 Luciana Oliveira1
Luciana Oliveira1 Simone Appenzeller
Simone Appenzeller Timothy B. Niewold
Timothy B. Niewold- 1Rheumatology Unit, Department of Medicine, Faculty of Medical Science, State University of Campinas, Campinas, Brazil
- 2Mayo Clinic, Division of Rheumatology, Department of Immunology, Rochester, MN, USA
In the autoimmune disease systemic lupus erythematosus (SLE), our normal antiviral defenses are inappropriately activated, resulting in over-activity of the type I interferon (IFN) pathway. This increased activity of the type I IFN pathway is an important primary pathogenic factor in the disease. Emerging evidence has implicated the antiviral helicases in this process. The antiviral helicases normally function as nucleic acid receptors in viral immunity. Genetic variations in antiviral helicase genes have been associated with SLE, supporting the idea that helicase pathways are involved in the primary pathogenesis of SLE. Studies have documented functional consequences of these genetic variations within the type I IFN pathway in human cell lines and SLE patients. In this review, we summarize the function of helicases in the anti-viral immune response, and how this response is dysregulated in SLE patients. In particular, we will focus on known functional genetic polymorphisms in the IFIH1 (MDA5) and mitochondrial antiviral signaling protein genes which have been implicated in human SLE. These data provide fascinating evidence for dysregulation of helicase-mediated innate immunity in SLE, and may support novel therapeutic strategies in the disease.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by multisystem inflammation commonly including the skin, kidneys, and joints, and other systems. While the pathogenesis of SLE is not completely understood, it seems likely that both genetic and environmental factors contribute to the disease. A number of genetic factors have been associated with SLE in recent years (Harley et al., 2008; Ghodke-Puranik and Niewold, 2013), providing a window into human disease pathogenesis. Interestingly, many of these genetic variations associated with risk of SLE have function within the type I interferon (IFN) pathway (Ghodke-Puranik and Niewold, 2013). Type I IFN is a classical anti-viral molecule which causes activation of antigen presenting cells within the innate immune system and increased expression of MHC and co-stimulatory molecules (Pestka et al., 2004).
Many lines of evidence support the idea that type I IFN plays a primary role in SLE pathogenesis (Niewold, 2011). Circulating type I IFN levels are elevated in many SLE patients (Weckerle et al., 2011), and this elevation is also observed in unaffected members of SLE families, suggesting that high IFN levels are a heritable risk factor for SLE (Niewold et al., 2007). Familial aggregation has been observed with other cytokines in SLE, such as tumor necrosis factor alpha and IL-10 (Grondal et al., 1999; Mangale et al., 2013), but in these cases unrelated family members such as spouses shared the trait as well, suggesting a contribution from environmental factors. Subsequent study of SLE-associated genetic factors has confirmed that SLE-risk genes contribute to the high IFN levels observed in SLE (Kariuki et al., 2008; Kariuki and Niewold, 2010; Agik et al., 2012; Niewold et al., 2012), and it seems that the high IFN trait is significantly polygenic (Harley et al., 2010; Kariuki et al., 2010; Koldobskaya et al., 2012; Jensen et al., 2013). The genetic data all support the concept that common gain-of-function variations in the type I IFN pathway are associated with SLE pathogenesis. Additionally, rare variants in the TREX1 gene have been described that are strongly associated with a SLE and Aicardi-Goutieres syndrome, a rare condition characterized by alterations in type I IFN and neurologic symptoms (Lee-Kirsch et al., 2007; Namjou et al., 2011). Recombinant human type I IFN has been administered as a therapeutic to treat some malignancies and chronic viral infection, and in some cases de novo SLE has developed (Ronnblom et al., 1990), which typically resolves when the type I IFN is stopped (Niewold and Swedler, 2005). These data taken together support the idea that type I IFN is a primary pathogenic factor in human SLE. While there are significant differences in SLE incidence between men and women (9:1 female), type I IFN pathway activation seems to be equal between men and women with SLE (Niewold et al., 2008a; Weckerle and Niewold, 2011). There is an increased incidence of SLE in African-Americans as compared to European-American populations (4:1), and in this case is seems that there are some differences in the way the pathway is activated, but high IFN is clearly seen in both populations (Ko et al., 2012, 2013). Thus type I IFN is a common pathway to SLE susceptibility, and it follows that molecules operating upstream of type I IFN production would play a role in disease. A large body of work has supports the relevance of the endosomal Toll-like receptors in SLE pathogenesis (Lovgren et al., 2004; Salloum and Niewold, 2011). In this review, we will focus on emerging data which implicates RNA helicases in type I IFN pathway dysregulation in human SLE. These data may also be relevant to other autoimmune diseases, as a number of conditions have been associated with increased type I IFN, including dermatomyositis, Sjogren’s syndrome, neuromyelitis optica, and others (Niewold et al., 2008b, 2011; Sweiss et al., 2011; Feng et al., 2012; Mavragani et al., 2013). In particular, we will focus on known functional genetic polymorphisms in the IFIH1 (MDA5) and mitochondrial antiviral signaling protein (MAVS) genes which function in helicase pathways, and have been implicated in human SLE.
