 Sofia Francia1,2*
Sofia Francia1,2*- 1IFOM – FIRC Institute of Molecular Oncology, Milan, Italy
- 2Istituto di Genetica Molecolare, Consiglio Nazionale delle Ricerche, Pavia, Italy
Chromatin conformation shapes the environment in which our genome is transcribed into RNA. Transcription is a source of DNA damage, thus it often occurs concomitantly to DNA damage signaling. Growing amounts of evidence suggest that different types of RNAs can, independently from their protein-coding properties, directly affect chromatin conformation, transcription and splicing, as well as promote the activation of the DNA damage response (DDR) and DNA repair. Therefore, transcription paradoxically functions to both threaten and safeguard genome integrity. On the other hand, DNA damage signaling is known to modulate chromatin to suppress transcription of the surrounding genetic unit. It is thus intriguing to understand how transcription can modulate DDR signaling while, in turn, DDR signaling represses transcription of chromatin around the DNA lesion. An unexpected player in this field is the RNA interference (RNAi) machinery, which play roles in transcription, splicing and chromatin modulation in several organisms. Non-coding RNAs (ncRNAs) and several protein factors involved in the RNAi pathway are well known master regulators of chromatin while only recent reports show their involvement in DDR. Here, we discuss the experimental evidence supporting the idea that ncRNAs act at the genomic loci from which they are transcribed to modulate chromatin, DDR signaling and DNA repair.
Introduction
Genetic information is transmitted as DNA, yet is functional as RNA in cellular organisms. Genome integrity and as a consequence transcription fidelity is continuously harmed by DNA lesions. The cascade of events that starts with the detection of DNA lesions proceeds through signaling pathways and eventually triggers repair is known as the DNA damage response (DDR; Ciccia and Elledge, 2010). Among the different kinds of lesions, DNA double-strand breaks (DSBs) are the most deleterious and must be accurately repaired. It has been evident for several years that RNA is an important player in the regulation of chromatin and transcription (Holoch and Moazed, 2015) but only recently has RNA been shown to directly participate in preserving genome integrity (Sabin et al., 2013; d’Adda di Fagagna, 2014). This occurs in different ways: not only do RNA transcripts promote DDR signaling when DNA damage arises at their genetic loci (Francia et al., 2012) and guide homology-directed DNA repair (Wei et al., 2012; Gao et al., 2014) but also provide an intact copy of corrupted genetic information to be used as a template for DSB repair (Storici et al., 2007; Keskin et al., 2014). These studies reveal that transcripts can play an active role in preserving the integrity of the genome from which they are generated, raising the paradox that transcription can lead to DNA damage while transcripts are beneficial for efficient DNA damage signaling and repair at the loci from which they are transcribed.
The generation of a DSB causes the appearance of transcripts mapping to sequences flanking the site of damage (Francia et al., 2012; Michalik et al., 2012; Wei et al., 2012) referred to as DNA damage-response RNAs (DDRNAs; Francia et al., 2012) or double-strand break-induced RNAs (diRNAs; Wei et al., 2012). Importantly, transcripts map extremely close to DNA lesion and form also if DNA damage is generated at a locus devoid of promoter sequences and positioned away from other transcription units, excluding the possibility that transcripts are generated passively (Francia et al., 2012). This novel phenomenon appears to be evolutionarily conserved since it has been reported in plants (Wei et al., 2012), insects (Michalik et al., 2012), mouse and human cells (Francia et al., 2012; Gao et al., 2014). However, these findings are contradictory to the more commonly accepted view that sees transcription as an inherently mutagenic process (Bermejo et al., 2012; Kim and Jinks-Robertson, 2012; Hamperl and Cimprich, 2014; Sollier et al., 2014) and DDR activation antagonistic to transcription (Shanbhag et al., 2010). A unifying model that reconciles these contradictory theories is missing.
The use of deep sequencing technology lead to a revolution in our understanding of epigenetic mechanisms behind eukaryotic genome functions by uncovering the existence of ncRNAs which act as master chromatin regulators (Peschansky and Wahlestedt, 2014). DDRNAs are ncRNAs required for activation of the DDR (Francia et al., 2012; Wei et al., 2012), but the mechanism by which they function is still under investigation. Because DDRNAs are active at very low abundance (Francia et al., 2012), a characteristic consistent with other epigenetic regulatory transcripts (Wang et al., 2008; Gupta et al., 2010; Sado and Brockdorff, 2013), it is tempting to speculate that they control DDR activation and DNA repair by modulating chromatin. Here, we discuss how ncRNAs and factors of the RNAi pathway are known to influence chromatin and transcription and we comment about the influence that DDRNAs might have on chromatin during DDR activation and DNA repair.
Long ncRNAs in Chromatin and Transcription Regulation
Non-coding RNAs are generally classified based on their length: long ncRNAs (lncRNAs) are considered all non-coding transcript longer then 200 base-pairs up to several kilobases; while below this arbitrary threshold ncRNAs are generally referred to as small ncRNAs (sncRNAs). However, this gross classification only defines two overarching categories, each of which includes several different families based on ncRNA biogenesis, localization and function (St Laurent et al., 2015). Apart from the well known big classes of ribosomal RNAs, transfer RNAs, heterogeneous nuclear RNAs, small nucleolar RNAs and ribozymes, several novel lncRNAs have been identified and are often involved in the epigenetic regulation of the eukaryotic genome. Indeed, lncRNAs have the ability to recruit chromatin modifying complexes to defined genomic loci through the concomitant function of protein-binding domains, and sequence specificity (Cech and Steitz, 2014). A paradigm for lncRNA-directed site-specific chromatin modulation comes from the characterization of X-chromosome inactivation (XCI) for dosage compensation. XCI is achieved by the cells through the deposition of H3K27me3 repressive histone marks by the Polycomb repressive complex PRC2, which is recruited to only one X chromosome via a direct interaction with lncRNA transcript from the X inactive-specific transcript (Xist) locus (Plath et al., 2003; Galupa and Heard, 2015). Acting locally as a sequence-specific scaffold, Xist lncRNA is active at a very low copy number per cells (Zhao et al., 2008). Intriguingly, few studies reported the interactions of Xist lncRNA with BRCA1, a DNA repair protein required for homologous recombination (HR) which has also been proposed to participate in XCI (Ganesan et al., 2002, 2004). Nevertheless, the putative involvement of BRCA1 in XCI have been challenged in other reports (Xiao et al., 2007). Similarly to Xist, other chromatin bound ncRNA act in cis as few copies per cell to repress transcription. One example is the ncRNACCND1 whose expression is induced upon DNA damage at the 5′ regulatory regions of CCND1. ncRNACCND1 allosterically interacts with the RNA binding protein FUS/TLS and represses the expression of CCND1 by inhibiting the activity of p300 histone acetyltransferase locally (Wang et al., 2008). A number of other lncRNAs are induced upon DNA damage, often in a TP53-dependent manner. Two examples are the long intergenic non-coding RNA-p21 (lincRNA-p21; Huarte et al., 2010) and the lncRNA named p21 associated ncRNA DNA damage activated (PANDA; Hung et al., 2011), both transcribed upstream of the cell cycle regulator p21. LincRNAp21 binds to the transcription factor hnRNP-K and controls the proper silencing of TP53-repressed genes (Huarte et al., 2010) while PANDA binds to NF-YA, PRC1 and PRC2 to modulate the expression of pro-apoptotic and senescence genes upon DNA damage (Puvvula et al., 2014; Zhang and Peng, 2015).
Long non-coding RNA with antisense orientation control the expression of complementary transcripts in cis. Intriguing examples are the p15AS (p15 antisense transcript), which is expressed in antisense orientation to the cyclin-dependent kinase inhibitor p15 and controls its silencing by heterochromatin formation in a DICER-dependent manner (Yu et al., 2008), and the KCNQ1 antisense transcripts (KCNQ1ot1) which represses KCNQ1 by recruiting the H3K9 histone methyltransferase G9a and the PRC2 complex increasing the level of H3K9me3 and H3K27me3 in cis to its locus (Pandey et al., 2008). Similarly, the monoallelically expressed ncRNA AIR represses Slc22a3, Slc22a2, and Igf2r genes in cis by interacting and recruiting G9a (Nagano et al., 2008).
However, not all ncRNAs act by modulating chromatin in cis. The long intergenic ncRNA HOX transcript antisense RNA (HOTAIR), which originates from the HOXC locus, represses transcription of the HOXD locus in trans by recruiting PRC2 (Rinn et al., 2007; Gupta et al., 2010; Spitale et al., 2011). HOTAIR can directly interact with both PRC2 and the LSD1/CoREST/REST histone de-methylase complex, thereby inactivating transcription at target sites by coupling H3K27 methylation with de-methylation of H3K4 (Tsai et al., 2010). HOTAIR misregulation has been observed in a variety of cancers and might affect the expression of genes involved in apoptosis, growth and metastasis (Yu and Li, 2015). Another lncRNA relevant for cancer is ANRIL that, by interacting with the PRC1-component CBX7, contributes to repress the INK4b/ARF/INK4a locus and therefore limits senescence (Yap et al., 2010).
Non-coding RNA scaffolds not only induce repressive chromatin conformation but also positively influence transcription. This is the case of HOTTIP, a lincRNA transcribed from the 5′ tip of the HOXA locus that, by targeting WDR5/MLL complexes across HOXA and driving histone H3K4 trimethylation, coordinates the activation of HOXA genes at specific timing in vivo (Wang et al., 2011a). Chromosomal looping and high order structure necessary for gene activation have been proposed to be guided by HOTTIP lncRNA (Wang et al., 2011a), similar to what occurs for the ncRNAs with enhancer functions (eRNAs), other activating lncRNAs whose transcription stimulates the expression of neighboring genes (Orom et al., 2010; Lai et al., 2013; Andersson et al., 2014). Another way in which RNA changes the architecture of chromatin is by binding multiple proteins, as demonstrated by the PCGEM1 lncRNA that interacts with the androgen receptor (AR) and the activating chromatin effector Pygopus homolog 2 (Pygo2), thereby enhancing selective looping of AR-bound enhancers to target gene promoters (Yang et al., 2013).
Overall, a unifying theme in ncRNA-directed chromatin modification is the use of transcripts as a scaffold to achieve locus specific chromatin modification a well characterized mechanism that raises the apparent paradox that RNA-guided transcriptional silencing requires on-going transcription of the same locus.
An additional mechanism proposed for the targeting of these lncRNA to the correct locus is via lncRNA:DNA triplex formation. One example is the recently identified MEG3 lncRNA which represses genes of TGF-beta pathway by recruiting EZH2 via RNA:DNA triplex formation (Mondal et al., 2015). Previously lncRNA:DNA triplex formation was proposed in the case of the promoter associated pRNA, ncRNA complementary to the rDNA promoter that, by recruiting the DNA methyltransferase DNMT3b, mediates de novo CpG methylation and silencing of rRNA gene promoters (Schmitz et al., 2010).
In Table 1 are listed the lncRNAs discussed.
