 Giulia Romano
Giulia Romano Dario Veneziano
Dario Veneziano Giovanni Nigita
Giovanni Nigita Serge P. Nana-Sinkam
Serge P. Nana-Sinkam- 1Internal Medicine “Division of Pulmonary and Critical Care Medicine”, Virginia Commonwealth University Health System, Richmond, VA, United States
- 2Department of Cancer Biology and Genetics, The Ohio State University, Columbus, OH, United States
Nearly all classes of coding and non-coding RNA undergo post-transcriptional modification, as more than 150 distinct modification types have been reported. Since RNA modifications were first described over 50 years ago, our understanding of their functional relevance in cellular control mechanisms and phenotypes has truly progressed only in the last 15 years due to advancements in detection and experimental techniques. Specifically, the phenomenon of RNA methylation in the context of ncRNA has emerged as a novel process in the arena of epitranscriptomics. Methylated ncRNA molecules may indeed contribute to a potentially vast functional panorama, from regulation of post-transcriptional gene expression to adaptive cellular responses. Recent discoveries have uncovered novel dynamic mechanisms and new layers of complexity, paving the way to a greater understanding of the role of such phenomena within the broader molecular cellular context of human disease.
Introduction
Up until recently, the central dogma (Crick, 1970) had supported primary focus on the molecular contributions of DNA and protein to human disease. The inability to detect and evaluate RNA with the necessary molecular resolution and precision has limited our understanding of the spectrum of RNA modifications that may drive disease.
Following the discovery of pseudouridine (Davis and Allen, 1957), nine additional modifications were identified in 1965 (Holley et al., 1965b). Finally, modification events in nucleotides of mRNA molecules were also uncovered in the 1970s (Desrosiers et al., 1974; Adams and Cory, 1975; Dubin and Taylor, 1975; Perry et al., 1975). Gradually, the “static” interpretation of the cellular role of RNA started to be challenged (Gilbert, 1986). With the discovery of novel species of non-coding RNA (ncRNA) and their mechanisms further investigated (Lee et al., 1993; Fire et al., 1998; Eddy, 2001), RNA biology came to the forefront (Todd and Karbstein, 2007). Along with advancements in experimental and transcriptomics techniques, which enabled a more detailed investigation of the translational control of cellular responses and phenotypes (Chan et al., 2010), interest in RNA modifications also grew, resulting in significant progress in the last 15 years. Recent discoveries, such as the first and second mRNA m6A demethylases FTO (Jia et al., 2011) and ALKBH5 (Zheng et al., 2013), as well as the identification of the METTL3/METTL14 methyltransferase complex (Liu et al., 2014), have triggered renewed interest in RNA modifications.
To date, a total of 163 post-transcriptional RNA modifications have been uncovered across all living organisms (Boccaletto et al., 2017) and are among the most evolutionarily conserved properties of RNAs (Li and Mason, 2014), revealing a “novel,” complex layer of biological regulation known as the epitranscriptome (Saletore et al., 2012). The functional diversity provided by these phenomena can indeed affect RNA structure, play a fundamental role in their interactions with other molecules and in regulatory networks, such as metabolic changes (Lewis et al., 2017), thus affecting every aspect of cellular physiology.
RNA modifications have been categorized as reversible and non-reversible. Among non-reversible modifications, we find well-studied phenomena such as RNA editing and pseudouridylation (Meier, 2011). Nonetheless, recent focus has shifted to reversible modifications, such as cytosine and adenosine methylations (Klungland et al., 2016). However, this classic distinction is being reassessed, in light of the discovery of “erasers” such as FTO and ALKBH5.
The importance of modifications in novel classes of ncRNA transcripts is also becoming relevant. Well-characterized chemical modifications in traditional classes of RNAs such as transfer (tRNAs) and ribosomal (rRNA) RNA, novel detection technologies and deep sequencing analysis (Veneziano et al., 2015, 2016), have paved the way for a fuller assessment of these molecular events also in regulatory ncRNAs, such as microRNA (Alarcon et al., 2015b) and long ncRNAs (Patil et al., 2016).
RNA Methylation
RNA methylation is a reversible, post-transcriptional RNA modification, affecting several biological processes, such as RNA stability and mRNA translation (Ji and Chen, 2012; Wang et al., 2014, 2015; Dev et al., 2017), through a variety of RNA methyltransferases, often using distinct catalytic strategies. Furthermore, recent studies have shown how the deregulation of proteins implicated in these modification phenomena is associated to disease (Supplementary Table 1A). In this section, we will review the main types and functions of methylation in ncRNAs (Figure 1).
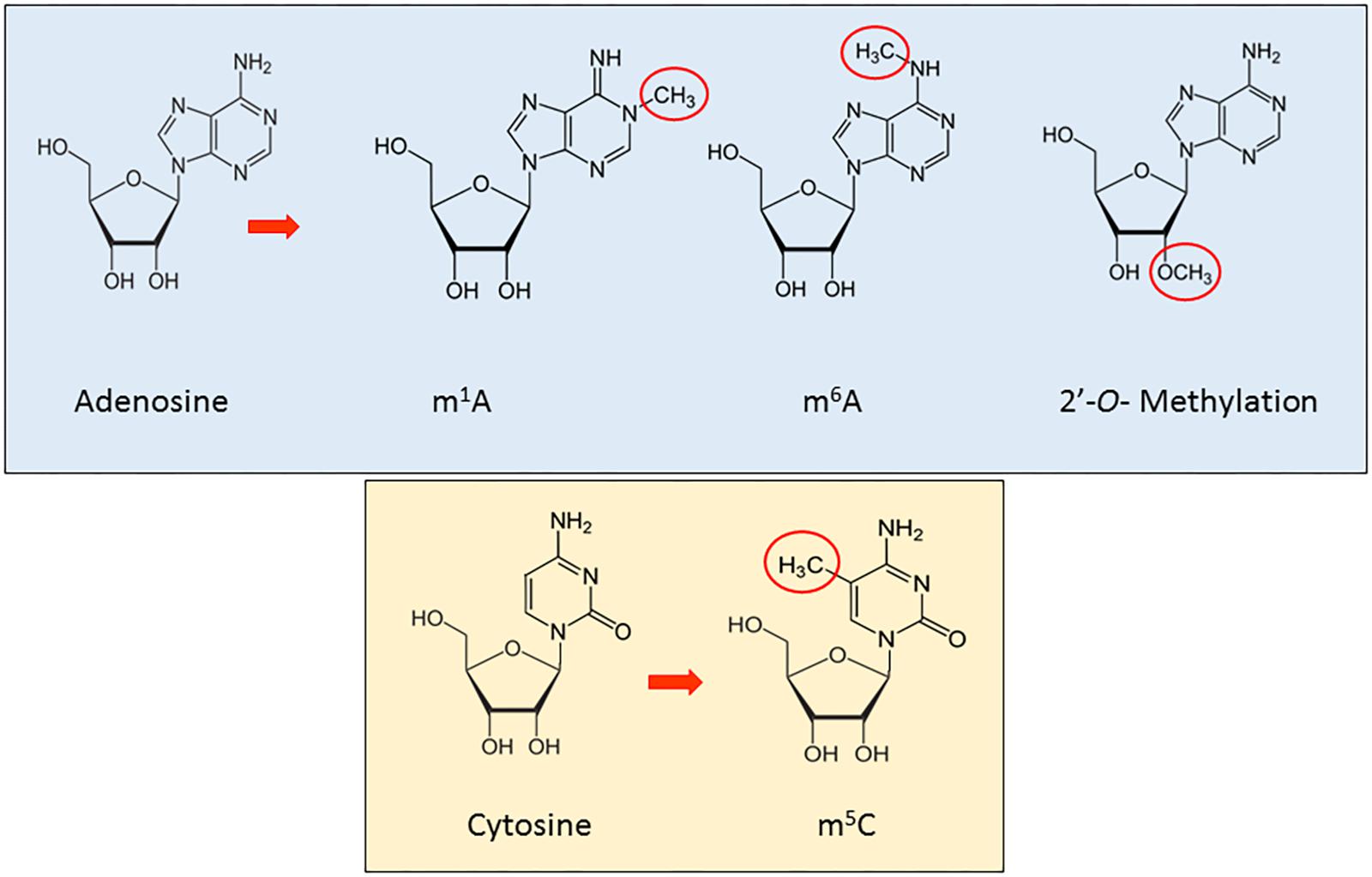
FIGURE 1. A schematic representation of the principal methylation modifications in eukaryotic RNA.
