
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 04 April 2018
Sec. Livestock Genomics
Volume 9 - 2018 | https://doi.org/10.3389/fgene.2018.00105
This article is part of the Research Topic Why Livestock Genomics for Developing Countries offers Opportunities for Success View all 25 articles
 Anahit Nazari-Ghadikolaei1
Anahit Nazari-Ghadikolaei1 Hassan Mehrabani-Yeganeh1*Seyed R. Miarei-Aashtiani1
Hassan Mehrabani-Yeganeh1*Seyed R. Miarei-Aashtiani1 Elizabeth A. Staiger2Amir Rashidi3
Elizabeth A. Staiger2Amir Rashidi3 Heather J. Huson2*
Heather J. Huson2*The Markhoz goat provides an opportunity to study the genetics underlying coat color and mohair traits of an Angora type goat using genome-wide association studies (GWAS). This indigenous Iranian breed is valued for its quality mohair used in ceremonial garments and has the distinction of exhibiting an array of coat colors including black, brown, and white. Here, we performed 16 GWAS for different fleece (mohair) traits and coat color in 228 Markhoz goats sampled from the Markhoz Goat Research Station in Sanandaj, Kurdistan province, located in western Iran using the Illumina Caprine 50K beadchip. The Efficient Mixed Model Linear analysis was used to identify genomic regions with potential candidate genes contributing to coat color and mohair characteristics while correcting for population structure. Significant associations to coat color were found within or near the ASIP, ITCH, AHCY, and RALY genes on chromosome 13 for black and brown coat color and the KIT and PDGFRA genes on chromosome 6 for white coat color. Individual mohair traits were analyzed for genetic association along with principal components that allowed for a broader perspective of combined traits reflecting overall mohair quality and volume. A multitude of markers demonstrated significant association to mohair traits highlighting potential candidate genes of POU1F1 on chromosome 1 for mohair quality, MREG on chromosome 2 for mohair volume, DUOX1 on chromosome 10 for yearling fleece weight, and ADGRV1 on chromosome 7 for grease percentage. Variation in allele frequencies and haplotypes were identified for coat color and differentiated common markers associated with both brown and black coat color. This demonstrates the potential for genetic markers to be used in future breeding programs to improve selection for coat color and mohair traits. Putative candidate genes, both novel and previously identified in other species or breeds, require further investigation to confirm phenotypic causality and potential epistatic relationships.
Throughout history, goats have played a vital role in the livelihood of humans, being a main source of meat, milk, fiber, and hides, especially in harsh environmental conditions. Archeological evidence supports goat domestication around 10,000 years ago in the Zagros Mountain region in Iran (Zeder and Hesse, 2000) with the UN Food and Agriculture Organization (FAO) estimating 25 million goats in the country today (FAOSTAT, 2008). Many goat breeds, including the Iranian Markhoz breed, have adapted to climates with extremely high temperatures, low rainfall, and low humidity. Yet it is the mohair or long, silky hair of the Markhoz goat that make this indigenous breed unique. Mohair is generally associated with the popular Angora breed and as such, the Markhoz goat is oftentimes considered an Angora goat. The Markhoz goat is the only mohair-producing breed within the Kurdistan province in the west of Iran. A prominent feature of this breed is the coat color variation, which can be dark to light brown, black, gray, or white. This coat color variation is unique among Angora goats which are predominantly selected for a white coat color (Rashidi et al., 2006). The mohair, particularly the brown color from the Markhoz goats, is often used to make clothing for important cultural ceremonies, especially weddings. Unfortunately, due to a reduction in population size, a genetic bottleneck has been observed in the Markhoz population reducing diversity (Rashidi et al., 2015). Cross breeding and inbreeding are additional concerns for all local breeds as inbreeding is likely increasing as population sizes diminish and innate characteristics of the indigenous breeds may be lost during admixture with other breeds (Hanotte et al., 2010).
While the Markhoz goat is indigenous to Iran, Angora goats are now found predominantly in South Africa, the United States, and Argentina, with smaller herds in other countries like Turkey, Australia, and New Zealand (Mohair South Africa, 2017). Mohair production is unique to these animals in that they are the only single coated breeds in which the primary and secondary hair follicles produce the same fiber (Mohair South Africa, 2017). Adult Angora goats are typically shorn twice a year in the aforementioned major production countries providing 2–2.5 kg of mohair per goat (Mohair South Africa, 2017). Mohair is a type of fiber collected from Markhoz and Angora goats that is exceptionally soft, has a high luster, and is used worldwide in the textile and clothing industries. The softness of mohair and other quality traits are determined based on the hair’s diameter, kemp, and medullated and greasy fiber content. Kemp is an undesirable fiber characteristic that causes irregular dying properties and a coarse appearance due to the medullated hair fibers having a core of air-filled cells and course medulla. The less kemp, the better the mohair quality and true fiber percentage of the fleece. Increased greasy fiber percentage or kemp percentage is unfavorable for industry purposes, yet both qualities are important adaptive mechanisms which protect the mohair against humidity and environmental contamination such as dirt and vegetation (Mohair South Africa, 2017). A softer hair fiber has a smaller diameter with less kemp, and less medullated and greasy fiber content. True fiber is considered pure fiber without kemp while fiber efficiency equates clean fleece without any grease content.
There are few genome-wide studies for production and disease related traits in goats as compared to other mammals (Zidi et al., 2014; Becker et al., 2015; Lan et al., 2015; Reber et al., 2015; Martin P. et al., 2016; Martin P.M. et al., 2016; Menzi et al., 2016). To date, goats are not included in a Quantitative Trait Loci (QTL) database (Hu et al., 2016). However, linkage analysis has identified QTL regions for coefficient of variation of fiber diameter, kemp fiber, discontinuous medullated fiber, staple length, fleece weight, fiber diameter, and comfort factor spinning fineness in Angora goats and for fleece yield in Cashmere goats (Cano et al., 2007, 2009; Visser et al., 2011; Roldan et al., 2014). The keratin (KRT) and keratin associated protein (KRTAP) family genes located on chromosomes 1 and 5 were highlighted as candidate genes potentially responsible for diameter and kemp traits in Angora goats and have been previously confirmed in sheep (Parsons et al., 1994; Cano et al., 2007). More recently, whole-genome sequences of two Chinese breeds of cashmere goats were compared for signatures of selection and identified genes and biological pathways potentially related to cashmere production (Li et al., 2017). Gene editing of the fibroblast growth factor 5 (FGF5) gene in goat embryos resulted in an increased number of second hair follicles and longer fiber length which would suggest greater cashmere production (Wang et al., 2016).
The vast majority of Markhoz goats have a brown coat color likely due to the higher value of this color fiber for cultural events. Coat color pigmentation is a polygenic trait with genes often having epistatic interactions (Sturm et al., 2001). Well-known genes involved in coat color include melanocyte-stimulating hormone receptor (MC1R) and agouti signaling protein (ASIP or Agouti) which have a consistent effect across many species. The MC1R gene plays a key role in melanin color theme synthesis and the concentration of eumelanin or pheomelanin, which can lead to the black/brown or red/yellow phenotype, respectively. There are several studies that have investigated MC1R in cattle and sheep for coat color patterns (Klungland et al., 1995; Joerg et al., 1996; Våge et al., 1999; Fontanesi et al., 2009a; Switonski et al., 2013). Similarly, mutations in the MC1R gene have been associated with various coat color patterns in the Girgentana, Maltese, Derivata di Siria, Murciano-Granadina, Camosciata delle Alpi and Saanen goats as well (Fontanesi et al., 2009a). ASIP has an epistatic effect with MC1R and can reduce MC1R activity to generate more pheomelanin by preventing cAMP production. Yellow or pheomelanin pigmentation is a result of a dominant allele (A) at the ASIP locus, while recessive allele (a) produces eumelanin resulting in the black/brown phenotype (Adalsteinsson et al., 1994). In Saanen goats, the dominant AWt (white/tan) allele seems to be responsible for the white coat color (Martin P.M. et al., 2016). In sheep, a gene duplication in the ASIP gene is responsible for white and black phenotypes (Norris and Whan, 2008). Another gene for coat color is proto-oncogene receptor tyrosine kinase (KIT) which plays a key role for different white color patterns in the pig, cat, cow, mouse, horse, rabbit, dog, and camel (Geissler et al., 1988; Marklund et al., 1998; Pielberg et al., 2002; Haase et al., 2009, 2015; Fontanesi et al., 2010a,b, 2014; Wong et al., 2013; David et al., 2014; Durig et al., 2017; Holl et al., 2017).
The aim of this study was to characterize mohair and coat color phenotypes in Markhoz goats, and identify candidate genes likely influencing these characteristics by exploring the entire genome. Traits included true fiber percentage, grease percentage, kemp percentage, efficiency, diameter, staple length, mature fleece weight and yearling fleece weight. Additionally, we performed principal component analysis (PCA) of the mohair traits to combine qualities into a single variable. The use of PCA generated two new traits with PC1 reflecting fiber quality and PC2 reflecting fiber volume. Candidate genes were identified through genome-wide association of genetically characterized goats using the Illumina Caprine 50K beadchip (Tosser-Klopp et al., 2014). Genome-wide association studies (GWAS) were conducted with significant associations identifying putative candidate genes including ASIP and KIT genes for brown/black and white coat color, respectively. These studies support the established roles of well-known genes such as ASIP and KIT in coat color, and identify novel genes such as melanoregulin (MREG), which potentially influence mohair quality. Additionally, specific genetic markers and haplotypes are identified which show potential for use in genetic selection schemes for coat color. This provides a foundation to further investigate the biological pathways and causative mutations influencing industry-valued qualities of mohair and the biological implication to animal adaptation.
Coat color and seven fleece traits from a total of 228 Markhoz goats (44 males and 184 females) were sampled at the Markhoz goat Research Station in Sanandaj, Kurdistan province, located in western Iran. All animal procedures were approved by the Cornell University Institutional Animal Care and Use Committee prior to sampling (protocol #2014-0121), and were conducted in a manner to minimize animal stress and handling. Goats ranged in age from 1 to 7 years old. Fleece traits included diameter, kemp percentage, staple length, true fiber percentage, efficiency, grease percentage, and fleece weight. True fiber is considered pure fiber without kemp. Fiber efficiency is equivalent to the clean fleece with no grease and can be calculated by subtracting clean fleece weight from greasy fleece weight and dividing this total by the greasy fleece weight. Coat color was classified into three different categories: brown (n = 168), black (n = 26), or white (n = 26; Figure 1). Coat colors have been routinely recorded by the research station since 1992. Photographs were taken of each animal at sample collection according to the USDA_AGIN Goat Sample Protocol (USDA, 2016).

FIGURE 1. Coat color variation in Markhoz goats used for a genome-wide association study. (A) Brown (n = 168), (B) black (n = 26), (C) white (n = 26).
All animals were assessed for data quality and completeness. Statistical analysis was performed using proc GLM in SAS studio university edition (SAS Institute Inc., Cary, NC, United States) for each fleece trait to determine their relationship to variables such as sex, age (from 1 to 7 years), dam’s age (2 to 8 years), type of birth (single, twin, or triplet) and color (black, brown, or white) for subsequent inclusion as covariates in the GWAS. The specific number of animals used in each GWAS is denoted in Table 1 and varies based on the number of animals with quality trait information and the GWAS model chosen. We also performed least square means and used the Tukey method for comparing means of males and females for yearling fleece weight and comparing coat colors (black, brown, and white) for fiber volume. PCA was performed on seven of the fiber traits for animals with complete records across all fiber traits using JMP PRO 12 (SAS Institute Inc., Cary, NC, United States). The correlation matrix was applied due to the wide variation in quantitative measures of each trait. PCA was used to generate single quantitative variables combining the seven mohair traits with principal components retained for interpretation and analysis if the eigenvalue score was greater than 1.0.