Pattern Recognition Receptors Involved in Anti-Viral Responses and SLE
Several families of receptors that recognize pathogen-associated molecular patterns (PAMPs) have been described, such as the Toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I)-like receptor (RLRs). TLRs are transmembrane receptors expressed in specific immune cells, such as dendritic cells and macrophages. TLR7, 8, and 9 are expressed in the endosomal membrane, and can recognize viral nucleic acid. In anti-viral immunity, viral immune complexes are taken up via Fc receptors, and then delivered to the endosome resulting in TLR activation. RLRs, on the other hand, are cytosolic proteins that can recognize viral nucleic acid in the cytosol. Activation of either TLRs or RLRs results in IFN production and an anti-viral response (Figure 1).
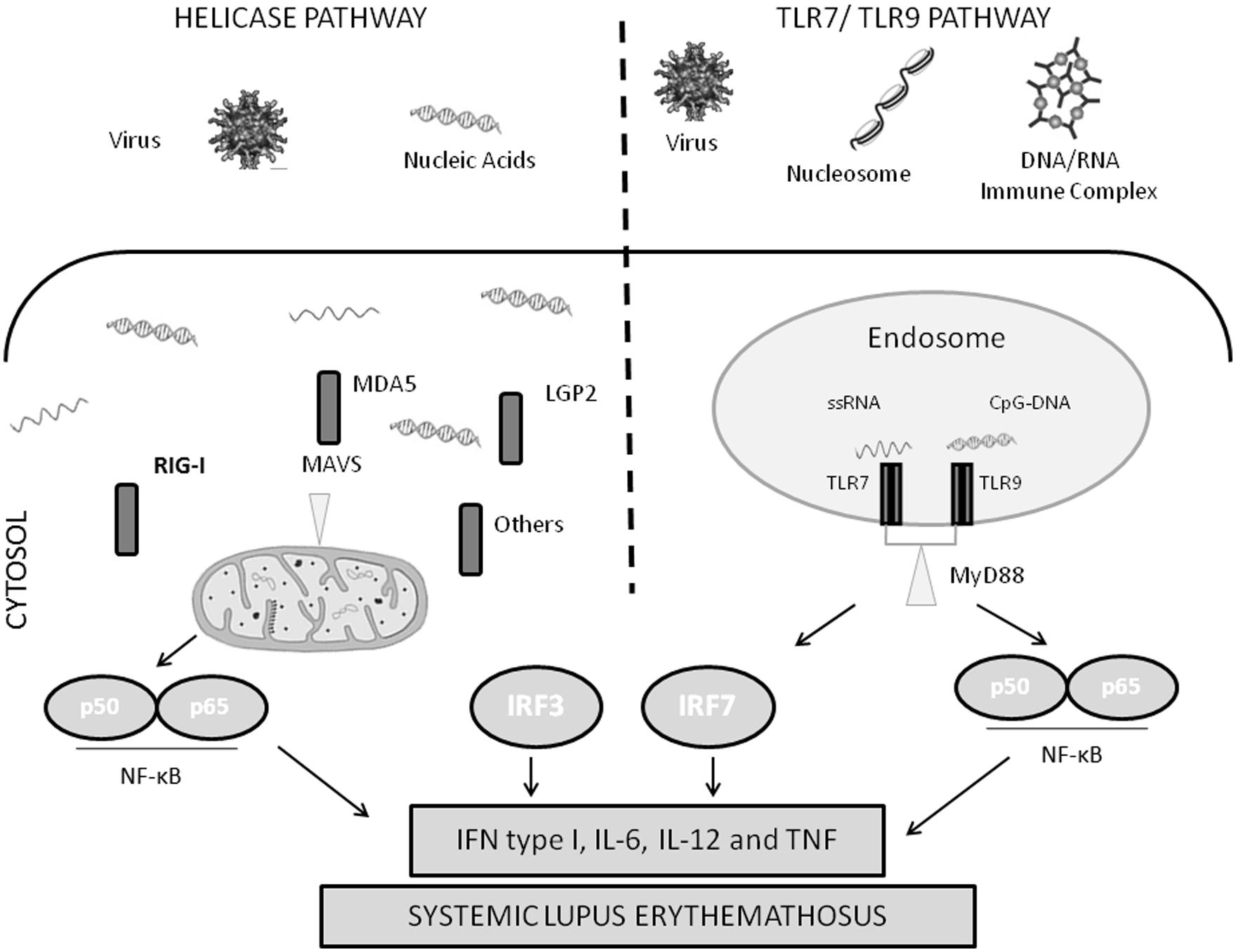
FIGURE 1. Diagram of nucleic acid sensing antiviral immune response pathways. IFN, type I interferon; IL, interleukin; IRF, interferon regulatory factor; LGP2, laboratory of genetics and physiology 2; MAVS, mitochondrial antiviral signaling protein; MDA5, melanoma differentiation-associated gene 5; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor-κB; RIG-I, retinoic acid-inducible gene I; TLR, Toll-like receptor; TNF, tumor necrosis factor; TRIF, Toll/IL-1R-domain-containing adaptor inducing interferon β.
