 Hussain Bahbahani1*
Hussain Bahbahani1* Abdulfatai Tijjani2,3
Abdulfatai Tijjani2,3 Christopher Mukasa4
Christopher Mukasa4 David Wragg5
David Wragg5 Faisal Almathen6
Faisal Almathen6 Oyekanmi Nash3Gerald N. Akpa7Mary Mbole-Kariuki2
Oyekanmi Nash3Gerald N. Akpa7Mary Mbole-Kariuki2 Sunir Malla8
Sunir Malla8 Mark Woolhouse9
Mark Woolhouse9 Tad Sonstegard10
Tad Sonstegard10 Curtis Van Tassell11
Curtis Van Tassell11 Martin Blythe8
Martin Blythe8 Heather Huson11
Heather Huson11 Olivier Hanotte2,12
Olivier Hanotte2,12- 1Department of Biological Sciences, Faculty of Science, Kuwait University, Kuwait, Kuwait
- 2School of Life Sciences, University of Nottingham, Nottingham, United Kingdom
- 3Centre for Genomics Research and Innovation, National Biotechnology Development Agency, Abuja, Nigeria
- 4National Animal Genetic Resource Centre and Data Bank, Entebbe, Uganda
- 5Centre for Tropical Livestock Genetics and Health, Roslin Institute, Edinburgh, United Kingdom
- 6Department of Veterinary Public Health and Animal Husbandry, College of Veterinary Medicine, King Faisal University, Al-Hasa, Saudi Arabia
- 7Department of Animal Science, Ahmadu Bello University, Zaria, Nigeria
- 8Deep Seq Department, University of Nottingham, Nottingham, United Kingdom
- 9Ashworth Laboratories, Centre for Immunity, Infection and Evolution, University of Edinburgh, Edinburgh, United Kingdom
- 10Recombinetics, Inc., St. Paul, MN, United States
- 11Animal Genomics and Improvement Laboratory, United States Department of Agriculture, Agricultural Research Service, Beltsville, MD, United States
- 12International Livestock Research Institute (ILRI), Addis Ababa, Ethiopia
The East African Shorthorn Zebu (EASZ) cattle are ancient hybrid between Asian zebu × African taurine cattle preferred by local farmers due to their adaptability to the African environment. The genetic controls of these adaptabilities are not clearly understood yet. Here, we genotyped 92 EASZ samples from Kenya (KEASZ) with more than 770,000 SNPs and sequenced the genome of a pool of 10 KEASZ. We observe an even admixed autosomal zebu × taurine genomic structure in the population. A total of 101 and 165 candidate regions of positive selection, based on genome-wide SNP analyses (meta-SS, Rsb, iHS, and ΔAF) and pooled heterozygosity (Hp) full genome sequence analysis, are identified, in which 35 regions are shared between them. A total of 142 functional variants, one novel, have been detected within these regions, in which 30 and 26 were classified as of zebu and African taurine origins, respectively. High density genome-wide SNP analysis of zebu × taurine admixed cattle populations from Uganda and Nigeria show that 25 of these regions are shared between KEASZ and Uganda cattle, and seven regions are shared across the KEASZ, Uganda, and Nigeria cattle. The identification of common candidate regions allows us to fine map 18 regions. These regions intersect with genes and QTL associated with reproduction and environmental stress (e.g., immunity and heat stress) suggesting that the genome of the zebu × taurine admixed cattle has been uniquely selected to maximize hybrid fitness both in terms of reproduction and survivability.
Introduction
Domestic cattle are classified, based on phenotypes, into humpless taurine (Bos taurus) and humped zebu (B. indicus) cattle. They originated from different auroch ancestral populations B. primigenius primigenius and B. p. namadicus at two separate domestication centers; the Near East and the Indus Valley—Northern part of the Indian subcontinent, respectively (Epstein, 1971; Loftus et al., 1994; Troy et al., 2001; Chen et al., 2010). These two cattle types are considered as distinct species or subspecies with mitochondrial DNA (Loftus et al., 1994) and microsatellite (MacHugh et al., 1997) analyses suggesting a common ancestry about 200,000 and 700,000 years ago, respectively.
Cattle populations of zebu × African taurine ancestries are common in Africa and in particularly on the eastern part of Africa, Southern Africa, and along the Sahelian belt. According to their phenotypes, they are either referred to as African zebu, e.g., East African Shorthorn Zebu (EASZ) in Kenya, Adamawa Gudali in Nigeria, and Karamojong zebu in Uganda, African sanga, e.g., Ankole in Uganda, or African zenga e.g., Nganda in Uganda (Epstein, 1971; Rege, 1999; Rege et al., 2001). These cattle originated from the earlier migration of B. taurus and B. indicus to the continent from their putative centers of domestication followed by subsequent hybridization between them (Loftus et al., 1994; Chen et al., 2010; Gifford-Gonzalez and Hanotte, 2011). An African auroch influence in their genome has been postulated (Decker et al., 2014) but this remains until now speculative. The cattle colonization of the African continent started first with the arrival of taurine cattle, ~7,000 years ago (Gifford-Gonzalez and Hanotte, 2011), followed by the zebu type in two waves, ~4,000 and ~1,300 years ago, according to pictorial, archeological and genetic evidences (Epstein, 1971; Hanotte et al., 2002). The later was through the Horn of Africa and it was likely linked to the development of the Swahili civilization (Hanotte et al., 2002). Zebu × taurine hybrids are therefore of ancient origin on the African continent. Given the sole presence of the taurine type mtDNA in African cattle, a male-mediated introgression of zebu cattle to the native African taurine has been proposed (Loftus et al., 1994; Bradley et al., 1996). Microsatellite analysis (Hanotte et al., 2002) and more recently genome-wide single nucleotide polymorphism (SNP) analyses (Decker et al., 2014) show that the indicine ancestry peaks in the Horn of Africa gradually declining toward the western and the southern parts of the continent.
The small EASZ is the main type of African zebu cattle populating East Africa (Rege et al., 2001). As for other indigenous cattle populations, EASZ are more preferred by the local farmers over the pure exotic highly productive taurine breeds due to their superior adaptability to their local environment, which is characterized by a warm climate (20–23°C), high humidity (60–80%) and high pathogenes challenges (e.g., Theileria parva, Ehrlichia ruminantium, and Haemonchus placei; de Clare Bronsvoort et al., 2013). These cattle show a degree of resistance to Rhipicephalus appendiculatus tick, the vector of East Coast Fever (ECF) protozoan parasite T. parva (Latif et al., 1991; Latif and Pegram, 1992), as well as tolerance to poor forage and water scarcity (Western and Finch, 1986). A mortality rate of ~16% has been observed mainly attributed to ECF, haemonchosis, and heartwater in an EASZ population of western Kenya under the traditional management system (de Clare Bronsvoort et al., 2013; Thumbi et al., 2014). The genetic structure of this cattle population has recently been investigated by Mbole-Kariuki et al. (2014) using mid-density genome-wide SNP data. This study showed that the EASZ is a stabilized admixed population of zebu and African taurine ancestries, with an average genome proportions of 0.84 ± 0.009 and 0.16 ± 0.009, respectively. However, some of the EASZ animal showed recent European taurine introgression likely following artificial insemination program aiming to improve indigenous cattle productivity (Mbole-Kariuki et al., 2014). This European introgression has been linked to increased vulnerability of EASZ to infectious diseases (Murray et al., 2013).
The genomes of several livestock species have now been intensively explored for signatures of positive selection, e.g., chicken (Rubin et al., 2010; Zhang et al., 2012), pigs (Rubin et al., 2012), sheep (Kijas et al., 2012), and cattle (Gautier et al., 2009; Gautier and Naves, 2011; Kemper et al., 2014; Bahbahani et al., 2015). In cattle, the main genomic tool used for this purpose is the commercially available genome-wide SNP chip (e.g., Illumina BovineSNP50 BeadChip and Illumina BovineHD BeadChip). Although the lower-density SNP chip has been widely used to detect signatures of selection on the genome of tropically adapted cattle (Gautier et al., 2009; Gautier and Naves, 2011; Flori et al., 2012, 2014) and commercial dairy and beef breeds (Flori et al., 2009; Qanbari et al., 2011; Khayatzadeh et al., 2016), it has been associated with two main drawbacks; (i) the limited coverage of the bovine genome with an average markers gap of 49.4 kb, (ii) the SNP ascertainment bias toward European taurine breeds (Matukumalli et al., 2009). These two issues were partly solved upon the development of the higher-density SNP chip (Illumina BovineHD BeadChip; Rincon et al., 2011). This recently developed tool has shown its usefulness in detecting signatures of selection in dairy and beef cattle breeds (Utsunomiya et al., 2013; Kemper et al., 2014; Perez O'Brien et al., 2014; Xu et al., 2015; Chen et al., 2016) and in tropical-adapted cattle (Porto-Neto et al., 2014; Xu et al., 2015). More specifically, Utsunomiya et al. (2013) have reported for the first time in cattle the use of a composite mapping index “Meta-analysis of Selection Signals (meta-SS),” using the Z-transformation method “Stouffer's method” (Stouffer et al., 1949), to define footprints of positive selection related to meat and milk production traits.
Full genome sequencing has also been used in livestock species to detect signatures of positive selection, e.g., in chicken (Rubin et al., 2010), pig (Rubin et al., 2012; Frantz et al., 2015), sheep (Liu et al., 2016), and cattle (Liao et al., 2013; Qanbari et al., 2014; Choi et al., 2015). In Gir cattle, assessing the pooled heterozygosity of sliding windows has defined footprints of selection on genes associated with heat tolerance, and innate and adaptive immunity (Liao et al., 2013). In Fleckvieh cattle, genes associated with coat color and sensory perception (e.g., olfaction and taste) have also shown signatures of positive selection (Qanbari et al., 2014). The PPP1R12A gene involved in intramuscular fat content was also considered as a candidate of positive selection upon analyzing the full genome re-sequence of Hanwoo cattle (Choi et al., 2015).
Recently, we explored the genome of an indigenous EASZ from western Kenya population using genome-wide SNP data from the Illumina BovineSNP50 BeadChip v.1, and identified 24 candidate genome regions harboring signatures of positive selection (Bahbahani et al., 2015). However, given the restricted genome coverage of this tool (Matukumalli et al., 2009), these likely represent only a subset of the positively selected regions. We now analyze the autosomes of the same population for signatures of positive selection using two highly informative genome-wide dataset; genome-wide SNP genotypes obtained from the Illumina BovineHD BeadChip and full genome re-sequencing data. Furthermore, we include zebu × taurine populations from Uganda and Nigeria in our SNP genotyping analysis to assess the presence of commonly selected regions across these populations. Our aim is to provide a complete picture of the genome landscape of positive selection footprints in EASZ and in particular attend to untangle, at genome level, selection for the environmental challenges and the two components of animals fitness; reproduction and survivability. The identified regions were further explored to define possible causative variants responsible for the associated signatures of selection.
Materials and Methods
Cattle Populations, SNP Genotyping, and Quality Control
Details of the cattle populations studied are presented at Table 1. They include 92 non-European introgressed small EASZ from the Western and Nyanza provinces of Kenya (KEASZ) as well as cattle from Uganda, Nigeria, Guinea, Europe, and India. They were genotyped for 777,962 SNPs mapped to the UMD3.1 bovine reference genome using the Illumina BovineHD Genotyping BeadChip.
