 Siegfried Hekimi
Siegfried Hekimi Ying Wang
Ying Wang Alycia Noë
Alycia Noë
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 14 September 2016
Sec. Genetics of Aging
Volume 7 - 2016 | https://doi.org/10.3389/fgene.2016.00161
This article is part of the Research TopicInterorganelle communication in agingView all 7 articles
It has become clear that mitochondrial reactive oxygen species (mtROS) are not simply villains and mitochondria the hapless targets of their attacks. Rather, it appears that mitochondrial dysfunction itself and the signaling function of mtROS can have positive effects on lifespan, helping to extend longevity. If events in the mitochondria can lead to better cellular homeostasis and better survival of the organism in ways beyond providing ATP and biosynthetic products, we can conjecture that they act on other cellular components through appropriate signaling pathways. We describe recent advances in a variety of species which promoted our understanding of how changes of mtROS generation are part of a system of signaling pathways that emanate from the mitochondria to impact organism lifespan through global changes, including in transcriptional patterns. In unraveling this, many old players in cellular homeostasis were encountered. Among these, maybe most strikingly, is the intrinsic apoptotic signaling pathway, which is the conduit by which at least one class of mtROS exercise their actions in the nematode Caenorhabditis elegans. This is a pathway that normally contributes to organismal homeostasis by killing defective or otherwise unwanted cells, and whose various compounds have also been implicated in other cellular processes. However, it was a surprise that that appropriate activation of a cell killing pathway can in fact prolong the lifespan of the organism. In the soma of adult C. elegans, all cells are post-mitotic, like many of our neurons and possibly some of our immune cells. These cells cannot simply be killed and replaced when showing signs of dysfunction. Thus, we speculate that it is the ability of the apoptotic pathway to pull together information about the functional and structural integrity of different cellular compartments that is the key property for why this pathway is used to decide when to boost defensive and repair processes in irreplaceable cells. When this process is artificially stimulated in mutants with elevated mtROS generation or with drug treatments it leads to lifespan prolongations beyond the normal lifespan of the organism.
Superoxide () is generated when an extra electron is donated to molecular oxygen, and hydrogen peroxide (H2O2) is generated in a variety of ways, including by the action of superoxide dismutases (SODs) on superoxide. Peroxide in turn can be broken down by catalase and other enzymatic processes. The chemical reactivity of reactive oxygen species (ROS) means that they can be toxic. For example, excessive ROS can inflict various oxidative damage upon macromolecules (nucleic acids, lipids, and proteins), thus compromising the integrity and viability of the cell. By reacting with lipids in the membrane and inducing lipid peroxidation, ROS can cause structural and functional changes of the plasma and intracellular organelle membranes. Oxidation of protein cysteine (Cys) residues can lead to the formation of sulfenic acid (-SOH), which can form disulfide bonds (S-S) with nearby Cys residues or undergo further oxidation to sulfinic acid (-SO2H) or sulfonic acid (-SO3H). These and other oxidative modifications to proteins may cause conformational changes, cross-linking, and peptide backbone breakage that ultimately alter protein functionality, including loss of biological activity (Friguet, 2006; Roos and Messens, 2011). In regards to oxidative damage to nucleic acids, ROS most frequently modify bases, inducing strand crosslinking and breakage (Jena, 2012). Guanine is the base most susceptible to oxidation, products of which can mispair with adenine during DNA synthesis yielding G:C to T:A transversions (Perillo et al., 2008). They may also stall DNA replication, resulting in single or double strand breaks (Cheng et al., 1992).
But ROS are also signaling molecules that impinge on classic signal transduction pathways, including via irreversible modification of macromolecules (Finkel, 2012; Holmstrom and Finkel, 2014). The best characterized examples of ROS signaling involve the oxidation of cysteine residues in redox-sensitive proteins. These oxidative modifications are reversible by the action of glutaredoxin (GRX) and thioredoxin (TRX), making them ideal for participation in signaling (Reczek and Chandel, 2015). PTEN, a tumor suppressor that regulates cell migration and growth, is a well-characterized example of a redox-sensitive protein. Analysis of cysteine mutants of PTEN revealed that H2O2 exposure causes Cys71 to form a disulfide bond with the essential Cys124 in the active site, thus inactivating it (Lee et al., 2002). ROS also affect the activation of mitogen-activated protein kinases, like p38 and ERK1/2. Treatment of rat adrenal medulla (PC12) cells with 500 μM peroxynitrite (a strong oxidant resulting from the interaction of superoxide and nitric oxide) increased the phosphorylation levels of both p38 and ERK1/2 within minutes (Jope et al., 2000). ROS also regulate apoptosis and necrosis (Shen and Liu, 2006) and stimulate autophagy (Filomeni et al., 2015). Even more unexpectedly, the modification of DNA via ROS is a requirement for the transcriptional activation of some genes. It has been shown that exposure of cells to estrogen causes the formation of 8-oxoguanine (8-oxoG) which is recognized by the DNA glycosylase OGG1. Excision of the 8-oxoG lesion by OGG1 creates single-strand breaks (SSBs) which then recruits topoisomerase IIβ to estrogen-responsive DNA elements in the promoter region of estrogen-responsive genes. This allows DNA to bend and thereby allows for the promoter to be brought in close proximity with the transcriptional initiation complex (Nathan and Cunningham-Bussel, 2013).
The potential toxicity of ROS, in particular that of ROS originating from mitochondria (mtROS), has led to the formulation of the oxidative stress theory of aging, which suggested that accumulation of oxidative damage to macromolecules is at the heart of the aging process (Ku et al., 1993). Recently, however, we and others have proposed alternative interpretations of some of the observations that led to the formulation of the theory (Blagosklonny, 2008; Lapointe and Hekimi, 2010). For example, we have proposed that ROS damage might not be causally involved in the aging process but that ROS levels are correlated with the aged phenotype because they modulate signal transduction pathways that are specifically involved in responding to the type of cellular stresses that are brought about by aging (Hekimi et al., 2011). In other words, ROS increase with age because they are involved in combating the consequences of aging, not because they cause aging. This hypothesis is supported by observations we review below in a variety of organisms, in particular in Caenorhabditis elegans.
If ROS cause aging then ROS generation and detoxification could not be uncoupled from effects on lifespan. But, for example, we and others have been able to show that knockdown and knockout of superoxide dismutases and consequent elevations in oxidative damage are not associated with decreased lifespan (Yang et al., 2007; Doonan et al., 2008; Yen et al., 2009; Van Raamsdonk and Hekimi, 2012), even when all five C. elegans SODs are lost simultaneously and the organism has become dramatically hypersensitive to experimental elevation of superoxide (Van Raamsdonk and Hekimi, 2012). In fact, loss of SOD-2 is even associated with increased lifespan (Van Raamsdonk and Hekimi, 2009; see below for further discussion). This uncoupling also works the other way round: elevating superoxide dismutase activity with SOD mimetics does not prolong lifespan (Keaney et al., 2004). In fact, the review of much of the literature about ROS and aging in C. elegans does not reveal any tight association between the two (Van Raamsdonk and Hekimi, 2010). Moreover, lifespan extension in some SOD overexpression models is likely unrelated to antioxidant activity (Cabreiro et al., 2011).
In seeking to investigate the association between ROS and aging, or the lack thereof, we found that, although aging was not tightly associated with ROS levels, lifespan could be lengthened rather than shortened by conditions and treatments that were generally believed to be deleterious to mitochondria. For example, over 20 years ago, the C. elegans clk-1 mutants were among the very first long-lived mutants to be described (Wong et al., 1995). clk-1 was subsequently shown to encode a hydroxylase necessary for ubiquinone (UQ; coenzyme Q, CoQ) biosynthesis (Ewbank et al., 1997) and, as a consequence for normal mitochondrial function (Felkai et al., 1999). Most strikingly in regard to aging, the mitochondria of clk-1 mutants lose the ability to accumulate an inner membrane potential-dependent dye at a younger age than the wild type despite their lengthened lifespan (Felkai et al., 1999).
In addition, purified clk-1 mitochondria show elevated superoxide levels and oxidative stress (Yang and Hekimi, 2010a). A subsequent genetic screen for clk-1-like mutants uncovered several additional long-lived mutants that, like clk-1, displayed a slow phenotype, maternal rescue, low energy metabolism and long life, but not decrease in overall oxidative damage (de Jong et al., 2004; Van Raamsdonk et al., 2010). Interestingly, the screen to identify these mutants was based on their slow phenotype and not their long life. Yet, all mutants identified in this way showed increased lifespan, suggesting that a hypo-metabolic phenotype is sufficient for increased lifespan.
The best studied genes among the clk-1-like genes all encode proteins whose activity is broad, affecting several, more or less independent processes: CLK-1 is necessary for UQ synthesis which is present as an electron transporter in most cellular membranes, carrying out diverse and independent functions in different locations (Wang and Hekimi, 2016); CLK-2 affects telomeres in C. elegans (Benard et al., 2001) and is a regulator of the stability of PI3K-related protein kinases, including the mammalian target of rapamycin (mTOR; Takai et al., 2007); TPK-1 is necessary for thiamine metabolism and therefore affects the activity of all thiamine-dependent enzymes (de Jong et al., 2004).
To identify mutations whose effects on energy metabolism and ROS were more specific, we screened for further clk-1-like mutants but without requiring the presence of a maternal effect (phenotypic rescue of a homozygous mutant animal by a wild type allele in the mother). This yielded two point mutations in subunits of the electron transport chain: isp-1(qm150) which alters one amino acid in the vicinity of the iron-sulfur center in the Rieske Iron Sulfur Protein of mitochondrial complex III (Feng et al., 2001), and nuo-6(qm200) which affects NDUFB4, a little studied subunit of complex I (Yang and Hekimi, 2010b) that is a preferential target of nitration by peroxynitrite in bovine heart (Murray et al., 2003). Both mutants are very long-lived and their effects on lifespan are not additive: that is, the double mutants are long-lived but live no longer than the single mutants (Yang and Hekimi, 2010b). The study of these mutants yielded the most unequivocal evidence connecting increased generation of mitochondrial ROS (mtROS) to increased lifespan. Indeed, purified mitochondria from both mutants show elevated superoxide generation (Yang and Hekimi, 2010a), their increased lifespan is suppressed by antioxidant treatment (N-acetyl cysteine, or vitamin C; Yang and Hekimi, 2010a), and their effect on lifespan can be phenocopied by treatment with very low level (0.1 mM) of the pro-oxidant paraquat (PQ; Yang and Hekimi, 2010a; Hwang et al., 2014).
