
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 27 October 2015
Sec. Computational Genomics
Volume 6 - 2015 | https://doi.org/10.3389/fgene.2015.00322
This article is part of the Research Topic Systems biology of transcription regulation View all 14 articles
 Joshua L. Payne1,2*
Joshua L. Payne1,2* Andreas Wagner1,2,3
Andreas Wagner1,2,3Robustness is the invariance of a phenotype in the face of environmental or genetic change. The phenotypes produced by transcriptional regulatory circuits are gene expression patterns that are to some extent robust to mutations. Here we review several causes of this robustness. They include robustness of individual transcription factor binding sites, homotypic clusters of such sites, redundant enhancers, transcription factors, redundant transcription factors, and the wiring of transcriptional regulatory circuits. Such robustness can either be an adaptation by itself, a byproduct of other adaptations, or the result of biophysical principles and non-adaptive forces of genome evolution. The potential consequences of such robustness include complex regulatory network topologies that arise through neutral evolution, as well as cryptic variation, i.e., genotypic divergence without phenotypic divergence. On the longest evolutionary timescales, the robustness of transcriptional regulation has helped shape life as we know it, by facilitating evolutionary innovations that helped organisms such as flowering plants and vertebrates diversify.
Robustness is the invariance of a phenotype in the face of environmental or genetic change. The phenotypes of living systems exhibit robustness at multiple scales of organization, ranging from the structural properties of macromolecules (Bloom et al., 2005; Wagner, 2008) to the preferred carbon sources of entire metabolisms (Samal et al., 2010). An immense body of work has focused on elucidating the mechanisms of robustness in living systems (reviewed in de Visser et al., 2003; Kitano, 2004; Stelling et al., 2004; Wagner, 2005; Masel and Siegal, 2009). Here we highlight a subset of this work, specifically those studies that have addressed the mechanisms of mutational robustness in transcriptional regulation.
Transcriptional regulation is fundamental to the control of gene expression. It allows cells to respond to environmental signals (Ptashne and Gann, 2002), such as hormones or sugars, and it drives fundamental behavioral and developmental processes, such as mating in yeast (Tsong et al., 2006) and embryonic patterning in fruit flies (Lawrence, 1992). Transcriptional regulation is largely carried out by transcription factors (TFs), proteins that bind short DNA sequences—TF binding sites—in the promoters or enhancers of genes. Such binding may induce or repress gene expression by promoting or inhibiting the recruitment of RNA polymerase. Given the fundamental importance of when and where genes are expressed, it is crucial that transcriptional regulation is robust to perturbation.
Genetic perturbations that may affect transcriptional regulation occur in both cis and in trans. They include point mutations in TF binding sites, which may impact transcriptional regulation by changing the affinity of a binding site for its cognate TF. They also include the insertion or deletion of large segments of DNA within promoters or enhancers, which may add or remove one or more regulatory interactions from a regulatory circuit. And they include changes to the amino acid sequence of the activation or DNA binding domains of a TF, which may alter the entire binding repertoire of the TF. Such perturbations can be deleterious, as shown by the numerous disease-associated mutations within gene regulatory regions and within genes that encode TFs (Vaquerizas et al., 2009; Maurano et al., 2012; Lee and Young, 2013).
Transcriptional regulation is not only subject to a litany of genetic insults, it is also remarkably robust to these insults (Weirauch and Hughes, 2010). Gene expression phenotypes are often insensitive to mutations in TF binding sites (Kasowski et al., 2010; Kwasnieski et al., 2012), to the turnover of regulatory control from one TF to another (Ludwig et al., 2000; Odom et al., 2007), to variation in gene expression levels (Garfield et al., 2013), and even to the rewiring of entire transcriptional regulatory circuits (Tsong et al., 2006; Isalan et al., 2008; Swanson et al., 2011). Here, we review the mechanisms that underlie this mutational robustness (Figure 1). Reviews of the equally important topic of robustness to environmental perturbations can be found elsewhere (Eldar et al., 2004; Alon, 2007; Macneil and Walhout, 2011; Silva-Rocha and de Lorenzo, 2010), as can primary literature on the contribution of post-transcriptional regulation to robust gene expression (McManus et al., 2014).
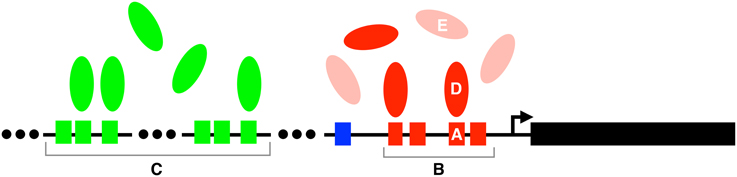
Figure 1. Mechanisms of mutational robustness in transcriptional regulation. Robustness can be conferred by (A) individual transcription factor binding sites, (B) homotypic clusters of such sites, (C) redundant enhancers, (D) individual transcription factors, and (E) redundant transcription factors. Small colored boxes represent transcription factor binding sites, ellipsoids represent transcription factors, and the arrow represents the transcription start site of the gene indicated by the large black rectangle. The lightly shaded ellipses in (E) represent paralogs of the transcription factors (red ellipses) in (D). Both the red and green transcription factors regulate the expression of the black gene. These regulatory interactions are part of a larger regulatory network, whose structural properties can also influence the robustness of transcriptional regulation.
TF binding sites are short DNA sequences (6–12 base pairs) that bind TFs to regulate gene expression. On the one hand, mutations in TF binding sites can be deleterious, as shown by their involvement in human disease (Pomerantz et al., 2009; Musunuru et al., 2010; Harismendy et al., 2011), including cancer (Khurana et al., 2013; Weinhold et al., 2014; Katainen et al., 2015; Melton et al., 2015). For instance, of 2931 disease-associated single nucleotide polymorphisms located within regulatory DNA, 93.2% fall within TF binding sites (Maurano et al., 2012). On the other hand, cross-species comparisons of regulatory regions often uncover variation in TF binding sites without obvious differences in the gene expression patterns that are driven by these sites (Ludwig et al., 2000; Odom et al., 2007). In addition, within-species variation in TF binding sites is common (Garfield et al., 2012; Spivakov et al., 2012; Arbiza et al., 2013; Khurana et al., 2013; Zheng et al., 2011), and such inter-individual differences often do not affect the expression level of target genes (Kasowski et al., 2010; Zheng et al., 2010).
The simplest cause of such mutational robustness is that individual binding sites are themselves robust to mutation. That is, they can often tolerate mutations without losing the ability to bind their cognate TFs. This results from two properties of TFs: (1) They typically bind dozens, if not hundreds of distinct DNA sequences (Sengupta et al., 2002; Berger et al., 2006; Badis et al., 2009; Wong et al., 2013) and (2) these sequences are almost always organized as large genotype networks in the space of all possible binding sites (Payne and Wagner, 2014). In such a genotype network, nodes represent DNA sequences that bind a particular TF and edges connect nodes if their corresponding sequences differ by a single small DNA mutation. Genotype networks confer robustness, because a mutation to any site in a TF's binding site repertoire is likely to yield another site that is also in the repertoire, thus preserving binding. Moreover, the binding affinities of neighboring sites in a genotype network are strongly correlated, indicating that a site's affinity for a TF is also robust to mutation. This is important, because mutations that affect binding affinity may impact the expression of a TF's target genes (Kasowski et al., 2010; Shultzaberger et al., 2010; Sharon et al., 2012). In addition, it is worth highlighting that the very short length of TF binding sites itself confers mutational robustness: Even though longer sites may offer greater specificity, they are also more susceptible to mutational disruption (Stewart et al., 2012).
