 Han B. Lee
Han B. Lee Brynn N. Sundberg
Brynn N. Sundberg Ashley N. Sigafoos
Ashley N. Sigafoos Karl J. Clark
Karl J. Clark- 1Neurobiology of Disease Graduate Program, Mayo Graduate School, Rochester, MN, USA
- 2Department of Biochemistry and Molecular Biology, Mayo Clinic, Rochester, MN, USA
Recent advancement in genome engineering technology is changing the landscape of biological research and providing neuroscientists with an opportunity to develop new methodologies to ask critical research questions. This advancement is highlighted by the increased use of programmable DNA-binding agents (PDBAs) such as transcription activator-like effector (TALE) and RNA-guided clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated (Cas) systems. These PDBAs fused or co-expressed with various effector domains allow precise modification of genomic sequences and gene expression levels. These technologies mirror and extend beyond classic gene targeting methods contributing to the development of novel tools for basic and clinical neuroscience. In this Review, we discuss the recent development in genome engineering and potential applications of this technology in the field of neuroscience.
Introduction
The relationship between genotype and phenotype is often not direct and one-to-one (Hirschhorn and Daly, 2005; Manolio et al., 2009). In clinical settings, analyzing patients' whole genome sequencing data and triaging the significance of a variant remain a challenging task (MacArthur et al., 2014; Shendure, 2014; Richards et al., 2015). This is especially the case for neural or behavioral phenotypes. Many neuropsychiatric disorders are multi-genic or multi-factorial involving complex gene-gene or gene-environment interactions (Franke et al., 2009; Sullivan et al., 2012; Dunn et al., 2015). Disease manifestations of neuronal diseases, whether neuropsychiatric or neurodegenerative, are often circuit- or neuronal population-specific making circuit-level understanding indispensable in neuroscience (Shin and Liberzon, 2009; Russo and Nestler, 2013; Shepherd, 2013). Neuronal subtypes are diverse, and each type may show distinct structural, functional, electrophysiological, and connectional properties. Different methods estimate the number of neuronal subtypes in the brain from ~100 to 1000 (Stevens, 1998; Masland, 2001, 2004; Nelson et al., 2006). Different epigenetic landscapes endow a distinct identity to each neuronal subtype that arises from a common genome within each organism. This epigenetic encoding could be the substrate in which the gene-environment interaction is consolidated (Vialou et al., 2013). Interrogating such complexity in the nervous system requires sophisticated methodologies that can manipulate each component while minimizing the perturbation of the whole system. Recently developed genome engineering technologies allow genomic perturbations with surgical precision, and these technologies are well-suited for advancing neuroscience.
There are several advantages to genome engineering technologies. First, programmable DNA-binding agents (PDBAs) can be targeted to any locus in the genome. Particularly, transcription activator-like effector (TALE) and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated (Cas) systems can be targeted at nucleotide resolution. Second, a variety of effectors can be fused to the PDBA as a module. PDBAs fused to endonucleases make breaks in the DNA that can introduce random or targeted DNA changes when co-delivered with exogenous DNA sequences. With transcription activators and repressors, or epigenetic modifiers, gene expression can be up- or down-regulated. Using fluorescent proteins as an effector, specific genomic loci can be visualized in live cells or animals allowing for structural and organizational investigations of chromatin. Third, PDBAs can be used for therapeutic purposes with applications ranging from correcting mutations in monogenic disorders to modulating gene expression. In this Review, we discuss recent developments in genome engineering and their potential applications in the field of neuroscience. We summarize the use of PDBAs as (1) genome editing tools and (2) gene expression modulators, and discuss (3) other considerations in applying these systems in neuroscience.
Genome Editing with Customizable Endonucleases
The field of genome engineering has developed over the past several decades. Classic gene targeting that has established mutant mouse models was the first to show that biological functions of the genes can be perturbed and studied in vivo (Evans and Kaufman, 1981; Smithies et al., 1985; Thomas and Capecchi, 1986). Around the same time, investigations into nonhomologous end joining (NHEJ) and homologous recombination (HR) were defining and uncovering DNA repair pathways (Roth and Wilson, 1985; Lehman et al., 1993; Phillips and Morgan, 1994; Hagmann et al., 1996). The biology of gene targeting and DNA repair mechanisms were gradually elucidated, uncovering critical variables for genome engineering such as linear donor DNA is more recombinogenic than circular ones (Orr-Weaver et al., 1981; Folger et al., 1982; Rong and Golic, 2001) or a DNA double-strand break (DSB) in the genome increases the rate of HR (Puchta et al., 1993; Rouet et al., 1994; Choulika et al., 1995; Smih et al., 1995). The structural studies and engineering of zinc finger protein led to the development of first programmable DNA binding agents (Pavletich and Pabo, 1991; Desjarlais and Berg, 1992; Choo and Klug, 1994; Wu et al., 1995; Kim et al., 1996; Smith et al., 2000).
Zinc Finger Nucleases (ZFNs)
The zinc finger nuclease (ZFN) was introduced in 1996 to address the need for more specific restriction endonucleases (Kim et al., 1996). Repeated zinc finger DNA-binding domains were fused to a FokI endonuclease domain to allow for programmable DNA sequence binding specificity as each finger motif moves along the major groove of the DNA double helix and stabilizes upon binding three matching base pairs (Kim et al., 1996).
In the early 1990s it was shown that HR increased by two- to four- orders of magnitude in plant and mammalian cells when a double-strand break (DSB) was introduced to the genome by the rare-cutting endonuclease, I-SceI (Puchta et al., 1993; Rouet et al., 1994; Choulika et al., 1995; Smih et al., 1995). Exploiting this, a collaboration between Chandrasegaran and Carroll labs showed that HR could be significantly increased when ZFNs made DSBs in the genome (Bibikova et al., 2001).
Subsequent refinement of ZFNs provided a template for innovation of next generation genome engineering tools–TALENs and CRISPR/Cas9. For example, modified FokI catalytic domains, which increased specificity or efficiency, were first developed for ZFN applications and later adapted for TALENs. Obligate heterodimeric versions of FokI significantly decreased off-target effects and genome toxicity caused by unintended homodimerization (Miller et al., 2007; Szczepek et al., 2007). Guo et al. used directed evolution to select for increased FokI activity, improving mutagenic efficiency by three- to six-fold (Guo et al., 2010). A zinc finger nickase system had been shown to favor homology-directed repair (HDR) without activating NHEJ repair, minimizing opportunities for errors and off-target effects (Kim et al., 2012; Ramirez et al., 2012). A nickase commonly refers to an endonuclease that generates a single-strand DNA break, nick.
ZFNs have been widely used to create animal models and are currently the only genome editing tool being utilized in clinical settings. Studies addressing diseases such as Huntington's disease (Garriga-Canut et al., 2012) and Hurler and Hunter syndromes (Sharma et al., 2015) are in pre-clinical development, and HIV treatments have reached early clinical trials (Perez et al., 2008; Wilen et al., 2011; Tebas et al., 2014; for a review, see Jo et al., 2015). ZFN research is now focused on improving delivery methods. Despite the pioneering role of ZFNs, difficulty of designing and optimizing the ZFN reagents (Greisman and Pabo, 1997) led to rapid adaptation of first TALEN and then CRISPR/Cas9 systems.
Transcription Activator-Like Effector Nucleases (TALENs)
The DNA-binding mechanism of TALEs was elucidated in 2009 (Boch et al., 2009; Moscou and Bogdanove, 2009), years after the discovery of TALEs as secreted proteins from the bacterial plant pathogen Xanthomonas (Bai et al., 2000; Yang and White, 2004; Gu et al., 2005; Kay et al., 2007; Sugio et al., 2007). Native TALEs are comprised of an N-terminal domain, central repeats of the DNA binding domain, a C-terminal segment that incorporates nuclear localization signals, and a transcriptional activation domain (Bogdanove et al., 2010). The DNA-binding domain consists of 33–35 amino acid repeats with differences at residues 12 and 13 (repeat variable di-residue; RVD). A specific RVD in the DNA-binding domain recognizes a base in the target locus, providing a structural feature to assemble predictable DNA-binding domains. The DNA binding domains of a TALE are fused to the catalytic domain of a type IIS FokI endonuclease to make a targetable TALE nuclease (TALEN; Figure 1A). To induce site-specific mutation, two individual TALEN arms, separated by a 14–20 base pair spacer region, bring FokI monomers in close proximity to dimerize and produce a targeted DSB (Figure 1A).
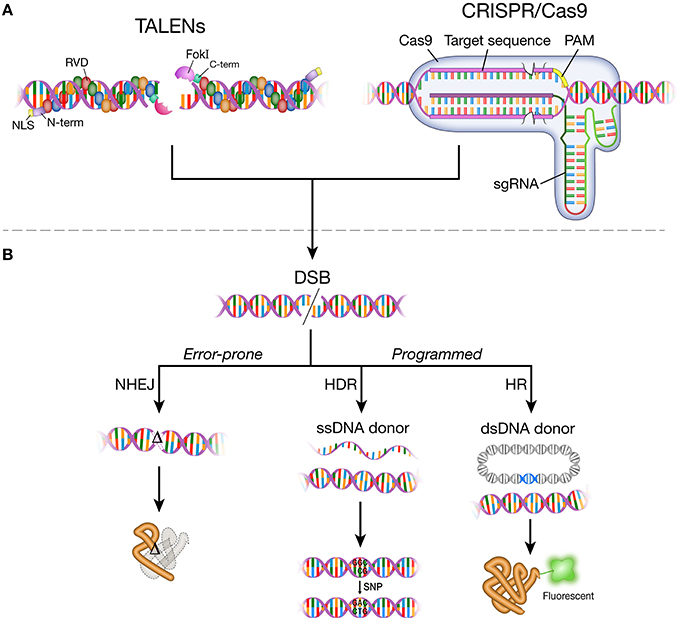
Figure 1. DNA-binding agents, DNA double-strand break (DSB) and repair pathways. (A) A pair of TALEN monomers bound to double-strand DNA. A TALEN monomer comprises N-terminal domain, nuclear localization signal, modular repeats that contain two highly variable amino acid residues (RVDs), C-terminal domain, and FokI endonuclease. TALENs bind to target DNA in the major groove. A pair of TALEN monomers targeted closely enable FokI dimerization to make DNA double-strand break. Cas9 protein and sgRNA form a complex. The sgRNA-Cas9 complex bind to target DNA through Watson-Crick base-pairing between the target recognition region of the sgRNA and the target DNA sequence (protospacer). The target recognition region (pink rectangle) is the RNA sequence in the sgRNA that matches the target DNA sequence (20 nt). The PAM (protospacer adjacent motif) sequence varies depending on the type of Cas protein. Streptococcus pyogenes Cas9 protein recognizes 5′-NGG-3′ sequence. (B) Targetable TALEN and CRISPR/Cas9 systems generate DNA double-strand break (DSB) at the target locus in the genome. DSBs are repaired by non-homologous end joining (NHEJ), homology-directed repair (HDR), or homologous recombination (HR). Error-prone NHEJ results in non-functional proteins or loss of protein through frameshift mutations introduced to open reading frame. HDR and HR mediate faithful repair of DNA break by incorporating exogenously provided DNA templates in either single strand DNA oligos (ssDNA donor) or plasmids (dsDNA donor). HDR and HR can be used to change small sequences or tag the target gene with a reporter (e.g., green fluorescent protein). TALENs, transcription activator-like effector nucleases; NLS, nuclear localization signal; RVD, repeat variable di-residue; FokI, FokI endonuclease; N-term, N-terminal domain; C-term, C-terminal domain; CRISPR/Cas9, clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9; sgRNA, single guide RNA; PAM, protospacer adjacent motif.
Several modifications to the native TALE composition and initial TALEN design have improved binding and cleavage specificity and efficiency (Christian et al., 2010; Hockemeyer et al., 2011; Li et al., 2011; Tesson et al., 2011). In an effort to improve TALEN efficiency, improved TALEN scaffolds incorporated optimized nuclear localization sequences, amino-terminal, and carboxy-terminal truncations, and assorted FokI nuclease linkers (Mussolino and Cathomen, 2012). In vitro scaffolding modifications have been studied by fusing variable C-terminal truncations to the catalytic domain of FokI to determine which variants enable efficient cleavage, the final modification of which retained only 28 or 63 of the 278 original C-terminal residues (Miller et al., 2011). Different TALEN scaffolds and native TALEs from other bacterial species continue to be tested and optimized to achieve a higher and more consistent activity of targeting genetic modifications. Our lab has used the GoldyTALEN scaffold to improve in vivo efficacy with a messenger RNA expression vector backbone (pT3TS). When using the GoldyTALEN scaffold, there was a six-fold increase in somatic gene modification, and germline modification rate increased from 17 to 71% (Bedell et al., 2012). Achieving a higher level of TALEN activity can depend on the length (base pairs) of the spacer between the two individual TALEN binding sites and the specificity of DNA binding (Ma A. C. et al., 2013a).
Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR Associated (CRISPR/Cas)
First identified in Streptococcus pyogenes, the native type II CRISPR/Cas system is a bacterial adaptive immune response and the simplest among the three CRISPR/Cas systems that requires only four Cas proteins. During exposure to foreign DNA (the acquisition phase), the type II CRISPR/Cas system incorporates a short foreign DNA sequence (protospacer), using Cas1 and Cas2 proteins, into the bacterial genome between short palindromic repeats, thus forming a CRISPR array. The expression phase of the immune response occurs when the CRISPR locus is transcribed into a long non-coding pre-CRISPR RNA (pre-crRNA), along with its partial complement, the trans-activating CRISPR RNA (tracrRNA). The tracrRNA forms base pairs with the repeat sequences of pre-crRNA, and host RNAIII cleaves the repeat sequence to produce mature crRNAs. For DNA cleavage to occur (the interference phase), the crRNA:tracrRNA:Cas9 complex scans the invading DNA for the PAM sequence and, upon Watson-Crick base pairing between crRNA and foreign DNA, neutralizes it via a DSB (for a review, see Sorek et al., 2008; Horvath and Barrangou, 2010; Jiang and Doudna, 2015; Wright et al., 2016).
Since the CRISPR sequence was discovered in 1987 (Ishino et al., 1987), the utility of CRISPR/Cas for targeted DNA cleavage was first demonstrated in 2012 in vitro and in bacterial cells (Gasiunas et al., 2012; Jinek et al., 2012). Unique from ZFNs and TALENs, which use protein domains to recognize specific DNA sequences, the CRISPR/Cas system uses RNA-based sequence recognition (Figure 1A). The crRNA and tracrRNA in the native system were simplified into a single guide RNA (sgRNA) of approximately 100 nucleotides with a generic tetraloop secondary structure to improve ease-of-use and customizability for engineering (Jinek et al., 2012). Since the application of CRISPR/Cas systems in mammalian cells (Cho et al., 2013; Cong et al., 2013; Jinek et al., 2013; Mali et al., 2013b), the technology has rapidly evolved: multiplexing in the CRISPR/Cas system (Cong et al., 2013), developing a paired Cas9 nickase system (Shen et al., 2014), dimerizing the dCas9 (dead Cas9; catalytically inactive) system fused to FokI endonuclease (Tsai et al., 2014a), adding a DNA-binding domain for improved target specificity (Bolukbasi et al., 2015), or splitting Cas9 into two components for improved packaging and delivery in a viral vector and temporal control (Truong et al., 2015; Wright et al., 2015; Zetsche et al., 2015b). Other efforts focus on reducing targeting limitations posed by the strict PAM recognition sequence. Different variants of Cas proteins have been shown to reduce targeting limitations (Kleinstiver et al., 2015); for example, a Cas9 ortholog, Cpf1, uses a T-rich PAM sequence (Zetsche et al., 2015a).
With these successive innovations, the CRISPR/Cas9 system has become widely adapted for genome engineering. Some of the reasons for its popularity are the constructs to produce sgRNA can be easily prepared with one cloning process, the Cas9 construct can be re-used without any modification, and targeting multiple loci (multiplexing) is easily achieved by introducing several sgRNAs. The simplicity of sgRNA production has made it possible to generate libraries of knockout guide RNAs for either human or mouse genes that are useful for forward genetic screens (Koike-Yusa et al., 2013; Shalem et al., 2014).
DNA Double-Strand Break Repair in Neurons
DNA double-strand breaks (DSBs) induced by targetable endonucleases are only the first step in mutagenesis (Figure 1B). The perturbation of the genomic DNA sequence is useful in genome engineering only because cellular DNA damage signals and repair machineries are activated and fix the damaged sequence. It is the repair process that we exploit to isolate desired disease alleles or to revert mutations to wild-type sequences.