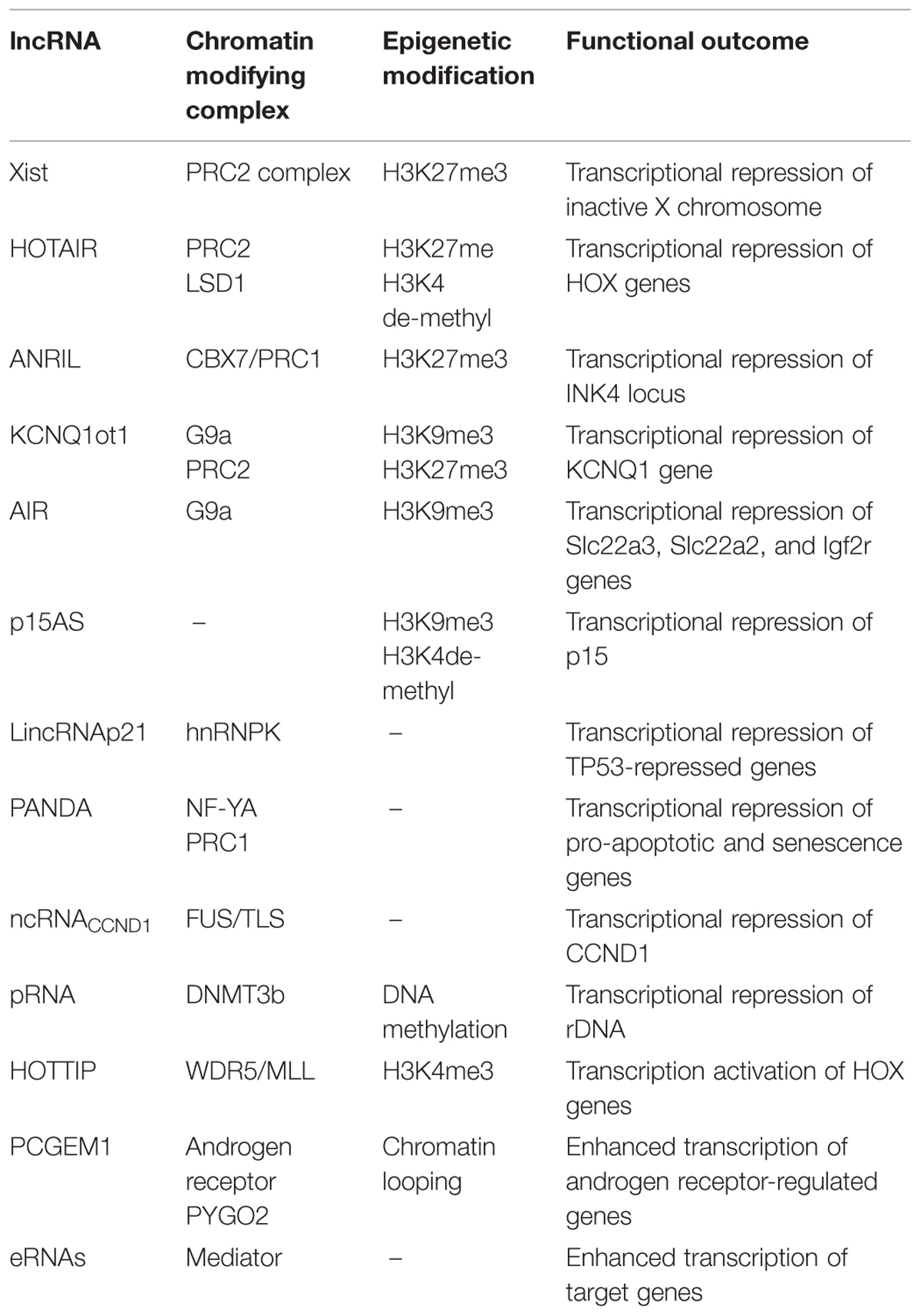
TABLE 1. List of lncRNAs discussed in the text. For each of them the interacting protein-complexes, the epigenetic modification and/or the related functional outcome are specified.
Small Non-coding RNA and the RNA Interference Machinery in DDR and Chromatin Modulation
Small non-coding RNAs (sncRNAs) are also divided in several different classes. Among a number of sncRNAs families, the more characterized are microRNAs (miRNAs or miR): DICER- and DROSHA-dependent sncRNAs which control the expression of more than 30% of human coding transcripts both at post-transcriptional and transcriptional level (Pasquinelli, 2015). Certainly, also DNA damage is a stimulus that results in altered expression of several miRNA families (Wouters et al., 2011). In particular, a group of miRNA promoters are targeted by the DDR effector and transcription factors TP53. Most relevant examples are the miR34a-c family and the miR29 family (Hermeking, 2012) which are induced in cells exposed to different genotoxic stress, and miR200c which increases following oxidative stress and triggers apoptosis and senescence by targeting ZEB1 (Magenta et al., 2011). The miR34a-c family represses the mRNA transcripts of several genes involved in cell proliferation and survival, such as BCL2, Cyclin D1 and E2 and cyclin-dependent kinases (CDK) 4 and 6, and therefore controls both apoptosis and senescence (Chang et al., 2007; Raver-Shapira et al., 2007; Tazawa et al., 2007; Yamazaki et al., 2012). The TP53 dependent up-regulation of the miR29 family, which represses the TP53 inhibitor Wip1 phosphatase, ultimately leads to TP53 induction and contributes to a positive feed-back loop during aging or chronic DDR activation (Ugalde et al., 2011). Other miRNAs induced upon DNA damage in a TP53-dependent manner and relevant for cancer are miR192, miR194, and miR215 (Braun et al., 2008). Intriguingly, mutant TP53 expressed in cancer not only loses its oncosuppressive properties but also regulates the expression of a different cohort of miRNAs responsible for its oncogenic activity (Liao et al., 2014). Moreover, TAp63, another member of the TP53 family, binds to DICER gene promoter and trans-activates it, directly controlling the processing of all miRNAs (Su et al., 2010).
DNA damage and DDR activation can also regulate miRNA expression by modulating the miRNA processing at maturation steps. Following DNA damage, the main DDR kinase Ataxia telangiectasia mutated ATM phosphorylates the KH-type splicing regulatory protein KSRP (Zhang et al., 2011), a multifunctional RNA-binding protein that interacts with both DROSHA and DICER (Trabucchi et al., 2009, 2011). KSRP was reported to interact with guanosine-rich regions in the terminal loop of miRNA precursors (pre-miRNA), and its ATM-dependent phosphorylation significantly enhanced the maturation of a cohort of pri-miRNA including pri-let 7, whereas KSRP mutations in the ATM-dependent phosphorylation sites impair its miRNA-regulating activity (Trabucchi et al., 2009, 2011; Gherzi et al., 2014). It was also shown that ATM dependent phosphorylation of exportin-5, which mediates the nuclear export of miRNAs, enhances its interaction with nuclear pores after DNA damage, defining an additional level of miRNA modulation by DNA-damage signaling (Wan et al., 2013).
Transcripts coding for key factors involved in DDR and chromatin modulation are also targeted by specific miRNAs at the post-transcriptional level following DNA damage. For example, one of the earliest events in the DDR is the ATM-dependent phosphorylation of the histone variant H2AX (Ciccia and Elledge, 2010), the coding mRNA of which is targeted by both miR24 (Lal et al., 2009; Brunner et al., 2012) and miR138 (Wang et al., 2011b; Yang et al., 2015a). Indeed, the overexpression of these miRNAs increases sensitivity to DNA damaging agents (Lal et al., 2009). ATM mRNA itself is also targeted by some miRNAs up-regulated in cancer, such as the N-myc-induced miR421 (Hu et al., 2010; Mansour et al., 2013), but also miR101 (Yan et al., 2010) and miR100 (Ng et al., 2010) whose overexpression enhances radiosensitivity by down-regulating ATM. Similarly, miR182 down regulates BRCA1 levels in human breast cancer cells, thus impeding repair by HR and sensitizing cell to treatment with PARP inhibitor and ionizing radiation (Moskwa et al., 2011; Krishnan et al., 2013).
However, a different group of miRNA has a positive impact on DDR activation. The recently described miR339-5p, and miR542-3p, indirectly stabilize TP53 by repressing MDM2 (Wang et al., 2014; Zhang et al., 2015). Overall, there is complex network of mutual modulations between DDR factors, DDR activation and miRNA biogenesis.
Transcripts originating from DNA breaks are processed by two ribonucleases of the RNAi machinery DROSHA and DICER, into sncRNAs (Francia et al., 2012; Michalik et al., 2012; Wei et al., 2012) and have been proposed to act in HR associated with the RNA-induced silencing complex (RISC) component ARGONAUTE 2 (AGO 2; Gao et al., 2014). RNA-processing enzymes have been linked to DDR activation in yeast as well (Manfrini et al., 2015). In agreement to the well-known function of RNAi in regulating gene expression by post-transcriptional gene silencing, DDRNAs have been shown to act as endogenous siRNA against truncated transcripts accumulating as a consequence of lesion generation (Michalik et al., 2012). However, sncRNA processed by the RNAi pathway are also known to silence gene expression at transcriptional level by modulating chromatin (Guang et al., 2008; Fagegaltier et al., 2009; Cernilogar et al., 2011). Indeed sncRNAs can act as guiding molecules for chromatin modifier enzymes in lower organisms (Fukagawa et al., 2004; Buhler et al., 2006; Castel and Martienssen, 2013; Keller et al., 2013). In mammals, RNAi factors such as DICER and AGO, historically considered merely cytoplasmatic, have been shown to localize in the nucleus of mammalian cells while maintaining activity (Janowski et al., 2006; Meister, 2013; Du Toit, 2014; Gagnon et al., 2014; Gao et al., 2014; White et al., 2014) and DICER-dependent, sequence-specific sncRNAs among which some miRNAs have been shown to control transcription at chromatin level in different contexts (Janowski et al., 2006; Kim et al., 2006; Gonzalez et al., 2008). This process is even enhanced in senescent cells, likely to suppress pro-proliferative genes (Benhamed et al., 2012). SncRNA-dependent chromatin modulation in mammals also occurs during deposition of heterochromatin at repetitive sequences (Giles et al., 2010) such as telomeres (Cao et al., 2009), centromeres (Halic and Moazed, 2010; Marasovic et al., 2013) and ribosomal DNA (Sinkkonen et al., 2010), as well as gene termination sites (Skourti-Stathaki et al., 2014). DICER-dependent sncRNAs associated with AGO1 and AGO2 have also been shown to modulate RNA polymerase II (RNAPII) elongation rate in mammalian cells by locally inducing chromatin compaction, thereby influencing splicing choices (Ameyar-Zazoua et al., 2012). A similar effect can be obtained at specific splicing sites via transfection of exogenous siRNA (Allo et al., 2009).
Although in all of these contexts sncRNAs recruit chromatin repressive complexes that induce silencing of target loci, DICER-dependent sncRNAs have also been proposed to play the opposite role of restraining the spreading of constitutive heterochromatin at the boundaries of centromeres (Keller et al., 2013) suggesting that their function might not be limited to heterochromatin formation and transcriptional silencing. Indeed, sequence-specific RNAs, often in complex with AGO2, have been show to map to transcriptional start sites (TSS-RNA) in human (Zamudio et al., 2014), mouse, chicken and fruit flies (Chitwood and Timmermans, 2010; Czech and Hannon, 2011) and small promoter-associated transcripts (PROMPTs) predicted to activate transcription through changes in chromatin structure have been described (Preker et al., 2008; Seila et al., 2008). Also, sncRNA-AGO2 complexes were shown to control transcription through reduction of H3K9 levels at sequence-specific promoter of a target gene, thereby inducing its activation (Li et al., 2006).
Increasing amounts of evidence also indicate that RNAi protein factors regulate gene expression co-transcriptionally through interaction with the transcriptional machinery (Castel and Martienssen, 2013). In Drosophila melanogaster Dicer 2 and Ago 2 interact with RNAPII and are required for positioning the transcription machinery on gene promoters in euchromatin (Cernilogar et al., 2011), while in mammals AGO1 associates with RNAPII and induces gene silencing at siRNA-targeted gene promoters (Kim et al., 2006). DROSHA and the DROSHA-containing complex known as microprocessor, have recently been proposed to control gene expression co-transcriptionally in two different ways: first acting at gene promoters by binding to hairpins of nascent transcripts, thus stabilizing them (Gromak et al., 2013), and second by cleaving stem-loops present in specific transcripts to induce transcription termination (Dhir et al., 2015). At sites of DNA damage, AGO2-associated sncRNAs influence the DNA repair pathway by recruiting the HR-factor Rad51 to actively transcribed regions (Gao et al., 2014). This is in agreement with the observation that transcriptionally active chromatin is preferentially repaired by HR (Aymard et al., 2014). Overall, several different examples exist of sequence-specific and RNAi-dependent sncRNAs acting at the level of chromatin in different organisms.
