 Alessandra Brambati
Alessandra Brambati Arianna Colosio1†
Arianna Colosio1† Luca Zardoni
Luca Zardoni Lorenzo Galanti
Lorenzo Galanti Giordano Liberi
Giordano Liberi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet. , 28 April 2015
Sec. Cancer Genetics
Volume 6 - 2015 | https://doi.org/10.3389/fgene.2015.00166
This article is part of the Research Topic DNA and RNA metabolism meet at chromatin to control genome stability View all 8 articles
DNA replication and transcription are vital cellular processes during which the genetic information is copied into complementary DNA and RNA molecules. Highly complex machineries required for DNA and RNA synthesis compete for the same DNA template, therefore being on a collision course. Unscheduled replication–transcription clashes alter the gene transcription program and generate replication stress, reducing fork speed. Molecular pathways and mechanisms that minimize the conflict between replication and transcription have been extensively characterized in prokaryotic cells and recently identified also in eukaryotes. A pathological outcome of replication–transcription collisions is the formation of stable RNA:DNA hybrids in molecular structures called R-loops. Growing evidence suggests that R-loop accumulation promotes both genetic and epigenetic instability, thus severely affecting genome functionality. In the present review, we summarize the current knowledge related to replication and transcription conflicts in eukaryotes, their consequences on genome stability and the pathways involved in their resolution. These findings are relevant to clarify the molecular basis of cancer and neurodegenerative diseases.
DNA replication and transcription are vital processes in all living organisms during which specialized polymerases copy the genetic information into complementary DNA and RNA molecules. Both processes must be completed with high fidelity to preserve genetic information and cell functionality. The DNA duplex, which is packed into chromatin, must be separated into two DNA single strands (ssDNA) before being replicated or transcribed, thus generating positive supercoils ahead the polymerases. DNA and RNA polymerases act in coordination with multiple enzymes and accessory factors, which include helicases that open the DNA duplex and topoisomerases that solve DNA topological constrains. Replication and transcription machineries are assembled at precise genomic locations, called origins, and promoters, respectively, and can travel for several DNA kilobases in 5′–3′ direction before being dismantled at termination sites. During transcription, several RNA polymerases transcribe one DNA strand, while the other remains transiently in single stranded conformation at the transcription bubble. During DNA synthesis, two replication machineries, called replisomes, move in opposite directions from one origin and duplicate both lagging and leading strands in a coordinated fashion, only once in each S-phase of the cell cycle. Replication and transcription compete for the same DNA template and can therefore interfere with each other. Transcription can arrest DNA synthesis and compromise replication fork stability, thus causing replication stress. Since the polarity of DNA and RNA synthesis is the same, a replication fork encounters the transcription machinery head-on on the lagging strand template and codirectionally on the leading strand. Although both types of collisions can disrupt or arrest replication forks in vivo (Deshpande and Newlon, 1996; Azvolinsky et al., 2009; Dutta et al., 2011; Merrikh et al., 2011; Alzu et al., 2012), several lines of evidence indicate that frontal clashes between replication and transcription mainly affect genome stability. The organization of bacterial genomes imparts in fact a co-orientation bias of replication and transcription of highly expressed and/or essential genes, thus avoiding deleterious head-on conflicts (Rocha, 2008). A preference for co-orientation of replication and transcription was also observed in human genome (Huvet et al., 2007). Moreover, head-on replication–transcription collisions are prevented by specific fork barriers at highly expressed ribosomal DNA (rDNA) in eukaryotic organisms (Kobayashi, 2014). From bacteria to humans, actively transcribed genes exhibit elevated spontaneous mutation and recombination rates, which are stimulated by replication (Prado and Aguilera, 2005; Kim et al., 2007; Gottipati et al., 2008; Paul et al., 2012). Transcription-associated recombination (TAR) and transcription-associated mutagenesis (TAM) increase when the lagging strand template is the transcribed strand (Prado and Aguilera, 2005; Kim et al., 2007; Paul et al., 2012), suggesting that head-on replication–transcription collisions are more detrimental to fork stability than codirectional ones. Protein–protein clashes on the lagging strand template, which contains ssDNA loop, would be particularly dangerous for fork integrity. Some evidence indeed suggests that the replisome may contact the transcription machinery (Mirkin and Mirkin, 2005). However, it is also possible that positive supercoils generated by polymerases moving toward each other, prevent a direct clash between them. In this case, fork arrest, and DNA damage may rather result from DNA topological constrains formation (Olavarrieta et al., 2002).
The replication fork has to face both nascent RNA and proteins, when it encounters the transcription machinery. RNA biogenesis proteins that co-transcriptionally process nascent RNA, including the splicing factor ASF/SF2, the THO/TREX mRNA export complex and the mRNA cleavage and polyadenylation machinery, prevent the re-hybridization of RNA to the transcribed DNA strand and therefore the formation of dangerous R-loop structures that could affect fork progression (Li and Manley, 2005; Gomez-Gonzalez et al., 2011; Wahba et al., 2011; Stirling et al., 2012). R-loop formation is favored by negative supercoiled DNA (Drolet, 2006), which accumulates behind the advancing RNA polymerase according to the twin-supercoiled domain model (Liu and Wang, 1987). It is therefore likely that the excess of positive supercoiled DNA accumulating in head-on encounters between the replisome and the transcription machinery may contribute to R-loop stabilization at the transcription bubble (Bermejo et al., 2012).
R-loops are physiological intermediates of several biological processes, including eukaryotic and prokaryotic immune responses or transcription termination (Skourti-Stathaki and Proudfoot, 2014). However, several studies from bacteria to humans suggest that uncontrolled accumulation of R-loops can affect genome integrity and proper chromatin organization, most likely by interfering with DNA synthesis.