Such DNA repair pathways are rapidly mobilized after damage because maintaining genomic integrity is critical for cell survival and function. DNA damage can occur in many ways. Cell metabolism generates oxidative DNA damage, ultraviolet radiation results in single- or double-strand breaks, and cross-linking agents lead to intra- or inter-strand linkages (Lindahl, 1993; Rao, 1993; Brooks, 2002; Caldecott, 2008; McKinnon, 2013; Pan et al., 2014). Among the many forms of DNA damage, double-strand breaks (DSBs) are considered the most detrimental and are repaired by three major pathways: homologous recombination (HR), homology-directed repair (HDR), and non-homologous end joining (NHEJ) (Figure 1B; Lieber, 2010; Chiruvella et al., 2013; Jasin and Rothstein, 2013). The HR pathway allows an error-free repair and requires an extensive sequence homology from which the sequences of the damaged DNA can be faithfully copied. The sequence homology is provided by the sister chromatid, and thus HR is operational during the S and early G2 phases of the cell cycle during which the sister chromatid is available (Potter et al., 1987; Maryon and Carroll, 1991; Benjamin and Little, 1992; Kadyk and Hartwell, 1992). NHEJ is an error-prone process in which the broken DNA ends are directly ligated through the end processing proteins, Ku70:Ku80 [a part of DNA dependent protein kinase (DNA-PK)], catalytic subunit of DNA-PK (DNA-PKsc), and ligase IV (Critchlow and Jackson, 1998). During this end-joining process, small-scale nucleotide insertions and deletions (indels) are commonly involved (Heidenreich et al., 2003). Since NHEJ does not require a homology sequence, it can occur throughout the cell cycle. HDR broadly defines a wide array of homology-dependent pathways of the DNA repair including single-strand annealing (SSA) and alternative NHEJ, also called microhomology-mediated end joining (MMEJ; He et al., 2015). MMEJ utilizes small homologous sequences (1–25 nt) within either end of the broken DNA (Liang et al., 2008; McVey and Lee, 2008). Like NHEJ, most HDR processes can occur throughout the cell cycle. On the basis that both HDR and HR use homologous sequences, HR can be considered part of HDR (Lieber, 2010). Yet, HR possesses processes unfound in HDR or NHEJ, such as extensive processing of the 5′-end of the DNA lesion and strand invasion by the Rad51-bound 3′-end of the lesion (Maryon and Carroll, 1991; Shinohara et al., 1992; Van Dyck et al., 1999; Jasin and Rothstein, 2013). Since DNA repair is a critical process for cell survival, the various repair pathways can be activated by different damage signals. Although there are proteins dedicated to a particular pathway, many of the signaling and repair molecules are redundant rather than exclusive to a single pathway (Takata et al., 1998; Madabhushi et al., 2014).
DNA repair pathways are utilized in two distinct phases in neurons. During the development of an organism, the highly proliferative neural tissues mainly use HR. At this stage, failure to faithfully repair the genome leads to apoptosis (Waser et al., 1979; Sugo et al., 2000; Orii et al., 2006). Since a single error in progenitor cells can lead to severe failures in a large number of cells, either a complete repair or abortive apoptosis is adaptive for the survival of the organism. Once neurons develop, the mature neurons are post-mitotic and their DNA is no longer replicated. In these mature neurons, the sister chromatid with sequence homology is not available and therefore HR is rare. NHEJ is considered to be the major pathway for DNA repair in adult neurons and apoptosis is not promoted (Sonoda et al., 2006; Fortini and Dogliotti, 2010; Iyama and Wilson, 2013). Whereas global repair activities are down-regulated in post-mitotic neurons, transcription-dependent DNA repair pathways are highly active, and transcribed genes are actively repaired regardless of the type of DNA damage (e.g., single strand or double-strand breaks; Nouspikel and Hanawalt, 2000, 2002). Although there is mounting evidence that connects DNA DSBs to neurodegeneration (Hochegger et al., 2006; Wang et al., 2013; Rulten et al., 2014), it is still not clear how these DSBs lead to neurodegeneration in mature neurons (for a review, see McKinnon, 2013; Madabhushi et al., 2014). Also uncertain is whether it is the type or number of errors that contributes to neurodegeneration.
Recently, it was found that DNA DSBs occur as part of normal neuronal physiology, and these DSBs are closely tied to learning, memory, and novel experience-dependent synaptic changes (Suberbielle et al., 2013; Madabhushi et al., 2015). These DSBs have been demonstrated to be required in the promoter regions of early-response genes—responsible for memory formation and synaptic consolidations—in primary neuron culture and hippocampal slices of Swiss Webster mice, and also in an in vivo training paradigm for contextual fear conditioning in 6-month-old mice (Madabhushi et al., 2015). In an exploration of novel environment learning paradigm, the DSBs in hippocampal regions in 4-7-month-old mice are repaired within 24 h of the occurrence (Suberbielle et al., 2013). The pathogenic protein in Alzheimer's disease, amyloid-beta, made DSBs persist beyond the 24-h period in 4-7-month-old human amyloid precursor protein (hAPP) transgenic mice (Suberbielle et al., 2013). In addition, DSB repair protein BRCA1 is depleted in a mouse model of Alzheimer's disease, and the depletion is correlated with cognitive decline in mice compared to wild-type mice (Suberbielle et al., 2015). These findings indicate that functional DSB repair is associated with normal neuronal physiology and impaired DSB repair with pathophysiology.
The distinctive DNA repair mechanisms and physiologic utility of DSBs in neurons warrant further investigation on the consequences of using targetable endonucleases in the nervous system. The majority of studies on the functions of PDBAs and the outcomes of PDBA applications were conducted with dividing cell lines such as immortalized cells (Cermak et al., 2011; Christian et al., 2012; Garg et al., 2012; Cho et al., 2013; Cong et al., 2013; Jinek et al., 2013) or stem cells (Hockemeyer et al., 2011; Cong et al., 2013; Ding et al., 2013a,b; Osborn et al., 2013; Mali et al., 2013b). The result of PDBA-induced DSBs is likely to be different in post-mitotic neurons compared with mitotic cells. For example, a faithful sequence correction might be more difficult in nervous tissues since the incorporation of exogenous DNA might be favorably mediated by HDR or NHEJ, both of which are more error-prone repair processes than HR. Or, it could be that neurons might activate the HR pathway if given exogenous DNA templates. These questions need to be empirically determined. Once detrimental mutations occur by DSBs, the nervous tissue would be more susceptible to degeneration since the restoration of the cell population through cell divisions is unlikely. These considerations need to be taken into account when targeting PDBAs in the nervous system as somatic therapeutic agents. Developing an experimental system with which nervous tissue-specific perturbations can be tested will be invaluable to spur therapeutic PDBA development targeting neuronal disorders.
NHEJ: Knock-Outs
Since indels in the NHEJ pathway can lead to frameshift mutations producing nonfunctional truncated proteins and loss of protein, NHEJ has been the predominant mode of PDBA-mediated mutagenesis. In earlier applications of PDBA-mediated targeted NHEJ in neuroscience, neuronal genes are targeted in dividing cells to demonstrate that mutations can be introduced at the target loci. Cong et al. targeted EMX1, Th, and PVALB implicated in neurodevelopment or neurodegeneration using a Cas9 system in HEK293FT cells (Cong et al., 2013; Table 1). PDBA-mediated NHEJ enabled an important advance in rapid modeling of human diseases in animals and cell lines. With PDBAs, virtually any genetic make-up with a well-defined genetic basis (e.g., monogenic) can be produced to model a disease in terms of genomic sequences, if we set aside for another discussion the caveats of biologically replicating the phenotypes, genetic compensation in germline mutants, and embryonic lethality in essential genes. Several investigations have introduced mutations to neuronal loci in zebrafish using ZFNs (Schmid et al., 2013), TALENs (Bedell et al., 2012; Huang et al., 2013), and a Cas9 system (Hwang et al., 2013), in rats using TALENs (Ferguson et al., 2013), and in mice and rats using a Cas9 system (Li et al., 2013; Tables 1, 2). PDBA-mediated NHEJ has made it possible to model neuropsychological and neurodevelopmental disorders in animals that were previously not possible due to lack of germline competent embryonic stem cells (ESCs). Using TALENs, two groups reported successful modification of the MECP2 locus implicated in Rett syndrome in non-human primate models (Liu H. et al., 2014; Liu Z. et al., 2014). In these two investigations, only one group saw successful birth of injected animals (Liu H. et al., 2014). The fact that the other group could not maintain the surrogate mother's pregnancy of injected zygotes indicates that mosaic F0 animals might not be viable when the mutagenic efficiency of PDBAs is high. Another group targeted PPARG—implicated in neurodegeneration—with a Cas9 system in cynomolgus monkeys (Macaca fascicularis; Niu et al., 2014). These genetic perturbations have not been fully investigated yet for their phenotypes, nor the degree to which they model human diseases. More follow-up investigations are expected in which the biological relevance of the models to human diseases is reported.
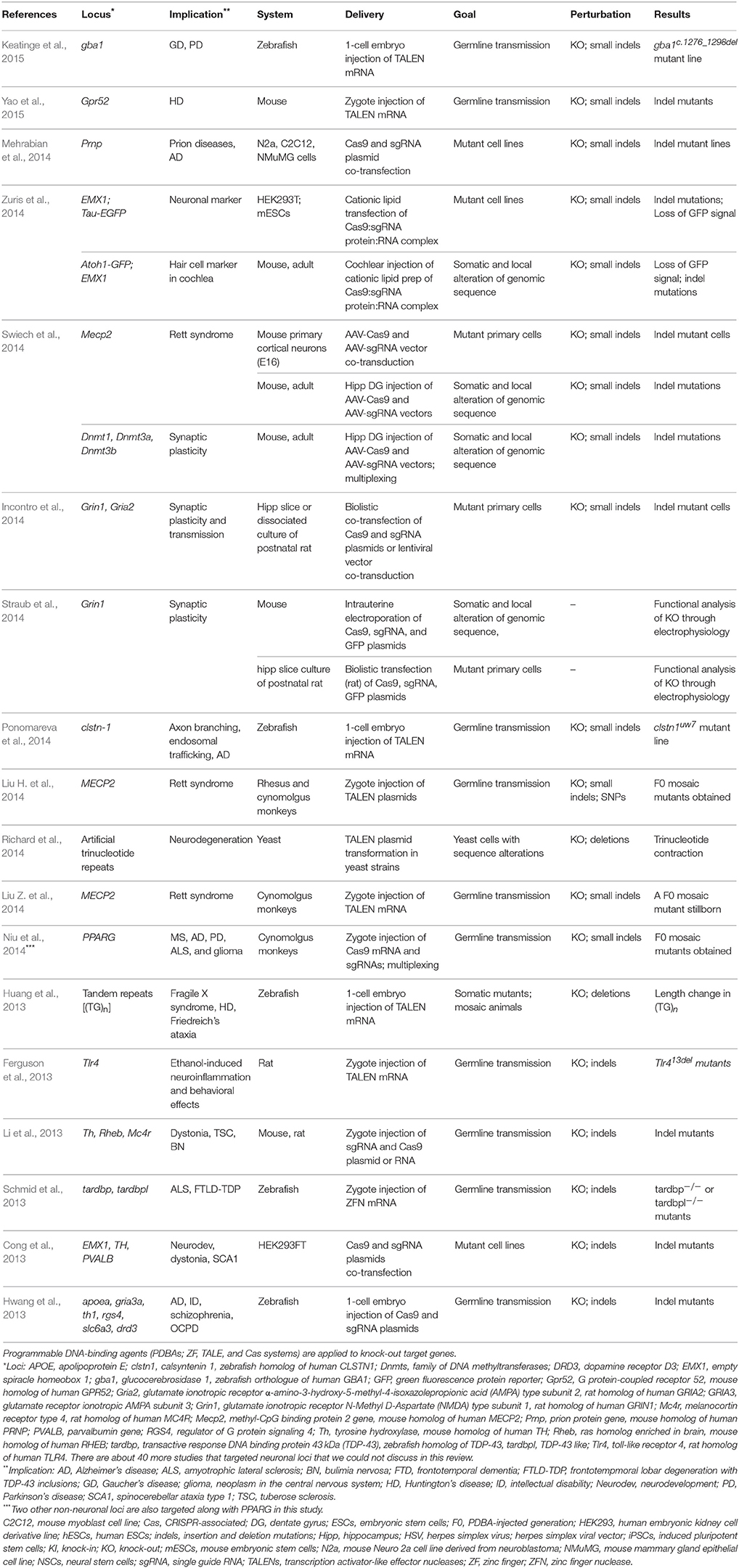
Table 1. Neuronal target loci: knock-out.
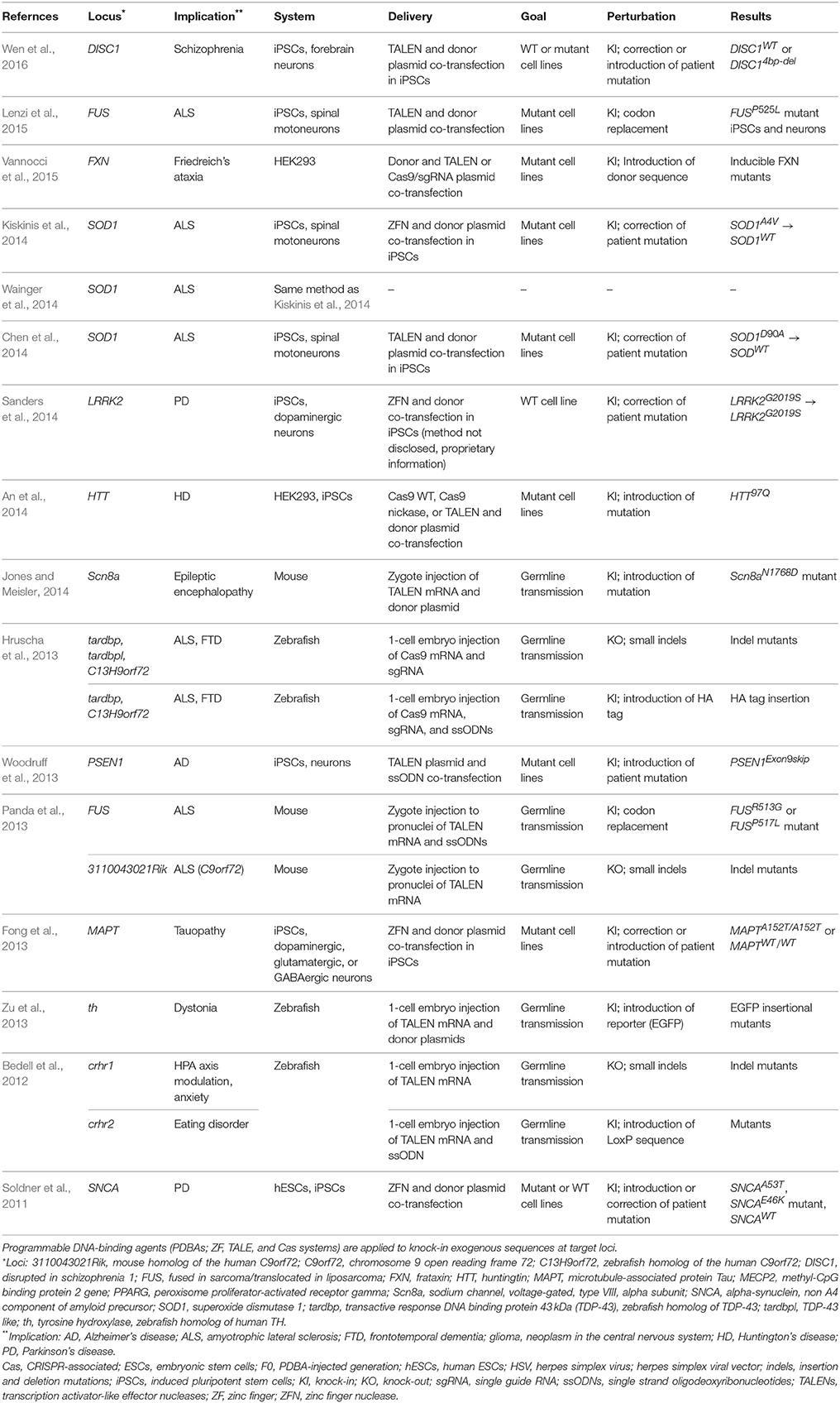
Table 2. Neuronal target loci: knock-in.
Moving beyond dividing cells and germline mutations in mutant animals, four groups directly applied Cas9 systems in post-mitotic neurons in which two synaptic proteins, an NMDA receptor subunit (Incontro et al., 2014; Straub et al., 2014) and an AMPA receptor subunit (Incontro et al., 2014), are targeted (Table 1). Straub et al. locally altered the genomic sequence in a Grin1 locus (NMDA receptor subunit 1) in vivo in E15 mouse hippocampi by delivering Cas9 and sgRNA plasmids through in-utero electroporation, as well as in hippocampal slice culture through biolistic transfection (Straub et al., 2014). The resulting neurons showed altered electrophysiological profiles providing functional evidence that the target gene, Grin1, is knocked down. However, mutations were not directly sequenced. Incontro et al. knocked out either Grin1 or Gria2 (AMPA subunit 2) in hippocampal slice culture from 6 to 11-day-old rats by delivering Cas9 and sgRNA plasmids through biolistic transfection (14-day expression of the construct before electrophysiology; Incontro et al., 2014). Incontro et al. showed that electrophysiological profiles were altered in the knock-out cells. They also reported that >90% of the mutations in Grin1 had indel mutations that were out-of-frame and that the function of the target protein was 100% altered in all cells in which Cas9 expression was confirmed (Incontro et al., 2014). The findings by Incontro et al. imply that NHEJ could be the dominant mode of DNA DSB repair in post-mitotic neurons and that mutagenesis in neural cells could be much higher compared to other somatic cells because their post-mitotic nature does not allow the dilution of PDBAs.