Table 1. The studied cattle populations.
Quality control (QC) analyses for 735,297 autosomal SNPs were conducted through the check. marker function implemented in the GenABEL package (Aulchenko et al., 2007) for R software version 2.15.1 (R development Core Team, 2012). SNPs with a minor allele frequency (MAF) <0.05 (n = 68,731) or call rate <95% (n = 18,667) were filtered out from the entire dataset. These include 1,712 SNPs that failed both criteria. In total, 649,611 SNPs were therefore retained. The ancestral allelic state for 373,005 SNPs [mean gap size = 6.7 kb and standard deviation (SD) = 12.1 kb] has been reported previously by Utsunomiya et al. (2013) following genotyping of three non-cattle Bovinae species: two B. gaurus (gaur), six Bubalus bubalis (water buffalo), and two B. grunniens (yak), with the fixed allele in the three species considered as ancestral. Only these SNPs were included in the downstream analyses.
Additional QC criteria included a minimum sample call rate of 95% and a maximum pairwise identity-by-state (IBS) of 95%, with the lower call rate animal eliminated from the high IBS pair. One ZS sample was excluded for having a low call rate, whilst 15 samples (two GIR, one NEL, four HOL, four JER, two AG, one AZ, and one WD) were excluded following the IBS criterion.
Principle Component Analysis (PCA)
PCA were conducted using the prcomp function implemented in GenABEL package for R software. These analyses were carried out in three levels; (i) all cattle populations, (ii) all cattle populations excluding the European taurine cattle, and (iii) only the KEASZ, Uganda (UGN) and Nigeria (NGR) cattle.
Estimation of Asian Zebu Autosomal Ancestry Proportion in African Zebu Cattle (Admixture Analysis)
Admixture analysis using ADMIXTURE 1.23 software (Alexander et al., 2009) with cross-validation and 200 bootstraps for K = 3 was conducted on the whole dataset to determine the European taurine, Asian zebu, and African taurine ancestries at genome-wide level and for each autosome separately. The output files were graphically displayed by the ggplot2 package (Wickham, 2009) for R software.
Extended Haplotype Homozygosity (EHH)—Based Statistics (Rsb and iHS)
Rsb analyses (Tang et al., 2007) were conducted between each of the KEASZ, combined UGN cattle populations (AO, KR, NG, ZS) and combined NGR cattle populations (AG, AZ, BJ, OR, SO, WD, YK; Tijjani, 2013; Mbole-Kariuki et al., 2014) with the combined reference cattle populations (NEL, GIR, NDM, MT, HOL, JER) using the rehh package (Gautier and Vitalis, 2012) for R software. The standardized Rsb-values were normally distributed (Supplementary Figure 1), so a Z-test was applied to identify statistically significant SNPs under selection on KEASZ, UGN, and NGR cattle populations. One-sided upper-tail P-values were derived as 1-Φ(Rsb) from the Gaussian cumulative density function Φ. Candidate regions were defined as having five adjacent SNPs, not separated by more than 500 kb, passing the threshold of −log10 P = 4.
iHS analyses (Voight et al., 2006) were conducted on KEASZ, combined UGN cattle and combined NGR cattle populations using the rehh package for the R software. This statistic was calculated for SNPs that passed the QC criteria and exhibited a within-population MAF of at least 0.05, since the algorithm of iHS has a limited power to calculate the statistic for fixed SNPs. As with Rsb, the standardized iHS-values followed a normal distribution (Supplementary Figure 1), so a two-tailed Z-test was applied to identify statistically significant SNPs under selection with either an unusual extended haplotype of ancestral or derived alleles relative to the genome. Two-sided P-values were derived as 1-2|Φ(iHS)-0.5| from the Gaussian cumulative density function Φ. Candidate regions were defined as in Rsb.
As a prerequisite to the Rsb and iHS analyses, fastPHASE 1.4 (Scheet and Stephens, 2006) was used to phase the genotyped SNPs into the corresponding haplotypes using K10 and T10 criteria. Population label information was used to estimate the phased haplotype background.
Inter-Population Change in SNP Allele Frequency (ΔAF)
ΔAF analysis investigates absolute allele frequency difference between two populations (Carneiro et al., 2014). In this analysis, the mean frequency of the first allele was estimated for KEASZ, combined UGN cattle and combined NGR cattle populations, separately (AFpop1). Likewise, the mean frequency of the first allele for each SNP was calculated for the combined reference cattle populations (AFpop2). The standardized values of the ΔAF (AFpop1–AFpop2) were normally distributed (Supplementary Figure 1), therefore a Z-test was applied to identify statistically significant SNPs showing higher allele frequency in the first population. Two-sided P-values were derived as 1-2|Φ (standardized ΔAF)-0.5| from the Gaussian cumulative density function Φ. Candidate regions were defined as in iHS and Rsb.
Meta-Analysis of Selection Signals (Meta-SS)
The Stouffer method was used to combine the P-values obtained from Rsb, iHS, and ΔAF, for each analyzed set of populations, i.e., KEASZ, UGN, and NGR, in meta-SS analyses (Whitlock, 2005; Utsunomiya et al., 2013). Each value, for every SNP in each test, was transformed to a Z-score, Φ−1 (1-P-value), where Φ−1 is the inverse cumulative distribution function for a standard normal distribution. Then, the SNP-specific Z-scores were combined together according to the following equation: Zi = (ZRsb + ZiHS + ZΔAF)/, where i and k are numbers of SNPs and tests, respectively. The resulting Z-scores were referred back to the standard normal distribution to obtain combined P-values [P-value = 1- Φ(Zi)], where is the cumulative distribution function for a standard normal distribution. Candidate regions were defined as having five adjacent SNPs not separated by more than 500 kb passing the threshold of −log10 P = 4.
KEASZ Whole Genome Sequencing Analysis
A single pool of 10 unrelated KEASZ DNA samples were sequenced using an ABI SOLiD 4 genetic analyser (Supplementary Table 1). SOLiD 2 × 50 bp mate-paired libraries were constructed and sequenced in the Deep Seq facility at the University of Nottingham according to the manufacturer's instructions. Reads were mapped to the UMD3.1 bovine reference genome assembly (Elsik et al., 2009) using the LifeScope Genomic Analysis software 2.5.1 re-sequencing mapping pipeline (http://www.lifetechnologies.com/lifescope). SNPs and indels (insertions/deletions) were called using the diBayes package implemented in LifeScope. A minimum coverage of two uniquely mapped reads and two non-reference allele counts were required to call a variant. Additionally, a minimum read mapping quality of 20 (MAPQ20) and base quality of 20 were also implemented.
Pooled heterozygosity Hp of SNPs detected in the pooled 10 KEASZ full genome SOLiD sequences were calculated on 100 kb sliding windows with 10 kb incremental steps. Window sizes were extended by the number of uncovered bases to improve the accuracy of the calculation and consistency across windows. For each SNP in the window, the number of reads for the most and least frequent allele was counted (nMAJ and nMIN, respectively). Hp-values were calculated using the following formula: Hp = 2 ∑ nMAJ ∑ nMIN/(∑ nMAJ + ∑ nMIN)2 (Rubin et al., 2010). The autosomal Hp-values were Z-transformed (ZHp = Hp–mean Hp/SD Hp) and a ZHp ≤ −4 was applied as a threshold to specify windows carrying selective sweep as in Liao et al. (2013). Overlapping candidate windows were merged into a single region.
KEASZ Exome Enrichment and Sequencing
The Agilent SureSelectXT target enrichment kit (cat no. G7530-90004) was used for bovine exome sequence enrichment. It covers a total of ~45 Mb of the bovine sequence that composed of the coding regions in the UMD3.1 reference genome (coding regions from Refseq and Ensembl, no UTR “untranslated regions”) and microRNA. The exomes of a further 10 KEASZ samples (Supplementary Table 1) were sequenced in the Deep Seq facility at the University of Nottingham using an ABI SOLiD 5500 genetic analyser. Using the LifeScope Enrichment Sequencing Pipeline, the generated 75 bp reads were mapped to the UMD3.1 bovine reference genome. Supplementary Table 2 summarizes the number of aligned reads, average depth of coverage, and percentage of reference exome for each sample. The same variants calling criteria used in the KEASZ whole genome sequence were implemented for the exome analysis except that reads with MAPQ ≥ 30 and base quality of 30 were used.
African Taurine (N'Dama and Muturu) Genome Sequence Analysis
To infer about the origin of the variants identified in the KEASZ selected regions, we analyzed 10 N'Dama cattle full genome sequences from Guinea available at the GenBank with the Bioproject accession number PRJNA312138 (Kim et al., 2017), and 10 Muturu cattle genome sequences from South East Nigeria. For the later, genomic DNA was extracted using Macherey-Nagel NucleoSpin® Tissue DNA extraction kit, according to manufacturer's protocol, from 10 ear tissue samples. One hundred and fifty bp paired-end libraries were constructed and sequenced using Illumina Hiseq2500 platform (Illumina, USA). Sequence reads were mapped to the UMD3.1 bovine reference genome assembly using the bwa-mem option of Burrows-Wheeler Alignment tool (BWA) version 0.7.5a (Li and Durbin, 2010; Supplementary Table 3). Genome Analysis Toolkits (GATK) version 3.4.0 (McKenna et al., 2010) was used for base quality recalibration, indel realignment, PCR duplicate removal and variants (SNPs and indels) calling, using Haplotypecaller function, according to GATK best practices recommendations (DePristo et al., 2011; Van der Auwera et al., 2013). A minimum read mapping quality of 50 (MAPQ50) and base quality of 30 were set as two criteria in variant calling.
Candidate Selected Regions Characterization
Protein-coding and RNA genes mapped within the candidate regions, based on the UMD3.1 bovine reference genome annotation, were processed using the functional annotation tool implemented in DAVID Bioinformatics resources 6.7 to determine the over-represented (enriched) functional terms (Huang da et al., 2009a,b). An enrichment score of 1.3, which is equivalent to the Fisher exact test P = 0.05, was used as a threshold to define the significantly enriched functional terms in comparison to the whole bovine reference genome background. The list of genes mapped on the UMD3.1 reference bovine genome was obtained from the Ensembl Genes 86 database (Flicek et al., 2013) using the BioMart tool (Kinsella et al., 2011). The bovine Quantitative Trait Loci (QTL) and their UMD3.1 genome coordinates were downloaded from the cattle QTL database (http://www.animalgenome.org/cgi-bin/QTLdb/BT/index). The intersectBed function from the BedTools software was used to overlap these QTL with the identified KEASZ candidate regions (Quinlan and Hall, 2010). Candidate regions lacking annotated genes (protein-coding, RNA, and pseudogenes) based on UMD3.1 bovine reference genome were also identified. The intersectBed function from the BedTools software was also used to overlap these candidate regions with transcription factors binding sites identified previously on the bovine reference genome by Bickhart and Liu (2013).
Variants (SNPs and indels) in the genes within the genome-wide SNP and Hp overlapping candidate regions were annotated using the variant effect predictor tool (McLaren et al., 2016) based on Ensembl variants 86 databases. Comparisons with the previously discovered bovine variants listed in the dbSNP database (Sherry et al., 2001) classified these variants into KEASZ-specific (novel) and general bovine variants. The identified missense, splice region SNPs, frameshift and splice region indels were cross-checked with variants identified in the KEASZ exome data and with N'Dama and Muturu full genome sequences.