Regulatory regions often contain multiple binding sites for the same TF (Johnson et al., 1979; Giniger and Ptashne, 1988; Carey et al., 1990; Thanos and Maniatis, 1995; Wasserman and Fickett, 1998; Krivan and Wasserman, 2001; Berman et al., 2002; Ezer et al., 2014). Such homotypic clusters of binding sites are common in both prokaryotic and eukaryotic organisms, including bacteria (Gama-Castro et al., 2011), fruit flies (Lifanov et al., 2003), and humans (Gotea et al., 2010). For example, in humans, 62% of promoters and roughly 40% of 487 experimentally-validated developmental enhancers contain such clusters (Gotea et al., 2010). The benefits of homotypic clusters include threshold-dependent (Lebrecht et al., 2005) and graded (Giogetti et al., 2010) transcriptional responses to input signals.
An additional benefit of homotypic clusters is mutational robustness. Experiments with high-throughput promoter screens show that increasing the number of binding sites within a homotypic cluster has a saturating effect on gene expression, such that increasing the number of sites beyond a threshold results in no further impact on gene expression (Sharon et al., 2012; Smith et al., 2013). This apparent redundancy of a subset of a cluster's binding sites can provide robustness to mutation. For example, the promoter of the mouse HTF9 genes contains a homotypic cluster of binding sites for the TF Sp1, and deletion of all but one of these sites has no effect on the expression of HTF9 genes (Somma et al., 1991). Similarly, mutations in a binding site of the human TF PU.1 are less likely to impact gene expression if a second, non-mutated site is nearby (Kilpinen et al., 2013). This finding echoes earlier observations made in an analysis of polymorphic TF binding sites in Drosophila melanogaster, which found that sites were more likely to tolerate deleterious mutations if they were located nearby other sites for the same TF (Spivakov et al., 2012).
Enhancers are DNA sequences (50–1500 base pair) that bind one or more TFs to activate the transcription of genes, often in a cell-specific manner (Banerji et al., 1981; de Villiers et al., 1982; Gillies et al., 1983; Small et al., 1996; Levine et al., 2014; Shlyueva et al., 2014). Enhancers often target genes across long chromosomal distances, but typically within well-defined structural units called topologically associating domains (Dixon et al., 2012). Many genes are regulated by more than one enhancer, as exemplified by the gap genes in Drosophila, which control anterior-posterior patterning in the developing embryo. For example, the gap genes hunchback, Kruppel, and knirps are each regulated by two distinct enhancers that work together to produce bands of gene expression in the presumptive head, thorax, and abdomen (Perry et al., 2011). More generally, a genome-wide analysis of enhancer activity in Drosophila S2 cells found that 434 genes are regulated by at least two enhancers, and 203 of these genes are regulated by more than five enhancers (Arnold et al., 2013). For many genes, all of the gene's enhancers are necessary to drive appropriate expression. For example, both of the enhancers that regulate the gap gene hunchback are necessary to ensure the gene's correct expression in the developing embryo (Perry et al., 2011). In some genes, however, enhancers appear to be functionally redundant: Under normal growth conditions, only one of a gene's multiple enhancers are necessary to drive correct expression (Frankel et al., 2010; Perry et al., 2010).
Redundant enhancers—sometimes referred to as shadow enhancers (Hong et al., 2008)—provide not only robustness to environmental perturbations (Frankel et al., 2010; Perry et al., 2010), but also robustness to mutations. This is because deletion of one enhancer is often insufficient to disrupt normal gene expression, even if the enhancers are only partially redundant. For example, the Drosophila gene snail—a key determinant of dorsal-ventral patterning—is regulated by two enhancers, and deletion of either of these enhancers does not alter the gene's expression pattern in the presumptive mesoderm under normal growth conditions (Perry et al., 2010). Redundant enhancers can also provide robustness to mutations that affect the expression level of their cognate TFs (Frankel et al., 2010; Perry et al., 2010). For example, the two enhancers of snail drive a normal pattern of expression upon reduction of the expression level of Dorsal, an activator of snail, whereas deletion of one of these enhancers yields erratic patterns of snail expression in response to this genetic perturbation (Perry et al., 2010).
We note that shadow enhancers do not always provide mutational robustness. For example, the Drosophila gene shavenbaby is regulated by three primary enhancers and two shadow enhancers (Frankel et al., 2010). While the shadow enhancers are not necessary to drive the gene's epidermal expression pattern under normal growth conditions, their presence does not compensate for the inactivation of any one of the three primary enhancers (McGregor et al., 2007).
Transcription factors are also to some extent robust to mutations, including those that change the amino acid sequence of the protein's DNA binding domain. There are at least two causes of this robustness. First, amino acid substitutions in a TF's DNA binding domain may have little or no effect on the TF's binding specificity. For example, the human helix-loop-helix transcription factor Max contacts DNA at five residues, and amino acid substitutions in three of these residues have no effect on binding specificity (Maerkl and Quake, 2009). Second, transcription factors often bind DNA cooperatively, and the presence of cofactors may ameliorate the effects of amino acid substitutions that impair binding specificity. For example, the binding specificity of Matα1, a regulator of cell-type specification in ascomycete fungi, has diverged so extensively among S. cerevisiae and C. albicans that the sequences recognized by these proteins appear unrelated by bioinformatic criteria (Baker et al., 2011). Nonetheless, Matα1 controls the same set of core genes in these two species, because its recognition sequences evolved along with it. This was most likely facilitated by a protein-protein interaction with Mcm1, which is conserved among S. cerevisiae and C. albicans, and may have helped stabilize Matα1 while its interaction with DNA slowly changed.
Despite these examples, it should be emphasized that mutations in a transcription factor's DNA binding domain often do affect binding specificity and that cofactors cannot always compensate for such changes. Because transcription factors typically regulate the expression of multiple genes, such mutations are often deleterious. This is demonstrated both by the common implication of such mutations in disease (Lee and Young, 2013) and by the high level of conservation of one-to-one transcription factor orthologs across highly diverse species (Nitta et al., 2015).
Gene duplication, which creates paralogous genes within the same genome, is a driving force in evolution. In eukaryotes, for instance, gene duplicates are estimated to arise at a rate of 0.01 per gene per million years (Lynch and Conery, 2000), and between 30 and 65 percent of a typical eukaryote's genes have paralogs (Zhang, 2003). Because gene duplicates are often functionally redundant at their time of origin, it is possible that they play compensatory roles, acting as a backup if one of the paralogs is functionally compromised. This possibility has led to a large body of research on redundant genes as a source of mutational robustness (e.g., Conant and Wagner, 2003; Gu et al., 2003).
Gene duplication has played an important role in the evolution of transcriptional regulatory systems. For example, an estimated 68% of TFs in yeast (Teichmann and Babu, 2003) and 73% of TFs in Escherichia coli (Madan Babu and Teichmann, 2003) are the result of gene duplication. Many of these paralogous transcription factors appear fully or partially redundant in function, because they recognize the same sets of binding sites in vitro (Weirauch et al., 2014) and bind to some of the same genomic regions in vivo. For example, genome-wide binding profiles of three ETS TFs in human T cells revealed that nearly 10% of 17,000 promoters bound more than two of the three TFs, and probably at the same binding site (Hollenhorst et al., 2007). A broader view of redundant TFs is provided by enhanced yeast one-hybrid assays (Reece-Hoyes et al., 2011), which have facilitated a test of nearly 400,000 putative binding events among 1086 human TFs and 360 enhancers (Fuxman Bass et al., 2015). This analysis found that human enhancers often bind multiple TFs that typically belong to the same TF family. Moreover, the greater the number of enhancers that a pair of TFs shares, the more likely it is that these factors are coexpressed, and the less likely it is that each factor is essential for viability (Fuxman Bass et al., 2015), providing additional support for their compensatory roles. Indeed, even distant paralogs may compensate for one another, at least in part (Kafri et al., 2005; He and Zhang, 2006; Tischler et al., 2006).
