC_059976 may act as a regulatory module by enhancing the expression of CNTFR and affecting the secretion of milk proteins.
The mammary gland is the most important organ for the synthesis and secretion of milk and milk proteins. Milk protein synthesis is influenced by thousands of molecules, including lncRNAs. Until now, little is known about the bovine lncRNA transcriptome in mammary gland. In this study, we conducted a preliminary investigation of lncRNA expression profiles in mammary glands of Chinese Holstein cows to assess the potential regulators of milk protein during the lactation period. Consequently, the present work provides an important resource of lncRNAs in mammary gland for future studies.
A total of 6,450 multiple-exon lncRNA transcripts were identified corresponding to 5,256 lncRNA genes, including 2,359 known and 4,091 novel lncRNAs transcripts from six mammary samples of Chinese Holstein cows. Most of the detected lncRNAs were expressed at low levels, which implies that lncRNAs and mRNAs have many differences in their biogenesis, processing, stability and spatial-temporal expression patterns (Zhou et al., 2014). The sizes of lncRNAs are smaller than mRNAs probably due to incomplete assembly. RNAs lack an ORF of 360 nt or longer have been classified as putative non-coding RNAs (Dinger et al., 2008). However, 4% of the ORF lengths of mRNAs are observed to be shorter than 120 AAs, which suggest that these short mRNAs may be much more prevalent than previously thought. The low GC content of lncRNAs indicate that they might contain fewer stable base-paired structures, making their primary sequence more accessible for interacting with cellular factors (Niazi and Valadkhan, 2012). PhastCons scores and NONCODE database searches demonstrate that lncRNAs have a less sequence conservation compared to protein-coding genes, although they are much more conserved than random sequences. This moderate conservation for lncRNAs will provide the central argument for lncRNA functionality (Guttman et al., 2009). In general, compared to protein-coding genes, the lncRNAs identified in the present study exhibit significantly lower expression levels, fewer exons, shorter transcripts and ORF lengths, and lower sequence conservation than mRNAs, which is consistent with studies in other species (Cabili et al., 2011; Pauli et al., 2012; Young et al., 2012).
lncRNAs may exert their regulatory functions by producing or interacting with small RNAs. For example, lncRNA-H19 hosts miR-675 in its first exon (Cai and Cullen, 2007; Keniry et al., 2012). Our study revealed that 13 lncRNAs contained 8 miRNA precursors, indicating that some lncRNAs could be processed into miRNAs, at least in bovine. Although only 8 miRNAs precursors were identified by our examination, lncRNAs might be an important resource for the identification of novel miRNAs. Whether lncRNAs exert functions by themselves or as precursors of miRNAs is required to be further explored. The target prediction supported a functional network between lncRNAs and miRNAs, and identifying well-established miRNAs that bind to lncRNAs may help infer the functions of lncRNAs. It has been reported that some miRNA may act as important regulators in the process of milk protein synthesis, such as miR-15a (Li H.M. et al., 2012), miR-486 (Li et al., 2015), miR-135 (Ji et al., 2015), miR-101a (Tanaka et al., 2009), miR-152 (Wang et al., 2014), and miR-139 (Cui et al., 2017). Here, 206 lncRNAs were identified having target sites with above miRNAs, implying these lncRNAs may involve in milk protein synthesis. miR-152 can reduce global DNA methylation and the activity of DNMT to reactivate the lactation signal transduction genes AKT and PPARγ (Wang et al., 2014). miR-139 can suppress β-casein synthesis and proliferation in bovine mammary epithelial cells by targeting the GHR and IGF1R signaling pathways (Cui et al., 2017). XLOC_059976 was predicted to be targeted by both miR-152 and miR-139, implying it might be involved in milk protein metabolism, although further specific validations are required to be confirmed.