N6-Methyladenosine (m6A)
N6-methyladenosine (m6A) is the most abundant internal modification detected to date in mRNA (Roundtree et al., 2017). Discovered in the 1970s, its function has been thoroughly investigated only in the last decade (Rottman et al., 1974; Wang and He, 2014). This was driven by the recent discovery and characterization of evolutionarily conserved proteins able to encode (writers), decode (readers), and remove (erasers) methylation (Lewis et al., 2017). Since 1994, different writers have been identified, including METTL3 and METTL14, proven to regulate the circadian clock, differentiation of embryonic stem cells and primary miRNA processing (Dominissini et al., 2012; Wang et al., 2014; Alarcon et al., 2015b). These enzymes work in complex with proteins essential to the correct processing of RNA methylation (Schwartz et al., 2014): Wilms tumor 1-associated protein (WTAP), RNA-binding motif protein 15 (RBM15) and Protein virilizer homolog (KIAA1429). Additionally, the discovery of ALKBH5 and FTO has revealed the dynamic dimension of this modification phenomenon for cellular metabolism (Jia et al., 2011; Zheng et al., 2013). Recently, the YTH domain family proteins (YTHDF1–3) and YTH domain-containing protein 1 (YTHDC1) have been characterized as m6A readers, providing the first functional evidence of m6A (Wang et al., 2014).
The methyl group in m6A does not affect the Watson–Crick base-pairing (Liu and Jia, 2014), is highly conserved between human and mice and located in 5′ UTRs, 3′ UTRs, around stop codons, long internal and alternatively spliced exons (Dominissini et al., 2016; Li et al., 2016a; Lewis et al., 2017). It is also found in tRNA, rRNA, and small nuclear RNA (snRNA) as well as several long non-coding RNA, such as Xist (Dominissini et al., 2012). While not completely understood, m6A has been shown to play critical roles in the biological regulation of mRNA and ncRNA (Liu and Jia, 2014), particularly splicing, stability, turnover, nuclear export, and mediation of cap-independent translation (Meyer et al., 2015). Recently, Sun et al. (2016) have integrated all m6A sequencing data into a novel database, RMBase, identifying ∼200,000 N6-Methyladenosines (m6A) sites in human and mouse. Finally, Linder et al. (2015) mapped m6A and m6Am at single-nucleotide resolution and identified small nucleolar RNAs (snoRNAs) as a new class of m6A-containing non-coding RNAs (ncRNAs).
N1-Methyladenosine (m1A)
Although the first studies on N1-methyladenosine (m1A) in total RNA date back more than 50 years (Dunn, 1961), only one study in the last decade has shed substantial light on function. m1A is a dynamic methylation event at the N1 position of adenosine, comprising the addition of a methyl group and a positive charge in the base, specifically in the Watson–Crick interface, obviously altering RNA-protein interaction and RNA secondary structures through electrostatic effects (Roundtree et al., 2017). m1A is abundant in tRNA and rRNA (El Yacoubi et al., 2012; Sharma et al., 2013) exercising major influence on structure and function (Anderson, 2005). Two groups recently found a strong conservation of the m1A pattern in several human and murine cell lines as well as in yeast, affirming the important role of this modification along the evolutionary chain. In particular, m1A has been shown to have a role in mRNA translation, via unique localization near the translation start site and first splice site (Dominissini et al., 2016; Li et al., 2016a; Roundtree et al., 2017) and by facilitating non-canonical binding of the exon–exon junction complex (Cenik et al., 2017).
2′-O-Methylation (2′OMe/Nm)
2′OMe is a very common RNA modification in abundant RNAs (rRNA, snRNA, tRNA) (Schibler and Perry, 1977; Borges and Martienssen, 2015; Roundtree et al., 2017) as well as in microRNA and it is fundamental for the biogenesis and function of these molecules (Ji and Chen, 2012). It was initially detected at the second and third nucleotide in many mRNA (Schibler and Perry, 1977). Further, it was observed that in rRNA, the loss of an individual modification had no apparent effect, while the deletion of 2–3 modifications in A and P site regions impairs translation and strongly delays pre-rRNA processing (Liang et al., 2009).
2′-O-methylation occurs in 3′ termini and is found to be important in plant biogenesis of small RNA, inter alia miRNA and siRNAs (Yu et al., 2005). Furthermore 2′-O-methylation plays an important role in protecting against 3′–5′ degradation and 3′ uridylation of some small RNAs as piRNAs in animals and Ago2-associated small RNAs in Drosophila (Ji and Chen, 2012). It has been found to be catalyzed by HUA-ENHANCER-1/piwi-methyltransferase (HEN1/piMET) enzyme.
5-Methylcytosine (m5C)
5-Methylcytosine (m5C) is an epitranscriptomic modification that involves the 5th carbon atom of cytosine as a target for methylation in poly(A) RNA, rRNA, tRNA, snRNA, and lncRNA (Amort et al., 2013, 2017; Lewis et al., 2017). While some of the proteins regulating m5C in different RNA have been identified, the biological function remains unclear (Nachtergaele and He, 2017). NOL1/NOP2/Sun domain family member 2 (NSUN2) together with DNA methyltransferase-like protein 2 (DNMT2) have been shown to be the writers of m5C, although to date no erasers or readers have been discovered (Lewis et al., 2017), though recently, investigators identified ALYREF as a potential reader of m5C (Yang et al., 2017). Several roles have been suggested for m5C, from the stabilizing of tRNA secondary structure and prevention of degradation or cleavage, to playing a role in translation when in rRNA and increasing the stability of mRNA transcripts (Esteller and Pandolfi, 2017).
Methodologies for the Detection and Profiling of RNA Methylation
The recent advent of more sensitive and robust sequencing technologies (Li et al., 2016b), coupled with novel biochemical techniques (Song and Yi, 2017), has greatly improved the characterization and understanding of RNA modifications (Frye et al., 2016). This has allowed us to address challenges such as limitations with reverse transcription (RT) signatures and low transcript expression, as is the case with mRNA and lncRNA. Major advances in high-throughput sequencing methods (Helm and Motorin, 2017) have indeed allowed for the systematic identification of RNA modifications at single-nucleotide resolution, effectively distinguishing their distribution patterns in a transcriptome-wide manner.