In prokaryotic cells, DNA synthesis starts at single origins of replication and since highly transcribed and/or essential genes are located on the leading strand template, harmful head-on conflicts between replication and transcription are prevented by genome organization (Rocha, 2008). Nevertheless, bacteria have evolved different strategies to resolve replication–transcription conflicts. These strategies relay on both auxiliary DNA helicases of the replisome that remove proteins and/or R-loops and transcription regulators that rescue stalled/backtracked RNA polymerases (Merrikh et al., 2012). Eukaryotic chromosomes are replicated from multiple origins differentially selected for firing, thus increasing the complexity of the replication–transcription interference.
Growing evidence suggests that the Ataxia telangiectasia mutated and Rad3-related (ATR) checkpoint kinase and downstream factors play a central role in coordinating replication with transcription. In mammals, the ATR pathway controls the stability of both common fragile sites (CFSs) and early replicating fragile sites (ERFSs), specific genomic regions prone to rearrangements under replication stress (Casper et al., 2002; Barlow et al., 2013). Some of these fragile elements correspond to R-loop accumulating long genes or highly transcribed genes (Helmrich et al., 2011; Barlow et al., 2013). Recent studies in budding yeast have suggested some mechanisms by which ATR pathway coordinates replication with transcription (Figure 1). The stress-activated protein kinase Hog1 phosphorylates Mrc1, a downstream component of the ATR pathway, and Mrc1 phosphorylation is crucial to slow down fork progression and to prevent TAR due to collisions with transcription (Duch et al., 2013). Moreover, the temporarily inhibition of transcription at fork passage mediated by the ATR pathway is a preferred mechanism to prevent replication–transcription collisions at both RNA Polymerase III (RNAPIII)- and RNA Polymerase II (RNAPII)-transcribed genes. ATR pathway actively controls the disassembly of the pre-initiation complex at tRNA genes (Nguyen et al., 2010) and assists fork progression and stability at RNAPII transcribed genes by inhibiting topological constrains caused by gene gating (Bermejo et al., 2011). This process, which is mediated by THO/TREX and TREX-2 complexes and nucleoporins, couples RNAPII transcription to mRNA export through the nuclear envelope (Blobel, 1985; Bermejo et al., 2012). Although required for gene expression, gene gating might aggravate transcription-associated topological problems, contributing to fork instability and it is therefore inhibited by ATR-dependent phosphorylation of nucleoporins (Bermejo et al., 2011). It has been proposed that, upon checkpoint-dependent gene gating inhibition, R-loop accumulation is favored at the twin supercoiled domain by head-on encounters between transcription and replication (Alzu et al., 2012; Bermejo et al., 2012). In this scenario, Topoisomerases I could be crucial to restrain R-loop accumulation by counteracting DNA negative supercoils (Tuduri et al., 2009).
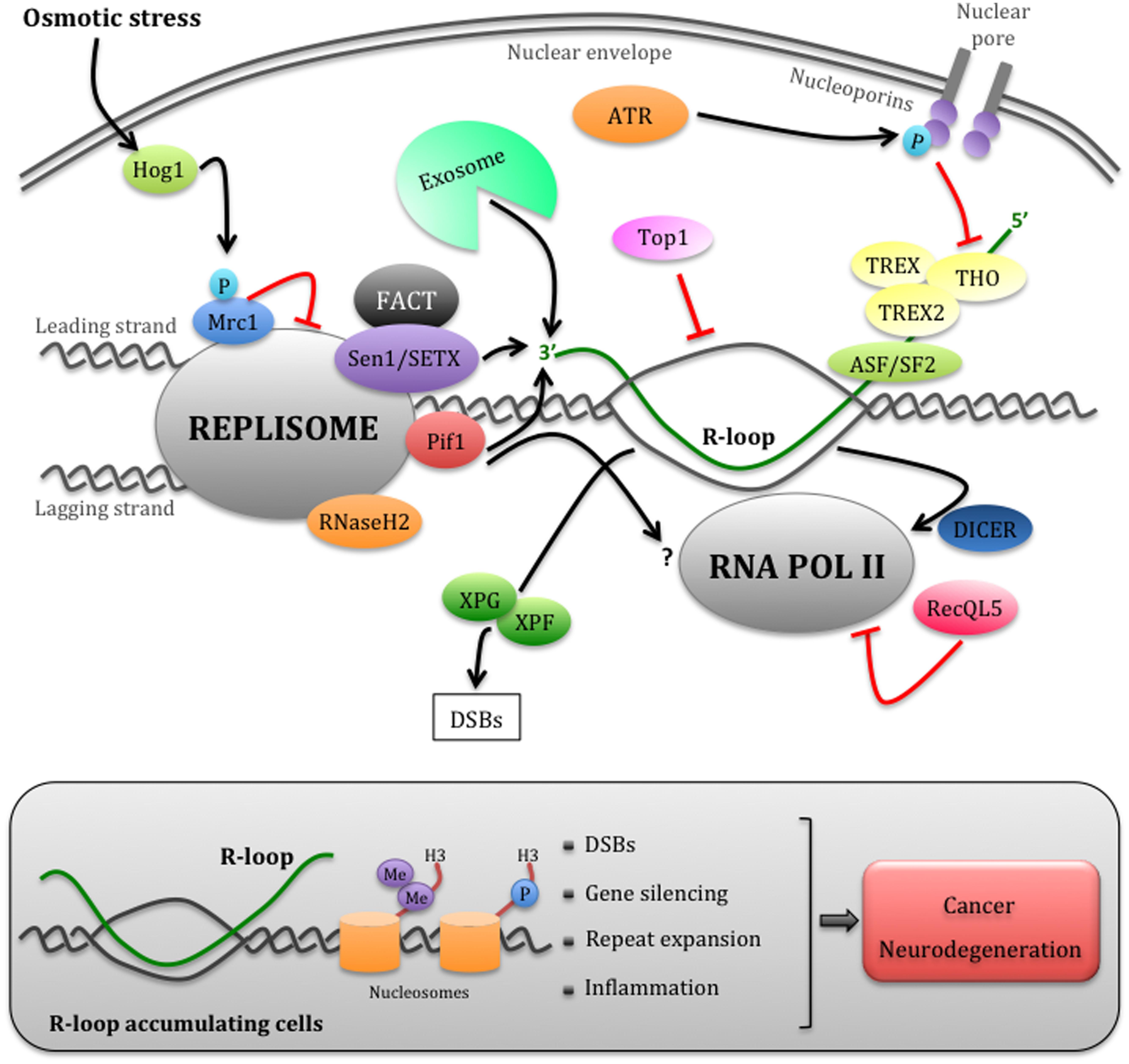
FIGURE 1. Eukaryotic mechanisms that manage replication– transcription conflicts. Schematic representation of a head-on encounter between the replisome and RNAPII. The cotrascriptional processing of nascent RNA, including its export through the nuclear envelope mediated by the THO/TREX and TREX2 complexes, can impede replication forks progression. The ATR checkpoint pathway temporarily inhibits RNA export by phosphorylating nucleoporins, thus allowing fork advancement. However, this process may generate harmful R-loop structures, more likely in head-on replication–transcription encounters. Multiple factors, including the accessory DNA/RNA helicases of the replisome Sen1/SETX and Pif1, the RNA exosome, RNaseH2, and Toposomerase I, may cooperate in limiting R-loop accumulation at the fork. The FACT complex, which interacts with SETX, could be involved in the re-establishment of chromatin status upon replication–transcription collisions. R-loops can be also processed into DSBs by XPG and XPF endonucleases. Hog1-dependent Mrc1 phosphorylation and RecQL5 modulate the speed of the replisome or RNAPII, respectively, while Dicer dislodges RNAPII at fork passage. Failure to promptly remove R-loops (gray box) causes not only DSBs, but also chromatin condensation through the accumulation of H3S10P and H3K9me2 markers, which contributes to fork arrest and gene silencing. Unrestrained R-loop accumulation has been also linked to repeats expansion and inflammation events, thus contributing to cancer and neurodegeneration (refer to text for further details).