Recent studies have indicated that some lncRNAs may act in trans and cis to regulate the expression of genes (Orom et al., 2010; Cabili et al., 2011; Luo et al., 2013). In this study, we found 31 differentially expressed lncRNAs significantly correlated with 11,161 mRNAs, indicating these lncRNAs may participate in various biological processes in mammary gland. A strong correlation was detected between lncRNAs and their neighboring genes. Taken together, these findings suggest that several lncRNAs may act in cis to regulate their neighboring gene expression. After merging the trans-acting correlation and neighbor cis results, we found 867 lncRNAs expression were significantly correlated with their 979 neighboring genes. Notably, GO functional annotation of trans-acting correlation and cis results show that the potential target genes of lncRNAs are enriched in transcription regulation, mammary gland development, tyrosine phosphorylation of Stat5 protein and peptide catabolic process. All these terms are related to milk protein synthesis. The important enriched pathways including EIF2 signaling, JAK/Stat signaling, GnRH signaling, PRL signaling, mTOR signaling and growth hormone signaling, are involved in milk protein synthesis. Overall, lncRNAs may affect the synthesis of milk proteins through regulating protein-coding genes in trans or cis.
Therefore, based on pairs from cis/trans target prediction of the differentially expressed lncRNAs combined with gene functions, pathways, milk protein QTL regions plus GWAS results, we are able to identify promising pairs and candidate lncRNAs relevant to milk protein synthesis, transport and metabolism. 30 lncRNAs were predicted to regulate 34 genes affecting milk protein synthesis. PRL is known to activate the phospholipase C-protein kinase C pathway, as well as the phosphorylation of STAT5 proteins, which can enhance the rate of transcription of several milk protein genes (Waters and Rillema, 1989; Wakao et al., 1992). Three DEGs (FOS, IRF2 and SOCS2) were found be involved in PRL signaling pathway (Li C. et al., 2016). It has been reported that the PRL stimulation of protein kinase C may be causally related to the PRL stimulation of FOS mRNA accumulation, and protein kinase C activation is essential for all of PRL’s actions on milk product synthesis and mitogenesis (Rillema and Rowady, 1996). The expression of XLOC_2427074, XLOC_2500996, XLOC_2938516 and XLOC_593004 significantly correlated with FOS gene imply their important role in milk protein synthesis. It is well-known that insulin and insulin-like growth factor can affect milk protein expression through the activation of STAT5 or regulating amount of translation via mTOR pathway (Burgos and Cant, 2010; Menzies et al., 2010). The CT/GT haplotype of IGF2 has been proved to be significantly associated with milk protein trait (Bagnicka et al., 2010). The expression of IGF2 is regulated by six IGF binding proteins (IGFBP-1 to -6) (Pfaffl et al., 2002). Interestingly, we found IGF2, IGFBP2, and IGFBP5 were differentially expressed between HP and LP group (Li C. et al., 2016). IGFBP2 was predicted to correlate with XLOC_1186672, XLOC_1243232, XLOC_1284424 and XLOC_2273208, while IGFBP5 was predicted to correlate with XLOC_051681, XLOC_059976 and XLOC_2280878. Based on these, we guess that the lncRNA- IGFBP2/IGFBP5 pairs may be involved in insulin-like growth factor pathway and affect milk protein synthesis.
As a novel milk protein associated gene, CNTFR is capable of activating multiple downstream signaling pathways, such as AMPK, Jak2-Stat5, MAPK and PI3K-AKT (Weis et al., 1999; Li C. et al., 2016), and their expression appears to be strongly correlated with XLOC_059976. We previously revealed that XLOC_059976 may affect milk protein synthesis by interacting with miR-152 and miR-139. Another possible function of this lncRNA is to regulate the transcription of protein-coding transcripts through binding regulatory region (Rinn and Chang, 2012). Here, we observed a specific binding site for XLOC_059976 located in the 5′ upstream region of CNTFR. Based on this specific interaction site, we speculate that XLOC_059976 may enhance CNTFR functions through recruiting transcription factors or directly initiating the transcription process, which can affect the downstream process of milk protein secretion. CNTFR is the ligand-specific component of a tripartite receptor for ciliary neurotrophic factor (CNTF), which can activate multiple downstream signaling pathways, such as AMPK, Jak2-Stat5, MAPK and PI3K-AKT. All these pathways are critical to milk protein synthesis (Bionaz and Loor, 2011; Tanos et al., 2012). Therefore, XLOC_059976 may play a significant role in the synthesis of milk proteins, and could be a key candidate marker for the selection of milk protein traits. Notably, multiple genes associated with milk secretion could be controlled by single lncRNA. In particular, XLOC_051681 was observed to be correlated with the expression of 16 genes. By contrast, one gene associated with milk protein traits could be regulated by several lncRNAs. For example, EIF2S2 is believed to be simultaneously targeted by XLOC_051681, XLOC_112491, XLOC_1261332, XLOC_240033, XLOC_2500996, XLOC_890495, and XLOC_929036 (Li C. et al., 2016). The functional roles of lncRNAs are highly complex and diverse. Therefore, our ongoing effort will focus on the function of their target genes, with the expectation that more fundamental information contributing to our understanding of milk protein synthesis and secretion on a molecular level will be obtained.