Compounds such as PQ (1,1′-dimethyl-4,4′-bipyridinium dichloride) can accept electrons from an electron carrier in the ETC and transfer them to molecular oxygen to produce superoxide () and are used to increase cellular, and in particular mitochondrial, oxidative stress (Bus et al., 1976; Cocheme and Murphy, 2008; Robb et al., 2015). In fact, mitochondria are likely the major site of superoxide generation by the action of PQ (Cocheme and Murphy, 2008). More specifically, it is believed that PQ2+ is reduced by electrons from mitochondrial complex I to form a radical cation (PQ•+), which reacts with O2 to produce superoxide.
The study of the effect of PQ was particularly instructive as it was found that it was not additive to the lifespan effect of the isp-1 and nuo-6 mutations, although it was additive to the effect of other previously identified long-lived mutants [e.g., eat-2 (which lives long because of calorie restriction) and daf-2 (which has reduced insulin-like signaling activity); Yang and Hekimi, 2010a]. In addition, the effect of PQ was not always, or very incompletely, suppressed by a variety of loss-of-function mutations in genes believed to function downstream of previously defined longevity pathways (e.g., daf-2, aak-2, jnk-1, wwp-1, skn-1, hsf-1, hif-1; Yang and Hekimi, 2010a).
We determined the dose-response relationship between the concentration of PQ and lifespan of the wild type and found to be in the form of an inverted U-shape, with an optimum concentration of 0.1 mM under the conditions used (Van Raamsdonk and Hekimi, 2012; Figure 1). This suggests that increased mitochondrial ROS generation increases lifespan (by signaling mechanisms discussed below), but that the effect becomes gradually weakened at higher concentrations by lifespan-shortening effects due to the toxicity of abnormally high ROS generation. Alternatively, it is possible that the cell type (unknown) or the sub-cellular site (likely mitochondria) where PQ acts preferentially has its optimum effect at a particular concentration of ROS, without implying that slightly higher concentrations necessarily produce unmanageable amounts of additional oxidative damage.
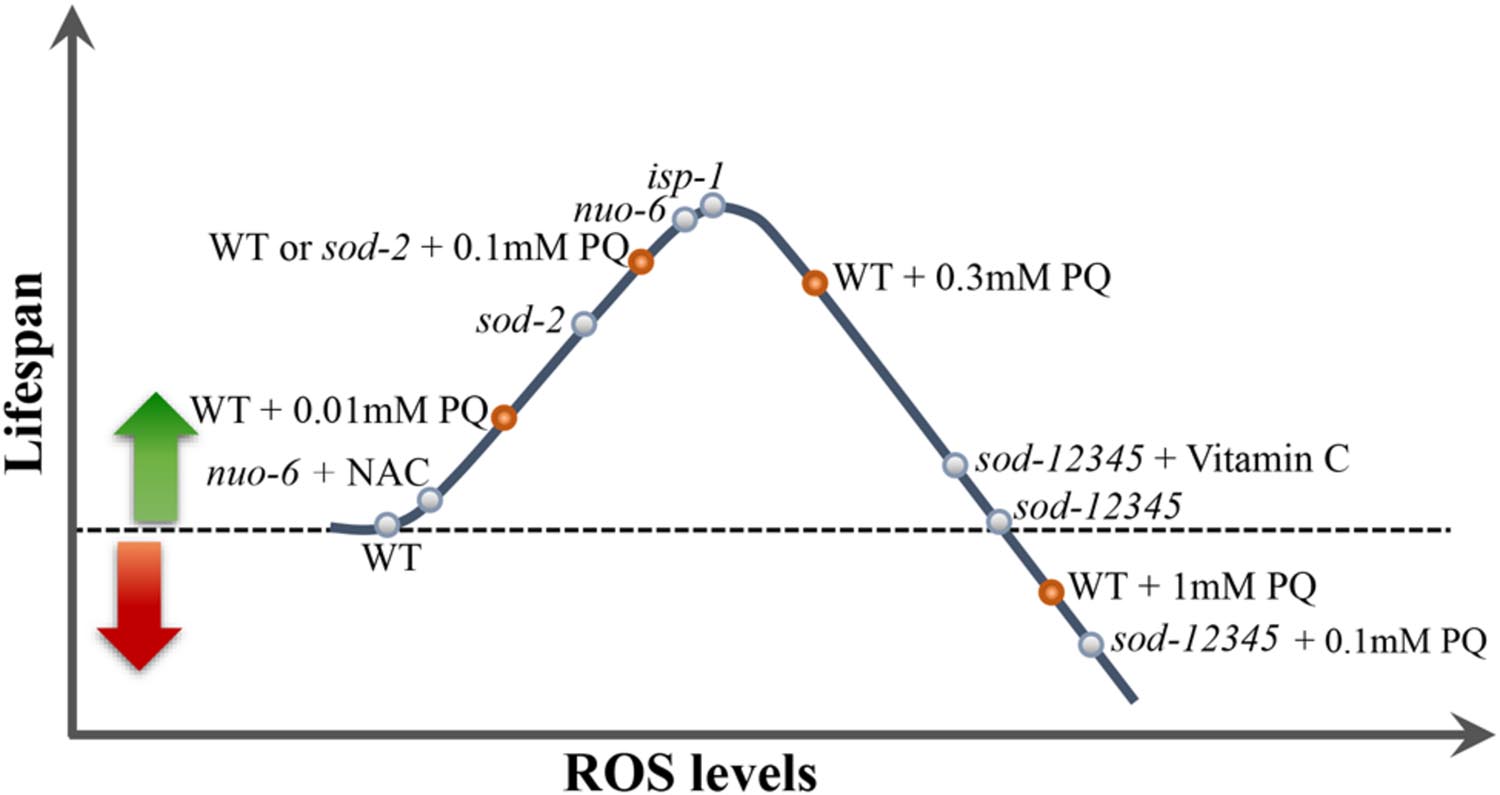
FIGURE 1. The inverted U-shaped relationship between ROS and lifespan. Increasing ROS levels in animals in which ROS levels are lower than the optimum for longevity results in an increase of lifespan. Beyond that optimal level, further increases have less effect and finally decrease animal lifespan when ROS become too elevated. Note that such a relationship implies that both pro-oxidants and antioxidants can increase or decrease lifespan depending on the genotype of the animals, but possibly depending also on the exact cellular or sub-cellular target of any particular redox-active compound. See Figure 2 for schematic representation of the lifespan data of some of the mutants plotted on the curve.
The idea of an inverted U-shaped relationship between ROS and lifespan suggested by the effects of PQ allowed to conceptualize previous results. For example, it suggests that the level of mitochondrial ROS generation in isp-1 and nuo-6 mutants is close to that produced by the optimum concentration of PQ (0.1 mM), explaining why 0.1 mM is not additive to the effect of the mutations, and why treatment with antioxidants shorten lifespan (Figures 2A,B). In addition, we tested PQ on a mutant strain (abbreviated sod-12345) which lacks all five C. elegans sod genes (Van Raamsdonk and Hekimi, 2012). The lifespan of these animals is wild type (Figure 2C), but, not surprisingly, they are hypersensitive to PQ and no concentration of PQ is capable of increasing their lifespan. Rather, even the lowest concentrations are lifespan-shortening. This could be due to the toxicity of excessive superoxide in the absence of detoxification. Alternatively, it is generally believed that peroxide (which results from the action of SODs on superoxide) is the main intracellular ROS messenger, due to its greater stability and capacity to cross membranes (Holmstrom and Finkel, 2014). Thus the absence of adequate peroxide generation because of the absence of SODs could shorten lifespan by preventing normal, beneficial, ROS signaling. This possibility is reinforced by the finding that the lifespan increase produced by the optimum concentration of PQ (0.1 mM) is abolished by loss of SOD-3 (Figure 2D), the minor mitochondrial SOD, but not by loss of SOD-2 (Yee et al., 2014). Note that the two hypotheses (toxicity of excessive superoxide and insufficient peroxide signaling) are not mutually exclusive to explain the lack of beneficial effect of PQ on sod-12345 mutants.