The transcriptional regulatory networks of organisms as different as bacteria and humans exhibit strikingly similar structural properties, including a heavy-tailed degree distribution, a modular organization, and non-random assortativity (Barabási and Oltvai, 2004; Boyle et al., 2014; Sorrells and Johnson, 2015). Each of these properties may confer mutational robustness in transcriptional regulation.
Many biological networks, including transcriptional regulatory networks, exhibit a heavy-tailed degree distribution (Aldana et al., 2007). Such networks are characterized by a preponderance of nodes with few connections and a small number of nodes with many connections. This topological property can endow a network with robustness to random gene deletion, because such deletions are more likely to affect low-degree nodes than high-degree nodes, and are therefore unlikely to disrupt the structure of a network (Albert et al., 2000). Simulations of model regulatory networks with heavy-tailed degree distributions show that such networks exhibit stable dynamical behavior over a broader range of parameter values than networks with a homogeneous degree distribution (Aldana and Cluzel, 2003). They are also more robust to both gene duplication (Aldana et al., 2007) and edge rewiring (Greenbury et al., 2010).
Transcriptional regulatory networks are modular. They can be decomposed into subnetworks of genes that are coregulated in response to different conditions and that are involved in distinct functions (Ihmels et al., 2002; Segal et al., 2003; Peter and Davidson, 2009). For example, an analysis of gene expression data in yeast uncovered 85 partially overlapping modules that participate in distinct cellular processes, including sporulation and rRNA processing (Ihmels et al., 2002). Similarly, the regulatory network controlling embryogenesis in the sea urchin has been decomposed into several modules that each perform distinct functions in patterning the pre-gastrular embryo, such as restricting gene expression to specific subdomains (Peter and Davidson, 2009). Such modularity may serve to contain damage, limiting the propagation of a mutation's effects to those genes that are also part of the module. For example, the yeast TF Ypl230w drives the expression of a module of hundreds of genes during entry to stationary phase. Analysis of differential gene expression upon deletion of Ypl230w found that differentially expressed genes were enriched within the module, indicating that the effect of the perturbation was largely contained (Segal et al., 2003). Similar observations have been made in simulations of model regulatory networks (Poblanno-Balp and Gershenson, 2011). It is therefore conceivable that modularity confers mutational robustness (Wagner et al., 2007), although in the context of transcriptional regulation, we currently have very little empirical evidence to support this possibility.
Assortativity is the propensity of nodes in a network to connect to other nodes with similar properties (Newman, 2002). For instance, in a network that is assortative with respect to the number of neighbors that a node (TF) has, nodes with many neighbors tend to connect to other nodes with many neighbors, and nodes with few neighbors tend to connect to nodes with few neighbors. Simulations of model transcriptional regulatory networks suggest that degree assortativity can confer robustness to mutations in regulatory regions (Pechenick et al., 2012) and to gene duplications (Pechenick et al., 2013). The transcriptional regulatory networks of 41 distinct human cell and tissue types exhibit such an assortativity signature (Pechenick et al., 2014), raising the possibility that this structural property confers robustness to transcriptional regulation in humans.
There are at least three possible origins of mutational robustness (de Visser et al., 2003): (1) Mutational robustness may itself be an adaptation to mutations, i.e., it may exist because it provides a selective advantage; (2) It may be a byproduct of other adaptations, such as environmental robustness; or, (3) It may be neither a direct adaptation nor an indirect by-product of an adaptation, and thus a non-adaptive result of biophysical principles or non-adaptive evolutionary forces.
The first, adaptive view can be traced to at least the early 1990s, when genetic studies first showed that many genes, including genes encoding TFs, are duplicated (Thomas, 1993). This observation raised the question whether such gene redundancy exists to protect genes against otherwise deleterious mutations, and lead to modeling work addressing this question (Clark, 1994; Nowak et al., 1997; Wagner, 1999, 2000; Lynch et al., 2001; O'Hely, 2006). Such models apply in principle not only to redundant genes, but also to binding site clusters with redundant sites and to redundant enhancers.
Redundancy is not the only route to adaptive robustness. In the context of transcriptional regulation, this became clear once it became possible to analyze the structure of genotype spaces of model transcriptional regulatory circuits. In such spaces, one finds that circuits with a given gene expression pattern usually form large and connected genotype networks, where differences between neighboring genotypes (circuits) can be caused by small genetic changes, such as alterations of single regulatory interactions (Ciliberti et al., 2007; Cotterell and Sharpe, 2010; Payne et al., 2014). Individual circuits in such a network can change their regulatory interactions without changing their expression pattern. Because these circuits also vary considerably in their mutational robustness, they can evolve increased robustness via a series of small mutations that maintain their expression phenotype. Empirical data on TF binding sites demonstrate that such sites show a similar organization in the space of DNA sequences (Payne and Wagner, 2014). In consequence, their mutational robustness could in principle increase through gradual genetic change (e.g., point mutations) that preserve transcription factor binding.
Despite these observations, robustness is unlikely to confer a sufficiently strong advantage in a binding site, regulatory circuit, or a redundant regulatory element to be maintained by natural selection in most evolving populations. The reason is that its selective advantage is small, i.e., on the order of the mutation rate μ, because selection of increased robustness is effective only when a population of organisms (binding sites, circuits, etc.) are polymorphic for robustness. Elementary population genetics dictates that this will be the case only when the product of the effective population size N and the mutation rate μ is much greater than one (Nμ ≫ 1) (van Nimwegen et al., 1999; Wagner, 2000). Especially for small mutational targets, this requires huge population sizes and very large mutation rates. Therefore, although robustness may sometimes be an adaptation, this is likely the exception rather than the rule.
Mutational robustness may also arise as a byproduct of selection for other traits, most notably robustness to environmental change (Wagner, 1997; Meiklejohn and Hartl, 2002). This is particularly relevant for transcriptional regulation, which is frought with noise, including stochastic fluctuations in signaling molecules and variable temperatures (Macneil and Walhout, 2011). Such noise can be viewed as incessant change in the molecular environment where transcriptional regulation operates. Shadow enhancers provide a useful example. As we mentioned in Section 2.3, the regulatory region of the Drosophila gene snail comprises two enhancers. Either of them is sufficient to drive wild-type gene expression patterns under normal growth conditions (Perry et al., 2010), which provides a source of mutational robustness. Under extreme temperatures, however, deletion of either of the enhancers results in aberant gene expression patterns, suggesting that the primary function of the shadow enhancer is to provide robustness to the destabilizing effects of sub-optimal temperatures, as is also the case for the two shadow enhancers associated with the Drosophila gene shavenbaby (Frankel et al., 2010). Additional support for the origin of mutational robustness as a byproduct of environmental robustness is found in model transcriptional regulatory circuits, which exhibit a positive correlation between mutational and environmental robustness (Ciliberti et al., 2007), such that selection for environmental robustness facilitates mutational robustness.
Finally, mutational robustness may also be a consequence of biophysical principles underlying transcriptional regulation, or of non-adaptive forces of genome evolution, i.e., genetic drift, mutation, and recombination.