The endosomal TLRs have been implicated in SLE pathogenesis in a number of previous studies. Genetic variations in TLR7 are associated with SLE susceptibility in humans (Deng et al., 2013). Mice which carry a duplication of the endosomal TLR region of the X-chromosome have enhanced susceptibility to an SLE-like disease (Subramanian et al., 2006). Interestingly, the autoantibodies which are characteristically produced in SLE target components of the cell nucleus, for example antibodies against double-stranded DNA and nuclear RNA-binding proteins. These SLE-associated autoantibodies form immune complexes that contain RNA and DNA, and the immune complexes can result in activation of the TLR system with subsequent type I IFN production (Lovgren et al., 2004, 2006). Thus, the anti-nuclear immune response that characterizes SLE produces immune complexes that are viral mimics, subverting normal viral immunity. These autoantibodies are frequently high titer and continuously present in SLE, and the antigens are ubiquitous, resulting in consistent inappropriate activation of the type I IFN pathway in SLE (Salloum and Niewold, 2011). Supporting this idea, one of the accepted and effective treatments for SLE, hydroxychloroquine, seems to interrupt endosomal TLR signaling (Kuznik et al., 2011; Sacre et al., 2012).
RNA Helicases
Ribonucleic acid helicases are involved in almost all cellular processes involving RNA (Steimer and Klostermeier, 2012). These enzymes use ATP to bind or remodel RNA and RNA–protein complexes (Linder and Jankowsky, 2011). Based on their shared conserved motifs and three dimensional core structures, RNA helicases are grouped into families and superfamilies (Linder and Jankowsky, 2011; Steimer and Klostermeier, 2012). The majority of RNA helicases belong to superfamily 2 (SF2; Pyle, 2008; Steimer and Klostermeier, 2012). DEAD box proteins are the largest family of helicases in SF2, and in humans these helicases have essential physiological roles in cellular RNA metabolism (Linder and Jankowsky, 2011). The DEAD box helicases work by destabilizing short RNA duplexes close to the binding site of the helicase. In contrast, the DExH group of helicases work in a progressive fashion, unwinding longer stretches of RNA (Pyle, 2008). DEAD box helicases frequently play a role in viral immunity by acting as sensors cytosolic viral nucleic acids. Besides the RLR DEAD box helicases which include RIG-I, MDA5, and LGP2, other DEAD box helicases likely perform this role as well, including DDX1, DDX3, DDX36, DDX41, DDX60, and others (Fullam and Schroder, 2013). In addition to sensing nucleic acid, some of these functions may be helicases further downstream in the pattern recognition receptor signaling pathways, potentially playing roles in transcriptional regulation (Fullam and Schroder, 2013). Interestingly, it appears that some RNA helicases are important for viral replication, suggesting that viruses have adopted this cellular mechanism to their own advantage in some cases (Fullam and Schroder, 2013).
Retinoic acid-inducible gene-I and MDA5 are proteins encoded by the DDX58 and IFIH1 genes, respectively. These RLRs are induced by type I IFN, and each recognize specific types of viruses (Yoneyama and Fujita, 2008). RIG-I and MDA5 recognize distinct viral RNA structures containing 5′ triphosphate in single and double-stranded RNA (Shrivastav and Niewold, 2013). These two RLRs demonstrate some specificity in the types of nucleic acids they recognize: while MDA5 senses picornavirus-derived nucleic acid, RIG-I senses other viral nucleic acids, such as those derived from influenza A (Yoneyama and Fujita, 2008). This differential recognition is based on the distinct RNA patterns generated by different viruses (Yoneyama and Fujita, 2008).