Similarly, Swiech et al. targeted Mecp2 or DNA methyltransferases (Dnmt1, Dnmt3a, and Dnmt3b; via multiplexing) in post-mitotic neurons in vitro and in vivo by stereotactically delivering Cas9 and sgRNA in adeno-associated viral vectors to the hippocampal dentate gyrus in adult mice (Swiech et al., 2014; Table 1). In primary cortical neuron culture, ~70% of transduced cells showed knock-out of Mecp2 by immunocytochemistry, western blot, and next generation sequencing. The in vivo knock-out efficiency was ~68% in purified nuclei of transduced cells (in vivo transduction efficiency of the vector was ~80%) from dissected brain tissue. The Mecp2 mutations led to altered performance in a contextual fear-conditioning paradigm, reinforcing that the in vivo knockdown of Mecp2 in hippocampal cells has neuronal and behavioral consequences. Swiech et al. also demonstrated that somatic Mecp2 knockdown affects the functions of neural circuits by showing altered primary visual cortex (V1) function when Mecp2 was targeted in the superficial layers of V1. Multiplexed targeting in vivo created mutations in Dnmt3a, Dnmt1, and Dnmt3b loci with ~75, ~75, and ~50% indel rates, respectively, when the three loci were targeted in a single transduction in the hippocampus (Incontro et al., 2014). Swiech et al. demonstrated that Cas9-mediated multiplexed gene targeting in the nervous system can be accomplished in vitro, in vivo, and in localized somatic application as well as through zygotic injections. Most recently, Zuris et al. demonstrated non-viral delivery of PDBAs in in vitro and in vivo post-mitotic neuronal cells (Zuris et al., 2014; Table 1). Zuris et al. delivered a Cas9:sgRNA protein:RNA complex in cationic lipid to the cochlear and altered the EMX1 locus with ~20% efficiency in neuronal cells (the outer hair cell) in 1-day-old mice. These four cases represent diverse Cas9 systems delivered with DNA plasmids, viral vectors, or cationic lipids, which are directly applicable to post-mitotic neurons in vitro and in vivo. These work demonstrate the feasibility of neuronal genome engineering and provide evidence that the mutagenic efficiency is comparable to or higher than that in dividing cells.
In addition to neuronal applications of PDBA-mediated NHEJ, understanding and exploiting the properties of NHEJ are underway in many investigations. Beyond out-of-frame knock-outs, in-frame indel mutations also occur and may be useful to study the function of specific amino acid residues or putative protein domains (Sung et al., 2013). Microhomology-mediated end joining (MMEJ) often results in deletions or translocations (He et al., 2015). First shown in mammalian cells in 1986, MMEJ has become a useful tool to bias deletions toward frameshift mutations (Roth and Wilson, 1986). The property of MMEJ that allows researchers to better predict mutagenic outcomes might be particularly useful for neuroscientists since NHEJ–resulting in random mutations–appears to be more active compared to dividing cells. Several software options provide microhomology-based prediction functions to design TALENs and CRISPR/Cas9 gRNAs that are more likely to induce frameshift mutations (Neff et al., 2013; Bae et al., 2014a).
HDR and HR: Knock-Ins
Unlike the NHEJ repair pathway, HR and some versions of HDR utilize an external DNA donor and repair the DNA damage without introducing erroneous sequences. A single-stranded oligo with short sequence homology (~50 nt) to the genomic DNA can act as a template for HDR, and a donor DNA construct with extended homology arms (hundreds to thousands of bps) can act as a template in HR. Applications of PDBA-mediated knock-ins in neuroscience have focused on engineering induced pluripotent stem cells (iPSCs) and generating animal models (Table 2). First, it has been a widely-used and effective approach to engineer patient-derived iPSCs with PDBAs to revert a disease-causing allele to wild-type via HDR and to differentiate these engineered and control (without modification) iPSCs to neurons. The genome engineered iPSCs and neurons are identical to the patients' cells except the corrected DNA sequences and a certain level of single nucleotide polymorphisms (SNPs) inherent to all stem cell models. Patient-derived iPSCs have been used to investigate altered cellular functions and establish cell-autonomous disease models in Parkinson's disease (PD), tauopathy, amyotrophic lateral sclerosis (ALS), Alzheimer's disease, and Huntington's disease (HD) using ZFN (Soldner et al., 2011; Fong et al., 2013; Kiskinis et al., 2014; Sanders et al., 2014), TALEN (Woodruff et al., 2013; Chen et al., 2014; Lenzi et al., 2015; Wen et al., 2016), and Cas9 systems (An et al., 2014; Table 2).
Whereas a knock-in approach had been challenging and time-consuming in animal models, PDBAs have spurred the generation of knock-in cell and animal models based on increased efficiencies of HR. When considering the rates of classic HR to be ~1 × 10−3 and 1 × 10−6 (Smithies et al., 1985; Thomas and Capecchi, 1986; Puchta et al., 1993; Rouet et al., 1994; Choulika et al., 1995; Rong and Golic, 2001), PDBAs can increase donor sequence integration at a rate of ~1–45% by generating DSBs in the genomic target locus (Urnov et al., 2005; Beumer et al., 2008; Greenwald et al., 2010; Li et al., 2013; Zu et al., 2013; Gratz et al., 2014; Krentz et al., 2014; Platt et al., 2014; Shin et al., 2014a; Xie F. et al., 2014a; Karakikes et al., 2015; Lee et al., 2015; Wang et al., 2016). Although direct comparison is nearly impossible because of the differences in the knock-in system, including the size and design of the donor, selection systems, and target loci in the genome, targeted DSBs with PDBAs are viewed as improving HR rates by 3–6 orders of magnitude. Bedell et al. first showed in vivo knock-in of modified LoxP sequences in a neuronal locus, crhr2, by zygote injection of TALEN mRNA and a single-stranded oligonucleotide in zebrafish (Bedell et al., 2012; Table 2). Currently, patient allelic variants of many neuronal disorders have been introduced and modeled in mice and zebrafish using TALEN (Panda et al., 2013; Jones and Meisler, 2014) and Cas9 systems (Hruscha et al., 2013) ranging from epilepsy to schizophrenia (Table 2).
Directly knocking-in exogenous DNA sequences with PDBAs in post-mitotic neurons has not been reported to our knowledge. It will be an important area of investigation when developing therapeutics, and the mechanism of foreign DNA knock-in is being pursued to improve future therapeutic applications. It is important to recognize that the NHEJ, HDR, or HR pathways all can be used to introduce exogenous DNA sequences into the genome. A linear piece of double-stranded DNA may be incorporated into the genome even during NHEJ repair (Lin and Waldman, 2001; Miller et al., 2004; Gabriel et al., 2011). This NHEJ process is still prone to errors at the break site and the mechanism is unclear. Inactivating key components of the NHEJ pathway (e.g., ligase IV) has been demonstrated to bias DSB repair away from NHEJ, in one example increasing HR efficiency by 20–65% in Drosophila (Beumer et al., 2008). Another way to circumvent the NHEJ pathway is by generating single-strand overhangs with nickases and providing a double-stranded oligo with complementary overhangs on either side (Ran et al., 2013). Albeit with lower efficiency, HR-based knock-ins with short or long homology arms allow many experimental manipulations—including the introduction of promoters, reporters, recombination sites, and wild-type alleles—with high accuracy and are utilized in neuroscience research (Liu et al., 2011; Corti et al., 2012).
Epigenetic Modification and Transcriptional Control
There are estimated to be hundreds to thousands of types of neuronal cell populations (Stevens, 1998; Shin et al., 2014b) in the mammalian cortex, suggesting equally complex epigenetics in the nervous system. Development, physiological function (such as memory consolidation), and pathological changes in neuropsychiatric and neurodegenerative disorders are closely tied to epigenetic changes (for a review, see Tsankova et al., 2007; Morris and Monteggia, 2014; Reul, 2014). In recent years, we have significantly broadened our understanding of the three-dimensional structures and organizations of chromatin in the nucleus (Takizawa and Meshorer, 2008; Lieberman-Aiden et al., 2009; Phillips-Cremins et al., 2013; Rao et al., 2014). Similar to how the deluge of genomic sequencing data has urged the functional study of disease-associated and candidate genes, we are in need of functional understanding of higher chromatin structures and their connection to neuronal diseases. The ability to perturb epigenetic states, gene expression patterns, and higher order chromatin structures in a targeted manner has been largely unavailable. Although it is still at a fledging state, we will discuss multiple investigations that have reported success in direct modulation of gene expression, epigenetic modulation, building synthetic circuits, and modifying regulatory elements in mammalian cells, yeasts, and prokaryotes using PDBAs. The majority of the studies were conducted in dividing cells, and only a small portion of the studies targeted neuronal loci. Still, these early studies demonstrate the feasibility of controlling regulatory elements with PDBAs and are worth a discussion for their future adoption in the nervous system.
Regulation of Gene Expression
As the name implies, TALEs were first identified and adapted as transcription factors (Bai et al., 2000; Gu et al., 2005; Boch et al., 2009; Moscou and Bogdanove, 2009). Applying this inherent functionality, TALEs have been used to up-regulate the expression of specific genes at targeted loci (Zhang et al., 2011; Figure 2A). The application of single TALEs showed a moderate effect of two to five-fold increases in target gene expression in endogenous loci (SOX2, KLF4) in HEK293T cells, but the fold increase could be augmented from 50- to 10,000-fold by applying multiple TALEs targeting an endogenous promoter (Perez-Pinera et al., 2013b; Maeder et al., 2013c). Replacing the TALE's native activation domain with a stronger activation domain (VP16 and later VP64) increased activation by 10 times in a reporter system (Geiβler et al., 2011). In addition, TALEs have been shown to repress gene expression when fused to a repressor domain such as the EAR-repression domain (SRDX) in plants (Mahfouz et al., 2011) and the Krüppel-associated box (KRAB) or Mad interaction domain (SID) in human cells (Cong et al., 2012). Yet, the modulatory effects of TALEs are inconsistent. Some TALEs show a higher activity while some fail to modulate the transcription of the target locus. Although this ineffectiveness was attributed to repressed regions of chromatin (Zhang et al., 2011; Bultmann et al., 2012), other studies showed that TALEs can activate transcription even in repressed chromatin regions (Scott et al., 2014b). One explanation for this discrepancy is the location of the TALE binding site in relation to nucleosome binding. In particular, the activity of TALEs was higher when targeting DNA sequences that are accessible in the nucleosome, while not as efficient when targeted to sites covered by the dyad of the nucleosomes (Scott et al., 2014a). Three studies using TALENs reported modulation of gene expression in neuronal loci (Table 3). Cong et al. showed ~5- and three-fold up-regulation of mRNA in CACNA1C target loci 1 and 2 with TALE-VP64 in HEK293FT cells (Cong et al., 2012). Targeting NTF3 in HEK293 cells, Miller et al. reported a ~30-fold increase in mRNA expression with TALE-VP16 (Miller et al., 2011) whereas Maeder et al. saw only a modest increase in the same gene with TALE-VP64 (though significant; Maeder et al., 2013c). These two cases show that, even in the same locus, the efficacy of TALE-mediated gene expression may be affected by multiple variables such as target loci, design and concentrations of TALE constructs, cell types, delivery methods, and the genetic make-up of transfected cells or injected animals. Similarly, different types of neurons in varying animal models could respond differently to the application of TALEs.
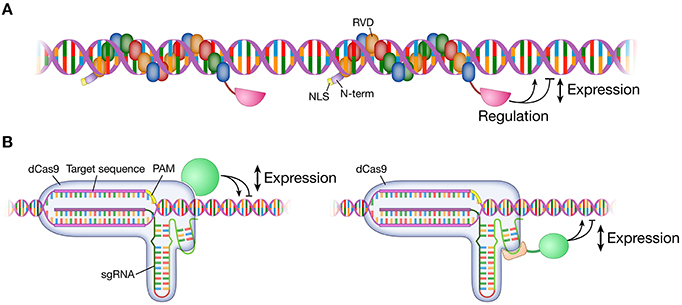
Figure 2. Modulation of gene expression. Targetable TALE and dCas9 fused to various effector domains modulate gene expression at the target locus in the genome. Effector domains can be transcription activator domains (e.g., VP64) or repressor domains (e.g., KRAB). A TALE monomer/sgRNA or multiple TALEs/sgRNAs are targeted to a locus for a range of gene expression modulation. (A) An effector domain is fused to the C-terminus of a TALE. (B) An effector domain is fused to or interacts with either dCas9 protein or sgRNA. TALE, transcription activator-like effector; dCas9, dead Cas9; catalytically inactivated Cas9 nuclease; sgRNA, single guide RNA; VP64, tetrameric repeat of herpes simplex virus activation domain (VP16); KRAB, Krüppel-associated box repression domain.
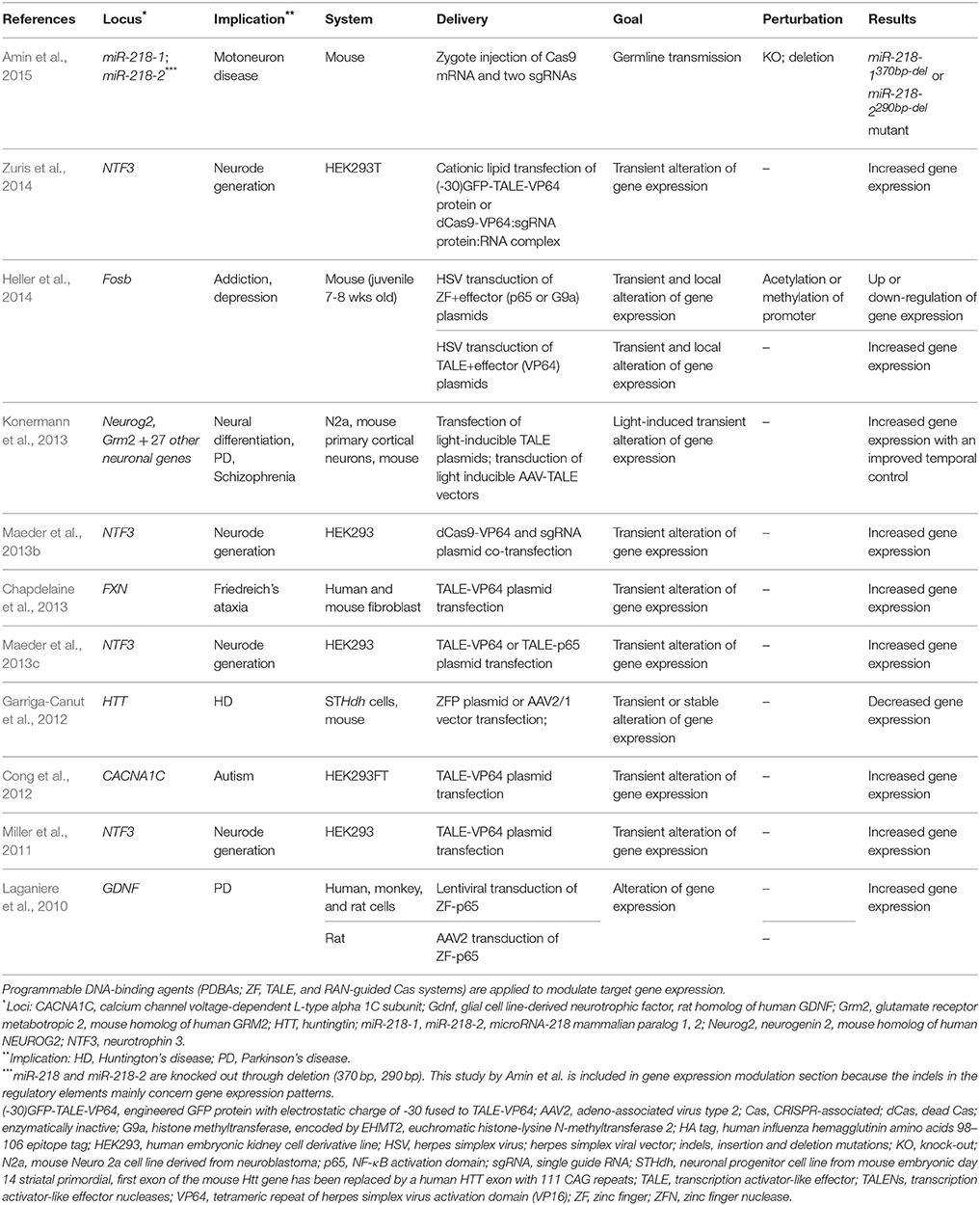
Table 3. Neuronal target loci: gene expression.
Although it is useful to be able to modulate gene expression levels at target loci by simply introducing TALEs to the system, these applications act constitutively and lack spatiotemporal control. Inducible TALEs and TALEs with regulated expression systems were developed to provide further spatiotemporal control. Allowing for increased temporal regulation, TALEs bound to a ligand-binding domain could modulate gene expression in response to light (Konermann et al., 2013), endogenous signals (Li et al., 2012), or exogenous ligands (Mercer et al., 2014). The study by Konermann et al. is noteworthy since the authors not only modulated two neuronal targets in an inducible system in dividing cells, primary neuron culture, and in vivo, but also profiled a total of 28 TALE-VP64 systems in primary neuron culture, each of which individually targeted one locus (Konermann et al., 2013). Konermann et al. built a two-hybrid system that allows TALE-mediated gene activation only when blue light (466 nm) is applied. One component of the two-hybrid is a TALE monomer bound to cryptochrome 2 protein (CRY2) effector domain that is sensitive to light and the other comprises a CRY2's interacting partner (C1B1 protein), nuclear localization signal, and VP64. With light stimulation, Neurog2 mRNA is significantly increased from 30 min onward until about 3 h after stopping the stimulation in N2a cells (mouse neuroblastoma cell line), reaching a peak induction of 20-fold compared to that of unstimulated cells. After AAV-mediated delivery of inducible constructs targeting a Grm2 locus in mouse primary cortical neurons and mice, Konermann et al. observed ~four- to six-fold increase in Grm2 mRNA after light stimulation in neuronal culture and ~3.5-fold increase in vivo (Table 3). In a light-inducible repressor system, Konermann et al. fused CRY2 with mSin3 interacting domains that repress gene expression through histone deacetylation and observed two-fold reduction in Grm2 mRNA expression in primary neuron culture (Konermann et al., 2013). Non-inducible TALE-VP64 constructs were tested in neurons as well by individually targeting 28 neuronal loci with a significant fold increase ranging from ~1.5 (Grin2a) to ~33 (Slc6a4).