Here, we performed a comprehensive analysis of mammary lncRNAs in Chinese Holstein cattle. Bioinformatics approaches were applied to predict the target genes and the potential functions of the differentially expressed lncRNAs. We also sought to explore their roles in milk protein synthesis. The structural characteristics of the identified lncRNAs in bovine are similar to those in other mammalian species. An integrated interpretation of differential lncRNA and mRNA expression reveals that 30 lncRNAs potentially regulate 34 genes affecting milk protein synthesis. As the role of lncRNAs in the bovine mammary gland have not been fully elucidated, our study provides a valuable starting point for future analyses. Moreover, the putative lncRNA XLOC_059976 could be a key candidate biomarker for the prediction of milk protein composition phenotypes, and deserve to be further explored.
The sequencing data have been submitted to the NCBI SRA, and are accessible through the accession number PRJNA416150.
SZ conceived and designed the study, and revised the manuscript. WC performed the RNA related experiments, data analysis, and drafted the manuscript. CL participated in the experimental design, mammary gland biopsy and samples collection. SL conducted the qPCR validation. CZ and HY participated in mammary gland biopsy and samples collection. JS and QZ participated in the result interpretation and paper revision. All authors read and approved the final manuscript.
This work is supported by the 863 project (2013AA102504), the National Science and Technology Programs of China (2011BAD28B02), National Key Technologies R&D Program (2012BAD12B01), Beijing Dairy Industry Innovation Team, China Agricultural Research System (CARS-37), and Xinjiang Province Key Technology Integration and Demonstration Program (201230116). We are deeply grateful to all donors who participated in this program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all contributors of the present study. We also thank Dr. Dengpan Pu and Dr. Meng Zhao for assistance in biopsy operation.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00281/full#supplementary-material
FIGURE S1. The distribution of reads aligning to genome. (a–c) The distribution of reads for HP group in genome. (d–f) The distribution of reads for LP group in genome.
FIGURE S2. The number of different class codes for Cufflinks, Scripture, StringTie and Transcomb. The class codes represented the transcripts falls different regions based on the reference annotation you provided. The class code definition (http://cole-trapnell-lab.github.io/cufflinks/cuffcompare/).
FIGURE S3. The predicted secondary structures of lncRNAs transcripts and miRNAs sequences. The secondary structure was created by Vienna RNA package RNAfold web (http://rna.tbi.univie.ac.at/).
FIGURE S4. Seven lncRNAs located in the casein genes cluster on bovine genome. Blue and red box represented lncRNA transcripts and the casein genes, respectively. TCONS_0484809 located in the first intron of CSN3.
FIGURE S5. The expression boxplot for neighbor genes of lncRNAs. The red and blue boxplots represent the gene expression within 100 and 10 k upstream/downstream of lncRNAs, respectively. The green boxplot is the expression of all protein-coding genes.
FIGURE S6. Validation of XLOC_059976 and target gene (CNTFR) by qRT-PCR. GAPDH gene was used as housekeeping internal control. Transcript expression was quantified relative to the expression level of housekeeping using the comparative cycle threshold (ΔCT) method. The data were presented as the mean ± SE (n = 3).
FIGURE S7. The combine sites between XLOC_059976 and CNTFR.
TABLE S1. The phenotype information of six Chinese Holstein cattle.