Activation of RIG-I and MDA5 by nucleic acid leads to binding of the mitochondrial adaptor IFN β promoter stimulater 1 (IPS-1) also known as MAVS (Reikine et al., 2014). As the name suggests, MAVS is found in the mitochondrial membrane, and is critical to signal transduction via MDA5 and RIG-I. MAVS forms large multimeric polymers on the mitochondrial membrane in combination with RIG-I and MDA5 bound to target nucleic acids, forming an active signaling complex (Reikine et al., 2014). This leads to activation of NF-κB, IRF3, and IRF7 (Reikine et al., 2014). These transcription factors are involved in IFN and interferon-stimulated gene expression, and the production of type-1 IFN and pro-inflammatory cytokines (Shrivastav and Niewold, 2013; Figure 1).
MDA5/IFIH1
IFIH1 is the gene that encodes MDA5, and a common coding-change polymorphism in the IFIH1 gene has been associated with risk of SLE and other autoimmune diseases in humans (Smyth et al., 2006; Sutherland et al., 2007; Harley et al., 2008; Gateva et al., 2009; Strange et al., 2010; Molineros et al., 2013). This A946T polymorphism in IFIH1 was identified in case-control genetic studies of SLE (Harley et al., 2008; Gateva et al., 2009), and interestingly this polymorphism was the major finding in a recent admixture-mapping genetic screen to identify genes associated with SLE in African-Americans (Molineros et al., 2013), supporting relevance of this polymorphism across multiple ancestral backgrounds. This polymorphism appears to be gain-of-function in nature, being associated with increased IFIH1 mRNA expression (Downes et al., 2010), increased sensitivity to type I IFN and increased IFN-induced gene expression in circulating blood cells from SLE patients (Robinson et al., 2011), and modulation of inflammation- and apoptosis-related gene expression (Molineros et al., 2013). These studies support the general idea that over-activity of the anti-viral helicases would result in greater type I IFN signaling and risk of SLE. Rare loss-of-function variations in IFIH1 have been discovered in the IFIH1 gene, and interestingly these loss-of-function variants appear to be protective against autoimmune disease (Nejentsev et al., 2009), further supporting the idea that increased function of IFIH1/MDA5 is associated with risk of autoimmune disease.
Studies in mice have also supported this hypothesis. A recent study demonstrated that a single coding-change mutation in IFIH1 (Gly821Ser) generated by N-ethyl-N-nitrosourea (ENU) resulted in constitutive activation of MDA5 (Funabiki et al., 2014). Mice with this mutation developed a systemic autoimmune disease similar to lupus, with nephritis characterized by lymphocyte infiltration as well as deposition of immunoglobulin and complement, systemic inflammation in the heart and lung, and increased tumor necrosis factor alpha, IL-6 and type I IFN (Funabiki et al., 2014). This gain-of-function mutation in the mouse line has not been observed in humans, but it supports the general concept that gain of function in IFIH1 is associated with autoimmunity.
MAVS
Genetic studies in human SLE have also identified a functional coding-change polymorphism in MAVS, a key adapter of both the RIG-I and MDA5 helicases. The C97F polymorphism in MAVS substantially reduced the expression of type I IFN and other proinflammatory mediators in human cell lines (Pothlichet et al., 2011). Interestingly, this variation was almost exclusively found in the African-American population, with a frequency of 10.2% in controls (Pothlichet et al., 2011). In African–American patients with SLE, the C79F allele was associated with low type I IFN and was more than twice as common (22.4% frequency) in SLE patients who lacked autoantibodies to RNA-binding proteins. This study demonstrated that a coding-change genetic variation in the gene encoding MAVS has a functional impact on the antiviral IFN pathway in humans, and is associated with a serologic subgroup of SLE patients (Pothlichet et al., 2011). These studies in both MAVS and IFIH1 demonstrate the importance of variations in these genes upon immune function and autoimmune disease risk in human populations.