To generate controlled and coordinated gene circuits, TALEs that interact with regulatory elements were developed, including TALEs that control synthetic promoters or TALEs that are expressed under the control of an artificial regulatory element. Multiple synthetic promoters that contain a TALE-binding site simultaneously activated gene expression in multiple loci in plants (Brückner et al., 2015), which showed the potential for a coordinated control of multiple genes. A similar system was developed to allow for a temporal regulation of TALE expression and circuit-level control of multiple genes in prokaryotes (Rai et al., 2015). RiboTALEs incorporate riboswitches upstream of the TALE-encoding sequences to regulate the time at which TALEs are expressed. Once expressed, TALEs repress gene expression of target loci that are engineered to have a TALE-binding site (Rai et al., 2015). Improved spatiotemporal control was also demonstrated in mammalian cells by combining the well-established tetracycline-response element (TRE) and Gal4-UAS system with TALEs fused to a repressor domain (Li Y. et al., 2015). Combining a TALE-repressor with other regulatory systems—in this case, TRE and Gal4-UAS—produced a synthetic circuit via a two-hybrid regulatory system.
Similar modulations of gene expression were achieved by engineered CRISPR/Cas systems. Unlike TALEs, the innate function of the CRISPR/Cas system is to make DNA DSBs as a bacterial adaptive immune system in nature (Barrangou et al., 2007; Garneau et al., 2010). To co-opt the CRISPR/Cas system as a transcription factor system, the innate endonuclease function of Cas9 is catalytically inactivated (dCas9; dead Cas9) by introducing D10A and H841A mutations within the nuclease domains, RuvC1 and HNH, respectively (Jinek et al., 2012; Figure 2B). The resulting dCas9 without any fused effector domains tends to block transcription in endogenous loci in prokaryotes and a reporter system in human cells, presumably through steric hindrance to the binding or elongation of the RNA polymerase (RNAP; Qi et al., 2013). dCas9 fused to a repression domain (KRAB) effectively silenced gene expression both in endogenous loci (CD71 and CXCR4) and in a reporter system. When fused to an activating domain (VP64), dCas9 increased GFP expressions in a reporter system (Gilbert et al., 2013). It was not reported if there was an activation in an endogenous locus (Gilbert et al., 2013). A neuronal endogenous locus was targeted with a dCas9-VP64 system showing a moderate (significant) level of increase in NTF3 mRNA (~three-fold) at a concentration of 200 ng sgRNA and 250 ng dCAs9-VP64 and a high increase (~5–25) at a concentration of 500 ng sgRNA and 500 ng dCas9-VP64 (Maeder et al., 2013b). This study indicates that applying an optimal concentration without toxicity is an important factor. Robust activation of endogenous gene expression (ranging from 8- to 2000-fold) in human cells (Cheng et al., 2013; Maeder et al., 2013b; Mali et al., 2013a; Perez-Pinera et al., 2013a) and in mouse zygotes (Cheng et al., 2013) was later reported, yet a combination of multiple sgRNAs for each locus was necessary to see effective activation of the target gene in all studies. Mali et al. engineered sgRNA to include MS2 bacteriophage coat protein-binding RNA stem loop at the 3′ end and function as a effector carrying molecule (Mali et al., 2013a). This approach still required multiple sgRNAs to significantly activate an endogenous gene. This is consistent with reports that multiple TALEs function better at increasing the transcription of target locus from two- to five-fold of a single TALE to 50- to 10,000-fold in multiple TALEs.
The inability of the CRISPR/dCas9 system to activate gene expression with one sgRNA was recently amended with an alternative approach. Instead of linking effector domains to dCas9, two groups independently engineered the sgRNA secondary structure allowing the binding of effector domains to an sgRNA motif similar to the approach by Mali et al. (Figure 2B). Zalatan et al. incorporated viral RNA sequences of MS2, PP7, and com into sgRNA (this RNA guide is referred to as scaffold RNA; scRNA) that are recognized by the MCP, PCP, and Com RNA-binding proteins, respectively (Zalatan et al., 2015). The MCP, PCP, and Com proteins are fused to an activation (VP64) or repression (KRAB) domain and operate as an RNA-binding adaptor for the effector. This system expanded the two-factor system of sgRNA and dCas9 to a three-factor system of scRNA, dCas9, and adaptor-effector. Utilizing this system, each scRNA activated or repressed a different target locus mediated by different combinations of the adaptor and effector complex, maximizing the orthogonal control of gene regulation (Zalatan et al., 2015). Konermann et al. engineered sgRNA to incorporate dimerized MS2-binding sequences, made an adaptor-effector complex comprising MS2-p65-HSF1, and kept the fused VP64 activation domain on dCas9 (this system is currently referred to as a synergistic activation mediator; SAM; Konermann et al., 2015). Each group showed that one gRNA-mediated targeting could increase gene expression in endogenous loci from ~5-fold (Zalatan et al.) to ~15-fold (Konerman et al.). Zalatan et al. demonstrated orthogonal regulation by simultaneously activating and silencing gene expression, and Konerman et al. introduced an optimized system in activating gene expression.
Besides immortalized human cell lines, the CRISPR/Cas system was tested for gene activation and silencing in prokaryotes (Bikard et al., 2013) and human pluripotent stem cells (hPSCs; Kearns et al., 2013). In Escherichia coli, dCas9, fused to an RNAP omega subunit that stabilizes RNAP at the proximal promoter, increased β-galactosidase activity in a reporter system by ~three-fold (Bikard et al., 2013). In hPSCs, one sgRNA increased endogenous gene expression (SOX17) by 287-fold (Kearns et al., 2013), which was the highest fold increase with one sgRNA in CRISPR/dCas9 applications reported to date in any human cell line or prokaryote. This finding might implicate that distinct regulatory landscapes and mechanisms in different cell lines determine the performance of dCas9 and that CRISPR/dCas9 may be useful to differentiate hPSCs to various types of somatic cells.
Still, when considering effective gene regulation, maximizing the inherent function of each reagent appears to be the best route. In all studies that we have reviewed as of 2015, excepting the report by Kearns et al. TALEs showed a better efficiency in activating endogenous gene expression than did CRISPR/dCas9 in both one and multiple TALE-monomer/sgRNA applications. Gao et al. investigated the efficiency of gene activation between a TALE construct and four different constructs of dCas9 which targeted enhancer regions in Oct4 and Nanog loci (Gao et al., 2014). The report showed that a TALE monomer activated either reporter or endogenous genes significantly better than did any construct of dCas9 with one sgRNA (Gao et al., 2014). Importantly, Gao et al. increased the rigor of the experiment from reporter expression to endogenous gene expression and to cellular reprograming differentiating mouse embryonic fibroblasts (MEFs) to iPSCs. dCas9s could not produce any reprogrammed cell colonies even though they could increase the reporter and endogenous gene expression, whereas TALEs produced reprogrammed iPSC colonies (Gao et al., 2014). On the other hand, dCas9 constructs had equivalent or better activity in silencing gene expression than did a TALE monomer (Gao et al., 2014). Gao et al. speculate that the innate steric hindrance that dCas9 shows at the target locus might interfere with the endogenous transcription factor machinery, limiting the capacity of dCas9 to activate gene expression.
Such strengths of dCas9 in silencing gene expression had been shown in an early study in which dCas9 without a repressor domain could override the effect of doxycycline-inducible rtTA (tetracycline transactivator) targeting the same tet operator sequence as the sgRNA (Gilbert et al., 2013). The potent gene repression by dCas9 has been applied to modify the lac regulatory pathway in E. coli (Qi et al., 2013), to reprogram complex biological pathways in yeasts (Zalatan et al., 2015), and to build two-hybrid regulatory circuits with the Gal4 system in mammalian cells (Kiani et al., 2014).
Modification of Regulatory Elements and Epigenetic Environment
In addition to the direct regulation of transcription, one can target and modify epigenetic environments and chromatin organizations to investigate important mechanisms of neuronal function. For example, TALEs fused to LSD1 histone demethylase demethylated histones at endogenous enhancer loci and subsequently down-regulated expression of the genes under the control of the targeted enhancer in human cell lines (Mendenhall et al., 2013). In another example, investigations from the Gersbach group using dCas9 fused to an effector domain showed robust modification of histone epigenetic markers. Hilton et al. fused dCas9 to the catalytic domain of human acetyltransferase p300 and observed robust up-regulation of H3K27 acetylation (four- to eight-fold) and of target gene expression (5- to 265-fold; Hilton et al., 2015). Thakore et al. investigated effects of KRAB repressor in epigenetic landscape and found enriched H3K9 trimethylation specifically at the target locus (HS2 enhancer) and consequent gene silencing of multiple globin genes under control of HS2 (Thakore et al., 2015). In addition to histone modifications, the methylation status of DNA contributes to the epigenetic environment. TET1 hydroxylase is responsible for the first step of 5-methylcytosine demethylation, which often activates a promoter (Shin et al., 2014b). This, too, can be modified with PDBAs, and targeted demethylation of CpGs in the promoter by TALEs fused to a TET1 hydroxylase catalytic domain increased the expression in endogenous target loci in human cells (Maeder et al., 2013a).
The genome has higher organizational structures beyond localized epigenetic environments. The multigene complex is one of the higher organizations and represents the locus on the genome where multiple genes on the same or different chromosomes associate with RNA polymerase (RNAP). The transcription of these multiple genes can be synchronously controlled at this multigene complex. Introducing mutations to a multigene complex using TALENs disrupted gene loop formation and significantly decreased gene expression (Fanucchi et al., 2013). Targeting Cas9 with two sgRNAs to the regulatory sequences, Li et al. demonstrated that efficient inversions and duplications of the regulatory elements and gene clusters can be induced in human cells and mice by inducing 2–4 DNA DSBs separated by a few tens of bases to several hundred kilo bases (Li J. et al., 2015). This perturbation was transmitted to offspring and led to the discovery of a new regulatory role of the protocadherin gamma gene cluster.
Visualization
Besides specific manipulation of genomic sequences, PDBAs can be adapted to visualize the native organization of the chromatin. Alterations in DNA structures are implicated in multiple trinucleotide repeat disorders such as Huntington's disease and spinocerebellar ataxia (Kovtun et al., 2001) as well as hexanucleotide (GGGGCC) repeats in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD; Haeusler et al., 2014). Using new PDBA reagents, genome organization in these disorders may be visualized in live cells. Using TALEs fused to fluorescent proteins (TALE-FP), Ma et al. imaged repetitive DNA sequences in the telomere of live or fixed human cells (U2OS cells; Ma, H. et al., 2013b). A specific chromosome was identified using centromeric repeat sequences specific to the particular chromosome (Ma, H. et al., 2013b). Miyanari et al. used TALE-FP to show that paternal and maternal chromosomes could be differentially labeled and visualized in live mouse cells and embryos using single nucleotide polymorphisms (SNPs) in the chromosomes due to the extreme sequence specificity of TALEs (Miyanari et al., 2013). Using a dCas9 system fused to fluorescent proteins and capitalizing on the strength of the CRISPR/dCas9 system for multiplexing, Chen et al. visualized non-repetitive genomic sequences by “tiling” sgRNA along the target locus (Chen et al., 2013). All three studies imaged changing chromosomal organizations at each cell cycle. It is not known whether and how post-mitotic neurons change genomic conformation. The three studies indicate that the structural and organizational understanding of the genome in neurons could be done in vivo.
Off-Target Effects
In each application of PDBAs, a strategy to identify off-target effects needs to be in place. Despite the precision that the PDBA systems have shown, it is imprudent to expect any PDBA to bind to one unique locus in the genome. Plant pathogenic bacteria in the genus Xanthomonas evolved type III secretion system delivering TALEs into plant cells to induce expression of the host genes beneficial to their survival (Yang and White, 2004; Gu et al., 2005). Several off-targets in DNA binding would not have compromised the purpose of the system. When bacteria detect foreign viral DNA sequences registered in their CRISPR sequences (Ishino et al., 1987; Barrangou et al., 2007; Garneau et al., 2010), a few mismatches might be tolerated as far as the exogenous DNAs are effectively cleaved in the same way the restriction endonuclease system has evolved in bacteria. Thus, the specificity of targeting a locus in the genome is a primary concern that arises with the adaptation of the biological systems to human needs, in particular, for therapeutic purposes. In most research systems, off-target effects of PDBAs can be ruled out by generating two independent mutant alleles in which a genotype-phenotype relationship is confirmed. In animal models, potentially undetected off-target effects can be diluted out through chromosomal shuffling during meiosis. In cell culture models, two clones with the same mutation can be chosen to ensure the phenotype appears due to the mutation. With therapeutic and clinical applications, the system needs to be highly specific. If a PDBA system is used somatically to alter DNA sequences in differentiated adult cells, the change is permanent in the individual. In germ-line applications, the changes to the genetic material are permanent and heritable. These serious implications in clinical applications need to be considered, but it is equally important to see that there are different levels of stringency for precision depending on the nature of the experiments planned. Therefore, for researchers who plan to use a PDBA system, their experimental purposes are the most important consideration when choosing a PDBA system. For genome engineering as a discipline, researchers have most intensely investigated two areas as they aim to develop therapies with PDBAs: one to improve the specificity of the system and the other to detect off-target sites and effects.
Statistically, a CRISPR/Cas or TALE (monomer with 15-RVDs) system can incur off-target effects (cleavage or altered expression), although a TALEN (15-RVDs × 2) system may offer unique binding even in large mammalian genomes including the human's. An sgRNA in the CRISPR/Cas system can encounter a binding sequence by chance every 412 bases (~1.5 × 107) to 414 bases (~2.5 × 108), taking into account two specific nucleotides in the PAM sequence (nGG) and 10–12 nt of seed sequences immediately upstream of the PAM sequence within a 20-nt sgRNA. This results in 11–179 potential binding sites in a human haploid genomic sequence (~3 gigabases) assuming random genomic sequences. Mismatches are tolerated even in the seed sequences in the sgRNA, which increases the chance of off-target binding of Cas9 further. A 15-RVD based and dimerized TALEN system would encounter a binding site every 430 bases (~1.0 × 1018), which suggests the possibility of a unique target site in the human haploid genome. However, since the binding of each RVD is not completely exclusive to one nucleotide, TALENs also have detectable off-target effects (Hockemeyer et al., 2011; Fine et al., 2014).
Statistical probability notwithstanding, experimentally-determined data on off-target effects of the CRISPR/Cas system have been different from that of bioinformatics predictions. Methods to identify off-target effects have evolved from sequence-based computational prediction to genome-wide identification of Cas9-binding sites with chromatin immunoprecipitation (ChIP), and later to genome-wide interrogation of DNA cleavage (for a review, see Ishida et al., 2015; Koo et al., 2015; O'Geen and Yu, 2015). Sequenced genomes allow direct comparison of the nucleotide sequences of designed sgRNAs, identifying predictable off-target binding sites. This bioinformatic identification of off-target sites was the basis of early studies in which these selected sites were monitored with reporter systems and sequenced (Cradick et al., 2013; Hsu et al., 2013; Pattanayak et al., 2013; Mali et al., 2013a; Cho et al., 2014; Fu et al., 2014). The off-target binding and DNA cleavage resulting from these studies varied widely—depending on the target site and sgRNAs—from no off-target sites to ~10 sites for each sgRNA. It is not clear if this is a site-specific epigenetic environmental effect, sgRNA sequence effect, or something else. After this selected investigation of off-target effects, more unbiased approaches were developed. These investigations utilized RNA sequencing (Gilbert et al., 2013; Perez-Pinera et al., 2013a) or ChIP followed by sequencing (Cencic et al., 2014; Kuscu et al., 2014; Wu et al., 2014; Polstein et al., 2015). To investigate the binding of Cas9 protein to the target locus, a modified version of dCas9 was used with ChIP-seq applications. It was hypothesized that since dCas9 does not cleave the DNA and stays bound, it would generate unbiased genome-wide snapshots unmasked by potentially varying efficiency of DNA repair in different cell lines. The majority of the studies found a highly enriched binding to the target sites and highly specific gene expression patterns (0 or 1 off-target binding sites), yet there was identifiable off-target binding (up to a few hundred sites) for some sgRNAs as well. These methods presented two challenges. First, it was difficult to functionally determine if the binding of dCas9 changed gene expression since sequence mutations were absent. A threshold has to be arbitrarily set to interpret if the changes in gene expression were significant. Second, this method also captured transient binding of dCas9 to the genomic DNA. The mechanism of Cas9-DNA binding suggests that Cas9 scans the genome for the PAM sequence (5′-NGG-3′) and that there is transient binding depending on the degree of sequence matching between the sgRNA and genomic DNA sequence (Anders et al., 2014). Thus, the off-target binding of dCas9 might not be off-target events but merely the locus of dCas9 at the moment of the assay.