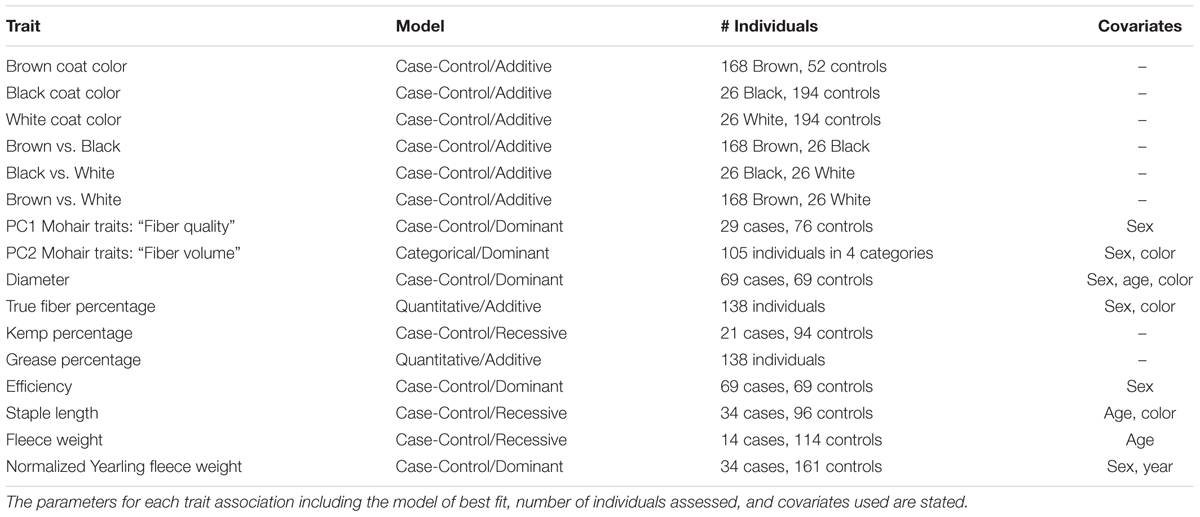
TABLE 1. Genome-wide association studies conducted for coat color and mohair traits in Markhoz goats.
Whole blood (5 ml) was obtained via the jugular vein into vacutainers with the anticoagulant K2EDTA for subsequent DNA extraction. Genomic DNA was extracted following a standard Phenol-Chloroform extraction protocol (Sambrook and Russell, 2006). All samples were genotyped on the Illumina Caprine 50K beadchip (Illumina, Inc., San Diego, CA, United States) at VHL Genetics (VHL Genetics, Wageningen, Netherlands). The initial 53,347 SNPs were assessed for quality and removed if they had a call rate less than 0.9 (n = 624) and a minor allele frequency less than 0.03 (n = 2540). An additional 419 SNPs, unassigned to a chromosome position, were removed, leaving 49,764 SNPs for analysis. Five samples were subsequently removed with a genotyping call rate less than 0.9. To evaluate population structure and relatedness, we used an identity-by-state (IBS) similarity matrix to calculate genome-wide identity-by-descent (IBD) estimates. Three animals were removed due to having an estimated IBS score greater than 0.90 denoting substantial relatedness. Genotype quality control was conducted using Golden Helix SVS v8.3.4 (Golden Helix, Bozeman, MT, United States).
Multiple genome-wide tests were performed for coat color and mohair traits (Table 1) using Golden Helix SVS v8.3.4 (Golden Helix, Bozeman, MT, United States). Two hundred and twenty individuals were included in the coat color GWAS and 138 individuals were included in the mohair trait GWAS including 179 females and 41 males, and 115 females and 23 males, respectively. Quantitative or case-control associations were used in an Efficient Mixed Model Linear analysis (EMMAX) (Kang et al., 2010) to correct for remaining population structure and relatedness by including genomic relationship matrix as a random effect in a model. Coat color GWAS were performed in a case-control study design comparing the identified coat color to all other coat colors combined, including brown compared to black and white, black compared to brown and white and white compared to black and brown. Additional GWAS were evaluated with smaller sample sizes to compare single coat colors to one another (i.e., brown compared to black). Variation in brown coat color was considered but small sample size and insufficient differentiation between color variations precluded an association analysis. Association studies with the covariates of sex (male or female), age (1–7 years old), dam’s age (2–8 years old), and type of birth (single or twins) were considered in additive, dominant, and recessive inheritance models. Quantile–quantile (QQ) plots were used to determine the model of best fit for each trait. Quantitative measures were used in the GWAS for true fiber, grease percentages, and yearling fleece weight. For the remaining mohair traits in which no statistically significant loci were observed using a quantitative variable, a case/control model was then applied using a threshold based on the median or quartile values to compare representatives demonstrating the greatest degree of phenotypic variation within the group. For traits that did not surpass an adjusted Bonferroni significance cutoff or an adjusted false discovery rate (FDR) significance cutoff of 0.05, we investigated significance using adaptive permutation for the model of best fit using PLINK v1.9 (Chang et al., 2015). Adaptive permutation evaluates the genomic dataset more quickly in that it discards SNPs which are not demonstrating association from further permutations, while continuing to analyze associated SNPs to the set threshold. Adaptive permutation output provides both the number of permutations achieved and corresponding P-value. Parameters used in the adaptive permutation testing included a minimum of five permutations performed but no more than 1,000,000 to determine significance using a confidence interval of 0.0001, alpha threshold of 0, intercept interval for pruning of 1, and slope interval of 0.001 for pruning (Che et al., 2014). Linkage disequilibrium (LD) structure and haplotype analysis was examined between associated markers using HAPLOVIEW v4.2 to assist in candidate gene identification (Barrett et al., 2005). Haplotypes blocks were defined using the algorithm from Gabriel et al. (2002). Putative candidate gene(s) within one million base pairs up or down stream or within LD blocks of significantly associated SNPs were identified based on the GCF-001704415.1(ARS1) assembly in Genome Data Viewer on National Center for Biotechnology Information (NCBI) (Bickhart et al., 2017).
Descriptive statistics of mean, standard deviation, minimum, and maximum for the seven fleece traits and yearling fleece weight are shown in Table 2. Covariate usage was determined using proc GLM in SAS for each GWAS (Table 1). No significant covariates were identified for kemp percentage or greasy fleece percentage. Sex was significant for diameter and true fiber percentage with a P-value < 0.0001, and for efficiency with a P-value < 0.05. Age was significant as a covariate for diameter and staple length with a P-value < 0.05, and for fleece weight with a P-value < 0.0001. Color was significant for diameter, true fiber, staple length and fleece weight with a P < 0.05. Least square means for yearling fleece weight in males was 392.4 ± 25.53, and 298.29 ± 18.69 in females (Tukey adjusted P-value < 0.0001). We normalized yearling fleece weight based on yearling body weight due to the positive correlation between these two traits.
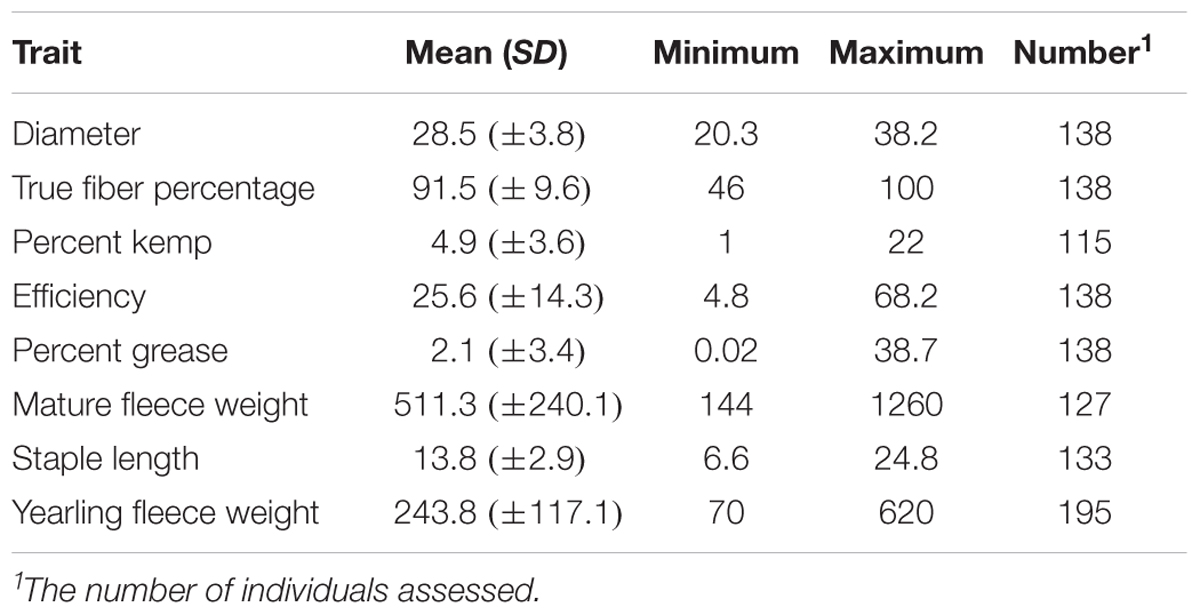
TABLE 2. Descriptive statistics for the mohair traits evaluated.
To further investigate the level of variation of fiber traits in the Markhoz goats, we performed PCA. Principal components 1 and 2 cumulatively accounted for 56.1% of the total variance and were retained for further analysis. The specific fiber traits with absolute loading values greater than 0.4 were used to broadly describe the variation represented by each principal component. Principal component 1 (PC1) accounted for 29.9% of the total variance with the fiber traits for diameter and kemp percentage loading strongly on the positive side, while true fiber percentage loaded negatively (Figure 2A). PC1 broadly describes individual fiber quality with negatively scoring individuals having desirable mohair traits such as increased true fiber percentage and efficiency as the fiber diameter decreases and kemp percentage is reduced. Principal component 2 (26.2%) generally describes fiber volume, with fleece weight and staple length loading positively in contrast to kemp percentage loading negatively (Figure 2B). Animals with a positive PC2 score will have longer, thicker fibers supporting greater fleece weight. Coat color was significantly associated with PC2 (P-value < 0.05). Least square means for PC2 scores in black animals was 11.45 ± 2.03, in brown animals was 11.22 ± 2.05, and in white animals was 12.13 ± 2.11 (P-value < 0.0001). White animals, particularly in comparison to brown animals, demonstrated a higher value of PC2 (Tukey adjusted P-value < 0.05) which correlated to white animals producing a greater volume of fiber than colored animals.