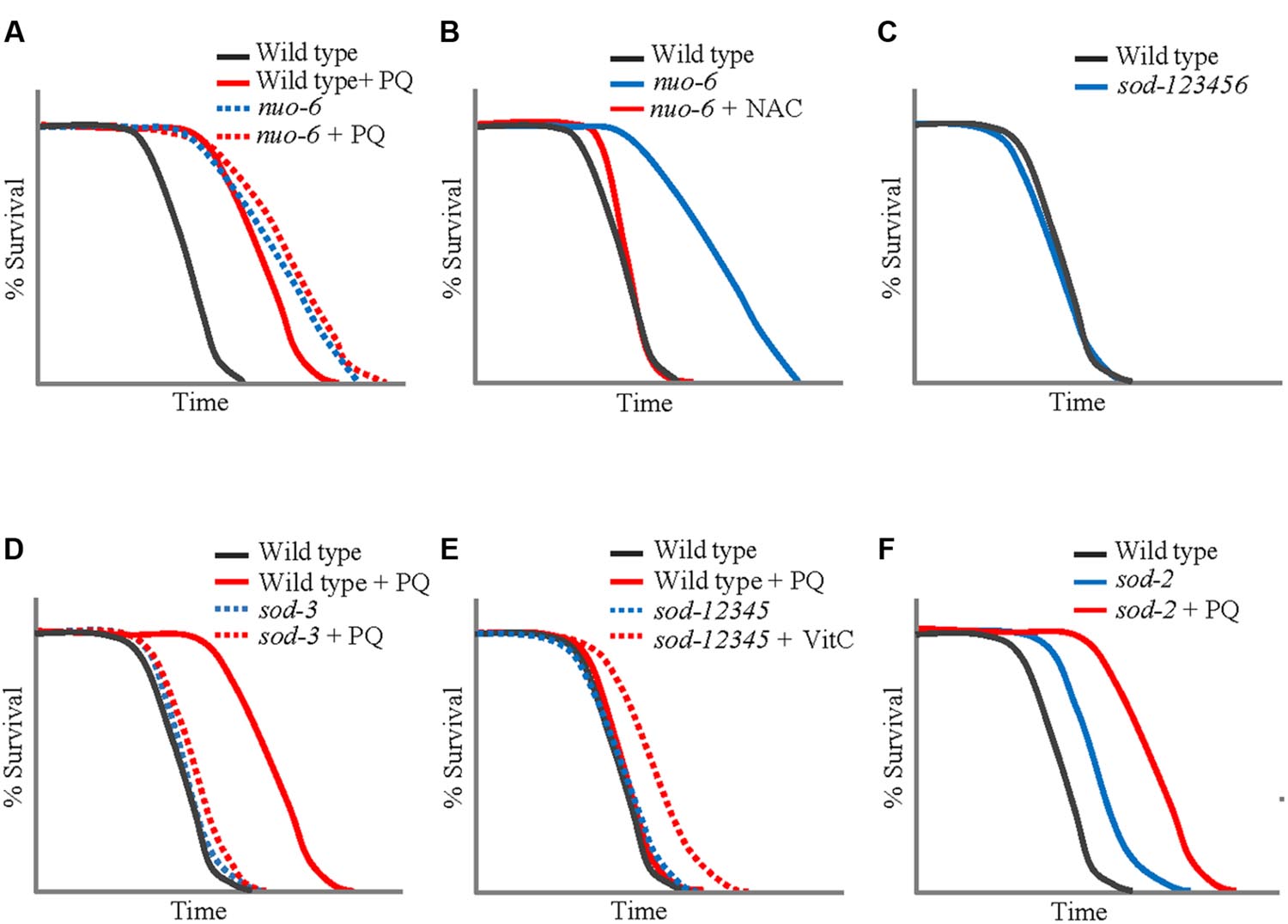
FIGURE 2. Lifespans of wild type Caenorhabditis elegans and mutants treated with PQ (0.1 mM) or antioxidants. (A) The PQ treatment has no effect on the lifespan of nuo-6(qm200) mutant (Yang and Hekimi, 2010a). (B) Antioxidant N-acetylcysteine (NAC) fully eliminates the extended longevity of nuo-6(qm200) (Yang and Hekimi, 2010a). (C) sod-12345 worms that lack all five sod genes live a normal lifespan (Van Raamsdonk and Hekimi, 2012). (D) The PQ treatment lengthens the lifespan of the wild type but is without effect on the longevity of sod-3(tm760) knockout mutant (Yee et al., 2014). (E) The lifespan of sod-12345 worms can be increased by the treatment with vitamin C (VitC; Van Raamsdonk and Hekimi, 2012). (F) Treatment with PQ further increases the lifespan of sod-2 mutant to that of the wide-type under the same treatment condition (Yang and Hekimi, 2010a). All figures are redrawn from the published work cited.
The wild type lifespan of the sod-12345 mutant strain allowed for a further test of the inverted U-shaped relationship between ROS and lifespan. As mentioned, these mutants have a normal lifespan that cannot by lengthened by PQ treatment. This suggest that the mutants’ ROS levels produce an exact balance between beneficial effects and toxicity. In other words, the mutants live on the far side of the ∩ (Figure 1). We treated these mutants and the wild type with the antioxidant vitamin C (VitC) as well as with Mn++, the co-factor of MnSODs, which has superoxide dismutase activity by itself (Archibald and Fridovich, 1981). Both treatment lengthened the lifespan of the mutant strain but were without effect on the wild type (Van Raamsdonk and Hekimi, 2012). This created a striking situation in which a strain devoid of superoxide detoxification lives longer than the wild type under the same conditions (Figure 2E).
The inverted U-shaped relationship of PQ and lifespan has been used to identify and characterize other activities that might modulate the lifespan effects of ROS. In particular, the activities of AMP kinase and HIF-1 have been found to be able to alter the placement of the ∩ along both the lifespan and dosage axes (Hwang et al., 2014). This suggests that there might be complex feedback mechanisms which respond to change of intracellular ROS levels and, via an additional level of ROS modulation, regulate downstream effector pathways and ultimately survival. PQ treatment also allows for testing of the involvement of beneficial ROS signaling in the long lifespan of clk-1 and sod-2 mutants (Figure 2F). Indeed, isp-1;clk-1 and isp-1;sod-2 double mutants are lethal or very sick, respectively (Van Raamsdonk and Hekimi, 2009), which makes them poor tools. However, we found that the action of PQ on lifespan was additive to the effect of sod-2 and clk-1 mutations (in contrast to the lack of additivity on isp-1 and nuo-6). How clk-1 affects lifespan in C. elegans is not well-understood and thus might not involve beneficial ROS signaling, even if an alteration in ROS metabolism is part of the phenotype of these mutants. However, it is difficult to envision that the action of sod-2 mutations would not be via an increase in mitochondrial superoxide generation. This suggests that there might be more than one mechanisms of lifespan extension that involves mitochondrial ROS generation (see below the discussion of the apoptotic pathway signaling for additional evidence for this). Furthermore, by the logic of additivity, there might be three distinct mechanisms as clk-1 and sod-2 are in fact additive (Van Raamsdonk and Hekimi, 2009). Although, as for sod-2 mutants, there is no direct proof that the altered ROS metabolism of clk-1 mutants is responsible for their longevity.
An number of additional studies have also been exploring the role of ROS in mitochondrial longevity, and a number of additional regulatory molecules, in particular transcription factors, have been implicated in parts of the response of C. elegans to increased ROS generation, including following PQ treatment (An and Blackwell, 2003; Curtis et al., 2006; Lee et al., 2010; Paek et al., 2012; Baruah et al., 2014; Hwang et al., 2014). These include CEP-1(p53), SKN-1(Nrf-1/2), and DAF-16(FOXO). They all appear to function downstream of the mtROS signal and none is absolutely required for the longevity response (Yang and Hekimi, 2010a; Hwang et al., 2014). SKN-1 and DAF-16 in particular appear to be important for resistance to ROS damage (Honda and Honda, 1999; An and Blackwell, 2003).
A number of studies including several genome-wide screening experiments have demonstrated that RNAi knockdown of ETC subunits or components of the mitochondrial translation machinery is sufficient to extend lifespan (Dillin et al., 2002; Lee et al., 2003; Hamilton et al., 2005; Chen et al., 2007; Curran and Ruvkun, 2007; Rea et al., 2007; Copeland et al., 2009; Yang and Hekimi, 2010b; Durieux et al., 2011; Houtkooper et al., 2013). In C. elegans, the most robust effect was seen with RNAi clones against the ETC genes nuo-2, nuo-6, isp-1, cyc-1, cco-1, and apt-3. An intriguing finding involving RNAi experiments is that at least for some ETC genes, e.g., isp-1 and nuo-6, the mechanism behind the effect of RNAi knockdowns is distinct from the mechanism that allows the point mutants to live a markedly extended lifespan. First, for both nuo-6 and isp-1 genes, when the mutant strain was treated with the RNAi against the other gene, lifespan was further lengthened, resulting in dramatically long lifespans (Yang and Hekimi, 2010b). These additive interactions suggest that the allele and RNAi treatment involved affect survival via separate downstream mechanisms. Another possible explanation would be that different interventions at the two distinct sites (for example, isp-1 mutation and nuo-6 RNAi) act via the same mechanism and in combination they act additively simply because each one by itself has a submaximal effect. However, we consider this unlikely, since more severe impairment of ETC induced by either a higher dose of RNAi or double mutations failed to achieve more prolonged lifespans (Yang and Hekimi, 2010b). Moreover, consistent with different mechanisms of action, the phenotypes of isp-1(qm150) and nuo-6(qm200) mutants and those of RNAi knockdown were found to be distinct in almost every ways, regarding oxygen consumption and ATP levels, patterns of growth and fertility, behavioral rates, resistance to paraquat, autophagy, as well as expression levels of SODs and other gene expression markers (Yang and Hekimi, 2010b).
Thus, it appears that, although genetic mutations and RNAi knockdown of specific ETC subunits extend lifespan in worms, this is by totally different mechanisms. As will be brought up further in a section below, ETC gene mutations act via the effect of altered mtROS levels on apoptotic signaling, while the longevity of RNAi-treated worms is not sensitive to loss of apoptotic signaling (Yee et al., 2014). Finally, in this regard, it has been proposed that induction of mitochondrial unfolded protein response (UPRmt) promotes longevity and is directly responsible for the lifespan extension from ETC inhibition (Yang and Hekimi, 2010b; Durieux et al., 2011). However, the results have since become controversial (Pujol et al., 2013; Rauthan et al., 2013; Bennett et al., 2014). More recently, it was discovered that knockdown of the complex IV subunit 1 (cco-1) is associated with chromatin restructuring that is required for the full activation of UPRmt and is partially accountable for the lifespan-extending effect (Tian et al., 2016). Whether, other types of ETC alternations induce such changes is yet to be seen.