TABLE S2. The concentration and quality information of RNA samples.
TABLE S3. The 12 previous GWAS studies for milk protein traits.
TABLE S4. Summary of sequence reads alignment.
TABLE S5. The annotation of lncRNAs with gtf format.
TABLE S6. The annotation of novel genes with gtf format.
TABLE S7. lncRNA sequences blast against NONCODE database.
TABLE S8. The results of differentially expressed lncRNAs, novel genes and protein-coding genes. (a) The differentially expressed lncRNAs results. (b) The differentially expressed novel gene results.
TABLE S9. lncRNAs harboring complete precursors of miRNAs.
TABLE S10. The target prediction between lncRNAs and miRNAs. (a) The target prediction between all lncRNAs and all miRNAs. (b) The target prediction between lncRNAs and milk protein miRNAs.
TABLE S11. Pearson correlations between protein-coding genes and differentially expressed lncRNAs.
TABLE S12. The protein-coding genes within 10 and 100 k upstream and downstream of an lncRNA. (a) The protein-coding genes within 10 and 100 k upstream and downstream of lncRNAs. (b) The prediction of lncRNA- mRNA pairs by both cis and trans. (c) The expression patterns for differentially expressed lncRNA and their differentially expressed neighboring genes.
TABLE S13. Functional enrichment analysis of the protein-coding genes of lncRNAs in cis and trans. (a) The Gene Ontology results of cis and trans target genes using DAVID. (b) The pathway results of cis and trans target genes using IPA.
TABLE S14. The integrate study for differential lncRNA-mRNA pairs.
ANXA2, annexin A2; ATF4, activating transcription factor 4; BLAST, basic local alignment search tool; CNS2, casein beta; CNTFR, ciliary neurotrophic factor receptor; CSN1S1, casein alpha s1; CSN1S2, casein alpha s2; CSN3, casein kappa; CYP1A1, cytochrome P450 family 1 subfamily A member 1; DEGs, differentially expressed genes; DHI, dairy herd improvement; EIF4G1, eukaryotic translation initiation factor 4 gamma 1; ENPP5, ectonucleotide pyrophosphatase/phosphodiesterase 5; ERBB2, receptor tyrosine-protein kinase erbB-2 precursor; FPKM, fragments per kilobase of transcript per million mapped reads; FOS, fos proto-oncogene, AP-1 transcription factor subunit; GALE, UDP-galactose-4-epimerase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GO, gene ontology; GWAS, genome wide association study; HP, high milk protein; HSPA8, heat shock protein family A member 8; IDH1, isocitrate dehydrogenase (NADP(+)) 1; IGF2, insulin like growth factor 2; IGFBP2, insulin like growth factor binding protein 2; IGFBP5, insulin like growth factor binding protein; ilncRNAs, intronic lncRNAs; IRF2, interferon regulatory factor 2; KEGG, kyoto encyclopedia of genes and genomes; LALBA, lactalbumin alpha; LARP4B, la ribonucleoprotein domain family member 4B; LGB, progestagen-associated endometrial protein; lincRNAs, intergenic lncRNAs; LncNATs, natural antisense lncRNAs; LP, low milk protein; MARVELD1, MARVEL domain containing 1; NGS, next generation sequencing; ORF, open reading frame; PRL, prolactin; qRT-RCR, real-time quantitative reverse transcript polymerase chain reaction; QTL, quantitative trait locus; RIN, RNA integrity number; SNP, single nucleotide polymorphism; SOCS2, suppressor of cytokine signaling 2; ssRNA-seq, strand-specific RNA sequencing; TRIB3, tribbles pseudokinase 3.
Andersson, L. (2009). Genome-wide association analysis in domestic animals: a powerful approach for genetic dissection of trait loci. Genetica 136, 341–349. doi: 10.1007/s10709-008-9312-4
Auldist, M. J., Johnston, K. A., White, N. J., Fitzsimons, W. P., and Boland, M. J. (2004). A comparison of the composition, coagulation characteristics and cheesemaking capacity of milk from Friesian and Jersey dairy cows. J. Dairy Res. 71, 51–57.