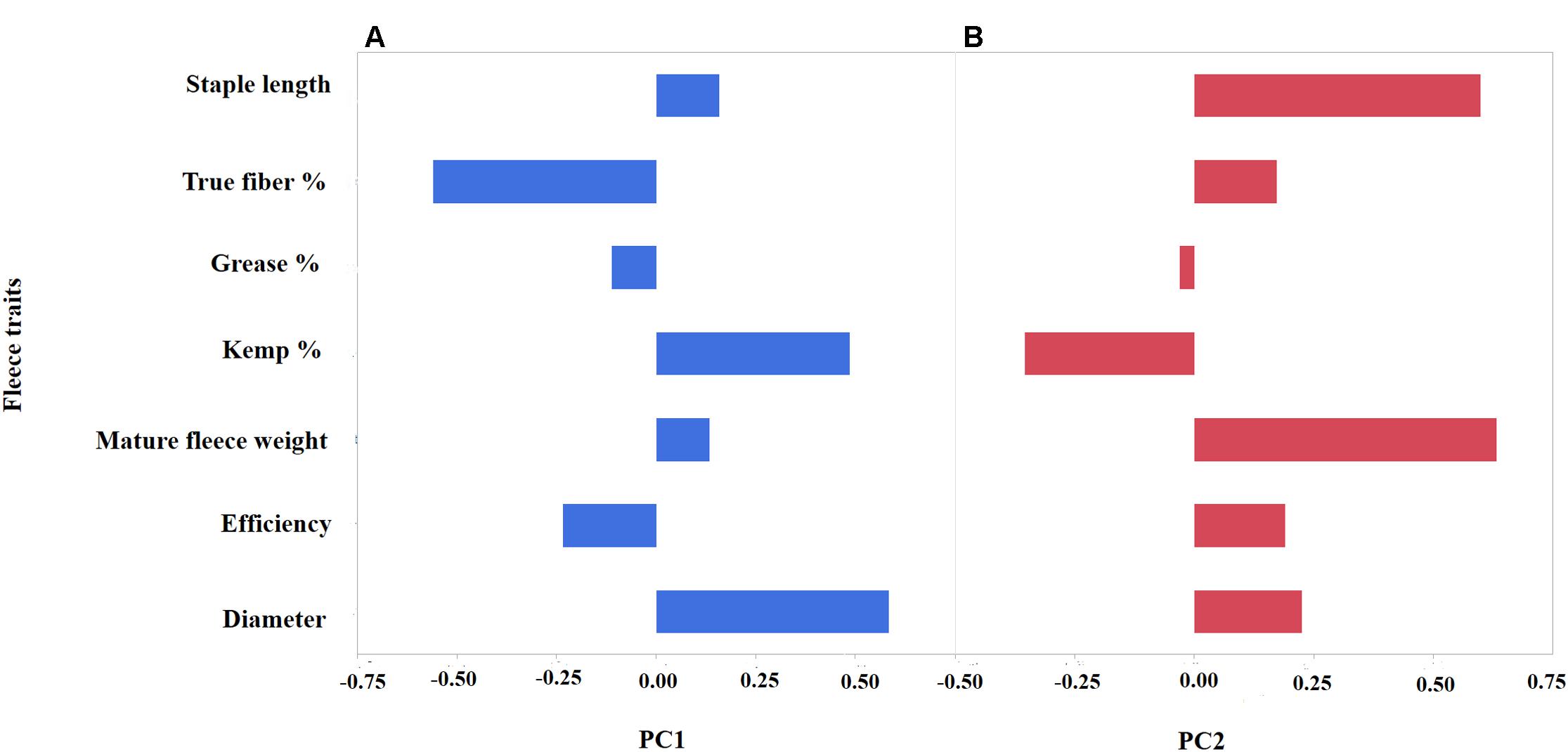
FIGURE 2. Principal Component Analysis (PCA) depicting the loading values (x-axis) of seven mohair traits (y-axis) for PC1 (A) and PC2 (B). PC1 (blue) broadly describes “mohair quality” as indicated by the positive correlation between diameter and kemp percentage, while having negative correlation to true fiber percentage. PC2 (red) reflects fleece volume with a positive correlation between increased staple length and fleece weight.
Genome-wide association studies were performed for the coat colors of black, brown, and white utilizing an additive inheritance case-control model for each (Figures 3A–C and Supplementary Figures S1, S2). Utilizing the broader approach of comparing the coat color of interest (case) to all individuals not presenting that coat color (control), QTL were identified with proposed candidate genes (Figures 3A–C). Pairwise comparisons of each coat color also identified significantly associated markers despite the low sample sizes with results supporting the broader analysis (Supplementary Figure S2 and Supplementary Table S2). Significant regions associated with black coat color were located on chromosomes 1, 6, 13, 18, 19, and 25, with EMMAX P-values ranging from 1.20 × 10-05 to 9.62 × 10-15 (Supplementary Table S1). Loci for brown coat color were identified on chromosomes 13, 19, and 25, with EMMAX P-values ranging from 1.62 × 10-06 to 5.18 × 10-09 (Supplementary Table S1). The assessment of white coat color produced the greatest number of results with significant associations on 11 different chromosomes (passing both Bonferroni and FDR cutoffs) with EMMAX P-values ranging from 9.68 × 10-05 to 5.12 × 10-12 (Supplementary Table S1).
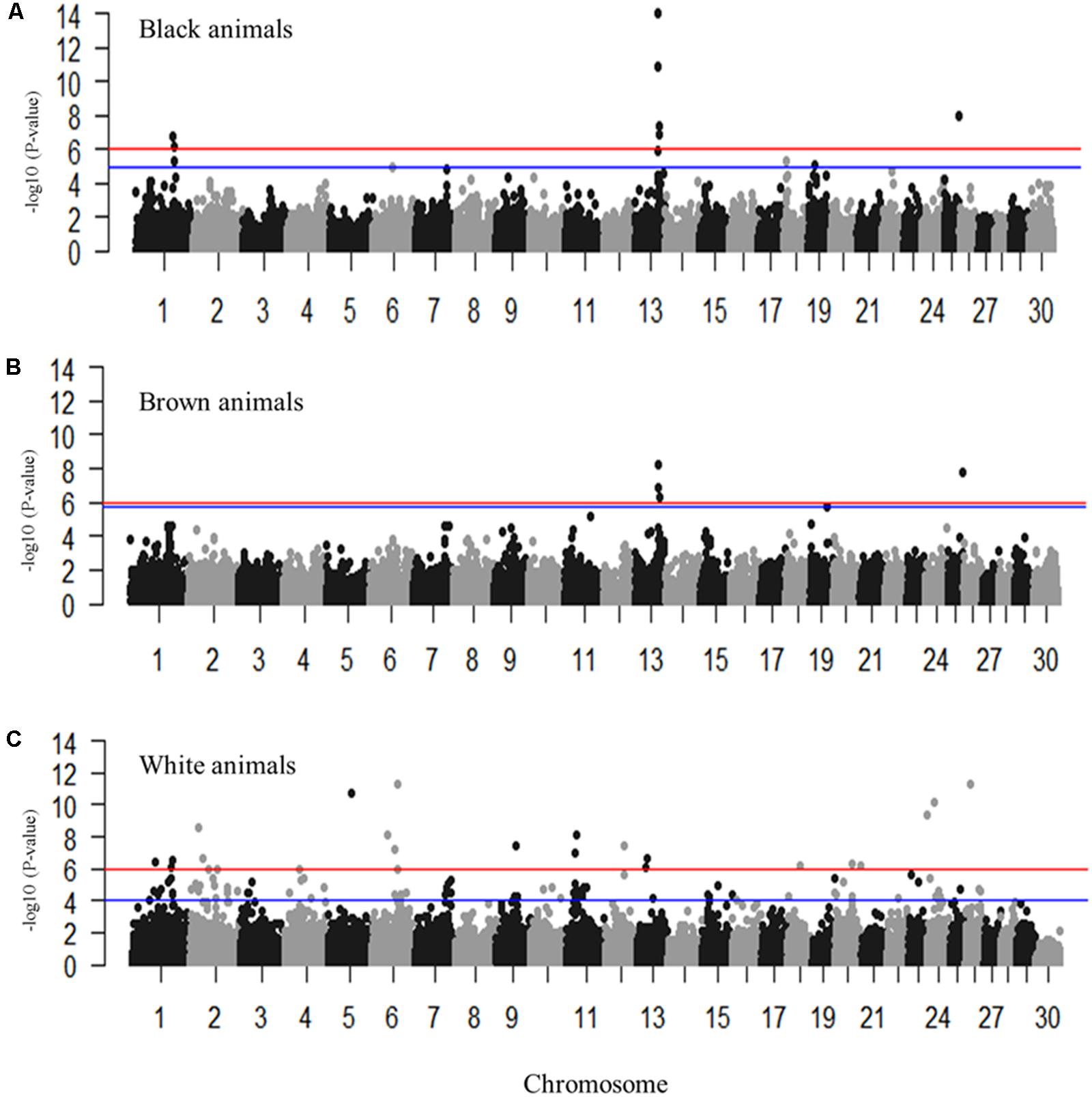
FIGURE 3. Manhattan plots for the coat colors of black, brown and white in Markhoz goats. (A) Black coat color contrasted to all non-black coat colors. (B) Brown coat color contrasted to all non-brown coat colors. (C) White coat color contrasted to all non-white coat colors. The red and blue horizontal lines indicate Bonferroni and FDR corrected P-values of 0.05, respectively.
The most significantly associated SNP for both the black and brown GWAS (Figures 3A,B) is located on chromosome 13 (snp55189-scaffold849-226217) within the Adenosylhomocysteinase (AHCY) gene. This SNP is within a 465 Kb block of LD encompassing AHCY, ASIP, and RALY heterogeneous nuclear ribonucleoprotein (RALY) genes, among others (Figures 4A,B). The second most associated SNP for black and third associated SNP for brown is also located on chromosome 13 (snp55186-scaffold849-83968) but within the itchy E3 ubiquitin protein ligase (ITCH) gene, also found in this same LD block (Figures 4A,B). The SNP on chromosome 25 falls within the sidekick cell adhesion molecule1 (SDK1) gene for both brown and black animals. The minor alleles and minor allele frequencies associated with these SNPs are presented in Supplementary Table S1. The pairwise comparison of black to brown supported these findings with variation in allele frequencies highlighting the same QTLs on chromosomes 13 and 25 (Supplementary Figure S2A and Supplementary Table S2). A new QTL was highlighted on chromosome 7 which narrowly missed the FDR threshold in the broader black comparison.