Overcoming these limitations, unbiased DNA cleavage event-based methods were developed. These methods integrate DNA double-strand break (DSB) events with insertion of reporters or capturing of the broken fragments with adaptors. IDLV (integrase-defective lentiviral vector), GUIDE (genome-wide, unbiased identification of DSBs enabled by sequencing), and BLESS (direct in situ breaks labeling, enrichment on streptavidin and next-generation sequencing) methods make use of lentivirus integration, double-strand oligo deoxyribonucleotide (dsODN), or streptavidin as a DSB capture device, respectively (Tsai et al., 2014b; Ran et al., 2015; Wang et al., 2015). While the strength of these methods is that DNA DSBs are captured, their shortcoming is that it might capture DSB events that are independent of PDBA-induced breaks. More recently, two technologies provided an improved way of identifying PDBA activities. O'Geen et al. co-opted a ChIP-seq based method to enrich significant dCas9-binding signal by designing capture probes (O'Geen et al., 2015). Kim et al. developed a system to identify off-targets without any capture devices by re-digesting Cas9-treated genomic DNA with Cas9 in vitro (Kim et al., 2015). These unbiased approaches find that CRISPR/Cas systems are specific, making one off-target cut in the whole genome when appropriately designed sgRNAs were used (O'Geen et al., 2015). For an sgRNA to be deemed appropriately designed, the sgRNA has to be unique in the genome by more than four nucleotides compared to potential off-target sites. However, there are cases of sgRNAs that led to several tens off-target sites despite the apparent soundness of the design (Tsai et al., 2014b; Kim et al., 2015; Ran et al., 2015; Wang et al., 2015). In addition, these methods confirmed that computational sequence predictions are different from actual off-target cleavage sites and that dCas9-binding sites are different from actual off-target cleavage sites. Some reasons for the differences identified are sequence variation in the sample (Yang et al., 2014) and DNA or sgRNA bulging at the target sites (Lin et al., 2014). These studies demonstrate that while CRISPR/Cas systems have clear off-targeting effects, they rarely have the spurious off-target sites that bioinformatic methods predicted. However, further optimization is absolutely required to adapt CRISPR/Cas systems for therapeutic purposes, as zero off-target sites is the expectation for clinical applications.
Efforts to increase the specificity of CRISPR/Cas9 systems have attempted various strategies. Optimized sgRNA designs such as shorter sgRNAs (17–18 nts) truncated at the 5′-end of conventional 20 nt sgRNAs (Fu et al., 2014) or addition of two guanine nucleotides at the 5′-end of sgRNAs (Cho et al., 2014) significantly decreased off-target effects. Cas9 has been engineered to make Cas9 variants with either endonuclease domain, RuvC or HNH, inactivated (Jinek et al., 2012). A pair of Cas9 nickases targeted to adjacent sites on opposite strands results in DNA DSBs with significantly reduced off-target effects (Ran et al., 2013; Mali et al., 2013a; Cho et al., 2014; Frock et al., 2014; Shen et al., 2014). In a similar pairing scheme, a dCas9 protein with nuclease domains inactivated is fused to FokI endonuclease, as FokI dimerization is required to make DNA DSBs. The dCas9-FokI systems showed significantly improved specificity (Tsai et al., 2014a; Guilinger et al., 2014b; Wyvekens et al., 2015). A disadvantage of nickase and dCas9-FokI systems is that the pairing requirement decreases the number of available target sites in a genome since two closely located PAM sequences are needed.
Most recently, two groups independently reported engineered S. pyogenes Cas9 (SpCas9) systems that show significantly decreased off-target effects using current unbiased genome-wide off-target screening technologies. Both groups optimized the energetics during the formation of target DNA-guide RNA-SpCas9 complex by decreasing non-specific interactions between SpCas9 protein and target DNA while maintaining specific interactions between guide RNA and the complementary strand of target DNA (Kleinstiver et al., 2016; Slaymaker et al., 2016). Slaymaker et al. focused on the non-specific electrostatic interactions between the negatively charged phosphate backbone of the non-complementary strand of target DNA and the positively charged amino acid residues in SpCas9 protein in the groove between two nuclease domains (Slaymaker et al., 2016). Three engineered SpCas9 variants [SpCas9 (K855A), eSpCas9(1.0) (K810A/K1003A/R1060A), and eSpCas9(1.1) (K848A/K1003A/R1060A)] showed undetectable off-target effects at three previously identified off-target sites using next generation sequencing in human cell cultures (Slaymaker et al., 2016). When all known off-target sites were investigated, eSpCas9(1.0) and eSpCas9(1.1) showed a significantly decreased cleavage rate (<0.2% indel) in 22 out of 24 sites. In BLESS assessments for unbiased genome-wide off-target screening, there were still noticeable indels while reduction in off-target cleavage was clear compared to wild-type Cas9. BLESS only detected 1.0 and 0.7% indels at two EMX1(1) off-target sites (OTs) and 2.5, 6.3, 14.7, and 25.5% at four VEGFA(1) OTs for SpCas9 (K855A). eSpCas9(1.1) showed no discernable indels at EMX1(1) and 0.3 and 27.0% indels at two VEGFA(1) OTs. The three SpCas9 variants [SpCas9 (K855A), eSpCas9(1.0), and eSpCas9(1.1)] maintained on-target indel efficiency comparable (30–160%) to wild-type SpCas9 assessed with 24 sgRNAs in 10 endogenous loci (Slaymaker et al., 2016).
While Slaymaker et al. focused on non-complementary DNA strand and electrostatic interactions, Kleinstiver et al. engineered the Cas9 amino acid residues that make non-specific contact with complementary DNA strand to sgRNA through hydrogen bonds (Kleinstiver et al., 2016). Kleinstiver et al. abrogated hydrogen bonding between the Cas9 residues (N497, R661, Q695, and Q926) and the phosphate backbone of the complementary strand of target DNA by replacing those residues with alanine residues. When assessed with GUIDE-seq for unbiased genome-wide off-target detection, the Cas9 variant, SpCas9-HF1, with all four residues replaced with alanine, showed a complete absence of off-target effects with seven sgRNAs in four endogenous loci in human cells and only one detectable off-target effect. Subsequent next generation sequencing of 36 wild-type Cas9 OTs identified in GUIDE-seq showed that SpCas9-HF1 had only a background level of indels at 34 out of 36 OTs and negligible indel rates of 0.049 and 0.037% at the other two OTs (Kleinstiver et al., 2016). SpCas9-HF1 maintained on-target mutagenic efficiency comparable (70–140% excluding one sgRNA with no activity) to wild-type Cas9 with 12 sgRNAs in five endogenous loci (Kleinstiver et al., 2016).
The differences in off-target effects between the two structure-guided protein engineering studies might come from varying sensitivity of genome-wide screening technologies (BLESS vs. GUIDE-seq). Importantly, these two studies demonstrated that rational protein engineering can improve specificity in CRISPR/Cas systems. Along with off-target detecting techniques and engineered Cas9 proteins, sgRNA designing software is evolving, which may further decrease off-target effects through design of higher-specificity sgRNAs (Bae et al., 2014b; Montague et al., 2014; Xie S. et al., 2014b; Moreno-Mateos et al., 2015).
Although statistics and bioinformatics predict that TALENs are highly specific, TALENs also have off-target effects. With optimization of the system, the number of off-target sites have significantly decreased (Osborn et al., 2013; Ousterout et al., 2013; Guilinger et al., 2014a). A study showed that the 5′-side of TALEN-binding site (N-term side of TALENs) is more influential in determining TALEN specificity and found that for higher activity and specificity, the first two bases should not be adenine-adenine or cytosine-adenine (Meckler et al., 2013; Juillerat et al., 2014). The degree of individual RVD specificity was also studied. All classic RVDs (NI:A, HD:C, NN:G, and NG:T) do not bind exclusively to one base; rather, RVDs have degrees of discriminating power among the four bases rather than an RVD exclusively binds to a base. For example, HD conventionally binds to cytosine (C) and discriminates C from adenine, guanine, and thymine with scores of 0.06, 0.80, and 0.47, respectively (where 1.00 represents perfect discrimination; Juillerat et al., 2015). This limited specificity led to the development of non-conventional RVDs to improve the specificity of TALENs targeting the same locus. In a different approach using modified phage display and protein evolution systems, Hubbard et al. developed a directed protein evolution system in which the DNA binding domain of TALENs can be rapidly optimized for an improved specificity (Hubbard et al., 2015). It is now critical to apply unbiased genome-wide screening developed for CRISPR/Cas9 systems to TALEN off-target screening to validate that directed protein evolution and non-conventional RVDs do indeed decrease off-target effects.
Acknowledging that none of the PDBAs are absolutely specific is important because it will facilitate investigations to improve specificity and detect off-target effects. As a practical guideline, any investigation using PDBAs should use an alternative design (two target sites against a locus) to show the genotype-phenotype relationship beyond doubt. In addition, large-scale projects should be mandated to employ at least one unbiased off-target screening method to show that the PDBA used is reasonably specific without any spurious off-target effects. Lastly, if the system is an animal model, F2 or later generations rather than F1s should be used to ensure the dilution of non-specific mutagenesis. If enough cautions are used, PDBAs are already precise enough to accommodate most (if not all) of research needs. For therapeutic applications, any PDBA systems will need to be further optimized. However, if the absence of off-target effects is shown in cell culture systems and animal models, the particular PDBA design that targets a specific locus may be tested in human subjects.
Conclusions and Perspectives
New generations of programmable DNA-binding agents (PDBAs) such as TALE and CRISPR/Cas systems have rapidly materialized highly efficient genome engineering systems. PDBAs can now target nearly any locus in the genome, introduce or correct mutations, modulate gene expression, and change epigenetic environments with a controllable level of off-target effects, which provides unprecedented opportunities to understand neuronal and neuropsychiatric disorders. These PDBA technologies are being incrementally introduced in neuroscience, and there are only a small number of investigations that focus on the effects and outcomes of PDBA application in the nervous system. Considering distinctive mechanisms of DNA double-strand break repair and epigenetic regulation in neurons, we need more studies to complete our understanding of the functions and effects of applying PDBAs in the nervous system. To access the central nervous system, delivery mechanisms will need to be further engineered (Holkers et al., 2014; Ain et al., 2015; Chen and Gonçalves, 2015). To achieve cell-line specific inducible PDBA systems, neural populations and cell-line specific promoters need to be better characterized to avoid false outcomes (Forni et al., 2006; Galichet et al., 2010). In spite of these outstanding challenges, PDBAs will be a pivotal technology that will lead to new understanding and drive innovations in neuroscience.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The National Institutes of Health (Grants: GM63904, DK84567), Mayo Graduate School, the Mayo Clinic Center for Individualized Medicine, Sanford Research, and the Mayo Foundation for Medical Education and Research provided funding and support for our work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank J. Mark Curry in Division of Creative Media at the Mayo Clinic for artistic rendition of the figures and Julie A. Swenson at the Mayo Clinic Libraries for library service.
References
Ain, Q. U., Chung, J. Y., and Kim, Y.-H. (2015). Current and future delivery systems for engineered nucleases: ZFN, TALEN and RGEN. J. Control. Release 205, 120–127. doi: 10.1016/j.jconrel.2014.12.036
Amin, N. D., Bai, G., Klug, J. R., Bonanomi, D., Pankratz, M. T., Gifford, W. D., et al. (2015). Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science 350, 1525–1529. doi: 10.1126/science.aad2509
An, M. C., O'Brien, R. N., Zhang, N., Patra, B. N., De La Cruz, M., Ray, A., et al. (2014). Polyglutamine disease modeling: epitope based screen for homologous recombination using CRISPR/Cas9 system. PLoS Curr. 6:ecurrents.hd.0242d2e7ad72225efa72f6964589369a. doi: 10.1371/currents.hd.0242d2e7ad72225efa72f6964589369a
Anders, C., Niewoehner, O., Duerst, A., and Jinek, M. (2014). Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573. doi: 10.1038/nature13579
Bae, S., Kweon, J., Kim, H. S., and Kim, J.-S. (2014a). Microhomology-based choice of Cas9 nuclease target sites. Nat. Meth. 11, 705–706. doi: 10.1038/nmeth.3015
Bae, S., Park, J., and Kim, J.-S. (2014b). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. doi: 10.1093/bioinformatics/btu048
Bai, J., Choi, S.-H., Ponciano, G., Leung, H., and Leach, J. E. (2000). Xanthomonas oryzae pv. oryzae avirulence genes contribute differently and specifically to pathogen aggressiveness. Mol. Plant Microbe Interact. 13, 1322–1329. doi: 10.1094/MPMI.2000.13.12.1322
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. doi: 10.1126/science.1138140
Bedell, V. M., Wang, Y., Campbell, J. M., Poshusta, T. L., Starker, C. G., Krug, R. G., et al. (2012). In vivo genome editing using a high-efficiency TALEN system. Nature 491, 114–118. doi: 10.1038/nature11537
Benjamin, M. B., and Little, J. B. (1992). X rays induce interallelic homologous recombination at the human thymidine kinase gene. Mol. Cell. Biol. 12, 2730–2738. doi: 10.1128/MCB.12.6.2730
Beumer, K. J., Trautman, J. K., Bozas, A., Liu, J.-L., Rutter, J., Gall, J. G., et al. (2008). Efficient gene targeting in Drosophila by direct embryo injection with zinc-finger nucleases. Proc. Natl. Acad. Sci. U.S.A. 105, 19821–19826. doi: 10.1073/pnas.0810475105
Bibikova, M., Carroll, D., Segal, D. J., Trautman, J. K., Smith, J., Kim, Y.-G., et al. (2001). Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell. Biol. 21, 289–297. doi: 10.1128/MCB.21.1.289-297.2001
Bikard, D., Jiang, W., Samai, P., Hochschild, A., Zhang, F., and Marraffini, L. A. (2013). Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41, 7429–7437. doi: 10.1093/nar/gkt520
Boch, J., Scholze, H., Schornack, S., Landgraf, A., Hahn, S., Kay, S., et al. (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512. doi: 10.1126/science.1178811
Bogdanove, A. J., Schornack, S., and Lahaye, T. (2010). TAL effectors: finding plant genes for disease and defense. Curr. Opin. Plant Biol. 13, 394–401. doi: 10.1016/j.pbi.2010.04.010
Bolukbasi, M. F., Gupta, A., Oikemus, S., Derr, A. G., Garber, M., Brodsky, M. H., et al. (2015). DNA-binding-domain fusions enhance the targeting range and precision of Cas9. Nat. Meth. 12, 1150–1156. doi: 10.1038/nmeth.3624