For example, homotypic clusters of TF binding sites may evolve simply because there are more ways to build a regulatory region using many low-affinity sites than there are with few high-affinity sites (He et al., 2012). The reason is that there are many more distinct DNA sequences that bind TFs with low affinity than with high affinity (Badis et al., 2009). In addition, such clusters could simply result from the inefficiency of selection at removing insertions, such that insertions containing TF binding sites accumulate over time (Lynch, 2007), or they may be a byproduct of recombination within regulatory regions (Lynch, 2007; Paixao and Azevedo, 2010). Moreover, the spatial organization of homotypic clusters may reflect a mutational bias toward deletions, as such mutations are more likely to bring different sites closer together than farther apart (Lusk and Eisen, 2000).
Similarly, robustness-conferring topological properties, such as heavy-tailed degree distributions, can originate as a by-product of biophysical principles. For example, a biophysical model of protein-protein interactions shows that this distribution can emerge if the number of surface-exposed hydrophobic amino acids on a protein follows a simple random distribution (Deeds et al., 2006). In addition, evolutionary forces other than natural selection can enhance the robustness of regulatory networks. For instance, heavy-tailed degree distributions (Lynch, 2007), a modular organization (Wagner et al., 2007), and the enrichment of particular circuit motifs (Artzy-Randrup et al., 2004; Cordero and Hogeweg, 2006; Sorrells and Johnson, 2015) can all emerge through random genetic drift.
Mutational robustness in transcriptional regulation has several consequences that emerge on evolutionary timescales. First, the mutational robustness of regulatory regions permits their evolutionary divergence without a corresponding divergence in the gene expression patterns they control. This phenomenon is often observed among closely-related species (Weirauch and Hughes, 2010). During such divergence, substantial binding site turnover may occur, such that different sets of TFs may regulate orthologous genes in different species (Moses et al., 2006; Borneman et al., 2007; Schmidt et al., 2010). Binding site turnover can even occur among activating and repressing TFs and can alter the architecture of a regulatory circuit, all without altering its gene expression phenotype (Tanay et al., 2005; Tsong et al., 2006; Swanson et al., 2011). A well-known practical consequence of this divergence is that regulatory regions are exceptionally difficult to align.
A related consequence of mutational robustness is that regulatory regions can accumulate genetic diversity within a population. Such diversity is often referred to as cryptic, because it does not generate phenotypic variation (Gibson and Dworkin, 2004; McGuigan and Sgro, 2009). However, cryptic diversity may generate phenotypic variation upon environmental or genetic perturbation (Rutherford and Lindquist, 1998; Queitsch et al., 2002). Cryptic diversity is commonly observed in DNA sequences regulating transcription (Rockman and Wray, 2002), including TF binding sites (Balhoff and Wray, 2005; Kasowski et al., 2010; Spivakov et al., 2012; Arbiza et al., 2013). Computational models of transcriptional regulatory circuits hint that such diversity may generate phenotypic variation in response to genetic or environmental perturbations (Siegal and Bergman, 2002; Bergman and Siegal, 2003). However, we currently have no experimental evidence that standing cryptic diversity in gene regulatory regions contributes to adaptation in transcriptional regulation.
Yet another consequence of mutational robustness is that it permits regulatory interactions to originate that do not contribute to gene regulation at the time of their origin. Over time, the accumulation of such non-functional interactions can give rise to dense, highly-interconnected transcriptional regulatory networks (Sorrells and Johnson, 2015). This is especially true if binding sites are short, regulatory regions are long, and TF binding specificities are low. Evidence exists that each of these conditions are met, especially in eukaryotes, where binding sites are on average merely ten nucleotides long (Stewart et al., 2012), regulatory regions comprise promoters and enhancers that span thousands of nucleotides (The ENCODE Project Consortium, 2012), and the average information content per nucleotide of binding sites is roughly 65% of the maximum, indicating modest specificity (Stewart et al., 2012). Taken together with evidence that synthetically-added regulatory interactions rarely impact phenotype (Isalan et al., 2008), these observations suggest that mutational robustness may contribute to the apparent complexity of transcriptional regulatory networks. What is more, non-functional regulatory interactions may form the substrate of subsequent adaptations (Isalan et al., 2008), implicating mutational robustness in the evolution of novel transcriptional regulatory programs.
A final consequence of robustness emerges from the duplication of transcription factor genes. By providing a back-up gene for any one essential molecular function, gene duplication facilitates the evolution of genes with novel functions (Ohno, 1970; Hahn, 2009; Innan and Kondrashov, 2010; Rensing, 2014), such as TFs with altered binding site repertoires that can take on novel regulatory roles (Pérez et al., 2014). Over long evolutionary time scales, this ability can have profound consequences. For example, gene and genome duplications that created novel homeobox TF genes have been implicated in the diversification of the vertebrate body plan (Carroll et al., 2001), and duplication of genes encoding MADS box TFs has played an important role in the diversification of flowering plants (De Bodt et al., 2003; Irish, 2003). In other words, robust transcriptional regulation has helped shape life as we know it.
JP and AW conceived of and wrote the paper.
JP acknowledges support through the Ambizione program of the Swiss National Science Foundation. AW acknowledges support through Swiss National Science Foundation grant 31003A_146137, as well as through the University Priority Research Program in Evolutionary Biology at the University of Zurich.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Albert, R., Jeong, H., and Barabási, A.-L. (2000). Error and attack tolerance of complex networks. Nature 406, 378–382. doi: 10.1038/35019019
Aldana, M., Balleza, E., Kauffman, S., and Resendiz, O. (2007). Robustness and evolvability in genetic regulatory networks. J. Theor. Biol. 245, 433–448. doi: 10.1016/j.jtbi.2006.10.027
Aldana, M., and Cluzel, P. (2003). A natural class of robust networks. Proc. Natl. Acad. Sci. U.S.A. 100, 8710–8714. doi: 10.1073/pnas.1536783100
Alon, U. (2007). Network motifs: theory and experimental approaches. Nat. Rev. Genet. 8, 450–461. doi: 10.1038/nrg2102
Arbiza, L., Gronau, I., Aksoy, B. A., Hubisz, M. J., Gulko, B., Keinan, A., et al. (2013). Genome-wide inference of natural selection on human transcription factor binding sites. Nat. Genet. 45, 723–729. doi: 10.1038/ng.2658
Arnold, C. D., Gerlach, D., Stelzer, C., Boryń, L. M., Rath, M., and Stark, A. (2013). Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339, 1074–1077. doi: 10.1126/science.1232542
Artzy-Randrup, Y., Fleishman, S. J., Ben-Tal, N., and Stone, L. (2004). Comment on “network motifs: simple building blocks of complex networks” and “superfamilies of evolved and designed networks.” Science 305, 1107c. doi: 10.1126/science.1099334
Badis, G., Berger, M. F., Philippakis, A. A., Talukder, S., Gehrke, A. R., Jaeger, S. A., et al. (2009). Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723. doi: 10.1126/science.1162327
Baker, C. R., Tuch, B. B., and Johnson, A. D. (2011). Extensive DNA-binding specificity divergence of a conserved transcription regulator. Proc. Natl. Acad. Sci. U.S.A. 108, 7493–7498. doi: 10.1073/pnas.1019177108
Balhoff, J. P., and Wray, G. A. (2005). Evolutionary analysis of the well characterized endo16 promoter reveals substantial variation within functional sites. Proc. Natl. Acad. Sci. U.S.A. 102, 8591–8596. doi: 10.1073/pnas.0409638102
Banerji, J., Rusconi, S., and Schaffner, W. (1981). Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 199–308. doi: 10.1016/0092-8674(81)90413-X
Barabási, A.-L., and Oltvai, Z. N. (2004). Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 5, 101–113. doi: 10.1038/nrg1272
Berger, M. F., Philippakis, A. A., Qureshi, A. M., He, F. S., Estep, P. W. III., and Bulyk, M. L. (2006). Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 24, 1429–1435. doi: 10.1038/nbt1246
Bergman, A., and Siegal, M. L. (2003). Evolutionary capacitance as a general feature of complex gene networks. Nature 424, 549–552. doi: 10.1038/nature01765
Berman, B. P., Nibu, Y., Pfeiffer, B. D., Tomancak, P., Celniker, S. E., Levine, M., et al. (2002). Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the drosophila genome. Proc. Natl. Acad. Sci. U.S.A. 99, 757–762. doi: 10.1073/pnas.231608898
Bloom, J. D., Silberg, J. J., Wilke, C. O., Drummond, D. A., Adami, C., and Arnold, F. H. (2005). Thermodynamic prediction of protein neutrality. Proc. Natl. Acad. Sci. U.S.A. 102, 606–611. doi: 10.1073/pnas.0406744102
Borneman, A. R., Gianoulis, T. A., Zhang, Z. D., Yu, H., Rozowsky, J., Serringhaus, M. R., et al. (2007). Divergence of transcription factor binding sites across related yeast species. Science 317, 815–819. doi: 10.1126/science.1140748
Boyle, A. P., Araya, C. L., Brdlik, C., Cayting, P., Cheng, C., Cheng, Y., et al. (2014). Comparative analysis of regulatory information and circuits across distant species. Nature 512, 453–456. doi: 10.1038/nature13668
Carey, M., Lin, Y.-S., Green, M. R., and Ptashne, M. (1990). A mechanism for synergistic activation of a mammalian gene by GAL4 derivatives. Nature 345, 361–364. doi: 10.1038/345361a0
Carroll, S. B., Grenier, J. K., and Weatherbee, S. D. (2001). From DNA to Diversity. Molecular Genetics and the Evolution of Animal Design. Malden, MA: Blackwell.