Conclusion
Dysregulation of anti-viral helicase immune responses represent a primary pathogenic factor in human SLE. This is demonstrated by the presence of coding-change polymorphisms in both the IFIH1 and MAVS genes which modulate function of the type I IFN pathway and risk of SLE in humans. While immune complexes formed by SLE autoantibodies and nuclear material seem to be the likely trigger for endosomal TLR stimulation in SLE, the exact triggers of the cytosolic anti-viral helicases in human SLE are somewhat less clear. Viruses may stimulate some of the early events in SLE patients, as strong epidemiological data implicates Epstein–Barr virus infection in the initial pathogenesis of SLE (James et al., 1997; Poole et al., 2006). It is possible that this represents a gene – environment interaction in human SLE – a hypersensitive or overactive anti-viral helicase system coupled with a viral trigger, such as Epstein–Barr virus infection, which then results in an exaggerated type I IFN response and subsequent misdirection of the adaptive immune response against self-antigens. It is also possible that viral-like elements, such as LINE elements, may also play a role in the chronic stimulation of these cytosolic nucleic acid receptors (Crow, 2010). Both Epstein-Barr virus and LINE-1 RNAs could potentially be seen as “foreign” by the cytosolic helicases. The fact that both the TLR and cytosolic pathways of viral recognition are involved in human SLE and the convergence of these pathways upon type I IFN and anti-viral responses is striking, suggesting that over-active anti-viral immunity represents a major common pathogenic pathway in human SLE. A number of therapeutics have been developed to target type I IFN in SLE, including monoclonal antibodies against IFN-α (Merrill et al., 2011; Kalunian et al., 2012; McBride et al., 2012), as well as a vaccination strategy aimed at inducing antibodies against IFN-α (Lauwerys et al., 2013). These studies are in early stages, phase I to phase II, and thus far the data generally support relative safety and proof-of-mechanism. It is too early to make conclusions about efficacy, but some of the larger phase II studies have reported potential subset effects within the overall SLE patient groups (Kalunian et al., 2012). This would suggest that anti-IFN therapies may not work for every patient, but the genetics data we discuss in this review may also suggest this outcome. It seems that the IFN pathway is impacted by a number of genetic factors, and these factors will not be shared by all patients. Therapeutics targeting the RNA helicases could be potentially interesting in SLE, and further understanding of the specific dysregulation of the helicase pathways in human SLE such as the work summarized in this review could help to determine optimal points of intervention in the pathway and in which group of patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Simone Appenzeller – Conselho Nacional de Desenvolvimento Cientifico e Tecnologico de Brasil, Visiting Researcher Project Grant, Program Ciencia Sem Frontieras; Timothy B. Niewold – Research grants from the NIH (AR060861, AI083790, AI071651), Rheumatology Research Foundation, the Mayo Clinic Foundation, the Lupus Foundation of Minnesota, and Conselho Nacional de Desenvolvimento Cientifico e Tecnologico de Brasil, Visiting Researcher Project Grant, Program Ciencia Sem Frontieras.
References
Agik, S., Franek, B. S., Kumar, A. A., Kumabe, M., Utset, T. O., Mikolaitis, R. A.,et al. (2012). The autoimmune disease risk allele of UBE2L3 in African American patients with systemic lupus erythematosus: a recessive effect upon subphenotypes. J. Rheumatol. 39, 73–78. doi: 10.3899/jrheum.110590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crow, M. K. (2010). Long interspersed nuclear elements (LINE-1): potential triggers of systemic autoimmune disease. Autoimmunity 43, 7–16. doi: 10.3109/08916930903374865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deng, Y., Zhao, J., Sakurai, D., Kaufman, K. M., Edberg, J. C., Kimberly, R. P.,et al. (2013). MicroRNA-3148 modulates allelic expression of toll-like receptor 7 variant associated with systemic lupus erythematosus. PLoS Genet. 9:e1003336. doi: 10.1371/journal.pgen.1003336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Downes, K., Pekalski, M., Angus, K. L., Hardy, M., Nutland, S., Smyth, D. J.,et al. (2010). Reduced expression of IFIH1 is protective for type 1 diabetes. PLoS ONE 55:e12646. doi: 10.1371/journal.pone.0012646
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feng, X., Reder, N. P., Yanamandala, M., Hill, A., Franek, B. S., Niewold, T. B.,et al. (2012). Type I interferon signature is high in lupus and neuromyelitis optica but low in multiple sclerosis. J. Neurol. Sci. 313, 48–53. doi: 10.1016/j.jns.2011.09.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fullam, A., and Schroder, M. (2013). DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim. Biophys. Acta 1829, 854–865. doi: 10.1016/j.bbagrm.2013.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Funabiki, M., Kato, H., Miyachi, Y., Toki, H., Motegi, H., Inoue, M.,et al. (2014). Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 40, 199–212. doi: 10.1016/j.immuni.2013.12.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gateva, V., Sandling, J. K., Hom, G., Taylor, K. E., Chung, S. A., Sun, X.,et al. (2009). A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 41, 1228–1233. doi: 10.1038/ng.468