Candidate genes within the overlapping genome-wide SNP and Hp candidate regions were selected based on their biological function, e.g., immunity, reproduction, fertility, heat tolerance and anatomical development. Variants (SNP and indels) within these genes were defined and the biological effect of the missense SNPs were predicted by the online tool PolyPhen-2 (Adzhubei et al., 2010).
Estimation of Excess-Deficiency in Asian Zebu Ancestry at Candidate Regions
LAMP software version 2.4 (Sankararaman et al., 2008) was used to estimate the Asian zebu and African taurine ancestry proportions of the high-density genotyped SNPs in the KEASZ samples. The genome-wide autosomal zebu ancestry proportion of 70% was obtained from the admixture proportions α of the ADMIXTURE analyses. An estimated number of 500 generations was set for the beginning of the zebu-taurine admixture in light of our current knowledge of zebu arrival on the continent, assuming a generation time of 6 years (Keightley and Eyre-Walker, 2000). A uniform recombination rate of 1 cM = 1 Mb was assumed. The average excess/deficiency in Asian zebu ancestry at each SNP (ΔAZ) was calculated by subtracting the average estimated Asian zebu ancestry of the SNP from the average estimated Asian zebu ancestry of all SNPs. The median ΔAZ for the significant SNPs of KEASZ meta-SS analysis within candidate regions was considered.
Results
The Admixed Genome Structure of African Cattle Populations
The PCA plot on all cattle populations using the filtered autosomal SNPs dataset (Figure 1A) reveals the already described triangle-like 2-dimentional global organization of cattle genetic diversity (Gautier et al., 2010). The first component, which accounts for 20.6% of the total variation, separates the European (HOL and JER) and African (NDM and MT) taurine populations from the Asian zebu cattle (NEL and GIR). The second component, which accounts for 4.9% of the total variation, separates the African taurine cattle from the other cattle populations. All the East (KEASZ and UGN) and West (NGR) African cattle populations analyzed are positioned between the African taurine and Asian zebu cattle supporting zebu × taurine genome admixture.
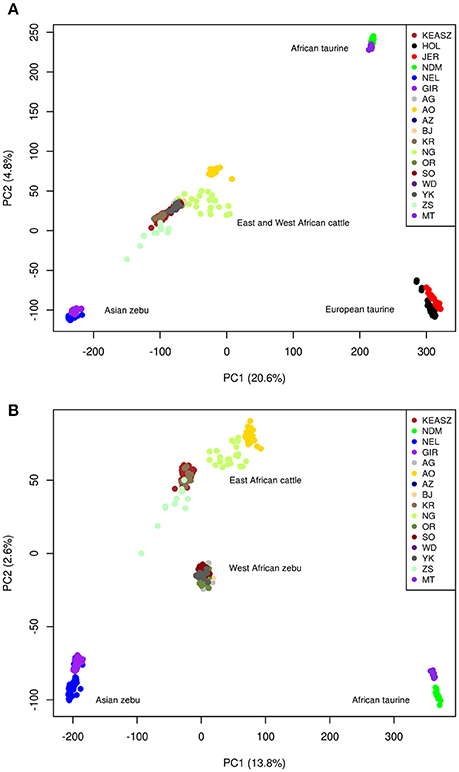
Figure 1. Plots of the highest two principle components resulted by analyzing autosomal SNPs in (A) all cattle populations included in this study, and (B) all cattle excluding European taurine populations.
The PCA conducted on Asian zebu (NEL and GIR), African taurine (NDM and MT), East African (KEASZ and UGN) and West African (NGR) cattle populations (Figure 1B) shows genetic differentiation within the East African cattle (KEASZ and UGN) cattle and between the East and West African (NGR) cattle. The first component, which explains 13.9% of the total variation, separates Asian zebu from African taurine. Whilst, the second component, which explains 2.6% of the total variation, separate West African zebu cattle (NGR) and the East African (KEASZ and UGN) cattle. A PCA on KEASZ, UGN, and NGR cattle only, separates first the East (KEASZ and UGN) and West (NGR) cattle populations (PC1 = 2.99%), while PC2 (2%) reveal a much higher genetic heterogeneity within and across East African cattle populations compared to the West African ones (Supplementary Figure 2).
The admixture analysis indicates a relatively even admixed genome of Asian zebu and African taurine ancestries across KEASZ animals with an estimated genetic proportion of 0.7 ± 0.01 SD and 0.30 ± 0.01 SD, respectively. The same even admixed proportion is present across the Karamajong zebu cattle from Uganda, while the Serere zebu has an estimated 0.74 ± 0.04 SD and 0.24 ± 0.04 SD zebu × taurine genetic proportions, with minor European taurine ancestry (0.02 ± 0.01 SD). The genome of the Ankole cattle has a larger African taurine genetic proportion (0.48 ± 0.01 SD). Nganda cattle has an admixed genome of 0.57 ± 0.04 SD Asian zebu and 0.36 ± 0.03 SD African taurine ancestries, it also carries low level of European taurine ancestry (0.07 ± 0.05 SD) (Figure 2). This pattern of admixture is also observed in the cattle of Nigeria but with higher average African taurine ancestry (0.35 ± 0.01 SD) and lower zebu ancestry (0.65 ± 0.01 SD) than the East African cattle, except Ankole. No European introgression was detected in the West African zebu cattle analyzed.
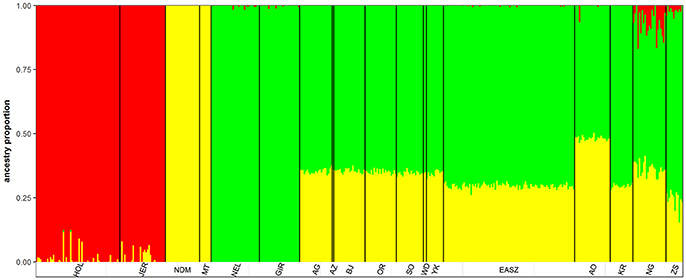
Figure 2. ADMIXTURE bar plot of the whole dataset at K = 3.
Variation in Asian zebu ancestry is observed among the autosomes of the East (KEASZ and UGN) and West African (NGR) cattle populations (Supplementary Figure 3, Supplementary Table 4) with increase or decrease (mean ± one SD) in Asian zebu ancestry proportions (Table 2). A strong correlation in the zebu ancestry autosomal variation is observed when comparing KEASZ with East (UGN) and West (NGR) cattle populations (P < 0.00001; Supplementary Figure 4).
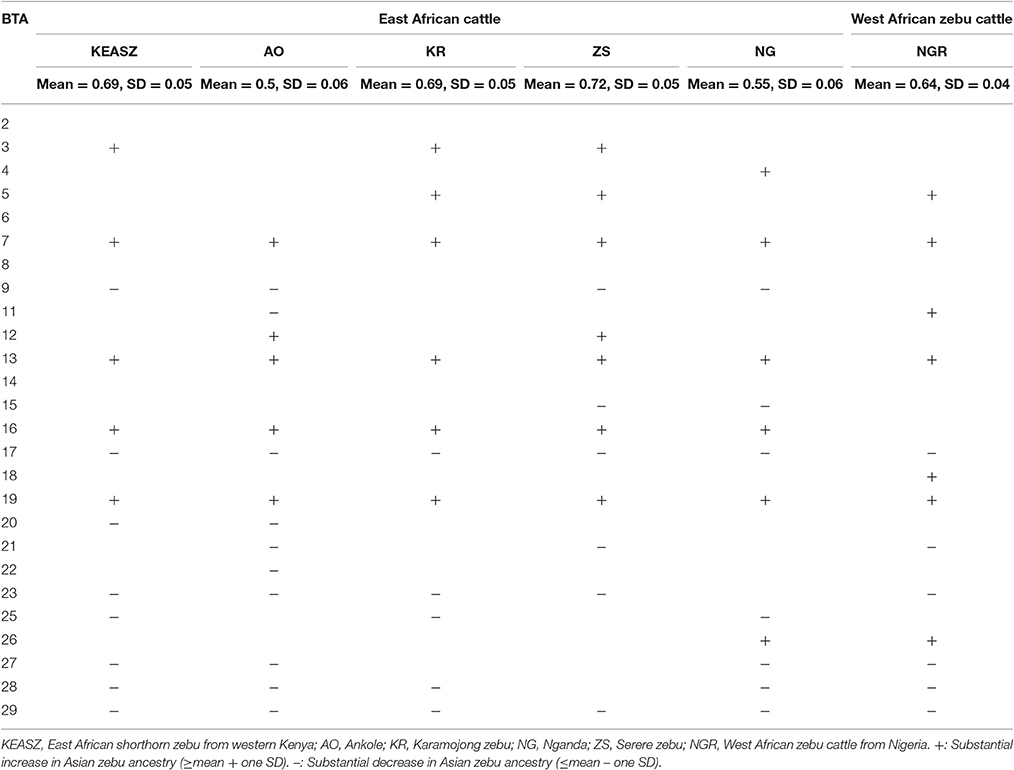
Table 2. Autosomes with substantial increase or decrease in Asian zebu ancestry among the East and West African cattle populations.
Signatures of Positive Selection
High Density Genome-Wide SNP Analysis
The iHS, Rsb, and ΔAF analyses conducted on KEASZ reveal one, 19 and six autosomal candidate regions harboring signatures of positive selection, respectively (Figures 3A–C, Supplementary Table 5). As the tests follow normal distributions (Supplementary Figure 1) and their genome-wide average P-values are weakly correlated (Pearson correlation coefficient r ≤ 0.228; Supplementary Table 6), the P-values of each SNP for the three tests were combined in a meta-SS analysis revealing 98 candidate regions (Figure 3D, Supplementary Table 5). All the candidate regions defined by each individual test are also included as candidate regions by the meta-SS analysis, with the exception of three regions on BTA 13 identified by ΔAF analysis only. Seventy-seven new candidate regions are detected after combining the P-values of the three tests.
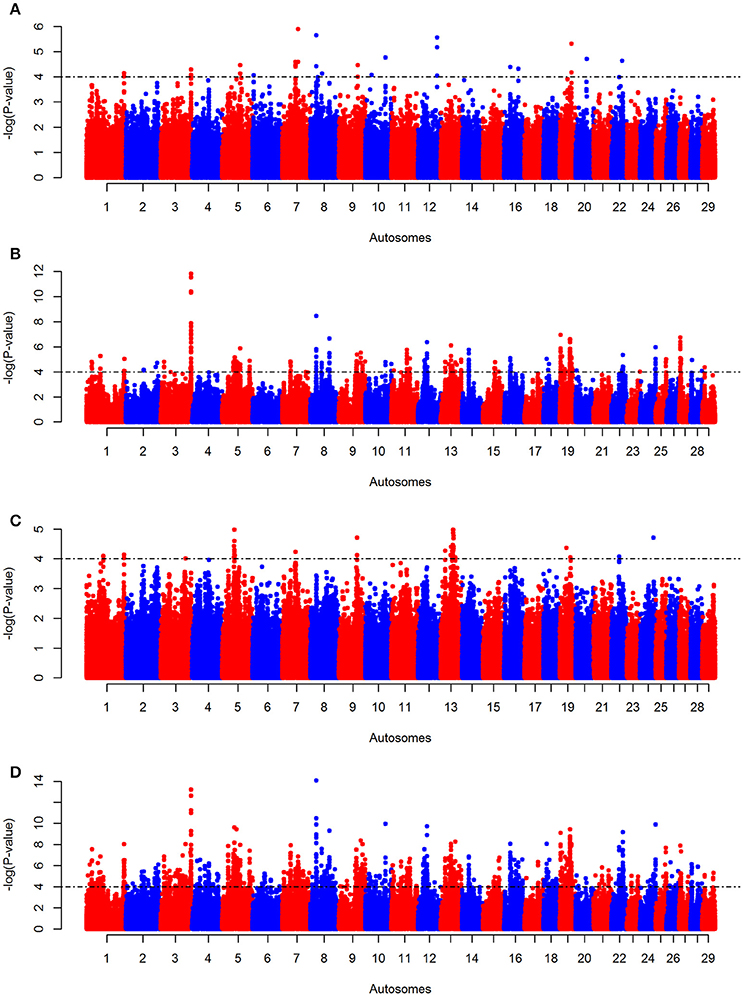
Figure 3. Manhattan plots for autosomal (A) KEASZ iHS, (B) Rsb, (C) ΔAF, and (D) meta-SS analyses between KEASZ and combined reference populations (Holstein-Friesian, Jersey, N'Dama, Muturu, Nelore and Gir). Threshold is set at–log10 P = 4.