Traditional biophysical targeted approaches for the detection and quantification of RNA modifications have further matured and provided the foundation for nearly all current high-throughput techniques (Vandivier and Gregory, 2017). Earlier methodologies relied on chromatography applied to direct sequencing, providing the very first evidence of modifications in RNA (Desrosiers et al., 1974). As these techniques only allowed detection of global patterns of modification, they were soon improved with the application of electrophoresis (Gupta and Randerath, 1979; Sprinzl and Vassilenko, 2005) and mass spectrometry (McCloskey and Nishimura, 1977; Kowalak et al., 1993) attaining for the first time base resolution. Recently other strategies, such as high-resolution melting (Golovina et al., 2014), have been implemented to narrow resolution. Nonetheless, an important strategy on which several high-throughput techniques were later developed, is based on the detection of variation in RT signatures (Brownlee and Cartwright, 1977; Motorin et al., 2007). As RNA modifications may interfere with the RT enzyme, inducing its arrest and/or the misincorporation of non-complementary deoxyribonucleoside triphosphates (dNTPs), this provided the foundation to several current methodologies exclusively RT-based as well as leveraging on chemical treatment of the RNA pool or the use of antibodies for the enrichment of modified RNA populations. Such is the case of techniques employing methyl RIP-seq (MeRIP-seq) (Mishima et al., 2015; Dominissini et al., 2016; Li et al., 2016a) coupled with various crosslinking techniques to improve the resolution window. For instance, in m1A-ID-seq, employ demethylases to generate a m1A-depleted control library for validation (Li et al., 2016a). In alternative techniques, such as m1A-seq, RNA pools undergo Dimroth rearrangement under alkaline conditions, converting m1A residues to m6A, thus producing different RT signatures that can validate the MeRIP data (Dominissini et al., 2016). Indeed, certain RNA modifications, such as m6A and m5C, are RT-silent. Despite simple antibody pulldown methods have satisfactorily mapped m6A sites (Dominissini et al., 2012; Meyer et al., 2012) and antibodies highly specific to methylated RNA bases have also been employed (Linder et al., 2015; Li et al., 2016a), most antibody-based methods do not provide nucleotide resolution. For this reason, more recent global approaches have paired antibody binding to covalent crosslinking at specific RNA sites, resulting in RT signatures able to improve resolution (Linder et al., 2015). For instance, after transcripts fragmentation in MeRIP protocols, antibodies forming non-covalent complexes with modified residues are further cross-linked to reactive residues nearby via UV light at distinct frequencies according to the specific techniques (i.e., miCLIP and PA-m6A-seq) for m6A detection (Chen K. et al., 2015). Such induced covalent crosslinks are then the sites at which RT stalls, yielding approximate or precise single-nucleotide resolution. Recently, an innovative detection technique has precisely elucidated m6A distributions across unknown regions via an antibody-independent strategy able to produce abortive cDNA signatures at m6A sites, greatly increasing resolution (Hong et al., 2018). In the case of m5C, bisulfite sequencing has yielded satisfactory results, although posing a few challenges. As unmodified cytosines are converted to inosines as a result of bisulfite treatment, m5C residues remain unaffected, providing a signature in cDNA. While this has been effective for highly abundant ncRNA populations (i.e., tRNA and rRNA) (Militello et al., 2014), degradation issues (due to higher pH conditions during treatment) and read mapping challenges have yielded poor results for low-abundance RNA species (Squires et al., 2012; Hussain et al., 2013; Jeltsch et al., 2017). An alternative approach termed “suicide enzyme trap” has been employed to characterize substrates of m5C-methyltransferases (m5C -MTases) NSUN2 and NSUN4 (Metodiev et al., 2014; Van Haute et al., 2016). By mutating m5C-MTases to form irreversible covalent bonds with target residues, the resulting stable enzyme–RNA complexes are suitable for immunoprecipitation and mapping. Such is also the case of the AZA-seq methodology formalized by Khoddami and Cairns (2014) in which “suicide inhibitor” nucleotide analog 5-azacytidine is incorporated into cellular RNA and “traps” m5C-MTases for pulldown and sequencing.
Finally, 2′OMe too can be detected at base resolution via differential RT profiles, with or without chemical treatment. The RiboMeth-seq methodology (Birkedal et al., 2015; Krogh et al., 2016; Marchand et al., 2016, 2017) for instance, leverages on the ability of 2′OMe to preserve adjacent phosphodiester bonds from alkaline cleavage and produces a high-throughput coverage profile of under-represented positions at the extremes of reads. Nonetheless, chemical treatment is not strictly necessary. Indeed, earlier methods relied on the natural ability of 2′OMe to interrupt RT at low dNTP concentrations (Maden et al., 1995). Such principle was recently employed in the development of a high-throughput protocol proven to be more sensitive and specific than methods based on alkaline hydrolysis. These methodologies have specifically been assessed on 2′OMe modifications occurring in ribosomal and transfer RNA, while not as efficiently identifying such phenomena in low abundance RNA molecules such as mRNA and several ncRNAs. To address such deficiency, the recently published Nm-seq protocol leverages on the ability of 2′OMe to confer resistance to oxidation by sodium periodate to the ribose backbone of RNA molecules, thus allowing the enrichment and mapping of reads originating from RNA fragments whose internal 2′OMe have been exposed at the 3′ end via the elimination of non-modified nucleotides. Such technique has provided a sensitive and precise 2′OMe detection method for rare RNA classes (Dai et al., 2017).
Due to the time-consuming and labor-intensive nature of such techniques, many transcriptomes and potentially novel modifications remain unexplored. For this reason, computational methods have also been developed for the accurate evaluation of modifications events (Zhang et al., 2015; Liu et al., 2017). Moreover, given the error-prone nature of high-throughput techniques, it is strongly suggested that modification sites predicted from big data not be considered as candidates if not validated with at least one additional methodology (Helm and Motorin, 2017). All methodologies described above are summarized in Supplementary Table 1B.
ncRNA Species and RNA Methylation: Functional Associations
tRNA
tRNA methylations were first identified concurrently with the initial sequencing of the clover-shaped molecule (Holley et al., 1965a). Initially, it was suggested that such phenomena was probably the result of a network of diverse enzymes (Hurwitz et al., 1964). It is now clear that tRNA methylation is highly conserved and that tRNAs are the RNA class containing the majority of modified nucleosides among all discovered RNA species. With a total of more than 90 modified nucleosides identified (MODOMICS) (Boccaletto et al., 2017), all tRNA molecules from the three domains of life contain 13 methylated nucleosides out of 18 shared (Marck and Grosjean, 2002; Jackman and Alfonzo, 2013). Originally, it was thought that tRNA modifications in general were a straightforward, static process occurring on specific sites of distinct tRNA species. Given the recent characterization of major tRNA modification pathways, along with their associated tRNA methyltransferase enzyme families (Hori, 2014), a relevant diversity has emerged among living organisms. The presence of catalytic interactions, distinct RNA substrate recognition mechanisms and diverse chemical processes, all suggest a complex functional panorama. Generally, four functional categories can be attributed to tRNA methylation phenomena: preservation of secondary and tertiary structures (Helm and Attardi, 2004; Voigts-Hoffmann et al., 2007); thermodynamic stability (Yokoyama et al., 1987); protection from degradation and rapid tRNA decay (Kadaba et al., 2004; Alexandrov et al., 2006; Guy et al., 2014); translation control and fidelity (Anderson et al., 1998, 2000; Chan et al., 2010, 2012). It is thus evident that tRNA methylation contributes to RNA quality control systems, cellular localization (Kaneko et al., 2003), response to stress stimuli (Schaefer et al., 2010; Becker et al., 2012; Muller et al., 2013), proliferation and many other processes (Phizicky and Hopper, 2015). Most importantly, disruption of energy and amino acid metabolism pathways (i.e., depletion of methionine, necessary for methylation) can damage downstream the RNA modification system, resulting in partially modified tRNAs and thus translational errors (explaining why living organisms use the methionine codon as the initiation codon for protein synthesis) (Hori, 2014). Recently, researchers discovered the first tRNA demethylase, ALKBH1, as a novel post-transcriptional gene expression regulation mechanism (Liu et al., 2016). Finally, tRNA methylations and their enzymes may cooperate collectively in functional networks in order to support adaptive cellular responses (Chan et al., 2010; Tomikawa et al., 2010; Ishida et al., 2011).