Bagnicka, E., Siadkowska, E., Strzałkowska, N., Żelazowska, B., Flisikowski, K., Krzyżewski, J., et al. (2010). Association of polymorphisms in exons 2 and 10 of the insulin-like growth factor 2 (IGF2) gene with milk production traits in polish Holstein-Friesian cattle. J. Dairy Res. 77, 37–42. doi: 10.1017/S0022029909990197
Bionaz, M., and Loor, J. J. (2011). Gene networks driving bovine mammary protein synthesis during the lactation cycle. Bioinform. Biol. Insights 5, 83–98. doi: 10.4137/BBI.S7003
Borodina, T., Adjaye, J., and Sultan, M. (2011). A strand-specific library preparation protocol for RNA sequencing. Methods Enzymol. 500, 79–98. doi: 10.1016/B978-0-12-385118-5.00005-0
Burgos, S., and Cant, J. (2010). IGF-1 stimulates protein synthesis by enhanced signaling through mTORC1 in bovine mammary epithelial cells. Domest. Anim. Endocrinol. 38, 211–221. doi: 10.1016/j.domaniend.2009.10.005
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazon-Vega, B., Regev, A., et al. (2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927. doi: 10.1101/gad.17446611
Cai, X., and Cullen, B. R. (2007). The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA 13, 313–316.
Canovas, A., Rincon, G., Islas-Trejo, A., Wickramasinghe, S., and Medrano, J. F. (2010). SNP discovery in the bovine milk transcriptome using RNA-Seq technology. Mamm. Genome 21, 592–598. doi: 10.1007/s00335-010-9297-z
Cesana, M., Cacchiarelli, D., Legnini, I., Santini, T., Sthandier, O., Chinappi, M., et al. (2011). A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147, 358–369. doi: 10.1016/j.cell.2011.09.028
Cui, Y., Sun, X., Jin, L., Yu, G., Li, Q., Gao, X., et al. (2017). MiR-139 suppresses beta-casein synthesis and proliferation in bovine mammary epithelial cells by targeting the GHR and IGF1R signaling pathways. BMC Vet. Res. 13:350. doi: 10.1186/s12917-017-1267-1
Derrien, T., Johnson, R., Bussotti, G., Tanzer, A., Djebali, S., Tilgner, H., et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. doi: 10.1101/gr.132159.111
Dinger, M. E., Pang, K. C., Mercer, T. R., and Mattick, J. S. (2008). Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput. Biol. 4:e1000176. doi: 10.1371/journal.pcbi.1000176
Esteve-Codina, A., Kofler, R., Palmieri, N., Bussotti, G., Notredame, C., and Perez-Enciso, M. (2011). Exploring the gonad transcriptome of two extreme male pigs with RNA-seq. BMC Genomics 12:552. doi: 10.1186/1471-2164-12-552
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Finn, R. D., Tate, J., Mistry, J., Coggill, P. C., Sammut, S. J., Hotz, H. R., et al. (2008). The Pfam protein families database. Nucleic Acids Res. 36, D281–D288.
Georges, M., Nielsen, D., Mackinnon, M., Mishra, A., Okimoto, R., Pasquino, A. T., et al. (1995). Mapping quantitative trait loci controlling milk production in dairy cattle by exploiting progeny testing. Genetics 139, 907–920.