Endogenous Sources of Long Double-stranded RNA, Precursors of sncRNAs
SncRNAs mostly act as single stranded RNA (ssRNA) molecules, however, DICER is able to cleave only double stranded RNA (dsRNA) precursors (Holoch and Moazed, 2015). How such longer double stranded precursors are generated in different contexts is often unclear. The accepted model of miRNA biogenesis in which a longer ssRNA is folded into a stem-loop secondary structure (Kim et al., 2009) does not always apply to non-miRNA loci. A recently proposed source of dsRNA precursors is oppositely oriented transcription of two independent genetic unit, leading to the formation of partially complementary transcripts able to anneal with each other and form long dsRNA molecules (Gullerova and Proudfoot, 2012; Skourti-Stathaki et al., 2014). Increasing evidence points to the fact that dsRNA generated by overlapping transcripts form relatively frequently in vivo. In fact, DICER inactivation leads to increased levels of dsRNAs and consequent activation of the interferon response, in agreement with the idea that transcripts able to form long dsRNAs are fairly abundant and are indeed substrate for DICER processing (White et al., 2014). In support of this notion, overlapping, inversely oriented transcripts originating at regions with high gene density, as well as bidirectional transcription at neighboring transcription units, are not uncommon in the genome of vertebrates (Adachi and Lieber, 2002; Okamura et al., 2008; Orekhova and Rubtsov, 2013). It has been proposed that a common feature of promoters in eukaryotic organisms is bidirectionality (Seila et al., 2009). However, this model is still controversial and other studies have proposed instead that promoters are intrinsically uni-directional (Duttke et al., 2015). An example of widespread and ubiquitous antisense transcripts is the recently characterized class of Natural Antisense Transcripts (NATs), regulatory RNA molecules influencing the expression of the complementary gene (Faghihi and Wahlestedt, 2009; Khorkova et al., 2014). These antisense transcripts may have been largely elusive because they are unstable and rapidly processed, similar to what happens with the recently identified nascent transcripts generated by divergent transcription initiation at promoter and enhancer (Weingarten-Gabbay and Segal, 2014).
Another interesting source of dsRNA has been proposed at human promoters where an engaged RNAPII is loaded in an opposite orientation respect to the gene and synthesizes complementary RNAs of few hundred bases but does not elongate beyond the promoter (Core and Lis, 2008; Core et al., 2008). In support of this model, phosphorylation of Tyrosine 1 on the RNAPII C-terminal domain (CTD) has been associated with antisense transcription at promoters and enhancers of mammalian cells (Descostes et al., 2014). Moreover, at RNAPII pausing sites upstream to promoters dsRNAs are known to accumulate (Flynn et al., 2011). Therefore, it has been proposed that a paused promoter-proximal RNAPII can synthesize both RNA strands that then anneal forming a dsRNA, which can be processed by DICER (Du Toit, 2014). Bound to AGO, these sncRNAs are believed to influence the local chromatin conformation, thus fine-tuning the switch between paused and elongating RNAPII. A similar mechanism has been described at transcription termination sites where RNAPII pausing is caused by R-loops (Skourti-Stathaki et al., 2014) and sncRNAs associated with AGO proteins are believed to recruit chromatin modifier enzymes and increase local chromatin compaction. Likewise, RNAPII is known to pause at alternative splice sites (Allo et al., 2009; Saint-Andre et al., 2011) where RNAi-dependent sncRNAs accumulate, further suggesting that paused RNAPII may generate antisense transcription and therefore dsRNA molecules. It is therefore plausible that in the physiological context of gene transcription, DNA damage and DDR signaling may induce RNAPII pausing and consequent synthesis of complementary transcripts. Once annealed, these transcripts may provide a dsRNA precursor for DICER-dependent DDRNA biogenesis that, in turn, may stimulate DDR activation and guide-chromatin modulation (see Figure 1 for a schematic representation).
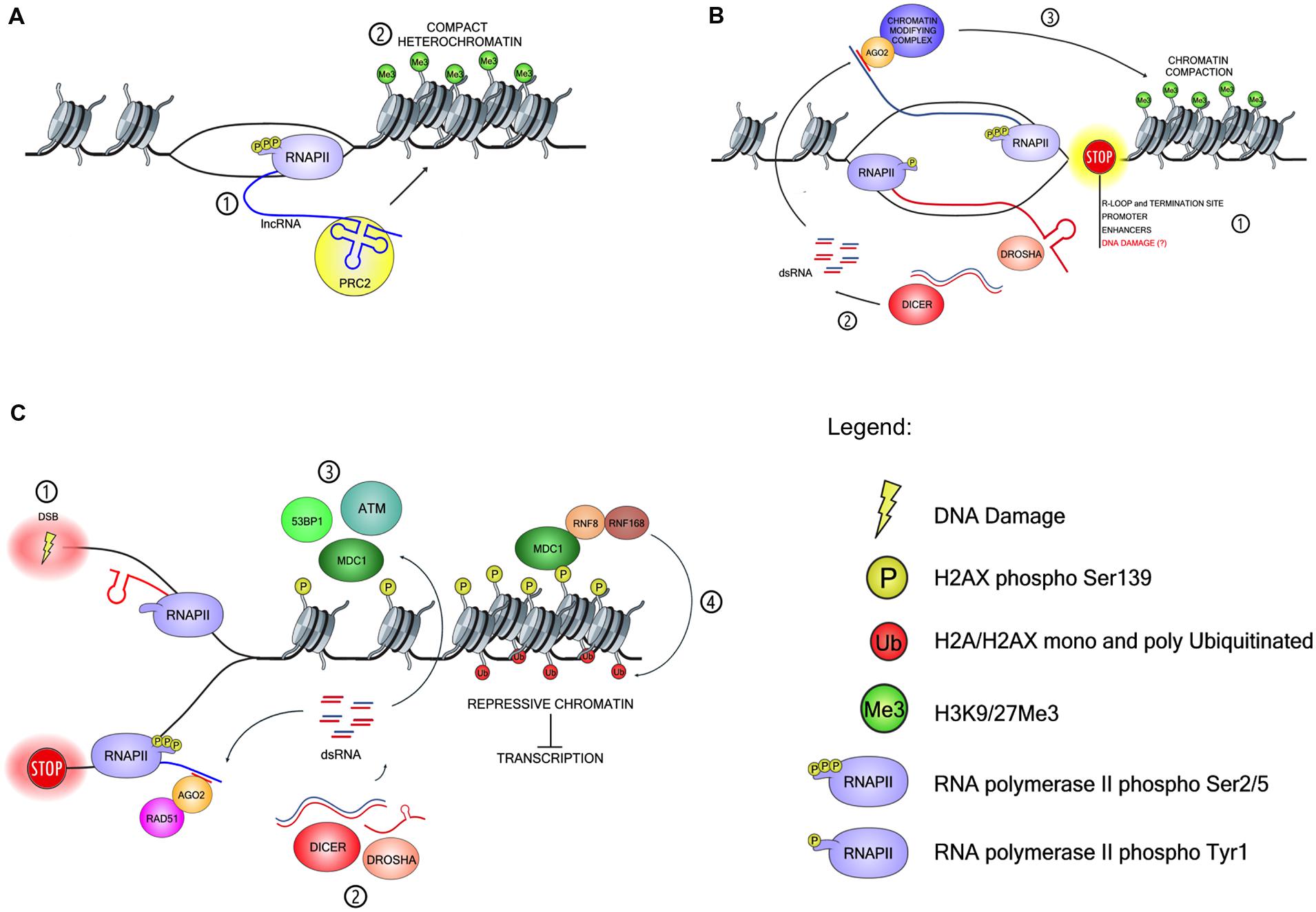
FIGURE 1. Models of ncRNA-mediated site-specific chromatin modification and DDR modulation. (A) Schematic representation of long ncRNA (lncRNA) in transcriptional repression. (1) RNAPII dependent transcripts fold in protein-binding domains and (2) recruit histone-modifying complexes such as PRC2 to chromatin in cis. (B) Schematic model of small ncRNAs (sncRNAs) generation and chromatin modulation at RNAPII pausing sites. (1) Pausing of RNAPII is induced in physiological condition at promoter and enhancer sequences and at transcription-termination sites prone to form R-loops, but could also occur in the presence of DNA lesions. (2) RNAPII pausing stimulates the loading of another RNAPII in opposite orientation on the complementary DNA template and generates antisense transcripts. DsRNA precursors are then processed by the RNAi-machinery into sncRNA. (3) ARGONAUTE 2 (AGO2) forms a complex with such sequence-specific sncRNA and guide chromatin-modifying enzymes to the pausing site via nascent RNA:sncRNA interaction. (C) Schematic model for sncRNAs generation and function at site of DNA damage. (1) Double-strand break (DSB) generation induces stalling of RNAPII and synthesis of partially complementary transcripts. (2) Stem-loop like secondary structures of the transcripts or long dsRNAs precursors are processed by DROSHA and DICER into sncRNA, DDRNA. (3) DDRNA stimulates DDR activation via an unknown mechanism. Initially, DDR activation leads to chromatin de-compaction, which may increase transcription and favors ATM activation. In turn, ATM induces mono- and poly-ubiquitination of H2A/H2AX by RNF8/RNF168 ubiquitin ligases, consequent chromatin compaction and transcriptional silencing (4). Chromatin compaction might promote reciprocal interactions between DDR proteins through their increased density, further boosting DDR activation in the absence of additional transcription.
In plants and yeasts an additional source of dsRNA precursors is provided by RNA dependent RNA polymerases (RdRP), able to synthesize the complementary strand of a single stranded RNA template (Ahlquist, 2002). Most higher eukaryotes also encode putative RdRPs, but in mammals RdRP activity associated with RNAi has remained elusive (Maida and Masutomi, 2011). Recently, it has been proposed that mammalian RNAPII retains RdRP activity similar to that of budding yeast RNAPII (Lehmann et al., 2007) and is involved in generating complementary strands of non-coding transcripts to target them for degradation (Wagner et al., 2013). A similar RdRP activity has been postulated for human telomerase TERT, which was proposed to produce telomeric dsRNAs then processed into sncRNA in a DICER-dependent manner (Maida et al., 2009; Maida and Masutomi, 2011) suggesting that TERT may contribute to amplifying sncRNA biogenesis from telomeric sequences. However, the putative RdRP activity of TERT is still under debate (Martinez and Blasco, 2011). At site of DNA damage, diRNA biogenesis requires RdRP activity in plants (Wei et al., 2012), suggesting that a residual RdRP activity of RNAPII could be involved in DDRNA biogenesis in mammalian cells too.
Chromatin Compaction and Relaxation at Sites of DNA Damage
Both condensation and relaxation have been show to occur at damaged chromatin. Recently, it has been shown that chromatin compaction after DNA damage is a secondary event that follows an initial chromatin relaxation (Burgess et al., 2014). Adjacent to DSBs, local chromatin de-condensation as well as histone reorganization and eviction have been previously observed in mammalian cells (Kruhlak et al., 2006; Ziv et al., 2006; Berkovich et al., 2007; Price and D’Andrea, 2013). It is also well established that important chromatin remodeling complexes such as SWI/SNF (Smith-Roe et al., 2015), CHD (Stanley et al., 2013), INO80 (Kashiwaba et al., 2010), SMARCA5 (Smeenk et al., 2013), ISWI (Aydin et al., 2014) and the poly (ADP-ribosyl)ation dependent, nucleosome-repositioning enzyme ALC1 (Ahel et al., 2009), are recruited to DSBs. The putative purpose of this recruitment is to remove nucleosomes from damaged DNA, shift back the position of nucleosomes and substitute histone variants (Panier and Durocher, 2013; Price and D’Andrea, 2013; Smeenk et al., 2013). Moreover, well-known histone marks characteristic of open chromatin are deposited after DNA damage in a DDR-dependent manner: histones H2A and H4 acetylation by the histone acetyl transferases TIP60 (Sun et al., 2009) and MOF (Sharma et al., 2010) and the monoubiquitination of histone H2B by RNF20/RNF40 (Miller and Jackson, 2012), have been extensively shown to be deposited at site of DNA damage. Interestingly, both MOF (Akhtar and Becker, 2000) and RNF20/40 are well known transcription activators (Shiloh et al., 2011) that also modulate chromatin de-condensation at transcribed regions, suggesting that similar chromatin modifications might be required for DDR activation.