Ciliberti, S., Martin, O. C., and Wagner, A. (2007). Robustness can evolve gradually in complex regulatory gene networks with varying topology. PLoS Comput. Biol. 3:e15. doi: 10.1371/journal.pcbi.0030015
Clark, A. G. (1994). Invasion and maintenance of a gene duplication. Proc. Natl. Acad. Sci. U.S.A. 91, 2950–2954. doi: 10.1073/pnas.91.8.2950
Conant, G. C., and Wagner, A. (2003). Duplicate genes and robustness to transient gene knock-downs in Caenorhabditis elegans. Proc. R. Soc. Lond. B, 271, 89–96. doi: 10.1098/rspb.2003.2560
Cordero, O. X., and Hogeweg, P. (2006). Feed-forward loop circuits as a side effect of genome evolution. Mol. Biol. Evol. 23, 1931–1936. doi: 10.1093/molbev/msl060
Cotterell, J., and Sharpe, J. (2010). An atlas of gene regulatory networks reveals multiple three-gene mechanisms for interpreting morphogen gradients. Mol. Syst. Biol. 6, 425. doi: 10.1038/msb.2010.74
De Bodt, S., Raes, J., Van de Peer, Y., and Theissen, G. (2003). And then there were many: MADS goes genomic. Trends Plant Sci. 8, 475–483. doi: 10.1016/j.tplants.2003.09.006
de Villiers, J., Olson, L., Tyndall, C., and Schaffner, W. (1982). Transcriptional ‘enhancers’ from sv40 and polyoma virus show a cell type preference. Nucleic Acids Res. 10, 7965–7976. doi: 10.1093/nar/10.24.7965
de Visser, J. A. G. M., Hermisson, J., Wagner, G. P., Ancel Meyers, L., Bagheri-Chaichian, H., Blankchard, J. L., et al. (2003). Evolution and detection of genetic robustness. Evolution 57, 1959–1972. doi: 10.1111/j.0014-3820.2003.tb00377.x
Deeds, E. J., Ashenberg, O., and Shakhnovich, E. I. (2006). A simple physical model for scaling in protein-protein interaction networks. Proc. Natl. Acad. Sci. U.S.A. 103, 311–316. doi: 10.1073/pnas.0509715102
Dixon, J. R., Selvaraj, S., Yue, F., Kim, A., Li, Y., Shen, Y., et al. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. doi: 10.1038/nature11082
Eldar, A., Shilo, B.-Z., and Barkai, N. (2004). Elucidating mechanisms underlying robustness of morphogen gradients. Curr. Opin. Genet. Dev. 14, 435–439. doi: 10.1016/j.gde.2004.06.009
Ezer, D., Zabet, N. R., and Adryan, B. (2014). Homotypic clusters of transcription factor binding sites: a model system for understanding the physical mechanics of gene expression. Comput. Struct. Biotechnol. J. 10, 63–69. doi: 10.1016/j.csbj.2014.07.005
Frankel, N., Davis, G. K., Vargas, D., Wang, S., Payre, F., and Stern, D. L. (2010). Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466, 490–493. doi: 10.1038/nature09158
Fuxman Bass, J. I., Sahni, N., Shrestha, S., Garcia-Gonzalez, A., Mori, A., Bhat, N., et al. (2015). Human gene-centered transcription factor networks for enhancers and disease variants. Cell 161, 661–673. doi: 10.1016/j.cell.2015.03.003
Gama-Castro, S., Salgado, H., Peralta-Gil, M., Santos-Zavaleta, A., Muniz, R. L., Solano-Lira, H., et al. (2011). RegulonDB version 7.0: transcriptional regulation of Escherichia coli K-12 integrated wihin genetic sensory response units (gensor units). Nucleic Acids Res. 39, D98–D105. doi: 10.1093/nar/gkq1110
Garfield, D., Haygood, R., Nielsen, W. J., and Wray, G. A. (2012). Population genetics of cis-regulatory sequences that operate during embryonic development in the sea urchin Stronglyocentrotus purpuratus. Evol. Dev. 14, 152–167. doi: 10.1111/j.1525-142X.2012.00532.x
Garfield, D. D., Runcie, D. E., Babbitt, C. C., Haygood, R., Nielsen, W. J., and Wray, G. A. (2013). The impact of gene expression variation on the robustness and evolvability of a developmental gene regulatory network. PLoS Biol. 11:e1001696. doi: 10.1371/journal.pbio.1001696
Gibson, G., and Dworkin, I. (2004). Uncovering cryptic genetic variation. Nat. Rev. Genet. 5, 681–690. doi: 10.1038/nrg1426
Gillies, S. D., Morrison, S. L., Oi, V. T., and Tonegawa, S. (1983). A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell 33, 717–728. doi: 10.1016/0092-8674(83)90014-4
Giniger, E., and Ptashne, M. (1988). Cooperative DNA binding of the yeast transcriptional activator GAL4. Proc. Natl. Acad. Sci. U.S.A. 85, 382–386. doi: 10.1073/pnas.85.2.382
Giogetti, L., Siggers, T., Tiana, G., Caprara, G., Notarbartolo, S., Corona, T., et al. (2010). Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded trancriptional responses to environmental inputs. Mol. Cell 37, 418–428. doi: 10.1016/j.molcel.2010.01.016
Gotea, V., Visel, A., Westlund, J. M., Nobrega, M. A., Pennacchio, L. A., and Ovcharenko, I. (2010). Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res. 20, 565–577. doi: 10.1101/gr.104471.109
Greenbury, S. F., Johnson, I. G., Smith, M. A., Doye, J. P. K., and Louis, A. A. (2010). The effect of scale-free topology on the robustness and evolvability of genetic regulatory networks. J. Theor. Biol. 267, 48–61. doi: 10.1016/j.jtbi.2010.08.006
Gu, Z., Steinmetz, L. M., Gu, X., Scharfe, C., Davis, R. W., and Li, W.-H. (2003). Role of duplicate genes in genetic robustness against null mutations. Nature 421, 63–66. doi: 10.1038/nature01198