Guil, S., and Esteller, M. (2012). Cis-acting noncoding RNAs: friends and foes. Nat. Struct. Mol. Biol. 19, 1068–1075. doi: 10.1038/nsmb.2428
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D., et al. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227. doi: 10.1038/nature07672
Guttman, M., Garber, M., Levin, J. Z., Donaghey, J., Robinson, J., Adiconis, X., et al. (2010). Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 28, 503–510. doi: 10.1038/nbt.1633
He, Y., Ding, Y., Zhan, F., Zhang, H., Han, B., Hu, G., et al. (2015). The conservation and signatures of lincRNAs in Marek’s disease of chicken. Sci. Rep. 5:15184. doi: 10.1038/srep15184
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2008). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Ji, Z., Dong, F., Wang, G., Hou, L., Liu, Z., Chao, T., et al. (2015). MiR-135a targets and regulates prolactin receptor gene in goat mammary epithelial cells. DNA Cell Biol. 34, 534–540. doi: 10.1089/dna.2015.2904
Keniry, A., Oxley, D., Monnier, P., Kyba, M., Dandolo, L., Smits, G., et al. (2012). The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat. Cell Biol. 14, 659–665. doi: 10.1038/ncb2521
Khalil, A. M., Guttman, M., Huarte, M., Garber, M., Raj, A., Rivea Morales, D., et al. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11667–11672. doi: 10.1073/pnas.0904715106
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Kornienko, A. E., Guenzl, P. M., Barlow, D. P., and Pauler, F. M. (2013). Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 11:59. doi: 10.1186/1741-7007-11-59
Kozak, M. (1986). Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 44, 283–292. doi: 10.1016/0092-8674(86)90762-2
Krämer, A., Green, J., Pollard, J. Jr., and Tugendreich, S. (2013). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. doi: 10.1093/bioinformatics/btt703
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Lai, F., and Shiekhattar, R. (2014). Where long noncoding RNAs meet DNA methylation. Cell Res. 24, 263–264. doi: 10.1038/cr.2014.13
Li, C., Cai, W., Zhou, C., Yin, H., Zhang, Z., Loor, J. J., et al. (2016). RNA-Seq reveals 10 novel promising candidate genes affecting milk protein concentration in the Chinese Holstein population. Sci. Rep. 6:26813. doi: 10.1038/srep26813
Li, R., Dudemaine, P. L., Zhao, X., Lei, C., and Ibeagha-Awemu, E. M. (2016). Comparative analysis of the miRNome of Bovine milk fat. Whey and cells. PLoS One 11:e0154129. doi: 10.1371/journal.pone.0154129
Li, D., Xie, X., Wang, J., Bian, Y., Li, Q., Gao, X., et al. (2015). MiR-486 regulates lactation and targets the PTEN gene in cow mammary glands. PLoS One 10:e0118284. doi: 10.1371/journal.pone.0118284
Li, H. M., Wang, C. M., Li, Q. Z., and Gao, X. J. (2012). MiR-15a decreases bovine mammary epithelial cell viability and lactation and regulates growth hormone receptor expression. Molecules 17, 12037–12048. doi: 10.3390/molecules171012037
Li, T., Wang, S., Wu, R., Zhou, X., Zhu, D., and Zhang, Y. (2012). Identification of long non-protein coding RNAs in chicken skeletal muscle using next generation sequencing. Genomics 99, 292–298. doi: 10.1016/j.ygeno.2012.02.003
Liu, J., Yu, T., Jiang, T., and Li, G. (2016). Transcomb: genome-guided transcriptome assembly via combing junctions in splicing graphs. Genome Biol. 17:213. doi: 10.1186/s13059-016-1074-1
Liu, X., Yang, J., Zhang, Q., and Jiang, L. (2017). Regulation of DNA methylation on EEF1D and RPL8 expression in cattle. Genetica 145, 387–395. doi: 10.1007/s10709-017-9974-x
Luo, M., Li, Z., Wang, W., Zeng, Y., Liu, Z., and Qiu, J. (2013). Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Lett. 333, 213–221. doi: 10.1016/j.canlet.2013.01.033
McGinnis, S., and Madden, T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25. doi: 10.1093/nar/gkh435
Menzies, K. K., Lee, H. J., Lefèvre, C., Ormandy, C. J., Macmillan, K. L., and Nicholas, K. R. (2010). Insulin, a key regulator of hormone responsive milk protein synthesis during lactogenesis in murine mammary explants. Funct. Integr. Genomics 10, 87–95. doi: 10.1007/s10142-009-0140-0
Merry, C. R., Forrest, M. E., Sabers, J. N., Beard, L., Gao, X. H., Hatzoglou, M., et al. (2015). DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum. Mol. Genet. 24, 6240–6253. doi: 10.1093/hmg/ddv343