miRNAs
From transcription to decay, the multi-level process of the biogenesis of miRNAs is regulated by two main actors: processing enzymes such as DROSHA, DICER, and AGO proteins (Ha and Kim, 2014); and post-transcriptional modifications. Established RNA modifications, such as RNA editing events, have been shown to dynamically alter the sequence and/or the structure of miRNAs (Nigita et al., 2015; Nishikura, 2016) and consequently, in some cases, their function (Kawahara et al., 2007; Nigita et al., 2016). Recently, this has been also investigated in the context of miRNAs and RNA methylation.
2′OMe has been detected at the 3′-end of miRNAs (only in plants) and found to confer stability and protection from 3′-uridylation and degradation (Backes et al., 2012; Borges and Martienssen, 2015). m6A within 3′ UTRs has been generally associated with the presence of miRNA binding sites; roughly 2/3 of mRNAs containing an m6A site within their 3′ UTR also have at least one microRNA binding site (Meyer et al., 2012). In another study, Alarcon et al. (2015b) described how miRNAs can undergo N6-adenosine methylation (m6A) as a result of the intervention of METTL3 during pri-miRNA processing. The same authors also showed that m6A marks in pri-miRNAs allow for the RNA-binding protein DGCR8 to identify its specific substrates, promoting the beginning of miRNA biogenesis. Alarcon et al. (2015a) have further hypothesized that the RNA-binding protein HNRNPA2B1 could function as nuclear reader of the m6A mark, binding to m6A marks in pri-miRNAs, thus promoting pri-miRNA processing. Additionally, the effects of RNA demethylation on miRNA expression have also been investigated. Berulava et al. (2015) reported significant miRNA expression dysregulation as a result of knocking down m6A demethylase FTO, providing indirect evidence of co-transcriptional processing in the methylation of mRNAs and miRNAs. Finally, Chen T. et al. (2015) discovered that miRNAs positively regulate m6A installment on mRNAs via a sequence pairing mechanism. Methylation events in miRNAs add a new layer of complexity in the regulation of post-transcriptional gene expression and warrant future studies in order to fully elucidate the roles and functions of modified miRNAs.
Long ncRNA
Although the majority of focus has been recently devoted to modifications in mRNA, 1000s of lncRNA transcripts have been detected containing a substantial number of modifications (Shafik et al., 2016). Evidence associating methylation with the most established lncRNA transcripts are just starting to be recognized. MALAT1 has been shown to bind with the m6A writer METTL16 at its 3′-triple-helical RNA stability element (Brown et al., 2016) specifically in its A-rich portion, after it was previously proven that MALAT1 can carry m6A (Liu et al., 2013). The presence of m6A has been further shown to destabilize the hairpin stems in the transcript, making them more flexible and solvent-accessible (Zhou et al., 2016) as well as more accessible for protein binding (Liu et al., 2015). Several putative m5C sites have also been detected in MALAT1 (Squires et al., 2012), but no enzymes have been identified. lncRNA HOTAIR (Khoddami and Cairns, 2013) possesses a specific m5C site which has been verified with a 100% modification rate (Amort et al., 2013). Finally, m6A events have been associated to XIST-mediated transcriptional repression (Patil et al., 2016) while m5C sites can prevent XIST-protein interactions, although it may not be a conserved mechanism (Amort et al., 2013). More detailed information can be found in Jacob et al. (2017).
Author Contributions
GR wrote and set up the manuscript. GN and DV wrote and reviewed the content. SN-S supervised and reviewed the manuscript writing and development.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00243/full#supplementary-material
References
Abbasi-Moheb, L., Mertel, S., Gonsior, M., Nouri-Vahid, L., Kahrizi, K., Cirak, S., et al. (2012). Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 90, 847–855. doi: 10.1016/j.ajhg.2012.03.021
Adams, J. M., and Cory, S. (1975). Modified nucleosides and bizarre 5′-termini in mouse myeloma mRNA. Nature 255, 28–33. doi: 10.1038/255028a0
Alarcon, C. R., Goodarzi, H., Lee, H., Liu, X., Tavazoie, S., and Tavazoie, S. F. (2015a). HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell 162, 1299–1308. doi: 10.1016/j.cell.2015.08.011
Alarcon, C. R., Lee, H., Goodarzi, H., Halberg, N., and Tavazoie, S. F. (2015b). N6-methyladenosine marks primary microRNAs for processing. Nature 519, 482–485. doi: 10.1038/nature14281
Alexandrov, A., Chernyakov, I., Gu, W., Hiley, S. L., Hughes, T. R., Grayhack, E. J., et al. (2006). Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell 21, 87–96. doi: 10.1016/j.molcel.2005.10.036
Amort, T., Rieder, D., Wille, A., Khokhlova-Cubberley, D., Riml, C., Trixl, L., et al. (2017). Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol. 18:1. doi: 10.1186/s13059-016-1139-1
Amort, T., Souliere, M. F., Wille, A., Jia, X. Y., Fiegl, H., Worle, H., et al. (2013). Long non-coding RNAs as targets for cytosine methylation. RNA Biol. 10, 1003–1008. doi: 10.4161/rna.24454
Anderson, J., Phan, L., Cuesta, R., Carlson, B. A., Pak, M., Asano, K., et al. (1998). The essential Gcd10p-Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev. 12, 3650–3662. doi: 10.1101/gad.12.23.3650
Anderson, J., Phan, L., and Hinnebusch, A. G. (2000). The Gcd10p/Gcd14p complex is the essential two-subunit tRNA(1-methyladenosine) methyl transferase of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 97, 5173–5178. doi: 10.1073/pnas.090102597
Anderson, J. T. (2005). RNA turnover: unexpected consequences of being tailed. Curr. Biol. 15, R635–R638. doi: 10.1016/j.cub.2005.08.002
Backes, S., Shapiro, J. S., Sabin, L. R., Pham, A. M., Reyes, I., Moss, B., et al. (2012). Degradation of host microRNAs by poxvirus poly(A) polymerase reveals terminal RNA methylation as a protective antiviral mechanism. Cell Host Microbe 12, 200–210. doi: 10.1016/j.chom.2012.05.019
Becker, M., Muller, S., Nellen, W., Jurkowski, T. P., Jeltsch, A., and Ehrenhofer-Murray, A. E. (2012). Pmt1, a Dnmt2 homolog in Schizosaccharomyces pombe, mediates tRNA methylation in response to nutrient signaling. Nucleic Acids Res. 40, 11648–11658. doi: 10.1093/nar/gks956
Begley, U., Sosa, M. S., Avivar-Valderas, A., Patil, A., Endres, L., Estrada, Y., et al. (2013). A human tRNA methyltransferase 9-like protein tumour growth by regulating LIN9 and HIF1-α. EMBO Mol. Med. 5, 366–383. doi: 10.1002/emmm.201201161