Mitochondria have been long considered to be important for the life-extending effect of dietary restriction (DR). It was initially proposed that DR induces a hypo-metabolic state and in turn lowers the rate of mtROS production and decreases ROS damage which is what extends lifespan (Sohal and Weindruch, 1996; Sohal et al., 2009). However, how DR affects metabolic rate and mitochondrial physiology is still highly controversial with many studies showing mixed findings (Sohal et al., 1994; Gredilla et al., 2001; Lambert et al., 2004). Specifically, several more recent studies have reported that at least some DR regimens do not suppress the rate of mitochondrial respiration, but instead increases it (Lin et al., 2002; Nisoli et al., 2005; Bishop and Guarente, 2007; Zid et al., 2009). In a yeast model of growth in low glucose a metabolic shift toward increased respiration was observed, and inhibition of ETC function severely attenuated lifespan increase (Lin et al., 2002). Similar findings were also demonstrated in the fruit fly (Zid et al., 2009; Bahadorani et al., 2010). Furthermore, in some rodent DR models, an induction in mitochondrial biogenesis was described (Nisoli et al., 2005; Lopez-Lluch et al., 2006; Lanza et al., 2012). In line with the growing recognition of ROS as signaling molecules, it has been proposed that DR promotes an increase in intracellular ROS that activates a program of responses to stress, thereby leading to prolonged lifespan. Additional evidence came from the findings that, under a particular DR regimen (imposed by glycolysis inhibitor 2-deoxy-D-glucose), worms show overall enhanced respiration rate and increased overall ROS formation (measured indirectly by DCF fluorescence) and that upon treatment with antioxidants the lifespan increase in the presence of 2-deoxy-D-glucose was diminished (Schulz et al., 2007). However, it is of note that no elevation of mtROS was detected in eat-2 mutants, a widely acknowledged genetic mimic of DR. This, added to the observation that PQ can further extend the lifespan of eat-2 mutants (Yang and Hekimi, 2010a), suggests that at least some DR models do not involve elevated ROS.
Reduced insulin/IGF-1-like signaling (IIS) also prolong lifespan in multiple different species (Kenyon, 2005). In C. elegans, the IIS pathway is activated through DAF-2, the solo insulin/IGF-1 receptor. It ultimately down-regulates DAF-16, a FOXO transcription factor, which regulates a wide variety of genes associated with growth, stress resistance, metabolism and survival (Ogg et al., 1997). Zarse et al. (2012) reported that daf-2 mutants have increased mitochondrial metabolism compared to the wild type but reduced overall ROS levels. On the other hand, the same study showed an early transient increase of ROS levels following daf-2 RNAi in adult worms (Zarse et al., 2012). The importance of this transient ROS increase was further underlined by the observation that inhibition of this ROS signal eliminated a part of the lifespan extension afforded by daf-2 RNAi (Zarse et al., 2012). Furthermore, it is of note that elevated superoxide production (but not overall ROS levels) was detected in daf-2 mutants (Yang and Hekimi, 2010a) and exogenous antioxidants have a shortening effect on their lifespan (Yang and Hekimi, 2010a; Zarse et al., 2012), also indicating that some of the mutant longevity phenotype is mediated by a ROS-dependent mechanism.
Lastly, such observations are not limited to C. elegans. There is growing data suggesting mtROS signaling can favor longevity in other organisms (Table 1). Decreased mTOR signaling has been demonstrated to extend lifespan in multiple organisms, the mechanisms of which appear to be exceedingly complex and are not fully understood (Johnson et al., 2013). Among them, a role of mtROS downstream of mTOR signaling was implied by the findings that reduced mTOR signaling in the yeast tor1Δ mutants enhanced mitochondrial respiration and mtROS generation during growth and that diminishing mtROS levels in the mutants significantly eliminated extended chronological lifespan (CLS; Bonawitz et al., 2007; Pan et al., 2011). Furthermore, deletion of sod2 or treating a wild type strain during growth with the superoxide generator menadione was shown to be sufficient to lengthen CLS, furthering support for the conclusion of a role for mtROS as a key mediator of yeast CLS extension (Pan et al., 2011; Schroeder et al., 2013). The group further searched for factors that are required for the observed CLS extension resulting from menadione-generated ROS. Intriguingly, they identified activation of the Tel1p and Rad53p DNA-damage response kinases and the downstream Rph1p-regulated subtelomeric silencing as a key outcome of mtROS signaling that ultimately prolongs the survival in stationary phase (i.e., extension of CLS; Schroeder et al., 2013).
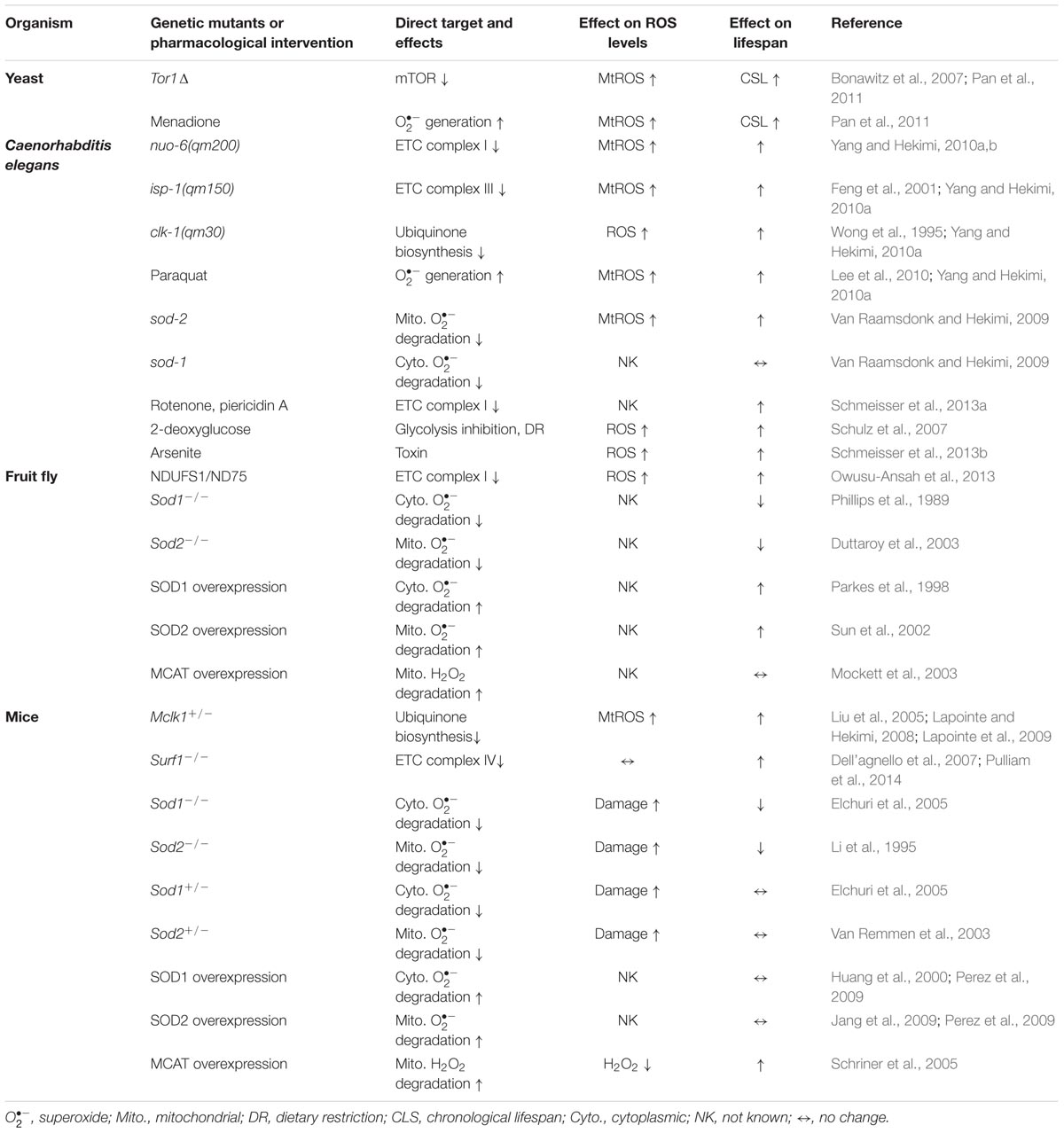
TABLE 1. Lifespan effect of mutations and treatments that affect ROS metabolism.
In metazoans such as fruit flies and mice, mutations and strong knockdown of ETC subunits or other essential mitochondria components are embryonic lethal or severely limit lifespan (Larsson et al., 1998; Trifunovic et al., 2004; Kujoth et al., 2005; Liu et al., 2007; Vempati et al., 2008; Hughes and Hekimi, 2011; Burman et al., 2014). However, there are a few studies that do suggest a possible benefit of high mtROS. In fruit flies, mild knockdown of muscle NDUFS1/ND75, a component of ETC complex I, increases ROS levels and prolongs lifespan (Owusu-Ansah et al., 2013). And, the effect on longevity can be abrogated by overexpression of an antioxidant enzyme (catalase or glutathione peroxidase 1), indicating a ROS-dependent mechanism (Owusu-Ansah et al., 2013). Heterozygous deletion of the gene Mclk1 (mouse clk-1) or homozygous knockout of the cytochrome c assembly factor Surf1 extends lifespan in mice (Liu et al., 2005; Dell’agnello et al., 2007; Lapointe et al., 2009). Mclk1+/- heterozygous mice have a mild UQ deficiency in the mitochondrial inner membrane associated with increased mitochondrial oxidative stress (Lapointe and Hekimi, 2008). They also exhibit a higher HIF-1α level, elevated expression of inflammatory cytokines, and an enhanced immune response (Wang et al., 2010, 2012). HIF-1α is an important regulator of immune and inflammatory responses and is known to be stabilized by mtROS (Chandel et al., 2000). Conceivably, high mtROS in the mutant may positively modulate some cellular responses, such as activation of HIF-1α, which would benefit survival and account for at least a part of the longevity phenotype. On the other hand, no change of mitochondrial ROS production was detected in isolated mitochondria from Surf1-/- mice (Pulliam et al., 2014). Some of the proteins involved in UPRmt were found to be elevated in muscles, which was postulated to be a possible mechanism for the observed pro-longevity effect (Pulliam et al., 2014).