Moreover, factors that remove transcription blocks and nascent RNA, similarly to what described for bacteria, could be also crucial to promote fork progression. Several helicases involved in replication fork stability maintenance, including HARP, WRN, BLM, Sen1, and Pif1, can remove RNA:DNA hybrids in vitro (Kim et al., 1999; Boule and Zakian, 2007; Popuri et al., 2008; Chakraborty and Grosse, 2011; Grierson et al., 2012; Kassavetis and Kadonaga, 2014). For yeast Pif1 and Sen1 DNA/RNA helicases a role in preventing replication–transcription interference has been also ascertained in vivo. In particular, members of the Pif1 helicase family assist fork progression through several type of natural barriers, including transcription blocks (Ivessa et al., 2003; Azvolinsky et al., 2009; Paeschke et al., 2011), while Sen1 is specifically required to prevent RNA:DNA hybrids accumulation at the fork in head-on encounters with RNAPII transcribed genes (Alzu et al., 2012). In human, both Senataxin/SETX, the ortholog of Sen1, and Aquarius, an helicase structurally related to Senataxin, prevent DNA damages caused by RNA:DNA hybrids accumulation, suggesting that this function has been evolutionarily conserved (Yuce and West, 2013; Sollier et al., 2014). Another suggested distinct role of Sen1/Senataxin is to promote transcription termination by removing R-loops (Mischo et al., 2011; Skourti-Stathaki et al., 2011). It is therefore possible that specific termination factors are engaged at fork passage to inhibit transcription. The above scenario is also consistent with the finding that the exosome, a multi-protein complex that degrades aberrant RNA molecules and is involved in transcription termination, co-localizes with Senataxin at R-loop-dependent nuclear foci in response to replication stress (Richard et al., 2013). The exosome could cooperate with Senataxin in removing R-loops by degrading RNA moiety (Richard et al., 2013).
RNaseH proteins, a class of enzymes that specifically degrades RNA in RNA:DNA hybrids, are also likely crucial to prevent replication–transcription interference. In budding yeast, RNaseH1 and RNaseH2 show specificity for R-loop resolution and RNaseH2 could have uniquely access to replication-associated RNA:DNA hybrids by interacting with the replisome (Wahba et al., 2011; Chon et al., 2013). Moreover, a yeast genome-wide study reported RNA:DNA hybrids accumulation at long or short genes in cells inactivated for RNaseH1/2 or Sen1, respectively (Chan et al., 2014). This data suggests that different anti-R-loop pathways could act at different genomic locations, even though how this is achieved remains unclear.
Finally, modulators of RNA polymerase activity have been involved in preventing replication–transcription collisions in eukaryotic cells. The human RecQL5 is a member of RecQ DNA helicases family that interacts with RNAPII (Aygun et al., 2008). RecQL5 acts as elongator factor to modulate RNAPII speed and to prevent chromosomal rearrangements across transcribed genes and certain CFSs (Saponaro et al., 2014). A recent study in fission yeast suggests another mechanism involving Dicer, a component of the RNA interference pathway (RNAi), in preventing transcription at putative sites of collisions with replication (Castel et al., 2014). Dicer, independently from the other components of the canonical RNAi pathway, promotes the release of RNAPII from the 3′ end of highly transcribed genes and from antisense transcribed rDNA regions. Transcription inhibition prevents the loss of rDNA repeats through homologous recombination. This specific function of Dicer at transcribed genes resembles the one of the RNAi pathway at pericentromeric regions, where it coordinates replication with transcription. In this way, RNAi pathway protects stalled forks from unscheduled homologous recombination, whose engagement interferes with the proper establishment of epigenetic modifications (Zaratiegui et al., 2011).