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghodke-Puranik, Y., and Niewold, T. B. (2013). Genetics of the type I interferon pathway in systemic lupus erythematosus. Int. J. Clin. Rheumtol. 8, 657–669. doi: 10.2217/ijr.13.58
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grondal, G., Kristjansdottir, H., Gunnlaugsdottir, B., Arnason, A., Lundberg, I., Klareskog, L.,et al. (1999). Increased number of interleukin-10-producing cells in systemic lupus erythematosus patients and their first-degree relatives and spouses in Icelandic multicase families. Arthritis Rheum. 42, 1649–1654. doi: 10.1002/1529-0131(199908)42:8<1649::AID-ANR13>3.0.CO;2-D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harley, J. B., Alarcon-Riquelme, M. E., Criswell, L. A., Jacob, C. O., Kimberly, R. P., Moser, K. L.,et al. (2008). Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 40, 204–210. doi: 10.1038/ng.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harley, T. W. I., Niewold, T. B., Stormont, R. M., Kaufman, K. M., Glenn, S. B., Franek, B. S.,et al. (2010). The role of genetic variation near interferon-kappa in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010:706825. doi: 10.1155/2010/706825
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
James, J. A., Kaufman, K. M., Farris, A. D., Taylor-Albert, E., Lehman, T. J., and Harley, J. B. (1997). An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J. Clin. Invest. 100, 3019–3026. doi: 10.1172/JCI119856
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jensen, M. A., Patterson, K. C., Kumar, A. A., Kumabe, M., Franek, B. S., and Niewold, T. B. (2013). Functional genetic polymorphisms in ILT3 are associated with decreased surface expression on dendritic cells and increased serum cytokines in lupus patients. Ann. Rheum. Dis. 72, 596–601. doi: 10.1136/annrheumdis-2012-202024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kalunian, K., Merrill, J. T., Maciuca, R., Ouyang, W., McBride, J. M., Townsend, M. J.,et al. (2012). Efficacy and safety of rontalizumab (Anti-Interferon Alpha) in SLE subjects with restricted immunosuppressant use: results of a randomized, double-blind, placebo-controlled phase 2 study. Arthritis Rheum. 64:2622. doi: 10.1002/art.40353
Kariuki, S. N., Crow, M. K., and Niewold, T. B. (2008). The PTPN22 C1858T polymorphism is associated with skewing of cytokine profiles toward high interferon-alpha activity and low tumor necrosis factor alpha levels in patients with lupus. Arthritis Rheum. 58, 2818–2823. doi: 10.1002/art.23728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kariuki, S. N., Franek, B. S., Kumar, A. A., Arrington, J., Mikolaitis, R. A., Utset, T. O.,et al. (2010). Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res. Ther. 12:R151. doi: 10.1186/ar3101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kariuki, S. N., and Niewold, T. B. (2010). Genetic regulation of serum cytokines in systemic lupus erythematosus. Transl. Res. 155, 109–117. doi: 10.1016/j.trsl.2009.08.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ko, K., Franek, B. S., Marion, M., Kaufman, K. M., Langefeld, C. D., Harley, J. B.,et al. (2012). Genetic ancestry, serum interferon-alpha activity, and autoantibodies in systemic lupus erythematosus. J. Rheumatol. 39, 1238–1240. doi: 10.3899/jrheum.111467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ko, K., Koldobskaya, Y., Rosenzweig, E., and Niewold, T. B. (2013). Activation of the interferon pathway is dependent upon autoantibodies in African-American SLE patients, but Not in European-American SLE patients. Front. Immunol. 4:309. doi: 10.3389/fimmu.2013.00309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koldobskaya, Y., Ko, K., Kumar, A. A., Agik, S., Arrington, J., Kariuki, S. N.,et al. (2012). Gene-expression-guided selection of candidate loci and molecular phenotype analyses enhance genetic discovery in systemic lupus erythematosus. Clin. Dev. Immunol. 2012:682018. doi: 10.1155/2012/682018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuznik, A., Bencina, M., Svajger, U., Jeras, M., Rozman, B., and Jerala, R. (2011). Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J. Immunol. 186, 4794–4804. doi: 10.4049/jimmunol.1000702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lauwerys, B. R., Hachulla, E., Spertini, F., Lazaro, E., Jorgensen, C., Mariette, X.,et al. (2013). Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon alpha-kinoid. Arthritis Rheum. 65, 447–456. doi: 10.1002/art.37785