To explain beneficial effects of a small increase in ROS levels, it has been suggested that it could result from an increase in ROS defenses in response to ROS-dependent damage, a mechanism sometimes called mitohormesis (Ristow, 2014; Yun and Finkel, 2014). To test this, we have examined patterns of gene expression linked to mtROS-dependent longevity. The effect on lifespan of the isp-1 and nuo-6 are not additive (Yang and Hekimi, 2010b), nor is the effect of PQ on either mutant (Yang and Hekimi, 2010a). We reasoned that changes in gene expression that are common to both mutants and PQ treatment of the wild type should reveal genes that participate in implementing ROS-dependent lifespan extension. Using Affimetrix gene arrays we observed a great amount of overlap between the effect of each of the three conditions (isp-1, nuo-6, and PQ treatment) compared to the untreated wild type (Yee et al., 2014). However, no pattern pointing to a particular mechanism of longevity was obvious in the list of altered genes. There was also no indication of an upregulation of ROS detoxification or repair of oxidative damage, whether considering the genes whose expression is altered in all three conditions, or even when considering all genes changed in either of the three conditions. Please note that the very idea that hormesis is the basis of ROS-dependent longevity is somewhat paradoxical (or circular). Indeed, it implies that ROS cause aging, as this is a necessary premise if one wants to argue that increased protection from ROS (in response to ROS) increases lifespan. Rather we believe our observations indicate that constitutively increased mtROS generation leads to increased longevity because general mechanisms of stress resistance and damage repair are triggered and amplified in the absence of actual damage or stress.
Above we have presented ample evidence that suggests that mtROS signaling can increase lifespan, possibly by more than one pathway. One signaling pathway that is known to include mtROS sensors is the mitochondrial intrinsic apoptosis signaling pathway (Circu and Aw, 2010). The pathway is called ‘intrinsic’ in contradistinction to the ‘extrinsic’ apoptotic pathway which involves interactions with other cells and receptors at the plasma membrane. Apoptosis per se is a highly controlled process that in vertebrates is sensitive to mitochondrial function, including mtROS, via the intrinsic pathway (Wang and Youle, 2009). In C. elegans a number of programmed apoptotic cell deaths occur during development. Defects in apoptosis are scored by counting apoptotic nuclei in embryos or by counting supernumerary neurons in the pharynx (the feeding organ, which has only few neurons). The intrinsic apoptotic machinery of C. elegans consists of the BH3-only protein EGL-1 (BH3 stands for Bcl2 homology domain 3), CED-9 (Bcl2-homolog), CED-4 (Apaf1-homolog), and CED-3 (Casp9-homolog). CED-9 is tethered to the outer mitochondrial membrane and is believed to bind CED-4. Upon interaction with EGL-1, CED-9 undergoes structural rearrangements leading to the release of CED-4, which re-localizes to perinuclear membranes where it assembles into active apoptosomes that activate the caspase CED-3. However, in contrast to vertebrates, there is no evidence for any role for mtROS or cytochrome c release in regulating apoptosis in C. elegans.
Interestingly, the individual proteins of the apoptotic signaling machinery appear to be able to carry out apoptosis-independent functions. For example, EGL-1 and CED-9 affect mitochondrial dynamics (Lu et al., 2011), CED-4 and CED-3 promote neuronal regeneration (Pinan-Lucarre et al., 2012), CED-4 appears to be involved in hypoxic pre-conditioning (Dasgupta et al., 2007) and S-phase checkpoint regulation (Zermati et al., 2007). These and similar findings in other organisms (Galluzzi et al., 2008; Dick and Megeney, 2013) suggest that the individual proteins of the intrinsic apoptotic pathway have bona fide signal transduction activities in other processes.
In fact, we recently provided evidence that mtROS act by signaling through the apoptotic pathway (Yee et al., 2014). Our study showed that mutations in the core apoptosis signaling pathway, which each abolishes apoptosis per se, also suppress the longevity of isp-1 and nuo-6 mutants, and that induced by PQ. However, these suppressions are entirely independent of apoptosis (meaning actual cell death) as they are independent of the BH3-only protein EGL-1, which is required for all apoptosis in C. elegans (Nehme and Conradt, 2008). In contrast, the mutants’ lifespan is suppressed by loss of CED-13, the only other C. elegans BH3-only protein and which is not required for apoptosis (Schumacher et al., 2005). In vertebrates, BH3-only proteins have been shown to be sensors of the cellular dysfunctions that trigger apoptotic programs (Galluzzi et al., 2014). We hypothesize that CED-13 similarly senses dysfunctional cellular states to trigger an appropriate stress response. Interestingly, treatment with PQ can bypass the need for CED-13, suggesting that mitochondrial ROS activates the pathway directly, possibly by acting on CED-9, which is physically associated with mitochondria. The epistatic relationships hold very clearly. For example, although PQ does not affect the lifespan of isp-1 mutants, the shortened lifespan of isp-1;ced-13 double mutants is lengthened back to the longevity of isp-1 by treatment with PQ (Yee et al., 2014).
The longevity suppression by loss of the core apoptotic machinery provides an additional tool to characterize the specificity of the mechanisms of ROS signaling under study. First, loss of ced-4 did not suppress the longevity of clk-1 and sod-2, confirming that there seems to be more than one mtROS-dependent pathway (see above). Furthermore, loss of ced-4 also does not suppress the longevity of mutants in daf-2, which encodes an insulin receptor-like protein, and in the longevity of which mtROS have also been implicated (Zarse et al., 2012). Finally, loss of ced-4 also does not suppress mutants that characterize other pathways, including eat-2 for calorie restriction (Lakowski and Hekimi, 1998), and glp-1 for germline signaling (Hsin and Kenyon, 1999; Berman and Kenyon, 2006).
Interestingly, in addition to increased longevity, the mitochondrial mutants also display a slowing of processes that consume energy, such as development and behavior. Loss of apoptotic signaling suppresses these phenotypes with the same epistatic patterns as for longevity, but it does not suppress the primary defects of low oxygen consumption and low ATP levels. This suggests that ATP-consuming processes are actively inhibited by mtROS signaling, possibly to protect from mitochondrial dysfunction and/or because ATP is diverted to protective processes. Unfortunately, the study of gene expression did not reveal what these processes are, which remains to be determined.
In a post-mitotic organism such as C. elegans damaged somatic cells cannot simply be removed as there is no mechanism to replace them. Instead, the involvement of the apoptotic pathway in longevity suggests that the ability of this pathway to sense cellular dysfunction is used to stimulate a program of stress resistance and repair rather than of cell death. When the pathway is triggered in the absence of damage, such as in the mitochondrial mutants or following low level PQ treatment, its activity results in increased lifespan. In vertebrates there are also cells that cannot simply be replaced when they are injured. Neurons present a dramatic example, as they have very limited regenerative capacities, if any, and neuronal death in both the brain and the periphery results in cognitive and motor impairments. Thus the organism must use other mechanisms than simple apoptotic elimination to ensure that the neurons that sustain molecular injury survive and carry out their functions. Such mechanisms might be more costly than removal of damaged cells followed by replacement from stem cells (or more dangerous, if potentially cancerous cells survive), which is why it is self-destruction and renewal that is commonly used for most cell types. We predict that in vertebrates, as in nematodes, the ability of the apoptotic pathway to sense and integrate cellular dysfunction (Galluzzi et al., 2014) is used to trigger mechanisms of stress-resistance and repair in cells that are difficult to regenerate or are irreplaceable (Figure 3). Hopefully, it will be possible to harness such a pathway to promote health and combat age-dependent diseases.
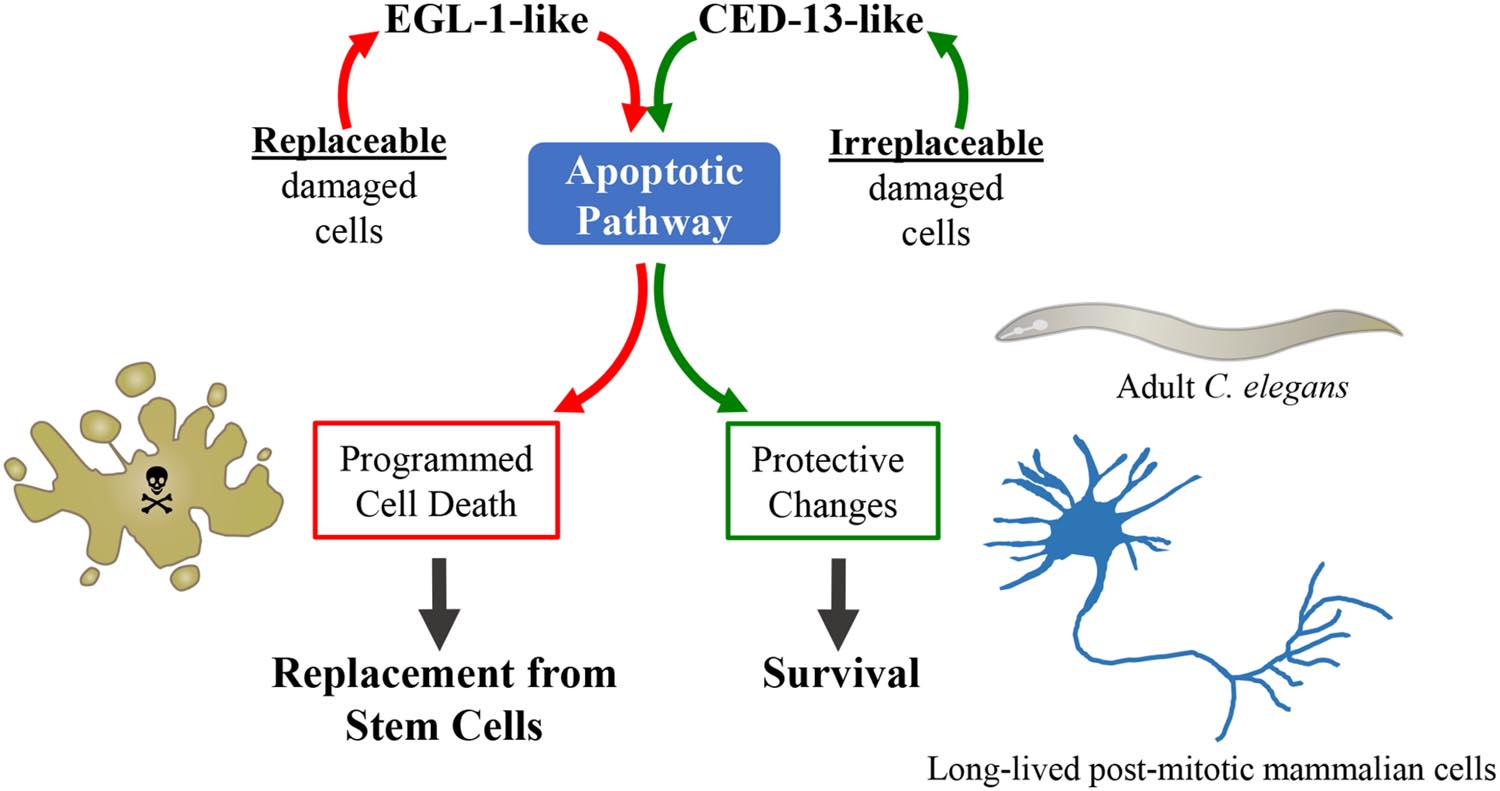
FIGURE 3. Non-apoptotic protective function of the intrinsic apoptotic pathway. In vertebrates, a key function of the apoptotic pathway is to eliminate unneeded, damaged or potentially dangerous cells. Removal of damaged cells by apoptosis followed by normal cell replacement from stem cells is essential for the maintenance of tissue homeostasis. The adult C. elegans is post-mitotic and has very few cells, with each cell having a unique function. Thus, the function of the apoptotic pathway in the soma of adult worm cannot be elimination of damaged cells, which, damaged or not, are irreplaceable. One interpretation of the pathway we have uncovered is therefore that C. elegans uses the apoptotic pathway under the control of CED-13 to judge the state of disrepair of a cell, not in order to decide to kill it or not, but to determine how strongly to turn on protective changes that help to preserve cellular and, consequently, organismal viability. It is exciting to speculate that there might be a CED-13-like pathway in vertebrate post-mitotic cells such as neurons, cardiomyocytes, or memory T-cells, where such a pathway could be harnessed to stimulate non-apoptotic protective processes.