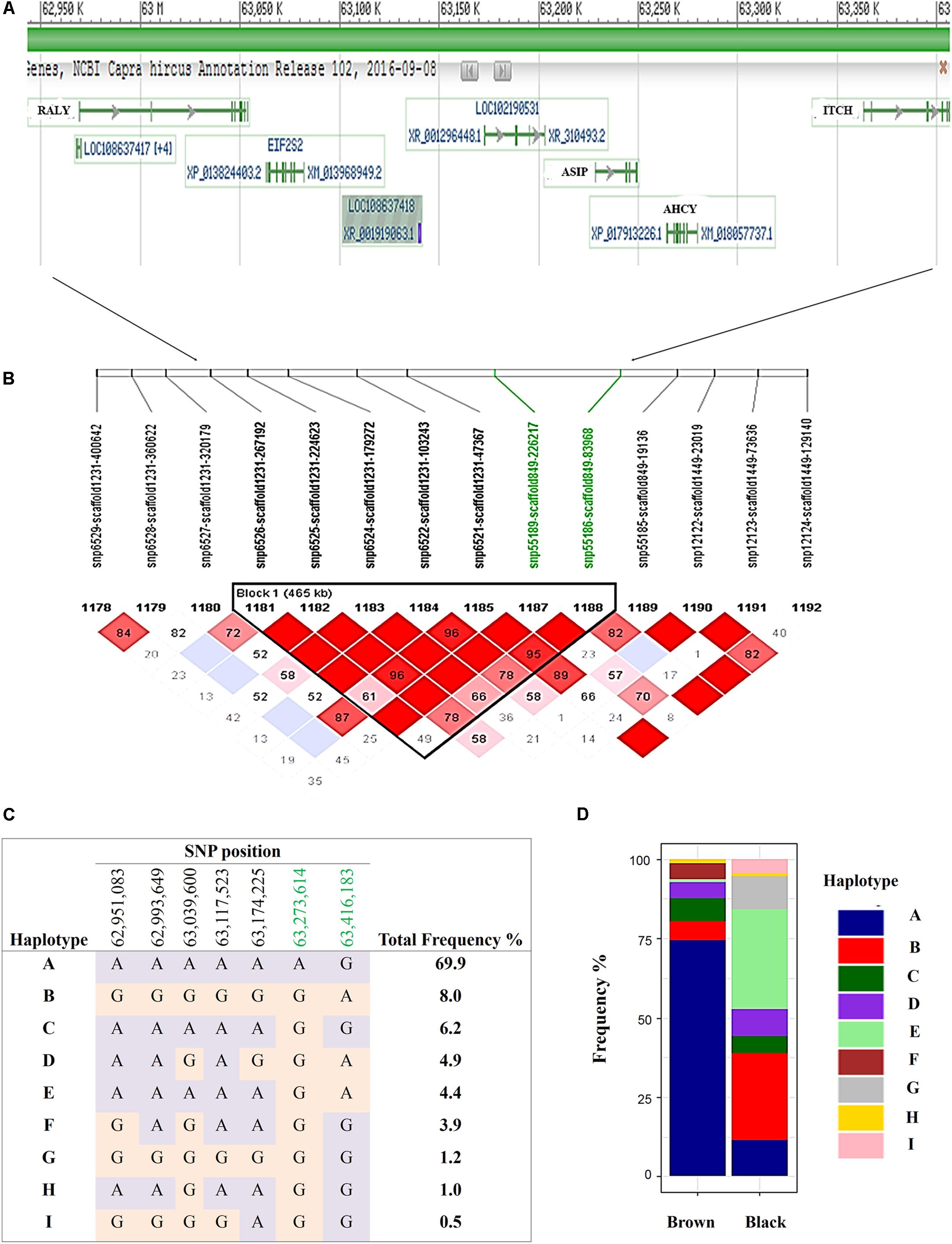
FIGURE 4. Haplotypes within a single 465 Kb LD block on chromosome 13 for brown and black coat color. (A) Genes incorporated in the (B) 465 Kb LD block (C) defined 9 haplotypes within the sampled Markhoz goat population and (D) the frequency of the haplotypes in black and brown goats. Gene track images are from NCBI Genome Data Viewer (https://www.ncbi.nlm.nih.gov/genome/gdv/browser/?acc=GCF_001704415.1&context=genome), accessed 10/11/2017).
Despite the common association of multiple SNPs for both brown and black coat color, genotypic frequencies and haplotype association reflect specific color descriptions. Nine haplotypes, incorporating seven SNPs, were identified within the 465 Kb LD block which included the above mentioned SNPs on chromosome 13 (Figure 4C). In total, five of the haplotypes were significantly associated with both black and brown coat color. Of the associated haplotypes, four of these were observed at a higher frequency in black animals while one was seen at a higher frequency in brown animals. A sixth haplotype was only associated and found at a higher frequency in brown animals. The percentage of each haplotype frequency found within black or brown coat color animals is depicted in Figure 4D and Supplementary Table S3. Genotypic frequencies for the individually associated SNP on chromosome 25 similarly reflects coat color variation between black and brown (Figure 5A).
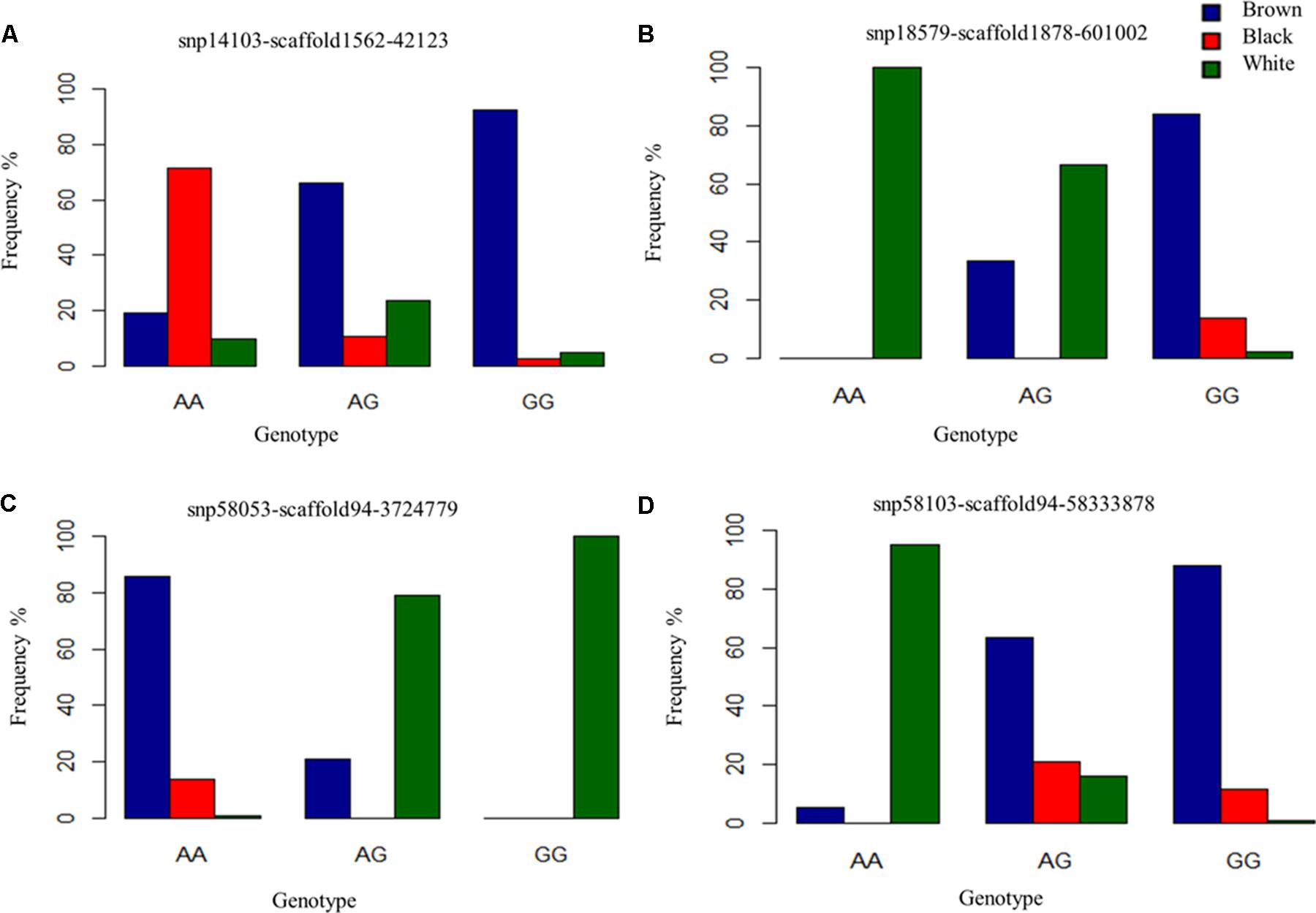
FIGURE 5. Genotype frequencies of prominently associated SNPs for each coat color. (A) Identifies a commonly associated SNP for black and brown coat color demonstrating variation in genotypic frequency. (B–D) Depict genotypic frequency distribution for three SNPs associated with white coat color.
For white animals, significant SNPs were detected on chromosomes 1, 2, 5, 6, 11, 12, 13, 18, 20, 26, and 24 with EMMAX P-values ranging from 9.68 × 10-05 to 5.12 × 10-12 (Supplementary Table S1). The most associated SNP (snp18579-scaffold1878-601002) is located on chromosome 26 (Figure 3C). However, the second associated SNP (snp58053-scaffold94-3724779) is on chromosome 6, relatively close (15,450 bp upstream) to the RAS like family 11 member B (RASL11B) gene but potentially more importantly, it is 1.6 Mb from the KIT gene (Supplementary Table S1). Association to the KIT gene is also supported by the association of snp58103-scaffold94-5833878, which is only 370 Kb downstream (Supplementary Table S1). Genotypic frequencies for these three SNPs are presented in Figures 5B–D. Pairwise comparison of white to black and white to brown (Supplementary Figures S2B,C, respectively) largely reflect the multiple signals found in the broader analysis of white coat color. This is particularly evident in the comparison of the brown to white individuals. However, the comparison of black to white individuals identified many new SNPs primarily on the same chromosomes and near the same regions. Novel QTL were identified on chromosomes 8 and 28 when comparing black to white individuals but yielded no candidate genes whereas the broader white comparison showed unique QTL on chromosomes 10, 11, 18, and 25.
We performed eight GWASs for the mohair traits. Only GWAS for true fiber, efficiency, grease percentage and yearling fleece weight could surpass our Bonferroni or FDR corrected P-value of less than 0.05 (Supplementary Table S1 and Supplementary Figure S1). GWAS for PC1, PC2, diameter, staple length and mature fleece weight did not surpass the Bonferroni or FDR cutoff, but following one million permutations, candidate regions were identified for each trait (Supplementary Table S1 and Supplementary Figure S1).
The GWAS for PC1 representing fiber quality identified nine loci on chromosomes 1, 6, 8, 15, and 18 with permutated P-values ranging from 3.18 × 10-05 to 1.53 × 10-06 (Figure 6A and Supplementary Table S1). SNPs related to true fiber percentage were identified on chromosome 24 and X with the EMMAX P-values ranging from 8.78 × 10-6 to 3.55 × 10-8 (Figure 6B). After one million permutations, four SNPs associated to fiber diameter (Figure 6C) were identified on chromosomes 13 and 27 passing one million permutations and additional SNPs on chromosome 1 and 6 reaching 999,991 and 826,000 permutations with the P-value from 4.36 × 10-5 to 1.00 × 10-6 (Figure 6C), respectively. SNPs on chromosomes 6 and 13 were located within the secreted protein acidic and cysteine rich (SPARC) and solute carrier family 24 member 3 (SLC24A3) genes (Supplementary Table S1). There were only two significantly associated SNPs on chromosome 7 and 9 for kemp percentage with EMMAX P-values of 7.41 × 10-07 and 7.83 × 10-08 P-values, respectively, (Figure 6D).