In East African cattle populations from Uganda (UGN), the three tests (iHS, Rsb and ΔAF) reveal six, 25 and three autosomal candidate regions, respectively (Supplementary Figures 5A–C, Supplementary Table 7). The same analyses on the zebu cattle populations from Nigeria (NGR) identify four, 22 and four autosomal candidate regions, respectively (Supplementary Figures 6A–C, Supplementary Table 8). After combining the tests P-values, 86 and 97 regions are considered as candidate regions for positive selection in UGN and NGR cattle populations, respectively (Supplementary Figures 5D, 6D, Supplementary Tables 7, 8).
Comparison with African Cattle from Uganda and Nigeria
A total of 32 KEASZ candidate regions are shared with the UGN cattle populations. We classify them as East African candidate regions. Twenty-two regions are also shared across the three comparisons and they are classified as East and West African candidate regions (Table 3). The cross-validation of candidate regions between the three sets of zebu × taurine admixed cattle populations (KEASZ, UGN, and NGR) allow us to fine map the size of 18 candidate regions. As indicated in Table 4, this approach allows us to narrow the size of these candidate regions to 94–894 kb, with the largest reduction in size (~1.9 Mb) observed for a candidate region on BTA 12 (Table 4).
Table 3. Shared candidate regions obtained by the genome-wide SNP analyses.
Table 4. Fine mapping of KEASZ candidate regions.
Ten of the East African candidate regions intersect with regions under positive selection identified previously (Gautier et al., 2009; Gautier and Naves, 2011; Larkin et al., 2012; Kemper et al., 2014; Xu et al., 2015). Nine of these regions were identified in tropical-adapted cattle populations, such as Creole, Borgou, and Guzerat. Whilst four were found to be under positive selection in commercial cattle breeds, e.g., Holstein, Shorthorn, and Charolais.
For the 22 shared East and West African candidate regions, five were found to be under selection in tropical-adapted cattle (e.g., Gir, Creole, and Borgou) and four in commercial breeds (e.g., Angus, Holstein, and Shorthorn; Table 3).
Functional Characterization of High Density Genome-Wide SNP Candidate Regions
The 101 KEASZ candidate regions included 1,024 genes based on the UMD3.1 bovine reference genome annotation (Supplementary Table 9). These genes cluster into 110 functional clusters following DAVID functional term clusters enrichment analysis. Six of these clusters are significantly enriched relative to the bovine genome as indicated in Table 5. Candidate regions shared between KEASZ and UGN cattle populations harbor 309 genes (Supplementary Table 9). They are grouped into 32 functional term clusters, in which three are significantly enriched (Table 5). For candidate regions shared across East (KEASZ and UGN) and West (NGR) African cattle populations, 87 genes are identified. They are grouped into 10 functional term clusters, in which a single cluster, associated with immune response to bacterial infection, is significantly enriched (Table 5).
Table 5. Significantly enriched functional term clusters in KEASZ, East African (KEASZ and UGN), and East and West African (KEASZ, UGN, and NGR) candidate regions.
Seven KEASZ candidate regions are classified as gene desert regions. Two of these regions are also identified in UGN cattle populations and none with NGR cattle (Supplementary Table 10). No transcription factors binding sites identified on cattle genome by Bickhart and Liu (2013) are overlapping with any of these gene desert regions.
KEASZ Full Genome Sequencing Analysis
The 10 pooled KEASZ full genome sequences generated a total of 615,413,240 reads with MAPQ ≥ 20 (0.1% probability of incorrect alignment) mapped on the UMD3.1 bovine reference autosomes. These MAPQ20 reads covered ~97% of the reference autosomes with an average of 11 times depth of coverage. SNP calling using LifeScope diBayes package identified a total of 10,466,699 SNPs (8,114,664 heterozygotes and 2,352,035 homozygotes).
Regions with signatures of selective sweep were defined by assessing the pooled SNPs heterozygosity Hp of 100 kb windows, as in Liao et al. (2013), incremented by 10 kb. Supplementary Figure 7 shows the distribution of SNPs in the 100 kb autosomal windows with a mean of 297 SNPs per window. The mean Hp-value is 0.42 (SD = 0.025). Out of the total 250,930 windows, 1,825 (~0.73%) have a ZHp score of ≤ −4 merged into 165 autosomal candidate sweep regions (Figure 4, Supplementary Table 11). The largest region, ~2 Mb in size, is on BTA 7 (51.4–53.4 Mb). This region contains windows with the lowest ZHp-value (ZHp = −16.6).
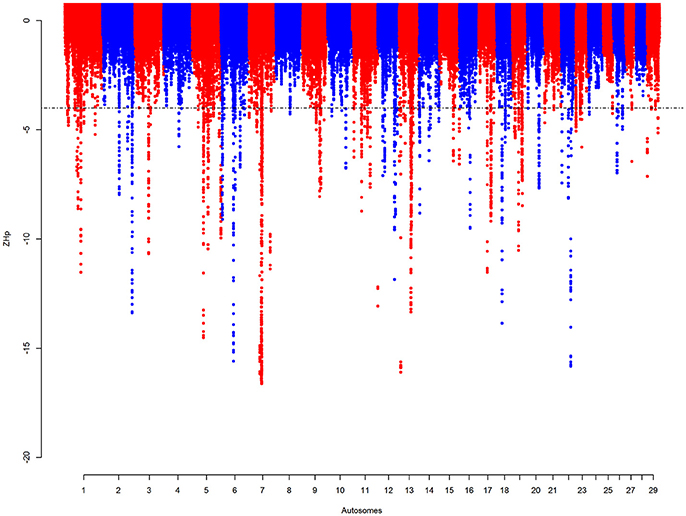
Figure 4. Manhattan plot for the autosomal Hp analyses on KEASZ. Each point represents a 100 kb window. The significant threshold is set at ZHp = −4.
Based on the annotated UMD3.1 bovine reference genome, 518 genes are found within 133 of these sweep regions (Supplementary Table 12). DAVID analyses were conducted in two levels including: (i) genes within KEASZ Hp candidate regions and (ii) genes within all KEASZ candidate regions from the SNPs and Hp analyses combined together. The first DAVID analysis identifies 57 functional term clusters with six significantly enriched clusters (Table 6). Whilst, the second one defines 148 clusters with six significantly enriched clusters (Table 6).
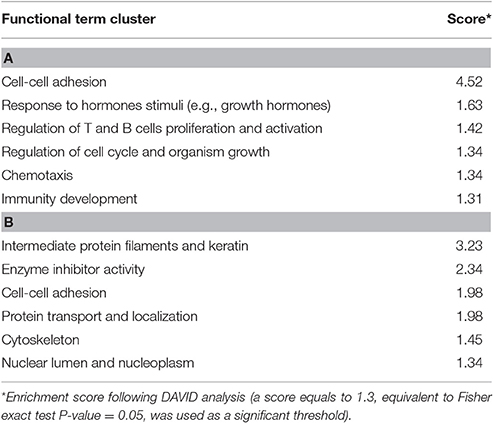
Table 6. Significantly enriched functional term clusters of the genes mapped within (A) Hp candidate sweep regions. (B) Combined KEASZ candidate regions (SNPs and Hp analyses).
A total of 32 candidate regions are gene deserts (Supplementary Table 10). Five of these regions were present in UGN cattle and one in NGR cattle (genome-wide HD SNPs analysis). None of transcription factors binding sites, identified by Bickhart and Liu (2013), are overlapping with the gene desert regions identified here.
Overlapping Candidate Sweep Regions between Genome-Wide HD SNP and Hp Full Genome Sequence Analyses
Among the 165 candidate Hp sweep regions, 35 regions overlap with the genome-wide HD SNP candidate regions of KEASZ. These include 25 regions also revealed by the SNP meta-SS analysis in UGN cattle populations, in which seven regions were also shared between the East (KEASZ and UGN) and West African (NGR) cattle populations (Table 7). Also, our genome sequence analysis reveals 101 candidate regions not previously identified to be under positive selection in other studied cattle populations (Supplementary Table 11).
Table 7. The overlapping candidate sweep regions between KEASZ Hp and genome-wide HD SNP analyses.
Within the 35 overlapping candidate regions, 185 genes are identified (Supplementary Table 13). DAVID analysis indicates 23 functional clusters with two significantly enriched functional clusters: response to hormone stimulus and signaling pathway (enrichment score = 2.07), and transcription regulation (enrichment score = 1.3). Also worth mentioning is a functional cluster associated with the immune system development and regulation, although it does not reach the 1.3 threshold (enrichment score = 1.24).
For the 25 East African overlapping sweep regions, 103 genes are found (Supplementary Table 13). They are grouped into four functional clusters: GTPase regulator activity (enrichment score = 1.17), protein complexes assembly (enrichment score = 0.76), regulation of transcription (enrichment score = 0.45), and nucleotides and ribonucleotides binding (enrichment score = 0.13), but none are significant. A total of 24 genes are within the seven overlapping regions across the East (KEASZ and UGN) and West African (NGR) populations (Supplementary Table 13). These genes are grouped into a single cluster associated with ion binding (enrichment score = 0.14).
A total of 11,915 SNPs in 148 genes and 484 indels in 96 genes are identified within the 35 overlapping genome-wide SNP and Hp candidate regions. These variants are either located on the coding region (missense, synonymous SNPs, and frameshift indels), or non-coding regions (intronic, splice regions, and 3′ and 5′ UTR SNPs and indels; Supplementary Table 14). A total of 261 SNPs in 50 genes and eight indels in seven genes have not been reported yet in the dbSNP database “novel variants.” Among all the variants, 88 SNPs in 49 genes (one novel) are missense, 50 SNPs in 37 genes (none novel) are on splice regions, two indels in two genes (none novel) are frameshift and two indels in two genes (none novel) are on splice regions (Supplementary Table 14).