A number of other processes that promote chromatin relaxation and are required for proper DDR activation have been reported, such as the mobilization of heterochromatin-associated factors HP1 and KAP-1 (Ziv et al., 2006; Ayoub et al., 2008; Noon et al., 2010; Bolderson et al., 2012) away from damaged loci and the re-localization of DNA breaks at the periphery of heterochromatin or inside euchromatin (Chiolo et al., 2011; Jakob et al., 2011). Collectively, these reports prove that active chromatin de-compaction at sites of DNA damage is a controlled process, rather than a side effect given by DNA breakage, raising questions about the possible mechanism by which both chromatin relaxation and re-compaction are modulated by DDR. Both events seem to play a functional role in DDR since the loss of the initial chromatin de-compaction but also the induction of persistent de-compaction on damaged chromatin impair DDR signaling (Burgess et al., 2014) and repair (Kakarougkas et al., 2014; Kalousi et al., 2015). On the other hand, forced chromatin compaction can also induce ATM signaling in the absence of physical DNA damage (Kaidi and Jackson, 2013). Overall, not only chromatin compaction but also local chromatin relaxation is intimately associated with DDR signaling at damaged chromatin. Antagonistic signals during DDR signaling have been implicated in providing boundaries to its spread along chromatin (Panier and Durocher, 2013). Thus, the fact that these two apparently contradictory phenomena coexist at site of damage may suggest that they finely tune each other.
During DDR activation two consecutive steps can be distinguished: the first consists of a primary, direct recognition of DNA lesions by DDR sensors, while the second consists of an amplification phase mediated by chromatin modifications that recruit and retain DDR mediators and kinases in close proximity, promoting their interaction (Fernandez-Capetillo et al., 2003; Bekker-Jensen and Mailand, 2010). A distinct, yet not alternative scenario envisages that the initial recognition of a DNA lesion requires chromatin de-compaction to allow the binding of DDR-sensors to damaged DNA, while the following DDR signal amplification might be enhanced by compaction, which increase the local signal and favors the reciprocal interaction among DDR proteins. Additionally, the initial chromatin de-compaction could indeed promote local transcription that generates RNA precursors for DDRNA biogenesis, in turn required to boost DDR signaling and thus repair (Figure 1), (Francia et al., 2012; d’Adda di Fagagna, 2014).
The Intricate Relationship between DDR Signaling and Transcription
A genomic locus is repaired more efficiently when actively transcribed than when kept silent (Goodarzi et al., 2008; Chaurasia et al., 2012; Fong et al., 2013), an observation suggesting that on-going transcription can play a positive role in DNA repair. Along the same line, RNAPII and the transcription machinery have been proposed to act as genome scanner, detecting and signaling the presence of DNA damage (Lindsey-Boltz and Sancar, 2007; Montecucco and Biamonti, 2013; Winsor et al., 2013). In this scenario, the recent understanding that most of our genome is pervasively transcribed at a basal level (Kapranov et al., 2007; Berretta and Morillon, 2009; Clark et al., 2011) emphasizes how prominent the role of transcribing RNAPII could be in the detection of damaged DNA and activation of consequent repair. More intriguingly, inhibition of RNAPII elongation by specific antibodies induces the ATM dependent phosphorylation of the tumor protein TP53 in the absence of DNA damage and in a replication-independent manner (Ljungman et al., 2001; Derheimer et al., 2007), highlighting that DDR might be activated upon inhibition of RNAPII elongation.
One related fascinating topic is how transcription is modulated around DNA breaks. It has been shown that DNA breaks can cause transcription inhibition of adjacent genes via two complementary mechanisms: by surrounding the damaged locus with repressive chromatin modifications and/or by excluding elongating RNAPII from damaged sites. An elegant study in a conveniently engineered cellular system shows that a cluster of DSBs generated by a restriction enzyme at a repetitive locus stimulates DDR-mediated chromatin silencing that spreads for kilobases from the damaged locus (Shanbhag et al., 2010). It is now well established that DNA damage-induced ATM activation recruits Ring Finger protein 8 (RNF8) and RNF168 E3-ubiquitin ligases that ubiquitinate histones H2A/H2AX in chromatin surrounding the breaks (Jackson and Durocher, 2013; Brown and Jackson, 2015). This event is believed to cause chromatin compaction and has been linked to repression of transcription units positioned a few kilobases downstream from the cluster of DSBs (Shanbhag et al., 2010). Importantly, transcription inhibition induced by DSB generation can be reversed by ATM inhibition or RNF8 and RNF168 co-depletion while still in the presence of DNA damage (Shanbhag et al., 2010), suggesting that transcription is not physically impeded by the DNA lesions itself, but is instead actively controlled by DDR-activation. In the same cellular system it has been shown that the chromatin remodeling complex Polybromo BRG1 (Brahma Related Gene 1) Associated Factor (PBAF) is phosphorylated by ATM and, together with the Polycomb repressive complexes PRC1 and PRC2, is required for transcription repression in cis to the DSB cluster (Kakarougkas et al., 2014).
Interestingly, the down-regulation of transcriptional units due to a nearby damaged locus correlates with a reduced presence of the elongating RNAPII isoform but not of total RNAPII at the γH2AX-positive domain (Shanbhag et al., 2010), suggesting that RNAPII might be initially pausing at damaged chromatin rather than being excluded from it. This hypothesis is in line with the recent observation that cells treated with DSB-inducing agents show an increase of RNAPII association with chromatin, rather than a decrease (Britton et al., 2014). Several other reports suggest the opposite, showing the exclusion of RNAPII from γH2AX positive chromatin when laser micro-irradiation is used to deliver a high dose of DNA damage in a confined area (Chou et al., 2010; Miller et al., 2010; Seiler et al., 2011; Polo et al., 2012).
Repression of transcription adjacent to a DNA lesion is also suggested by the fact that several heterochromatin markers and heterochromatinization-inducing enzymes are present at sites of DSBs. Some examples are HP1 (Baldeyron et al., 2011), the histone methyltransferases SUV3-9 and PRDM2, responsible for the deposition of the H3K9me3 (Ayrapetov et al., 2014), the histone deacetylases HDAC1 and HDAC2 (Luijsterburg et al., 2009; Zarebski et al., 2009; Miller et al., 2010) and the histone methyltransferase MMSET (Pei et al., 2011). Moreover, chromatin-repressive complexes, such as PBAF, Polycomb and the Nucleosome Remodeling Deacetylase NuRD complex, associate with damaged chromatin (Chou et al., 2010; Kakarougkas et al., 2014; Leung et al., 2014). Overall, it is well established that DDR activation induces a signaling pathway that results in repression of adjacent genes, a phenomenon that might be useful to counteract the accumulation of corrupted or truncated transcripts and to avoid collisions between transcription and repair intermediates.
Conversely, it has been proposed that when a single DSB is generated inside a gene, RNAPII elongation rate is only transiently arrested by DNAPKcs-dependent DDR signaling (Pankotai et al., 2012). DNA-PKcs inhibition allows transcription elongation to take place, regardless of the presence of the break, and possibly even across the break (Pankotai et al., 2012). This provocative interpretation is in line with the in vitro observation that RNA polymerases can bypass different types of DNA lesions, including DNA breaks, by mis-incorporating ribonucleotides, a process known as transcriptional mutagenesis (Doetsch, 2002; Saxowsky and Doetsch, 2006; Pankotai and Soutoglou, 2013; Xu et al., 2014). Thus, a possible model to reconcile these contradictory observations predicts that DDR activation modifies chromatin to induce transcriptional repression at heavily damaged chromosomes, while a single DNA break causes only a transient RNAPII pausing, thus enabling a rapid restart after DNA repair.
RNAi Pathway and Innate Immunity against Invading Genetic Elements
sncRNAs are known to take part in other essential events linking chromatin and transcription modulation with genome instability, such as genome defense from invading nucleic acids and transposable elements (TE; Fagegaltier et al., 2009; Cam, 2010). In this context, RNAi-dependent sncRNAs guide the deposition of repressive histone marks at invading genetic element through base pairing with nascent transcripts.
It is easy to imagine that complementary transcripts might originate from the expression of repetitive elements such as Long Interspersed Nuclear Elements (LINE) and Short Interspersed Nuclear Elements (SINE), which derive from retrotransposons and Alu repeats. LINE and SINE are present in millions of copies in our genome occupying respectively 17% and 11% of it. Indeed, by inducing transcriptional gene silencing of repetitive sequences, RNAi represents one of the main forms of innate immunity against viruses and invading genetic elements in all organisms (Obbard et al., 2009). From archaebacteria, the RNAi related CRISPR/Cas immune pathway exploits an RNA-guide to direct sequence specific DNA cleavage against invading phages or plasmids (Rimer et al., 2014). It has been recently demonstrated that DNA target recognition involves its transcription and that cleavage by Cas can occur both on DNA and RNA complementary molecules in the case of Type III CRISPR-Cas system (Samai et al., 2015).
In plants, RNAi is a major source of immunity functioning through induction of exogenous DNA silencing via methylation (Kim and Zilberman, 2014), while in animals the evolution of a protein-based adaptive immune response has partially reduced the need of antiviral RNAi activity. Still, TEs and viruses have invaded and profoundly shaped the mammalian genome (Buckley and Lis, 2014). It has been estimated that almost 40% of our genome is composed of invading genetic elements such as integrated viruses and TEs, which, once activated, are potent inducers of DNA damage (Goodier and Kazazian, 2008). In human cells, DICER has been shown to play an important role in counteracting Alu-dependent dsRNA accumulation and toxicity (Kaneko et al., 2011).
RNA-mediated control of transposon activation is particularly active in the mammalian germ line where a devoted class of small-interfering RNA, PIWI-interacting RNA (piRNA), has been linked to both epigenetic and post-transcriptional gene silencing of retrotransposons (Siomi et al., 2011; Castel and Martienssen, 2013). piRNAs are not expressed in somatic cells but are aberrantly re-expressed in cancer cells, suggesting that TE could be reactivated by the large amount of DNA damage experienced by cancer cells and in turn cause additional deleterious mutations promoting cellular transformation (Cheng et al., 2011). To protect themselves, viruses and TEs suppress RNAi and try to escape RNAi control by evolving rapidly. It has been proposed that genes encoding for the RNAi factors have co-evolved quickly with the viruses that they counteract (Obbard et al., 2006).
Quelling: An Ancient Link between RNAi and Genome Instability
Another interesting phenomenon linking RNAi-guided gene silencing and control of genome stability is quelling in Neurospora crassa (Pickford et al., 2002; Fulci and Macino, 2007; Lee et al., 2009) a post transcriptional gene silencing triggered by multiple copies of a transgene, similar to co-suppression in Caenorhabditis elegans (Pickford et al., 2002; Fulci and Macino, 2007; Lee et al., 2009). During quelling, ‘abortive’ transcripts from the transgene are synthesized by the DNA-dependent RNA polymerase QDE-1 and amplified by the RdRP activity of QDE-1 itself (Cogoni and Macino, 1999a,b). DsRNA is then processed by dicing enzymes, generating sncRNAs necessary for post transcriptional gene silencing of the transgene itself (Catalanotto et al., 2004). Both sncRNAs and the Argonaute-related protein, QDE-2, whose mutation yields DNA damage sensitivity, are overexpressed upon DNA damage (Lee et al., 2009), a fact suggesting that quelling might be involved in DDRNAs biogenesis in N. crassa. Similarly to what occur during quelling, DNA breaks in endogenous epetitive sequences might induce the expression of complementary transcript processed into sncRNAs, which in turn stimulate repair and silencing of the repetitive DNA (Yang et al., 2015b).