Brooks, P. J. (2002). DNA repair in neural cells: basic science and clinical implications. Mutat. Res. 509, 93–108. doi: 10.1016/S0027-5107(02)00222-1
Brückner, K., Schäfer, P., Weber, E., Grützner, R., Marillonnet, S., and Tissier, A. (2015). A library of synthetic transcription activator-like effector-activated promoters for coordinated orthogonal gene expression in plants. Plant J. 82, 707–716. doi: 10.1111/tpj.12843
Bultmann, S., Morbitzer, R., Schmidt, C. S., Thanisch, K., Spada, F., Elsaesser, J., et al. (2012). Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer TALEs and inhibition of epigenetic modifiers. Nucleic Acids Res. 40, 5368–5377. doi: 10.1093/nar/gks199
Caldecott, K. W. (2008). Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631. doi: 10.1038/nrg2380
Cencic, R., Miura, H., Malina, A., Robert, F., Ethier, S., Schmeing, T. M., et al. (2014). Protospacer adjacent motif (PAM)-distal sequences engage CRISPR Cas9 DNA target cleavage. PLoS ONE 9:e109213. doi: 10.1371/journal.pone.0109213
Cermak, T., Doyle, E. L., Christian, M., Wang, L., Zhang, Y., Schmidt, C., et al. (2011). Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, e82. doi: 10.1093/nar/gkr739
Chapdelaine, P., Coulombe, Z., Chikh, A., Gérard, C., and Tremblay, J. P. (2013). A potential new therapeutic approach for friedreich ataxia: induction of frataxin expression with TALE proteins. Mol. Ther. Nucleic Acids 2, e119. doi: 10.1038/mtna.2013.41
Chen, B., Gilbert, L. A., Cimini, B. A., Schnitzbauer, J., Zhang, W., Li, G.-W., et al. (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491. doi: 10.1016/j.cell.2013.12.001
Chen, H., Qian, K., Du, Z., Cao, J., Petersen, A., Liu, H., et al. (2014). Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 14, 796–809. doi: 10.1016/j.stem.2014.02.004
Chen, X., and Gonçalves, M. A. F. V. (2015). Engineered viruses as genome editing devices. Mol. Ther. 24, 447–457. doi: 10.1038/mt.2015.164
Cheng, A. W., Wang, H., Yang, H., Shi, L., Katz, Y., Theunissen, T. W., et al. (2013). Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 23, 1163–1171. doi: 10.1038/cr.2013.122
Chiruvella, K. K., Liang, Z., and Wilson, T. E. (2013). Repair of double-strand breaks by end joining. Cold Spring Harb. Perspect. Biol. 5:a012757. doi: 10.1101/cshperspect.a012757
Cho, S. W., Kim, S., Kim, J. M., and Kim, J.-S. (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232. doi: 10.1038/nbt.2507
Cho, S. W., Kim, S., Kim, Y., Kweon, J., Kim, H. S., Bae, S., et al. (2014). Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24, 132–141. doi: 10.1101/gr.162339.113
Choo, Y., and Klug, A. (1994). Toward a code for the interactions of zinc fingers with DNA: selection of randomized fingers displayed on phage. Proc. Natl. Acad. Sci. U.S.A. 91, 11163–11167. doi: 10.1073/pnas.91.23.11163
Choulika, A., Perrin, A., Dujon, B., and Nicolas, J. F. (1995). Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol. Cell. Biol. 15, 1968–1973. doi: 10.1128/MCB.15.4.1968
Christian, M., Cermak, T., Doyle, E. L., Schmidt, C., Zhang, F., Hummel, A., et al. (2010). Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186, 757–761. doi: 10.1534/genetics.110.120717
Christian, M. L., Demorest, Z. L., Starker, C. G., Osborn, M. J., Nyquist, M. D., Zhang, Y., et al. (2012). Targeting G with TAL effectors: a comparison of activities of TALENs constructed with NN and NK repeat variable di-residues. PLoS ONE 7:e45383. doi: 10.1371/journal.pone.0045383
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi: 10.1126/science.1231143
Cong, L., Zhou, R., Kuo, Y.-C., Cunniff, M., and Zhang, F. (2012). Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat. Commun. 3, 968. doi: 10.1038/ncomms1962
Corti, S., Nizzardo, M., Simone, C., Falcone, M., Nardini, M., Ronchi, D., et al. (2012). Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci. Transl. Med. 4, 165ra162. doi: 10.1126/scitranslmed.3004108
Cradick, T. J., Fine, E. J., Antico, C. J., and Bao, G. (2013). CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 41, 9584–9592. doi: 10.1093/nar/gkt714
Critchlow, S. E., and Jackson, S. P. (1998). DNA end-joining: from yeast to man. Trends Biochem. Sci. 23, 394–398. doi: 10.1016/S0968-0004(98)01284-5
Desjarlais, J. R., and Berg, J. M. (1992). Toward rules relating zinc finger protein sequences and DNA binding site preferences. Proc. Natl. Acad. Sci. U.S.A. 89, 7345–7349. doi: 10.1073/pnas.89.16.7345
Ding, Q., Lee, Y.-K., Schaefer, E. A. K., Peters, D. T., Veres, A., Kim, K., et al. (2013a). A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 12, 238–251. doi: 10.1016/j.stem.2012.11.011
Ding, Q., Regan, S. N., Xia, Y., Oostrom, L. A., Cowan, C. A., and Musunuru, K. (2013b). Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12, 393–394. doi: 10.1016/j.stem.2013.03.006
Dunn, E. C., Brown, R. C., Dai, Y., Rosand, J., Nugent, N. R., Amstadter, A. B., et al. (2015). Genetic determinants of depression: recent findings and future directions. Harv. Rev. Psychiatry 23, 1–18. doi: 10.1097/HRP.0000000000000054
Evans, M. J., and Kaufman, M. H. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156. doi: 10.1038/292154a0
Fanucchi, S., Shibayama, Y., Burd, S., Weinberg, M. S., and Mhlanga, M. M. (2013). Chromosomal contact permits transcription between coregulated genes. Cell 155, 606–620. doi: 10.1016/j.cell.2013.09.051
Ferguson, C., McKay, M., Harris, R. A., and Homanics, G. E. (2013). Toll-like receptor 4 (Tlr4) knockout rats produced by transcriptional activator-like effector nuclease (TALEN)-mediated gene inactivation. Alcohol 47, 595–599. doi: 10.1016/j.alcohol.2013.09.043
Fine, E. J., Cradick, T. J., Zhao, C. L., Lin, Y., and Bao, G. (2014). An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res. 42, e42. doi: 10.1093/nar/gkt1326
Folger, K. R., Wong, E. A., Wahl, G., and Capecchi, M. R. (1982). Patterns of integration of DNA microinjected into cultured mammalian cells: evidence for homologous recombination between injected plasmid DNA molecules. Mol. Cell. Biol. 2, 1372–1387. doi: 10.1128/MCB.2.11.1372
Fong, H., Wang, C., Knoferle, J., Walker, D., Balestra, M. E., Tong, L. M., et al. (2013). Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Rep. 1, 226–234. doi: 10.1016/j.stemcr.2013.08.001
Forni, P. E., Scuoppo, C., Imayoshi, I., Taulli, R., Dastrù, W., Sala, V., et al. (2006). High levels of Cre expression in neuronal progenitors cause defects in brain development leading to microencephaly and hydrocephaly. J. Neurosci. 26, 9593–9602. doi: 10.1523/JNEUROSCI.2815-06.2006
Fortini, P., and Dogliotti, E. (2010). Mechanisms of dealing with DNA damage in terminally differentiated cells. Mutat. Res. 685, 38–44. doi: 10.1016/j.mrfmmm.2009.11.003
Franke, B., Neale, B. M., and Faraone, S. V. (2009). Genome-wide association studies in ADHD. Hum. Genet. 126, 13–50. doi: 10.1007/s00439-009-0663-4
Frock, R. L., Hu, J., Meyers, R. M., Ho, Y.-J., Kii, E., and Alt, F. W. (2014). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 33, 179–186. doi: 10.1038/nbt.3101
Fu, Y., Sander, J. D., Reyon, D., Cascio, V. M., and Joung, J. K. (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 32, 279–284. doi: 10.1038/nbt.2808
Gabriel, R., Lombardo, A., Arens, A., Miller, J. C., Genovese, P., Kaeppel, C., et al. (2011). An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 29, 816–823. doi: 10.1038/nbt.1948
Galichet, C., Lovell-Badge, R., and Rizzoti, K. (2010). Nestin-cre mice are affected by hypopituitarism, which is not due to significant activity of the transgene in the pituitary gland. PLoS ONE 5:e11443. doi: 10.1371/journal.pone.0011443
Gao, X., Tsang, J. C. H., Gaba, F., Wu, D., Lu, L., and Liu, P. (2014). Comparison of TALE designer transcription factors and the CRISPR/dCas9 in regulation of gene expression by targeting enhancers. Nucleic Acids Res. 42, e155. doi: 10.1093/nar/gku836
Garg, A., Lohmueller, J. J., Silver, P. A., and Armel, T. Z. (2012). Engineering synthetic TAL effectors with orthogonal target sites. Nucleic Acids Res. 40, 7584–7595. doi: 10.1093/nar/gks404
Garneau, J. E., Dupuis, M.-È., Villion, M., Romero, D. A., Barrangou, R., Boyaval, P., et al. (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. doi: 10.1038/nature09523
Garriga-Canut, M., Agustín-Pavón, C., Herrmann, F., Sánchez, A., Dierssen, M., Fillat, C., et al. (2012). Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc. Natl. Acad. Sci. U.S.A. 109, E3136–E3145. doi: 10.1073/pnas.1206506109
Gasiunas, G., Barrangou, R., Horvath, P., and Siksnys, V. (2012). Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A. 109, E2579–E2586. doi: 10.1073/pnas.1208507109
Geiβler, R., Scholze, H., Hahn, S., Streubel, J., Bonas, U., Behrens, S.-E., et al. (2011). Transcriptional activators of human genes with programmable DNA-specificity. PLoS ONE 6:e19509. doi: 10.1371/journal.pone.0019509
Gilbert, L. A., Larson, M. H., Morsut, L., Liu, Z., Brar, G. A., Torres, S. E., et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. doi: 10.1016/j.cell.2013.06.044
Gratz, S. J., Ukken, F. P., Rubinstein, C. D., Thiede, G., Donohue, L. K., Cummings, A. M., et al. (2014). Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics 196, 961–971. doi: 10.1534/genetics.113.160713
Greenwald, D. L., Cashman, S. M., and Kumar-Singh, R. (2010). Engineered zinc finger nuclease–mediated homologous recombination of the human rhodopsin gene. Invest. Ophthalmol. Vis. Sci. 51, 6374–6377. doi: 10.1167/iovs.10-5781
Greisman, H. A., and Pabo, C. O. (1997). A general strategy for selecting high-affinity zinc finger proteins for diverse DNA target sites. Science 275, 657–661. doi: 10.1126/science.275.5300.657
Gu, K., Yang, B., Tian, D., Wu, L., Wang, D., Sreekala, C., et al. (2005). R gene expression induced by a type-III effector triggers disease resistance in rice. Nature 435, 1122–1125. doi: 10.1038/nature03630
Guilinger, J. P., Pattanayak, V., Reyon, D., Tsai, S. Q., Sander, J. D., Joung, J. K., et al. (2014a). Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Meth. 11, 429–435. doi: 10.1038/nmeth.2845
Guilinger, J. P., Thompson, D. B., and Liu, D. R. (2014b). Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 32, 577–582. doi: 10.1038/nbt.2909
Guo, J., Gaj, T., and Barbas, C. F. (2010). Directed evolution of an enhanced and highly efficient FokI cleavage domain for zinc finger nucleases. J. Mol. Biol. 400, 96–107. doi: 10.1016/j.jmb.2010.04.060
Haeusler, A. R., Donnelly, C. J., Periz, G., Simko, E. A. J., Shaw, P. G., Kim, M.-S., et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200. doi: 10.1038/nature13124
Hagmann, M., Adlkofer, K., Pfeiffer, P., Bruggmann, R., Georgiev, O., Rungger, D., et al. (1996). Dramatic changes in the ratio of homologous recombination to nonhomologous DNA-end joining in oocytes and early embryos of Xenopus laevis. Biol. Chem. Hoppe Seyler 377, 239–250. doi: 10.1515/bchm3.1996.377.4.239
He, M.-D., Zhang, F.-H., Wang, H.-L., Wang, H.-P., Zhu, Z.-Y., and Sun, Y.-H. (2015). Efficient ligase 3-dependent microhomology-mediated end joining repair of DNA double-strand breaks in zebrafish embryos. Mutat. Res. 780, 86–96. doi: 10.1016/j.mrfmmm.2015.08.004
Heidenreich, E., Novotny, R., Kneidinger, B., Holzmann, V., and Wintersberger, U. (2003). Non−homologous end joining as an important mutagenic process in cell cycle−arrested cells. EMBO J. 22, 2274–2283. doi: 10.1093/emboj/cdg203
Heller, E. A., Cates, H. M., Peña, C. J., Sun, H., Shao, N., Feng, J., et al. (2014). Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat. Neurosci. 17, 1720–1727. doi: 10.1038/nn.3871
Hilton, I. B., D'Ippolito, A. M., Vockley, C. M., Thakore, P. I., Crawford, G. E., Reddy, T. E., et al. (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 33, 510–517. doi: 10.1038/nbt.3199
Hirschhorn, J. N., and Daly, M. J. (2005). Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 6, 95–108. doi: 10.1038/nrg1521
Hochegger, H., Dejsuphong, D., Fukushima, T., Morrison, C., Sonoda, E., Schreiber, V., et al. (2006). Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 25, 1305–1314. doi: 10.1038/sj.emboj.7601015
Hockemeyer, D., Wang, H., Kiani, S., Lai, C. S., Gao, Q., Cassady, J. P., et al. (2011). Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 29, 731–734. doi: 10.1038/nbt.1927
Holkers, M., Maggio, I., Henriques, S. F. D., Janssen, J. M., Cathomen, T., and Gonçalves, M. A. F. V. (2014). Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat. Meth. 11, 1051–1057. doi: 10.1038/nmeth.3075
Horvath, P., and Barrangou, R. (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. doi: 10.1126/science.1179555
Hruscha, A., Krawitz, P., Rechenberg, A., Heinrich, V., Hecht, J., Haass, C., et al. (2013). Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140, 4982–4987. doi: 10.1242/dev.099085
Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832. doi: 10.1038/nbt.2647
Huang, W., Zheng, J., He, Y., and Luo, C. (2013). Tandem repeat modification during double-strand break repair induced by an engineered TAL effector nuclease in zebrafish genome. PLoS ONE 8:e84176. doi: 10.1371/journal.pone.0084176
Hubbard, B. P., Badran, A. H., Zuris, J. A., Guilinger, J. P., Davis, K. M., Chen, L., et al. (2015). Continuous directed evolution of DNA-binding proteins to improve TALEN specificity. Nat. Meth. 12, 939–942. doi: 10.1038/nmeth.3515
Hwang, W. Y., Fu, Y., Reyon, D., Maeder, M. L., Tsai, S. Q., Sander, J. D., et al. (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229. doi: 10.1038/nbt.2501
Incontro, S., Asensio, C. S., Edwards, R. H., and Nicoll, R. A. (2014). Efficient, complete deletion of synaptic proteins using CRISPR. Neuron 83, 1051–1057. doi: 10.1016/j.neuron.2014.07.043
Ishida, K., Gee, P., and Hotta, A. (2015). Minimizing off-target mutagenesis risks caused by programmable nucleases. IJMS 16, 24751–24771. doi: 10.3390/ijms161024751
Ishino, Y., Shinagawa, H., Makino, K., Amemura, M., and Nakata, A. (1987). Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 169, 5429–5433.
Iyama, T., and Wilson, D. M. III (2013). DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst). 12, 620–636. doi: 10.1016/j.dnarep.2013.04.015
Jasin, M., and Rothstein, R. (2013). Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 5:a012740. doi: 10.1101/cshperspect.a012740
Jiang, F., and Doudna, J. A. (2015). The structural biology of CRISPR-Cas systems. Curr. Opin. Struct. Biol. 30, 100–111. doi: 10.1016/j.sbi.2015.02.002
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Jinek, M., East, A., Cheng, A., Lin, S., Ma, E., and Doudna, J. (2013). RNA-programmed genome editing in human cells. Elife 2:e00471. doi: 10.7554/eLife.00471
Jo, Y.-I., Kim, H., and Ramakrishna, S. (2015). Recent developments and clinical studies utilizing engineered zinc finger nuclease technology. Cell. Mol. Life Sci. 72, 3819–3830. doi: 10.1007/s00018-015-1956-5
Jones, J. M., and Meisler, M. H. (2014). Modeling human epilepsy by TALEN targeting of mouse sodium channel Scn8a. Genesis 52, 141–148. doi: 10.1002/dvg.22731
Juillerat, A., Dubois, G., Valton, J., Thomas, S., Stella, S., Maréchal, A., et al. (2014). Comprehensive analysis of the specificity of transcription activator-like effector nucleases. Nucleic Acids Res. 42, 5390–5402. doi: 10.1093/nar/gku155
Juillerat, A., Pessereau, C., Dubois, G., Guyot, V., Maréchal, A., Valton, J., et al. (2015). Optimized tuning of TALEN specificity using non-conventional RVDs. Sci. Rep. 5, 8150–8157. doi: 10.1038/srep08150
Kadyk, L. C., and Hartwell, L. H. (1992). Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 132, 387–402.