Hahn, M. W. (2009). Distinguishing among evolutionary models for the maintenance of gene duplicates. J. Hered. 100, 605–617. doi: 10.1093/jhered/esp047
Harismendy, O., Notani, D., Song, X., Rahim, N. G., Tanasa, B., Heintzman, N., et al. (2011). 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature 470, 264–268. doi: 10.1038/nature09753
He, X., Duque, T. S. P. C., and Sinha, S. (2012). Evolutionary origins of transcription factor binding sites. Mol. Biol. Evol. 29, 1059–1070. doi: 10.1093/molbev/msr277
He, X., and Zhang, J. (2006). Transcriptional reprogramming and backup between duplicate genes: is it a genomewide phenomenon? Genetics 172, 1363–1367. doi: 10.1534/genetics.105.049890
Hollenhorst, P. C., Shah, A. A., Hopkins, C., and Graves, B. J. (2007). Genome-wide analysis reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev. 21, 1882–1894. doi: 10.1101/gad.1561707
Hong, J. W., Hendrix, D. A., and Levine, M. S. (2008). Shadow enhancers as a source of evolutionary novelty. Science 321, 1314. doi: 10.1126/science.1160631
Ihmels, J., Friedlander, G., Bergmann, S., Sarig, O., Ziv, Y., and Barkai, N. (2002). Revealing modular organization in the yeast transcriptional network. Nat. Genet. 31, 370–377. doi: 10.1038/ng941
Innan, H., and Kondrashov, F. (2010). The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11, 97–108. doi: 10.1038/nrg2689
Irish, V. F. (2003). The evolution of floral homeotic gene function. BioEssays 25, 637–646. doi: 10.1002/bies.10292
Isalan, M., Lemerle, C., Michalodimitrakis, K., Horn, C., Beltrao, P., Raineri, E., et al. (2008). Evolvability and hierarchy in rewired bacterial gene networks. Nature 452, 840–846. doi: 10.1038/nature06847
Johnson, A. D., Meyer, B. J., and Ptashne, M. (1979). Interactions between DNA-bound repressors govern regulation by the λ phage repressor. Proc. Natl. Acad. Sci. U.S.A. 76, 5061–5065. doi: 10.1073/pnas.76.10.5061
Kafri, R., Bar-Even, A., and Pilpel, Y. (2005). Transcription control reprogramming in genetic backup circuits. Nat. Genet. 37, 295–299. doi: 10.1038/ng1523
Kasowski, M., Grubert, F., Heffelfinger, C., Hariharan, M., Asabere, A., Waszak, S. M., et al. (2010). Variation in transcription factor binding among humans. Science 328, 232–235. doi: 10.1126/science.1183621
Katainen, R., Dave, K., Pitknen, E., Palin, K., Kivioja, T., Vlimki, N., et al. (2015). CTCF/cohesin-binding sites are frequently mutated in cancer. Nat. Genet. 47, 818–821. doi: 10.1038/ng.3335
Khurana, E., Fu, Y., Colonna, V., Mu, X. J., Kang, H. M., Lappalainen, T., et al. (2013). Integrative annotation of variants from 1092 humans: application to cancer genomics. Science 342, 84. doi: 10.1126/science.1235587
Kilpinen, H., Waszak, S. M., Gschwind, A. R., Raghav, S. K., Witwicki, R. M., Orioli, A., et al. (2013). Coordinated effects of sequence variation on dna binding, chromatin structure, and transcription. Science 342, 744–747. doi: 10.1126/science.1242463
Krivan, W., and Wasserman, W. W. (2001). A predictive model for regulatory sequences directing liver-specific transcription. Genome Res. 11, 1559–1566. doi: 10.1101/gr.180601
Kwasnieski, J. C., Mogno, I., Meyers, C. A., Corbo, J. C., and Cohen, B. A. (2012). Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc. Natl. Acad. Sci. U.S.A. 109, 19498–19503. doi: 10.1073/pnas.1210678109
Lawrence, P. A. (1992). The Making of a Fly: The Genetics of Animal Design. Oxford: Blackwell Science Ltd.
Lebrecht, D., Foehr, M., Smith, E., Lopes, F. J. P., Vanario-Alonso, C. E., Reinitz, J., et al. (2005). Bicoid cooperative DNA binding is critical for embryonic patterning in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 102, 13176–13181. doi: 10.1073/pnas.0506462102
Lee, T. I., and Young, R. A. (2013). Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251. doi: 10.1016/j.cell.2013.02.014
Levine, M., Cattoglio, C., and Tijan, R. (2014). Looping back to leap forward: transcription enters a new era. Cell 157, 13–25. doi: 10.1016/j.cell.2014.02.009
Lifanov, A. P., Makeev, V. J., Nazina, A. G., and Papatsenko, D. A. (2003). Homotypic regulatory clusters in Drosophila. Genome Res. 13, 579–588. doi: 10.1101/gr.668403
Ludwig, M. Z., Bergman, C., Patel, N. H., and Kreitman, M. (2000). Evidence for stabilizing selection in a eukaryotic enhancer element. Nature 403, 564–567. doi: 10.1038/35000615
Lusk, R. W., and Eisen, M. B. (2000). Evolutionary mirages: selection on binding site composition creates the illusion of conserved grammars in Drosophila enhancers. PLoS Genet. 6:e1000829. doi: 10.1371/journal.pgen.1000829
Lynch, M. (2007). The evolution of genetic networks by non-adaptive processes. Nat. Rev. Gen. 8, 803–813. doi: 10.1038/nrg2192
Lynch, M., and Conery, J. S. (2000). The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155. doi: 10.1126/science.290.5494.1151
Lynch, M., O'Hely, M., Walsh, B., and Force, A. (2001). The probability of preservation of a newly arisen gene duplicate. Genetics 159, 1789–1804.