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Research in Hekimi lab is funded by grants from the Canadian Institutes of Health Research: MOP-114891, MOP-123295 and MOP-97869, as well as by McGill University. SH is Strathcona Chair of Zoology and Campbell Chair of Developmental Biology.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
An, J. H., and Blackwell, T. K. (2003). SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 17, 1882–1893. doi: 10.1101/gad.1107803
Archibald, F. S., and Fridovich, I. (1981). Manganese and defenses against oxygen toxicity in Lactobacillus plantarum. J. Bacteriol. 145, 442–451.
Bahadorani, S., Hur, J. H., Lo, T. Jr., Vu, K., and Walker, D. W. (2010). Perturbation of mitochondrial complex V alters the response to dietary restriction in Drosophila. Aging Cell 9, 100–103. doi: 10.1111/j.1474-9726.2009.00537.x
Baruah, A., Chang, H., Hall, M., Yuan, J., Gordon, S., Johnson, E., et al. (2014). CEP-1, the Caenorhabditis elegans p53 homolog, mediates opposing longevity outcomes in mitochondrial electron transport chain mutants. PLoS Genet. 10:e1004097. doi: 10.1371/journal.pgen.1004097
Benard, C., McCright, B., Zhang, Y., Felkai, S., Lakowski, B., and Hekimi, S. (2001). The C. elegans maternal-effect gene clk-2 is essential for embryonic development, encodes a protein homologous to yeast Tel2p and affects telomere length. Development 128, 4045–4055.
Bennett, C. F., Vander Wende, H., Simko, M., Klum, S., Barfield, S., Choi, H., et al. (2014). Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 5, 3483. doi: 10.1038/ncomms4483
Berman, J. R., and Kenyon, C. (2006). Germ-cell loss extends C. elegans life span through regulation of DAF-16 by kri-1 and lipophilic-hormone signaling. Cell 124, 1055–1068. doi: 10.1016/j.cell.2006.01.039
Bishop, N. A., and Guarente, L. (2007). Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature 447, 545–549. doi: 10.1038/nature05904
Bonawitz, N. D., Chatenay-Lapointe, M., Pan, Y., and Shadel, G. S. (2007). Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 5, 265–277. doi: 10.1016/j.cmet.2007.02.009
Burman, J. L., Itsara, L. S., Kayser, E. B., Suthammarak, W., Wang, A. M., Kaeberlein, M., et al. (2014). A Drosophila model of mitochondrial disease caused by a complex I mutation that uncouples proton pumping from electron transfer. Dis. Model Mech. 7, 1165–1174. doi: 10.1242/dmm.015321
Bus, J. S., Aust, S. D., and Gibson, J. E. (1976). Paraquat toxicity: proposed mechanism of action involving lipid peroxidation. Environ. Health Perspect. 16, 139–146. doi: 10.1289/ehp.7616139
Cabreiro, F., Ackerman, D., Doonan, R., Araiz, C., Back, P., Papp, D., et al. (2011). Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic. Biol. Med. 51, 1575–1582. doi: 10.1016/j.freeradbiomed.2011.07.020
Chandel, N. S., McClintock, D. S., Feliciano, C. E., Wood, T. M., Melendez, J. A., Rodriguez, A. M., et al. (2000). Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 275, 25130–25138. doi: 10.1074/jbc.M001914200
Chen, D., Pan, K. Z., Palter, J. E., and Kapahi, P. (2007). Longevity determined by developmental arrest genes in Caenorhabditis elegans. Aging Cell 6, 525–533. doi: 10.1111/j.1474-9726.2007.00305.x
Cheng, K. C., Cahill, D. S., Kasai, H., Nishimura, S., and Loeb, L. A. (1992). 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions. J. Biol. Chem. 267, 166–172.
Circu, M. L., and Aw, T. Y. (2010). Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 48, 749–762. doi: 10.1016/j.freeradbiomed.2009.12.022
Cocheme, H. M., and Murphy, M. P. (2008). Complex I is the major site of mitochondrial superoxide production by paraquat. J. Biol. Chem. 283, 1786–1798. doi: 10.1074/jbc.M708597200
Copeland, J. M., Cho, J., Lo, T. Jr., Hur, J. H., Bahadorani, S., Arabyan, T., et al. (2009). Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598. doi: 10.1016/j.cub.2009.08.016
Curran, S. P., and Ruvkun, G. (2007). Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 3:e56. doi: 10.1371/journal.pgen.0030056
Curtis, R., O’Connor, G., and DiStefano, P. S. (2006). Aging networks in Caenorhabditis elegans: AMP-activated protein kinase (aak-2) links multiple aging and metabolism pathways. Aging Cell 5, 119–126. doi: 10.1111/j.1474-9726.2006.00205.x
Dasgupta, N., Patel, A. M., Scott, B. A., and Crowder, C. M. (2007). Hypoxic preconditioning requires the apoptosis protein CED-4 in C. elegans. Curr. Biol. 17, 1954–1959. doi: 10.1016/j.cub.2007.10.017
de Jong, L., Meng, Y., Dent, J., and Hekimi, S. (2004). Thiamine pyrophosphate biosynthesis and transport in the nematode Caenorhabditis elegans. Genetics 168, 845–854. doi: 10.1534/genetics.104.028605
Dell’agnello, C., Leo, S., Agostino, A., Szabadkai, G., Tiveron, C., Zulian, A., et al. (2007). Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 16, 431–444. doi: 10.1093/hmg/ddl477
Dick, S. A., and Megeney, L. A. (2013). Cell death proteins: an evolutionary role in cellular adaptation before the advent of apoptosis. Bioessays 35, 974–983. doi: 10.1002/bies.201300052
Dillin, A., Hsu, A. L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A. G., et al. (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401. doi: 10.1126/science.1077780
Doonan, R., McElwee, J. J., Matthijssens, F., Walker, G. A., Houthoofd, K., Back, P., et al. (2008). Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 22, 3236–3241. doi: 10.1101/gad.504808
Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. doi: 10.1016/j.cell.2010.12.016
Duttaroy, A., Paul, A., Kundu, M., and Belton, A. (2003). A Sod2 null mutation confers severely reduced adult life span in Drosophila. Genetics 165, 2295–2299.
Elchuri, S., Oberley, T. D., Qi, W., Eisenstein, R. S., Jackson Roberts, L., Van Remmen, H., et al. (2005). CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 24, 367–380. doi: 10.1038/sj.onc.1208207
Ewbank, J. J., Barnes, T. M., Lakowski, B., Lussier, M., Bussey, H., and Hekimi, S. (1997). Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science 275, 980–983. doi: 10.1126/science.275.5302.980
Felkai, S., Ewbank, J. J., Lemieux, J., Labbe, J. C., Brown, G. G., and Hekimi, S. (1999). CLK-1 controls respiration, behavior and aging in the nematode Caenorhabditis elegans. EMBO J. 18, 1783–1792. doi: 10.1093/emboj/18.7.1783
Feng, J., Bussiere, F., and Hekimi, S. (2001). Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 1, 633–644. doi: 10.1016/S1534-5807(01)00071-5
Filomeni, G., De Zio, D., and Cecconi, F. (2015). Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 22, 377–388. doi: 10.1038/cdd.2014.150
Finkel, T. (2012). Signal transduction by mitochondrial oxidants. J. Biol. Chem. 287, 4434–4440. doi: 10.1074/jbc.R111.271999
Friguet, B. (2006). Oxidized protein degradation and repair in ageing and oxidative stress. FEBS Lett. 580, 2910–2916. doi: 10.1016/j.febslet.2006.03.028
Galluzzi, L., Bravo-San Pedro, J. M., and Kroemer, G. (2014). Organelle-specific initiation of cell death. Nat. Cell Biol. 16, 728–736. doi: 10.1038/ncb3005
Galluzzi, L., Joza, N., Tasdemir, E., Maiuri, M. C., Hengartner, M., Abrams, J. M., et al. (2008). No death without life: vital functions of apoptotic effectors. Cell Death Differ. 15, 1113–1123. doi: 10.1038/cdd.2008.28
Gredilla, R., Sanz, A., Lopez-Torres, M., and Barja, G. (2001). Caloric restriction decreases mitochondrial free radical generation at complex I and lowers oxidative damage to mitochondrial DNA in the rat heart. FASEB J. 15, 1589–1591.