Karakikes, I., Stillitano, F., Nonnenmacher, M., Tzimas, C., Sanoudou, D., Termglinchan, V., et al. (2015). Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 6, 1–10. doi: 10.1038/ncomms7955
Kay, S., Hahn, S., Marois, E., Hause, G., and Bonas, U. (2007). A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science 318, 648–651. doi: 10.1126/science.1144956
Kearns, N. A., Genga, R. M. J., Enuameh, M. S., Garber, M., Wolfe, S. A., and Maehr, R. (2013). Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 141, 219–223. doi: 10.1242/dev.103341
Keatinge, M., Bui, H., Menke, A., Chen, Y.-C., Sokol, A. M., Bai, Q., et al. (2015). Glucocerebrosidase 1 deficient Danio reriomirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum. Mol. Genet. 24, 6640–6652. doi: 10.1093/hmg/ddv369
Kiani, S., Beal, J., Ebrahimkhani, M. R., Huh, J., Hall, R. N., Xie, Z., et al. (2014). CRISPR transcriptional repression devices and layered circuits in mammalian cells. Nat. Publishing Group 11, 723–726. doi: 10.1038/nmeth.2969
Kim, D., Bae, S., Park, J., Kim, E., Kim, S., Yu, H. R., et al. (2015). Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Meth. 12, 237–243. doi: 10.1038/nmeth.3284
Kim, E., Kim, S., Kim, D. H., Choi, B.-S., Choi, I.-Y., and Kim, J.-S. (2012). Precision genome engineering with programmable DNA-nicking enzymes. Genome Res. 22, 1327–1333. doi: 10.1101/gr.138792.112
Kim, Y.-G., Cha, J., and Chandrasegaran, S. (1996). Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. U.S.A. 93, 1156–1160. doi: 10.1073/pnas.93.3.1156
Kiskinis, E., Sandoe, J., Williams, L. A., Boulting, G. L., Moccia, R., Wainger, B. J., et al. (2014). Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795. doi: 10.1016/j.stem.2014.03.004
Kleinstiver, B. P., Pattanayak, V., Prew, M. S., Tsai, S. Q., Nguyen, N. T., Zheng, Z., et al. (2016). High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495. doi: 10.1038/nature16526
Kleinstiver, B. P., Prew, M. S., Tsai, S. Q., Nguyen, N. T., Topkar, V. V., Zheng, Z., et al. (2015). Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 33, 1293–1298. doi: 10.1038/nbt.3404
Koike-Yusa, H., Li, Y., Tan, E.-P., Velasco-Herrera, M. D. C., and Yusa, K. (2013). Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 32, 267–273. doi: 10.1038/nbt.2800
Konermann, S., Brigham, M. D., Trevino, A. E., Hsu, P. D., Heidenreich, M., Cong, L., et al. (2013). Optical control of mammalian endogenous transcription and epigenetic states. Nature 500, 472–476. doi: 10.1038/nature12466
Konermann, S., Brigham, M. D., Trevino, A. E., Joung, J., Abudayyeh, O. O., Barcena, C., et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. doi: 10.1038/nature14136
Koo, T., Lee, J., and Kim, J.-S. (2015). Measuring and reducing off-target activities of programmable nucleases including CRISPR-Cas9. Mol. Cells 38, 475–481. doi: 10.14348/molcells.2015.0103
Kovtun, I. V., Goellner, G., and McMurray, C. T. (2001). Structural features of trinucleotide repeats associated with DNA expansion. Biochem. Cell Biol. 79, 325–336. doi: 10.1139/o01-101
Krentz, N. A. J., Nian, C., and Lynn, F. C. (2014). TALEN/CRISPR-mediated eGFP knock-in add-on at the OCT4 locus does not impact differentiation of human embryonic stem cells towards endoderm. PLoS ONE 9:e114275. doi: 10.1371/journal.pone.0114275
Kuscu, C., Arslan, S., Singh, R., Thorpe, J., and Adli, M. (2014). Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 32, 677–683. doi: 10.1038/nbt.2916
Laganiere, J., Kells, A. P., Lai, J. T., Guschin, D., Paschon, D. E., Meng, X., et al. (2010). An engineered zinc finger protein activator of the endogenous glial cell line-derived neurotrophic factor gene provides functional neuroprotection in a rat model of parkinson's disease. J. Neurosci 30, 16469–16474. doi: 10.1523/JNEUROSCI.2440-10.2010
Lee, J. S., Kallehauge, T. B., Pedersen, L. E., and Kildegaard, H. F. (2015). Site-specific integration in CHO cells mediated by CRISPR/Cas9 and homology-directed DNA repair pathway. Sci. Rep. 5, 8572–8511. doi: 10.1038/srep08572
Lehman, C. W., Clemens, M., Worthylake, D. K., Trautman, J. K., and Carroll, D. (1993). Homologous and illegitimate recombination in developing Xenopus oocytes and eggs. Mol. Cell. Biol. 13, 6897–6906. doi: 10.1128/MCB.13.11.6897
Lenzi, J., De Santis, R., de Turris, V., Morlando, M., Laneve, P., Calvo, A., et al. (2015). ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis. Model. Mech. 8, 755–766. doi: 10.1242/dmm.020099
Li, D., Qiu, Z., Shao, Y., Chen, Y., Guan, Y., Liu, M., et al. (2013). Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 31, 681–683. doi: 10.1038/nbt.2661
Li, J., Shou, J., Guo, Y., Tang, Y., Wu, Y., Jia, Z., et al. (2015). Efficient inversions and duplications of mammalian regulatory DNA elements and gene clusters by CRISPR/Cas9. J. Mol. Cell Biol. 7, 284–298. doi: 10.1093/jmcb/mjv016
Li, T., Huang, S., Jiang, W. Z., Wright, D., Spalding, M. H., Weeks, D. P., et al. (2011). TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 39, 359–372. doi: 10.1093/nar/gkq704
Li, Y., Jiang, Y., Chen, H., Liao, W., Li, Z., Weiss, R., et al. (2015). Modular construction of mammalian gene circuits using TALE transcriptional repressors. Nat. Chem. Biol. 11, 207–213. doi: 10.1038/nchembio.1736
Li, Y., Moore, R., Guinn, M., and Bleris, L. (2012). Transcription activator-like effector hybrids for conditional control and rewiring of chromosomal transgene expression. Sci. Rep. 2, 1–7. doi: 10.1038/srep00897
Liang, L., Deng, L., Nguyen, S. C., Zhao, X., Maulion, C. D., Shao, C., et al. (2008). Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 36, 3297–3310. doi: 10.1093/nar/gkn184
Lieber, M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211. doi: 10.1146/annurev.biochem.052308.093131
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. doi: 10.1126/science.1181369
Lin, Y., Cradick, T. J., Brown, M. T., Deshmukh, H., Ranjan, P., Sarode, N., et al. (2014). CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 42, 7473–7485. doi: 10.1093/nar/gku402
Lin, Y., and Waldman, A. S. (2001). Promiscuous patching of broken chromosomes in mammalian cells with extrachromosomal DNA. Nucleic Acids Res. 29, 3975–3981. doi: 10.1093/nar/29.19.3975
Lindahl, T. (1993). Instability and decay of the primary structure of DNA. Nature 362, 709–715. doi: 10.1038/362709a0
Liu, G.-H., Suzuki, K., Qu, J., Sancho-Martinez, I., Yi, F., Li, M., et al. (2011). Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell 8, 688–694. doi: 10.1016/j.stem.2011.04.019
Liu, H., Chen, Y., Niu, Y., Zhang, K., Kang, Y., Ge, W., et al. (2014). TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell 14, 323–328. doi: 10.1016/j.stem.2014.01.018
Liu, Z., Zhou, X., Zhu, Y., Chen, Z.-F., Yu, B., Wang, Y., et al. (2014). Generation of a monkey with MECP2 mutations by TALEN-based gene targeting. Neurosci. Bull. 30, 381–386. doi: 10.1007/s12264-014-1434-8
Ma, A. C., Lee, H. B., Clark, K. J., and Ekker, S. C. (2013a). High efficiency in vivo genome engineering with a simplified 15-RVD GoldyTALEN design. PLoS ONE 8:e65259. doi: 10.1371/journal.pone.0065259
Ma, H., Reyes-Gutierrez, P., and Pederson, T. (2013b). Visualization of repetitive DNA sequences in human chromosomes with transcription activator-like effectors. Proc. Natl. Acad. Sci. U.S.A. 110, 21048–21053. doi: 10.1073/pnas.1319097110
MacArthur, D. G., Manolio, T. A., Dimmock, D. P., Rehm, H. L., Shendure, J., Abecasis, G. R., et al. (2014). Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476. doi: 10.1038/nature13127
Madabhushi, R., Gao, F., Pfenning, A. R., Pan, L., Yamakawa, S., Seo, J., et al. (2015). Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161, 1592–1605. doi: 10.1016/j.cell.2015.05.032
Madabhushi, R., Pan, L., and Tsai, L.-H. (2014). DNA damage and its links to neurodegeneration. Neuron 83, 266–282. doi: 10.1016/j.neuron.2014.06.034
Maeder, M. L., Angstman, J. F., Richardson, M. E., Linder, S. J., Cascio, V. M., Tsai, S. Q., et al. (2013a). Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol. 31, 1137–1142. doi: 10.1038/nbt.2726
Maeder, M. L., Linder, S. J., Cascio, V. M., Fu, Y., Ho, Q. H., and Joung, J. K. (2013b). CRISPR RNA-guided activation of endogenous human genes. Nat. Meth. 10, 977–979. doi: 10.1038/nmeth.2598
Maeder, M. L., Linder, S. J., Reyon, D., Angstman, J. F., Fu, Y., Sander, J. D., et al. (2013c). Robust, synergistic regulation of human gene expression using TALE activators. Nat. Meth. 10, 243–245. doi: 10.1038/nmeth.2366
Mahfouz, M. M., Li, L., Piatek, M., Fang, X., Mansour, H., Bangarusamy, D. K., et al. (2011). Targeted transcriptional repression using a chimeric TALE-SRDX repressor protein. Plant Mol. Biol. 78, 311–321. doi: 10.1007/s11103-011-9866-x
Mali, P., Aach, J., Stranges, P. B., Esvelt, K. M., Moosburner, M., Kosuri, S., et al. (2013a). CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838. doi: 10.1038/nbt.2675
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013b). RNA-guided human genome engineering via Cas9. Science 339, 823–826. doi: 10.1126/science.1232033
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
Maryon, E., and Carroll, D. (1991). Involvement of single-stranded tails in homologous recombination of DNA injected into Xenopus laevis oocyte nuclei. Mol. Cell. Biol. 11, 3268–3277. doi: 10.1128/MCB.11.6.3268
Masland, R. H. (2001). Neuronal diversity in the retina. Curr. Opin. Neurobiol. 11, 431–436. doi: 10.1016/S0959-4388(00)00230-0
Masland, R. H. (2004). Neuronal cell types. Curr. Biol. 14, R497–R500. doi: 10.1016/j.cub.2004.06.035
McKinnon, P. J. (2013). Maintaining genome stability in the nervous system. Nat. Rev. Neurosci. 16, 1523–1529. doi: 10.1038/nn.3537
McVey, M., and Lee, S. E. (2008). MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 24, 529–538. doi: 10.1016/j.tig.2008.08.007
Meckler, J. F., Bhakta, M. S., Kim, M.-S., Ovadia, R., Habrian, C. H., Zykovich, A., et al. (2013). Quantitative analysis of TALE-DNA interactions suggests polarity effects. Nucleic Acids Res. 41, 4118–4128. doi: 10.1093/nar/gkt085
Mehrabian, M., Brethour, D., MacIsaac, S., Kim, J. K., Gunawardana, C. G., Wang, H., et al. (2014). CRISPR-Cas9-based knockout of the prion protein and its effect on the proteome. PLoS ONE 9:e114594. doi: 10.1371/journal.pone.0114594
Mendenhall, E. M., Williamson, K. E., Reyon, D., Zou, J. Y., Ram, O., Joung, J. K., et al. (2013). Locus-specific editing of histone modifications at endogenous enhancers. Nat. Biotechnol. 31, 1133–1136. doi: 10.1038/nbt.2701
Mercer, A. C., Gaj, T., Sirk, S. J., Lamb, B. M., and Barbas, C. F. III (2014). Regulation of endogenous human gene expression by ligand-inducible TALE transcription factors. ACS Synth. Biol. 3, 723–730. doi: 10.1021/sb400114p
Miller, D. G., Petek, L. M., and Russell, D. W. (2004). Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 36, 767–773. doi: 10.1038/ng1380
Miller, J. C., Holmes, M. C., Wang, J., Guschin, D. Y., Lee, Y.-L., Rupniewski, I., et al. (2007). An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 25, 778–785. doi: 10.1038/nbt1319
Miller, J. C., Tan, S., Qiao, G., Barlow, K. A., Wang, J., Xia, D. F., et al. (2011). A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 29, 143–148. doi: 10.1038/nbt.1755
Miyanari, Y., Ziegler-Birling, C., and Torres-Padilla, M.-E. (2013). Live visualization of chromatin dynamics with fluorescent TALEs. Nat. Struct. Mol. Biol. 20, 1321–1324. doi: 10.1038/nsmb.2680
Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M., and Valen, E. (2014). CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 42, W401–W407. doi: 10.1093/nar/gku410
Moreno-Mateos, M. A., Vejnar, C. E., Beaudoin, J.-D., Fernandez, J. P., Mis, E. K., Khokha, M. K., et al. (2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Meth. 12, 982–988. doi: 10.1038/nmeth.3543
Morris, M. J., and Monteggia, L. M. (2014). Role of DNA methylation and the DNA methyltransferases in learning and memory. Dialogues Clin. Neurosci. 16, 359–371.
Moscou, M. J., and Bogdanove, A. J. (2009). A simple cipher governs DNA recognition by TAL effectors. Science 326, 1501–1501. doi: 10.1126/science.1178817
Mussolino, C., and Cathomen, T. (2012). TALE nucleases: tailored genome engineering made easy. Curr. Opin. Biotechnol. 23, 644–650. doi: 10.1016/j.copbio.2012.01.013
Neff, K. L., Argue, D. P., Ma, A. C., Lee, H. B., Clark, K. J., and Ekker, S. C. (2013). Mojo Hand, a TALEN design tool for genome editing applications. BMC Bioinform. 14:1. doi: 10.1186/1471-2105-14-1
Nelson, S. B., Sugino, K., and Hempel, C. M. (2006). The problem of neuronal cell types: a physiological genomics approach. Trends Neurosci. 29, 339–345. doi: 10.1016/j.tins.2006.05.004
Niu, Y., Shen, B., Cui, Y., Chen, Y., Wang, J., Wang, L., et al. (2014). Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156, 836–843. doi: 10.1016/j.cell.2014.01.027
Nouspikel, T., and Hanawalt, P. C. (2000). Terminally differentiated human neurons repair transcribed genes but display attenuated global DNA repair and modulation of repair gene expression. Mol. Cell. Biol. 20, 1562–1570. doi: 10.1128/MCB.20.5.1562-1570.2000
Nouspikel, T., and Hanawalt, P. C. (2002). DNA repair in terminally differentiated cells. DNA Repair (Amst). 1, 59–75. doi: 10.1016/S1568-7864(01)00005-2
O'Geen, H., Henry, I. M., Bhakta, M. S., and Meckler, J. F. (2015). A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 43, 3389–3404. doi: 10.1093/nar/gkv137
O'Geen, H., and Yu, A. S. (2015). How specific is CRISPR/Cas9 really? Curr. Opin. Chem. Biol. 29, 72–78. doi: 10.1016/j.cbpa.2015.10.001
Orii, K. E., Lee, Y., Kondo, N., and McKinnon, P. J. (2006). Selective utilization of nonhomologous end-joining and homologous recombination DNA repair pathways during nervous system development. Proc. Natl. Acad. Sci. U.S.A. 103, 10017–10022. doi: 10.1073/pnas.0602436103
Orr-Weaver, T. L., Szostak, J. W., and Rothstein, R. J. (1981). Yeast transformation: a model system for the study of recombination. Proc. Natl. Acad. Sci. U.S.A. 78, 6354–6358. doi: 10.1073/pnas.78.10.6354
Osborn, M. J., Starker, C. G., McElroy, A. N., Webber, B. R., Riddle, M. J., Xia, L., et al. (2013). TALEN-based gene correction for epidermolysis bullosa. Mol. Ther. 21, 1151–1159. doi: 10.1038/mt.2013.56
Ousterout, D. G., Perez-Pinera, P., Thakore, P. I., Kabadi, A. M., Brown, M. T., Qin, X., et al. (2013). Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol. Ther. 21, 1718–1726. doi: 10.1038/mt.2013.111
Pan, L., Penney, J., and Tsai, L.-H. (2014). Chromatin regulation of DNA damage repair and genome integrity in the central nervous system. J. Mol. Biol. 426, 3376–3388. doi: 10.1016/j.jmb.2014.08.001
Panda, S. K., Wefers, B., Ortiz, O., Floss, T., Schmid, B., Haass, C., et al. (2013). Highly efficient targeted mutagenesis in mice using TALENs. Genetics 195, 703–713. doi: 10.1534/genetics.113.156570
Pattanayak, V., Lin, S., Guilinger, J. P., Ma, E., Doudna, J. A., and Liu, D. R. (2013). High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 31, 839–837. doi: 10.1038/nbt.2673
Pavletich, N. P., and Pabo, C. O. (1991). Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science 252, 809–817. doi: 10.1126/science.2028256
Perez, E. E., Wang, J., Miller, J. C., Jouvenot, Y., Kim, K. A., Liu, O., et al. (2008). Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 26, 808–816. doi: 10.1038/nbt1410
Perez-Pinera, P., Kocak, D. D., Vockley, C. M., Adler, A. F., Kabadi, A. M., Polstein, L. R., et al. (2013a). RNA-guided gene activation by CRISPR-Cas9–based transcription factors. Nat. Meth. 10, 973–976. doi: 10.1038/nmeth.2600
Perez-Pinera, P., Ousterout, D. G., Brunger, J. M., Farin, A. M., Glass, K. A., Guilak, F., et al. (2013b). Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat. Meth. 10, 239–242. doi: 10.1038/nmeth.2361
Phillips, J. W., and Morgan, W. F. (1994). Illegitimate recombination induced by DNA double-strand breaks in a mammalian chromosome. Mol. Cell. Biol. 14, 5794–5803. doi: 10.1128/MCB.14.9.5794
Phillips-Cremins, J. E., Sauria, M. E. G., Sanyal, A., Gerasimova, T. I., Lajoie, B. R., Bell, J. S. K., et al. (2013). Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295. doi: 10.1016/j.cell.2013.04.053
Platt, R. J., Chen, S., Zhou, Y., Yim, M. J., Swiech, L., Kempton, H. R., et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. doi: 10.1016/j.cell.2014.09.014
Polstein, L. R., Perez-Pinera, P., Kocak, D. D., Vockley, C. M., Bledsoe, P., Song, L., et al. (2015). Genome-wide specificity of DNA binding, gene regulation, and chromatin remodeling by TALE- and CRISPR/Cas9-based transcriptional activators. Genome Res. 25, 1158–1169. doi: 10.1101/gr.179044.114
Ponomareva, O. Y., Holmen, I. C., Sperry, A. J., Eliceiri, K. W., and Halloran, M. C. (2014). Calsyntenin-1 regulates axon branching and endosomal trafficking during sensory neuron development in vivo. J. Neurosci. 34, 9235–9248. doi: 10.1523/JNEUROSCI.0561-14.2014
Potter, T. A., Zeff, R. A., Frankel, W., and Rajan, T. V. (1987). Mitotic recombination between homologous chromosomes generates H-2 somatic cell variants in vitro. Proc. Natl. Acad. Sci. U.S.A. 84, 1634–1637. doi: 10.1073/pnas.84.6.1634
Puchta, H., Dujon, B., and Hohn, B. (1993). Homologous recombination in plant cells is enhanced by in vivo induction of double strand breaks into DNA by a site-specific endonuclease. Nucleic Acids Res. 21, 5034–5040. doi: 10.1093/nar/21.22.5034
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., et al. (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. doi: 10.1016/j.cell.2013.02.022
Rai, N., Ferreiro, A., Neckelmann, A., Soon, A., Yao, A., Siegel, J., et al. (2015). RiboTALE: a modular, inducible system for accurate gene expression control. Nat. Publishing Group 5, 1–11. doi: 10.1038/srep10658
Ramirez, C. L., Certo, M. T., Mussolino, C., Goodwin, M. J., Cradick, T. J., McCaffrey, A. P., et al. (2012). Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res. 40, 5560–5568. doi: 10.1093/nar/gks179
Ran, F. A., Cong, L., Yan, W. X., Scott, D. A., Gootenberg, J. S., Kriz, A. J., et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191. doi: 10.1038/nature14299
Ran, F. A., Hsu, P. D., Lin, C.-Y., Gootenberg, J. S., Konermann, S., Trevino, A. E., et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389. doi: 10.1016/j.cell.2013.08.021
Rao, K. S. (1993). Genomic damage and its repair in young and aging brain. Mol. Neurobiol. 7, 23–48. doi: 10.1007/BF02780607
Rao, S. S. P., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Reul, J. M. H. M. (2014). Making memories of stressful events: a journey along epigenetic, gene transcription, and signaling pathways. Front. Psychiatry 5:5. doi: 10.3389/fpsyt.2014.00005
Richard, G. F., Viterbo, D., Khanna, V., Mosbach, V., Castelain, L., and Dujon, B. (2014). Highly specific contractions of a single CAG/CTG trinucleotide repeat by TALEN in yeast. PLoS ONE 9:e95611. doi: 10.1371/journal.pone.0095611
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423. doi: 10.1038/gim.2015.30
Rong, Y. S., and Golic, K. G. (2001). A targeted gene knockout in Drosophila. Genetics 157, 1307–1312.