Seventy-five missense SNPs in 44 genes were also identified in the KEASZ exome data spanning the overlapping regions. Twenty-three of these SNPs, in 13 genes, were not present in African taurine (Muturu and N'Dama), they are likely of zebu origin in KEASZ. Whilst, 15 missense SNPs in 12 genes were also identified in the two African taurine cattle populations examined and therefore they may be of taurine origin. Among the 50 splice regions SNPs, 44 in 32 genes are identified in the KEASZ exome data, in which seven SNPs in six genes may be of zebu origin and ten SNPs in nine genes may be of taurine origin (Supplementary Table 15). Only a single indel classified as frameshift and splice region indel has been identified in the KEASZ exome data. This indel has also been found in the genome of N'Dama and Muturu cattle and may be of taurine origin (Supplementary Table 15).
Twelve genes within the overlapping genome-wide SNP and Hp candidate regions were selected as examples of interesting candidates of positive selection in KEASZ following their biological roles (Table 8). These genes have functional roles linked to traits for adaptation to the African environment and/or reproductive fitness, e.g., immunological-related traits (e.g., disease challenges) and reproduction-related traits (e.g., fertility).
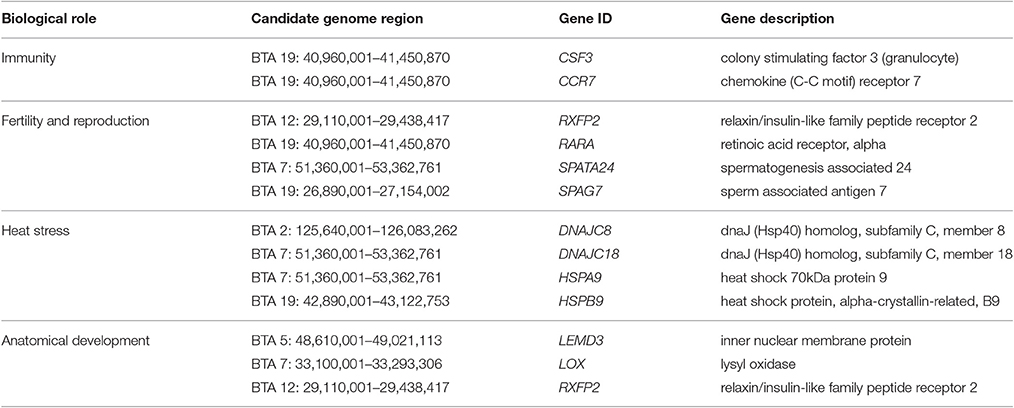
Table 8. Candidate genes within the KEASZ overlapping genome-wide SNP and Hp candidate signatures of selection regions.
Polymorphisms were identified in all these genes with the exception of the HSPB9 gene, which was found monomorphic. A total of 668 SNPs and 32 indels were detected (Supplementary Table 16). In particular, four missense variants on three genes, one splice region SNP on one gene and one frameshift indel on one gene were identified (Table 9), in which all have been reported previously in the dbSNP database (Sherry et al., 2001). Both of the two missense variants in RXFP2 are considered as of probable zebu origin. Whilst the splice region SNP on SPATA24 is considered as of probable taurine origin (Table 9, Supplementary Table 15).

Table 9. Missense, splice region SNPs, and frameshift indels within KEASZ candidate genes under positive selection.
Estimation of Excess-Deficiency in Asian Zebu Ancestry
LAMP software 2.4 (Sankararaman et al., 2008) estimated the mean Asian zebu ancestry proportion for all autosomal SNPs in KEASZ to be 0.76 (SD = 0.14). Based on this estimation, the mean ΔAZ for autosomal SNPs is 0 (SD = 0.14). The majority of the candidate sweep regions show high zebu ancestry proportion, but similar to the mean Asian zebu ancestry proportion (within one SD from the mean). For the 32 East African candidate regions (Table 3), eight regions reveal substantial ΔAZ, a single region shows deficiency and seven show excesses (more than ± one SD from the mean ΔAZ). Moreover, six East and West African candidate regions (Table 3) demonstrate substantial ΔAZ. Two of these regions show deficiencies and four show excesses of Asian zebu ancestry. When the genome-wide SNP and Hp analyses overlapping KEASZ candidate regions were considered, 13 regions demonstrate substantial ΔAZ. One region, which is shared between East and West African cattle, show deficiency in Asian zebu ancestry. Whilst 12 regions show excess in Asian zebu ancestry, in which six are specific to East African cattle and one is shared between East and West African cattle (Table 7).
Discussion
In this study, we unravel the autosomal zebu × taurine admixed genome structure of African indigenous cattle populations from the eastern (Kenya and Uganda) and western (Nigeria) part of the African continent using genome-wide high density SNP data. Also, for the first time, both genome-wide SNP and full genome sequence data have been utilized to identify candidate signatures of positive selection in the genome of an African cattle, the EASZ from Kenya, based on meta-analysis of selection signals (meta-SS) and pooled heterozygosity (Hp) analysis. These regions were then further characterized to identify candidate causative variants and to assess their probable zebu or African taurine origins.
Genetic Structure of East and West African Cattle Populations
Archaeological and genetic evidences so far indicate that the history of African zebu cattle started with the introgression of Asian zebu to the native African taurine populations ~4,000 and 1,300 years ago (Epstein, 1971; Hanotte et al., 2002). The coordinates of the African cattle samples (KEASZ, UGN, and NGR) in the PCA plots (Figures 1A,B) indicate zebu × taurine admixture level in their genome. This zebu introgression appears even across animals within population on the autosomes of KEASZ and also in West African cattle from Nigeria as well as in Ankole and Karamojong zebu cattle from Uganda (Figure 2). At the contrary, uneven European taurine introgression, likely of recent origin following ongoing crossbreeding of indigenous African cattle populations with exotic cattle breeds to improve their productivity (Mwai et al., 2015), is observed in the genome of Nganda and Serere zebu cattle from Uganda (Figure 2).
Interestingly, we also observe positive significant correlation in Asian zebu ancestry autosomal proportion between the KEASZ and the other East and West African cattle populations examined. This is in agreement with the known history of zebu cattle on the continent (Hanotte et al., 2002) and it supports a common ancestry for the African cattle examined here. Our results also support an East African origin for the West African zebu cattle as previously showed in Hanotte et al. (2002) with migration of admixed zebu × taurine cattle populations.
Candidate Genomics Regions under Positive Selection
This study is the first to our knowledge that exhaustively investigated the genome of an indigenous African cattle population for signatures of positive selection using both high density genome-wide SNP and full genome sequence information. The outputs of these analyses represent a follow up of our previous identifications of signatures of selection on KEASZ genome using the lower density Illumina BovineSNP50 BeadChip v.1 (Bahbahani et al., 2015). Here, we have validated 14 candidate regions out of 24 regions previously identified to be targeted by positive selection in KEASZ (Supplementary Table 17; Bahbahani et al., 2015). The remaining 10 regions might have been false positives. Indeed, in our previous study, we used a genomic tool characterized by high European taurine ascertainment bias and low genome coverage. This issue has been addressed in this study by using high-density HD SNP array and full genome sequence data. Moreover, we previously selected candidate regions based on only two SNPs above the significant threshold. Here we have been using much more stringent criteria with a minimum of five SNPs above the threshold to define a candidate region under positive selection (see Materials and Methods Section).
We also used a different strategy to increase the power of detecting genomic signatures of positive selection. First, instead of analyzing regions defined by each genome-wide SNP analyses (Rsb, iHS, and ΔAF) separately, a composite statistical approach “meta-SS analysis” was conducted at the autosomal level as in Utsunomiya et al. (2013) to combine the SNP-specific P-values for each test into a single index.
Simulation data have shown that combining the signals from different tests into a single statistic increase the power of defining genomic regions under selection (Grossman et al., 2010). Most importantly, coalescent simulations and accurate calibrated demographic models, which are lacking in African cattle, are not required by meta-SS in comparison to the original Composite of Multiple Signals (CMS) method proposed by Grossman et al. (2010). Secondly, the reference cattle populations were pooled into a single population as in Bahbahani et al. (2015). As suggested by Gautier and Naves (2011), this pooling approach increases the haplotype diversity in the reference populations and it breakdown population-specific linkage disequilibrium (LD) which may result from genetic drift. Thirdly, information from both genome-wide SNP genotypes and full genome sequence were used to adequately cover the KEASZ genome, as well as, to address any breed ascertainment bias associated with the commercially available SNP arrays (Matukumalli et al., 2009). Overlapping candidate regions from these two approaches further support the identification of the candidate regions.
The Candidate Regions: A Result of Positive Selection or Genetic Drift?
Both selection and genetic drift may have shaped the genome of the KEASZ and the other populations examined here. Distinction between the two is difficult, and fixation or near fixation of allele through genetic drift may lead to false positive candidate regions for signatures of positive selection (Qanbari and Simianer, 2014). Comparison of the results between different cattle populations, the unraveling of the zebu or taurine origin of the selected regions and the function of genes within selected regions may here provide further information.
In this study, we observe a small number of candidate regions for positive selection shared between KEASZ HD SNP and/or genome sequencing information and the East and West African cattle population examined here (Tables 3, 7). While it may be argued that these overlapping candidate regions for positive selection are a consequence of common genetic backgrounds, the low number of shared candidate regions as well as the identification of the same regions in tropical-adapted cattle populations with different population histories, e.g., Creole cattle from Guadeloupe (Gautier and Naves, 2011), zebu × taurine admixed cattle from West Africa (Gautier et al., 2009; Flori et al., 2014; Xu et al., 2015), Brahman (Ramey et al., 2013; Xu et al., 2015) and Gir (Liao et al., 2013; Perez O'Brien et al., 2014) cattle (Tables 3, 7, and Supplementary Table 11) strongly supports the role of selection rather genetic drift for these shared regions. It underlines the importance that these genome regions may play a role in the adaptive traits of African tropical-adapted admixed cattle.
Examining the probable ancestral origin of the candidate regions reveals a subset of candidate regions with substantial ΔAZ (Tables 3, 7). Most of these regions show excesses of Asian zebu ancestry, e.g., BTA 7: 51.4–53.4 Mb and BTA 13: 47.5–48.1 Mb, indicating that the indicine haplotypes are more likely to be under selection in the African admixed cattle populations than the taurine. This is perhaps not surprising considering the predominant zebu genomic background in EASZ (Mbole-Kariuki et al., 2014). However, we also observed candidate regions showing substantial excess of African taurine ancestry, e.g., BTA 3: 76.1–76.4 Mb and BTA 19: 3.3–3.8 Mb (Table 3). These are present in chromosomes with overall low level of African taurine ancestry adding further support for selective pressure rather than genetic drift for their presence (Table 2).
Biological Functions of the Genes Present in Signatures of Selection Regions in EASZ
Examining the potential biological functions under positive selection reveal several different significantly enriched biological pathways likely under selection in KEASZ given their importance for a cattle population living in a challenging tropical environment (e.g., bovine adaptive and innate immunity, response to hormone stimuli, intermediate filaments, and keratins pathways). Additionally, genes and QTL related to regulation of bovine immunity, fertility and reproduction, anatomical development, and heat stress have also been found within the identified KEASZ candidate regions (Supplementary Tables 9, 12, 18).