While it is evident that multiple pathways are involved in dealing with replication–transcription conflicts in eukaryotic cells (Figure 1), further studies are required to dissect their interconnections and the checkpoint-mediated regulation. These studies will be relevant to understand the causes of unrestrained R-loop accumulation that triggers both genetic and epigenetic instability in replication–transcription interference.
Strong evidence from bacteria to humans indicates that transcription damage DNA by arresting replication fork progression. Indeed stalled replication forks accumulate ssDNA gaps and become prone to unscheduled recombination events and DNA double strand breaks (DSBs) formation. R-loops are thought to contribute to fork arrest, although the mechanism involved is unclear. An acknowledged model suggests that the RNA:DNA hybrid in the R-loop hampers fork progression, although R-loop bypass mechanisms could also be envisaged. Supporting this idea and consistent with in vitro observations in bacterial system, mRNA mediated re-priming of DNA synthesis at the leading strand has been observed during codirectional collisions with transcription (Pomerantz and O’Donnell, 2008). Moreover, another possible by-pass mechanism could relay on the uncoupling of leading and lagging strand synthesis and replication across the non-transcribed strand (Alzu et al., 2012). This pathway may require post-replication repair mechanisms, such as template switching and/or translesion DNA synthesis, for DNA replication across RNA:DNA hybrids (Gomez-Gonzalez et al., 2009). However, these by-pass processes may contribute to TAR and TAM occurrence, thus causing genome instability. The R-loop structure, which resembles the one of a D-loop recombination intermediate, could be processed by specific endonucleases leading to DSBs. In agreement with this idea, a recent study showed that in cells lacking Senataxin, Aquarius or the splicing factor ASF/SF2, unscheduled R-loops are processed into DSBs by XPG and XPF endonucleases, together with the Cockayne syndrome group B protein belonging to the transcription-coupled nucleotide excision repair (TC-NER) pathway (Figure 1; Sollier et al., 2014). These data clearly suggest that uncontrolled R-loop formation causes DSBs, although how R-loop processing by TC-NER pathway is coordinated with replication remains to be elucidated.
Recent findings suggested that pathological accumulation of R-loops and/or RNA:DNA hybrids is also linked to complex genomic rearrangements, including quasi-palindrome-associated mutations (Kim et al., 2013) and nucleotide repeat expansions, which underline several human neurological disorders (Lin et al., 2010; Groh et al., 2014; Haeusler et al., 2014; Loomis et al., 2014; Rigby et al., 2014).
In addition, uncoordinated replication–transcription clashes may also interfere with proper chromatin markers deposition, causing epigenetic instability (Figure 1). Chromatin markers are maintained through DNA replication by coupling the deposition of recycled parental histones to newly synthesized histones on duplicated DNA (Alabert and Groth, 2012). Defective fork progression at natural replication barriers such as G4 forming sequences leads to accumulation of repressive chromatin markers and gene silencing (Paeschke et al., 2011; Schiavone et al., 2014). Moreover, accumulation of stalled replication forks induced by treatment with the anticancer drug doxorubicin causes gene repression (Im et al., 2014). R-loops and/or RNA:DNA hybrids also influence the chromatin status. For instance, R-loop formation associates with unmethylated CpG island promoters in human genome (Ginno et al., 2012) and is linked to RNAi-directed heterochromatin formation at mammalian gene terminators (Skourti-Stathaki et al., 2014) and at centromeres in fission yeast (Nakama et al., 2012). While in these contexts R-loops promote a proper chromatin organization, it has been shown that an excessive R-loop accumulation at certain DNA regions induces unscheduled chromatin condensation. A recent study demonstrated that human, worm, or yeast cells depleted for Senataxin, RNaseH or the THO complex accumulate at transcribed genes both R-loops and phosphorylated H3 at S10 (H3S10P), a mitotic marker of chromatin condensation (Castellano-Pozo et al., 2013). R-loops are also linked to increased levels of the heterochromatin marker H3 dimetylated K9 (H3K9me2) in nematode cells inactivated for the THO complex (Castellano-Pozo et al., 2013). Furthermore, colocalization of R-loops and H3K9me2 has been also reported at the FXN gene expanded in Fragile X syndrome, likely contributing to its transcriptional silencing (Groh et al., 2014). It has been proposed that R-loop-mediated chromatin compaction would not only prevent transcription, but also contribute to impair fork progression (Castellano-Pozo et al., 2013). The idea that replication–transcription conflicts impact on chromatin structure is also supported by additional recent findings on the chromatin remodeling FACT complex, which physically interacts with Senataxin (Yuce and West, 2013; Hill et al., 2014). The FACT complex seems to be crucial to re-establish proper chromatin status after replication fork passage throughout transcribed DNA regions (Herrera-Moyano et al., 2014).
Altogether, it appears that failure to coordinate replication with transcription not only damages DNA, but also prevents gene expression, thus seriously affecting cell functionality.
Replication stress is a hallmark of precancerous cells and is responsible for the gross chromosomal rearrangements observed in advanced tumors (Bartkova et al., 2006; Di Micco et al., 2006; Burrell et al., 2013). In human cells, transcription promotes oncogene-induced replication stress (Jones et al., 2012) and contributes to the expression of both ERFSs and CFSs, which match to regions of chromosomal abnormalities observed in cancer cells (Helmrich et al., 2011; Barlow et al., 2013). As mentioned above, recombinogenic RNA:DNA hybrids and/or R-loops are tightly connected to replication–transcription conflicts, suggesting that dysfunctions in R-loops metabolism contributes to cancer development (Table 1; Tuduri et al., 2010; Bermejo et al., 2012). This idea is supported by recent studies that have involved the well-characterized tumor suppressor genes BRCA1 and BRCA2 in R-loop processing (Bhatia et al., 2014; Hill et al., 2014; Hatchi et al., 2015). In particular, the finding that BRCA1 interacts with Senataxin and with factors crucial for fork recovery from replication stress, such as the FACT and the MMS22–TONSL complexes (Hill et al., 2014), raises the possibility that BRCA1 limits the replication–transcription conflicts.