Macneil, L. T., and Walhout, A. J. M. (2011). Gene regulatory networks and the role of robustness and stochasticity in the control of gene expression. Genome Res. 21, 645–657. doi: 10.1101/gr.097378.109
Madan Babu, M., and Teichmann, S. A. (2003). Evolution of transcription factors and the gene regulatory network of Escherichia coli. Nucleic Acids Res. 31, 1234–1244. doi: 10.1093/nar/gkg210
Maerkl, S. J., and Quake, S. R. (2009). Experimental determination of the evolvability of a transcription factor. Proc. Natl. Acad. Sci. U.S.A. 106, 18650–18655. doi: 10.1073/pnas.0907688106
Masel, J., and Siegal, M. L. (2009). Robustness: mechanisms and consequences. Trends Genet. 25, 395–403. doi: 10.1016/j.tig.2009.07.005
Maurano, M. T., Humbert, R., Rynes, E., Thurman, R. E., Haugen, E., Wang, H., et al. (2012). Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195. doi: 10.1126/science.1222794
McGregor, A. P., Orgogozo, V., Delon, I., Zanet, J., Srinivasan, D. G., Payre, F., et al. (2007). Morphological evolution through multiple cis-regulatory mutations at a single gene. Nature 448, 587–590. doi: 10.1038/nature05988
McGuigan, K., and Sgrò, C. M. (2009). Evolutionary consequences of cryptic genetic variation. Trends Ecol. Evol. 24, 305–311. doi: 10.1016/j.tree.2009.02.001
McManus, C. J., May, G. E., Spealman, P., and Shteyman, A. (2014). Ribosome profiling reveals post-transcriptional buffering of divergent gene expression in yeast. Genome Res. 24, 422–430. doi: 10.1101/gr.164996.113
Meiklejohn, C. D., and Hartl, D. L. (2002). A single mode of canalization. Trends Ecol. Evol. 17, 468–473. doi: 10.1016/S0169-5347(02)02596-X
Melton, C., Reuter, J. A., Spacek, D. V., and Snyder, M. (2015). Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat. Genet. 47, 710–716. doi: 10.1038/ng.3332
Moses, A. M., Pollard, D. A., Nix, D. A., Iyer, V. N., Li, X., Biggin, M. D., et al. (2006). Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput. Biol. 2:e130. doi: 10.1371/journal.pcbi.0020130
Musunuru, K., Strong, A., Frank-Kamenetsky, M., Lee, N. E., Ahfeldt, T., Sachs, K., et al. (2010). From noncoding variant to phenotype via sort1 at the 1p13 cholesterol locus. Nature 466, 714–719. doi: 10.1038/nature09266
Newman, M. E. J. (2002). Assortative mixing in networks. Phys. Rev. Lett. 89:208701. doi: 10.1103/PhysRevLett.89.208701
Nitta, K. R., Jolma, A., Yin, Y., Morgunova, E., Kivioja, T., Akhtar, J., et al. (2015). Conservation of transcription factor binding specificities across 600 million years of bilateria evolution. eLife 4:e04837. doi: 10.7554/eLife.04837
Nowak, M. A., Boerlijst, M. C., Cooke, J., and Maynard Smith, J. M. (1997). Evolution of genetic redundancy. Nature 388, 167–171. doi: 10.1038/40618
Odom, D. T., Dowell, R. D., Jacobsen, E. S., Gordon, W., Danford, T. W., MacIsaac, K. D., et al. (2007). Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat. Genet. 39, 730–732. doi: 10.1038/ng2047
O'Hely, M. (2006). A diffusion approach to approximating preservation probabilities for gene duplicates. J. Math. Biol. 53, 215–230. doi: 10.1007/s00285-006-0001-6
Ohno, S. (1970). Evolution by Gene Duplication. New York, NY: Blackwell Science Ltd; Springer. doi: 10.1007/978-3-642-86659-3
Paixão, T., and Azevedo, R. B. R. (2010). Redundancy and the evolution of cis-regulatory element multiplicity. PLoS Comput. Biol. 6:e10000848. doi: 10.1371/journal.pcbi.1000848
Payne, J. L., Moore, J. H., and Wagner, A. (2014). Robustness, evolvability, and the logic of genetic regulation. Artif. Life 20, 111–126. doi: 10.1162/ARTL_a_00099
Payne, J. L., and Wagner, A. (2014). The robustness and evolvability of transcription factor binding sites. Science 466, 714–719. doi: 10.1126/science.1249046
Pechenick, D. A., Moore, J. H., and Payne, J. L. (2013). The influence of assortativity on the robustness and evolvability of gene regulatory networks upon gene birth. J. Theor. Biol. 330, 26–36. doi: 10.1016/j.jtbi.2013.03.019
Pechenick, D. A., Payne, J. L., and Moore, J. H. (2012). The influence of assortativity on the robustness of signal-integration logic in gene regulatory networks. J. Theor. Biol. 296, 21–32. doi: 10.1016/j.jtbi.2011.11.029
Pechenick, D. A., Payne, J. L., and Moore, J. H. (2014). Phenotypic robustness and the assortativity signature of human transcription factor networks. PLoS Comput. Biol. 10:e1003780. doi: 10.1371/journal.pcbi.1003780
Pérez, J. C., Fordyce, P. M., Lohse, M. B., Hanson-Smith, V., DeRisi, J. L., and Johnson, A. D. (2014). How duplicated transcription regulators can diversify to govern the expression of nonoverlapping sets of genes. Genes Dev. 28, 1272–1277. doi: 10.1101/gad.242271.114
Perry, M. W., Boettiger, A. N., Bothma, J. P., and Levine, M. (2010). Shadow enhancers foster robustness of Drosophila gastrulation. Curr. Biol. 20, 1562–1567. doi: 10.1016/j.cub.2010.07.043
Perry, M. W., Boettiger, A. N., and Levine, M. (2011). Multiple enhancers ensure precision of gap gene-expression patterns in the drosophila embryo. Proc. Natl. Acad. Sci. U.S.A. 108, 13570–13575. doi: 10.1073/pnas.1109873108
Peter, I. S., and Davidson, E. H. (2009). Modularity and design principles in the sea urchin embryo gene regulatory network. FEBS Lett. 583, 3948–3958. doi: 10.1016/j.febslet.2009.11.060
Poblanno-Balp, R., and Gershenson, C. (2011). Modular random boolean networks. Artif. Life 17, 331–351. doi: 10.1162/artl_a_00042
Pomerantz, M. M., Ahmadiyeh, N., Jia, L., Herman, P., Verzi, M. P., Doddapaneni, H., et al. (2009). The 8q24 cancer risk variant rs6983267 shows long-range interaction with myc in colorectal cancer. Nat. Genet. 41, 882–884. doi: 10.1038/ng.403
Ptashne, M., and Gann, A. (2002). Genes & Signals. New York, NY: Cold Spring Harbor Laboratory Press.