Indeed, innate and adaptive immune genes may be expected to be primary targets of selection in African cattle that are exposed to a diversity of pathogens and associated physiological stresses in their surrounding environment, e.g., endoparasites, haemoparasites, and bacteria (de Clare Bronsvoort et al., 2013; Murray et al., 2013; Thumbi et al., 2014). Examples of candidate genes related to this category are: C-C chemokine receptor type 7 precursor (CCR7) and granulocyte macrophage-colony stimulating factor (CSF3). Upon binding to two chemoattractants: CCL19 and CCL21, CCR7 is involved in maturating dendritic cells and hence activate T lymphocytes (Marsland et al., 2005; Forster et al., 2008). This receptor has also demonstrated a role in regulating innate immunity by attracting macrophages to sites of infection (van Zwam et al., 2010). The multifunctional cytokine (CSF3) acts as a positive regulator for macrophages to induce their antimicrobial effects (Grabstein et al., 1986; Tarr, 1996). Interestingly, several trypanotolerance QTL, identified by Hanotte et al. (2003), overlap with the identified KEASZ candidate regions (Supplementary Table 19). These QTL might indicate an undocumented level of trypanotolerance in KEASZ, as it has already been shown in other East African cattle populations (e.g., Orma Boran, Sheko and Mursi cattle; Dolan, 1987; Mwangi et al., 1993; Bahbahani and Hanotte, 2015).
Our results also show that besides tolerance to disease challenges, fertility and reproduction traits have been also selected in the African zebu × taurine admixed cattle. Examples of these candidate genes are: the retinoic acid receptor α subunit (RARA), relaxin/insulin-like family peptide receptor 2 (RXFP2), spermatogenesis associated 24 (SPATA24), and sperm associated antigen 7 (SPAG7). The retinoic acid receptor, which is expressed in sertoli cells in the seminiferous tubules, plays a role in maintaining retinoic acid signal to induce spermatogonia differentiation (Wolgemuth and Chung, 2007). RXFP2, which has been found to be under selection in admixed Creole cattle (Gautier and Naves, 2011) and Gir cattle (Liao et al., 2013), plays a role in testicular descent development (Gorlov et al., 2002; Agoulnik, 2007; Park et al., 2008; Feng et al., 2009). Candidate genes such as spermatogenesis-associated 24 (SPATA24) and sperm-associated antigen 7 (SPAG7) can also be classified into the cattle fertility and sexual reproduction category due to their role in spermatogenesis. Such signals may be the legacy of selection for fertility in hybrid populations between two cattle lineages (zebu and taurine), with a common ancestry perhaps as old as half a million years ago (MacHugh et al., 1997). This requires further investigation. QTL related to bovine reproduction and fertility, e.g., sperm motility and calving ease, are also target of positive selection in KEASZ.
An important gene category identified within the candidate regions is the heat stress category. Genes and QTL within this category might be targeted by natural selection to adapt to the tropical environmental condition (Hansen, 2004). Two heat shock protein genes (HSPA9 and HSPB9) and two members of the DnaJ family (DNAJC8 and DNAJC18) are mapped within three of the identified candidate regions (Table 8). Heat shock proteins have critical roles in maintaining protein folding under heat stress (Parsell and Lindquist, 1994; Coleman et al., 1995). The members of DnaJ family act as cofactors for other heat shock proteins (Hsp70) to maintain protein folding (Kampinga and Craig, 2010).
Genes and QTL related to anatomical development were also identified within the KEASZ candidate regions (e.g., LEMD3, LOX, and RXFP2). These genes are important to maintain optimum growth and development. LEMD3 and LOX are associated with the development of different organs, such as heart (LEMD3), lung and blood vessels (LOX) (Maki et al., 2005; Ishimura et al., 2008). The candidate region harboring LEMD3 has also been found to be under selection in Brahman cattle (Ramey et al., 2013). The role of RXFP2 in testicular descent development (Gorlov et al., 2002; Agoulnik, 2007; Park et al., 2008; Feng et al., 2009) can classify this gene into the anatomical development, fertility and reproduction as well as the heat tolerance categories. This gene has also been associated with the horn phenotype in sheep (Johnston et al., 2011; Kijas et al., 2012) and a study by Johnston et al. (2013) has demonstrated an association between variants of this gene and reproductive success and survival rate in Soay sheep from St. Kilda.
In addition, various production traits QTL, e.g., marbling score, milk fat percentage, and milk fat yield have also been found within the KEASZ candidate regions. Given that several of the identified KEASZ candidate regions overlap with regions under positive selection in commercial dairy and beef cattle breeds (Tables 3, 7, Supplementary Table S11; Larkin et al., 2012; Kemper et al., 2014; Qanbari et al., 2014), this may illustrate possible human selection for some production traits, e.g., milk yield, has taken place in KEASZ at least in the past.
In parallel to candidate regions harboring genes, the 39 gene desert candidate regions on KEASZ are targets of further research to define their biological roles. Although these regions do not contain any of the bovine transcription factors binding sites identified by Bickhart and Liu (2013), they may still harbor unannotated regulatory elements and/or genes targeted by positive selection. These regions may also be transcribed to generate long non-coding RNA transcripts (≥200 nucleotides), which could be further validated by RNA sequencing. This type of RNA molecules has critical roles in regulating the expression of neighboring genes at transcriptional and post-transcriptional levels (Mercer et al., 2009; Wang and Chang, 2011).
Putative Causative Variants in Candidate Regions
Several genomic variants, in which some are unique so far to KEASZ, have been identified within the KEASZ overlapping genome-wide SNP and Hp candidate regions. A subset of these variants, such as missense SNPs, framshift indels, and splice region SNPs/indels, can be considered as primary target of positive selection due to their functional roles on the corresponding genes. Interestingly, within the selected candidate genes four missense SNPs, a splice region SNP and a frameshift indel have been identified. These variants need to be confirmed in larger sample size of tropical-adapted (e.g., EASZ) and non-tropical-adapted (e.g., European taurine) cattle to further support their role as causative variants under selection. Two of the missense variants are predicted to show non-benign effects on their genes calling for their effect to be validated, e.g., through gene editing approaches (Carlson et al., 2012; Wang et al., 2013). Moreover, variants on the other genes within the candidate regions or in non-coding regions following their possible roles in regulating gene expression may also be targets of selection.
Based on available sequences of KEASZ and African taurine cattle (N'Dama and Muturu), we were able to infer the possible ancestral origin of some of these functional variants (Asian zebu or African taurine origin) indicating the role of the zebu × taurine admixture as a selective force shaping the genome of KEASZ. However, this origin assignment was only partially successful due to the low number of sequenced samples and the unavailability of a full indicine de novo reference genome.
Conclusion
We reported here for the first time extensive exploring for candidate regions and putative causative variants under selection on the genome of an indigenous African cattle (East African shorthorn zebu) using both genome-wide SNP data and full genome sequence data. The possible ancestral origins of some of these variants have been inferred using exome data of EASZ from Kenya and full genome sequence of African taurine cattle. In this study we have defined three selective forces on the KEASZ genome; the external environmental pressures, the internal admixed genome pressure and possible ancient human selection for production traits. Our results can be considered as the first milestone in conserving the adaptive genetic resources of the indigenous African cattle and to further improve their breeding strategy. This will enhance the productivity of the indigenous African cattle populations and at the same time retain their African environment adaptability. Although in this study we have used two genomic tools and cattle populations from East and West of Africa, these results need to be further validated in larger number of cattle populations with different ancestries and environments.
Data Accessibility
The pooled full genome sequence and the 10 exome sequences of the East African shorthorn zebu, in addition to the Muturu full genome sequences, are publicly available from GenBank with the Bioproject accession number PRJNA386202. All identified SNPs are deposited in dbSNP under handel (UON_LAB_A100). The high density SNP genotyping data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.38jp6.
Ethics Statement
Standard techniques were used to collect blood. The procedure was reviewed and approved by the University of Edinburgh Ethics Committee (reference number OS 03-06) and also by the Institute Animal Care and Use Committee of the International Livestock Research Institute, Nairobi.
Author Contributions
HB and OH conceived, designed the experiment. HB and OH performed the experiment. HB, AT, and FA analysed the data. MB and DW helped in the bioinformatic analysis. TS, MW, HH, ON, AT, GA, CM, MM, and CV contributed in data. HB and OH wrote the manuscript. All authors have agreed on the contents of the manuscript.
Funding
We would like to extend our sincere gratitude to the Wellcome Trust (grant reference 07995) for financially supporting this project. USDA-ARS Animal Genome Improvement Laboratory provided funding through project 1265-31000-098-00.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MB and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
We would like to thank Geneseek veterinary diagnostics for providing invaluable technical assistance through the genotyping of the samples. Finally, we wish to acknowledge the grass root farmers of Western Kenya who participated fully and made this project a success. We would like to acknowledge the support of SRUL01/13 from Kuwait University for using their computer facility.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fgene.2017.00068/full#supplementary-material
Abbreviations
KEASZ, East African Shorthorn Zebu from Kenya; UGN, Uganda; NGR, Nigeria; AO, Ankole; KR, Karamojong zebu; NG, Nganda; ZS, Serere zebu; AG, Adamawa Gudali; AZ, Azawak; BJ, Bunaji; OR, Red bororo; SO, Sokoto Gudali; WD, Wadara; YK, Yakanaji.
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Agoulnik, A. I. (2007). Relaxin and related peptides in male reproduction. Adv. Exp. Med. Biol. 612, 49–64. doi: 10.1007/978-0-387-74672-2_5
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Aulchenko, Y. S., Ripke, S., Isaacs, A., and van Duijn, C. M. (2007). GenABEL: an R library for genome-wide association analysis. Bioinformatics 23, 1294–1296. doi: 10.1093/bioinformatics/btm108
Bahbahani, H., Clifford, H., Wragg, D., Mbole-Kariuki, M. N., Van Tassell, C., Sonstegard, T., et al. (2015). Signatures of positive selection in East African Shorthorn Zebu: a genome-wide single nucleotide polymorphism analysis. Sci. Rep. 5:11729. doi: 10.1038/srep11729
Bahbahani, H., and Hanotte, O. (2015). Genetic resistance: tolerance to vector-borne diseases, prospect and challenges of genomics. OIE Sci. Technol. Rev. 34, 185–197. doi: 10.20506/rst.34.1.2353
Bickhart, D. M., and Liu, G. E. (2013). Identification of candidate transcription factor binding sites in the cattle genome. Genomics Proteomics Bioinform. 11, 195–198. doi: 10.1016/j.gpb.2012.10.004
Bradley, D. G., MacHugh, D. E., Cunningham, P., and Loftus, R. T. (1996). Mitochondrial diversity and the origins of African and European cattle. Proc. Natl. Acad. Sci. U.S.A. 93, 5131–5135. doi: 10.1073/pnas.93.10.5131
Carlson, D. F., Tan, W., Lillico, S. G., Stverakova, D., Proudfoot, C., Christian, M., et al. (2012). Efficient TALEN-mediated gene knockout in livestock. Proc. Natl. Acad. Sci. U.S.A. 109, 17382–17387. doi: 10.1073/pnas.1211446109
Carneiro, M., Rubin, C. J., Di Palma, F., Albert, F. W., Alfoldi, J., Barrio, A. M., et al. (2014). Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science 345, 1074–1079. doi: 10.1126/science.1253714
Chen, M., Pan, D., Ren, H., Fu, J., Li, J., Su, G., et al. (2016). Identification of selective sweeps reveals divergent selection between Chinese Holstein and Simmental cattle populations. Genet. Select. Evol. 48:76. doi: 10.1186/s12711-016-0254-5
Chen, S., Lin, B. Z., Baig, M., Mitra, B., Lopes, R. J., Santos, A. M., et al. (2010). Zebu cattle are an exclusive legacy of the South Asia neolithic. Mol. Biol. Evol. 27, 1–6. doi: 10.1093/molbev/msp213
Choi, J. W., Choi, B. H., Lee, S. H., Lee, S. S., Kim, H. C., Yu, D., et al. (2015). Whole-genome resequencing analysis of hanwoo and yanbian cattle to identify genome-wide SNPs and signatures of selection. Mol. Cells 38, 466–473. doi: 10.14348/molcells.2015.0019
Coleman, J. S., Heckathorn, S. A., and Hallberg, R. L. (1995). Heat-shock proteins and thermotolerance: linking molecular and ecological perspectives. Trends Ecol. Evol. 10, 305–306. doi: 10.1016/S0169-5347(00)89112-0
DAGRIS (2007). Domestic Animal Genetic Resources Information System (DAGRIS). Addis Ababa: International Livestock Research Institute.