Berulava, T., Rahmann, S., Rademacher, K., Klein-Hitpass, L., and Horsthemke, B. (2015). N6-adenosine methylation in MiRNAs. PLoS One 10:e0118438. doi: 10.1371/journal.pone.0118438
Birkedal, U., Christensen-Dalsgaard, M., Krogh, N., Sabarinathan, R., Gorodkin, J., and Nielsen, H. (2015). Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chem. Int. Ed. Engl. 54, 451–455. doi: 10.1002/anie.201408362
Boccaletto, P., Machnicka, M. A., Purta, E., Piatkowski, P., Baginski, B., Wirecki, T. K., et al. (2017). MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307. doi: 10.1093/nar/gkx1030
Borges, F., and Martienssen, R. A. (2015). The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Biol. 16, 727–741. doi: 10.1038/nrm4085
Brown, J. A., Kinzig, C. G., Degregorio, S. J., and Steitz, J. A. (2016). Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. U.S.A. 113, 14013–14018. doi: 10.1073/pnas.1614759113
Brownlee, G. G., and Cartwright, E. M. (1977). Rapid gel sequencing of RNA by primed synthesis with reverse transcriptase. J. Mol. Biol. 114, 93–117. doi: 10.1016/0022-2836(77)90285-6
Cenik, C., Chua, H. N., Singh, G., Akef, A., Snyder, M. P., Palazzo, A. F., et al. (2017). A common class of transcripts with 5′-intron depletion, distinct early coding sequence features, and N(1)-methyladenosine modification. RNA 23, 270–283. doi: 10.1261/rna.059105.116
Chan, C. T., Dyavaiah, M., Demott, M. S., Taghizadeh, K., Dedon, P. C., and Begley, T. J. (2010). A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 6:e1001247. doi: 10.1371/journal.pgen.1001247
Chan, C. T., Pang, Y. L., Deng, W., Babu, I. R., Dyavaiah, M., Begley, T. J., et al. (2012). Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 3:937. doi: 10.1038/ncomms1938
Chen, K., Lu, Z., Wang, X., Fu, Y., Luo, G. Z., Liu, N., et al. (2015). High-resolution N6 -methyladenosine (m6 A) map using photo-crosslinking-assisted m6 A sequencing. Angew. Chem. Int. Ed. Engl. 54, 1587–1590. doi: 10.1002/anie.201410647
Chen, T., Hao, Y. J., Zhang, Y., Li, M. M., Wang, M., Han, W., et al. (2015). m6A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 16, 289–301. doi: 10.1016/j.stem.2015.01.016
Cui, Q., Shi, H., Ye, P., Li, L., Qu, Q., Sun, G., et al. (2017). m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 14, 2622–2634. doi: 10.1016/j.celrep.2017.02.059
Dai, Q., Moshitch-Moshkovitz, S., Han, D., Kol, N., Amariglio, N., Rechavi, G., et al. (2017). Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods 14, 695–698. doi: 10.1038/nmeth.4294
Davis, F. F., and Allen, F. W. (1957). Ribonucleic acids from yeast which contain a fifth nucleotide. J. Biol. Chem. 227, 907–915.
Desrosiers, R., Friderici, K., and Rottman, F. (1974). Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. U.S.A. 71, 3971–3975. doi: 10.1073/pnas.71.10.3971
Dev, R. R., Ganji, R., Singh, S. P., Mahalingam, S., Banerjee, S., and Khosla, S. (2017). Cytosine methylation by DNMT2 facilitates stability and survival of HIV-1 RNA in the host cell during infection. Biochem. J. 474, 2009–2026. doi: 10.1042/BCJ20170258
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. doi: 10.1038/nature11112
Dominissini, D., Nachtergaele, S., Moshitch-Moshkovitz, S., Peer, E., Kol, N., Ben-Haim, M. S., et al. (2016). The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 530, 441–446. doi: 10.1038/nature16998
Dubin, D. T., and Taylor, R. H. (1975). The methylation state of poly A-containing messenger RNA from cultured hamster cells. Nucleic Acids Res. 2, 1653–1668. doi: 10.1093/nar/2.10.1653
Dunn, D. B. (1961). The occurrence of 1-methyladenine in ribonucleic acid. Biochim. Biophys. Acta 46, 198–200. doi: 10.1016/0006-3002(61)90668-0
Eddy, S. R. (2001). Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2, 919–929. doi: 10.1038/35103511
El Yacoubi, B., Bailly, M., and De Crecy-Lagard, V. (2012). Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 46, 69–95. doi: 10.1146/annurev-genet-110711-155641
Elhardt, W., Shanmugam, R., Jurkowski, T. P., and Jeltsch, A. (2015). Somatic cancer mutations in the DNMT2 tRNA methyltransferase alter its catalytic properties. Biochimie 112, 66–72. doi: 10.1016/j.biochi.2015.02.022
Esteller, M., and Pandolfi, P. P. (2017). The epitranscriptome of noncoding RNAs in cancer. Cancer Discov. 7, 359–368. doi: 10.1158/2159-8290.CD-16-1292
Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., and Mello, C. C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. doi: 10.1038/35888
Fischer, J., Koch, L., Emmerling, C., Vierkotten, J., Peters, T., Brüning, J. C., et al. (2009). Inactivation of the Fto gene protects from obesity. Nature 458, 894–898. doi: 10.1038/nature07848
Frye, M., Jaffrey, S. R., Pan, T., Rechavi, G., and Suzuki, T. (2016). RNA modifications: what have we learned and where are we headed? Nat. Rev. Genet. 17, 365–372. doi: 10.1038/nrg.2016.47
Golovina, A. Y., Dzama, M. M., Petriukov, K. S., Zatsepin, T. S., Sergiev, P. V., Bogdanov, A. A., et al. (2014). Method for site-specific detection of m6A nucleoside presence in RNA based on high-resolution melting (HRM) analysis. Nucleic Acids Res. 42:e27. doi: 10.1093/nar/gkt1160
Gupta, R. C., and Randerath, K. (1979). Rapid print-readout technique for sequencing of RNA’s containing modified nucleotides. Nucleic Acids Res. 6, 3443–3458. doi: 10.1093/nar/6.11.3443
Guy, M. P., Young, D. L., Payea, M. J., Zhang, X., Kon, Y., Dean, K. M., et al. (2014). Identification of the determinants of tRNA function and susceptibility to rapid tRNA decay by high-throughput in vivo analysis. Genes Dev. 28, 1721–1732. doi: 10.1101/gad.245936.114
Ha, M., and Kim, V. N. (2014). Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 15, 509–524. doi: 10.1038/nrm3838
Helm, M., and Attardi, G. (2004). Nuclear control of cloverleaf structure of human mitochondrial tRNA(Lys). J. Mol. Biol. 337, 545–560. doi: 10.1016/j.jmb.2004.01.036
Helm, M., and Motorin, Y. (2017). Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet. 18, 275–291. doi: 10.1038/nrg.2016.169
Holley, R. W., Apgar, J., Everett, G. A., Madison, J. T., Marquisee, M., Merrill, S. H., et al. (1965a). Structure of a ribonucleic acid. Science 147, 1462–1465. doi: 10.1126/science.147.3664.1462
Holley, R. W., Everett, G. A., Madison, J. T., and Zamir, A. (1965b). Nucleotide sequences in the yeast alanine transfer ribonucleic acid. J. Biol. Chem. 240, 2122–2128.