TABLE 1. Eukaryotic factors that limit replication-transcription conflicts and/or R-loop accumulation.
It is interesting to note that the most characterized factors that counteract R-loop accumulation have been implicated in neurological disorders (Table 1). Senataxin is mutated in juvenile forms of Ataxia and amyotrophic lateral sclerosis (ALS) (Chen et al., 2004; Moreira et al., 2004), while RNaseH2 in the neuroinflammatory Aicardi–Goutières disorder (Crow et al., 2006). The observation that replication and homologous recombination do not occur in neurons raises the question of whether replication-associated and recombinogenic R-loops contribute to neurodegeneration. It is possible that unscheduled accumulation of R-loops impacts on the functionality of glial cells, a population of cycling cells that interacts with neurons and whose dysfunctions contribute to neurodegeneration (Lobsiger and Cleveland, 2007). Furthermore, uncontrolled R-loop accumulation during neurogenesis could have long-term effects on genome integrity and gene expression in mature neurons. Indeed persistent RNA:DNA hybrids are associated to repeats instability, transcriptional silencing and replication-dependent DSBs accumulation at GAA/TTC tracts, which characterize Frederich Ataxia and Fragile X neurological syndromes (Zhang et al., 2012; Groh et al., 2014; Loomis et al., 2014). Stable RNA:DNA hybrids are also linked to GGGGCC-expansion in C9ORF72, the most common genetic alteration in the neurodegenerative disorders ALS and frontotemporal dementia (Reddy et al., 2010; Haeusler et al., 2014).
Of interest, recent findings suggested that RNA:DNA hybrids containing viral or bacterial derived sequences can stimulate the innate immune system response (Kailasan Vanaja et al., 2014; Mankan et al., 2014; Rigby et al., 2014). The identification of RNA:DNA hybrids as activators of innate immunity has obvious implications for autoimmune diseases, including Aicardi–Goutières neuroinflammatory disorder. Moreover, if RNA:DNA hybrids directly contribute to trigger chronic inflammations status, this would have broad implications for the onset of cancer and neurodegenerative diseases (Figure 1; Amor et al., 2014; Hagerling et al., 2014).
Almost 30 years of pioneering works in simple model organisms and recent studies in human cells have established that uncoordinated replication–transcription conflicts and unscheduled R-loop accumulation significantly contribute to cause genetic and epigenetic instability associated to replication stress, a pathological condition that alters chromosomal structure and functionality. In humans increasing evidence links the inactivation of factors that limit replication–transcription interference and R-loop formation with cancer and/or neurodegenerative disorders onset (Table 1). An accurate dissection of the molecular mechanisms that prevent transcription-induced replication stress could therefore provide a future framework for understanding the molecular basis of cancer and neurodegeneration.
The Editor Alessandra Montecucco declares that, despite being affiliated to the same institution as the authors Alessandra Brambati, Arianna Colosio, Luca Zardoni, Lorenzo Galanti and Giordano Liberi, the review process was handled objectively and no conflict of interest exists. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to all authors whose relevant contributions could not be cited due to space limitation. Work in our lab is supported by research grants from the Associazione Italiana per la Ricerca sul Cancro (IG-13115) and Fondazione Cariplo (2013-0790). This manuscript is dedicated to the memory of our friend and colleague Dr. Massimo Serafini.
Alabert, C., and Groth, A. (2012). Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 13, 153–167. doi: 10.1038/nrm3288
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alzu, A., Bermejo, R., Begnis, M., Lucca, C., Piccini, D., Carotenuto, w., et al. (2012). Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 151, 835–846. doi: 10.1016/j.cell.2012.09.041
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amor, S., Peferoen, L. A., Vogel, D. Y., Breur, M., van der Valk, P., Baker, D., et al. (2014). Inflammation in neurodegenerative diseases–an update. Immunology 142, 151–166. doi: 10.1111/imm.12233
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aygun, O., Svejstrup, J., and Liu, Y. (2008). A RECQ5-RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc. Natl. Acad. Sci. U.S.A. 105, 8580–8584. doi: 10.1073/pnas.0804424105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Azvolinsky, A., Giresi, P. G., Lieb, J. D., and Zakian, V. A. (2009). Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol. Cell 34, 722–734. doi: 10.1016/j.molcel.2009.05.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barlow, J. H., Faryabi, R. B., Callen, E., Wong, N., Malhowski, A., Chen, H. T., et al. (2013). Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632. doi: 10.1016/j.cell.2013.01.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P., Kletsas, D., Issaeva, N., et al. (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637. doi: 10.1038/nature05268
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bermejo, R., Capra, T., Jossen, R., Colosio, A., Frattini, C., Carotenuto, W., et al. (2011). The replication checkpoint protects fork stability by releasing transcribed genes from nuclear pores. Cell 146, 233–246. doi: 10.1016/j.cell.2011.06.033
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bermejo, R., Lai, M. S., and Foiani, M. (2012). Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol. Cell 45, 710–718. doi: 10.1016/j.molcel.2012.03.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bhatia, V., Barroso, S. I., Garcia-Rubio, M. L., Tumini, E., Herrera-Moyano, E., and Aguilera, A. (2014). BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511, 362–365. doi: 10.1038/nature13374
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blobel, G. (1985). Gene gating: a hypothesis. Proc. Natl. Acad. Sci. U.S.A. 82, 8527–8529. doi: 10.1073/pnas.82.24.8527