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee-Kirsch, M. A., Gong, M., Chowdhury, D., Senenko, L., Engel, K., Lee, Y. A.,et al. (2007). Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 39, 1065–1067. doi: 10.1038/ng2091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Linder, P., and Jankowsky, E. (2011). From unwinding to clamping – the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 12, 505–516. doi: 10.1038/nrm3154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lovgren, T., Eloranta, M. L., Bave, U., Alm, G. V., and Ronnblom, L. (2004). Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 50, 1861–1872. doi: 10.1002/art.20254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lovgren, T., Eloranta, M. L., Kastner, B., Wahren-Herlenius, M., Alm, G. V., and Ronnblom, L. (2006). Induction of interferon-alpha by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjogren’s syndrome autoantigen-associated RNA. Arthritis Rheum. 54, 1917–1927. doi: 10.1002/art.21893
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mangale, D., Kariuki, S. N., Chrabot, B. S., Kumabe, M., Kelly, J. A., Harley, J. B.,et al. (2013). Familial aggregation of high tumor necrosis factor alpha levels in systemic lupus erythematosus. Clin. Dev. Immunol. 2013:267430. doi: 10.1155/2013/267430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mavragani, C. P., Niewold, T. B., Chatzigeorgiou, A., Danielides, S., Thomas, D., Kirou, K. A.,et al. (2013). Increased serum type I interferon activity in organ-specific autoimmune disorders: clinical, imaging, and serological associations. Front. Immunol. 4:238. doi: 10.3389/fimmu.2013.00238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McBride, J. M., Jiang, J., Abbas, A. R., Morimoto, A., Li, J., Maciuca, R.,et al. (2012). Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum. 64, 3666–3676. doi: 10.1002/art.34632
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merrill, J. T., Wallace, D. J., Petri, M., Kirou, K. A., Yao, Y., White, W. I.,et al. (2011). Lupus interferon skin activity study, safety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann. Rheum. Dis. 70, 1905–1913. doi: 10.1136/ard.2010.144485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Molineros, J. E., Maiti, A. K., Sun, C., Looger, L. L., Han, S., Kim-Howard, X.,et al. (2013). Admixture mapping in lupus identifies multiple functional variants within IFIH1 associated with apoptosis, inflammation, and autoantibody production. PLoS Genet. 9:e1003222. doi: 10.1371/journal.pgen.1003222
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Namjou, B., Kothari, P. H., Kelly, J. A., Glenn, S. B., Ojwang, J. O., Adler, A.,et al. (2011). Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 12, 270–279. doi: 10.1038/gene.2010.73
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nejentsev, S., Walker, N., Riches, D., Egholm, M., and Todd, J. A. (2009). Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324, 387–389. doi: 10.1126/science.1167728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niewold, T. B. (2011). Interferon alpha as a primary pathogenic factor in human lupus. J. Interferon Cytokine Res. 31, 887–892. doi: 10.1089/jir.2011.0071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niewold, T. B., Adler, J. E., Glenn, S. B., Lehman, T. J., Harley, J. B., and Crow, M. K. (2008a). Age- and sex-related patterns of serum interfeon-alpha activity in lupus families. Arthritis Rheum. 58, 2113–2119. doi: 10.1002/art.23619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niewold, T. B., Rivera, T. L., Buyon, J. P., and Crow, M. K. (2008b). Serum type I interferon activity is dependent on maternal diagnosis in anti-SSA/Ro-positive mothers of children with neonatal lupus. Arthritis Rheum. 58, 541–546. doi: 10.1002/art.23191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niewold, T. B., Hua, J., Lehman, T. J., Harley, J. B., and Crow, M. K. (2007). High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 8, 492–502. doi: 10.1038/sj.gene.6364408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niewold, T. B., Kelly, J. A., Kariuki, S. N., Franek, B. S., Kumar, A. A., Kaufman, K. M.,et al. (2012). IRF5 haplotypes demonstrate diverse serological associations which predict serum interferon alpha activity and explain the majority of the genetic association with systemic lupus erythematosus. Ann. Rheum. Dis. 71, 463–468. doi: 10.1136/annrheumdis-2011-200463
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niewold, T. B., and Swedler, W. I. (2005). Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin. Rheumatol. 24, 178–181. doi: 10.1007/s10067-004-1024-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niewold, T. B., Wu, S. C., Smith, M., Morgan, G. A., and Pachman, L. M. (2011). Familial aggregation of autoimmune disease in juvenile dermatomyositis. Pediatrics 127, e1239–e1246. doi: 10.1542/peds.2010-3022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pestka, S., Krause, C. D., and Walter, M. R. (2004). Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202, 8–32. doi: 10.1111/j.0105-2896.2004.00204.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Poole, B. D., Scofield, R. H., Harley, J. B., and James, J. A. (2006). Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 39, 63–70. doi: 10.1080/08916930500484849