Hong, T., Yuan, Y., Chen, Z., Xi, K., Wang, T., Xie, Y., et al. (2018). Precise antibody-independent m6A identification via 4SedTTP-Involved and FTO-assisted strategy at single-nucleotide resolution. J. Am. Chem. Soc. 140, 5886–5889. doi: 10.1021/jacs.7b13633
Hori, H. (2014). Methylated nucleosides in tRNA and tRNA methyltransferases. Front. Genet. 5:144. doi: 10.3389/fgene.2014.00144
Hurwitz, J., Gold, M., and Anders, M. (1964). The enzymatic methylation of ribonucleic acid and deoxyribonucleic acid. Iv. The properties of the soluble ribonucleic acid-methylating enzymes. J. Biol. Chem. 239, 3474–3482.
Hussain, S., Aleksic, J., Blanco, S., Dietmann, S., and Frye, M. (2013). Characterizing 5-methylcytosine in the mammalian epitranscriptome. Genome Biol. 14:215. doi: 10.1186/gb4143
Incarnato, D., Anselmi, F., Morandi, E., Neri, F., Maldotti, M., Rapelli, S., et al. (2017). High-throughput single-base resolution mapping of RNA 2′-O-methylated residues. Nucleic Acids Res. 45, 1433–1441. doi: 10.1093/nar/gkw810
Ishida, K., Kunibayashi, T., Tomikawa, C., Ochi, A., Kanai, T., Hirata, A., et al. (2011). Pseudouridine at position 55 in tRNA controls the contents of other modified nucleotides for low-temperature adaptation in the extreme-thermophilic eubacterium Thermus thermophilus. Nucleic Acids Res. 39, 2304–2318. doi: 10.1093/nar/gkq1180
Jackman, J. E., and Alfonzo, J. D. (2013). Transfer RNA modifications: nature’s combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA 4, 35–48. doi: 10.1002/wrna.1144
Jacob, R., Zander, S., and Gutschner, T. (2017). The dark side of the epitranscriptome: chemical modifications in long non-coding RNAs. Int. J. Mol. Sci. 18:E2387. doi: 10.3390/ijms18112387
Jeltsch, A., Ehrenhofer-Murray, A., Jurkowski, T. P., Lyko, F., Reuter, G., Ankri, S., et al. (2017). Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 14, 1108–1123. doi: 10.1080/15476286.2016.1191737
Ji, L., and Chen, X. (2012). Regulation of small RNA stability: methylation and beyond. Cell Res. 22, 624–636. doi: 10.1038/cr.2012.36
Jia, G., Fu, Y., Zhao, X., Dai, Q., Zheng, G., Yang, Y., et al. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887. doi: 10.1038/nchembio.687
Kadaba, S., Krueger, A., Trice, T., Krecic, A. M., Hinnebusch, A. G., and Anderson, J. (2004). Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 18, 1227–1240. doi: 10.1101/gad.1183804
Kaneko, T., Suzuki, T., Kapushoc, S. T., Rubio, M. A., Ghazvini, J., Watanabe, K., et al. (2003). Wobble modification differences and subcellular localization of tRNAs in Leishmania tarentolae: implication for tRNA sorting mechanism. EMBO J. 22, 657–667. doi: 10.1093/emboj/cdg066
Kawahara, Y., Zinshteyn, B., Sethupathy, P., Iizasa, H., Hatzigeorgiou, A. G., and Nishikura, K. (2007). Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315, 1137–1140. doi: 10.1126/science.1138050
Khoddami, V., and Cairns, B. R. (2013). Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 31, 458–464. doi: 10.1038/nbt.2566
Khoddami, V., and Cairns, B. R. (2014). Transcriptome-wide target profiling of RNA cytosine methyltransferases using the mechanism-based enrichment procedure Aza-IP. Nat. Protoc. 9, 337–361. doi: 10.1038/nprot.2014.014
Klungland, A., Dahl, J. A., Greggains, G., Fedorcsak, P., and Filipczyk, A. (2016). Reversible RNA modifications in meiosis and pluripotency. Nat. Methods 14, 18–22. doi: 10.1038/nmeth.4111
Kowalak, J. A., Pomerantz, S. C., Crain, P. F., and Mccloskey, J. A. (1993). A novel method for the determination of post-transcriptional modification in RNA by mass spectrometry. Nucleic Acids Res. 21, 4577–4585. doi: 10.1093/nar/21.19.4577
Krogh, N., Jansson, M. D., Hafner, S. J., Tehler, D., Birkedal, U., Christensen-Dalsgaard, M., et al. (2016). Profiling of 2′-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 44, 7884–7895. doi: 10.1093/nar/gkw482
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854. doi: 10.1016/0092-8674(93)90529-Y
Lewis, C. J., Pan, T., and Kalsotra, A. (2017). RNA modifications and structures cooperate to guide RNA-protein interactions. Nat. Rev. Mol. Cell Biol. 18, 202–210. doi: 10.1038/nrm.2016.163
Li, S., and Mason, C. E. (2014). The pivotal regulatory landscape of RNA modifications. Annu. Rev. Genomics Hum. Genet. 15, 127–150. doi: 10.1146/annurev-genom-090413-025405
Li, X., Xiong, X., Wang, K., Wang, L., Shu, X., Ma, S., et al. (2016a). Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat. Chem. Biol. 12, 311–316. doi: 10.1038/nchembio.2040
Li, X., Xiong, X., and Yi, C. (2016b). Epitranscriptome sequencing technologies: decoding RNA modifications. Nat. Methods 14, 23–31. doi: 10.1038/nmeth.4110
Li, Z., Weng, H., Su, R., Weng, X., Zuo, Z., Li, C., et al. (2017). FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-Methyladenosine RNA demethylase. Cancer Cell 31, 127–141. doi: 10.1016/j.ccell.2016.11.017
Liang, X. H., Liu, Q., and Fournier, M. J. (2009). Loss of rRNA modifications in the decoding center of the ribosome impairs translation and strongly delays pre-rRNA processing. RNA 15, 1716–1728. doi: 10.1261/rna.1724409
Lichinchi, G., Gao, S., Saletore, Y., Gonzalez, G. M., Bansal, V., Wang, Y., et al. (2016a). Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol. 1:16011. doi: 10.1038/nmicrobiol.2016.11
Lichinchi, G., Zhao, B. S., Wu, Y., Lu, Z., Qin, Y., He, C., et al. (2016b). Dynamics of human and viral RNA methylation during zika virus infection. Cell Host Microbe 20, 666–673. doi: 10.1016/j.chom.2016.10.002
Linder, B., Grozhik, A. V., Olarerin-George, A. O., Meydan, C., Mason, C. E., and Jaffrey, S. R. (2015). Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772. doi: 10.1038/nmeth.3453
Little, N. A., Hastie, N. D., and Davies, R. C. (2000). Identification of WTAP, a novel Wilms’ tumour 1-associating protein. Hum. Mol. Genet. 9, 2231–2239. doi: 10.1093/oxfordjournals.hmg.a018914
Liu, F., Clark, W., Luo, G., Wang, X., Fu, Y., Wei, J., et al. (2016). ALKBH1-mediated tRNA demethylation regulates translation. Cell 167, 816–828e816. doi: 10.1016/j.cell.2016.09.038
Liu, J., and Jia, G. (2014). Methylation modifications in eukaryotic messenger RNA. J. Genet. Genomics 41, 21–33. doi: 10.1016/j.jgg.2013.10.002
Liu, J., Yue, Y., Han, D., Wang, X., Fu, Y., Zhang, L., et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95. doi: 10.1038/nchembio.1432
Liu, L., Zhang, S. W., Huang, Y., and Meng, J. (2017). QNB: differential RNA methylation analysis for count-based small-sample sequencing data with a quad-negative binomial model. BMC Bioinformatics 18:387. doi: 10.1186/s12859-017-1808-4
Liu, N., Dai, Q., Zheng, G., He, C., Parisien, M., and Pan, T. (2015). N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564. doi: 10.1038/nature14234