Boule, J. B., and Zakian, V. A. (2007). The yeast Pif1p DNA helicase preferentially unwinds RNA DNA substrates. Nucleic Acids Res. 35, 5809–5818. doi: 10.1093/nar/gkm613
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Burrell, R. A., McClelland, S. E., Endesfelder, D., Groth, P., Weller, M. C., Shaikh, n., et al. (2013). Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496. doi: 10.1038/nature11935
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Casper, A. M., Nghiem, P., Arlt, M. F., and Glover, T. W. (2002). ATR regulates fragile site stability. Cell 111, 779–789. doi: 10.1016/S0092-8674(02)01113-3
Castel, S. E., Ren, J., Bhattacharjee, S., Chang, A. Y., Sanchez, M., Valbuena, A., et al. (2014). Dicer promotes transcription termination at sites of replication stress to maintain genome stability. Cell 159, 572–583. doi: 10.1016/j.cell.2014.09.031
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Castellano-Pozo, M., Santos-Pereira, J. M., Rondon, A. G., Barroso, S., Andujar, E., Perez-Alegre, M., et al. (2013). R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 52, 583–590. doi: 10.1016/j.molcel.2013.10.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chakraborty, P., and Grosse, F. (2011). Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair (Amst) 10, 654–665. doi: 10.1016/j.dnarep.2011.04.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chan, Y. A., Aristizabal, M. J., Lu, P. Y., Luo, Z., Hamza, A., Kobor, M. S., et al. (2014). Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS Genet. 10:e1004288. doi: 10.1371/journal.pgen.1004288
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, Y. Z., Bennett, C. L., Huynh, H. M., Blair, I. P., Puls, I., Irobi, J., et al. (2004). DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 74, 1128–1135. doi: 10.1086/421054
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chon, H., Sparks, J. L., Rychlik, M., Nowotny, M., Burgers, P. M., Crouch, R. J., et al. (2013). RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res. 41, 3130–3143. doi: 10.1093/nar/gkt027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Crow, Y. J., Leitch, A., Hayward, B. E., Garner, A., Parmar, R., Griffith, E., et al. (2006). Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat. Genet. 38, 910–916. doi: 10.1038/ng1842
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deshpande, A. M., and Newlon, C. S. (1996). DNA replication fork pause sites dependent on transcription. Science 272, 1030–1033. doi: 10.1126/science.272.5264.1030
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C., et al. (2006). Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642. doi: 10.1038/nature05327
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Drolet, M. (2006). Growth inhibition mediated by excess negative supercoiling: the interplay between transcription elongation, R-loop formation and DNA topology. Mol. Microbiol. 59, 723–730. doi: 10.1111/j.1365-2958.2005.05006.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duch, A., Felipe-Abrio, I., Barroso, S., Yaakov, G., Garcia-Rubio, M., Aguilera, A., et al. (2013). Coordinated control of replication and transcription by a SAPK protects genomic integrity. Nature 493, 116–119. doi: 10.1038/nature11675
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dutta, D., Shatalin, K., Epshtein, V., Gottesman, M. E., and Nudler, E. (2011). Linking RNA polymerase backtracking to genome instability in E. coli. Cell 146, 533–543. doi: 10.1016/j.cell.2011.07.034
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., and Chedin, F. (2012). R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 45, 814–825. doi: 10.1016/j.molcel.2012.01.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gomez-Gonzalez, B., Felipe-Abrio, I., and Aguilera, A. (2009). The S-phase checkpoint is required to respond to R-loops accumulated in THO mutants. Mol. Cell. Biol. 29, 5203–5213. doi: 10.1128/MCB.00402-09
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gomez-Gonzalez, B., Garcia-Rubio, M., Bermejo, R., Gaillard, H., Shirahige, K., Marin, A., et al. (2011). Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J. 30, 3106–3119.
Gottipati, P., Cassel, T. N., Savolainen, L., and Helleday, T. (2008). Transcription-associated recombination is dependent on replication in Mammalian cells. Mol. Cell. Biol. 28, 154–164. doi: 10.1128/MCB.00816-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grierson, P. M., Lillard, K., Behbehani, G. K., Combs, K. A., Bhattacharyya, S., Acharya, S., et al. (2012). BLM helicase facilitates RNA polymerase I-mediated ribosomal RNA transcription. Hum. Mol. Genet. 21, 1172–1183. doi: 10.1093/hmg/ddr545
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Groh, M., Lufino, M. M., Wade-Martins, R., and Gromak, N. (2014). R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 10:e1004318. doi: 10.1371/journal.pgen.1004318
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haeusler, A. R., Donnelly, C. J., Periz, G., Simko, E. A., Shaw, P. G., Kim, M. S., et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200. doi: 10.1038/nature13124
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hagerling, C., Casbon, A. J., and Werb, Z. (2014). Balancing the innate immune system in tumor development. Trends Cell Biol. 25, 214–220. doi: 10.1016/j.tcb.2014.11.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hatchi, E., Skourti-Stathaki, K., Ventz, S., Pinello, L., Yen, A., Kamieniarz-Gdula, K., et al. (2015). BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 57, 636–647. doi: 10.1016/j.molcel.2015.01.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Helmrich, A., Ballarino, M., and Tora, L. (2011). Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44, 966–977. doi: 10.1016/j.molcel.2011.10.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Herrera-Moyano, E., Mergui, X., Garcia-Rubio, M. L., Barroso, S., and Aguilera, A. (2014). The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev. 28, 735–748. doi: 10.1101/gad.234070.113
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hill, S. J., Rolland, T., Adelmant, G., Xia, X., Owen, M. S., Dricot, A., et al. (2014). Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes Dev. 28, 1957–1975. doi: 10.1101/gad.241620.114