Conclusion
It seems that sncRNAs play an evolutionarily conserved role in both chromatin dynamics and genome stability. Thus, the discoveries of DDRNAs and diRNAs highlight the existence of a consistent, yet previously unknown, sncRNA-mediated layer in the regulation of DDR signaling and DNA repair. This discovery raises the possibility of novel approaches in designing chemical or other pharmacological molecules that might act on, or through, the DDR pathway in hopes of identifying novel strategies for disease treatment (Pearl et al., 2015). The idea that DDRNAs control DDR activation is likely interconnected with the fact that such sncRNAs may also modulate chromatin around damaged DNA. However, the exact role played by DDRNA in controlling chromatin conformation is still poorly defined and needs further investigation.
Given the involvement of DDR in a number of physiological and pathological processes such as immunodeficiency, neurodegeneration, sterility and development (Jackson and Bartek, 2009), combined with the potential tumor suppressive functions of both DDR and chromatin mediated gene silencing (Sulli et al., 2012), the study of the molecular mechanisms by which DDRNAs act is of tremendous interest.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SF thanks C. Jones-Weinert for correcting and editing the manuscript and M. Cabrini for assembling Figure 1. SF thank S. Sharma, J. Vermezovic, F. d’Adda di Fagagna, G. Biamonti, and A. Montecucco for reading and commenting the manuscript and all member of F. d’Adda di Fagagna laboratory for stimulating discussions. SF is supported by Collegio Ghislieri and Fondazione Cariplo (Grant N. 2014-1215).
References
Adachi, N., and Lieber, M. R. (2002). Bidirectional gene organization: a common architectural feature of the human genome. Cell 109, 807–809. doi: 10.1016/S0092-8674(02)00758-4
Ahel, D., Horejsi, Z., Wiechens, N., Polo, S. E., Garcia-Wilson, E., Ahel, I., et al. (2009). Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325, 1240–1243. doi: 10.1126/science.1177321
Ahlquist, P. (2002). RNA-dependent RNA polymerases, viruses, and RNA silencing. Science 296, 1270–1273. doi: 10.1126/science.1069132
Akhtar, A., and Becker, P. B. (2000). Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell 5, 367–375. doi: 10.1016/S1097-2765(00)80431-1
Allo, M., Buggiano, V., Fededa, J. P., Petrillo, E., Schor, I., de la Mata, M., et al. (2009). Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat. Struct. Mol. Biol. 16, 717–724. doi: 10.1038/nsmb.1620
Ameyar-Zazoua, M., Rachez, C., Souidi, M., Robin, P., Fritsch, L., Young, R., et al. (2012). Argonaute proteins couple chromatin silencing to alternative splicing. Nat. Struct. Mol. Biol. 19, 998–1004. doi: 10.1038/nsmb.2373
Andersson, R., Gebhard, C., Miguel-Escalada, I., Hoof, I., Bornholdt, J., Boyd, M., et al. (2014). An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461. doi: 10.1038/nature12787
Aydin, O. Z., Vermeulen, W., and Lans, H. (2014). ISWI chromatin remodeling complexes in the DNA damage response. Cell Cycle 13, 3016–3025. doi: 10.4161/15384101.2014.956551
Aymard, F., Bugler, B., Schmidt, C. K., Guillou, E., Caron, P., Briois, S., et al. (2014). Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 21, 366–374. doi: 10.1038/nsmb.2796
Ayoub, N., Jeyasekharan, A. D., Bernal, J. A., and Venkitaraman, A. R. (2008). HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature 453, 682–686. doi: 10.1038/nature06875
Ayrapetov, M. K., Gursoy-Yuzugullu, O., Xu, C., Xu, Y., and Price, B. D. (2014). DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. U.S.A. 111, 9169–9174. doi: 10.1073/pnas.1403565111
Baldeyron, C., Soria, G., Roche, D., Cook, A. J., and Almouzni, G. (2011). HP1alpha recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J. Cell Biol. 193, 81–95. doi: 10.1083/jcb.201101030
Bekker-Jensen, S., and Mailand, N. (2010). Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair 9, 1219–1228. doi: 10.1016/j.dnarep.2010.09.010
Benhamed, M., Herbig, U., Ye, T., Dejean, A., and Bischof, O. (2012). Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nat. Cell Biol. 14, 266–275. doi: 10.1038/ncb2443
Berkovich, E., Monnat, R. J. Jr., and Kastan, M. B. (2007). Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 9, 683–690. doi: 10.1038/ncb1599
Bermejo, R., Lai, M. S., and Foiani, M. (2012). Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol. Cell 45, 710–718. doi: 10.1016/j.molcel.2012.03.001
Berretta, J., and Morillon, A. (2009). Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep. 10, 973–982. doi: 10.1038/embor.2009.181
Bolderson, E., Savage, K. I., Mahen, R., Pisupati, V., Graham, M. E., Richard, D. J., et al. (2012). Kruppel-associated Box (KRAB)-associated co-repressor (KAP-1) Ser-473 phosphorylation regulates heterochromatin protein 1beta (HP1-beta) mobilization and DNA repair in heterochromatin. J. Biol. Chem. 287, 28122–28131. doi: 10.1074/jbc.M112.368381
Braun, C. J., Zhang, X., Savelyeva, I., Wolff, S., Moll, U. M., Schepeler, T., et al. (2008). p53-responsive micrornas 192 and 215 are capable of inducing cell cycle arrest. Cancer Res. 68, 10094–10104. doi: 10.1158/0008-5472.CAN-08-1569
Britton, S., Dernoncourt, E., Delteil, C., Froment, C., Schiltz, O., Salles, B., et al. (2014). DNA damage triggers SAF-A and RNA biogenesis factors exclusion from chromatin coupled to R-loops removal. Nucleic Acids Res. 42, 9047–9062. doi: 10.1093/nar/gku601
Brown, J. S., and Jackson, S. P. (2015). Ubiquitylation, neddylation and the DNA damage response. Open Biol. 5, 150018. doi: 10.1098/rsob.150018
Brunner, S., Herndler-Brandstetter, D., Arnold, C. R., Wiegers, G. J., Villunger, A., Hackl, M., et al. (2012). Upregulation of miR-24 is associated with a decreased DNA damage response upon etoposide treatment in highly differentiated CD8(+) T cells sensitizing them to apoptotic cell death. Aging Cell 11, 579–587. doi: 10.1111/j.1474-9726.2012.00819.x
Buckley, M. S., and Lis, J. T. (2014). Imaging RNA Polymerase II transcription sites in living cells. Curr. Opin. Genet. Dev. 25, 126–130. doi: 10.1016/j.gde.2014.01.002
Buhler, M., Verdel, A., and Moazed, D. (2006). Tethering RITS to a nascent transcript initiates RNAi- and heterochromatin-dependent gene silencing. Cell 125, 873–886. doi: 10.1016/j.cell.2006.04.025
Burgess, R. C., Burman, B., Kruhlak, M. J., and Misteli, T. (2014). Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 9, 1703–1717. doi: 10.1016/j.celrep.2014.10.060
Cam, H. P. (2010). Roles of RNAi in chromatin regulation and epigenetic inheritance. Epigenomics 2, 613–626. doi: 10.2217/epi.10.46
Cao, F., Li, X., Hiew, S., Brady, H., Liu, Y., and Dou, Y. (2009). Dicer independent small RNAs associate with telomeric heterochromatin. RNA 15, 1274–1281. doi: 10.1261/rna.1423309
Castel, S. E., and Martienssen, R. A. (2013). RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet. 14, 100–112. doi: 10.1038/nrg3355
Catalanotto, C., Pallotta, M., ReFalo, P., Sachs, M. S., Vayssie, L., Macino, G., et al. (2004). Redundancy of the two dicer genes in transgene-induced posttranscriptional gene silencing in Neurospora crassa. Mol. Cell. Biol. 24, 2536–2545. doi: 10.1128/MCB.24.6.2536-2545.2004
Cech, T. R., and Steitz, J. A. (2014). The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94. doi: 10.1016/j.cell.2014.03.008
Cernilogar, F. M., Onorati, M. C., Kothe, G. O., Burroughs, A. M., Parsi, K. M., Breiling, A., et al. (2011). Chromatin-associated RNA interference components contribute to transcriptional regulation in Drosophila. Nature 480, 391–395. doi: 10.1038/nature10492
Chang, T. C., Wentzel, E. A., Kent, O. A., Ramachandran, K., Mullendore, M., Lee, K. H., et al. (2007). Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 26, 745–752. doi: 10.1016/j.molcel.2007.05.010
Chaurasia, P., Sen, R., Pandita, T. K., and Bhaumik, S. R. (2012). Preferential repair of DNA double-strand break at the active gene in vivo. J. Biol. Chem. 287, 36414–36422. doi: 10.1074/jbc.M112.364661
Cheng, J., Guo, J. M., Xiao, B. X., Miao, Y., Jiang, Z., Zhou, H., et al. (2011). piRNA, the new non-coding RNA, is aberrantly expressed in human cancer cells. Clin. Chim. Acta 412, 1621–1625. doi: 10.1016/j.cca.2011.05.015
Chiolo, I., Minoda, A., Colmenares, S. U., Polyzos, A., Costes, S. V., and Karpen, G. H. (2011). Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144, 732–744. doi: 10.1016/j.cell.2011.02.012
Chitwood, D. H., and Timmermans, M. C. (2010). Small RNAs are on the move. Nature 467, 415–419. doi: 10.1038/nature09351
Chou, D. M., Adamson, B., Dephoure, N. E., Tan, X., Nottke, A. C., Hurov, K. E., et al. (2010). A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. U.S.A. 107, 18475–18480. doi: 10.1073/pnas.1012946107
Ciccia, A., and Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. doi: 10.1016/j.molcel.2010.09.019
Clark, M. B., Amaral, P. P., Schlesinger, F. J., Dinger, M. E., Taft, R. J., Rinn, J. L., et al. (2011). The reality of pervasive transcription. PLoS Biol. 9:e1000625. doi: 10.1371/journal.pbio.1000625
Cogoni, C., and Macino, G. (1999a). Gene silencing in Neurospora crassa requires a protein homologous to RNA-dependent RNA polymerase. Nature 399, 166–169. doi: 10.1038/20215
Cogoni, C., and Macino, G. (1999b). Posttranscriptional gene silencing in Neurospora by a RecQ DNA helicase. Science 286, 2342–2344. doi: 10.1126/science.286.5448.2342
Core, L. J., and Lis, J. T. (2008). Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science 319, 1791–1792. doi: 10.1126/science.1150843
Core, L. J., Waterfall, J. J., and Lis, J. T. (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848. doi: 10.1126/science.1162228
Czech, B., and Hannon, G. J. (2011). Small RNA sorting: matchmaking for Argonautes. Nat. Rev. Genet. 12, 19–31. doi: 10.1038/nrg2916
d’Adda di Fagagna, F. (2014). A direct role for small non-coding RNAs in DNA damage response. Trends Cell Biol. 24, 171–178. doi: 10.1016/j.tcb.2013.09.008
Derheimer, F. A., O’Hagan, H. M., Krueger, H. M., Hanasoge, S., Paulsen, M. T., and Ljungman, M. (2007). RPA and ATR link transcriptional stress to p53. Proc. Natl. Acad. Sci. U.S.A. 104, 12778–12783. doi: 10.1073/pnas.0705317104