Nguyen, M., Boutinaud, M., Petridou, B., Gabory, A., Pannetier, M., Chat, S., et al. (2014). DNA methylation and transcription in a distal region upstream from the bovine alphaS1 casein gene after once or twice daily milking. PLoS One 9:e111556. doi: 10.1371/journal.pone.0111556
Niazi, F., and Valadkhan, S. (2012). Computational analysis of functional long noncoding RNAs reveals lack of peptide-coding capacity and parallels with 3’. UTRs. RNA 18, 825–843. doi: 10.1261/rna.029520.111
Orom, U. A., Derrien, T., Guigo, R., and Shiekhattar, R. (2010). Long noncoding RNAs as enhancers of gene expression. Cold Spring Harb. Symp. Quant. Biol. 75, 325–331. doi: 10.1101/sqb.2010.75.058
Pandey, R. R., Mondal, T., Mohammad, F., Enroth, S., Redrup, L., Komorowski, J., et al. (2008). Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 32, 232–246. doi: 10.1016/j.molcel.2008.08.022
Pauli, A., Valen, E., Lin, M. F., Garber, M., Vastenhouw, N. L., Levin, J. Z., et al. (2012). Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 22, 577–591. doi: 10.1101/gr.133009.111
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., and Salzberg, S. L. (2015). Stringtie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Pfaffl, M., Georgieva, T. M., Georgiev, I. P., Ontsouka, E., Hageleit, M., and Blum, J. (2002). Real-time RT-PCR quantification of insulin-like growth factor (IGF)-1. IGF-1 receptor, IGF-2, IGF-2 receptor, insulin receptor, growth hormone receptor, IGF-binding proteins 1, 2 and 3 in the bovine species. Domest. Anim. Endocrinol. 22, 91–102. doi: 10.1016/S0739-7240(01)00128-X
Ponting, C. P., Oliver, P. L., and Reik, W. (2009). Evolution and functions of long noncoding RNAs. Cell 136, 629–641. doi: 10.1016/j.cell.2009.02.006
Qu, Z., and Adelson, D. L. (2012). Bovine ncRNAs are abundant, primarily intergenic, conserved and associated with regulatory genes. PLoS One 7:e42638. doi: 10.1371/journal.pone.0042638
Ren, H., Wang, G., Chen, L., Jiang, J., Liu, L., Li, N., et al. (2016). Genome-wide analysis of long non-coding RNAs at early stage of skin pigmentation in goats (Capra hircus). BMC Genomics 17:67. doi: 10.1186/s12864-016-2365-3
Rillema, J., and Rowady, D. (1996). Characteristics of the prolactin stimulation of c-fos mRNA levels in mouse mammary gland explants. Proc. Soc. Exp. Biol. Med. 212, 142–146. doi: 10.3181/00379727-212-44001
Rinn, J. L., and Chang, H. Y. (2012). Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166. doi: 10.1146/annurev-biochem-051410-092902
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A., et al. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323. doi: 10.1016/j.cell.2007.05.022
Saremi, B., Sauerwein, H., Dänicke, S., and Mielenz, M. (2012). Identification of reference genes for gene expression studies in different bovine tissues focusing on different fat depots. J. Dairy Sci. 95, 3131–3138. doi: 10.3168/jds.2011-4803
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504.
Siepel, A., Bejerano, G., Pedersen, J. S., Hinrichs, A. S., Hou, M., Rosenbloom, K., et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050.
Singh, K., Molenaar, A. J., Swanson, K. M., Gudex, B., Arias, J. A., Erdman, R. A., et al. (2012). Epigenetics: a possible role in acute and transgenerational regulation of dairy cow milk production. Animal 6, 375–381. doi: 10.1017/S1751731111002564
Tanaka, T., Haneda, S., Imakawa, K., Sakai, S., and Nagaoka, K. (2009). A microRNA, miR-101a, controls mammary gland development by regulating cyclooxygenase-2 expression. Differentiation 77, 181–187. doi: 10.1016/j.diff.2008.10.001
Tanos, T., Rojo, L., Echeverria, P., and Brisken, C. (2012). ER and PR signaling nodes during mammary gland development. Breast Cancer Res. 14:210. doi: 10.1186/bcr3166
Tong, C., Chen, Q., Zhao, L., Ma, J., Ibeagha-Awemu, E. M., and Zhao, X. (2017). Identification and characterization of long intergenic noncoding RNAs in bovine mammary glands. BMC Genomics 18:468. doi: 10.1186/s12864-017-3858-4
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with tophat and cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Wakao, H., Schmitt-Ney, M., and Groner, B. (1992). Mammary gland-specific nuclear factor is present in lactating rodent and bovine mammary tissue and composed of a single polypeptide of 89 kDa. J. Biol. Chem. 267, 16365–16370.