Hamilton, B., Dong, Y., Shindo, M., Liu, W., Odell, I., Ruvkun, G., et al. (2005). A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 19, 1544–1555. doi: 10.1101/gad.1308205
Hekimi, S., Lapointe, J., and Wen, Y. (2011). Taking a good look at free radicals in the aging process. Trends Cell Biol. 21, 569–576. doi: 10.1016/j.tcb.2011.06.008
Holmstrom, K. M., and Finkel, T. (2014). Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421. doi: 10.1038/nrm3801
Honda, Y., and Honda, S. (1999). The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 13, 1385–1393.
Houtkooper, R. H., Mouchiroud, L., Ryu, D., Moullan, N., Katsyuba, E., Knott, G., et al. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. doi: 10.1038/nature12188
Hsin, H., and Kenyon, C. (1999). Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399, 362–366. doi: 10.1038/20694
Huang, T. T., Carlson, E. J., Gillespie, A. M., Shi, Y., and Epstein, C. J. (2000). Ubiquitous overexpression of CuZn superoxide dismutase does not extend life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 55, B5–B9. doi: 10.1093/gerona/55.1.B5
Hughes, B. G., and Hekimi, S. (2011). A mild impairment of mitochondrial electron transport has sex-specific effects on lifespan and aging in mice. PLoS ONE 6:e26116. doi: 10.1371/journal.pone.0026116
Hwang, A. B., Ryu, E. A., Artan, M., Chang, H. W., Kabir, M. H., Nam, H. J., et al. (2014). Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 111, E4458–E4467. doi: 10.1073/pnas.1411199111
Jang, Y. C., Perez, V. I., Song, W., Lustgarten, M. S., Salmon, A. B., Mele, J., et al. (2009). Overexpression of Mn superoxide dismutase does not increase life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 64, 1114–1125. doi: 10.1093/gerona/glp100
Jena, N. R. (2012). DNA damage by reactive species: mechanisms, mutation and repair. J. Biosci. 37, 503–517. doi: 10.1007/s12038-012-9218-2
Johnson, S. C., Rabinovitch, P. S., and Kaeberlein, M. (2013). mTOR is a key modulator of ageing and age-related disease. Nature 493, 338–345. doi: 10.1038/nature11861
Jope, R. S., Zhang, L., and Song, L. (2000). Peroxynitrite modulates the activation of p38 and extracellular regulated kinases in PC12 Cells. Arch. Biochem. Biophys. 376, 365–370. doi: 10.1006/abbi.2000.1728
Keaney, M., Matthijssens, F., Sharpe, M., Vanfleteren, J., and Gems, D. (2004). Superoxide dismutase mimetics elevate superoxide dismutase activity in vivo but do not retard aging in the nematode Caenorhabditis elegans. Free Radic. Biol. Med. 37, 239–250. doi: 10.1016/j.freeradbiomed.2004.04.005
Kenyon, C. (2005). The plasticity of aging: insights from long-lived mutants. Cell 120, 449–460. doi: 10.1016/j.cell.2005.02.002
Ku, H.-H., Brunk, U. T., and Sohal, R. S. (1993). Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian species. Free Radic. Biol. Med. 15, 621–627. doi: 10.1016/0891-5849(93)90165-Q
Kujoth, G. C., Hiona, A., Pugh, T. D., Someya, S., Panzer, K., Wohlgemuth, S. E., et al. (2005). Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484. doi: 10.1126/science.1112125
Lakowski, B., and Hekimi, S. (1998). The genetics of caloric restriction in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 95, 13091–13096. doi: 10.1073/pnas.95.22.13091
Lambert, A. J., Wang, B., Yardley, J., Edwards, J., and Merry, B. J. (2004). The effect of aging and caloric restriction on mitochondrial protein density and oxygen consumption. Exp. Gerontol. 39, 289–295. doi: 10.1016/j.exger.2003.12.009
Lanza, I. R., Zabielski, P., Klaus, K. A., Morse, D. M., Heppelmann, C. J., Bergen, H. R., et al. (2012). Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metab. 16, 777–788. doi: 10.1016/j.cmet.2012.11.003
Lapointe, J., and Hekimi, S. (2008). Early mitochondrial dysfunction in long-lived Mclk1+/- mice. J. Biol. Chem. 283, 26217–26227. doi: 10.1074/jbc.M803287200
Lapointe, J., and Hekimi, S. (2010). When a theory of aging ages badly. Cell Mol. Life Sci. 67, 1–8. doi: 10.1007/s00018-009-0138-8
Lapointe, J., Stepanyan, Z., Bigras, E., and Hekimi, S. (2009). Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived Mclk1+/- mice. J. Biol. Chem. 284, 20364–20374. doi: 10.1074/jbc.M109.006569
Larsson, N. G., Wang, J., Wilhelmsson, H., Oldfors, A., Rustin, P., Lewandoski, M., et al. (1998). Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 18, 231–236. doi: 10.1038/ng0398-231
Lee, S. J., Hwang, A. B., and Kenyon, C. (2010). Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 20, 2131–2136. doi: 10.1016/j.cub.2010.10.057
Lee, S. R., Yang, K. S., Kwon, J., Lee, C., Jeong, W., and Rhee, S. G. (2002). Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 277, 20336–20342. doi: 10.1074/jbc.M111899200
Lee, S. S., Lee, R. Y., Fraser, A. G., Kamath, R. S., Ahringer, J., and Ruvkun, G. (2003). A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33, 40–48. doi: 10.1038/ng1056
Li, Y., Huang, T. T., Carlson, E. J., Melov, S., Ursell, P. C., Olson, J. L., et al. (1995). Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 11, 376–381. doi: 10.1038/ng1295-376
Lin, S. J., Kaeberlein, M., Andalis, A. A., Sturtz, L. A., Defossez, P. A., Culotta, V. C., et al. (2002). Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418, 344–348. doi: 10.1038/nature00829
Liu, W., Gnanasambandam, R., Benjamin, J., Kaur, G., Getman, P. B., Siegel, A. J., et al. (2007). Mutations in cytochrome c oxidase subunit VIa cause neurodegeneration and motor dysfunction in Drosophila. Genetics 176, 937–946. doi: 10.1534/genetics.107.071688
Liu, X., Jiang, N., Hughes, B., Bigras, E., Shoubridge, E., and Hekimi, S. (2005). Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 19, 2424–2434. doi: 10.1101/gad.1352905
Lopez-Lluch, G., Hunt, N., Jones, B., Zhu, M., Jamieson, H., Hilmer, S., et al. (2006). Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. U.S.A. 103, 1768–1773. doi: 10.1073/pnas.0510452103
Lu, Y., Rolland, S. G., and Conradt, B. (2011). A molecular switch that governs mitochondrial fusion and fission mediated by the BCL2-like protein CED-9 of Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 108, E813–E822. doi: 10.1073/pnas.1103218108
Mockett, R. J., Bayne, A. C., Kwong, L. K., Orr, W. C., and Sohal, R. S. (2003). Ectopic expression of catalase in Drosophila mitochondria increases stress resistance but not longevity. Free Radic. Biol. Med. 34, 207–217. doi: 10.1016/S0891-5849(02)01190-5
Murray, J., Taylor, S. W., Zhang, B., Ghosh, S. S., and Capaldi, R. A. (2003). Oxidative damage to mitochondrial complex I due to peroxynitrite: identification of reactive tyrosines by mass spectrometry. J. Biol. Chem. 278, 37223–37230. doi: 10.1074/jbc.M305694200
Nathan, C., and Cunningham-Bussel, A. (2013). Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 13, 349–361. doi: 10.1038/nri3423
Nehme, R., and Conradt, B. (2008). egl-1: a key activator of apoptotic cell death in C. elegans. Oncogene 27(Suppl. 1), S30–S40. doi: 10.1038/onc.2009.41
Nisoli, E., Tonello, C., Cardile, A., Cozzi, V., Bracale, R., Tedesco, L., et al. (2005). Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310, 314–317. doi: 10.1126/science.1117728
Ogg, S., Paradis, S., Gottlieb, S., Patterson, G. I., Lee, L., Tissenbaum, H. A., et al. (1997). The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389, 994–999. doi: 10.1038/40194
Owusu-Ansah, E., Song, W., and Perrimon, N. (2013). Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155, 699–712. doi: 10.1016/j.cell.2013.09.021
Paek, J., Lo, J. Y., Narasimhan, S. D., Nguyen, T. N., Glover-Cutter, K., Robida-Stubbs, S., et al. (2012). Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab. 16, 526–537. doi: 10.1016/j.cmet.2012.09.007
Pan, Y., Schroeder, E. A., Ocampo, A., Barrientos, A., and Shadel, G. S. (2011). Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 13, 668–678. doi: 10.1016/j.cmet.2011.03.018
Parkes, T. L., Elia, A. J., Dickinson, D., Hilliker, A. J., Phillips, J. P., and Boulianne, G. L. (1998). Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat. Genet. 19, 171–174. doi: 10.1038/534
Perez, V. I., Van Remmen, H., Bokov, A., Epstein, C. J., Vijg, J., and Richardson, A. (2009). The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 8, 73–75. doi: 10.1111/j.1474-9726.2008.00449.x
Perillo, B., Ombra, M. N., Bertoni, A., Cuozzo, C., Sacchetti, S., Sasso, A., et al. (2008). DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319, 202–206. doi: 10.1126/science.1147674