Descostes, N., Heidemann, M., Spinelli, L., Schuller, R., Maqbool, M. A., Fenouil, R., et al. (2014). Tyrosine phosphorylation of RNA polymerase II CTD is associated with antisense promoter transcription and active enhancers in mammalian cells. Elife 3:e02105. doi: 10.7554/eLife.02105
Dhir, A., Dhir, S., Proudfoot, N. J., and Jopling, C. L. (2015). Microprocessor mediates transcriptional termination of long noncoding RNA transcripts hosting microRNAs. Nat. Struct. Mol. Biol. 22, 319–327. doi: 10.1038/nsmb.2982
Doetsch, P. W. (2002). Translesion synthesis by RNA polymerases: occurrence and biological implications for transcriptional mutagenesis. Mutat. Res. 510, 131–140. doi: 10.1016/S0027-5107(02)00258-0
Du Toit, A. (2014). RNA interference: nuclear Dicer makes the cut. Nat. Rev. Mol. Cell Biol. 15, 366–367. doi: 10.1038/nrm3813
Duttke, S. H., Lacadie, S. A., Ibrahim, M. M., Glass, C. K., Corcoran, D. L., Benner, C., et al. (2015). Human promoters are intrinsically directional. Mol. Cell 57, 674–684. doi: 10.1016/j.molcel.2014.12.029
Fagegaltier, D., Bouge, A. L., Berry, B., Poisot, E., Sismeiro, O., Coppee, J. Y., et al. (2009). The endogenous siRNA pathway is involved in heterochromatin formation in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 106, 21258–21263. doi: 10.1073/pnas.0809208105
Faghihi, M. A., and Wahlestedt, C. (2009). Regulatory roles of natural antisense transcripts. Nat. Rev. Mol. Cell Biol. 10, 637–643. doi: 10.1038/nrm2738
Fernandez-Capetillo, O., Celeste, A., and Nussenzweig, A. (2003). Focusing on foci: H2AX and the recruitment of DNA-damage response factors. Cell Cycle 2, 426–427. doi: 10.4161/cc.2.5.509
Flynn, R. A., Almada, A. E., Zamudio, J. R., and Sharp, P. A. (2011). Antisense RNA polymerase II divergent transcripts are P-TEFb dependent and substrates for the RNA exosome. Proc. Natl. Acad. Sci. U.S.A. 108, 10460–10465. doi: 10.1073/pnas.1106630108
Fong, Y. W., Cattoglio, C., and Tjian, R. (2013). The intertwined roles of transcription and repair proteins. Mol. Cell 52, 291–302. doi: 10.1016/j.molcel.2013.10.018
Francia, S., Michelini, F., Saxena, A., Tang, D., de Hoon, M., Anelli, V., et al. (2012). Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 488, 231–235. doi: 10.1038/nature11179
Fukagawa, T., Nogami, M., Yoshikawa, M., Ikeno, M., Okazaki, T., Takami, Y., et al. (2004). Dicer is essential for formation of the heterochromatin structure in vertebrate cells. Nat. Cell Biol. 6, 784–791. doi: 10.1038/ncb1155
Fulci, V., and Macino, G. (2007). Quelling: post-transcriptional gene silencing guided by small RNAs in Neurospora crassa. Curr. Opin. Microbiol. 10, 199–203. doi: 10.1016/j.mib.2007.03.016
Gagnon, K. T., Li, L., Chu, Y., Janowski, B. A., and Corey, D. R. (2014). RNAi factors are present and active in human cell nuclei. Cell Rep. 6, 211–221. doi: 10.1016/j.celrep.2013.12.013
Galupa, R., and Heard, E. (2015). X-chromosome inactivation: new insights into cis and trans regulation. Curr. Opin. Gen. Dev. 31, 57–66. doi: 10.1016/j.gde.2015.04.002
Ganesan, S., Silver, D. P., Drapkin, R., Greenberg, R., Feunteun, J., and Livingston, D. M. (2004). Association of BRCA1 with the inactive X chromosome and XIST RNA. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 123–128. doi: 10.1098/rstb.2003.1371
Ganesan, S., Silver, D. P., Greenberg, R. A., Avni, D., Drapkin, R., Miron, A., et al. (2002). BRCA1 supports XIST RNA concentration on the inactive X chromosome. Cell 111, 393–405. doi: 10.1016/S0092-8674(02)01052-8
Gao, M., Wei, W., Li, M. M., Wu, Y. S., Ba, Z., Jin, K. X., et al. (2014). Ago2 facilitates Rad51 recruitment and DNA double-strand break repair by homologous recombination. Cell Res. 24, 532–541. doi: 10.1038/cr.2014.36
Gherzi, R., Chen, C. Y., Ramos, A., and Briata, P. (2014). KSRP controls pleiotropic cellular functions. Semin. Cell Dev. Biol. 34, 2–8. doi: 10.1016/j.semcdb.2014.05.004
Giles, K. E., Ghirlando, R., and Felsenfeld, G. (2010). Maintenance of a constitutive heterochromatin domain in vertebrates by a Dicer-dependent mechanism. Nat. Cell Biol. 12, 94–99. doi: 10.1038/ncb2010
Gonzalez, S., Pisano, D. G., and Serrano, M. (2008). Mechanistic principles of chromatin remodeling guided by siRNAs and miRNAs. Cell Cycle 7, 2601–2608. doi: 10.4161/cc.7.16.6541
Goodarzi, A. A., Noon, A. T., Deckbar, D., Ziv, Y., Shiloh, Y., Lobrich, M., et al. (2008). ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 31, 167–177. doi: 10.1016/j.molcel.2008.05.017
Goodier, J. L., and Kazazian, H. H. Jr. (2008). Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135, 23–35. doi: 10.1016/j.cell.2008.09.022
Gromak, N., Dienstbier, M., Macias, S., Plass, M., Eyras, E., Caceres, J. F., et al. (2013). Drosha regulates gene expression independently of RNA cleavage function. Cell Rep. 5, 1499–1510. doi: 10.1016/j.celrep.2013.11.032
Guang, S., Bochner, A. F., Pavelec, D. M., Burkhart, K. B., Harding, S., Lachowiec, J., et al. (2008). An argonaute transports siRNAs from the cytoplasm to the nucleus. Science 321, 537–541. doi: 10.1126/science.1157647
Gullerova, M., and Proudfoot, N. J. (2012). Convergent transcription induces transcriptional gene silencing in fission yeast and mammalian cells. Nat. Struct. Mol. Biol. 19, 1193–1201. doi: 10.1038/nsmb.2392
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., et al. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076. doi: 10.1038/nature08975
Halic, M., and Moazed, D. (2010). Dicer-independent primal RNAs trigger RNAi and heterochromatin formation. Cell 140, 504–516. doi: 10.1016/j.cell.2010.01.019
Hamperl, S., and Cimprich, K. A. (2014). The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair 19, 84–94. doi: 10.1016/j.dnarep.2014.03.023
Hermeking, H. (2012). MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat. Rev. Cancer 12, 613–626. doi: 10.1038/nrc3318
Holoch, D., and Moazed, D. (2015). RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 16, 71–84. doi: 10.1038/nrg3863
Hu, H., Du, L., Nagabayashi, G., Seeger, R. C., and Gatti, R. A. (2010). ATM is down-regulated by N-Myc-regulated microRNA-421. Proc. Natl. Acad. Sci. U.S.A. 107, 1506–1511. doi: 10.1073/pnas.0907763107
Huarte, M., Guttman, M., Feldser, D., Garber, M., Koziol, M. J., Kenzelmann-Broz, D., et al. (2010). A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142, 409–419. doi: 10.1016/j.cell.2010.06.040
Hung, T., Wang, Y., Lin, M. F., Koegel, A. K., Kotake, Y., Grant, G. D., et al. (2011). Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 43, 621–629. doi: 10.1038/ng.848
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Jackson, S. P., and Durocher, D. (2013). Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807. doi: 10.1016/j.molcel.2013.01.017
Jakob, B., Splinter, J., Conrad, S., Voss, K. O., Zink, D., Durante, M., et al. (2011). DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 39, 6489–6499. doi: 10.1093/nar/gkr230
Janowski, B. A., Huffman, K. E., Schwartz, J. C., Ram, R., Nordsell, R., Shames, D. S., et al. (2006). Involvement of AGO1 and AGO2 in mammalian transcriptional silencing. Nat. Struct. Mol. Biol. 13, 787–792. doi: 10.1038/nsmb1140
Kaidi, A., and Jackson, S. P. (2013). KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature 498, 70–74. doi: 10.1038/nature12201
Kakarougkas, A., Ismail, A., Chambers, A. L., Riballo, E., Herbert, A. D., Kunzel, J., et al. (2014). Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 55, 723–732. doi: 10.1016/j.molcel.2014.06.028
Kalousi, A., Hoffbeck, A. S., Selemenakis, P. N., Pinder, J., Savage, K. I., Khanna, K. K., et al. (2015). The nuclear oncogene SET controls DNA repair by KAP1 and HP1 retention to chromatin. Cell Rep. 11, 149–163. doi: 10.1016/j.celrep.2015.03.005
Kaneko, H., Dridi, S., Tarallo, V., Gelfand, B. D., Fowler, B. J., Cho, W. G., et al. (2011). DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 471, 325–330. doi: 10.1038/nature09830
Kapranov, P., Cheng, J., Dike, S., Nix, D. A., Duttagupta, R., Willingham, A. T., et al. (2007). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488. doi: 10.1126/science.1138341
Kashiwaba, S., Kitahashi, K., Watanabe, T., Onoda, F., Ohtsu, M., and Murakami, Y. (2010). The mammalian INO80 complex is recruited to DNA damage sites in an ARP8 dependent manner. Biochem. Biophys. Res. Commun. 402, 619–625. doi: 10.1016/j.bbrc.2010.10.066
Keller, C., Kulasegaran-Shylini, R., Shimada, Y., Hotz, H. R., and Buhler, M. (2013). Noncoding RNAs prevent spreading of a repressive histone mark. Nat. Struct. Mol. Biol. 20, 994–1000. doi: 10.1038/nsmb1113-1340
Keskin, H., Shen, Y., Huang, F., Patel, M., Yang, T., Ashley, K., et al. (2014). Transcript-RNA-templated DNA recombination and repair. Nature 515, 436–439. doi: 10.1038/nature13682
Khorkova, O., Myers, A. J., Hsiao, J., and Wahlestedt, C. (2014). Natural antisense transcripts. Hum. Mol. Genet. 23, R54–R63. doi: 10.1093/hmg/ddu207
Kim, D. H., Villeneuve, L. M., Morris, K. V., and Rossi, J. J. (2006). Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat. Struct. Mol. Biol. 13, 793–797. doi: 10.1038/nsmb1142
Kim, M. Y., and Zilberman, D. (2014). DNA methylation as a system of plant genomic immunity. Trends Plant Sci. 19, 320–326. doi: 10.1016/j.tplants.2014.01.014
Kim, N., and Jinks-Robertson, S. (2012). Transcription as a source of genome instability. Nat. Rev. Genet. 13, 204–214. doi: 10.1038/nrg3152
Kim, V. N., Han, J., and Siomi, M. C. (2009). Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 10, 126–139. doi: 10.1038/nrm2632
Krishnan, K., Steptoe, A. L., Martin, H. C., Wani, S., Nones, K., Waddell, N., et al. (2013). MicroRNA-182-5p targets a network of genes involved in DNA repair. RNA 19, 230–242. doi: 10.1261/rna.034926.112
Kruhlak, M. J., Celeste, A., Dellaire, G., Fernandez-Capetillo, O., Muller, W. G., McNally, J. G., et al. (2006). Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 172, 823–834. doi: 10.1083/jcb.200510015
Lai, F., Orom, U. A., Cesaroni, M., Beringer, M., Taatjes, D. J., Blobel, G. A., et al. (2013). Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 494, 497–501. doi: 10.1038/nature11884
Lal, A., Pan, Y., Navarro, F., Dykxhoorn, D. M., Moreau, L., Meire, E., et al. (2009). miR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nat. Struct. Mol. Biol. 16, 492–498. doi: 10.1038/nsmb.1589