Decker, J. E., McKay, S. D., Rolf, M. M., Kim, J., Molina Alcala, A., Sonstegard, T. S., et al. (2014). Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 10:e1004254. doi: 10.1371/journal.pgen.1004254
de Clare Bronsvoort, B. M., Thumbi, S. M., Poole, E. J., Kiara, H., Auguet, O. T., Handel, I. G., et al. (2013). Design and descriptive epidemiology of the Infectious Diseases of East African Livestock (IDEAL) project, a longitudinal calf cohort study in western Kenya. BMC Vet. Res. 9:171. doi: 10.1186/1746-6148-9-171
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Dolan, R. B. (1987). Genetics and trypanotolerance. Parasitol. Today 3, 137–143. doi: 10.1016/0169-4758(87)90197-9
Elsik, C. G., Tellam, R. L., Worley, K. C., Gibbs, R. A., Muzny, D. M., Weinstock, G. M., et al. (2009). The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science 324, 522–528. doi: 10.1126/science.1169588
Epstein, H. (1971). The Origin of the Domestic Animals of Africa. New York, NY; London; Munich: Africana Publishing Corporation.
Feng, S., Ferlin, A., Truong, A., Bathgate, R., Wade, J. D., Corbett, S., et al. (2009). INSL3/RXFP2 signaling in testicular descent. Ann. N.Y. Acad. Sci. 1160, 197–204. doi: 10.1111/j.1749-6632.2009.03841.x
Flicek, P., Ahmed, I., Amode, M. R., Barrell, D., Beal, K., Brent, S., et al. (2013). Ensembl 2013. Nucleic Acids Res. 41, D48–D55. doi: 10.1093/nar/gks1236
Flori, L., Fritz, S., Jaffrezic, F., Boussaha, M., Gut, I., Heath, S., et al. (2009). The genome response to artificial selection: a case study in dairy cattle. PLoS ONE 4:e6595. doi: 10.1371/journal.pone.0006595
Flori, L., Gonzatti, M. I., Thevenon, S., Chantal, I., Pinto, J., Berthier, D., et al. (2012). A quasi-exclusive European ancestry in the Senepol tropical cattle breed highlights the importance of the slick locus in tropical adaptation. PLoS ONE 7:e36133. doi: 10.1371/journal.pone.0036133
Flori, L., Thevenon, S., Dayo, G. K., Senou, M., Sylla, S., Berthier, D., et al. (2014). Adaptive admixture in the West African bovine hybrid zone: insight from the Borgou population. Mol. Ecol. 23, 3241–3257. doi: 10.1111/mec.12816
Forster, R., Davalos-Misslitz, A. C., and Rot, A. (2008). CCR7 and its ligands: balancing immunity and tolerance. Nat. Rev. Immunol. 8, 362–371. doi: 10.1038/nri2297
Frantz, L. A. F., Schraiber, J. G., Madsen, O., Megens, H.-J., Cagan, A., Bosse, M., et al. (2015). Evidence of long-term gene flow and selection during domestication from analyses of Eurasian wild and domestic pig genomes. Nat. Genet. 47, 1141–1148. doi: 10.1038/ng.3394
Gautier, M., Flori, L., Riebler, A., Jaffrezic, F., Laloe, D., Gut, I., et al. (2009). A whole genome Bayesian scan for adaptive genetic divergence in West African cattle. BMC Genomics 10:550. doi: 10.1186/1471-2164-10-550
Gautier, M., Laloë, D., and Moazami-Goudarzi, K. (2010). Insights into the genetic history of French cattle from dense SNP data on 47 worldwide breeds. PLoS ONE 5:e13038. doi: 10.1371/journal.pone.0013038
Gautier, M., and Naves, M. (2011). Footprints of selection in the ancestral admixture of a New World Creole cattle breed. Mol. Ecol. 20, 3128–3143. doi: 10.1111/j.1365-294X.2011.05163.x
Gautier, M., and Vitalis, R. (2012). rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 28, 1176–1177. doi: 10.1093/bioinformatics/bts115
Gifford-Gonzalez, D., and Hanotte, O. (2011). Domesticating animals in Africa: implications of genetic and archaeological findings. J. World Prehist. 24, 1–23. doi: 10.1007/s10963-010-9042-2
Gorlov, I. P., Kamat, A., Bogatcheva, N. V., Jones, E., Lamb, D. J., Truong, A., et al. (2002). Mutations of the GREAT gene cause cryptorchidism. Hum. Mol. Genet. 11, 2309–2318. doi: 10.1093/hmg/11.19.2309
Grabstein, K. H., Urdal, D. L., Tushinski, R. J., Mochizuki, D. Y., Price, V. L., Cantrell, M. A., et al. (1986). Induction of macrophage tumoricidal activity by granulocyte-macrophage colony-stimulating factor. Science 232, 506–508. doi: 10.1126/science.3083507
Grossman, S. R., Shlyakhter, I., Karlsson, E. K., Byrne, E. H., Morales, S., Frieden, G., et al. (2010). A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 327, 883–886. doi: 10.1126/science.1183863
Hanotte, O., Bradley, D. G., Ochieng, J. W., Verjee, Y., Hill, E. W., and Rege, J. E. (2002). African pastoralism: genetic imprints of origins and migrations. Science 296, 336–339. doi: 10.1126/science.1069878
Hanotte, O., Ronin, Y., Agaba, M., Nilsson, P., Gelhaus, A., Horstmann, R., et al. (2003). Mapping of quantitative trait loci controlling trypanotolerance in a cross of tolerant West African N'Dama and susceptible East African Boran cattle. Proc. Natl. Acad. Sci. U.S.A. 100, 7443–7448. doi: 10.1073/pnas.1232392100
Hansen, P. J. (2004). Physiological and cellular adaptations of zebu cattle to thermal stress. Anim. Reprod. Sci. 82–83, 349–360. doi: 10.1016/j.anireprosci.2004.04.011
Huang da, W., Sherman, B. T., and Lempicki, R. A. (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. doi: 10.1093/nar/gkn923
Huang da, W., Sherman, B. T., and Lempicki, R. A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1186/s12711-016-0254-5
Ishimura, A., Chida, S., and Osada, S. (2008). Man1, an inner nuclear membrane protein, regulates left-right axis formation by controlling nodal signaling in a node-independent manner. Dev. Dyn. 237, 3565–3576. doi: 10.1002/dvdy.21663
Johnston, S. E., Gratten, J., Berenos, C., Pilkington, J. G., Clutton-Brock, T. H., Pemberton, J. M., et al. (2013). Life history trade-offs at a single locus maintain sexually selected genetic variation. Nature 502, 93–95. doi: 10.1038/nature12489
Johnston, S. E., McEwan, J. C., Pickering, N. K., Kijas, J. W., Beraldi, D., Pilkington, J. G., et al. (2011). Genome-wide association mapping identifies the genetic basis of discrete and quantitative variation in sexual weaponry in a wild sheep population. Mol. Ecol. 20, 2555–2566. doi: 10.1111/j.1365-294X.2011.05076.x
Kampinga, H. H., and Craig, E. A. (2010). The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592. doi: 10.1038/nrm2941
Keightley, P. D., and Eyre-Walker, A. (2000). Deleterious mutations and the evolution of sex. Science 290, 331–333. doi: 10.1126/science.290.5490.331
Kemper, K. E., Saxton, S. J., Bolormaa, S., Hayes, B. J., and Goddard, M. E. (2014). Selection for complex traits leaves little or no classic signatures of selection. BMC Genomics 15:246. doi: 10.1186/1471-2164-15-246
Khayatzadeh, N., Mészáros, G., Utsunomiya, Y. T., Garcia, J. F., Schnyder, U., Gredler, B., et al. (2016). Locus-specific ancestry to detect recent response to selection in admixed Swiss Fleckvieh cattle. Anim. Genet. 47, 637–646. doi: 10.1111/age.12470
Kijas, J., Lenstra, J., Hayes, B., Boitard, S., Porto Neto, L., San Cristobal, M., et al. (2012). Genome wide analysis of the world's sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 10:e1001258. doi: 10.1371/journal.pbio.1001258
Kim, J., Hanotte, O., Mwai, O. A., Dessie, T., Bashir, S., Diallo, B., et al. (2017). The genome landscape of indigenous African cattle. Genome Biol. 18:34. doi: 10.1186/s13059-017-1153-y
Kinsella, R. J., Kahari, A., Haider, S., Zamora, J., Proctor, G., Spudich, G., et al. (2011). Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database 2011:bar030. doi: 10.1093/database/bar030
Larkin, D. M., Daetwyler, H. D., Hernandez, A. G., Wright, C. L., Hetrick, L. A., Boucek, L., et al. (2012). Whole-genome resequencing of two elite sires for the detection of haplotypes under selection in dairy cattle. Proc. Natl. Acad. Sci. U.S.A. 109, 7693–7698. doi: 10.1073/pnas.1114546109
Latif, A. A., Nokoe, S., Punyua, D. K., and Capstick, P. B. (1991). Tick infestations on Zebu cattle in western Kenya: quantitative assessment of host resistance. J. Med. Entomol. 28, 122–126. doi: 10.1093/jmedent/28.1.122
Latif, A. A., and Pegram, R. G. (1992). Naturally acquired host resistance in tick control in Africa. Int. J. Trop. Insect Sci. 13, 505–513. doi: 10.1017/S1742758400016088
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Liao, X., Peng, F., Forni, S., McLaren, D., Plastow, G., and Stothard, P. (2013). Whole genome sequencing of Gir cattle for identifying polymorphisms and loci under selection. Genome 56, 592–598. doi: 10.1139/gen-2013-0082
Liu, Z., Ji, Z., Wang, G., Chao, T., Hou, L., and Wang, J. (2016). Genome-wide analysis reveals signatures of selection for important traits in domestic sheep from different ecoregions. BMC Genomics 17:863. doi: 10.1186/s12864-016-3212-2
Loftus, R. T., Machugh, D. E., Bradley, D. G., Sharp, P. M., and Cunningham, P. (1994). Evidence for 2 independent domestications of cattle. Proc. Natl. Acad. Sci. U.S.A. 91, 2757–2761. doi: 10.1073/pnas.91.7.2757
MacHugh, D. E., Shriver, M. D., Loftus, R. T., Cunningham, P., and Bradley, D. G. (1997). Microsatellite DNA variation and the evolution, domestication and phylogeography of taurine and zebu cattle (Bos taurus and Bos indicus). Genetics 146, 1071–1086.