Liu, N., Parisien, M., Dai, Q., Zheng, G., He, C., and Pan, T. (2013). Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19, 1848–1856. doi: 10.1261/rna.041178.113
Ma, J. Z., Yang, F., Zhou, C. C., Liu, F., Yuan, J. H., Wang, F., et al. (2017). METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary microRNA processing. Hepatology 65, 529–543. doi: 10.1002/hep.28885
Maden, B. E., Corbett, M. E., Heeney, P. A., Pugh, K., and Ajuh, P. M. (1995). Classical and novel approaches to the detection and localization of the numerous modified nucleotides in eukaryotic ribosomal RNA. Biochimie 77, 22–29. doi: 10.1016/0300-9084(96)88100-4
Marchand, V., Blanloeil-Oillo, F., Helm, M., and Motorin, Y. (2016). Illumina-based RiboMethSeq approach for mapping of 2′-O-Me residues in RNA. Nucleic Acids Res. 44:e135. doi: 10.1093/nar/gkw547
Marchand, V., Pichot, F., Thuring, K., Ayadi, L., Freund, I., Dalpke, A., et al. (2017). Next-generation sequencing-based RiboMethSeq protocol for analysis of tRNA 2′-O-methylation. Biomolecules 7:E13. doi: 10.3390/biom7010013
Marck, C., and Grosjean, H. (2002). tRNomics: analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA 8, 1189–1232. doi: 10.1017/S1355838202022021
Martinez, F. J., Lee, J. H., Lee, J. E., Blanco, S., Nickerson, E., Gabriel, S., et al. (2012). Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J. Med. Genet. 49, 380–385. doi: 10.1136/jmedgenet-2011-100686
McCloskey, J. A., and Nishimura, S. (1977). Modified nucleosides in transfer RNA. Acc. Chem. Res. 10, 403–410. doi: 10.1021/ar50119a004
Meier, U. T. (2011). Pseudouridylation goes regulatory. EMBO J. 30, 3–4. doi: 10.1038/emboj.2010.323
Metodiev, M. D., Spahr, H., Loguercio Polosa, P., Meharg, C., Becker, C., Altmueller, J., et al. (2014). NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet. 10:e1004110. doi: 10.1371/journal.pgen.1004110
Meyer, K. D., Patil, D. P., Zhou, J., Zinoviev, A., Skabkin, M. A., Elemento, O., et al. (2015). 5′ UTR m(6)A promotes cap-independent translation. Cell 163, 999–1010. doi: 10.1016/j.cell.2015.10.012
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3′. UTRs and near stop codons. Cell 149, 1635–1646. doi: 10.1016/j.cell.2012.05.003
Militello, K. T., Chen, L. M., Ackerman, S. E., Mandarano, A. H., and Valentine, E. L. (2014). A map of 5-methylcytosine residues in Trypanosoma brucei tRNA revealed by sodium bisulfite sequencing. Mol. Biochem. Parasitol. 193, 122–126. doi: 10.1016/j.molbiopara.2013.12.003
Mishima, E., Jinno, D., Akiyama, Y., Itoh, K., Nankumo, S., Shima, H., et al. (2015). Immuno-northern blotting: detection of RNA modifications by using antibodies against modified nucleosides. PLoS One 10:e0143756. doi: 10.1371/journal.pone.0143756
Motorin, Y., Muller, S., Behm-Ansmant, I., and Branlant, C. (2007). Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol. 425, 21–53. doi: 10.1016/S0076-6879(07)25002-5
Muller, S., Windhof, I. M., Maximov, V., Jurkowski, T., Jeltsch, A., Forstner, K. U., et al. (2013). Target recognition, RNA methylation activity and transcriptional regulation of the Dictyostelium discoideum Dnmt2-homologue (DnmA). Nucleic Acids Res. 41, 8615–8627. doi: 10.1093/nar/gkt634
Nachtergaele, S., and He, C. (2017). The emerging biology of RNA post-transcriptional modifications. RNA Biol. 14, 156–163. doi: 10.1080/15476286.2016.1267096
Nigita, G., Acunzo, M., Romano, G., Veneziano, D., Lagana, A., Vitiello, M., et al. (2016). microRNA editing in seed region aligns with cellular changes in hypoxic conditions. Nucleic Acids Res. 44, 6298–6308. doi: 10.1093/nar/gkw532
Nigita, G., Veneziano, D., and Ferro, A. (2015). A-to-I RNA editing: current knowledge sources and computational approaches with special emphasis on non-coding RNA molecules. Front. Bioeng. Biotechnol. 3:37. doi: 10.3389/fbioe.2015.00037
Nishikura, K. (2016). A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 17, 83–96. doi: 10.1038/nrm.2015.4
Patil, D. P., Chen, C. K., Pickering, B. F., Chow, A., Jackson, C., Guttman, M., et al. (2016). m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373. doi: 10.1038/nature19342
Perry, R. P., Kelley, D. E., Friderici, K., and Rottman, F. (1975). The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5′ terminus. Cell 4, 387–394. doi: 10.1016/0092-8674(75)90159-2
Phizicky, E. M., and Hopper, A. K. (2015). tRNA processing, modification, and subcellular dynamics: past, present, and future. RNA 21, 483–485. doi: 10.1261/rna.049932.115
Reitz, C., Tosto, G., Mayeux, R., and Luchsinger, J. A. (2012). NIA-LOAD/NCRAD family study group, Alzheimer’s disease neuroimaging initiative. genetic variants in the fat and obesity associated (FTO) gene and risk of Alzheimer’s disease. PLoS One 7:e50354. doi: 10.1371/journal.pone.0050354
Rottman, F., Shatkin, A. J., and Perry, R. P. (1974). Sequences containing methylated nucleotides at the 5′ termini of messenger RNAs: possible implications for processing. Cell 3, 197–199. doi: 10.1016/0092-8674(74)90131-7
Roundtree, I. A., Evans, M. E., Pan, T., and He, C. (2017). Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200. doi: 10.1016/j.cell.2017.05.045
Saletore, Y., Meyer, K., Korlach, J., Vilfan, I. D., Jaffrey, S., and Mason, C. E. (2012). The birth of the epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 13:175. doi: 10.1186/gb-2012-13-10-175
Schaefer, M., Pollex, T., Hanna, K., Tuorto, F., Meusburger, M., Helm, M., et al. (2010). RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 24, 1590–1595. doi: 10.1101/gad.586710
Schibler, U., and Perry, R. P. (1977). The 5′-termini of heterogeneous nuclear RNA: a comparison among molecules of different sizes and ages. Nucleic Acids Res. 4, 4133–4149. doi: 10.1093/nar/4.12.4133
Schwartz, S., Mumbach, M. R., Jovanovic, M., Wang, T., Maciag, K., Bushkin, G. G., et al. (2014). Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 8, 284–296. doi: 10.1016/j.celrep.2014.05.048
Shafik, A., Schumann, U., Evers, M., Sibbritt, T., and Preiss, T. (2016). The emerging epitranscriptomics of long noncoding RNAs. Biochim. Biophys. Acta 1859, 59–70. doi: 10.1016/j.bbagrm.2015.10.019
Sharma, S., Watzinger, P., Kotter, P., and Entian, K. D. (2013). Identification of a novel methyltransferase, Bmt2, responsible for the N-1-methyl-adenosine base modification of 25S rRNA in Saccharomyces cerevisiae. Nucleic Acids Res. 41, 5428–5443. doi: 10.1093/nar/gkt195
Song, J., and Yi, C. (2017). Chemical modifications to RNA: a new layer of gene expression regulation. ACS Chem. Biol. 12, 316–325. doi: 10.1021/acschembio.6b00960
Sprinzl, M., and Vassilenko, K. S. (2005). Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 33, D139–D140.