Lee, H. C., Chang, S. S., Choudhary, S., Aalto, A. P., Maiti, M., Bamford, D. H., et al. (2009). qiRNA is a new type of small interfering RNA induced by DNA damage. Nature 459, 274–277. doi: 10.1038/nature08041
Lehmann, E., Brueckner, F., and Cramer, P. (2007). Molecular basis of RNA-dependent RNA polymerase II activity. Nature 450, 445–449. doi: 10.1038/nature06290
Leung, J. W., Agarwal, P., Canny, M. D., Gong, F., Robison, A. D., Finkelstein, I. J., et al. (2014). Nucleosome acidic patch promotes RNF168- and RING1B/BMI1-dependent H2AX and H2A ubiquitination and DNA damage signaling. PLoS Genet. 10:e1004178. doi: 10.1371/journal.pgen.1004178
Li, L. C., Okino, S. T., Zhao, H., Pookot, D., Place, R. F., Urakami, S., et al. (2006). Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. U.S.A. 103, 17337–17342. doi: 10.1073/pnas.0607015103
Liao, J. M., Cao, B., Zhou, X., and Lu, H. (2014). New insights into p53 functions through its target microRNAs. J. Mol. Cell Biol. 6, 206–213. doi: 10.1093/jmcb/mju018
Lindsey-Boltz, L. A., and Sancar, A. (2007). RNA polymerase: the most specific damage recognition protein in cellular responses to DNA damage? Proc. Natl. Acad. Sci. U.S.A. 104, 13213–13214. doi: 10.1073/pnas.0706316104
Ljungman, M., O’Hagan, H. M., and Paulsen, M. T. (2001). Induction of ser15 and lys382 modifications of p53 by blockage of transcription elongation. Oncogene 20, 5964–5971. doi: 10.1038/sj.onc.1204734
Luijsterburg, M. S., Dinant, C., Lans, H., Stap, J., Wiernasz, E., Lagerwerf, S., et al. (2009). Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol. 185, 577–586. doi: 10.1083/jcb.200810035
Magenta, A., Cencioni, C., Fasanaro, P., Zaccagnini, G., Greco, S., Sarra-Ferraris, G., et al. (2011). miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 18, 1628–1639. doi: 10.1038/cdd.2011.42
Maida, Y., and Masutomi, K. (2011). RNA-dependent RNA polymerases in RNA silencing. Biol. Chem. 392, 299–304. doi: 10.1515/BC.2011.035
Maida, Y., Yasukawa, M., Furuuchi, M., Lassmann, T., Possemato, R., Okamoto, N., et al. (2009). An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 461, 230–235. doi: 10.1038/nature08283
Manfrini, N., Trovesi, C., Wery, M., Martina, M., Cesena, D., Descrimes, M., et al. (2015). RNA-processing proteins regulate Mec1/ATR activation by promoting generation of RPA-coated ssDNA. EMBO Rep. 16, 221–231. doi: 10.15252/embr.201439458
Mansour, W. Y., Bogdanova, N. V., Kasten-Pisula, U., Rieckmann, T., Kocher, S., Borgmann, K., et al. (2013). Aberrant overexpression of miR-421 downregulates ATM and leads to a pronounced DSB repair defect and clinical hypersensitivity in SKX squamous cell carcinoma. Radiother. Oncol. 106, 147–154. doi: 10.1016/j.radonc.2012.10.020
Marasovic, M., Zocco, M., and Halic, M. (2013). Argonaute and Triman generate dicer-independent priRNAs and mature siRNAs to initiate heterochromatin formation. Mol. Cell 52, 173–183. doi: 10.1016/j.molcel.2013.08.046
Martinez, P., and Blasco, M. A. (2011). Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 11, 161–176. doi: 10.1038/nrc3025
Meister, G. (2013). Argonaute proteins: functional insights and emerging roles. Nat. Rev. Genet. 14, 447–459. doi: 10.1038/nrg3462
Michalik, K. M., Bottcher, R., and Forstemann, K. (2012). A small RNA response at DNA ends in Drosophila. Nucleic Acids Res. 40, 9596–9603. doi: 10.1093/nar/gks711
Miller, K. M., and Jackson, S. P. (2012). Histone marks: repairing DNA breaks within the context of chromatin. Biochem. Soc. Trans. 40, 370–376. doi: 10.1042/BST20110747
Miller, K. M., Tjeertes, J. V., Coates, J., Legube, G., Polo, S. E., Britton, S., et al. (2010). Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 17, 1144–1151. doi: 10.1038/nsmb.1899
Mondal, T., Subhash, S., Vaid, R., Enroth, S., Uday, S., Reinius, B., et al. (2015). MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 6:7743. doi: 10.1038/ncomms8743
Montecucco, A., and Biamonti, G. (2013). Pre-mRNA processing factors meet the DNA damage response. Front. Genet. 4:102. doi: 10.3389/fgene.2013.00102
Moskwa, P., Buffa, F. M., Pan, Y., Panchakshari, R., Gottipati, P., Muschel, R. J., et al. (2011). miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell 41, 210–220. doi: 10.1016/j.molcel.2010.12.005
Nagano, T., Mitchell, J. A., Sanz, L. A., Pauler, F. M., Ferguson-Smith, A. C., Feil, R., et al. (2008). The air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322, 1717–1720. doi: 10.1126/science.1163802
Ng, W. L., Yan, D., Zhang, X., Mo, Y. Y., and Wang, Y. (2010). Over-expression of miR-100 is responsible for the low-expression of ATM in the human glioma cell line: M059J. DNA Repair 9, 1170–1175. doi: 10.1016/j.dnarep.2010.08.007
Noon, A. T., Shibata, A., Rief, N., Lobrich, M., Stewart, G. S., Jeggo, P. A., et al. (2010). 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 12, 177–184. doi: 10.1038/ncb2017
Obbard, D. J., Gordon, K. H., Buck, A. H., and Jiggins, F. M. (2009). The evolution of RNAi as a defence against viruses and transposable elements. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 99–115. doi: 10.1098/rstb.2008.0168
Obbard, D. J., Jiggins, F. M., Halligan, D. L., and Little, T. J. (2006). Natural selection drives extremely rapid evolution in antiviral RNAi genes. Curr. Biol. 16, 580–585. doi: 10.1016/j.cub.2006.01.065
Okamura, K., Balla, S., Martin, R., Liu, N., and Lai, E. C. (2008). Two distinct mechanisms generate endogenous siRNAs from bidirectional transcription in Drosophila melanogaster. Nat. Struct. Mol. Biol. 15, 581–590. doi: 10.1038/nsmb0908-998c
Orekhova, A. S., and Rubtsov, P. M. (2013). Bidirectional promoters in the transcription of mammalian genomes. Biochemistry (Mosc) 78, 335–341. doi: 10.1134/S0006297913040020
Orom, U. A., Derrien, T., Beringer, M., Gumireddy, K., Gardini, A., Bussotti, G., et al. (2010). Long noncoding RNAs with enhancer-like function in human cells. Cell 143, 46–58. doi: 10.1016/j.cell.2010.09.001
Pandey, R. R., Mondal, T., Mohammad, F., Enroth, S., Redrup, L., Komorowski, J., et al. (2008). Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 32, 232–246. doi: 10.1016/j.molcel.2008.08.022
Panier, S., and Durocher, D. (2013). Push back to respond better: regulatory inhibition of the DNA double-strand break response. Nat. Rev. Mol. Cell Biol. 14, 661–672. doi: 10.1038/nrm3659
Pankotai, T., Bonhomme, C., Chen, D., and Soutoglou, E. (2012). DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol. 19, 276–282. doi: 10.1038/nsmb.2224
Pankotai, T., and Soutoglou, E. (2013). Double strand breaks: hurdles for RNA polymerase II transcription? Transcription 4, 34–38. doi: 10.4161/trns.22879
Pasquinelli, A. E. (2015). MicroRNAs: heralds of the noncoding RNA revolution. RNA 21, 709–710. doi: 10.1261/rna.049981.115
Pearl, L. H., Schierz, A. C., Ward, S. E., Al-Lazikani, B., and Pearl, F. M. (2015). Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 15, 166–180. doi: 10.1038/nrc3891
Pei, H., Zhang, L., Luo, K., Qin, Y., Chesi, M., Fei, F., et al. (2011). MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 470, 124–128. doi: 10.1038/nature09658
Peschansky, V. J., and Wahlestedt, C. (2014). Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics 9, 3–12. doi: 10.4161/epi.27473
Pickford, A. S., Catalanotto, C., Cogoni, C., and Macino, G. (2002). Quelling in Neurospora crassa. Adv. Genet. 46, 277–303. doi: 10.1016/S0065-2660(02)46010-5
Plath, K., Fang, J., Mlynarczyk-Evans, S. K., Cao, R., Worringer, K. A., Wang, H., et al. (2003). Role of histone H3 lysine 27 methylation in X inactivation. Science 300, 131–135. doi: 10.1126/science.1084274
Polo, S. E., Blackford, A. N., Chapman, J. R., Baskcomb, L., Gravel, S., Rusch, A., et al. (2012). Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell. 45, 505–516. doi: 10.1016/j.molcel.2011.12.035
Preker, P., Nielsen, J., Kammler, S., Lykke-Andersen, S., Christensen, M. S., Mapendano, C. K., et al. (2008). RNA exosome depletion reveals transcription upstream of active human promoters. Science 322, 1851–1854. doi: 10.1126/science.1164096
Price, B. D., and D’Andrea, A. D. (2013). Chromatin remodeling at DNA double-strand breaks. Cell 152, 1344–1354. doi: 10.1016/j.cell.2013.02.011
Puvvula, P. K., Desetty, R. D., Pineau, P., Marchio, A., Moon, A., Dejean, A., et al. (2014). Long noncoding RNA PANDA and scaffold-attachment-factor SAFA control senescence entry and exit. Nat. Commun. 5:5323. doi: 10.1038/ncomms6323
Raver-Shapira, N., Marciano, E., Meiri, E., Spector, Y., Rosenfeld, N., Moskovits, N., et al. (2007). Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell. 26, 731–743. doi: 10.1016/j.molcel.2007.05.017
Rimer, J., Cohen, I. R., and Friedman, N. (2014). Do all creatures possess an acquired immune system of some sort? Bioessays 36, 273–281. doi: 10.1002/bies.201300124
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A., et al. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323. doi: 10.1016/j.cell.2007.05.022
Sabin, L. R., Delas, M. J., and Hannon, G. J. (2013). Dogma derailed: the many influences of RNA on the genome. Mol. Cell. 49, 783–794. doi: 10.1016/j.molcel.2013.02.010
Sado, T., and Brockdorff, N. (2013). Advances in understanding chromosome silencing by the long non-coding RNA Xist. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20110325. doi: 10.1098/rstb.2011.0325
Saint-Andre, V., Batsche, E., Rachez, C., and Muchardt, C. (2011). Histone H3 lysine 9 trimethylation and HP1gamma favor inclusion of alternative exons. Nat. Struct. Mol. Biol. 18, 337–344. doi: 10.1038/nsmb.1995
Samai, P., Pyenson, N., Jiang, W., Goldberg, G. W., Hatoum-Aslan, A., and Marraffini, L. A. (2015). Co-transcriptional DNA and RNA cleavage during Type III CRISPR-Cas immunity. Cell 161, 1164–1174. doi: 10.1016/j.cell.2015.04.027
Saxowsky, T. T., and Doetsch, P. W. (2006). RNA polymerase encounters with DNA damage: transcription-coupled repair or transcriptional mutagenesis? Chem. Rev. 106, 474–488. doi: 10.1021/cr040466q
Schmitz, K. M., Mayer, C., Postepska, A., and Grummt, I. (2010). Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 24, 2264–2269. doi: 10.1101/gad.590910
Seila, A. C., Calabrese, J. M., Levine, S. S., Yeo, G. W., Rahl, P. B., Flynn, R. A., et al. (2008). Divergent transcription from active promoters. Science 322, 1849–1851. doi: 10.1126/science.1162253