Roth, D. B., and Wilson, J. H. (1985). Relative rates of homologous and nonhomologous recombination in transfected DNA. Proc. Natl. Acad. Sci. U.S.A. 82, 3355–3359. doi: 10.1073/pnas.82.10.3355
Roth, D. B., and Wilson, J. H. (1986). Nonhomologous recombination in mammalian cells: role for short sequence homologies in the joining reaction. Mol. Cell. Biol. 6, 4295–4304. doi: 10.1128/MCB.6.12.4295
Rouet, P., Smih, F., and Jasin, M. (1994). Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 91, 6064–6068. doi: 10.1073/pnas.91.13.6064
Rulten, S. L., Rotheray, A., Green, R. L., Grundy, G. J., Moore, D. A. Q., Gómez-Herreros, F., et al. (2014). PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 42, 307–314. doi: 10.1093/nar/gkt835
Russo, S. J., and Nestler, E. J. (2013). The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 14, 609–625. doi: 10.1038/nrn3381
Sanders, L. H., Laganière, J., Cooper, O., Mak, S. K., Vu, B. J., Huang, Y. A., et al. (2014). LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson's disease patients: reversal by gene correction. Neurobiol. Dis. 62, 381–386. doi: 10.1016/j.nbd.2013.10.013
Schmid, B., Hruscha, A., Hogl, S., Banzhaf-Strathmann, J., Strecker, K., van der Zee, J., et al. (2013). Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proc. Natl. Acad. Sci. U.S.A. 110, 4986–4991. doi: 10.1073/pnas.1218311110
Scott, J. N. F., Kupinski, A. P., and Boyes, J. (2014a). Targeted genome regulation and modification using transcription activator-like effectors. FEBS J. 281, 4583–4597. doi: 10.1111/febs.12973
Scott, J. N. F., Kupinski, A. P., Kirkham, C. M., Tuma, R., and Boyes, J. (2014b). TALE proteins bind to both active and inactive chromatin. Biochem. J. 458, 153–158. doi: 10.1042/BJ20131327
Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelsen, T. S., et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. doi: 10.1126/science.1247005
Sharma, R., Anguela, X. M., Doyon, Y., Wechsler, T., DeKelver, R. C., Sproul, S., et al. (2015). In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 126, 1777–1784. doi: 10.1182/blood-2014-12-615492
Shen, B., Zhang, W., Zhang, J., Zhou, J., Wang, J., Chen, L., et al. (2014). Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Meth. 11, 399–402. doi: 10.1038/nmeth.2857
Shepherd, G. M. G. (2013). Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 14, 278–291. doi: 10.1038/nrn3469
Shin, J., Chen, J., and Solnica-Krezel, L. (2014a). Efficient homologous recombination-mediated genome engineering in zebrafish using TALE nucleases. Development 141, 3807–3818. doi: 10.1242/dev.108019
Shin, J., Ming, G. L., and Song, H. (2014b). DNA modifications in the mammalian brain. Philos. Trans. R. Soc. B Biol. Sci. 369, 20130512. doi: 10.1098/rstb.2013.0512
Shin, L. M., and Liberzon, I. (2009). The Neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology 35, 169–191. doi: 10.1038/npp.2009.83
Shinohara, A., Ogawa, H., and Ogawa, T. (1992). Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 69, 457–470. doi: 10.1016/0092-8674(92)90447-K
Slaymaker, I. M., Gao, L., Zetsche, B., Scott, D. A., Yan, W. X., and Zhang, F. (2016). Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88. doi: 10.1126/science.aad5227
Smih, F., Rouet, P., Romanienko, P. J., and Jasin, M. (1995). Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res. 23, 5012–5019. doi: 10.1093/nar/23.24.5012
Smith, J., Bibikova, M., Whitby, F. G., Reddy, A. R., Chandrasegaran, S., and Carroll, D. (2000). Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 28, 3361–3369. doi: 10.1093/nar/28.17.3361
Smithies, O., Gregg, R. G., Boggs, S. S., Koralewski, M. A., and Kucherlapati, R. S. (1985). Insertion of DNA sequences into the human chromosomal β-globin locus by homologous recombination. Nature 317, 230–234. doi: 10.1038/317230a0
Soldner, F., Laganière, J., Cheng, A. W., Hockemeyer, D., Gao, Q., Alagappan, R., et al. (2011). Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell 146, 318–331. doi: 10.1016/j.cell.2011.06.019
Sonoda, E., Hochegger, H., Saberi, A., Taniguchi, Y., and Takeda, S. (2006). Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair (Amst). 5, 1021–1029. doi: 10.1016/j.dnarep.2006.05.022
Sorek, R., Kunin, V., and Hugenholtz, P. (2008). CRISPR–a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 6, 181–186. doi: 10.1038/nrmicro1793
Stevens, C. F. (1998). Neuronal diversity: too many cell types for comfort? Curr. Biol. 8, R708–R710.
Straub, C., Granger, A. J., Saulnier, J. L., and Sabatini, B. L. (2014). CRISPR/Cas9-Mediated Gene Knock-Down in Post-Mitotic Neurons. PLoS ONE 9:e105584. doi: 10.1371/journal.pone.0105584
Suberbielle, E., Djukic, B., Evans, M., Kim, D. H., Taneja, P., Wang, X., et al. (2015). DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 6, 8897. doi: 10.1038/ncomms9897
Suberbielle, E., Sanchez, P. E., Kravitz, A. V., Wang, X., Ho, K., Eilertson, K., et al. (2013). Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 16, 613–621. doi: 10.1038/nn.3356
Sugio, A., Yang, B., Zhu, T., and White, F. F. (2007). Two type III effector genes of Xanthomonas oryzae pv. oryzae control the induction of the host genes OsTFIIAγ1 and OsTFX1 during bacterial blight of rice. Proc. Natl. Acad. Sci. U.S.A. 104, 10720–10725. doi: 10.1073/pnas.0701742104
Sugo, N., Aratani, Y., Nagashima, Y., Kubota, Y., and Koyama, H. (2000). Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J. 19, 1397–1404. doi: 10.1093/emboj/19.6.1397
Sullivan, P. F., Daly, M. J., and O'Donovan, M. (2012). Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat. Publishing Group 13, 537–551. doi: 10.1038/nrg3240
Sung, Y. H., Baek, I.-J., Kim, D. H., Jeon, J., Lee, J., Lee, K., et al. (2013). Knockout mice created by TALEN-mediated gene targeting. Nat. Biotechnol. 31, 23–24. doi: 10.1038/nbt.2477
Swiech, L., Heidenreich, M., Banerjee, A., Habib, N., Li, Y., Trombetta, J., et al. (2014). In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 33, 99–103. doi: 10.1038/nbt.3055
Szczepek, M., Brondani, V., Büchel, J., Serrano, L., and Cathomen, T. (2007). Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol. 25, 786–793. doi: 10.1038/nbt1317
Takata, M., Sasaki, M. S., Sonoda, E., Morrison, C., Hashimoto, M., Utsumi, H., et al. (1998). Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 17, 5497–5508. doi: 10.1093/emboj/17.18.5497
Takizawa, T., and Meshorer, E. (2008). Chromatin and nuclear architecture in the nervous system. Trends Neurosci. 31, 343–352. doi: 10.1016/j.tins.2008.03.005
Tebas, P., Stein, D., Tang, W. W., Frank, I., Wang, S. Q., Lee, G., et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 370, 901–910. doi: 10.1056/NEJMoa1300662
Tesson, L., Usal, C., Ménoret, S., Leung, E., Niles, B. J., Remy, S., et al. (2011). Knockout rats generated by embryo microinjection of TALENs. Nat. Biotechnol. 29, 695–696. doi: 10.1038/nbt.1940
Thakore, P. I., D'Ippolito, A. M., Song, L., Safi, A., Shivakumar, N. K., Kabadi, A. M., et al. (2015). Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Meth. 12, 1143–1149. doi: 10.1038/nmeth.3630
Thomas, K. R., and Capecchi, M. R. (1986). Introduction of homologous DNA sequences into mammalian cells induces mutations in the cognate gene. Nature 324, 34–38. doi: 10.1038/324034a0
Truong, D.-J. J., Kühner, K., Kühn, R., Werfel, S., Engelhardt, S., Wurst, W., et al. (2015). Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 43, 6450–6458. doi: 10.1093/nar/gkv601
Tsai, S. Q., Wyvekens, N., Khayter, C., Foden, J. A., Thapar, V., Reyon, D., et al. (2014a). Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 32, 569–576. doi: 10.1038/nbt.2908
Tsai, S. Q., Zheng, Z., Nguyen, N. T., Liebers, M., Topkar, V. V., Thapar, V., et al. (2014b). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197. doi: 10.1038/nbt.3117
Tsankova, N., Renthal, W., Kumar, A., and Nestler, E. J. (2007). Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 8, 355–367. doi: 10.1038/nrn2132
Urnov, F. D., Miller, J. C., Lee, Y.-L., Beausejour, C. M., Rock, J. M., Augustus, S., et al. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435, 646–651. doi: 10.1038/nature03556
Van Dyck, E., Stasiak, A. Z., Stasiak, A., and West, S. C. (1999). Binding of double-strand breaks in DNA by human Rad52 protein. Nature 398, 728–731. doi: 10.1038/19560
Vannocci, T., Faggianelli, N., Zaccagnino, S., Rosa, della, I., Adinolfi, S., and Pastore, A. (2015). A new cellular model to follow Friedreich's ataxia development in a time-resolved way. Dis. Model. Mech. 8, 711–719. doi: 10.1242/dmm.020545
Vialou, V., Feng, J., Robison, A. J., and Nestler, E. J. (2013). Epigenetic mechanisms of depression and antidepressant action. Annu. Rev. Pharmacol. Toxicol. 53, 59–87. doi: 10.1146/annurev-pharmtox-010611-134540
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S. W., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. doi: 10.1016/j.celrep.2014.03.019
Wang, J., DeClercq, J. J., Hayward, S. B., Li, P. W.-L., Shivak, D. A., Gregory, P. D., et al. (2016). Highly efficient homology-driven genome editing in human T cells by combining zinc-finger nuclease mRNA and AAV6 donor delivery. Nucleic Acids Res. 44, e30. doi: 10.1093/nar/gkv1121
Wang, W.-Y., Pan, L., Su, S. C., Quinn, E. J., Sasaki, M., Jimenez, J. C., et al. (2013). Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 16, 1383–1391. doi: 10.1038/nn.3514
Wang, X., Wang, Y., Wu, X., Wang, J., Wang, Y., Qiu, Z., et al. (2015). Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 33, 175–178. doi: 10.1038/nbt.3127
Waser, J., Hübscher, U., Kuenzle, C. C., and Spadari, S. (1979). DNA polymerase beta from brain neurons is a repair enzyme. Eur. J. Biochem. 97, 361–368. doi: 10.1111/j.1432-1033.1979.tb13122.x
Wen, Z., Nguyen, H. N., Guo, Z., Lalli, M. A., Wang, X., Su, Y., et al. (2016). Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418. doi: 10.1038/nature13716
Wilen, C. B., Wang, J., Tilton, J. C., Miller, J. C., Kim, K. A., Rebar, E. J., et al. (2011). Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 7:e1002020. doi: 10.1371/journal.ppat.1002020
Woodruff, G., Young, J. E., Martinez, F. J., Buen, F., Gore, A., Kinaga, J., et al. (2013). The Presenilin-1 deltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 5, 974–985. doi: 10.1016/j.celrep.2013.10.018
Wright, A. V., Nuñez, J. K., and Doudna, J. A. (2016). Biology and Applications of CRISPR Systems: Harnessing Nature's Toolbox for Genome Engineering. Cell 164, 29–44. doi: 10.1016/j.cell.2015.12.035
Wright, A. V., Sternberg, S. H., Taylor, D. W., Staahl, B. T., Bardales, J. A., Kornfeld, J. E., et al. (2015). Rational design of a split-Cas9 enzyme complex. Proc. Natl. Acad. Sci. U.S.A. 112, 2984–2989. doi: 10.1073/pnas.1501698112
Wu, H., Yang, W.-P. P., and Barbas, C. F. III (1995). Building zinc fingers by selection: toward a therapeutic application. Proc. Natl. Acad. Sci. U.S.A. 92, 344–348. doi: 10.1073/pnas.92.2.344
Wu, X., Scott, D. A., Kriz, A. J., Chiu, A. C., Hsu, P. D., Dadon, D. B., et al. (2014). Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 32, 670–676. doi: 10.1038/nbt.2889
Wyvekens, N., Topkar, V. V., Khayter, C., Joung, J. K., and Tsai, S. Q. (2015). Dimeric CRISPR RNA-guided FokI-dCas9 nucleases directed by truncated gRNAs for highly specific genome editing. Hum. Gene Ther. 26, 425–431. doi: 10.1089/hum.2015.084
Xie, F., Ye, L., Chang, J. C., Beyer, A. I., Wang, J., Muench, M. O., et al. (2014a). Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 24, 1526–1533. doi: 10.1101/gr.173427.114
Xie, S., Shen, B., Zhang, C., Huang, X., and Zhang, Y. (2014b). sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS ONE 9:e100448. doi: 10.1371/journal.pone.0100448
Yang, B., and White, F. F. (2004). Diverse members of the AvrBs3/PthA family of type III effectors are major virulence determinants in bacterial blight disease of rice. Mol. Plant Microbe Interact. 17, 1192–1200. doi: 10.1094/MPMI.2004.17.11.1192
Yang, L., Grishin, D., Wang, G., Aach, J., Zhang, C.-Z., Chari, R., et al. (2014). Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nat. Commun. 5, 5507. doi: 10.1038/ncomms6507
Yao, Y., Cui, X., Al-Ramahi, I., Sun, X., Li, B., Hou, J., et al. (2015). A striatal-enriched intronic GPCR modulates huntingtin levels and toxicity. Elife 4, 26714. doi: 10.7554/eLife.05449
Zalatan, J. G., Lee, M. E., Almeida, R., Gilbert, L. A., Whitehead, E. H., La Russa, M., et al. (2015). Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 160, 339–350. doi: 10.1016/j.cell.2014.11.052
Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., et al. (2015a). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 1–14. doi: 10.1016/j.cell.2015.09.038
Zetsche, B., Volz, S. E., and Zhang, F. (2015b). A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 33, 139–142. doi: 10.1038/nbt.3149
Zhang, F., Cong, L., Lodato, S., Kosuri, S., Church, G. M., and Arlotta, P. (2011). Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 29, 149–153. doi: 10.1038/nbt.1775
Zu, Y., Tong, X., Wang, Z., Liu, D., Pan, R., Li, Z., et al. (2013). TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat. Meth. 10, 329–331. doi: 10.1038/nmeth.2374
Keywords: ZFN, TALE, TALEN, CRISPR, Cas9, genome engineering, neuroscience
Citation: Lee HB, Sundberg BN, Sigafoos AN and Clark KJ (2016) Genome Engineering with TALE and CRISPR Systems in Neuroscience. Front. Genet. 7:47. doi: 10.3389/fgene.2016.00047
Received: 11 January 2016; Accepted: 16 March 2016;
Published: 06 April 2016.
Edited by:
Robert Gerlai, University of Toronto, CanadaCopyright © 2016 Lee, Sundberg, Sigafoos and Clark. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karl J. Clark, Y2xhcmsua2FybEBtYXlvLmVkdQ==