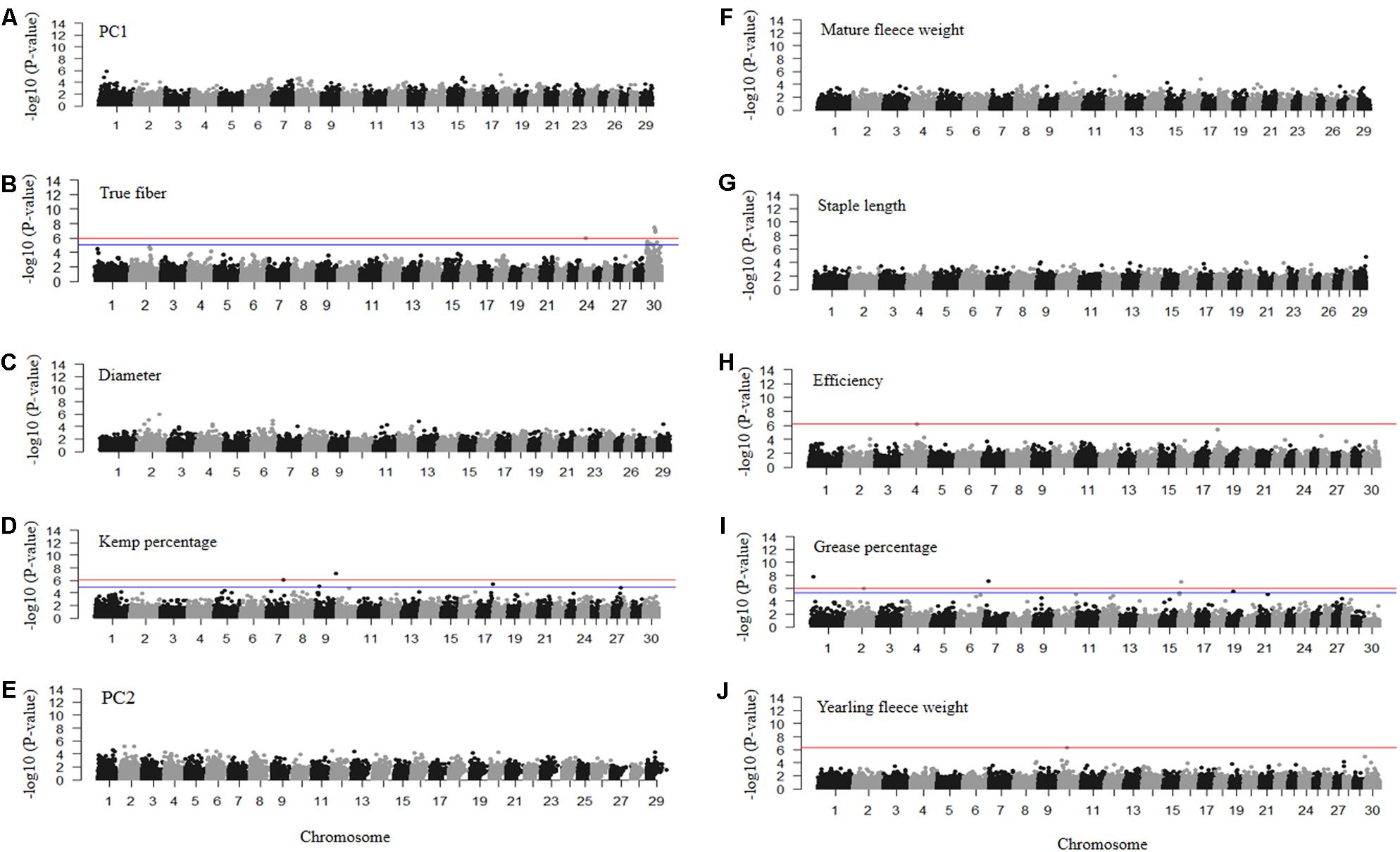
FIGURE 6. Manhattan plots of –log10 (P-values) for association of SNP loci for mohair traits (A) PC1 reflecting fiber quality, (B) true fiber percentage, (C) fiber diameter, (D) kemp percentage, (E) PC2 reflecting fiber volume, (F) mature fleece weight, (G) staple length, (H) efficiency, (I) grease percentage, and (J) yearling fleece weight. Traits with red and blue horizontal lines indicate Bonferroni and FDR corrected P-values of 0.05, respectively. Those with only a red horizontal line indicate P-values thresholds were the same for both Bonferroni and FDR. Those without horizontal lines reflect GWAS run using permutation testing.
Permutation testing of PC2 scores reflecting fiber volume identified six SNPs that passed one million permutations on chromosomes 1, 2, 6, and 12 with permutated P-values ranging from 3.79 × 10-05 to 6.00 × 10-06 (Figure 6E). The SNPs on chromosome 2 fell within the coiled-coil domain containing 148 (CCDC148) gene. SNPs for mature fleece weight, measured at the sampling age of the goats (1–8 years old), were unable to reach one million permutations, however, a SNP on chromosome 16 reached 734,732 permutations (Figure 6F) and is located within the uncharacterized LOC102169208 gene (Supplementary Table S1). Staple length produced a SNP on chromosome 29 with a permutated P-value of 1.58 × 10-05 after reaching 886,838 permutations (Figure 6G) that fell within the potassium voltage-gated channel subfamily Q member 1 (KCNQ1) gene. The GWAS for efficiency only identified one significantly associated SNP on chromosome 4 with an EMMAX P-value of 6.38 × 10-07 (Figure 6H) within the inner mitochondrial membrane peptidase subunit2 (IMMP2L) gene. Grease percentage had multiple significantly associated SNPs on chromosomes 1, 2, 7, 16, and 19 with EMMAX P-values ranging from 8.36 × 10-06 to 1.72 × 10-08 (Figure 6I and Supplementary Table S1). The GWAS for yearling fleece weight identified one associated SNP on chromosome 10 with EMMAX P-value 5.21 × 10-07 (Figure 6J) within the sorbitol dehydrogenase (SORD) gene (Supplementary Table S1).
Markhoz goats are one of the few mohair-producing breeds and, due to their important cultural and economic roles, have unique coat color diversity with selection toward the brown coat color as opposed to the traditionally white Angora mohair. Thus, it would be extremely valuable to identify genetic variants and the underlying genes related to coat color and mohair traits. Recently, the release of an improved goat genome assembly (Bickhart et al., 2017) and caprine 50K SNP beadchip have offered more opportunities to examine the genetics of economically important traits in the goat (Tosser-Klopp et al., 2014). Here, we have identified multiple QTL and several putative candidate genes associated with coat color and mohair traits through genome-wide association that warrant further investigation for causative effect and potential use for genomic selection.
Association mapping for the black and brown coat colors separately identified a major locus on chromosome 13 (Figures 3A,B). Based on LD, the target region expanded to include the AHCY, ASIP, RALY, and ITCH genes (Figures 4A,B). The ASIP gene is well known for its role in coat color across several species and for its epistatic interaction with MC1R gene (Graham et al., 1997). Genetic variations within ASIP have been associated with coat color variation in other goat breeds such as the Saanen breed. Fontanesi et al. (2009b) reported that the dominant AWt allele can lead to white color in Saanen goats while a comparison of eight different goat breeds identified numerous missense mutations in the ASIP gene and a copy number variant (CNV) in ASIP and AHCY genes (Fontanesi et al., 2009b). The CNV is suggested to be responsible for introducing the AWt allele into both the Girgentana and Saanen goat breeds in a similar manner as observed in sheep previously (Norris and Whan, 2008). A GWAS for pink and pink necks in Saanen goats identified a QTL near the ASIP gene as well (Martin P.M. et al., 2016). Given the dominant effect of the ASIP allele for light coat color in Saanen goats and sheep, we suspect the recessive ASIP allele may contribute to the darker coat color of the Markhoz goats, but further testing is needed.
While ASIP is likely the major contributor to brown and black coat color in the Markhoz goat, the strong LD within the region identified specific haplotypes for brown vs. black coat color and suggests the AHCY, RALY, and ITCH genes could play regulatory roles in coat color. Significantly associated SNPs fell within each of these genes. Norris and Whan (2008) reported that a duplication containing the coding regions of ASIP and AHCY, and the ITCH promoter site is responsible for white coat color in sheep. The ITCH gene plays a role in apoptosis in melanoma cells (Yang et al., 2010). Melanoma is also a well-known skin cancer related to skin pigmentation. Individuals with fairer skin color, lighter hair and eye color are at higher risk for melanoma (Bradford, 2009) which supports a potential relationship between the ITCH gene and coat color, potentially contributing to the brown color variation observed. In horses, a duplication of the syntaxin 17 (STX17) gene is responsible for the gray coat color and melanoma, with increased susceptibility to melanoma in horses homozygous for the recessive ASIP allele (Rosengren Pielberg et al., 2008), indicating it is plausible for the ITCH gene to have similar pleiotropic effects. While further study is required to unravel the molecular interactions of the region in the Markhoz goats, the haplotypes we have identified for coat color (Figures 4C,D) could be valuable today in the genetic selection of black and brown animals.
A prior study of brown coat color in goats identified a non-synonymous variant in the TYRP1 gene region on chromosome 8 (Becker et al., 2015). However, we were unable to identify this region within our own sample population. We did identify a SNP within the RALY gene that is present only in our black animals. The lethal yellow Ay allele of ASIP is known to disrupt the structure and expression of the RALY gene (Michaud et al., 1993). The RALY gene has also been associated with the saddle tan phenotype in the black and tan Basset hounds and Pembroke welsh corgis (Dreger et al., 2013). Therefore, we suspect that RALY together with ASIP gene could have a potential role in black coat color.
Association mapping for white coat color identified 98 significantly associated SNPs spanning 11 chromosomes which was substantially greater than results for either black or brown coat color. Retrospective analysis of the dataset showed that all 26 white animals also had wattles (Supplementary Figure S3). Wattles are a hair-covered appendage consisting of skin, blood vessels, muscle and core cartilage with an unclear biological function (Imagawa et al., 1994). None of our black or brown animals had wattles, therefore we cannot differentiate if our results are associated with a white coat color or with the presence of wattles. We suspect we have captured regions associated with both traits as genes within the regions have roles in keratinocyte differentiation, tissue morphogenesis, and coat color.
KIT and platelet derived growth factor receptor alpha (PDGFRA), both on chromosome 6, are the most promising genes we detected for white coat color (Figure 3C). To date, no study has associated the KIT gene with coat color in goats despite several studies in other species identifying KIT’s role in coat color (Marklund et al., 1998; Pielberg et al., 2002; Fontanesi et al., 2010a,b, 2014; Wong et al., 2013; David et al., 2014; Yan et al., 2014; Holl et al., 2017). In pigs, the PDGFRA gene is shown to be tightly associated with the dominant white coat color (Johansson et al., 1992; Johansson Moller et al., 1996). Within the same region as KIT and PDGFRA, is the KDR gene, well-known for playing a role in angiogenesis, vascular development, and hematopoiesis regulation (Risau, 1997; Gogat et al., 2004). The gene complex of KDR, KIT, and PDGFRA has been associated with the reddening coat color pattern in Angus cattle (Hanna et al., 2014). While KDR has been identified as a putative candidate for coat color in the cattle study, we suspect KDR is more likely contributing to the wattle vascular development in our goats based on its known role in angiogenesis. The fact that these three genes, including KIT and PDGFRA which are likely influencing white coat color and KDR which might contribute to wattle development, are in the same region could explain why wattles are only present in the white goats within our dataset.
Additional candidate genes functionally related to pigmentation, hair growth, and keratinocyte differentiation have a less obvious influence on white coat color and suggest either a complex regulation of the trait or are instead related to the development of wattles or increased fiber volume. Indeed, PC2 was directly linked to white animals which produced more fiber. This further complicates the interpretation of genetic signatures associated with white animals but provides some insight as to why the GWAS for white produced so many QTL.
The GWAS for white coat color and PC2 independently highlighted overlapping QTL on chromosome 2 for which the midpoints of the respective QTL were 312 Kb apart. Within this region were the MREG gene, which regulates melanosome transfer for which inhibition results in skin lightening (Wu et al., 2012), and the Abca12 ATP-binding cassette sub-family A (ABC1), member 12 (ABCA12) gene which regulates keratinocyte differentiation and epidermal lipid transportation (Akiyama, 2014). The fibronectin 1 (FN1) gene, which regulates tissue morphogenesis (Foolen et al., 2016), is also in this region but seems a more likely candidate for wattle development.