Squires, J. E., Patel, H. R., Nousch, M., Sibbritt, T., Humphreys, D. T., Parker, B. J., et al. (2012). Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 40, 5023–5033. doi: 10.1093/nar/gks144
Sun, W. J., Li, J. H., Liu, S., Wu, J., Zhou, H., Qu, L. H., et al. (2016). RMBase: a resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 44, D259–D265. doi: 10.1093/nar/gkv1036
Tan, A., Dang, Y., Chen, G., and Mo, Z. (2015). Overexpression of the fat mass and obesity associated gene (FTO) in breast cancer and its clinical implications. Int. J. Clin. Exp. Pathol. 8, 13405–13410.
Todd, G., and Karbstein, K. (2007). RNA takes center stage. Biopolymers 87, 275–278. doi: 10.1002/bip.20824
Tomikawa, C., Yokogawa, T., Kanai, T., and Hori, H. (2010). N7-Methylguanine at position 46 (m7G46) in tRNA from Thermus thermophilus is required for cell viability at high temperatures through a tRNA modification network. Nucleic Acids Res. 38, 942–957. doi: 10.1093/nar/gkp1059
Van Haute, L., Dietmann, S., Kremer, L., Hussain, S., Pearce, S. F., Powell, C. A., et al. (2016). Deficient methylation and formylation of mt-tRNA(Met) wobble cytosine in a patient carrying mutations in NSUN3. Nat. Commun. 7:12039. doi: 10.1038/ncomms12039
Vandivier, L. E., and Gregory, B. D. (2017). Reading the epitranscriptome: new techniques and perspectives. Enzymes 41, 269–298. doi: 10.1016/bs.enz.2017.03.004
Veneziano, D., Di Bella, S., Nigita, G., Lagana, A., Ferro, A., and Croce, C. M. (2016). Noncoding RNA: current deep sequencing data analysis approaches and challenges. Hum. Mutat. 37, 1283–1298. doi: 10.1002/humu.23066
Veneziano, D., Nigita, G., and Ferro, A. (2015). Computational approaches for the analysis of ncRNA through deep sequencing techniques. Front. Bioeng. Biotechnol. 3:77. doi: 10.3389/fbioe.2015.00077
Voigts-Hoffmann, F., Hengesbach, M., Kobitski, A. Y., Van Aerschot, A., Herdewijn, P., Nienhaus, G. U., et al. (2007). A methyl group controls conformational equilibrium in human mitochondrial tRNA(Lys). J. Am. Chem. Soc. 129, 13382–13383. doi: 10.1021/ja075520+
Wang, X., and He, C. (2014). Reading RNA methylation codes through methyl-specific binding proteins. RNA Biol. 11, 669–672. doi: 10.4161/rna.28829
Wang, X., Lu, Z., Gomez, A., Hon, G. C., Yue, Y., Han, D., et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. doi: 10.1038/nature12730
Wang, X., Zhao, B. S., Roundtree, I. A., Lu, Z., Han, D., Ma, H., et al. (2015). N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399. doi: 10.1016/j.cell.2015.05.014
Yang, X., Yang, Y., Sun, B. F., Chen, Y. S., Xu, J. W., Lai, W. Y., et al. (2017). 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 27, 606–625. doi: 10.1038/cr.2017.55
Yi, J., Gao, R., Chen, Y., Yang, Z., Han, P., Zhang, H., et al. (2017). Overexpression of NSUN2 by DNA hypomethylation is associated with metastatic progression in human breast cancer. Oncotarget 8, 20751–20765. doi: 10.18632/oncotarget.10612
Yokoyama, S., Watanabe, K., and Miyazawa, T. (1987). Dynamic structures and functions of transfer ribonucleic acids from extreme thermophiles. Adv. Biophys. 23, 115–147. doi: 10.1016/0065-227X(87)90006-2
Yu, B., Yang, Z., Li, J., Minakhina, S., Yang, M., Padgett, R. W., et al. (2005). Methylation as a crucial step in plant microRNA biogenesis. Science 307, 932–935. doi: 10.1126/science.1107130
Zhang, S., Zhao, B. S., Zhou, A., Lin, K., Zheng, S., Lu, Z., et al. (2017). m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by Sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31, 591–606. doi: 10.1016/j.ccell.2017.02.013
Zhang, Y. C., Zhang, S. W., Liu, L., Liu, H., Zhang, L., Cui, X., et al. (2015). Spatially enhanced differential RNA methylation analysis from affinity-based sequencing data with hidden markov model. Biomed Res. Int. 2015:852070. doi: 10.1155/2015/852070
Zheng, G., Dahl, J. A., Niu, Y., Fedorcsak, P., Huang, C. M., Li, C. J., et al. (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29. doi: 10.1016/j.molcel.2012.10.015
Keywords: RNA, methylation, epigenetics, non-coding RNAs, RNA methodologies
Citation: Romano G, Veneziano D, Nigita G and Nana-Sinkam SP (2018) RNA Methylation in ncRNA: Classes, Detection, and Molecular Associations. Front. Genet. 9:243. doi: 10.3389/fgene.2018.00243
Received: 05 March 2018; Accepted: 20 June 2018;
Published: 12 July 2018.
Edited by:
Florent Hubé, UMR 7216, Epigénétique et Destin Cellulaire, FranceReviewed by:
Erik Dassi, University of Trento, ItalyYuri Motorin, Université de Lorraine, France
Clement Carre, FR3631 Institut de Biologie Paris Seine, France
Copyright © 2018 Romano, Veneziano, Nigita and Nana-Sinkam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Serge P. Nana-Sinkam, UGF0cmljay5OYW5hLVNpbmthbUB2Y3VoZWFsdGgub3Jn