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huvet, M., Nicolay, S., Touchon, M., Audit, B., d’Aubenton-Carafa, Y., Arneodo, A., et al. (2007). Human gene organization driven by the coordination of replication and transcription. Genome Res. 17, 1278–1285. doi: 10.1101/gr.6533407
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Im, J. S., Keaton, M., Lee, K. Y., Kumar, P., Park, J., and Dutta, A. (2014). ATR checkpoint kinase and CRL1betaTRCP collaborate to degrade ASF1a and thus repress genes overlapping with clusters of stalled replication forks. Genes Dev. 28, 875–887. doi: 10.1101/gad.239194.114
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ivessa, A. S., Lenzmeier, B. A., Bessler, J. B., Goudsouzian, L. K., Schnakenberg, S. L., and Zakian, V. A. (2003). The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell 12, 1525–1536. doi: 10.1016/S1097-2765(03)00456-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jones, R. M., Mortusewicz, O., Afzal, I., Lorvellec, M., Garcia, P., Helleday, T., et al. (2012). Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 32, 3744–3753. doi: 10.1038/onc.2012.387
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kailasan Vanaja, S., Rathinam, V. A., Atianand, M. K., Kalantari, P., Skehan, B., Fitzgerald, K. A., et al. (2014). Bacterial RNA:DNA hybrids are activators of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 111, 7765–7770. doi: 10.1073/pnas.1400075111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kassavetis, G. A., and Kadonaga, J. T. (2014). The annealing helicase and branch migration activities of Drosophila HARP. PLoS ONE 9:e98173. doi: 10.1371/journal.pone.0098173
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, H. D., Choe, J., and Seo, Y. S. (1999). The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry 38, 14697–14710. doi: 10.1021/bi991470c
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, N., Abdulovic, A. L., Gealy, R., Lippert, M. J., and Jinks-Robertson, S. (2007). Transcription-associated mutagenesis in yeast is directly proportional to the level of gene expression and influenced by the direction of DNA replication. DNA Repair (Amst) 6, 1285–1296. doi: 10.1016/j.dnarep.2007.02.023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, N., Cho, J. E., Li, Y. C., and Jinks-Robertson, S. (2013). RNA:DNA hybrids initiate quasi-palindrome-associated mutations in highly transcribed yeast DNA. PLoS Genet. 9:e1003924. doi: 10.1371/journal.pgen.1003924
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kobayashi, T. (2014). Ribosomal RNA gene repeats, their stability and cellular senescence. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 90, 119–129. doi: 10.2183/pjab.90.119
Li, X., and Manley, J. L. (2005). Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 122, 365–378. doi: 10.1016/j.cell.2005.06.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, Y., Dent, S. Y., Wilson, J. H., Wells, R. D., and Napierala, M. (2010). R loops stimulate genetic instability of CTG.CAG repeats. Proc. Natl. Acad. Sci. U.S.A. 107, 692–697. doi: 10.1073/pnas.0909740107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, L. F., and Wang, J. C. (1987). Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. U.S.A. 84, 7024–7027. doi: 10.1073/pnas.84.20.7024
Lobsiger, C. S., and Cleveland, D. W. (2007). Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat. Neurosci. 10, 1355–1360. doi: 10.1038/nn1988
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Loomis, E. W., Sanz, L. A., Chedin, F., and Hagerman, P. J. (2014). Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 10:e1004294. doi: 10.1371/journal.pgen.1004294
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mankan, A. K., Schmidt, T., Chauhan, D., Goldeck, M., Honing, K., Gaidt, M., et al. (2014). Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. 33, 2937–2946. doi: 10.15252/embj.201488726
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Merrikh, H., Machon, C., Grainger, W. H., Grossman, A. D., and Soultanas, P. (2011). Co-directional replication-transcription conflicts lead to replication restart. Nature 470, 554–557. doi: 10.1038/nature09758
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Merrikh, H., Zhang, Y., Grossman, A. D., and Wang, J. D. (2012). Replication-transcription conflicts in bacteria. Nat. Rev. Microbiol. 10, 449–458. doi: 10.1038/nrmicro2800
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mirkin, E. V., and Mirkin, S. M. (2005). Mechanisms of transcription-replication collisions in bacteria. Mol. Cell. Biol. 25, 888–895. doi: 10.1128/MCB.25.3.888-895.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mischo, H. E., Gomez-Gonzalez, B., Grzechnik, P., Rondon, A. G., Wei, W., Steinmetz, L., et al. (2011). Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell 41, 21–32. doi: 10.1016/j.molcel.2010.12.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moreira, M. C., Klur, S., Watanabe, M., Nemeth, A. H., Le Ber, I., Moniz, J. C., et al. (2004). Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 36, 225–227. doi: 10.1038/ng1303ng1303
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakama, M., Kawakami, K., Kajitani, T., Urano, T., and Murakami, Y. (2012). DNA-RNA hybrid formation mediates RNAi-directed heterochromatin formation. Genes Cells 17, 218–233. doi: 10.1111/j.1365-2443.2012.01583.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nguyen, V. C., Clelland, B. W., Hockman, D. J., Kujat-Choy, S. L., Mewhort, H. E., and Schultz, M. C. (2010). Replication stress checkpoint signaling controls tRNA gene transcription. Nat. Struct. Mol. Biol. 17, 976–981. doi: 10.1038/nsmb.1857
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Olavarrieta, L., Martinez-Robles, M. L., Hernandez, P., Krimer, D. B., and Schvartzman, J. B. (2002). Knotting dynamics during DNA replication. Mol. Microbiol. 46, 699–707. doi: 10.1046/j.1365-2958.2002.03217.x