Seila, A. C., Core, L. J., Lis, J. T., and Sharp, P. A. (2009). Divergent transcription: a new feature of active promoters. Cell Cycle 8, 2557–2564. doi: 10.4161/cc.8.16.9305
Seiler, D. M., Rouquette, J., Schmid, V. J., Strickfaden, H., Ottmann, C., Drexler, G. A., et al. (2011). Double-strand break-induced transcriptional silencing is associated with loss of tri-methylation at H3K4. Chromosome Res. 19, 883–899. doi: 10.1007/s10577-011-9244-1
Shanbhag, N. M., Rafalska-Metcalf, I. U., Balane-Bolivar, C., Janicki, S. M., and Greenberg, R. A. (2010). ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981. doi: 10.1016/j.cell.2010.04.038
Sharma, G. G., So, S., Gupta, A., Kumar, R., Cayrou, C., Avvakumov, N., et al. (2010). MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell. Biol. 30, 3582–3595. doi: 10.1128/MCB.01476-09
Shiloh, Y., Shema, E., Moyal, L., and Oren, M. (2011). RNF20-RNF40: a ubiquitin-driven link between gene expression and the DNA damage response. FEBS Lett. 585, 2795–2802. doi: 10.1016/j.febslet.2011.07.034
Sinkkonen, L., Hugenschmidt, T., Filipowicz, W., and Svoboda, P. (2010). Dicer is associated with ribosomal DNA chromatin in mammalian cells. PLoS ONE 5:e12175. doi: 10.1371/journal.pone.0012175
Siomi, M. C., Sato, K., Pezic, D., and Aravin, A. A. (2011). PIWI-interacting small RNAs: the vanguard of genome defence. Nat. Rev. Mol. Cell Biol. 12, 246–258. doi: 10.1038/nrm3089
Skourti-Stathaki, K., Kamieniarz-Gdula, K., and Proudfoot, N. J. (2014). R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 516, 436–439. doi: 10.1038/nature13787
Smeenk, G., Wiegant, W. W., Marteijn, J. A., Luijsterburg, M. S., Sroczynski, N., Costelloe, T., et al. (2013). Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J. Cell Sci. 126, 889–903. doi: 10.1242/jcs.109413
Smith-Roe, S. L., Nakamura, J., Holley, D., Chastain, P. D. II, Rosson, G. B., Simpson, D. A., et al. (2015). SWI/SNF complexes are required for full activation of the DNA-damage response. Oncotarget 6, 732–745. doi: 10.18632/oncotarget.2715
Sollier, J., Stork, C. T., Garcia-Rubio, M. L., Paulsen, R. D., Aguilera, A., and Cimprich, K. A. (2014). Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 56, 777–785. doi: 10.1016/j.molcel.2014.10.020
Spitale, R. C., Tsai, M. C., and Chang, H. Y. (2011). RNA templating the epigenome: long noncoding RNAs as molecular scaffolds. Epigenetics 6, 539–543. doi: 10.4161/epi.6.5.15221
Stanley, F. K., Moore, S., and Goodarzi, A. A. (2013). CHD chromatin remodelling enzymes and the DNA damage response. Mutat. Res. 750, 31–44. doi: 10.1016/j.mrfmmm.2013.07.008
St Laurent, G., Wahlestedt, C., and Kapranov, P. (2015). The landscape of long noncoding RNA classification. Trends Genet. 31, 239–251. doi: 10.1016/j.tig.2015.03.007
Storici, F., Bebenek, K., Kunkel, T. A., Gordenin, D. A., and Resnick, M. A. (2007). RNA-templated DNA repair. Nature 447, 338–341. doi: 10.1038/nature05720
Su, X., Chakravarti, D., Cho, M. S., Liu, L., Gi, Y. J., Lin, Y. L., et al. (2010). TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 467, 986–990. doi: 10.1038/nature09459
Sulli, G., Di Micco, R., and d’Adda di Fagagna, F. (2012). Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 12, 709–720. doi: 10.1038/nrc3344
Sun, Y., Jiang, X., Xu, Y., Ayrapetov, M. K., Moreau, L. A., Whetstine, J. R., et al. (2009). Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 11, 1376–1382. doi: 10.1038/ncb1982
Tazawa, H., Tsuchiya, N., Izumiya, M., and Nakagama, H. (2007). Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc. Natl. Acad. Sci. U.S.A. 104, 15472–15477. doi: 10.1073/pnas.0707351104
Trabucchi, M., Briata, P., Filipowicz, W., Ramos, A., Gherzi, R., and Rosenfeld, M. G. (2011). KSRP promotes the maturation of a group of miRNA precuresors. Adv. Exp. Med. Biol. 700, 36–42. doi: 10.1007/978-1-4419-7823-3_4
Trabucchi, M., Briata, P., Garcia-Mayoral, M., Haase, A. D., Filipowicz, W., Ramos, A., et al. (2009). The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459, 1010–1014. doi: 10.1038/nature08025
Tsai, M. C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., et al. (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693. doi: 10.1126/science.1192002
Ugalde, A. P., Ramsay, A. J., de la Rosa, J., Varela, I., Marino, G., Cadinanos, J., et al. (2011). Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J. 30, 2219–2232. doi: 10.1038/emboj.2011.124
Wagner, S. D., Yakovchuk, P., Gilman, B., Ponicsan, S. L., Drullinger, L. F., Kugel, J. F., et al. (2013). RNA polymerase II acts as an RNA-dependent RNA polymerase to extend and destabilize a non-coding RNA. EMBO J. 32, 781–790. doi: 10.1038/emboj.2013.18
Wan, G., Zhang, X., Langley, R. R., Liu, Y., Hu, X., Han, C., et al. (2013). DNA-damage-induced nuclear export of precursor microRNAs is regulated by the ATM-AKT pathway. Cell Rep. 3, 2100–2112. doi: 10.1016/j.celrep.2013.05.038
Wang, K. C., Yang, Y. W., Liu, B., Sanyal, A., Corces-Zimmerman, R., Chen, Y., et al. (2011a). A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472, 120–124. doi: 10.1038/nature09819
Wang, Y., Huang, J. W., Li, M., Cavenee, W. K., Mitchell, P. S., Zhou, X., et al. (2011b). MicroRNA-138 modulates DNA damage response by repressing histone H2AX expression. Mol. Cancer Res. MCR 9, 1100–1111. doi: 10.1158/1541-7786.MCR-11-0007
Wang, X., Arai, S., Song, X., Reichart, D., Du, K., Pascual, G., et al. (2008). Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454, 126–130. doi: 10.1038/nature06992
Wang, Y., Huang, J. W., Castella, M., Huntsman, D. G., and Taniguchi, T. (2014). p53 is positively regulated by miR-542-3p. Cancer Res. 74, 3218–3227. doi: 10.1158/0008-5472.CAN-13-1706
Wei, W., Ba, Z., Gao, M., Wu, Y., Ma, Y., Amiard, S., et al. (2012). A Role for small RNAs in DNA double-strand break repair. Cell 149, 101–112. doi: 10.1016/j.cell.2012.03.002
Weingarten-Gabbay, S., and Segal, E. (2014). A shared architecture for promoters and enhancers. Nat. Genet. 46, 1253–1254. doi: 10.1038/ng.3152
White, E., Schlackow, M., Kamieniarz-Gdula, K., Proudfoot, N. J., and Gullerova, M. (2014). Human nuclear dicer restricts the deleterious accumulation of endogenous double-stranded RNA. Nat. Struct. Mol. Biol. 21, 552–559. doi: 10.1038/nsmb.2827
Winsor, T. S., Bartkowiak, B., Bennett, C. B., and Greenleaf, A. L. (2013). A DNA damage response system associated with the phosphoCTD of elongating RNA polymerase II. PLoS ONE 8:e60909. doi: 10.1371/journal.pone.0060909
Wouters, M. D., van Gent, D. C., Hoeijmakers, J. H., and Pothof, J. (2011). MicroRNAs, the DNA damage response and cancer. Mutat. Res. 717, 54–66. doi: 10.1016/j.mrfmmm.2011.03.012
Xiao, C., Sharp, J. A., Kawahara, M., Davalos, A. R., Difilippantonio, M. J., Hu, Y., et al. (2007). The XIST noncoding RNA functions independently of BRCA1 in X inactivation. Cell 128, 977–989. doi: 10.1016/j.cell.2007.01.034
Xu, L., Da, L., Plouffe, S. W., Chong, J., Kool, E., and Wang, D. (2014). Molecular basis of transcriptional fidelity and DNA lesion-induced transcriptional mutagenesis. DNA Repair 19, 71–83. doi: 10.1016/j.dnarep.2014.03.024
Yamazaki, H., Chijiwa, T., Inoue, Y., Abe, Y., Suemizu, H., Kawai, K., et al. (2012). Overexpression of the miR-34 family suppresses invasive growth of malignant melanoma with the wild-type p53 gene. Exp. Ther. Med. 3, 793–796.
Yan, D., Ng, W. L., Zhang, X., Wang, P., Zhang, Z., Mo, Y. Y., et al. (2010). Targeting DNA-PKcs and ATM with miR-101 sensitizes tumors to radiation. PLoS ONE 5:e11397. doi: 10.1371/journal.pone.0011397
Yang, H., Luo, J., Liu, Z., Zhou, R., and Luo, H. (2015a). MicroRNA-138 regulates DNA damage response in small cell lung cancer cells by directly targeting H2AX. Cancer Invest. 33, 126–136. doi: 10.3109/07357907.2015.1006329
Yang, Q., Ye, Q. A., and Liu, Y. (2015b). Mechanism of siRNA production from repetitive DNA. Genes Dev. 29, 526–537. doi: 10.1101/gad.255828.114
Yang, L., Lin, C., Jin, C., Yang, J. C., Tanasa, B., Li, W., et al. (2013). lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 500, 598–602. doi: 10.1038/nature12451
Yap, K. L., Li, S., Munoz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S., et al. (2010). Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38, 662–674. doi: 10.1016/j.molcel.2010.03.021
Yu, W., Gius, D., Onyango, P., Muldoon-Jacobs, K., Karp, J., Feinberg, A. P., et al. (2008). Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 451, 202–206. doi: 10.1038/nature06468
Yu, X., and Li, Z. (2015). Long non-coding RNA HOTAIR: a novel oncogene (Review). Mol. Med. Rep. 12, 5611–5618. doi: 10.3892/mmr.2015.4161
Zamudio, J. R., Kelly, T. J., and Sharp, P. A. (2014). Argonaute-bound small RNAs from promoter-proximal RNA polymerase II. Cell 156, 920–934. doi: 10.1016/j.cell.2014.01.041
Zarebski, M., Wiernasz, E., and Dobrucki, J. W. (2009). Recruitment of heterochromatin protein 1 to DNA repair sites. Cytometry A 75, 619–625. doi: 10.1002/cyto.a.20734
Zhang, C., Liu, J., Wang, X., and Feng, Z. (2015). The regulation of the p53/MDM2 feedback loop by microRNAs. RNA Dis. 2:e502.
Zhang, C., and Peng, G. (2015). Non-coding RNAs: an emerging player in DNA damage response. Mutat. Res. Rev. Mutat. Res. 763, 202–211. doi: 10.1016/j.mrrev.2014.11.003
Zhang, X., Wan, G., Berger, F. G., He, X., and Lu, X. (2011). The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol. Cell 41, 371–383. doi: 10.1016/j.molcel.2011.01.020
Zhao, J., Sun, B. K., Erwin, J. A., Song, J. J., and Lee, J. T. (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756. doi: 10.1126/science.1163045
Keywords: non-coding RNA, DNA-damage response, RNA interference, chromatin modulation, transcription
Citation: Francia S (2015) Non-Coding RNA: Sequence-Specific Guide for Chromatin Modification and DNA Damage Signaling. Front. Genet. 6:320. doi: 10.3389/fgene.2015.00320
Received: 21 May 2015; Accepted: 09 October 2015;
Published: 13 November 2015.
Edited by:
Rui Henrique, Portuguese Oncology Institute Porto, PortugalReviewed by:
Pascal Chartrand, Université de Montréal, CanadaStephan Gasser, National University of Singapore, Singapore
Kotb Abdelmohsen, National Institutes of Health, USA
Copyright © 2015 Francia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sofia Francia, c29maWEuZnJhbmNpYUBpZm9tLmV1