The following genes were highlighted in QTL for white coat color and retrospectively, wattles. The RAB11 family interacting protein 2 (RAB11FIP2) gene is thought to suppress the internalization of epidermal growth factor receptors (EGFR) (Cullis et al., 2002) which activate hair growth in both the mouse and human (Moore et al., 1981; Mak and Chan, 2003) while the Receptor type K (PTPRK) gene can be regulated by TGF-β pathway which decreases the EGFR activation in human primary keratinocytes (Xu et al., 2015). The nuclear factor of activated T cells 1 (NFACTC1) gene plays a role in skin tumorigenesis regulation via DMBA metabolism in which the loss of expression of this gene will decrease the skin tumorigenesis (Goldstein et al., 2015). The glutaredoxin and cysteine rich domain containing 1 (GRXCR1) gene is known to influence hair cell development with protein expression in the sensory epithelia in the inner ear. Mutations in the GRXCR1 have been linked to hearing loss in both mice and humans (Odeh et al., 2010).
When focusing solely on candidate genes plausible for wattle development, we identified the previously mentioned FN1, as well as the sarcoglycan gamma (SGCG), and iroquois homeobox 2 (IRX2). Mutations in SGCG have been associated with muscle degradation (El Kerch et al., 2014) and IRX2 plays a role in digit formation (Zulch et al., 2001). Thus, we hypothesized that these genes may influence wattle development due to their roles in tissue morphogenesis, muscle, and digit development, respectively. Our data did not reveal a QTL in the FMN1/GREM1 region on chromosome 10 which was previously associated with wattle formation in a genome-wide analysis of nine Swiss goat breeds (Reber et al., 2015). Breed variation or our confounding overlap of white coat color and fiber volume traits may have influenced the different results. In general, further studies are needed to decipher the roles of these genes for which their functional annotation suggests potential roles in coat color, wattle formation, and/or fiber volume.
Two different strategies were applied for mapping mohair traits in our goat population. First, PCA was used to group overlapping mohair qualities correlating to broader characteristics such as fiber quality and volume, which were then mapped using the resulting principal components 1 and 2, respectively. Second, we mapped each individual fiber trait. This mapping strategy was planned for three reasons: (1) to identify QTL and candidate genes related to broader characteristics of mohair quality and quantity which are economically important, (2) to compare the results from the PCA GWAS to individual trait mapping to look for potential overlapping regions, and (3) identify regions driving the regulation of specific mohair traits.
Principal component 1 described overall fiber quality, with true fiber scores being negatively correlated to increased kemp and larger diameter. Permutation mapping of PC1 identified a region on chromosome 1 near the POU class 1 homeobox 1(POU1F1) gene. This gene is known to play an important role in wool production in sheep and in greasy fiber percentage and staple length in cashmere goats (Lan et al., 2009; Zeng et al., 2011; Sun et al., 2013). Additionally, other studies have demonstrated that POU1F1 has some effect on growth and milk production in other mammals (Mura et al., 2012; Ozmen et al., 2014; Sadeghi et al., 2014). This is the first study to apply PCA to fiber traits for a broader perspective of the genetic regulation of overall mohair quality and quantity.
Association mapping for the individual traits of true fiber percentage and kemp percentage did not identify genes with obvious roles in hair or fiber characteristics. However, association studies on both PC1 and fiber diameter highlighted the same region on chromosome 1 close to the POU1F1 gene described above. With PC2, we were able to describe overall fleece volume related to fleece weight and staple length. Candidate genes such as MREG and ABCA12 were previously described as they overlapped with white coat color QTL. ABCA12 has a more plausible relationship with fiber volume as it regulates keratinocyte differentiation and epidermal lipid transportation. Laminin subunit beta 3 (LAMB3) and hydroxysteroid 11-beta dehydrogenase 1 (HSD11B1) genes, which influence hair morphogenesis and dermatitis in both mice and humans, respectively, were identified as the most likely candidates for influencing mature fleece weight within the associated QTL (Imanishi et al., 2014; Terao et al., 2016). While the GWAS for fiber efficiency highlighted the novel gene of IMMP2L, previously unassociated with fiber traits, the association mapping for greasy fleece percentage produced more intriguing results. This included the adhesion G protein-coupled receptor V1 (ADGRV1) gene which has a role in the development of auditory hair bundles in mice and is related to Usher syndrome, a highly heritable disease which consists of various symptoms including hearing loss and vision impairment (McGee et al., 2006; Kahrizi et al., 2014; Yang et al., 2016). It would be of interest to explore the role of auditory hairs, which collect wax within the ear canal, and the incidence of deafness among the goats. Ironically, both ADGRV1 and GRXCR1 are associated with auditory hair development and mutations are linked to hearing loss. Another gene includes the histamine N-methyltransferase (HNMT) gene, which has a link to skin lesions in mice (Furukawa et al., 2009). As these genes are involved in hair and skin disorders, they might influence fiber development and fiber traits both directly and indirectly via different pathways.
Lastly, the GWAS for yearling fleece weight, which is related to a finer mohair yield due to the younger age of the animals, was conducted. As the goats increase in age, the mohair fiber becomes more coarse as the fiber diameter increases (Mohair South Africa, 2017). The SORD, dual oxidase 1 (DUOX1), dual oxidase maturation factor 2 (DUOXA2), dual oxidase 2 (DUOX2), dual oxidase maturation factor 1(DUOXA1), and arginine-glycine aminotransferase (GATM) genes on chromosome 10 reside in the associated QTL for yearling fleece weight. The SORD gene is regulated by androgens and expressed in epithelial cells (Szabo et al., 2010). Coincidently, our data showed a positive correlation between increased yearling fleece weight and the male sex which we hypothesize may be due to differential regulation of the SORD gene. Studies in humans have reported that DUOX1 plays a role in the expression levels of normal keratinocytes (Choi et al., 2014). As keratinocytes produce keratin, the main protein for hair, nail, and skin synthesis, we hypothesize that this gene may be associated with additional fiber development. Although not related to fiber, a deficiency in the GATM gene is related to an autosomal-recessive disorder with varying symptoms including myopathy (Choe et al., 2013; Stockler-Ipsiroglu et al., 2015). The analysis of yearling fleece weight was normalized as it was positively correlated with yearly weight likely reflecting muscle mass.
In all, many of our QTL differed from previous mapping studies for fiber related traits (Cano et al., 2007, 2009; Mohammad Abadi et al., 2009; Visser et al., 2011; Roldan et al., 2014). Disagreement in our findings compared to these studies is likely due to breed differences as well as marker placement and density used in the analysis. Fine mapping and expression studies of some of the unique fiber quality related genes such as POU1F1. ADGRV1, MREG, LAMB3, and HSD11B1 may lead to new insight toward biological pathways influencing hair development and growth. In contrast, our QTL related to coat color highlighted candidate genes extensively documented for influencing color patterns in goats as well as a variety of other species. Future fine mapping of the identified regions, especially ASIP, RALY, KIT, and PDGFRA genes is needed to identify causal mutations or other structural phenomenon such as CNVs that are contributing to coat color of the Markhoz breed. Unexpectedly, we were also able to highlight genes potentially influencing the presence of wattles, which currently remain a biological mystery.
Despite advances in the genome assembly and tool development in several species over the last 10 years, there have been few genome scale studies for traits considered economically important in goats, particularly in Angora and Cashmere goats. This is the first genetic study to identify regions associated with coat color and fiber traits in the Markhoz breed as well as potential SNPs and haplotypes for genetic selection of coat color. The relatively small cohort of goats investigated was a limiting factor yet provided biological insight for these traits and a foundation to further genomic research on coat color and mohair traits in goats.
All SNP genotype data are available at the publically assessable Zenodo repository (https://zenodo.org/record/1198730#.WqlmbcPwZdg).
All authors were engaged in the development of the overall research plan and assisted with research advisement. AN-G was the lead researcher performing data collection, statistical and genomic analysis, result interpretation, and drafting of the manuscript. AR provided assistance with sample and data collection of the Markhoz goats. HM-Y and SM-A provided primary advisement during sample collection and laboratory support for DNA extraction. ES assisted with the genomic methodology, data analysis, result interpretation, and initial manuscript draft. HH managed the genotyping and directed the genomic data analysis, result interpretation, and was the primary editor of the manuscript.
Funding for the genomic research and analysis was supported by the laboratory of HH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Animal sampling was made possible through the cooperation of the Markhoz goat performance testing station staff and through the assistance of Mr. Jafari, Rashid, Kakehkhani and also Ms. Karimi for providing mohair traits.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00105/full#supplementary-material
Adalsteinsson, S., Sponenberg, D. P., Alexieva, S., and Russel, A. J. (1994). Inheritance of goat coat colors. J. Hered. 85, 267–272. doi: 10.1093/oxfordjournals.jhered.a111454
Akiyama, M. (2014). The roles of ABCA12 in epidermal lipid barrier formation and keratinocyte differentiation. Biochim. Biophys. Acta 1841, 435–440. doi: 10.1016/j.bbalip.2013.08.009
Barrett, J. C., Fry, B., Maller, J., and Daly, M. J. (2005). Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265. doi: 10.1093/bioinformatics/bth457
Becker, D., Otto, M., Ammann, P., Keller, I., Drogemuller, C., and Leeb, T. (2015). The brown coat colour of Coppernecked goats is associated with a non-synonymous variant at the TYRP1 locus on chromosome 8. Anim. Genet. 46, 50–54. doi: 10.1111/age.12240
Bickhart, D. M., Rosen, B. D., Koren, S., Sayre, B. L., Hastie, A. R., Chan, S., et al. (2017). Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 49, 643–650. doi: 10.1038/ng.3802
Bradford, P. T. (2009). Skin cancer in skin of color. Dermatology nursing/Dermatology Nurses’. Association 21, 170–178.
Cano, E. M., Daverio, S., Cáceres, M., Debenedetti, S., Costoya, S., Abad, M., et al. (2009). Detection of QTL affecting fleece traits on CHI 19 in angora goats. Trop. Subtrop. Agroecosyst. 11, 189–191.