Wang, D., Liang, G., Wang, B., Sun, H., Liu, J., and Luo Guan, L. (2016). Systematic microRNAome profiling reveals the roles of microRNAs in milk protein metabolism and quality: insights on low-quality forage utilization. Sci. Rep. 6:21194. doi: 10.1038/srep21194
Wang, J., Bian, Y., Wang, Z., Li, D., Wang, C., Li, Q., et al. (2014). MicroRNA-152 regulates DNA methyltransferase 1 and is involved in the development and lactation of mammary glands in dairy cows. PLoS One 9:e101358. doi: 10.1371/journal.pone.0101358
Wang, M., Hancock, T. P., Macleod, I. M., Pryce, J. E., Cocks, B. G., and Hayes, B. J. (2017). Putative enhancer sites in the bovine genome are enriched with variants affecting complex traits. Genet. Sel. Evol. 49:56. doi: 10.1186/s12711-017-0331-4
Waters, S., and Rillema, J. (1989). Role of protein kinase C in the prolactin-induced responses in mouse mammary gland explants. Mol. Cell. Endocrinol. 63, 159–166. doi: 10.1016/0303-7207(89)90092-0
Weikard, R., Hadlich, F., and Kuehn, C. (2013). Identification of novel transcripts and noncoding RNAs in bovine skin by deep next generation sequencing. BMC Genomics 14:789. doi: 10.1186/1471-2164-14-789
Weis, J., Schonrock, L. M., Zuchner, S. L., Lie, D. C., Sure, U., Schul, C., et al. (1999). CNTF and its receptor subunits in human gliomas. J. Neurooncol. 44, 243–253. doi: 10.1023/A:1006303221064
Wickramasinghe, S., Hua, S., Rincon, G., Islas-Trejo, A., German, J. B., Lebrilla, C. B., et al. (2011). Transcriptome profiling of bovine milk oligosaccharide metabolism genes using RNA-sequencing. PLoS One 6:e18895. doi: 10.1371/journal.pone.0018895
Young, R. S., Marques, A. C., Tibbit, C., Haerty, W., Bassett, A. R., Liu, J. L., et al. (2012). Identification and properties of 1,119 candidate lincRNA loci in the Drosophila melanogaster genome. Genome Biol. Evol. 4, 427–442. doi: 10.1093/gbe/evs020
Zhao, J., Sun, B. K., Erwin, J. A., Song, J. J., and Lee, J. T. (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756. doi: 10.1126/science.1163045
Zhao, X. Y., and Lin, J. D. (2015). Long noncoding RNAs: a new regulatory code in metabolic control. Trends Biochem. Sci. 40, 586–596. doi: 10.1016/j.tibs.2015.08.002
Keywords: long non-coding RNA, mammary gland, transcriptome, milk proteins, integrate study
Citation: Cai W, Li C, Liu S, Zhou C, Yin H, Song J, Zhang Q and Zhang S (2018) Genome Wide Identification of Novel Long Non-coding RNAs and Their Potential Associations With Milk Proteins in Chinese Holstein Cows. Front. Genet. 9:281. doi: 10.3389/fgene.2018.00281
Received: 02 February 2018; Accepted: 09 July 2018;
Published: 30 July 2018.
Edited by:
Tad Stewart Sonstegard, Recombinetics, United StatesReviewed by:
Eveline M. Ibeagha-Awemu, Agriculture and Agri-Food Canada (AAFC), CanadaCopyright © 2018 Cai, Li, Liu, Zhou, Yin, Song, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengli Zhang, emhhbmdzbGNhdUBjYXUuZWR1LmNu
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.