Paeschke, K., Capra, J. A., and Zakian, V. A. (2011). DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145, 678–691. doi: 10.1016/j.cell.2011.04.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Paul, S., Million-Weaver, S., Chattopadhyay, S., Sokurenko, E., and Merrikh, H. (2012). Accelerated gene evolution through replication-transcription conflicts. Nature 495, 512–515. doi: 10.1038/nature11989
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pomerantz, R. T., and O’Donnell, M. (2008). The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature 456, 762–766. doi: 10.1038/nature07527
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Popuri, V., Bachrati, C. Z., Muzzolini, L., Mosedale, G., Costantini, S., Giacomini, E., et al. (2008). The human RecQ helicases, BLM and RECQ1, display distinct DNA substrate specificities. J. Biol. Chem. 283, 17766–17776. doi: 10.1074/jbc.M709749200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Prado, F., and Aguilera, A. (2005). Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 24, 1267–1276. doi: 10.1038/sj.emboj.7600602
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reddy, K., Schmidt, M. H., Geist, J. M., Thakkar, N. P., Panigrahi, G. B., Wang, Y. H., et al. (2010). Processing of double-R-loops in (CAG).(CTG) and C9orf72 (GGGGCC).(GGCCCC) repeats causes instability. Nucleic Acids Res. 42, 10473–10487. doi: 10.1093/nar/gku658
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Richard, P., Feng, S., and Manley, J. L. (2013). A SUMO-dependent interaction between Senataxin and the exosome, disrupted in the neurodegenerative disease AOA2, targets the exosome to sites of transcription-induced DNA damage. Genes Dev. 27, 2227–2232. doi: 10.1101/gad.224923.113
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rigby, R. E., Webb, L. M., Mackenzie, K. J., Li, Y., Leitch, A., Reijns, M. A., et al. (2014). RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J. 33, 542–558. doi: 10.1002/embj.201386117
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rocha, E. P. (2008). The organization of the bacterial genome. Annu. Rev. Genet. 42, 211–233. doi: 10.1146/annurev.genet.42.110807.091653
Saponaro, M., Kantidakis, T., Mitter, R., Kelly, G. P., Heron, M., Williams, H., et al. (2014). RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 157, 1037–1049. doi: 10.1016/j.cell.2014.03.048
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schiavone, D., Guilbaud, G., Murat, P., Papadopoulou, C., Sarkies, P., Prioleau, M. N., et al. (2014). Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. EMBO J. 33, 2507–2520. doi: 10.15252/embj.201488398
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Skourti-Stathaki, K., Kamieniarz-Gdula, K., and Proudfoot, N. J. (2014). R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 516, 436–439. doi: 10.1038/nature13787
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Skourti-Stathaki, K., and Proudfoot, N. J. (2014). A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 28, 1384–1396. doi: 10.1101/gad.242990.114
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Skourti-Stathaki, K., Proudfoot, N. J., and Gromak, N. (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 42, 794–805. doi: 10.1016/j.molcel.2011.04.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sollier, J., Stork, C. T., Garcia-Rubio, M. L., Paulsen, R. D., Aguilera, A., and Cimprich, K. A. (2014). Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 56, 777–785. doi: 10.1016/j.molcel.2014.10.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stirling, P. C., Chan, Y. A., Minaker, S. W., Aristizabal, M. J., Barrett, I., Sipahimalani, P., et al. (2012). R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev. 26, 163–175. doi: 10.1101/gad.179721.111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tuduri, S., Crabbe, L., Conti, C., Tourriere, H., Holtgreve-Grez, H., Jauch, A., et al. (2009). Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 11, 1315–1324. doi: 10.1038/ncb1984
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tuduri, S., Crabbe, L., Tourriere, H., Coquelle, A., and Pasero, P. (2010). Does interference between replication and transcription contribute to genomic instability in cancer cells? Cell Cycle 9, 1886–1892. doi: 10.4161/cc.9.10.11539
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wahba, L., Amon, J. D., Koshland, D., and Vuica-Ross, M. (2011). RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell 44, 978–988. doi: 10.1016/j.molcel.2011.10.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yuce, O., and West, S. C. (2013). Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol. Cell. Biol. 33, 406–417. doi: 10.1128/MCB.01195-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zaratiegui, M., Castel, S. E., Irvine, D. V., Kloc, A., Ren, J., Li, F., et al. (2011). RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature 479, 135–138. doi: 10.1038/nature10501
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, Y., Shishkin, A. A., Nishida, Y., Marcinkowski-Desmond, D., Saini, N., Volkov, K. V., et al. (2012). Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Mol. Cell 48, 254–265. doi: 10.1016/j.molcel.2012.08.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: replication–transcription conflicts, R-loops, replication stress, cancer, neurodegeneration, genetic instability, epigenetic instability
Citation: Brambati A, Colosio A, Zardoni L, Galanti L and Liberi G (2015) Replication and transcription on a collision course: eukaryotic regulation mechanisms and implications for DNA stability. Front. Genet. 6:166. doi: 10.3389/fgene.2015.00166
Received: 27 February 2015; Paper pending published 27 March 2015;
Accepted: 12 April 2015 Published online: 28 April 2015.
Edited by:
Alessandra Montecucco, Consiglio Nazionale delle Ricerche, ItalyReviewed by:
Parvin Mehdipour, Tehran University of Medical Sciences, IranCopyright © 2015 Brambati, Colosio, Zardoni, Galanti and Liberi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giordano Liberi, Istituto di Genetica Molecolare del Consiglio Nazionale delle Ricerche, Via Abbiategrasso 207, 27100 Pavia, Italy,Z2lvcmRhbm8ubGliZXJpQGlnbS5jbnIuaXQ=
† These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.