Maki, J. M., Sormunen, R., Lippo, S., Kaarteenaho-Wiik, R., Soininen, R., and Myllyharju, J. (2005). Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am. J. Pathol. 167, 927–936. doi: 10.1016/S0002-9440(10)61183-2
Marsland, B. J., Battig, P., Bauer, M., Ruedl, C., Lassing, U., Beerli, R. R., et al. (2005). CCL19 and CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity 22, 493–505. doi: 10.1016/j.immuni.2005.02.010
Matukumalli, L. K., Lawley, C. T., Schnabel, R. D., Taylor, J. F., Allan, M. F., Heaton, M. P., et al. (2009). Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 4:e5350. doi: 10.1371/journal.pone.0005350
Mbole-Kariuki, M. N., Sonstegard, T., Orth, A., Thumbi, S. M., Bronsvoort, B. M., Kiara, H., et al. (2014). Genome-wide analysis reveals the ancient and recent admixture history of East African Shorthorn Zebu from Western Kenya. Heredity 113, 297–305. doi: 10.1038/hdy.2014.31
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R., Thormann, A., et al. (2016). The ensembl variant effect predictor. Genome Biol. 17:122. doi: 10.1186/s13059-016-0974-4
Mercer, T. R., Dinger, M. E., and Mattick, J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159. doi: 10.1038/nrg2521
Murray, G. G., Woolhouse, M. E., Tapio, M., Mbole-Kariuki, M. N., Sonstegard, T. S., Thumbi, S. M., et al. (2013). Genetic susceptibility to infectious disease in East African Shorthorn Zebu: a genome-wide analysis of the effect of heterozygosity and exotic introgression. BMC Evol. Biol. 13:246. doi: 10.1186/1471-2148-13-246
Mwai, O., Hanotte, O., Kwon, Y.-J., and Cho, S. (2015). African Indigenous cattle: unique genetic resources in a rapidly changing world. Asian-Australas J. Anim. Sci. 28, 911–921. doi: 10.5713/ajas.15.0002R
Mwangi, E. K., Stevenson, P., Gettinby, G., and Murray, M. (1993). “Variation in susceptibility to tsetse-borne trypanosomiasis among Bos indicus cattle breeds in East Africa,” in Towards Increased Use of Trypanotolerance: Current Research and Future Directions, eds J. Rowlands and A.J. Teale (Nairobi: ILRAD-ALCA), 81–86.
Park, J. I., Semyonov, J., Chang, C. L., Yi, W., Warren, W., and Hsu, S. Y. (2008). Origin of INSL3-mediated testicular descent in therian mammals. Genome Res. 18, 974–985. doi: 10.1101/gr.7119108
Parsell, D. A., and Lindquist, S. (1994). “Heat shock proteins and stress tolerance,” in The Biology of Heat Schock Proteins and Molecular Chaperones, eds R. I. Morimoto, A. Tissieres, and C. Georgopoulos (New York, NY: Cold Spring Harbor Laboratory Press), 457–494.
Perez O'Brien, A. M., Utsunomiya, Y. T., Meszaros, G., Bickhart, D. M., Liu, G. E., Van Tassell, C. P., et al. (2014). Assessing signatures of selection through variation in linkage disequilibrium between taurine and indicine cattle. Genet. Sel. Evol. 46:19. doi: 10.1186/1297-9686-46-19
Porto-Neto, L. R., Reverter, A., Prayaga, K. C., Chan, E. K., Johnston, D. J., Hawken, R. J., et al. (2014). The genetic architecture of climatic adaptation of tropical cattle. PLoS ONE 9:e113284. doi: 10.1371/journal.pone.0113284
Porto-Neto, L. R., Sonstegard, T. S., Liu, G. E., Bickhart, D. M., Da Silva, M. V. B., Machado, M. A., et al. (2013). Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genomics 14:876. doi: 10.1186/1471-2164-14-876
Qanbari, S., Gianola, D., Hayes, B., Schenkel, F., Miller, S., Moore, S., et al. (2011). Application of site and haplotype-frequency based approaches for detecting selection signatures in cattle. BMC Genomics 12:318. doi: 10.1186/1471-2164-12-318
Qanbari, S., Pausch, H., Jansen, S., Somel, M., Strom, T. M., Fries, R., et al. (2014). Classic selective sweeps revealed by massive sequencing in cattle. PLoS Genet. 10:e1004148. doi: 10.1371/journal.pgen.1004148
Qanbari, S., and Simianer, H. (2014). Mapping signatures of positive selection in the genome of livestock. Livest. Sci. 166, 133–143. doi: 10.1016/j.livsci.2014.05.003
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Ramey, H. R., Decker, J. E., McKay, S. D., Rolf, M. M., Schnabel, R. D., and Taylor, J. F. (2013). Detection of selective sweeps in cattle using genome-wide SNP data. BMC Genomics 14:382. doi: 10.1186/1471-2164-14-382
R development Core Team (2012). R: A Language and Environment for Statistical Computing. Vienna: R Development Core Team.
Rege, J. E. O. (1999). The state of African cattle genetic resources I. Classification framework and identification of threatened and extinct breeds. Anim. Genet. Resour. Inform. 25, 1–25. doi: 10.1017/S1014233900003448
Rege, J. E. O., Kahi, A., Okomo-Adhiambo, M., Mwacharo, J., and Hanotte, O. (2001). Zebu cattle of Kenya: Uses, Performance, Farmer Preferences and Measures of Genetic Diversity. Nairobi: International Livestock Reaserch Institute.
Rincon, G., Weber, K. L., Eenennaam, A. L., Golden, B. L., and Medrano, J. F. (2011). Hot topic: performance of bovine high-density genotyping platforms in Holsteins and Jerseys. J. Dairy Sci. 94, 6116–6121. doi: 10.3168/jds.2011-4764
Rubin, C. J., Megens, H. J., Martinez Barrio, A., Maqbool, K., Sayyab, S., Schwochow, D., et al. (2012). Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. U.S.A. 109, 19529–19536. doi: 10.1073/pnas.1217149109
Rubin, C. J., Zody, M. C., Eriksson, J., Meadows, J. R., Sherwood, E., Webster, M. T., et al. (2010). Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464, 587–591. doi: 10.1038/nature08832
Sankararaman, S., Sridhar, S., Kimmel, G., and Halperin, E. (2008). Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 82, 290–303. doi: 10.1016/j.ajhg.2007.09.022
Scheet, P., and Stephens, M. (2006). A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78, 629–644. doi: 10.1086/502802
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M., et al. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311. doi: 10.1093/nar/29.1.308
Stouffer, S. A., Suchman, E. A., DeVinney, L. C., Star, S. A., and Williams, R. M. (1949). The American Soldier, Vol. 1: Adjustment during Army Life. Princeton, NY: Princeton University Press.
Tang, K., Thornton, K. R., and Stoneking, M. (2007). A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 5:e171. doi: 10.1371/journal.pbio.0050171
Tarr, P. E. (1996). Granulocyte-macrophage colony-stimulating factor and the immune system. Med. Oncol. 13, 133–140. doi: 10.1007/B.F.02990841
Thumbi, S. M., Bronsvoort, B. M., Poole, E. J., Kiara, H., Toye, P. G., Mbole-Kariuki, M. N., et al. (2014). Parasite co-infections and their impact on survival of indigenous cattle. PLoS ONE 9:e76324. doi: 10.1371/journal.pone.0076324
Tijjani, A. (2013). Genome-Wide Characterization of Diversity and Admixture in African Cattle Breeds using High Density SNP Markers. Nottingham: University of Nottingham.
Troy, C. S., MacHugh, D. E., Bailey, J. F., Magee, D. A., Loftus, R. T., Cunningham, P., et al. (2001). Genetic evidence for Near-Eastern origins of European cattle. Nature 410, 1088–1091. doi: 10.1038/35074088
Utsunomiya, Y. T., Perez O'Brien, A. M., Sonstegard, T. S., Van Tassell, C. P., do Carmo, A. S., Meszaros, G., et al. (2013). Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS ONE 8:e64280. doi: 10.1371/journal.pone.0064280
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 11.10.11–33. doi: 10.1002/0471250953.bi1110s43
van Zwam, M., Wierenga-Wolf, A. F., Melief, M. J., Schrijver, B., Laman, J. D., and Boven, L. A. (2010). Myelin ingestion by macrophages promotes their motility and capacity to recruit myeloid cells. J. Neuroimmunol. 225, 112–117. doi: 10.1016/j.jneuroim.2010.04.021
Voight, B., Kudaravalli, S., Wen, X., and Pritchard, J. (2006). A map of recent positive selection in the human genome. PLoS Biol. 4:e72. doi: 10.1371/journal.pbio.0040072
Wang, H., Yang, H., Shivalila, C. S., Dawlaty, M. M., Cheng, A. W., Zhang, F., et al. (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918. doi: 10.1016/j.cell.2013.04.025
Wang, K. C., and Chang, H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914. doi: 10.1016/j.molcel.2011.08.018
Western, D., and Finch, V. (1986). Cattle and pastoralism: survival and production in arid lands. Hum. Ecol. 14, 77–94. doi: 10.1007/BF00889211
Whitlock, M. C. (2005). Combining probability from independent tests: the weighted Z-method is superior to Fisher's approach. J. Evol. Biol. 18, 1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x
Wolgemuth, D. J., and Chung, S. S. (2007). Retinoid signaling during spermatogenesis as revealed by genetic and metabolic manipulations of retinoic acid receptor alpha. Soc. Reprod. Fertil. Suppl. 63, 11–23.
Xu, L., Bickhart, D. M., Cole, J. B., Schroeder, S. G., Song, J., Tassell, C. P., et al. (2015). Genomic signatures reveal new evidences for selection of important traits in domestic cattle. Mol. Biol. Evol. 32, 711–725. doi: 10.1093/molbev/msu333
Keywords: African cattle, positive selection, environmental adaptation, hybrid fitness, meta-analysis of selection signals
Citation: Bahbahani H, Tijjani A, Mukasa C, Wragg D, Almathen F, Nash O, Akpa GN, Mbole-Kariuki M, Malla S, Woolhouse M, Sonstegard T, Van Tassell C, Blythe M, Huson H and Hanotte O (2017) Signatures of Selection for Environmental Adaptation and Zebu × Taurine Hybrid Fitness in East African Shorthorn Zebu. Front. Genet. 8:68. doi: 10.3389/fgene.2017.00068
Received: 04 March 2017; Accepted: 12 May 2017;
Published: 08 June 2017.
Edited by:
Riccardo Negrini, Università Cattolica del Sacro Cuore, ItalyReviewed by:
Mario Barbato, Università Cattolica del Sacro Cuore, ItalyShahin Eghbalsaied, Islamic Azad University, Isfahan, Iran
Copyright © 2017 Bahbahani, Tijjani, Mukasa, Wragg, Almathen, Nash, Akpa, Mbole-Kariuki, Malla, Woolhouse, Sonstegard, Van Tassell, Blythe, Huson and Hanotte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hussain Bahbahani, aC5iYWhiYWhhbmlAaG90bWFpbC5jb20=; aHVzc2Fpbi5iYWhiYWhhbmlAa3UuZWR1Lmt3