Cano, E. M., Marrube, G., Roldan, D. L., Bidinost, F., Abad, M., Allain, D., et al. (2007). QTL affecting fleece traits in Angora goats. Small Rumin. Res. 71, 158–164. doi: 10.1016/j.smallrumres.2006.06.002
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. doi: 10.1186/s13742-015-0047-8
Che, R., Jack, J. R., Motsinger-Reif, A. A., and Brown, C. C. (2014). An adaptive permutation approach for genome-wide association study: evaluation and recommendations for use. BioData Min 7:9. doi: 10.1186/1756-0381-7-9
Choe, C. U., Nabuurs, C., Stockebrand, M. C., Neu, A., Nunes, P., Morellini, F., et al. (2013). L-arginine:glycine amidinotransferase deficiency protects from metabolic syndrome. Hum. Mol. Genet. 22, 110–123. doi: 10.1093/hmg/dds407
Choi, H., Park, J. Y., Kim, H. J., Noh, M., Ueyama, T., Bae, Y., et al. (2014). Hydrogen peroxide generated by DUOX1 regulates the expression levels of specific differentiation markers in normal human keratinocytes. J. Dermatol. Sci. 74, 56–63. doi: 10.1016/j.jdermsci.2013.11.011
Cullis, D. N., Philip, B., Baleja, J. D., and Feig, L. A. (2002). Rab11-FIP2, an adaptor protein connecting cellular components involved in internalization and recycling of epidermal growth factor receptors. J. Biol. Chem. 277, 49158–49166. doi: 10.1074/jbc.M206316200
David, V. A., Menotti-Raymond, M., Wallace, A. C., Roelke, M., Kehler, J., Leighty, R., et al. (2014). Endogenous retrovirus insertion in the KIT oncogene determines white and white spotting in domestic cats. G3 (Bethesda) 4, 1881–1891. doi: 10.1534/g3.114.013425
Dreger, D. L., Parker, H. G., Ostrander, E. A., and Schmutz, S. M. (2013). Identification of a mutation that is associated with the saddle tan and black-and-tan phenotypes in Basset Hounds and Pembroke Welsh Corgis. J. Hered. 104, 399–406. doi: 10.1093/jhered/est012
Durig, N., Jude, R., Holl, H., Brooks, S. A., Lafayette, C., Jagannathan, V., et al. (2017). Whole genome sequencing reveals a novel deletion variant in the KIT gene in horses with white spotted coat colour phenotypes. Anim. Genet. 48, 483–485. doi: 10.1111/age.12556
El Kerch, F., Ratbi, I., Sbiti, A., Laarabi, F. Z., Barkat, A., and Sefiani, A. (2014). Carrier frequency of the c.525delT mutation in the SGCG gene and estimated prevalence of limb girdle muscular dystrophy type 2C among the Moroccan population. Genet. Test Mol. Biomark. 18, 253–256. doi: 10.1089/gtmb.2013.0326
FAOSTAT (2008). Available at: http://faostat.fao.org/default.aspx
Fontanesi, L., Beretti, F., Riggio, V., Dall’Olio, S., González, E. G., Finocchiaro, R., et al. (2009a). Missense and nonsense mutations in melanocortin 1 receptor (MC1R) gene of different goat breeds: association with red and black coat colour phenotypes but with unexpected evidences. BMC Genet. 10:47. doi: 10.1186/1471-2156-10-47
Fontanesi, L., Beretti, F., Riggio, V., Gomez Gonzalez, E., Dall’Olio, S., Davoli, R., et al. (2009b). Copy number variation and missense mutations of the agouti signaling protein (ASIP) gene in goat breeds with different coat colors. Cytogenet. Genome Res. 126, 333–347. doi: 10.1159/000268089
Fontanesi, L., D’Alessandro, E., Scotti, E., Liotta, L., Crovetti, A., Chiofalo, V., et al. (2010a). Genetic heterogeneity and selection signature at the KIT gene in pigs showing different coat colours and patterns. Anim. Genet. 41, 478–492. doi: 10.1111/j.1365-2052.2010.02054.x
Fontanesi, L., Tazzoli, M., Russo, V., and Beever, J. (2010b). Genetic heterogeneity at the bovine KIT gene in cattle breeds carrying different putative alleles at the spotting locus. Anim. Genet. 41, 295–303. doi: 10.1111/j.1365-2052.2009.02007.x
Fontanesi, L., Vargiolu, M., Scotti, E., Latorre, R., Faussone Pellegrini, M. S., Mazzoni, M., et al. (2014). The KIT gene is associated with the English spotting coat color locus and congenital megacolon in Checkered Giant rabbits (Oryctolagus cuniculus). PLoS One 9:e93750. doi: 10.1371/journal.pone.0093750
Foolen, J., Shiu, J. Y., Mitsi, M., Zhang, Y., Chen, C. S., and Vogel, V. (2016). Full-length fibronectin drives fibroblast accumulation at the surface of collagen microtissues during cell-induced tissue morphogenesis. PLoS One 11:e0160369. doi: 10.1371/journal.pone.0160369
Furukawa, F., Yoshimasu, T., Yamamoto, Y., Kanazawa, N., and Tachibana, T. (2009). Mast cells and histamine metabolism in skin lesions from MRL/MP-lpr/lpr mice. Autoimmun. Rev. 8, 495–499. doi: 10.1016/j.autrev.2008.12.016
Gabriel, S. B., Schaffner, S. F., Nguyen, H., Moore, J. M., Roy, J., Blumenstiel, B., et al. (2002). The structure of haplotype blocks in the human genome. Science 296, 2225–2229. doi: 10.1126/science.1069424
Geissler, E. N., Ryan, M. A., and Housman, D. E. (1988). The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell 55, 185–192. doi: 10.1016/0092-8674(88)90020-7
Gogat, K., Le Gat, L., Van Den Berghe, L., Marchant, D., Kobetz, A., Gadin, S., et al. (2004). VEGF and KDR gene expression during human embryonic and fetal eye development. Invest. Ophthalmol. Vis. Sci. 45, 7–14. doi: 10.1167/iovs.02-1096
Goldstein, J., Roth, E., Roberts, N., Zwick, R., Lin, S., Fletcher, S., et al. (2015). Loss of endogenous Nfatc1 reduces the rate of DMBA/TPA-induced skin tumorigenesis. Mol. Biol. Cell 26, 3606–3614. doi: 10.1091/mbc.E15-05-0282
Graham, A., Wakamatsu, K., Hunt, G., Ito, S., and Thody, A. J. (1997). Agouti protein inhibits the production of eumelanin and phaeomelanin in the presence and absence of alpha-melanocyte stimulating hormone. Pigment Cell Res. 10, 298–303. doi: 10.1111/j.1600-0749.1997.tb00689.x
Haase, B., Brooks, S. A., Tozaki, T., Burger, D., Poncet, P. A., Rieder, S., et al. (2009). Seven novel KIT mutations in horses with white coat colour phenotypes. Anim. Genet. 40, 623–629. doi: 10.1111/j.1365-2052.2009.01893.x
Haase, B., Jagannathan, V., Rieder, S., and Leeb, T. (2015). A novel KIT variant in an Icelandic horse with white-spotted coat colour. Anim. Genet. 46:466. doi: 10.1111/age.12313
Hanna, L. L., Sanders, J. O., Riley, D. G., Abbey, C. A., and Gill, C. A. (2014). Identification of a major locus interacting with MC1R and modifying black coat color in an F(2) Nellore-Angus population. Genet. Sel. Evol. 46:4. doi: 10.1186/1297-9686-46-4
Hanotte, O., Dessie, T., and Kemp, S. (2010). Time to tap africa’s livestock genomes. Science 328, 1640–1641. doi: 10.1126/science.1186254
Holl, H., Isaza, R., Mohamoud, Y., Ahmed, A., Almathen, F., Youcef, C., et al. (2017). A frameshift mutation in KIT is associated with white spotting in the Arabian camel. Genes (Basel.) 8:E102. doi: 10.3390/genes8030102
Hu, Z.-L., Park, C. A., and Reecy, J. M. (2016). Developmental progress and current status of the Animal QTLdb. Nucleic Acids Res. 44, D827–D833. doi: 10.1093/nar/gkv1233
Imagawa, T., Kon, Y., Kitagawa, H., Hashimoto, Y., Uehara, M., and Sugimura, M. (1994). Anatomical and histological re-examination of Appendices colli in the goat. Ann. Anat. 176, 175–179. doi: 10.1016/S0940-9602(11)80447-4
Imanishi, H., Tsuruta, D., Tateishi, C., Sugawara, K., Kobayashi, H., Ishii, M., et al. (2014). Spatial and temporal control of laminin-332 and -511 expressions during hair morphogenesis. Med. Mol. Morphol. 47, 38–42. doi: 10.1007/s00795-013-0040-1
Joerg, H., Fries, H. R., Meijerink, E., and Stranzinger, G. F. (1996). Red coat color in Holstein cattle is associated with a deletion in the MSHR gene. Mamm. Genome 7, 317–318. doi: 10.1007/s003359900090
Johansson, M., Ellegren, H., Marklund, L., Gustavsson, U., Ringmar-Cederberg, E., Andersson, K., et al. (1992). The gene for dominant white color in the pig is closely linked to ALB and PDGRFRA on chromosome 8. Genomics 14, 965–969. doi: 10.1016/S0888-7543(05)80118-1
Johansson Moller, M., Chaudhary, R., Hellmen, E., Hoyheim, B., Chowdhary, B., and Andersson, L. (1996). Pigs with the dominant white coat color phenotype carry a duplication of the KIT gene encoding the mast/stem cell growth factor receptor. Mamm. Genome 7, 822–830. doi: 10.1007/s003359900244
Kahrizi, K., Bazazzadegan, N., Jamali, L., Nikzat, N., Kashef, A., and Najmabadi, H. (2014). A novel mutation of the USH2C (GPR98) gene in an Iranian family with Usher syndrome type II. J. Genet. 93, 837–841. doi: 10.1007/s12041-014-0443-3
Kang, H. M., Sul, J. H., Service, S. K., Zaitlen, N. A., Kong, S. Y., and Freimer, N. B. (2010). Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354. doi: 10.1038/ng.548
Klungland, H., Vage, D. I., Gomez-Raya, L., Adalsteinsson, S., and Lien, S. (1995). The role of melanocyte-stimulating hormone (MSH) receptor in bovine coat color determination. Mamm. Genome 6, 636–639. doi: 10.1007/bf00352371
Lan, R., Zhu, L., and Yao, X.-R. (2015). Genome-wide association study of lambing number in goat. Acta Vet. Zootechn. Sin. 46, 549–554.