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pothlichet, J., Niewold, T. B., Vitour, D., Solhonne, B., Crow, M. K., and Si-Tahar, M. (2011). A loss-of-function variant of the antiviral molecule MAVS is associated with a subset of systemic lupus patients. EMBO Mol. Med. 3, 142–152. doi: 10.1002/emmm.201000120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pyle, A. M. (2008). Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys. 37, 317–336. doi: 10.1146/annurev.biophys.37.032807.125908
Reikine, S., Nguyen, J. B., and Modis, Y. (2014). Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front. Immunol. 5:342. doi: 10.3389/fimmu.2014.00342
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robinson, T., Kariuki, S. N., Franek, B. S., Kumabe, M., Kumar, A. A., Badaracco, M.,et al. (2011). Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-alpha and serologic autoimmunity in lupus patients. J. Immunol. 187, 1298–1303. doi: 10.4049/jimmunol.1100857
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ronnblom, L. E., Alm, G. V., and Oberg, K. E. (1990). Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J. Intern. Med. 227, 207–210. doi: 10.1111/j.1365-2796.1990.tb00144.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sacre, K., Criswell, L. A., and McCune, J. M. (2012). Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus . Arthritis Res. Ther. 14:R155. doi: 10.1186/ar3895
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Salloum, R., and Niewold, T. B. (2011). Interferon regulatory factors in human lupus pathogenesis. Transl. Res. 157, 326–331. doi: 10.1016/j.trsl.2011.01.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shrivastav, M., and Niewold, T. B. (2013). Nucleic acid sensors and type I interferon production in systemic lupus erythematosus. Front. Immunol. 4:319. doi: 10.3389/fimmu.2013.00319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smyth, D. J., Cooper, J. D., Bailey, R., Field, S., Burren, O., Smink, L. J.,et al. (2006). A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 38, 617–619. doi: 10.1038/ng1800
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steimer, L., and Klostermeier, D. (2012). RNA helicases in infection and disease. RNA Biol. 9, 751–771. doi: 10.4161/rna.20090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strange, A., Capon, F., Spencer, C. C., Knight, J., Weale, M. E., Allen, M. H.,et al. (2010). A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 42, 985–990. doi: 10.1038/ng.694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Subramanian, S., Tus, K., Li, Q. Z., Wang, A., Tian, X. H., Zhou, J.,et al. (2006). A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. U.S.A. 103, 9970–9975. doi: 10.1073/pnas.0603912103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sutherland, A., Davies, J., Owen, C. J., Vaikkakara, S., Walker, C., Cheetham, T. D.,et al. (2007). Genomic polymorphism at the interferon-induced helicase (IFIH1) locus contributes to Graves’ disease susceptibility. J. Clin. Endocrinol. Metab. 92, 3338–3341. doi: 10.1210/jc.2007-0173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sweiss, N. J., Zhang, W., Franek, B. S., Kariuki, S. N., Moller, D. R., Patterson, K. C.,et al. (2011). Linkage of type I interferon activity and TNF-alpha levels in serum with sarcoidosis manifestations and ancestry. PLoS ONE 6:e29126. doi: 10.1371/journal.pone.0029126.t001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weckerle, C. E., Franek, B. S., Kelly, J. A., Kumabe, M., Mikolaitis, R. A., Green, S. L.,et al. (2011). Network analysis of associations between serum interferon-alpha activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. 63, 1044–1053. doi: 10.1002/art.30187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weckerle, C. E., and Niewold, T. B. (2011). The unexplained female predominance of systemic lupus erythematosus: clues from genetic and cytokine studies. Clin. Rev. Allergy. Immunol. 40, 42–49. doi: 10.1007/s12016-009-8192-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoneyama, M., and Fujita, T. (2008). Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity 29, 178–181. doi: 10.1016/j.immuni.2008.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: antiviral helicase, systemic lupus erythematosus, interferon
Citation: Oliveira L, Sinicato NA, Postal M, Appenzeller S and Niewold TB (2014) Dysregulation of antiviral helicase pathways in systemic lupus erythematosus. Front. Genet. 5:418. doi: 10.3389/fgene.2014.00418
Received: 08 September 2014; Accepted: 10 November 2014;
Published online: 25 November 2014.
Edited by:
Fumiaki Uchiumi, Tokyo University of Science, JapanReviewed by:
Miriam Wittmann, University of Leeds, UKAnca Macovei, International Center for Genetic Engineering and Biotechnology, India
Copyright © 2014 Oliveira, Sinicato, Postal, Appenzeller and Niewold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy B. Niewold, Mayo Clinic, Division of Rheumatology, Department of Immunology, 200 1st Street South West, Guggenheim Building 3-42, Rochester, MN 55905, USA e-mail: niewold.timothy@mayo.edu