Queitsch, C., Sanger, T. A., and Lindquist, S. (2002). Hsp90 as a capacitor for phenotypic variation. Nature 417, 618–624. doi: 10.1038/nature749
Reece-Hoyes, J. S., Diallo, A., Lajoie, B., Kent, A., Shrestha, S., Kadreppa, S., et al. (2011). Enhanced yeast one-hybrid assays for high-throughput gene-centered regulatory network mapping. Nat. Methods 8, 1059–1064. doi: 10.1038/nmeth.1748
Rensing, S. A. (2014). Gene duplication as a driver of plant morphogenetic evolution. Curr. Opin. Plant Biol. 17, 43–48. doi: 10.1016/j.pbi.2013.11.002
Rockman, M. V., and Wray, G. A. (2002). Abundant raw material for cis-regulatory evolution in humans. Mol. Biol. Evol. 19, 1991–2004. doi: 10.1093/oxfordjournals.molbev.a004023
Rutherford, S. L., and Lindquist, S. (1998). Hsp90 as a capacitor for morphological evolution. Nature 396, 336–342. doi: 10.1038/24550
Samal, A., Matias Rodrigues, J. F., Jost, J., Martin, O. C., and Wagner, A. (2010). Genotype networks in metabolic reaction spaces. BMC Syst. Biol. 4:30. doi: 10.1186/1752-0509-4-30
Schmidt, D., Wilson, M. D., Ballester, B., Schwalie, P. C., Brown, G. D., Marshall, A., et al. (2010). Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328, 1036–1040. doi: 10.1126/science.1186176
Segal, E., Shapira, M., Regev, A., Pe'er, D., Botstein, D., Koller, D., et al. (2003). Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 34, 166–176. doi: 10.1038/ng1165
Sengupta, A. M., Djordjevic, M., and Shraiman, B. I. (2002). Specificity and robustness in transcription control networks. Proc. Natl. Acad. Sci. U.S.A. 99, 2072–2077. doi: 10.1073/pnas.022388499
Sharon, E., Kalma, Y., Sharp, A., Raveh-Sadka, T., Levo, M., Zeevi, D., et al. (2012). Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol. 30, 521–530. doi: 10.1038/nbt.2205
Shlyueva, D., Stampfel, G., and Stark, A. (2014). Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 15, 272–286. doi: 10.1038/nrg3682
Shultzaberger, R. K., Malashock, D. S., Kirsch, J. F., and Eisen, M. B. (2010). The fitness landscape fo cis-acting binding sites in different promoter and environmental contexts. PLoS Genet. 6:e1001042. doi: 10.1371/journal.pgen.1001042
Siegal, M. L., and Bergman, A. (2002). Waddington's canalization revisited: developmental stability and evolution. Proc. Natl. Acad. Sci. U.S.A. 99, 10528–10532. doi: 10.1073/pnas.102303999
Silva-Rocha, R., and de Lorenzo, V. (2010). Noise and robustness in prokaryotic regulatory networks. Annu. Rev. Microbiol. 64, 257–275. doi: 10.1146/annurev.micro.091208.073229
Small, S., Blair, A., and Levine, M. (1996). Regulation of two pair-rule stripes by a single enhancer in the Drosophila embryo. Dev. Biol. 175, 314–324. doi: 10.1006/dbio.1996.0117
Smith, R. P., Taher, L., Patwardhan, R. P., Kim, M. J., Inoue, F., Shendure, J., et al. (2013). Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat. Genet. 45, 1021–1028. doi: 10.1038/ng.2713
Somma, M. P., Pisano, C., and Lavia, P. (1991). The housekeeping promoter from the mouse CpG island HTF9 contains multiple protein-binding elements that are functionally redundant. Nucleic Acids Res. 19, 2817–2824. doi: 10.1093/nar/19.11.2817
Sorrells, T. R., and Johnson, A. D. (2015). Making sense of transcription networks. Cell 161, 714–723. doi: 10.1016/j.cell.2015.04.014
Spivakov, M., Akhtar, J., Kheradpour, P., Beal, K., Girardot, C., Koscielny, G., et al. (2012). Analysis of variation at transcription factor binding sites in Drosophila and humans. Genome Biol. 13, R49. doi: 10.1186/gb-2012-13-9-r49
Stelling, J., Sauer, U., Szallasi, Z., Doyle, F. J. III., and Doyle, J. (2004). Robustness of cellular functions. Cell 118, 675–685. doi: 10.1016/j.cell.2004.09.008
Stewart, A. J., Hannenhalli, S., and Plotkin, J. B. (2012). Why transcription factor binding sites are ten nucleotides long. Genetics 192, 973–985. doi: 10.1534/genetics.112.143370
Swanson, C. I., Schwimmer, D. B., and Barolo, S. (2011). Rapid evolutionary rewiring of a structurally constrained eye enhancer. Curr. Biol. 21, 1186–1196. doi: 10.1016/j.cub.2011.05.056
Tanay, A., Regev, A., and Shamir, R. (2005). Conservation and evolvability in regulatory networks: the evolution of ribosomal evolution in yeast. Proc. Natl. Acad. Sci. U.S.A. 102, 7203–7208. doi: 10.1073/pnas.0502521102
Teichmann, S. A., and Babu, M. M. (2003). Gene regulatory network growth by duplication. Nat. Genet. 36, 492–496. doi: 10.1038/ng1340
Thanos, D., and Maniatis, T. (1995). Virus induction of human ifnβ gene expression requires the assembly of an enhanceosome. Cell 83, 1091–1100. doi: 10.1016/0092-8674(95)90136-1
The ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Thomas, J. H. (1993). Thinking about genetic redundancy. Trends Genet. 9, 395–399. doi: 10.1016/0168-9525(93)90140-D
Tischler, J., Lehner, B., Chen, N., and Fraser, A. G. (2006). Combinatorial RNA interference in Caenorhabditis elegans reveals that redundancy between gene duplicates can be maintained for more than 80 million years of evolution. Genome Biol. 7:R69. doi: 10.1186/gb-2006-7-8-r69
Tsong, A. E., Tuch, B. B., Li, H., and Johnson, A. D. (2006). Evolution of alternative transcriptional circuits with identical logic. Nature 443, 415–420. doi: 10.1038/nature05099
van Nimwegen, E., Crutchfield, J. P., and Huynen, M. (1999). Neutral evolution of mutational robustness. Proc. Natl. Acad. Sci. U.S.A. 96, 9716–9720. doi: 10.1073/pnas.96.17.9716
Vaquerizas, J. M., Kummerfeld, S. K., Teichmann, S. A., and Luscombe, N. M. (2009). A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 10, 252–263. doi: 10.1038/nrg2538
Wagner, A. (1997). A population genetic theory of canalization. Evolution 51, 329–347. doi: 10.2307/2411105
Wagner, A. (1999). Redundant gene functions and natural selection. J. Evol. Biol. 12, 1–16. doi: 10.1046/j.1420-9101.1999.00008.x
Wagner, A. (2000). Robustness against mutations in genetic networks of yeast. Nat. Genet. 24, 355–361. doi: 10.1038/74174
Wagner, A. (2005). Robustness and evolvability in living systems. Princeton, NJ: Princeton University Press.
Wagner, A. (2008). Robustness and evolvability: a paradox resolved. Proc. Biol. Sci. 275, 91–100. doi: 10.1098/rspb.2007.1137
Wagner, G. P., Pavlicev, M., and Cheverud, J. M. (2007). The road to modularity. Nat. Rev. Genet. 8, 921–931. doi: 10.1038/nrg2267
Wasserman, W. W., and Fickett, J. W. (1998). Identification of regulatory regions which confer muscle-specific gene expression. J. Mol. Biol. 278, 167–181. doi: 10.1006/jmbi.1998.1700
Weinhold, N., Jacobsen, A., Schultz, N., Sander, C., and Lee, W. (2014). Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 46, 1160–1165. doi: 10.1038/ng.3101
Weirauch, M. T., and Hughes, T. R. (2010). Conserved expression without conserved regulatory sequence: the more things change, the more they stay the same. Trends Genet. 26, 66–74. doi: 10.1016/j.tig.2009.12.002
Weirauch, M. T., Yang, A., Albu, M., Cote, A. G., Montenegro-Montero, A., Drewe, P., et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. doi: 10.1016/j.cell.2014.08.009
Wong, K.-C., Chan, T.-M., Peng, C., Li, Y., and Zhang, Z. (2013). DNA motif elucidation using belief propagation. Nucleic Acids Res. 41, e153. doi: 10.1093/nar/gkt574
Zhang, J. (2003). Evolution by gene duplication: an update. Trends Ecol. Evol. 18, 292–298. doi: 10.1016/S0169-5347(03)00033-8
Zheng, W., Gianoulis, T. A., Karczewski, K. J., Zhao, H., and Snyder, M. (2011). Regulatory variation within and between species. Annu. Rev. Genomics Hum. Genet. 12, 327–346. doi: 10.1146/annurev-genom-082908-150139
Keywords: homotypic clusters, redundancy, regulatory networks, shadow enhancers, transcription factor binding sites
Citation: Payne JL and Wagner A (2015) Mechanisms of mutational robustness in transcriptional regulation. Front. Genet. 6:322. doi: 10.3389/fgene.2015.00322
Received: 11 September 2015; Accepted: 10 October 2015;
Published: 27 October 2015.
Edited by:
Ekaterina Shelest, Hans-Knoell Institute, GermanyReviewed by:
Bartek Wilczynski, University of Warsaw, PolandCopyright © 2015 Payne and Wagner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joshua L. Payne, am9zaHVhLnBheW5lQGlldS51emguY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.