Lan, X. Y., Shu, J. H., Chen, H., Pan, C. Y., Lei, C. Z., Wang, X., et al. (2009). A PstI polymorphism at 3′UTR of goat POU1F1 gene and its effect on cashmere production. Mol. Biol. Rep. 36, 1371–1374. doi: 10.1007/s11033-008-9322-4
Li, X., Su, R., Wan, W., Zhang, W., Jiang, H., Qiao, X., et al. (2017). Identification of selection signals by large-scale whole-genome resequencing of cashmere goats. Sci. Rep. 7:15142. doi: 10.1038/s41598-017-15516-0
Mak, K. K., and Chan, S. Y. (2003). Epidermal growth factor as a biologic switch in hair growth cycle. J. Biol. Chem. 278, 26120–26126. doi: 10.1074/jbc.M212082200
Marklund, S., Kijas, J., Rodriguez-Martinez, H., Ronnstrand, L., Funa, K., Moller, M., et al. (1998). Molecular basis for the dominant white phenotype in the domestic pig. Genome Res. 8, 826–833. doi: 10.1101/gr.8.8.826
Martin, P., Palhière, I., Tosser-Klopp, G., and Rupp, R. (2016). Heritability and genome-wide association mapping for supernumerary teats in French Alpine and Saanen dairy goats. J. Dairy Sci. 99, 8891–8900. doi: 10.3168/jds.2016-11210
Martin, P. M., Palhiere, I., Ricard, A., Tosser-Klopp, G., and Rupp, R. (2016). Genome wide association study identifies new loci associated with undesired coat color phenotypes in Saanen goats. PLoS One 11:e0152426. doi: 10.1371/journal.pone.0152426
McGee, J., Goodyear, R. J., McMillan, D. R., Stauffer, E. A., Holt, J. R., Locke, K. G., et al. (2006). The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J. Neurosci. 26, 6543–6553. doi: 10.1523/jneurosci.0693-06.2006
Menzi, F., Keller, I., Reber, I., Beck, J., Brenig, B., Schütz, E., et al. (2016). Genomic amplification of the caprine EDNRA locus might lead to a dose dependent loss of pigmentation. Sci. Rep. 6:28438. doi: 10.1038/srep28438
Michaud, E. J., Bultman, S. J., Stubbs, L. J., and Woychik, R. P. (1993). The embryonic lethality of homozygous lethal yellow mice (Ay/Ay) is associated with the disruption of a novel RNA-binding protein. Genes Dev. 7, 1203–1213. doi: 10.1101/gad.7.7a.1203
Mohair South Africa (2017). Available at: http://www.mohair.co.za/page/mohair_knowledge_and_information_database
Mohammad Abadi, M. R., Askari, N., Baghizadeh, A., and Esmailizadeh, A. K. (2009). A directed search around caprine candidate loci provided evidence for microsatellites linkage to growth and cashmere yield in Rayini goats. Small Rumin. Res. 81, 146–151. doi: 10.1016/j.smallrumres.2008.12.012
Moore, G. P., Panaretto, B. A., and Robertson, D. (1981). Effects of epidermal growth factor on hair growth in the mouse. J. Endocrinol. 88, 293–299. doi: 10.1677/joe.0.0880293
Mura, M. C., Daga, C., Paludo, M., Luridiana, S., Pazzola, M., Bodano, S., et al. (2012). Analysis of polymorphism within POU1F1 gene in relation to milk production traits in dairy Sarda sheep breed. Mol. Biol. Rep. 39, 6975–6979. doi: 10.1007/s11033-012-1525-z
Norris, B. J., and Whan, V. A. (2008). A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep. Genome Res. 18, 1282–1293. doi: 10.1101/gr.072090.107
Odeh, H., Hunker, K. L., Belyantseva, I. A., Azaiez, H., Avenarius, M. R., Zheng, L., et al. (2010). Mutations in Grxcr1 are the basis for inner ear dysfunction in the pirouette mouse. Am. J. Hum. Genet. 86, 148–160. doi: 10.1016/j.ajhg.2010.01.016
Ozmen, O., Kul, S., and Unal, E. O. (2014). Polymorphism of sheep POU1F1 gene exon 6 and 3′UTR region and their association with milk production traits. Iran J. Vet. Res. 15, 331–335.
Parsons, Y. M., Cooper, D. W., and Piper, L. R. (1994). Evidence of linkage between high-glycine-tyrosine keratin gene loci and wool fibre diameter in a Merino half-sib family. Anim. Genet. 25, 105–108. doi: 10.1111/j.1365-2052.1994.tb00088.x
Pielberg, G., Olsson, C., Syvanen, A. C., and Andersson, L. (2002). Unexpectedly high allelic diversity at the KIT locus causing dominant white color in the domestic pig. Genetics 160, 305–311.
Rashidi, A., Mokhtari, M. S., and Gutiérrez, J. P. (2015). Pedigree analysis and inbreeding effects on early growth traits and greasy fleece weight in Markhoz goat. Small Rumin. Res. 124, 1–8. doi: 10.1016/j.smallrumres.2014.12.011
Rashidi, A., Ramazanian, M., and Torshizi, R. V. (2006). Genetic Parameter Estimates for Growth Traits and Fleece Weight in Markhoz Goats. Minas Gerais: Instituto Prociência.
Reber, I., Keller, I., Becker, D., Flury, C., Welle, M., and Drogemuller, C. (2015). Wattles in goats are associated with the FMN1/GREM1 region on chromosome 10. Anim. Genet. 46, 316–320. doi: 10.1111/age.12279
Roldan, D., Debenedetti, S., Cano, E. M., Taddeo, H. R., and Poli, M. A. (2014). “Preliminar refined localization of QTL for fleece traits in five goat chromosomes using SNP markers in a backcross population,” in Proceedings of the, 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC, 885.
Rosengren Pielberg, G., Golovko, A., Sundstrom, E., Curik, I., Lennartsson, J., Seltenhammer, M. H., et al. (2008). A cis-acting regulatory mutation causes premature hair graying and susceptibility to melanoma in the horse. Nat. Genet. 40, 1004–1009. doi: 10.1038/ng.185
Sadeghi, M., Jalil-Sarghale, A., and Moradi-Shahrbabak, M. (2014). Associations of POU1F1 gene polymorphisms and protein structure changes with growth traits and blood metabolites in two Iranian sheep breeds. J. Genet. 93, 831–835. doi: 10.1007/s12041-014-0438-0
Sambrook, J., and Russell, D. W. (2006). Purification of nucleic acids by extraction with phenol:chloroform. CSH Protoc. 2006:pdb.prot4455. doi: 10.1101/pdb.prot4455
Stockler-Ipsiroglu, S., Apatean, D., Battini, R., DeBrosse, S., Dessoffy, K., Edvardson, S., et al. (2015). Arginine:glycine amidinotransferase (AGAT) deficiency: Clinical features and long term outcomes in 16 patients diagnosed worldwide. Mol. Genet. Metab. 116, 252–259. doi: 10.1016/j.ymgme.2015.10.003
Sturm, R. A., Teasdale, R. D., and Box, N. F. (2001). Human pigmentation genes: identification, structure and consequences of polymorphic variation. Gene 277, 49–62. doi: 10.1016/S0378-1119(01)00694-1
Sun, W., Ni, R., Yin, J. F., Musa, H. H., Ding, T., and Chen, L. (2013). Genome array of hair follicle genes in lambskin with different patterns. PLoS One 8:e68840. doi: 10.1371/journal.pone.0068840
Switonski, M., Mankowska, M., and Salamon, S. (2013). Family of melanocortin receptor (MCR) genes in mammals—mutations, polymorphisms and phenotypic effects. J. Appl. Genet. 54, 461–472. doi: 10.1007/s13353-013-0163-z
Szabo, Z., Hamalainen, J., Loikkanen, I., Moilanen, A. M., Hirvikoski, P., Vaisanen, T., et al. (2010). Sorbitol dehydrogenase expression is regulated by androgens in the human prostate. Oncol. Rep. 23, 1233–1239.
Terao, M., Itoi, S., Matsumura, S., Yang, L., Murota, H., and Katayama, I. (2016). Local glucocorticoid activation by 11beta-hydroxysteroid dehydrogenase 1 in keratinocytes: the role in hapten-induced dermatitis. Am. J. Pathol. 186, 1499–1510. doi: 10.1016/j.ajpath.2016.01.014
Tosser-Klopp, G., Bardou, P., Bouchez, O., Cabau, C., Crooijmans, R., Dong, Y., et al. (2014). Design and characterization of a 52K SNP chip for goats. PLoS One 9:e86227. doi: 10.1371/journal.pone.0086227
USDA (2016). Available at: https://www.ars.usda.gov/office-of-international-research-programs/ars-international-action-agin-iv/
Våge, D. I., Klungland, H., Lu, D., and Cone, R. D. (1999). Molecular and pharmacological characterization of dominant black coat color in sheep. Mamm. Genome 10, 39–43. doi: 10.1007/s003359900939
Visser, C., Van Marle-Köster, E., Bovenhuis, H., and Crooijmans, R. P. M. A. (2011). QTL for mohair traits in South African Angora goats. Small Rumin. Res. 100, 8–14. doi: 10.1016/j.smallrumres.2011.05.007
Wang, X., Cai, B., Zhou, J., Zhu, H., Niu, Y., Ma, B., et al. (2016). Disruption of FGF5 in cashmere goats using CRISPR/Cas9 results in more secondary hair follicles and longer fibers. PLoS One 11:e0164640. doi: 10.1371/journal.pone.0164640
Wong, A. K., Ruhe, A. L., Robertson, K. R., Loew, E. R., Williams, D. C., and Neff, M. W. (2013). A de novo mutation in KIT causes white spotting in a subpopulation of German Shepherd dogs. Anim. Genet. 44, 305–310. doi: 10.1111/age.12006
Wu, X. S., Masedunskas, A., Weigert, R., Copeland, N. G., Jenkins, N. A., and Hammer, J. A. (2012). Melanoregulin regulates a shedding mechanism that drives melanosome transfer from melanocytes to keratinocytes. Proc. Natl. Acad. Sci. U.S.A. 109, E2101–E2109. doi: 10.1073/pnas.1209397109
Xu, Y., Xue, S., Zhou, J., Voorhees, J. J., and Fisher, G. J. (2015). Notch and TGF-beta pathways cooperatively regulate receptor protein tyrosine phosphatase-kappa (PTPRK) gene expression in human primary keratinocytes. Mol. Biol. Cell 26, 1199–1206. doi: 10.1091/mbc.E14-12-1591
Yan, S. Q., Hou, J. N., Bai, C. Y., Jiang, Y., Zhang, X. J., Ren, H. L., et al. (2014). A base substitution in the donor site of intron 12 of KIT gene is responsible for the dominant white coat colour of blue fox (Alopex lagopus). Anim. Genet. 45, 293–296. doi: 10.1111/age.12105
Yang, F., Tay, K. H., Dong, L., Thorne, R. F., Jiang, C. C., Yang, E., et al. (2010). Cystatin B inhibition of TRAIL-induced apoptosis is associated with the protection of FLIP(L) from degradation by the E3 ligase itch in human melanoma cells. Cell Death Differ. 17, 1354–1367. doi: 10.1038/cdd.2010.29
Yang, J., Huang, X. F., Tong, Y., and Jin, Z. B. (2016). Targeted exome sequencing identified two novel truncation mutations in GPR98 causing Usher syndrome. Clin. Exp. Ophthalmol. 44, 197–199. doi: 10.1111/ceo.12664
Zeder, M. A., and Hesse, B. (2000). The initial domestication of goats (Capra hircus) in the Zagros mountains 10,000 years ago. Science 287, 2254–2257. doi: 10.1126/science.287.5461.2254
Zeng, X. C., Chen, H. Y., Jia, B., Zhao, Z. S., Hui, W. Q., Wang, Z. B., et al. (2011). Identification of SNPs within the sheep PROP1 gene and their effects on wool traits. Mol. Biol. Rep. 38, 2723–2728. doi: 10.1007/s11033-010-0416-4
Zidi, A., Abo-Shady, H., Molina Alcalá, A., Menendez-Buxadera, A., Snchez-Rodrguez, M. J., Daz, C., et al. (2014). “Genome wide association for heat stress tolerance/susceptibility in florida dairy goats,” in 10th World Congress on Genetics Applied to Livestock Production, Vancouver, BC.
Keywords: genome-wide association study, coat color, mohair, fleece, Markhoz, goat, Angora, wattles
Citation: Nazari-Ghadikolaei A, Mehrabani-Yeganeh H, Miarei-Aashtiani SR, Staiger EA, Rashidi A and Huson HJ (2018) Genome-Wide Association Studies Identify Candidate Genes for Coat Color and Mohair Traits in the Iranian Markhoz Goat. Front. Genet. 9:105. doi: 10.3389/fgene.2018.00105
Received: 27 November 2017; Accepted: 16 March 2018;
Published: 04 April 2018.
Edited by:
Max F. Rothschild, Iowa State University, United StatesReviewed by:
Tosso Leeb, Universität Bern, SwitzerlandCopyright © 2018 Nazari-Ghadikolaei, Mehrabani-Yeganeh, Miarei-Aashtiani, Staiger, Rashidi and Huson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hassan Mehrabani-Yeganeh, aG1laHJiYW5pQHV0LmFjLmly Heather J. Huson, aGpoM0Bjb3JuZWxsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.