Phillips, J. P., Campbell, S. D., Michaud, D., Charbonneau, M., and Hilliker, A. J. (1989). Null mutation of copper/zinc superoxide dismutase in Drosophila confers hypersensitivity to paraquat and reduced longevity. Proc. Natl. Acad. Sci. U.S.A. 86, 2761–2765. doi: 10.1073/pnas.86.8.2761
Pinan-Lucarre, B., Gabel, C. V., Reina, C. P., Hulme, S. E., Shevkoplyas, S. S., Slone, R. D., et al. (2012). The core apoptotic executioner proteins CED-3 and CED-4 promote initiation of neuronal regeneration in Caenorhabditis elegans. PLoS Biol. 10:e1001331. doi: 10.1371/journal.pbio.1001331
Pujol, C., Bratic-Hench, I., Sumakovic, M., Hench, J., Mourier, A., Baumann, L., et al. (2013). Succinate dehydrogenase upregulation destabilize complex I and limits the lifespan of gas-1 mutant. PLoS ONE 8:e59493. doi: 10.1371/journal.pone.0059493
Pulliam, D. A., Deepa, S. S., Liu, Y., Hill, S., Lin, A. L., Bhattacharya, A., et al. (2014). Complex IV-deficient Surf1(-/-) mice initiate mitochondrial stress responses. Biochem. J. 462, 359–371. doi: 10.1042/BJ20140291
Rauthan, M., Ranji, P., Aguilera Pradenas, N., Pitot, C., and Pilon, M. (2013). The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. Proc. Natl. Acad. Sci. U.S.A. 110, 5981–5986. doi: 10.1073/pnas.1218778110
Rea, S. L., Ventura, N., and Johnson, T. E. (2007). Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 5:e259. doi: 10.1371/journal.pbio.0050259
Reczek, C. R., and Chandel, N. S. (2015). ROS-dependent signal transduction. Curr. Opin. Cell Biol. 33, 8–13. doi: 10.1016/j.ceb.2014.09.010
Ristow, M. (2014). Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat. Med. 20, 709–711. doi: 10.1038/nm.3624
Robb, E. L., Gawel, J. M., Aksentijevic, D., Cocheme, H. M., Stewart, T. S., Shchepinova, M. M., et al. (2015). Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat. Free Radic. Biol. Med. 89, 883–894. doi: 10.1016/j.freeradbiomed.2015.08.021
Roos, G., and Messens, J. (2011). Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic. Biol. Med. 51, 314–326. doi: 10.1016/j.freeradbiomed.2011.04.031
Schmeisser, S., Priebe, S., Groth, M., Monajembashi, S., Hemmerich, P., Guthke, R., et al. (2013a). Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol. Metab. 2, 92–102. doi: 10.1016/j.molmet.2013.02.002
Schmeisser, S., Schmeisser, K., Weimer, S., Groth, M., Priebe, S., Fazius, E., et al. (2013b). Mitochondrial hormesis links low-dose arsenite exposure to lifespan extension. Aging Cell 12, 508–517. doi: 10.1111/acel.12076
Schriner, S. E., Linford, N. J., Martin, G. M., Treuting, P., Ogburn, C. E., Emond, M., et al. (2005). Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308, 1909–1911. doi: 10.1126/science.1106653
Schroeder, E. A., Raimundo, N., and Shadel, G. S. (2013). Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 17, 954–964. doi: 10.1016/j.cmet.2013.04.003
Schulz, T. J., Zarse, K., Voigt, A., Urban, N., Birringer, M., and Ristow, M. (2007). Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 6, 280–293. doi: 10.1016/j.cmet.2007.08.011
Schumacher, B., Schertel, C., Wittenburg, N., Tuck, S., Mitani, S., Gartner, A., et al. (2005). C. elegans ced-13 can promote apoptosis and is induced in response to DNA damage. Cell Death Differ. 12, 153–161. doi: 10.1038/sj.cdd.4401605
Shen, H.-M., and Liu, Z.-G. (2006). JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic. Biol. Med. 40, 928–939. doi: 10.1016/j.freeradbiomed.2005.10.056
Sohal, R. S., Ferguson, M., Sohal, B. H., and Forster, M. J. (2009). Life span extension in mice by food restriction depends on an energy imbalance. J. Nutr. 139, 533–539. doi: 10.3945/jn.108.100313
Sohal, R. S., Ku, H. H., Agarwal, S., Forster, M. J., and Lal, H. (1994). Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech. Ageing Dev. 74, 121–133. doi: 10.1016/0047-6374(94)90104-X
Sohal, R. S., and Weindruch, R. (1996). Oxidative stress, caloric restriction, and aging. Science 273, 59–63. doi: 10.1126/science.273.5271.59
Sun, J., Folk, D., Bradley, T. J., and Tower, J. (2002). Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics 161, 661–672.
Takai, H., Wang, R. C., Takai, K. K., Yang, H., and de Lange, T. (2007). Tel2 regulates the stability of PI3K-related protein kinases. Cell 131, 1248–1259. doi: 10.1016/j.cell.2007.10.052
Tian, Y., Garcia, G., Bian, Q., Steffen, K. K., Joe, L., Wolff, S., et al. (2016). Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 165, 1197–1208. doi: 10.1016/j.cell.2016.04.011
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. doi: 10.1038/nature02517
Van Raamsdonk, J. M., and Hekimi, S. (2009). Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 5:e1000361. doi: 10.1371/journal.pgen.1000361
Van Raamsdonk, J. M., and Hekimi, S. (2010). Reactive oxygen species and aging in Caenorhabditis elegans: causal or casual relationship? Antioxid. Redox Signal. 13, 1911–1953. doi: 10.1089/ars.2010.3215
Van Raamsdonk, J. M., and Hekimi, S. (2012). Superoxide dismutase is dispensable for normal animal lifespan. Proc. Natl. Acad. Sci. U.S.A. 109, 5785–5790. doi: 10.1073/pnas.1116158109
Van Raamsdonk, J. M., Meng, Y., Camp, D., Yang, W., Jia, X., Benard, C., et al. (2010). Decreased energy metabolism extends life span in Caenorhabditis elegans without reducing oxidative damage. Genetics 185, 559–571. doi: 10.1534/genetics.110.115378
Van Remmen, H., Ikeno, Y., Hamilton, M., Pahlavani, M., Wolf, N., Thorpe, S. R., et al. (2003). Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genomics 16, 29–37. doi: 10.1152/physiolgenomics.00122.2003
Vempati, U. D., Torraco, A., and Moraes, C. T. (2008). Mouse models of oxidative phosphorylation dysfunction and disease. Methods 46, 241–247. doi: 10.1016/j.ymeth.2008.09.008
Wang, C., and Youle, R. J. (2009). The role of mitochondria in apoptosis*. Annu. Rev. Genet. 43, 95–118. doi: 10.1146/annurev-genet-102108-134850
Wang, D., Malo, D., and Hekimi, S. (2010). Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1α in long-lived Mclk1+/- mouse mutants. J. Immunol. 184, 582–590. doi: 10.4049/jimmunol.0902352
Wang, D., Wang, Y., Argyriou, C., Carrière, A., Malo, D., and Hekimi, S. (2012). An enhanced immune response of Mclk1+/- mutant mice is associated with partial protection from fibrosis, cancer and the development of biomarkers of aging. PLoS ONE 7:e49606. doi: 10.1371/journal.pone.0049606
Wang, Y., and Hekimi, S. (2016). Understanding Ubiquinone. Trends Cell Biol. 26, 367–378. doi: 10.1016/j.tcb.2015.12.007
Wong, A., Boutis, P., and Hekimi, S. (1995). Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 139, 1247–1259.
Yang, W., and Hekimi, S. (2010a). A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 8:e1000556. doi: 10.1371/journal.pbio.1000556
Yang, W., and Hekimi, S. (2010b). Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 9, 433–447. doi: 10.1111/j.1474-9726.2010.00571.x
Yang, W., Li, J., and Hekimi, S. (2007). A measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics 177, 2063–2074. doi: 10.1534/genetics.107.080788
Yee, C., Yang, W., and Hekimi, S. (2014). The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell 157, 897–909. doi: 10.1016/j.cell.2014.02.055
Yen, K., Patel, H. B., Lublin, A. L., and Mobbs, C. V. (2009). SOD isoforms play no role in lifespan in ad lib or dietary restricted conditions, but mutational inactivation of SOD-1 reduces life extension by cold. Mech. Ageing Dev. 130, 173–178. doi: 10.1016/j.mad.2008.11.003
Yun, J., and Finkel, T. (2014). Mitohormesis. Cell Metab. 19, 757–766. doi: 10.1016/j.cmet.2014.01.011
Zarse, K., Schmeisser, S., Groth, M., Priebe, S., Beuster, G., Kuhlow, D., et al. (2012). Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 15, 451–465. doi: 10.1016/j.cmet.2012.02.013
Zermati, Y., Mouhamad, S., Stergiou, L., Besse, B., Galluzzi, L., Boehrer, S., et al. (2007). Nonapoptotic role for Apaf-1 in the DNA damage checkpoint. Mol. Cell. 28, 624–637. doi: 10.1016/j.molcel.2007.09.030
Keywords: ROS signaling, superoxide, intrinsic apoptotic pathway, mitochondrial ROS, longevity, aging
Citation: Hekimi S, Wang Y and Noë A (2016) Mitochondrial ROS and the Effectors of the Intrinsic Apoptotic Pathway in Aging Cells: The Discerning Killers! Front. Genet. 7:161. doi: 10.3389/fgene.2016.00161
Received: 30 June 2016; Accepted: 30 August 2016;
Published: 14 September 2016.
Edited by:
S. Michal Jazwinski, Tulane University, USAReviewed by:
Seung-Jae Lee, Pohang University of Science and Technology, South KoreaCopyright © 2016 Hekimi, Wang and Noë. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siegfried Hekimi, c2llZ2ZyaWVkLmhla2ltaUBtY2dpbGwuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.